 Qi Ke
Qi Ke- 1Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Inflammasome molecules make up a family of receptors that typically function to initiate a proinflammatory response upon infection by microbial pathogens. Dysregulation of inflammasome activity has been linked to unwanted chronic inflammation, which has also been implicated in certain autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, type 1 diabetes, systemic lupus erythematosus, and related animal models. Classical inflammasome activation-dependent events have intrinsic and extrinsic effects on both innate and adaptive immune effectors, as well as resident cells in the target tissue, which all can contribute to an autoimmune response. Recently, inflammasome molecules have also been found to regulate the differentiation and function of immune effector cells independent of classical inflammasome-activated inflammation. These alternative functions for inflammasome molecules shape the nature of the adaptive immune response, that in turn can either promote or suppress the progression of autoimmunity. In this review we will summarize the roles of inflammasome molecules in regulating self-tolerance and the development of autoimmunity.
Introduction
A functioning immune system is characterized by the capacity to distinguish between self-antigens versus microbial pathogens and foreign molecules. Several mechanisms are in place regulating both innate and adaptive immunity to establish persistent self-tolerance. These mechanisms maintain self-tolerance by limiting the activation and maturation of innate effectors such as monocytes, macrophages and dendritic cells (DC), while regulating self-specific T and B cells via intrinsic and extrinsic events. Immunoregulation is a dominant mechanism by which self-tolerance is established and maintained. Multiple subsets of self-specific T cells, including forkhead box P3 (FoxP3)-expressing regulatory CD4+ T cells (Foxp3+Treg), as well as regulatory B cells, mediate immunoregulation via 1) secretion of anti-inflammatory cytokines (e.g. TGF β1, IL-10) and modulatory factors, and 2) cognate interactions with T and B cells and/or DC and macrophages serving as antigen-presenting cells (APC) by engagement of various ligand-receptor molecules. Subsets of DC and macrophages also contribute to immunoregulation through secretion of cytokines and factors. Breakdown of self-tolerance leads to autoimmunity, typically characterized by chronic inflammation driven by autoreactive T and B cells, autoantibodies, and/or activated macrophages, DC and other innate effectors (1). Autoimmune diseases are characterized as: 1) organ-specific autoimmunity, such as multiple sclerosis (MS), rheumatoid arthritis (RA) and type 1 diabetes (T1D), or 2) systemic autoimmune diseases, such as systemic lupus erythematosus (SLE) (2). Events leading to the failure of self-tolerance are complex and influenced in a polygenic manner, while involving a host of ill-defined environmental factors including microbial infections, toxins, ultraviolet (UV) irradiation, diet, and dysbiosis of the gut microbiota.
The immune system has also evolved to detect and rapidly respond to invading pathogens via innate cell-driven events. This early inflammation leads to subsequent expansion and differentiation of effector T and B cells, typically resulting in clearance of the pathogen, and establishment of long-lasting immune protection. Recognition of an invading microbial pathogen is mediated by surface and cytoplasmic pattern recognition receptors (PRRs) which recognize: 1) conserved pathogen-associated molecular patterns (PAMPs), and 2) endogenous-derived danger-associated molecular patterns (DAMPs) induced by tissue damage and cellular activities mediated by microbial virulence factors (3).
Inflammasomes are oligomeric complexes that play an important role in initiating inflammation in response to PAMPs and DAMPs (4). Appropriately regulated activation of inflammasomes protects against microbial infection. However, aberrant inflammasome activity has been associated with severe inflammation-driven pathologies (5–7), as well as autoinflammatory and autoimmune diseases (8). Notably, inflammasome receptor molecules regulate the properties of different immune cell effectors as well as non-immune cell types that is independent of classical activation and inflammation-inducing events (9). This alternative function of inflammasome molecules has also been directly linked to autoimmunity and sterile inflammation. In this review, we will discuss how inflammasomes contribute to autoimmunity: 1) by inflammation driven by classical inflammasome activation, and 2) via alternative functions inflammasome molecules display.
Inflammasome-mediated inflammation- an overview
Inflammasome-driven inflammation in the context of innate immunity generally entails the production of proinflammatory cytokines such as IL-1β and IL-18, as well as induction of programmed cell death. The typical inflammasome complex consists of three components; namely 1) a sensor molecule such as a nucleotide oligomerization domain-like receptor (NLR), Absent in melanoma 2-like receptors (ALR) or pyrin, 2) the adaptor molecule apoptosis-associated speck-like protein (ASC) that contains a caspase activation and recruitment domain (CARD), and 3) pro-caspase-1 (Figure 1) (4). The assembled inflammasome provides a platform for cleavage of pro-caspase-1 (4). Once activated via an autolytic processing event, caspase-1 mediates maturation of pro-IL-1β and pro-IL-18 precursors, as well as initiating pyroptosis (4).
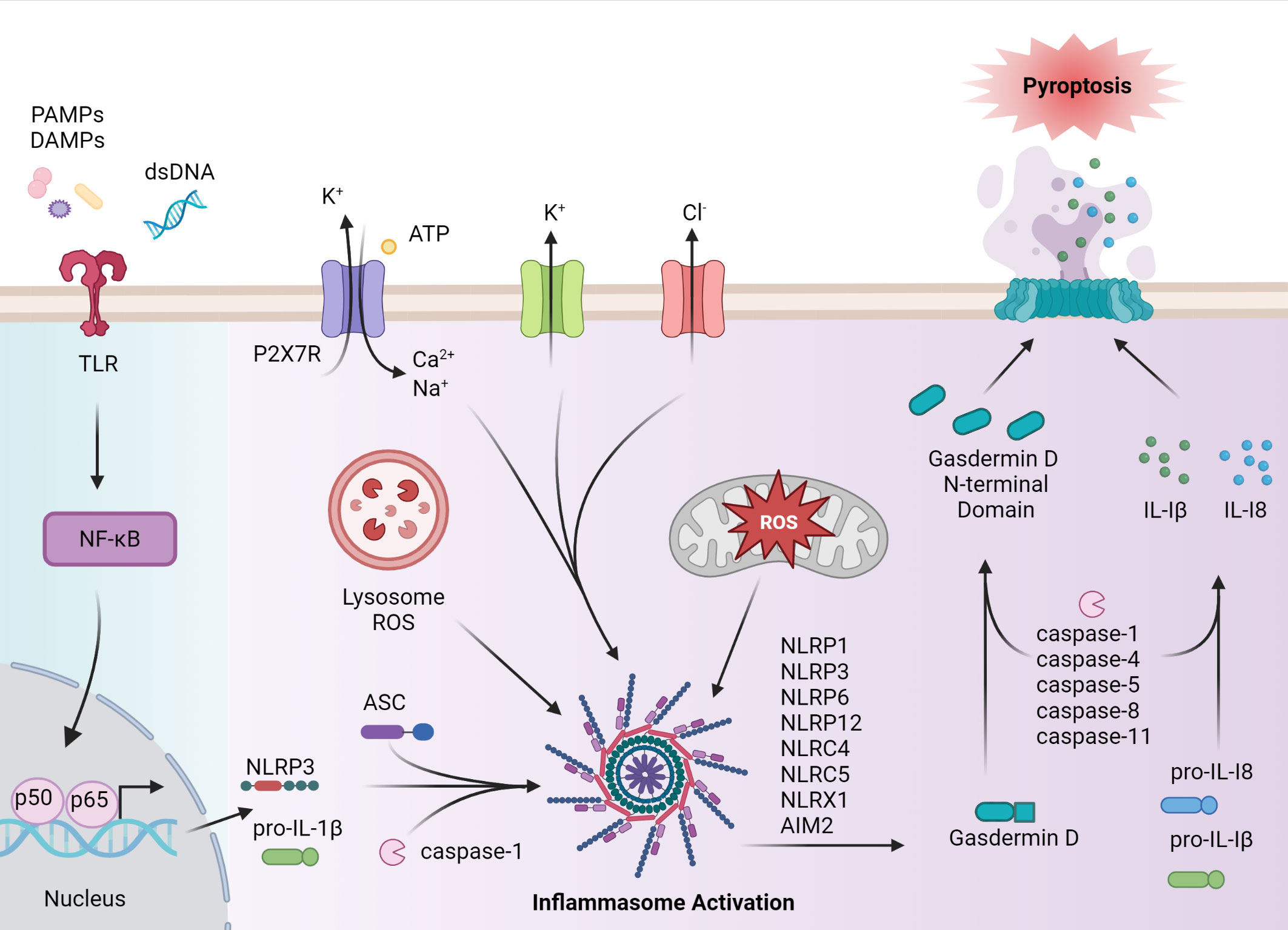
Figure 1 Inflammasome assembly and activation. Canonical activation of the inflammasome pathway begins with a primary signal, such as PAMPs, endogenous-derived DAMPs, or dsDNA, that are recognized by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs). PRR activation induces NF-κB and subsequent expression of NLRP, pro-IL-1β and pro-IL-18, and post-translational events. Formation of the inflammasome complex occurs when the sensor protein, such as NLRP3, binds to ASC, driving caspase activation and inflammasome assembly. Caspase enzymes cleave pro-IL-1β and pro-IL-18 as well as the C terminus from gasdermin D, allowing the gasdermin D N-terminal domain to form pores necessary for pyroptosis. IL-1β and IL-18, as well as cellular contents are released to establish a proinflammatory response. In autoimmune disease, inflammasome activation can occur via activation in a noncanonical matter including agonist-induced ion flux and lysosomal and mitochondrial reactive oxygen species (ROS). The figure was prepared using Biorender software licensed to the UNC Lineberger Comprehensive Cancer Center.
Pyroptosis, a lytic form of programmed cell death, is induced through caspase-1-mediated cleavage of gasdermin D (GSDMD), which removes the autoinhibitory C-terminus portion of the protein (10). Cleaved GSDMD also forms pores in the cell membrane, which facilitate the secretion of mature IL-1β and IL-18 (11). Cleavage of GSDMD and induction of pyroptosis is also achieved by a nonconical pathway in which murine caspase-11 or human caspase-4/5 are activated by cytosolic lipopolysaccharide (LPS), a gram-negative bacteria endotoxin (11, 12). In addition to pyroptosis, certain inflammasome molecules such as NLR family pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2), have been associated with PANoptosis-driven cell death in response to microbial infection and changes in cellular homeostasis (13). PANoptosis is regulated by the PANoptosome, which is a multimeric complex consisting in part of effector molecules involved in pyroptotic (caspase 1), apoptotic (caspase 8), and necroptotic (receptor-interacting protein kinase 1 (RIPK1), receptor-interacting protein kinase 3 (RIPK3)) cell death pathways (14). The composition of the PANoptosome varies with the nature of the stimulatory response, and complexes consisting of the ASC adaptor and NLRP3 or AIM2 sensor molecules have been identified (15).
Inflammasome activation is achieved in response to a broad range of stimuli derived from microbial infection, tissue damage, and/or dysregulation of metabolic events (Figure 1). The process of inflammasome activation typically entails two sets of signaling events that prime (signal 1), and activate (signal 2) the inflammasome (11). This multiple-step pathway ensures robust regulation of inflammasome activity. Signal one, induced by PRR (e.g. toll-like receptors (TLR)) primes inflammasome assembly via activation of NF-κB, upregulation of pro-IL-1β and pro-IL-18 expression, and induction of post-translational events that favor the formation of an inflammasome complex (11, 12). Signal two is specific for a given sensor molecule and induces inflammasome activation (12). Binding of an agonist to the leucine-rich repeat containing receptor (LRR) portion of the sensor protein leads to oligomerization via homotypic pyrin (PYD) interactions with the ASC adaptor molecule. ASC is important for linking the sensory protein with caspase-1 via CARD interactions (11, 12). Events driving caspase-1 activation, IL-1β and IL-18 maturation, and induction of pyroptosis and/or PANoptosis then follow (11, 12). To date, the role of inflammasomes in autoimmunity have largely focused on NLRP3 and AIM2, but other inflammasome molecules such as NLRP1, and NLR family CARD domain-containing protein 4 (NLRC4) have also been implicated in autoimmunity (16, 17). The respective inflammasomes are defined by the sensor protein.
NLRP3 has been the most extensively studied inflammasome, in general and in autoimmunity (18). NLRP3 agonists are structurally and chemically diverse: such agonists include 1) PAMPs expressed by bacteria, virus, and fungi, and 2) DAMPs including cholesterol, extracellular ATP, microbial pore-forming toxins, and particulate matter such as uric acid crystals (19). Consequently, it is believed that these agonists are indirectly sensed by NLRP3. Here, agonist-induced K+ and Cl- effluxes, Ca2+ fluxes, lysosomal damage, and mitochondrial damage and/or dysfunction coupled with the release of reactive oxygen species (ROS) are directly sensed by NLRP3 (20). For instance, noncanonical-induced activation of GSDMD results in K+ efflux, which activates NLRP3 and leads to caspase-1-mediated IL-1β and IL-18 production via the classical pathway (21–23). Gain of function variants in the NLRP3 gene resulting in aberrant NLRP3 inflammasome activation cause a family of diseases referred to cryopyrin-associated periodic syndromes (CAPS), which are marked by reoccurring systemic inflammation (20). NLRP3 activation has also been linked to diseases of the central nervous system (CNS) such as Alzheimer’s Disease (AD) (24, 25). In AD, the accumulation and subsequent uptake of amyloid-β by microglia residing in the brain results in lysosomal destabilization and NLRP3 activation (24). Production of IL-1β also has neurotoxic effects on microglia and astrocytes (25).
The process of NLRP1 activation is distinct from other inflammasomes (26). Here, motif-dependent ubiquitination followed by degradation of the N-terminal subunit by proteasome are required for activation of NLRP1 (27, 28). Various bacterial toxins and viral proteases have been reported to activate NLRP1 in mice and humans (29–33). However, since mice encode several NLRP1 orthologues with sequences that differ from the single human encoded NLRP1 gene, specific PAMPs and DAMPs triggering NLRP1 activation are variable and not fully defined among the species (34–37). The NLRC4 inflammasome is also distinct compared to other inflammasomes, in which the sensor protein functions as an agonist receptor. Instead, the NLRC4 protein associates with NLR family apoptosis inhibitory proteins (NAIPs) that act as cytosolic innate immune receptors, and which bind bacterial flagellin and type III secretion system components (T3SS) (38, 39). Gain-of-function variants in NLRC4 lead to periodic fever syndromes marked by increased systemic IL-18 (40).
AIM2 is responsive to cytosolic double-stranded DNA (dsDNA) from bacteria and DNA viruses. Notably, AIM2 binds both endogenous and microbe-derived dsDNA independent of nucleic acid sequence (41). Expression of AIM2 is upregulated by type I interferon (IFN), and the AIM2 inflammasome is key in host defense against bacterial and viral pathogens such as Francisella tularensis and Listeria monocytogenes, and vaccinia virus and cytomegalovirus, respectively (42). In addition, the AIM2 inflammasome promotes caspase-1-driven death of intestinal epithelial cells and hematopoietic bone marrow cells upon recognition of dsDNA breaks due to ionizing radiation or chemotherapeutic drugs (43).
The roles of IL-1β and IL-18 in inflammation
Inflammasome generated IL-1β and IL-18 enhances both innate and adaptive immunity against microbial pathogens. However, dysregulated production of these two cytokines by inflammasomes is also linked to chronic autoimmune diseases.
IL-1β is produced largely by monocytes, macrophages, and DC (44). Local release of IL-1β amplifies inflammation by inducing increased expression of 1) adhesion molecules and chemokines for recruitment of immune effectors, as well as 2) proinflammatory mediators such as cyclooxygenase type 2 (COX-2) and prostaglandin-E2 (PGE2) (44–46). IL-1β production can also lead to systemic inflammation via induction of the acute phase response, vasodilatation, angiogenesis, and leukocyte activation (44, 45).
T cell responses are also regulated both indirectly and directly by IL-1β. For instance, IL-1β enhances the stimulatory capacity of DC by driving maturation and upregulation of co-stimulatory molecules needed for efficient T cell activation and expansion (47). Increased IL-12 secretion by IL-1β stimulated DC favors differentiation of antigen-stimulated T cells towards a type 1 phenotype, marked by IFNγ production by CD4+ Th1 and CD8+ Tc1 cells (48). On the other hand, IL-1β has direct effects on CD4+ and CD8+ T cells, influencing expansion and subset differentiation depending on the extracellular milieu (49). In mice, IL-1β synergizes with IL-6, IL-21 and IL-23 to induce the differentiation of CD4+ T cells into IL-17-secreting Th17 cells (49). In humans, IL-1β has a more potent role in driving Th17 differentiation. Both Th1 and Th17 cells play key roles in several autoimmune diseases. Furthermore, IL-1β can suppress the function and/or reduce the stability of Foxp3+Treg (50, 51). Dysregulation of the Foxp3+Treg pool leading to skewed differentiation and pathogenic function of autoreactive effector T cells (Teff) is associated with a number of autoimmune diseases (52–56). CD8+ T cell expansion and differentiation are also regulated by IL-1β (57).
IL-1β has regulatory effects on the B cell compartment by enhancing B cell proliferation and antibody production (45). In addition, IL-1β increases proliferation and secretion of IL-4 and IL-21 by CD4+ T follicular helper cells (Tfh) (58). Tfh cells play a critical role in regulating antibody production by B cells and have also been implicated in the production of autoantibodies during autoimmunity (59).
IL-18 is expressed by a variety of cells such as Kupffer cells, macrophages, DC, and non-hematopoietic cells that include intestinal epithelial cells, keratinocytes and endothelial cells (60). Locally, IL-18 stimulates myeloid and endothelial cells to upregulate nitric oxide (NO) synthesis, and expression of cell adhesion molecules and chemokines to recruit and activate additional immune effectors at the site (60). In addition, IL-18 has potent regulatory effects on T cells and natural killer (NK) cells (60). IL-18 along with IL-12 drives the differentiation of Th1 cells and induces IFNγ production by CD8+ T cells and NK cells (60, 61). Furthermore, IL-18 stimulation upregulates 1) perforin- and Fas ligand (FasL)-dependent cytotoxicity in CD8+ T cells and NK cells, and 2) IL-17 secretion by γδ T cells (62). Not only is IL-18 linked to autoimmune diseases such as T1D and SLE, IL-18 has also been shown to play a key role in the maintenance of the intestinal epithelial barrier and regulation gut microbiota composition (63, 64). Dysbiosis of gut microbiota has been suggested as a risk factor for the development of autoimmunity (65, 66).
Classical inflammasome activation-dependent events in autoimmunity
In view of highly potent proinflammatory effects, it is not surprising that classical inflammasome activation is linked to a host of autoimmune diseases. Inflammasome activation is detected in innate and adaptive immune effectors thereby having indirect and direct effects that shape and maintain the proinflammatory response either locally and/or systemically in autoimmunity. In addition, inflammasome activation in non-immune cell types that makeup a given organ can initiate and/or exacerbate an autoimmune response. Finally, evidence indicates that inflammasome activation can have a protective role and contribute to maintenance of self-tolerance. In the following, we will describe the different roles classical inflammasome activation has in common tissue-specific and systemic autoimmune diseases (Table 1).
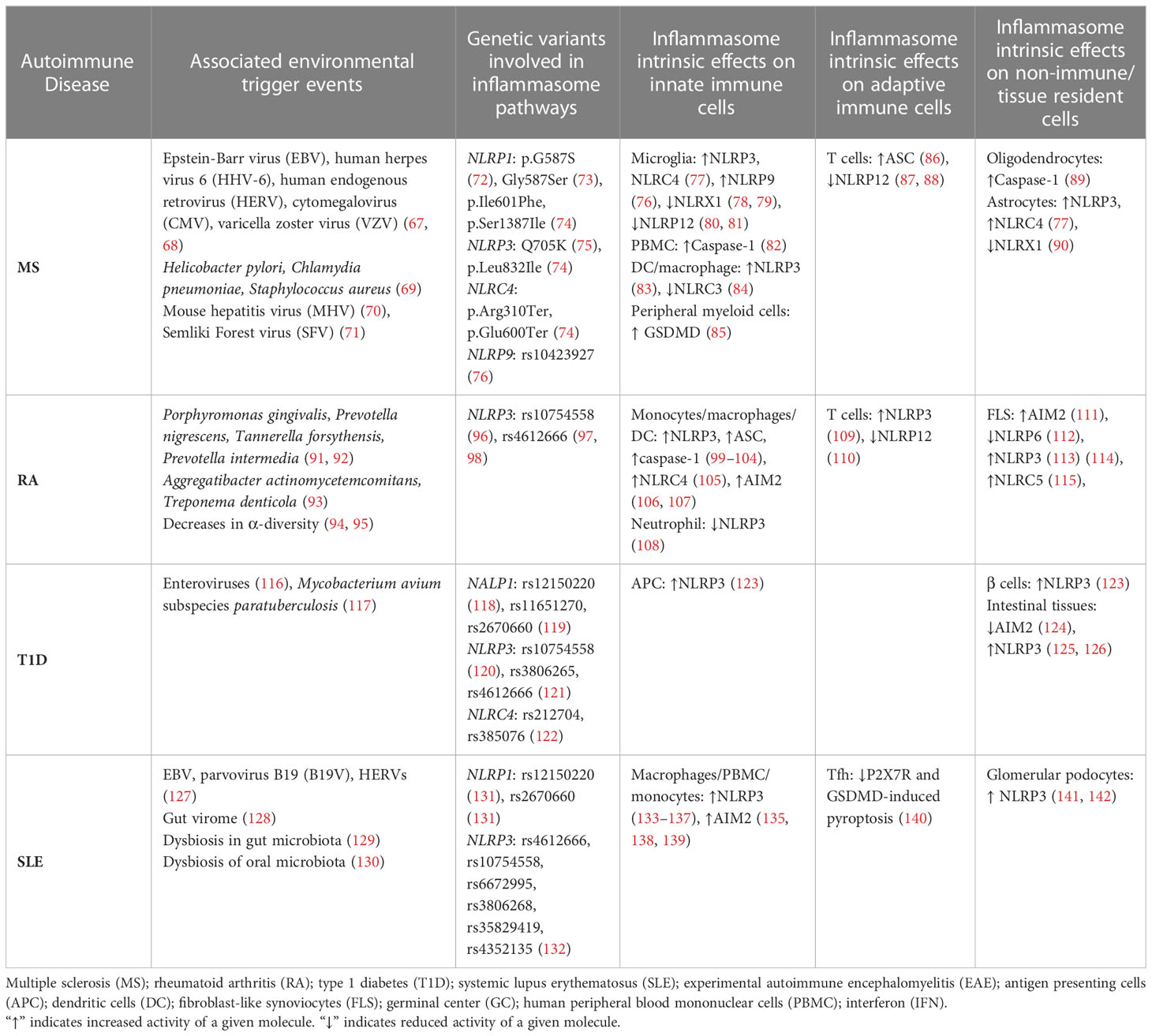
Table 1 Intrinsic-effects of classical inflammasome-mediated inflammation in autoimmunity.
Multiple sclerosis and inflammasome-mediated neuroinflammation
MS is a demyelinating autoimmune disease marked by chronic inflammation of the CNS, leading to variable neurological symptoms and heterogenous clinical outcomes (143, 144). MS susceptibility and disease progression are influenced by both genetic and environmental factors (145). Although ill-defined, the autoimmune response in MS is believed to be initiated in the periphery, involving stimulation of CD4+ and CD8+ T cells specific for myelin proteins (146, 147). Differentiation of the encephalitogenic CD4+ T cell pool is skewed towards Th1 and Th17 subsets. This pool coupled with CD8+ T cells and B cells migrate across the CNS microvascular endothelium and into the brain and spinal cord (148, 149). The CNS infiltrate includes peripheral monocytes/macrophages and DC that further amplify the autoimmune response. Upon activation, microglia, which are tissue-resident macrophages as well as resident astrocytes also contribute to inflammation (144, 150) by production of: 1) proinflammatory cytokines such as IL-1β, which has neurotoxic and immunomodulatory effects in the CNS, as well as 2) chemokines that promote recruitment of immune effector cells (151, 152).
Studies of MS patients and rodent experimental autoimmune encephalomyelitis (EAE), a model of MS, demonstrate that inflammasomes such as NLRP3, are associated with various aspects of the autoimmune process (153–155) (Figure 2). mRNA expression of NLRP3 and IL1B are detected in MS lesions as well as increased levels of IL-1β and IL-18 in blood and cerebrospinal fluid (CSF) (150, 156). Furthermore, the P2X7 purinergic receptor (P2X7R), a ligand-gated ion channel regulated by extracellular ATP that activates the NLRP3 inflammasome (157), is elevated in the spinal cord of MS patients. Indeed, increased extracellular levels of ATP and uric acid are found in the CSF and serum of MS patients (158, 159). ATP is normally abundant in the extracellular space of the CNS, where it functions as an excitatory neurotransmitter. Interestingly, various drugs used to clinically treat MS such as recombinant IFNβ, glatiramer acetate and natalizumab suppress NLRP3 mRNA expression, and decrease IL-1β in the blood and CSF of MS patients (160–162). In the brain lesions of MS patients, NLRP9 protein is also up-regulated in microglia but not astrocytes, suggesting a role for NLRP9 in modulating the encephalitogenic response (76).
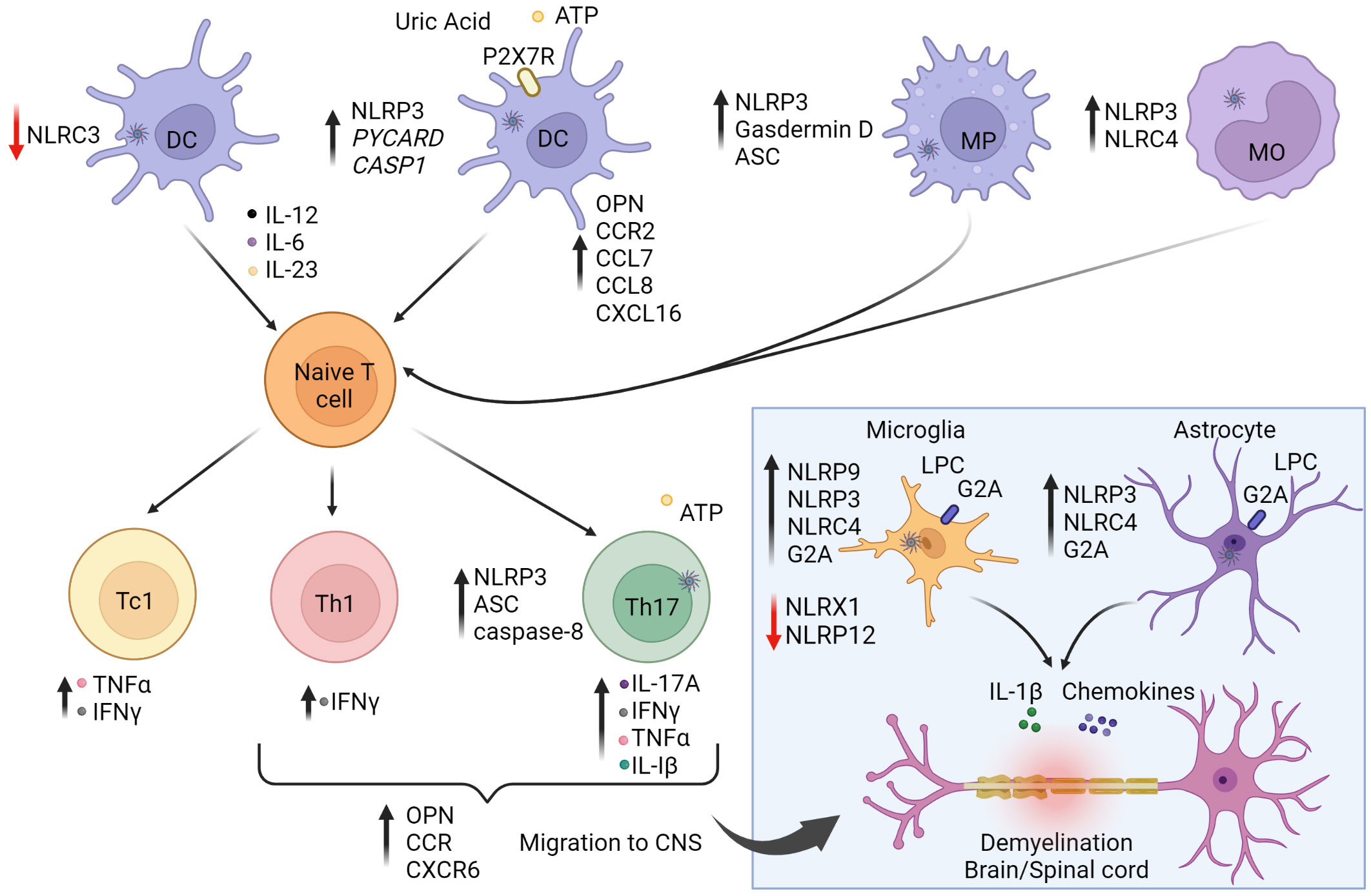
Figure 2 The role of inflammasomes in multiple sclerosis (MS) and experimental autoimmune encephalitis (EAE). The autoimmune response for MS is believed to begin in the periphery. Activation of NLRP3 and NLRC4 inflammasome pathways in antigen-presenting cells (APC) enhance stimulation and differentiation of pathogenic CD4+ Th1/Th17 and CD8+ Tc1 subsets. On the other hand, NLRC3 activation in dendritic cells (DC) is protective against disease by inhibiting DC maturation. Secretion of IL-1β and IL-18 increase T cell expression of osteopontin (OPN), CCR2 (binding CCL7/8), and CXCR6 (binding CXCL16) to promote infiltration to the central nervous system (CNS). Upon activation and differentiation, CD4+ and CD8+ T cells, and B cells migrate to the CNS. In the CNS, peripheral DC, macrophages (MP), and monocytes (MO) further amplify inflammation. CNS resident cells such as microglia and astrocytes also promote inflammation. Lysophosphatidylcholine (LPC) activates NLRP3 and NLRC4, causing secretion of IL-1β and chemokines, leading to further inflammation and demyelination. NLRP9 expression is increased in microglia. NLRX1 and NLRP12 serve to down-regulate neuroinflammation and provide protection against disease as indicated by the red arrows. Reduction of NLRX1 and NLRP12 can lead to exacerbated disease states. Purinergic receptor (P2X7R). The figure was prepared using Biorender software licensed to the UNC Lineberger Comprehensive Cancer Center.
The functional role of inflammasomes and inflammasome-related molecules has been investigated using EAE and other demyelinating rodent models. Earlier studies have shown that the progression and severity of EAE are reduced in mice deficient in NLRP3 (NLRP3-/-), ASC (ASC-/-) and to a lesser extent caspase-1 (Caspase1-/-) (83, 163). Attenuated EAE in NLRP3-/- and ASC-/- mice coincides with decreased infiltrates of Th1 and Th17 cells, macrophages and DC in the brain and spinal cord (83). This reduction in CNS infiltration is attributed to decreased production of IL-1β and IL-18 by APC (83). The latter are needed to adequately activate and upregulate T cell expression of osteopontin (OPN), and chemokine receptors CCR2, and CXCR6 for efficient migration into the CNS (83). In addition, lack of NLRP3 and ASC expression also limits DC and macrophages to upregulate matching receptor/ligands for OPN (α4β1 integrin), CCR2 (CCL7/CCL8), and CXCR6 (CXCL16) (83), resulting in aberrant APC migration into the CNS. These findings support a role for APC-expressed NLRP3 in mediating chemotactic recruitment of immune effectors to the CNS.
Peripheral APC also regulate the progression of EAE via inflammasome-mediated pyroptosis. EAE is attenuated in mice lacking GSDMD expression by peripheral myeloid cells (85). On the other hand, selective deletion of GSDMD in microglia has no effect on EAE, indicating that pyroptosis of CNS-resident APC may have only a limited role. The T cell stimulatory capacity of GSDMD-/- APC is reduced, which is marked by diminished numbers and effector function of Th1 and Th17 cells in the CNS. Notably, selectively blocking GSDMD-mediated pyroptosis with the inhibitor disulfiram, also attenuates EAE, demonstrating a direct role for pyroptosis (85). It is believed that pyroptosis of APC heightens local inflammation to promote efficient T cell activation, and subset differentiation needed to generate a robust encephalitogenic T cell pool.
In addition to APC, inflammasome activity intrinsic to T cells impacts EAE progression (Figure 2). Selective ASC-deficiency in T cells attenuates EAE marked by reduced infiltration of CD4+ T cells, B cells, and neutrophils (86). ASC-/- T cells are readily activated and undergo normal in vitro and in vivo differentiation into Th1, Th2, Th17 and Foxp3+Treg subsets. However, ASC-deficiency affects the properties of Th17 but not Th1 cells. ASC-/- Th17 exhibit reduced survival and pathogenicity reflected by decreased secretion of IL-17A, IFNγ, TNFα, as well as IL-1β. Here, IL-1β plays a key role in an autocrine manner, by enhancing the survival and effector function of Th17 cells residing in the CNS. Interestingly, cleavage of pro-IL-1β in Th17 cells is mediated via a noncanonical pathway involving caspase 8 activation. In this scenario, increased extracellular ATP levels due to release by stressed and dying cells drives activation of the NLRP3-ASC-caspase-8 complex, establishing a feed-forward loop promoting Th17 cell-mediated pathogenicity.
In addition to NLRP3, the activity of other inflammasome molecules in non-immune CNS resident cell-types have been found to promote neuroinflammation. Both NLRP3 and NLRC4 regulate the activity of microglia and astrocytes in a cuprizone model of inflammation-induced demyelination (77). Both cell types are known mediators of neuroinflammation through secretion of proinflammatory cytokines and chemokines. Cuprizone-induced pathology is prevented in NLRP3- and NLRC4-deficient mice characterized by microglia and astrocytes lacking IL-1β production, and exhibiting reduced expression of G2A, the receptor for lysophosphatidylcholine (LPC) (Figure 2). LPC, known for proinflammatory properties, is rapidly metabolized under homeostasis but accumulates under pathological conditions in the CNS (77). Following cuprizone treatment, LPC levels are increased, and LPC functioning as a DAMP, activates NLRP3 and NLRC4 expressed by microglia and astrocytes (77). In MS patients, expression of G2A and NLRC4 are increased, suggesting a role in the MS autoimmune response (77).
Interestingly, inflammasomes have also been shown to play a protective role in EAE. For instance, deficiency of NLRC3 exacerbates EAE (84). Lack of NLRC3 results in DC producing increased proinflammatory cytokines such as IL-12, IL-6, and IL-23, that in turn enhance differentiation of encephalitogenic Th1 and Th17 cells (84). NLRC3 negatively regulates DC maturation by inhibiting activation of the p38 signaling pathway (84). The ligand(s) regulating NLRC3 activity in DC is currently undefined (84). Also serving a protective function is NLR family member X1 (NLRX1), a more recently characterized NLR that is ubiquitously expressed and located in the mitochondria (78, 90). NLRX1 inhibits proinflammatory pathways, including type I IFN and TLR-mediated NF-κB signaling events, and may play a role in regulating mitochondria oxidative damage (78). Mice deficient of NLRX1 have increased T cell infiltration of the CNS, and consequently develop more severe EAE (79). Microglia exhibit a hyperactivated phenotype characterized by elevated expression of MHC class II molecules and production of IL-6 and chemokines, which in turn aid T cell recruitment and expansion (79). Accordingly, NLRX1 function is predicted to attenuate the proinflammatory properties of microglia. On the other hand, NLRX1-deficiency has no intrinsic effect on the pool of encephalitogenic T cells (79). NLRX1 may also play a protective function in astrocytes; NLRX1-/- astrocytes release excess glutamate in a Ca2+ dependent manner and contain reduced ATP levels compared to wild-type astrocytes, suggesting that NLRX1 promotes mitochondria ATP production (90). Furthermore, ROS levels in NLRX1 deficient astrocytes are increased compared to wild-type astrocytes, which may explain the reduced glutamate uptake (90). Recent evidence suggests that NLRX1 inhibits microglial activation in the early stages of EAE, which prevents activation of neurotoxic astrocytes (78).
NLRP12 has also been shown to regulate the progression and nature of CNS inflammation in EAE (87, 88, 153). NLRP12 mediates classical inflammasome driven inflammation in innate effector cells to certain microbes (164, 165), but also serves as a negative regulator of the NF-κB signaling pathway (80, 87, 88, 166, 167). In mice deficient of NLRP12, a more rapid and severe EAE develops (81). This exacerbated disease is characterized by increased mRNA levels encoding IL-1β and other proinflammatory molecules in the CNS, as well as activated microglia producing heightened levels of inducible NO synthase (iNOS), NO, TNFα, and IL-6 (81). A second study reported that EAE induction in NLRP12-/- mice results in neuroinflammation that promotes ataxia and poor balance, rather than the ascending paralysis that normally develops in wild-type mice (87). Furthermore, NLRP12-deficiency has intrinsic effects on T cells. In the absence of NLRP12 expression, T cells exhibit increased proliferation, and secretion of IFNγ, IL-17 and IL-4, that is in part due to hyperactivation of NF-κB (87). Therefore, NLRP12 negatively regulates various aspects of innate cell activation, as well as CD4+ T cell expansion and effector function via blocking NF-κB signaling (88).
Rheumatoid arthritis and inflammasome-mediated joint inflammation
RA is a chronic autoimmune disease characterized by the inflammation of the joints, leading to synovial tissue proliferation, cartilage erosion and joint destruction (168–170). Pathology is in part driven by Th1 and Th17 CD4+ T cells and B cells, as well as innate effectors such as monocytes, DC and neutrophils that traffick into the synovium (171–173). Joint-resident cells such fibroblast-like synoviocytes (FLS) also promote local inflammation (174). Normally, FLS play a key role in maintaining joint homeostasis via production of the extracellular matrix and matrix metalloproteinases (MMPs) (175).
The autoimmune response of RA also involves high levels of serum complement and the production of autoantibodies that target the Fc region of IgG (i.e. rheumatoid factor), cartilage components, nuclear proteins and proteins post-translationally modified by citrullination (176, 177). Key proinflammatory cytokines driving RA include IL-1β and IL-18, as well as IL-6 and TNFα (178). In addition to having immunomodulatory effects, IL-1β mediates cartilage erosion and prevents chondrocyte matrix formation (179). Furthermore, the severity of RA correlates with elevated serum IL-18 (180, 181). Moreover, during the early-stages of RA, FLS proliferate and differentiate into distinct subsets of activated synovial fibroblasts that produce inflammatory cytokines, matrix-degrading enzymes and proangiogenic factors which lead to the release of inflammatory mediators, bone destruction and angiogenesis (182–184). FLS also promote T cell survival, Tfh and Th17 cell differentiation, and can function as antigen presenters to autoreactive T cells (185–193). The etiology of RA is ill-defined but genetic and a host of environmental factors are known to influence disease susceptibility and progression. Evidence also suggests that inflammasomes likely have an important role in RA pathogenesis (Figure 3).
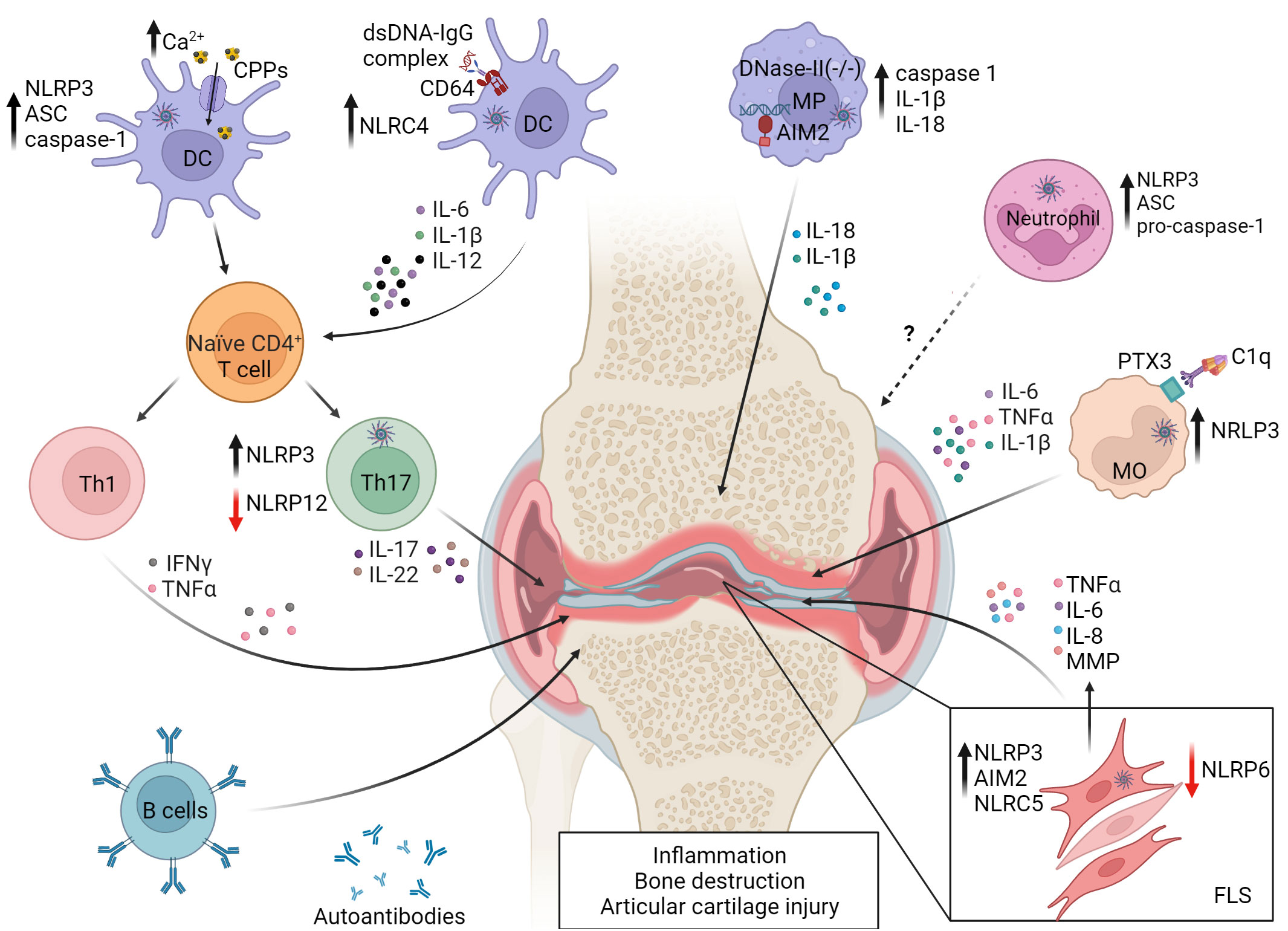
Figure 3 Events in dysregulated inflammasome activation in rheumatoid arthritis (RA). NLRP3 and NLRC4 activity are increased in monocytes (MO) and DC by Fc-receptor (FcR) binding of DNA-IgG immune complexes and complement component 1q (C1q) binding to pentraxin 3 (PTX3). Uptake of elevated levels of calciprotein particles (CPPs) in the joint by resident DC also leads to NLRP3 activation. Resulting pyroptosis and secretion of proinflammatory cytokines promote RA progression by favoring Th1 and Th17 differentiation and development of autoantibodies and RA factor producing plasma cells. NLRP3 activation is also increased in Th17 cells. Aberrant lysosomal processing of endocytosed dsDNA can lead to AIM2 activation in joint resident macrophages (MP). Neutrophils exhibit reduced expression of inflammasome molecules, which correlates with decreased disease severity. NLRP3, NLRC5 and AIM2 are associated with proinflammatory properties of fibroblast-like synoviocytes (FLS), while NLRP6 and NLRP12 serve protective roles, indicated by the red arrows. NLRP6 limits FLS cytokine production, and NLRP12 negatively regulates Th17 subset differentiation. Reduced expression of NLRP6 and NLRP12 leads to pathology. Matrix metalloproteinases (MMP). The figure was prepared using Biorender software licensed to the UNC Lineberger Comprehensive Cancer Center.
In RA patients, NLRP3 and NLRP3-inflammasome-related proteins are upregulated in a cell-specific manner among innate effectors. For instance, expression of NLRP3, ASC, and caspase-1 as well as IL-1β secretion is generally increased in monocytes, macrophages, and DC from RA patients (99–102) (Figure 3). CD4+ T cells from RA patients also exhibit increased NLRP3 expression, which correlates with elevated serum IL-17A concentrations and disease activity (109) (Figure 3). Notably, differentiation of Th17 cells is inhibited by NLRP3 knockdown (109), suggesting that NLRP3 regulates the proinflammatory activity of both innate and adaptive effectors in RA. Interestingly, NLRP3 activation in monocytes is mediated via multiple mechanisms in RA patients. C1q binding to pentraxin 3, a key regulator of complement activity and which is increased on the surface of RA CD14+ monocytes, leads to NLRP3 activation, enhanced IL-1β and IL-6 secretion, and GSDMD-induced pyroptosis (178). In addition, due to elevated extracellular Ca2+ in the joint and concomitant heightened activity of calcium-sensitive receptors, macropinocytosis of calciprotein particles (CPPs) is elevated by local monocytes (194). After uptake, CPPs disrupt lysosome integrity resulting in enhanced NLRP3 activation and IL-1β secretion (194).
Whereas NLRP3 and related inflammasome proteins are typically elevated in various innate and adaptive immune effectors, neutrophils from RA patients exhibit reduced NLRP3, ASC and pro-caspase-1 expression (108). Here, NLRP3 mRNA levels in neutrophils negatively correlate with disease severity (108). This suggests that NLRP3 may serve a protective role in the context of neutrophil function via an ill-defined mechanism (108).
Various inflammasome molecules, in addition to NLRP3, have been found to be involved with RA (Figure 3). NLRC4 activity is increased in DC residing in the synovial membrane of RA patients (105). These DC secrete elevated IL-1β, have increased expression of CD64, an IgG Fc receptor, and display an enhanced capacity to stimulate Th1 and Th17 subset differentiation (105). This capacity is due to a novel mechanism of upregulation of NLRC4 expression and activity. Here, dsDNA-IgG complexes bind to CD64, are internalized, and the combination of CD64 signaling and intracellular sensing of the dsDNA increases NLRC4 activity (105). AIM2 expression is increased in synovial tissue of RA patients, and knockdown of AIM2 mRNA inhibits in vitro proliferation of FLS derived from RA patients (111). On the other hand, NLRP6 levels are reduced in FLS from patients with RA versus osteoarthritis (112). Furthermore, increased ectopic expression of NLRP6 in RA patient-derived FLS blocks the production of inflammatory cytokines such as IL-1β, IL-6, and TNFα, as well as MMP via inhibition of the NF-κB pathway. The latter indicates that NLRP6 serves a protective role in RA (112), and is consistent with NLRP6 having a negative regulatory function in colitis (195).
Animal studies further support the notion that the role for inflammasomes in RA is complex, and that cell type-dependent, inflammasome molecules can have distinct effects on immune cells and effector molecules depending on the RA model (103, 196) (Figure 3). Mice deficient of ASC are resistant to collagen induced arthritis (CIA), in part due to a reduced T cell stimulatory capacity of ASC-/- DC (103). However, CIA develops in both NLRP3–/– and Caspase1–/– mice suggesting that ASC has caspase 1-independent effects in DC (103). On the other hand, NLRP3 and caspase-1 play a key role in the spontaneous polyarthritis that develops in mice in which the RA susceptibility gene A20/Tnfaip3 is selectively ablated in myeloid cells (A20myel-KO mice) (104). Here, macrophages lacking A20 have increased constitutive and LPS-induced expression of NLRP3 and pro-IL-1β. The latter is indicative of the established role A20 has as an inhibitor of NF-κB activation (197), which is needed for NLRP3 and pro-IL-1β transcription following inflammasome priming. Furthermore, activation of NLRP3 in A20-deficient macrophages results in enhanced caspase-1 activation, IL-1β secretion, and pyroptosis. Notably, pathology in A20myel-KO mice is blocked by ablation of NLRP3, caspase-1 and the IL-1 receptor (IL-1R), demonstrating a direct role for classical NLRP3 inflammasome activation in this spontaneous autoimmune model of cartilage destruction (104). NLRP3 is also associated with the proinflammatory properties of FLS. NLRP3 expression is increased in FLS isolated from mice with adjuvant-induced arthritis (AA) (113), and knockdown of Nlrp3 mRNA expression in FLS reduces disease severity in a monosodium urate-induced model of gout arthritis in rats (114).
AIM2 has also been shown to have a key role in joint inflammation. Mice deficient in expression of lysosomal endonuclease DNase II and type I IFN receptor (IFNαR) develop polyarthritis marked by production of autoantibodies, and macrophage secreted proinflammatory cytokines such as IL-1β, IL-6 and TNFα (106). Lack of lysosomal endonuclease DNase II results in aberrant processing of dsDNA in lysosomal compartments, and translocation of undigested DNA into the cytoplasm of macrophages (106, 107). AIM2-deficiency limits joint inflammation marked by reduced caspase-1 activity, IL-1β and IL-18 expression, and macrophage infiltration (106, 107). Notably, however, autoantibody production is unaffected by AIM2-ablation indicating a tissue-specific role for AIM2. Furthermore, AIM2-ablation has no effect on the transfer of arthritogenic serum from K/BxN mice (107). In this passive model, arthritis is induced by the deposition of immune complexes within the joint, leading to complement fixation and ensuing pathology (106, 107). Therefore, AIM2 regulates inflammation when cytosolic DNA is the key driving event. A contribution for NLRC5 in joint inflammation has been reported (115). NLRC5 expression is elevated in the synovium and FLS in rat AA (115), and knockdown of Nlrc5 mRNA blocks FLS proliferation and production of TNFα and IL-6, due to suppressed NF-κB activation (115).
Similar to NLRP6, NLRP12 has been shown to negatively regulate joint inflammation (110). The severity of antigen-induced arthritis in NLRP12-/- mice is increased, marked by elevated levels of joint infiltrating Th17 cells (110). Notably, in vitro Th17 cell differentiation is enhanced in NLRP12-/- CD4+ T cells marked by elevated IL-6-induced activation of signal transducer and activator of transcription (STAT) 3 (110).
Type 1 diabetes and inflammasome-mediated pancreatic islet inflammation
T1D is characterized by chronic inflammation of the pancreatic islets (insulitis) that results in the dysfunction and/or destruction of the insulin producing β cells (198–200). Despite life-long insulin therapy, T1D patients typically develop a variety of complications including retinopathy, neuropathy, and nephropathy related to hyperglycemia and inflammation. The autoimmune response involves islet infiltration of CD4+ and CD8+ T cells, B cells, macrophages, and DC. β cell-specific CD4+ and CD8+ T cells are generally believed to be the key drivers of pathology (198–200). Diabetogenic CD4+ and CD8+ T cells typically exhibit a type 1 effector phenotype, although Th17 cells are also implicated in the disease process (199). In addition to serving as APC, islet-infiltrating macrophages and DC, mediate β cell destruction through secretion of proinflammatory mediators and cytokines such as IL-1β, IFNγ and TNFα that have direct β cell-cytotoxic effects (199). The initiation and progression of T1D are influenced by genetic and poorly defined environmental factors (201–204). The latter include viral infections, and dysbiosis of gut microbiota, which are events that can be impacted by inflammasome activity (16, 201, 205).
Studies using murine models of T1D show that NLRP3 regulates the diabetogenic response (Figure 4). In non-obese diabetic (NOD) mice, which spontaneously develop β cell autoimmunity and overt diabetes, NLRP3 deficiency results in a reduced incidence of diabetes (123). This attenuated diabetes is due in part to NLRP3-/- APC having a decreased capacity to promote Th1 cell differentiation; Th17 cell differentiation, however, is unaffected. Importantly, NLRP3-/- β cells exhibit decreased production of IL-1β and chemokines such as CCL5, and CXCL10 (123). The latter limits migration into the islets by immune effectors including diabetogenic T cells (123) (Figure 4). Interestingly, limited IL-1β production leads to reduced activation of interferon regulatory factor 1 (IRF1) that is needed for β cell expression of CCL5 and CXCL10. Diminished IL-1β secretion by β cells is also expected to aid β cell viability and function, as well as enhance the maintenance and function of protective Foxp3+Treg in the islets. Notably, upregulation of NLRP3 and IL-1β is also detected in human β cells upon LPS and ATP stimulation in vitro (206). A regulatory function for NLRP3 in the disease process is also seen in a multiple low dose streptozotocin (MLD-STZ)-induced model of T1D. Here, progression of β cell autoimmunity is reduced in MLD-STZ treated C57BL/6 mice lacking NLRP3 expression (207). In this model NLRP3 is activated in macrophages residing in the draining pancreatic lymph nodes (PLN) by mitochondrial DNA (mtDNA) that is released following STZ treatment. NLRP3 activation results in increased caspase-1 activity, and IL-1β production, which drives expansion of pathogenic Th1 and Th17 cells and the induction of diabetes. The PLN are a key site for priming of diabetogenic CD4+ and CD8+ T cells. Interestingly, plasma levels of mtDNA are increased in T1D versus healthy subjects, which is expected to contribute to systemic inflammasome activation (208). Indeed, circulatory mtDNA induced by MLD-STZ in mice activates NLRP3 in endothelial cells via Ca2+ influx and mitochondrial ROS generation, which leads to endothelial dysfunction and vascular inflammation (208). Vascular inflammation is a key driver of complications that develop in T1D. Together these studies indicate that NLRP3 promotes pathological events driving β cell autoimmunity. Nevertheless, mechanisms by which NLRP3 mediate effects are likely to be complex and cell dependent. For instance, disease progression in NOD mice is unaffected by caspase-1 deficiency (209, 210), and only minimally affected by IL-1R ablation (211).
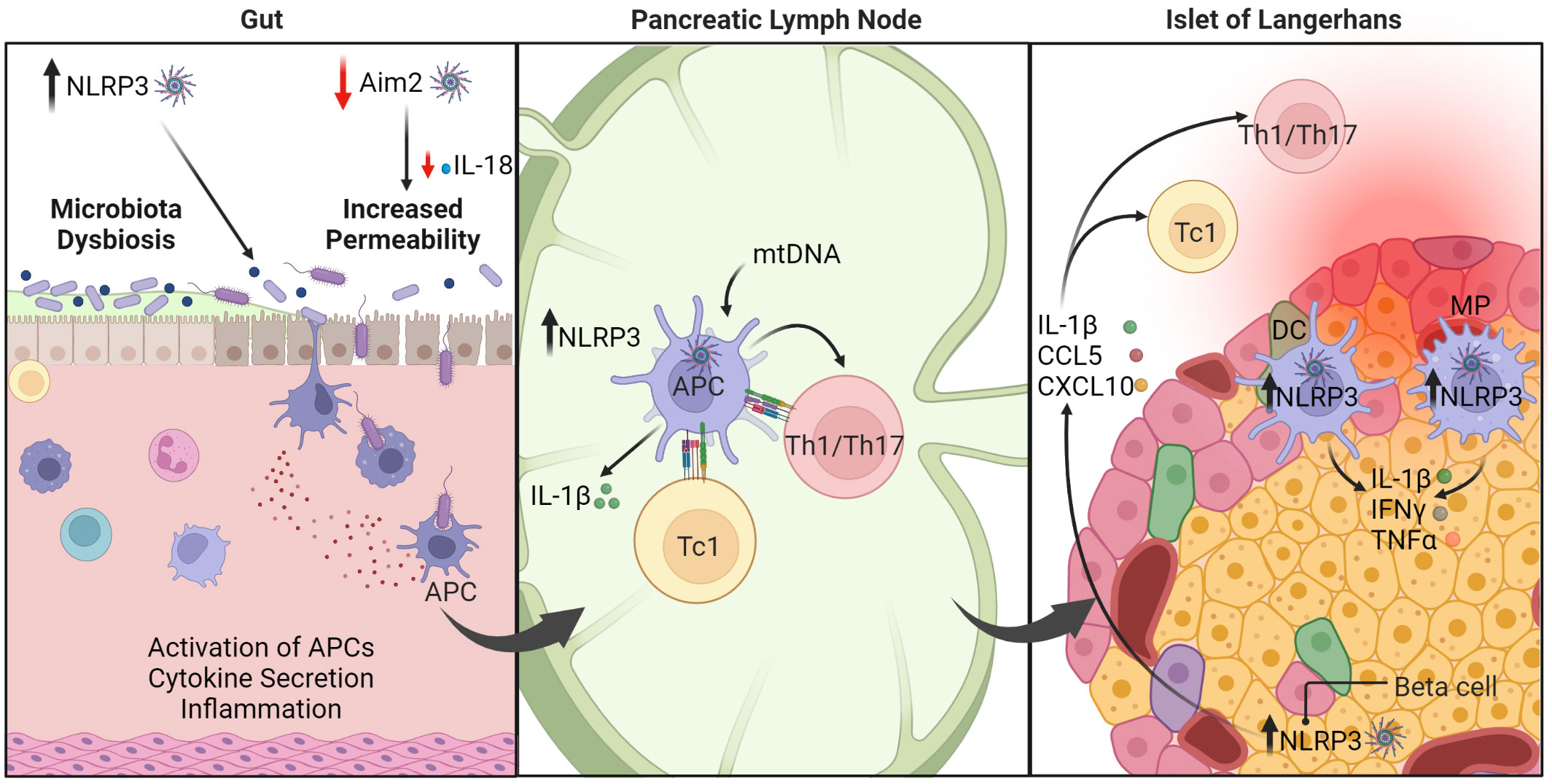
Figure 4 The roles of inflammasomes in type 1 diabetes (T1D). Under homeostasis, healthy intestinal epithelial cells maintain intestinal barrier function and regulate permeability to prevent passage of harmful elements such as microorganisms and toxins. AIM2 serves a protective function (indicated by the red arrow). Dysregulation of inflammasome function, such as AIM2 deficiency, leads to reduced production of IL-18, which is necessary for maintaining intestinal barrier function. Consequently, inflammasome dysregulation enhances intestinal permeability and triggers inflammation. On the other hand, NLRP3 is linked to dysbiosis within the gut microbiota, which can exacerbate T1D progression. In the pancreatic lymph node (PLN), upregulation of NLRP3 in APC promotes IL-1β production that ultimately drives differentiation of diabetogenic CD8+ Tc1, CD4+ Th1, and Th17 cells. In the pancreatic islets, NLRP3 hyperactivity in β cells induces release of cytokines and chemokines. These conditions combined with other immunomodulatory factors establish a positive feedback loop to further perpetuate pancreatic inflammation. Macrophage (MP), dendritic cell (DC), antigen-presenting cell (APC). The figure was prepared using Biorender software licensed to the UNC Lineberger Comprehensive Cancer Center.
In contrast to NLRP3-deficient C57BL/6 (207), MLD-STZ enhances diabetes development in AIM2-deficient C57BL/6 mice (124). Interestingly, disease exacerbation in the AIM2-/- mice is mediated by enhanced intestinal permeability, alterations in the gut microbiota, and increased bacterial translocation to the PLN where CD4+ Th1 and CD8+ Tc1 are readily expanded (Figure 4). Importantly, AIM2 deficiency results in decreased maturation of IL-18 which is needed to maintain intestinal barrier function (124). On the other hand, reduced NLRP3 expression in colonic NOD mouse tissue is associated with decreased microbiota dysbiosis, enhanced intestinal barrier function and diabetes prevention (125, 126). It is well established that dysbiosis within the gut microbiota significantly affects disease progression in NOD mice, and clinical findings suggest similar effects may also occur in T1D subjects (16, 205, 212–215). These studies provide evidence that inflammasomes may play a key role in regulating T1D progression in part via effects on gut microbiota and intestinal barrier function (16). Studies have reported that gut microbiota composition and/or intestinal barrier permeability are also influenced by other inflammasome molecules such as NLRP6 (216), NLRC4 (217), NLRX1 (218, 219), and NLRP12 (220, 221). Further investigation is necessary to elucidate the connection between inflammasomes, gut microbiota homeostasis, and autoimmunity.
Systemic lupus erythematosus and the role of inflammasome activity in widespread inflammation
SLE is a chronic autoimmune disease with diverse clinical manifestations. Development of SLE is influenced by genetic, hormonal, and environmental factors that lead to dysregulation of mechanisms of innate and adaptive-mediated self-tolerance. The autoimmune response is characterized by the generation of anti-nuclear autoantibodies, tissue deposition of immune complexes, increased type I IFN production, and inflammation in multiple organs with the kidneys being the most commonly affected (222). CD4+ T cells such as Tfh cells are key drivers of the autoantibody response, and Th17 cells, found infiltrating the kidneys and skin contribute to tissue damage (223). Innate effectors such as monocytes, macrophages, DC and neutrophils also play roles in mediating the systemic inflammation and tissue damage in SLE (223).
The etiology of SLE is not fully understood but evidence from humans and animal models indicate that inflammasomes contribute to disease progression (Figure 5). Inflammasome components are typically upregulated in kidney biopsies from SLE patients, and NLRP3, IL-1β and IL-18 are increased in SLE patient macrophages, peripheral blood mononuclear cells (PBMC), and serum (133, 134). A critical meditator of pathology in SLE are anti-nuclear autoantibodies (ANA) that target endogenous dsDNA and ribonucleoproteins (RNP) (224). Immune complexes (IC) of dsDNA upregulate NLRP3 and caspase-1 activity leading to increased IL-1β production by monocytes and macrophages of SLE patients (225). Here, the IC activates TLR9, a DNA sensor, which subsequently upregulates NF-κB and primes inflammasome assembly via increasing NLRP3 and pro-IL-1β (225). Upon IC binding, TLR9 also promotes mitochondrial ROS production and K+ efflux and subsequent NLRP3 activation. Notably, SLE monocytes stimulated with dsDNA-antibody complexes readily promote differentiation of Th17 cells, which is also seen in vivo in lupus-prone NZBW/F1 mice injected with anti-dsDNA autoantibodies from SLE patients (224). Similarly, autoantibody complexes of U1-small nuclear RNP (U1-snRNP) activate the NLRP3 inflammasome involving cytoplasmic RNA sensors TLR7 and TLR8 signaling in human monocytes (226). Antibody complexes of endogenous snRNP also induce production of macrophage migration inhibitory factor (MIF) in human monocytes, which enhances NLRP3 activation and IL-1β production (227). Interestingly, the context of nucleic acid uptake appears to determine the identity of the inflammasome molecule being engaged. For instance, unbound dsDNA, normally found at high levels in SLE patient serum, is taken up by monocytes via macropinocytosis, which activates AIM2 as well as NLRP3 (135). Uptake of free nucleic acid, however, requires antibody to be internalized by macropinocytosis but not Fc receptor (FcR) (135). On the other hand, internalization of dsDNA/snRNP autoantibody complexes via FcR may favor activation of NLRP3, and possibly NLRC4 as seen in RA (105). In each of the aforementioned scenarios, IL-1β and IL-18 are secreted to maintain/amplify inflammation. Furthermore, induced pyroptotic death and release of cellular and nuclear contents lead to the production of ANA to further fuel the autoimmune response (228, 229).
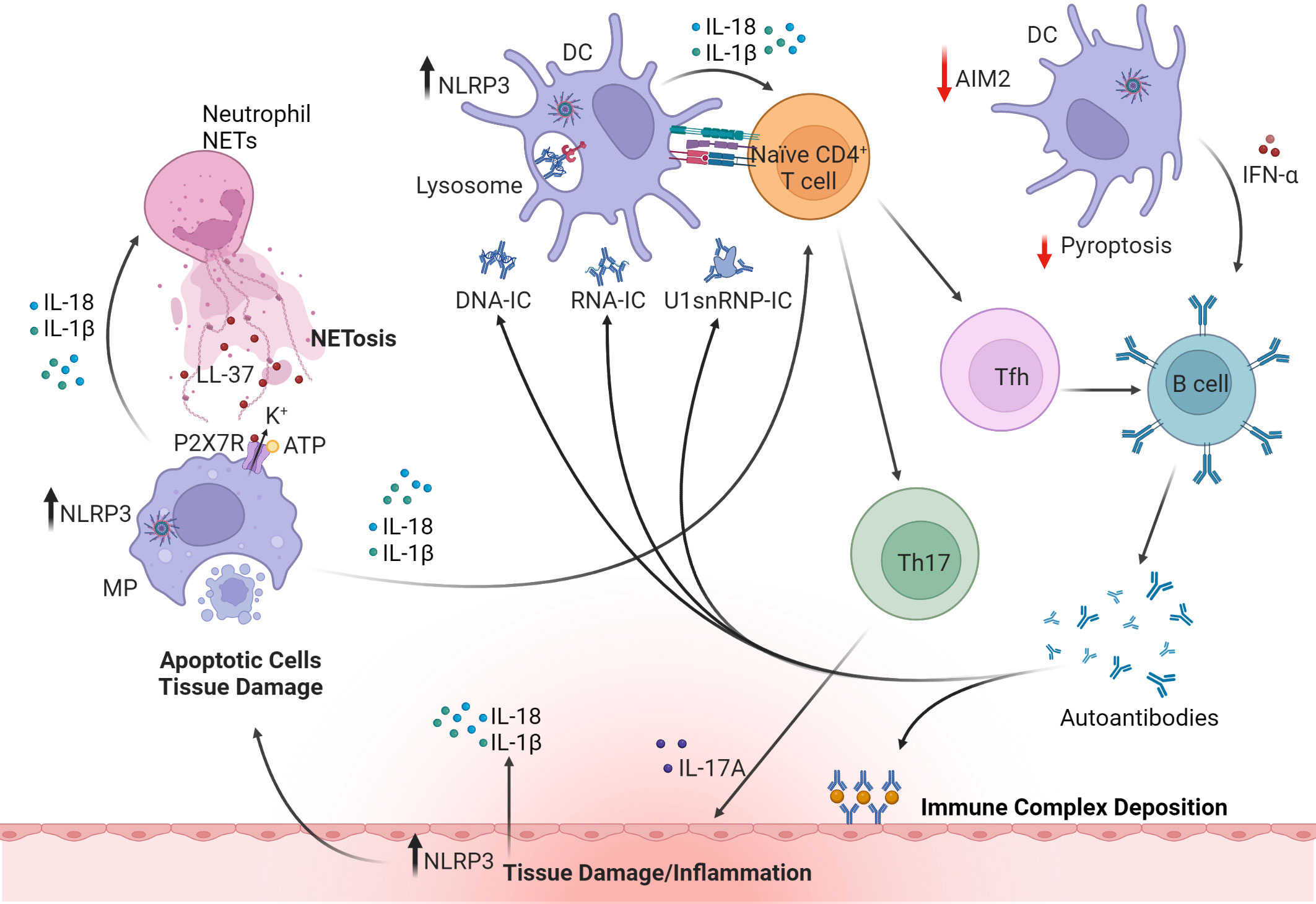
Figure 5 The roles of inflammasomes in systemic lupus erythematosus (SLE). Upregulation of NLRP3 inflammasome in macrophages (MP) and DC by DNA or RNA immune complexes (IC) or small nuclear ribonucleoprotein (snRNP) leads to release of proinflammatory cytokines such as IL-1β, IL-18 and IFNα. Dysregulation of inflammasomes in APC also promotes Th17 and Tfh cell differentiation. Tfh cells and IFNα facilitate B cell maturation and autoantibody production. However, production of IFNα is regulated by AIM2-mediated pyroptosis (indicated by red arrows). Deposition of IC, infiltrating Th17 cells, and production of autoantibodies and cytokines all contribute to tissue damage. IL-18 activates NETosis in neutrophils and in turn upregulates NLRP3 and IL-1β and IL-18 secretion in macrophages via cathelicidin antimicrobial peptide (LL37)-driven K+ efflux mediated by the P2X7 receptor (P2X7R). These cytokines further induce pyroptosis and release of cellular and nuclear contents, leading to the production of anti-nuclear autoantibodies and further amplifying systemic inflammation. Inflammasome activation in cells of target tissues, such as kidney resident podocytes also contributes to disease pathology by producing IL-1β. The figure was prepared using Biorender software licensed to the UNC Lineberger Comprehensive Cancer Center.
Aberrant clearance of neutrophil extracellular traps (NETs) is also linked with the pathogenesis of SLE and inflammasome activation (Figure 5). NETs are a network of chromatin fibers containing anti-microbial peptides such as LL37 and enzymes that participate in host defense (230). NETs are primarily released by activated neutrophils that undergo NETosis, a programmed cell-death mechanism (231). Notably, NETs activate NLRP3 inflammasome and IL-1β and IL-18 secretion in macrophages from SLE patients via LL37-driven K+ efflux mediated by the P2X7R (136). Furthermore, IL-18 activates NETs and promotes NETosis suggesting that a feed-forward loop exists that helps to maintain inflammation (136).
Monocytes from SLE patients versus healthy controls exhibit enhanced NLRP3 activation and IL-1β secretion (138, 139). This hyperactivity is attributed to chronic IFNα stimulation of monocytes. Elevated type I IFN-induced gene expression “signatures” correlate with the presence of autoantibodies, nephritis, and disease activity (232). Prolonged IFNα exposure in vivo induces NLRP3 hyperactivity by an IRF1 signaling pathway (138). However, consistent with other studies (233), short-term IFNα exposure of monocytes blocks NLRP3 activation (138). The latter, importantly, indicates that chronic type I IFN stimulation can have distinct effects on inflammasome activation.
The study of different murine lupus models provides further evidence that inflammasomes regulate SLE pathogenesis. Mice deficient in caspase-1 expression versus wild-type mice exhibit reduced autoantibody production, a limited IFN signature, as well as diminished NETosis and kidney pathology induced by pristane administration (136). In addition, blocking the P2X7R significantly impacts the development of spontaneous lupus in MRL/lpr mice. Here, limiting NLRP3 activation reduces the production of anti-dsDNA autoantibodies and IL-1β, and decreases Th17 cell expansion and the severity of nephritis (234). Furthermore, various drugs that inhibit NLRP3 inflammasome activation attenuate disease severity in different lupus mouse models (137, 235–237). On the other hand, nephritis induced by pristane treatment is exacerbated in mice in which myeloid cells selectively express a transgene encoding a hyperactive Nlrp3R258W mutant protein (238).
In addition to immune effector cell types, inflammasome activation in target tissues also contributes to disease pathology (Figure 5). Endothelial cells, basement membrane, and podocytes form a glomerular filtration barrier, which is essential for maintaining kidney function (239). In NZM2328 mice, which spontaneously develop lupus nephritis, severe proteinuria correlates with increased activation of NLRP3 and caspase-1 as well as IL-1β secretion by glomerular podocytes (141, 142). NZM2328 mice treated with MCC950, an NLRP3 inhibitor, exhibit reduced NLRP3 activation by podocytes, and attenuated renal tissue damage and proteinuria (141, 142).
Depending on the lupus model, inflammasome molecules have also been shown to play a protective role. In C57BL/6lpr/lpr mice, which develop mild lupus, deficiency of NLRP3 or ASC exacerbates pathology marked by an increase in activated macrophages and DC and production of proinflammatory cytokines, and T and B cell proliferation but no effect is seen on autoantibody production (240). This enhanced pathology is marked by reduced SMAD2/3 phosphorylation during TGF-β receptor signaling, and consistent with the role of TGF-β1 as a key regulator of immune homeostasis (240). In this scenario, it is likely that NLRP3 or ASC serve functions independent of classical inflammasome activation (see below), consistent with the observation that IL-1R- or IL-18-deficiency in C57BL/6lpr/lpr mice does not exacerbate pathology.
Studies have indicated that AIM2 may also serve a protective role in lupus by negatively regulating type I IFN production. In B6.Nba2 mice, which spontaneously develop lupus nephritis, p202, another IFN-inducible p200 family member is up-regulated (241, 242). Notably, p202 blocks AIM2 inflammasome assembly, and pyroptosis-mediated cell death. Consequently, p202 or other dsDNA sensors such as cyclic GMP-AMP synthase (cGAS), bind cytosolic DNA to promote prolonged type I IFN production that would be normally terminated by AIM2-induced pyroptosis (243). Regulation of pyroptosis has also been found to impact other aspects of the autoimmune response driving lupus nephritis. Pristane-induced lupus nephritis is exacerbated in mice lacking T cell expression of the P2X7R (140). Here, the P2X7R normally mediates GSDMD-driven pyroptosis of Tfh cells, which then limits differentiation of autoantibody secreting plasma cells in the germinal centers. Together these findings demonstrate the complexity of the roles inflammasomes have in both promoting and suppressing the autoimmune response of SLE.
Alternative roles of inflammasome molecule-mediated regulation
Classical inflammasome activation and induction of a proinflammatory response contributes to autoimmunity in a variety of ways as described above. It is becoming apparent, however, that inflammasome molecules also serve regulatory functions independent of typical inflammation-driving events (Table 2). Caspase-1 for instance, in addition to being involved in the maturation of IL-1β and IL-18, has been shown to modulate protein secretion, cell death, and lysosomal function in many cell types such as neurons, hepatocytes, epithelial cells, and cardiomyocytes (244–251). These alternative roles for inflammasome molecules have been linked to regulation of immune effector cells such as T and B cells, as well as non-immune tissue-resident cell types. Accordingly, some of these events have been reported to be directly involved in the progression of autoimmunity, and/or can be expected to contribute to an autoimmune response.
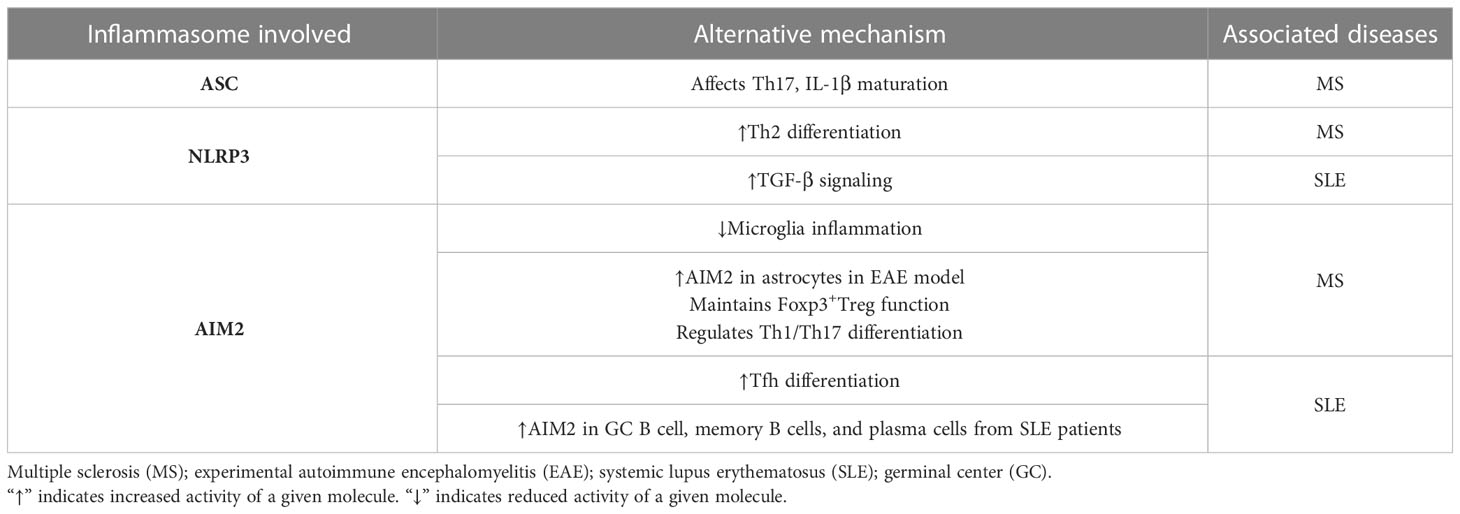
Table 2 Alternative functions of inflammasome molecules in autoimmunity.
ASC: A regulatory function in CD4+ T cells
ASC has a T cell intrinsic effect regulating the production of IL-1β needed to maintain CNS-resident Th17 cells in EAE. Recent findings indicate that ASC also regulates properties of murine CD4+ T cells independent of classical inflammasome activation and IL-1β maturation (252). ASC is constitutively expressed in naïve CD4+ T cells, and after anti-CD3/CD28 antibody stimulated TCR signaling, ASC is upregulated but no IL-1β or IL-18 secretion is detected (252). Naïve CD4+ T cells lacking ASC expression normally differentiate in vitro into Th1, Th2, Th17, Th9, and Foxp3+Treg subsets under polarizing conditions (252). Notably, recombination activation gene (Rag)-deficient mice develop more severe colitis after transfer of ASC-/- CD4+ T cells versus wildtype, NLRP3-/-, or Caspase1-/- CD4+ T cells (252). This increased pathogenic function of ASC-/- CD4+ T cells is marked by enhanced TCR signaling in vitro, elevated lymphopenic proliferation in vivo, and an increased metabolic state marked by higher glycolytic flux and increased glucose transporter 1 (Glut-1) surface expression (252). These findings suggest a negative regulatory function for ASC in CD4+ T cell TCR signaling, proliferation, and metabolism. The mechanism(s) by which ASC regulates these events still needs to be defined. Nevertheless, one could envision a scenario in which dysregulation of alternative ASC function enhances the pathogenic potential of autoreactive CD4+ (and possibly CD8+) T cells to aid autoimmune disease progression.
NLRP3 and Th2 cell differentiation
NLRP3 has also been found to have T cell-intrinsic effects independent of classical inflammasome activation. Specifically, NLRP3 positively regulates Th2 subset differentiation (253). Upon TCR stimulation by anti-CD3/CD28 antibody, expression of NLRP3 is increased in both Th1 and Th2 cells, due in part to IL-2 induced STAT5 activity (253). However, NLRP3-deficiency reduces Th2 but not Th1 cell differentiation (253). Importantly, ASC or caspase-1 deficiency has no effect on NLRP3-mediated Th2 lineage differentiation ruling out a role for classical NLRP3 inflammasome activity (253). Findings indicate that NLRP3 functions as a transcription factor regulating Il4 transcription (253). Here, NLRP3 forms a complex with the transcription factor IRF4, that enhances the binding of the IRF4 to the Il4 promoter; however, NLRP3 alone is insufficient to mediate Il4 transcription (253). Notably, induction of asthma, which is Th2 cell-dependent, is reduced in NLRP3-deficient mice (253). Furthermore, NLRP3-/- mice also more readily reject implanted B16F10 tumor cells due to an elevated Th1 cell response (253). In wildtype recipients, increased differentiation of Th2 cells permits the progression of B16F10 tumors (253). In the case of autoimmunity, aberrant Th2 cell differentiation has been associated with skewed development of Th1 and Th17 cells, which drive the pathology in MS, RA, T1D and SLE (254). Accordingly, aberrant expression and/or function of NLRP3 that is independent of inflammasome activity, may favor the development of pathogenic autoreactive Th1 and Th17 effectors. For instance, reduced IL-2 signaling and STAT5 activation, which is associated with T1D (255), would be expected to limit Nlrp3 transcription and Th2 cell differentiation.
Roles of AIM2 independent of inflammasome activation
Studies demonstrate that AIM2 displays a number of alternative functions independent of inflammasome activation in various cell types, that affect the progression of autoimmunity. Recently, AIM2 was shown to have a T cell-intrinsic role in regulating peripheral Foxp3+Treg (256). AIM2 is highly expressed in murine and human Foxp3+Treg, and AIM2 expression is upregulated by TGF-β1 stimulation (256). TGF-β1 is required for peripheral differentiation of CD4+ T cells into Foxp3+Treg (257). In AIM2-deficient C57BL/6 mice, MOG35-55-induced EAE is exacerbated characterized by increased Th1 and Th17 cell infiltration, and a reduction in the frequency of Foxp3+Treg in the CNS (256). A diminished local pool of Foxp3+Treg favors the expansion and effector function of encephalitogenic Teff (256, 257). Foxp3+Treg are unaffected by ASC-deficiency, indicating that the role for AIM2 is inflammasome-independent (256). Notably, AIM2 in Foxp3+Treg attenuates AKT activation, and downstream mTOR and MYC signaling that leads to glycolysis (256). Normal Foxp3+Treg differentiation and lineage maintenance is achieved under metabolic conditions favoring oxidative phosphorylation of lipids (256). On the other hand, glycolysis negatively impacts Foxp3+Treg stability and function (256). AIM2 serves to maintain Foxp3+Treg under proinflammatory conditions by forming a complex consisting of the adaptor protein receptor for activated C kinase 1 (RACK1), and the protein phosphatase 2 (PP2A) phosphatase that blocks AKT phosphorylation (256).
AIM2 has also been reported to regulate Tfh independent of inflammasome activation (258). Tfh from blood and skin lesions of SLE patients express elevated levels of AIM2. In mice in which AIM2 is conditionally ablated in T cells, the severity of pristane-induced lupus nephritis is reduced relative to control animals. The latter corresponds with a decreased Tfh pool. Notably, AIM2 regulates Tfh differentiation through an interaction with transcription factor c-MAF, that in turn is needed to promote Il21 gene transcription (258). Interestingly, Aim2 mRNA expression is upregulated by IL-21 stimulation suggesting that AIM2 participates in a feed-forward loop promoting Tfh differentiation and function.
In addition to T cells, AIM2 has been shown to have a B cell-intrinsic effect independent of inflammasome activation. SLE patients exhibit elevated AIM2 expression in germinal center (GC) B cells, memory B cells and antibody secreting plasma cells prepared from the tonsils, blood and/or skin lesions (259). Furthermore, pristane-induced lupus nephritis is attenuated in mice in which AIM2 is conditionally ablated in B cells. Limited disease is reflected by diminished numbers of GC B cells, and plasma cells. Findings suggest that AIM2 is an upstream regulator of the Blimp1-BCL6 transcriptional axis, which drives GC B cell and plasma cell differentiation (259).
AIM2 also serves a protective role in EAE by limiting the inflammatory properties of brain-resident microglia (151). Whereas ASC-deficiency in mice attenuates EAE as discussed above, AIM2-deficiency exacerbates EAE severity. Furthermore, selective ablation of AIM2 in microglia is sufficient to enhance the encephalitogenic response. In microglia, AIM2 negatively regulates a proinflammatory phenotype by suppressing the activity of DNA-dependent protein kinase (DNA-PK) and downstream activation of AKT3. Inhibition of AKT3 reduces phosphorylation of the key transcriptional factor IRF3, which blocks the production of chemokines, type I IFN, and the expression of antigen presentation molecules by microglia (151). AIM2 similarly inhibits DNA-PK and AKT activation in colon epithelial cells to protect mice from colitis and colon cancer (260). Interestingly, a recent study provides evidence that AIM2 has an alternative role in an EAE model independent of robust classical inflammasome activation (152). Through the use of a novel reporter mouse to track inflammasome activation in situ, AIM2 activation is seen to be prevalent in astrocytes but not CNS infiltrating monocytes and macrophages. Despite elevated AIM2 expression, no marked Il1b expression and cell death are detected in astrocytes (152). The role of AIM2 in this scenario needs to be further defined.
Targeting inflammasome molecules to prevent/treat autoimmunity
Inflammasome molecules offer an appealing target for immunotherapy and the treatment of autoimmunity. Several inhibitors targeting inflammasome-related molecules have been identified, developed, and tested in preclinical studies or clinical trials (Table 3). MCC950, a small-molecule inhibitor, specifically binds to the Walker B motif of the NACHT domain of NLRP3 to block function (287). Therapeutic efficacy and safety of MCC950 and analogs (Inzomelid and Somalix) have been assessed in several preclinical studies with promising results (288–294) (TrialTrovelID-368867; TrialTrovelID-360928). Nevertheless, a phase II clinical trial for RA showed that MCC950 has safety concerns related to elevated serum liver enzyme levels. Other NLRP3 inhibitors are currently being evaluated in animal studies of EAE (264, 266, 272, 279).
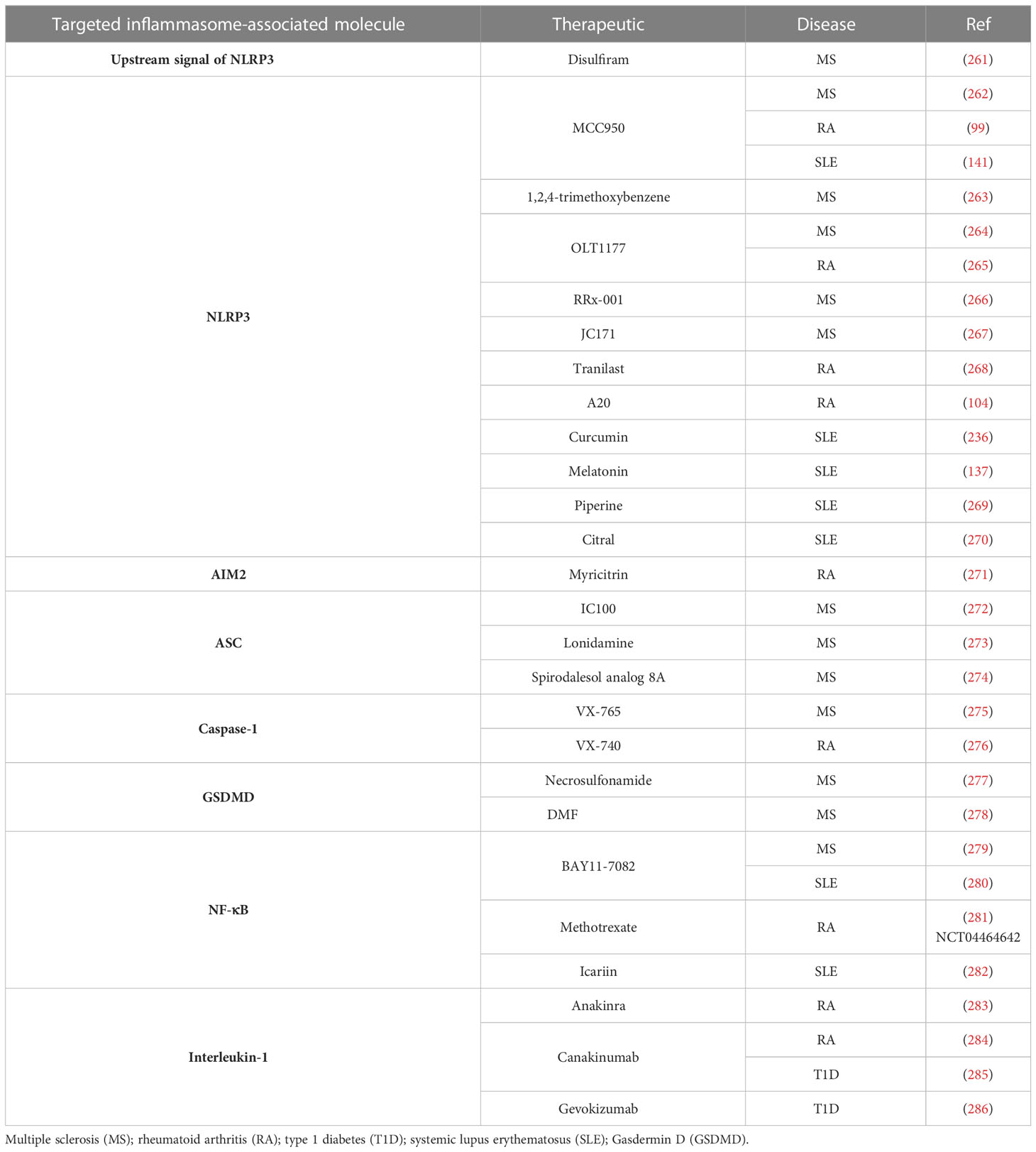
Table 3 Therapeutic strategies targeting inflammasomes for autoimmunity.
Caspase-1 is another key target for therapeutic intervention of autoimmunity. VX-765 (belnacasan), a caspase-1 inhibitor, blocks GSDMD-mediated pyroptosis, reduces inflammasome-associated proteins in the CNS, and attenuates EAE in mice (275). However, testing of the related caspase-1 inhibitor VX-740 was discontinued in a RA clinical trial due to the liver toxicity observed in animal models (295). Inhibiting GSDMD by necrosulfonamide reduces neuroinflammation and necroptosis in collagenase VII-induced mouse intracerebral hemorrhage model (277). In addition, dimethyl fumarate, an immunosuppressive drug used for the treatment of recurrent remission MS and plaque psoriasis promotes succination of GSDMD, which in turn disrupts the interaction with caspase-1 and blocks pyropotosis (278). Disulfiram, a drug used for alcohol addiction treatment, blocks pore formation by targeting Cys191/Cys192 in GSDMD (261).
IL-1β, which is associated with the pathogenesis of several autoimmune diseases, has been therapeutically targeted. Two FDA-approved biologics that block IL-1 activity have been clinically tested. Anakinra is a recombinant human IL-1R antagonist mainly applied for the treatment of RA. Due to a short half-life and low response rate compared to other treatments available, the usage of anakinra is limited, and efficacy is selective. For example, anakinra shows no efficacy for the treatment of T1D and Sjogren’s disease. Canakinumab is an anti-IL-1β neutralizing monoclonal antibody and has shown efficacy in RA and systemic juvenile idiopathic arthritis but no benefit for recent onset T1D patients (285, 296). IL-18 blockers have also been established but have not been applied for the treatment of autoimmunity.
Summary/conclusions
The evidence at hand establishes roles for classical inflammasome activated inflammation and alternative pathways regulated by inflammasome molecules in autoimmunity. Inflammasome molecules have been implicated in human MS, RA, T1D and SLE, and shown in corresponding disease models to override and/or maintain self-tolerance (Table 1). Intrinsic and extrinsic effects on both APC and other innate effectors as well as T and B cells enables inflammasome molecules to establish the nature and specificity of an autoimmune response. Similarly, inflammasome molecules have intrinsic and extrinsic effects that alter the cellular integrity of tissues, independent of immune effectors. In a given tissue, inflammasome activity can impact inflammation by initiating and/or further driving a local autoimmune response, which in turn may be influenced by induction of pyroptosis versus PANoptosis cell death pathways. Alternatively, dysregulated inflammasome function can have more broad effects. This is seen with aberrant inflammasome activity reducing intestinal barrier function, which results in shifts within the microbiota composition that can impact the production of systemically released metabolites and favor proinflammatory versus immunoregulatory events (214).
The key events that drive inflammasome molecule activity in autoimmunity are poorly understood. What is apparent, however, is that multiple pathways and mechanisms exist to induce activation, in part reflecting the specificity of different inflammasome molecules. Poorly understood environmental factors known to influence MS, T1D, RA and SLE are likely involved in inducing inflammasome molecule activity. Release of PAMPs due to microbial infections or DAMPs due to cytotoxic effects of drugs, toxins, or UV irradiation for example, are obvious candidates to engage classical inflammasome-mediated inflammation. Polymorphisms in various inflammasome genes may also contribute to the polygenic influence on the development of MS, T1D, RA and SLE. Genetic analyses show that single nucleotide polymorphisms (SNPs) in genes encoding sensor molecules (i.e. NLRP1, NLRP3, AIM2) and inflammasome-related proteins (i.e. PYCARD, CASP1) are linked with susceptibility to and/or response to therapy for MS, T1D, RA and SLE (75, 78, 98, 119, 120, 153, 297–303). However, whether the disease-linked SNPs override the normally tight regulation of gene expression and/or function of inflammasome molecules needs to be ascertained. Inflammasome activity is also the consequence of collateral damage induced by autoimmunity. Autoimmune-mediated cytotoxicity leads to the release of DAMPs and a proinflammatory milieu induces local cellular stress affecting metabolism and mitochondrial function for instance, that drive inflammasome molecule activity.
The relative contribution(s) inflammasome molecule activity has in autoimmunity is poorly understood. Questions of whether inflammasome molecules mediate initiating events and/or modulate the progression and severity of autoimmunity need to be addressed. Environmental insults have typically been proposed to initiate autoimmunity where inflammasome activation is likely to occur (Table 1). Alternatively, sterile inflammation driven by metabolically stressed cells may stimulate dysregulated inflammasome activity and initiate autoimmunity. Pancreatic β cells are susceptible to metabolic stress due high levels of insulin expression and secretion (304, 305) that may lead to NLRP3 activation, for example. Reports showing that inflammasome expression and activity are upregulated in MS, T1D, RA and SLE patients suggest a role in at least supporting disease progression. Feed-forward loops in which inflammasome molecule activity are self-sustaining as well as promoting autoimmune reactivity and vice versa have been described. The use of murine models of spontaneous autoimmunity coupled with cell-specific and inducible expression systems will be helpful in further defining the contribution in the disease process for a given inflammasome molecule.
Of keen interest moving forward is defining regulation of inflammasome molecule-mediated events that are independent of classical activation of inflammation (Table 2). A hint to the complexity that is involved is exemplified by AIM2. As discussed above AIM2 regulates peripheral Foxp3+Treg differentiation by blocking AKT signaling through a AIM2-RACK1-PP2A complex (256). On the other hand, AIM2 suppresses colon carcinoma by binding to and inhibiting DNA-PK and downstream AKT signaling events needed for colon epithelial cell transformation (260). Therefore, depending on the cell-type, AIM2 inhibits PI3K-AKT signaling but via distinct complexes and mechanisms. Furthermore, AIM2 is reported to interact with the c-MAF transcription factor to positively promote Tfh differentiation (258). The nature of the signaling events that stimulate alternative inflammasome molecule activity, and the outcome of that activity in immune and non-immune cell types are important issues that require continued investigation.
To date, the therapeutic benefit of inhibiting inflammasome activation has mostly been demonstrated in animal disease models with limited success in the clinic (Table 3). The general lack of efficacy may reflect the timing and relative contribution of an inflammasome molecule in a given autoimmune disease. For instance, inflammasome activation may play a prominent role early in a disease process. Therefore, targeting inflammasome activity once an autoimmune response is well established, which is typical in the clinic, may have only a minimal effect. There is the important concern that inhibiting a given inflammasome molecule, particularly long-term, may compromise immunity against pathogens. Therefore, both efficacy and safety may be enhanced by combining an inflammasome-based approach with other types of immunotherapies. For example, limiting ongoing inflammation by blocking inflammasome activity may enhance the efficacy of antigen-based immunotherapy and induction of protective Treg.
The etiology of MS, T1D, RA and SLE is highly complex, and ill-defined. Establishing the roles of inflammasome activity in autoimmunity will aid our understanding of the mechanisms that drive these disease processes, as well as provide the impetus for the development of novel strategies of immunotherapy for disease prevention and treatment.
Author contributions
QK, AG, VM, XJ and RT contributed to the preparation of the review article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institutes of Health grants R01DK100256, R01AI139475, R01AI141631, R21AI115752 (RT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Davidson A, Diamond B. Autoimmune diseases. N Engl J Med (2001) 345(5):340–50. doi: 10.1056/nejm200108023450506
2. Papp G, Boros P, Nakken B, Szodoray P, Zeher M. Regulatory immune cells and functions in autoimmunity and transplantation immunology. Autoimmun Rev (2017) 16(5):435–44. doi: 10.1016/j.autrev.2017.03.011
3. Davis BK, Wen H, Ting JP. The inflammasome NLRS in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
4. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157(5):1013–22. doi: 10.1016/j.cell.2014.04.007
5. Cordero MD, Alcocer-Gómez E, Ryffel B. Gain of function mutation and inflammasome driven diseases in human and mouse models. J Autoimmun (2018) 91:13–22. doi: 10.1016/j.jaut.2018.03.002
6. Drutman SB, Haerynck F, Zhong FL, Hum D, Hernandez NJ, Belkaya S, et al. Homozygous NLRP1 gain-of-Function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci USA (2019) 116(38):19055–63. doi: 10.1073/pnas.1906184116
7. Romberg N, Vogel TP, Canna SW. NLRc4 inflammasomopathies. Curr Opin Allergy Clin Immunol (2017) 17(6):398–404. doi: 10.1097/aci.0000000000000396
8. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
9. Deets KA, Vance RE. Inflammasomes and adaptive immune responses. Nat Immunol (2021) 22(4):412–22. doi: 10.1038/s41590-021-00869-6
10. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
11. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci (2017) 130(23):3955–63. doi: 10.1242/jcs.207365
12. Broz P, Dixit VM. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
13. Pandian N, Kanneganti TD. Panoptosis: A unique innate immune inflammatory cell death modality. J Immunol (2022) 209(9):1625–33. doi: 10.4049/jimmunol.2200508
14. Samir P, Malireddi RKS, Kanneganti TD. The panoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (Panoptosis). Front Cell Infect Microbiol (2020) 10:238. doi: 10.3389/fcimb.2020.00238
15. Gullett JM, Tweedell RE, Kanneganti TD. It's all in the PAN: Crosstalk, plasticity, redundancies, switches, and interconnectedness encompassed by PANoptosis underlying the totality of cell death-associated biological effects. Cells (2022) 11(9). doi: 10.3390/cells11091495
16. Pearson JA, Wong FS, Wen L. Inflammasomes and type 1 diabetes. Front Immunol (2021) 12:686956. doi: 10.3389/fimmu.2021.686956
17. Tartey S, Kanneganti TD. Inflammasomes in the pathophysiology of autoinflammatory syndromes. J Leukoc Biol (2020) 107(3):379–91. doi: 10.1002/jlb.3mir0919-191r
18. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
19. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol (2016) 13(2):148–59. doi: 10.1038/cmi.2015.95
20. Blevins HM, Xu Y, Biby S, Zhang S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front Aging Neurosci (2022) 14:879021. doi: 10.3389/fnagi.2022.879021
21. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
22. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10(2):128. doi: 10.1038/s41419-019-1413-8
23. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
24. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The Nalp3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
25. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol Neurodegener (2016) 11:23. doi: 10.1186/s13024-016-0088-1
26. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science (2019) 364(6435):eaau1330. doi: 10.1126/science.aau1330
27. Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1b inflammasome. Science (2019) 364(6435):82–5. doi: 10.1126/science.aau1208
28. Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, et al. The N-end rule ubiquitin ligase Ubr2 mediates NLRP1b inflammasome activation by anthrax lethal toxin. EMBO J (2019) 38(13):e101996. doi: 10.15252/embj.2019101996
29. Robert Hollingsworth L, David L, Li Y, Griswold AR, Ruan J, Sharif H, et al. Mechanism of filament formation in upa-promoted Card8 and NLRP1 inflammasomes. Nat Commun (2021) 12(1):189. doi: 10.1038/s41467-020-20320-y
30. Planès R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, et al. Human NLRP1 is a sensor of pathogenic coronavirus 3cl proteases in lung epithelial cells. Mol Cell (2022) 82(13):2385–400.e9. doi: 10.1016/j.molcel.2022.04.033
31. Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded rna. Science (2021) 371(6528):eabd0811. doi: 10.1126/science.abd0811
32. Tsu BV, Beierschmitt C, Ryan AP, Agarwal R, Mitchell PS, Daugherty MD. Diverse viral proteases activate the NLRP1 inflammasome. Elife (2021) 10:e60609. doi: 10.7554/eLife.60609
33. Robinson KS, Teo DET, Tan KS, Toh GA, Ong HH, Lim CK, et al. Enteroviral 3c protease activates the human NLRP1 inflammasome in airway epithelia. Science (2020) 370(6521):eaay2002. doi: 10.1126/science.aay2002
34. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of NLRP1 is required for activation of the inflammasome. PloS Pathog (2012) 8(3):e1002638. doi: 10.1371/journal.ppat.1002638
35. Mitchell PS, Sandstrom A, Vance RE. The NLRP1 inflammasome: New mechanistic insights and unresolved mysteries. Curr Opin Immunol (2019) 60:37–45. doi: 10.1016/j.coi.2019.04.015
36. Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, et al. Dpp8 and Dpp9 inhibition induces pro-Caspase-1-dependent monocyte and macrophage pyroptosis. Nat Chem Biol (2017) 13(1):46–53. doi: 10.1038/nchembio.2229
37. Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, et al. Human Dpp9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and fiind domain binding. J Biol Chem (2018) 293(49):18864–78. doi: 10.1074/jbc.RA118.004350
38. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type iii secretion apparatus through the NLRc4 inflammasome. Proc Natl Acad Sci USA (2010) 107(7):3076–80. doi: 10.1073/pnas.0913087107
39. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRc4 inflammasome receptors for bacterial flagellin and type iii secretion apparatus. Nature (2011) 477(7366):596–600. doi: 10.1038/nature10510
40. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRc4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
41. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-Activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
42. Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol (2016) 46(2):269–80. doi: 10.1002/eji.201545839
43. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science (2016) 354(6313):765–8. doi: 10.1126/science.aaf7532
44. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: Back to the future. Immunity (2013) 39(6):1003–18. doi: 10.1016/j.immuni.2013.11.010
45. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
46. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med (2020) 217(1):e20190314. doi: 10.1084/jem.20190314
47. Waisman A, Lukas D, Clausen BE, Yogev N. Dendritic cells as gatekeepers of tolerance. Semin Immunopathol (2017) 39(2):153–63. doi: 10.1007/s00281-016-0583-z
48. Zaghouani H, Miller MM. Early-life T-helper 1 immunity. Crit Rev Immunol (2020) 40(5):407–11. doi: 10.1615/CritRevImmunol.2020035106
49. Martynova E, Rizvanov A, Urbanowicz RA, Khaiboullina S. Inflammasome contribution to the activation of Th1, Th2, and Th17 immune responses. Front Microbiol (2022) 13:851835. doi: 10.3389/fmicb.2022.851835
50. Zhang S. The role of transforming growth factor B in T helper 17 differentiation. Immunology (2018) 155(1):24–35. doi: 10.1111/imm.12938
51. Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGFbeta-Induced Foxp3 expression. J Immunol (2014) 192(4):1449–58. doi: 10.4049/jimmunol.1300387
52. Zhu X, Zhu J. CD4 T helper cell subsets and related human immunological disorders. Int J Mol Sci (2020) 21(21). doi: 10.3390/ijms21218011
53. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of Foxp3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
54. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new Forkhead/Winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi: 10.1038/83784
55. Van Gool F, Nguyen MLT, Mumbach MR, Satpathy AT, Rosenthal WL, Giacometti S, et al. A mutation in the transcription factor Foxp3 drives T helper 2 effector function in regulatory T cells. Immunity (2019) 50(2):362–77.e6. doi: 10.1016/j.immuni.2018.12.016
56. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-Linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27(1):18–20. doi: 10.1038/83707
57. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med (2013) 210(3):491–502. doi: 10.1084/jem.20122006
58. Dolence JJ, Kobayashi T, Iijima K, Krempski J, Drake LY, Dent AL, et al. Airway exposure initiates peanut allergy by involving the IL-1 pathway and T follicular helper cells in mice. J Allergy Clin Immunol (2018) 142(4):1144–58 e8. doi: 10.1016/j.jaci.2017.11.020
59. Choi J, Crotty S. Bcl6-mediated transcriptional regulation of follicular helper T cells (TFH). Trends Immunol (2021) 42(4):336–49. doi: 10.1016/j.it.2021.02.002
60. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev (2018) 281(1):138–53. doi: 10.1111/imr.12616
61. Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18, more than a Th1 cytokine. Semin Immunol (2013) 25(6):439–48. doi: 10.1016/j.smim.2013.10.014
62. Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-gamma-inducing factor up-regulates fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol (1996) 157(9):3967–73.
63. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell (2015) 163(6):1444–56. doi: 10.1016/j.cell.2015.10.072
64. Somm E, Jornayvaz FR. Interleukin-18 in metabolism: From mice physiology to human diseases. Front Endocrinol (Lausanne) (2022) 13:971745. doi: 10.3389/fendo.2022.971745
65. Dagar S, Singh J, Saini A, Kumar Y, Chhabra S, Minz RW, et al. Gut bacteriome, mycobiome and virome alterations in rheumatoid arthritis. Front Endocrinol (Lausanne) (2022) 13:1044673. doi: 10.3389/fendo.2022.1044673
66. Davis-Richardson AG, Triplett EW. A model for the role of gut bacteria in the development of autoimmunity for type 1 diabetes. Diabetologia (2015) 58(7):1386–93. doi: 10.1007/s00125-015-3614-8
67. Donati D. Viral infections and multiple sclerosis. Drug Discovery Today Dis Models (2020) 32:27–33. doi: 10.1016/j.ddmod.2020.02.003
68. Karampoor S, Zahednasab H, Ramagopalan S, Mehrpour M, Etemadifar M, Alsahebfosoul F, et al. Cytomegalovirus and varicella zoster virus seropositivity of Iranian patients with multiple sclerosis: A population-based study. J Neuroimmunol (2017) 309:4–6. doi: 10.1016/j.jneuroim.2017.04.004
69. Marrodan M, Alessandro L, Farez MF, Correale J. The role of infections in multiple sclerosis. Mult Scler (2019) 25(7):891–901. doi: 10.1177/1352458518823940
70. Lane TE, Hosking MP. The pathogenesis of murine coronavirus infection of the central nervous system. Crit Rev Immunol (2010) 30(2):119–30. doi: 10.1615/critrevimmunol.v30.i2.20
71. Morris MM, Dyson H, Baker D, Harbige LS, Fazakerley JK, Amor S. Characterization of the cellular and cytokine response in the central nervous system following semliki forest virus infection. J Neuroimmunol (1997) 74(1-2):185–97. doi: 10.1016/s0165-5728(96)00786-2
72. Bernales CQ, Encarnacion M, Criscuoli MG, Yee IM, Traboulsee AL, Sadovnick AD, et al. Analysis of NOD-like receptor NLRP1 in multiple sclerosis families. Immunogenetics (2018) 70(3):205–7. doi: 10.1007/s00251-017-1034-2
73. Maver A, Lavtar P, Ristić S, Stopinšek S, Simčič S, Hočevar K, et al. Identification of rare genetic variation of NLRP1 gene in familial multiple sclerosis. Sci Rep (2017) 7(1):3715. doi: 10.1038/s41598-017-03536-9
74. Popplewell LF, Encarnacion M, Bernales CQ, Sadovnick AD, Traboulsee AL, Quandt JA, et al. Genetic analysis of nucleotide-binding leucine-rich repeat (NLR) receptors in multiple sclerosis. Immunogenetics (2020) 72(6-7):381–5. doi: 10.1007/s00251-020-01170-w
75. Soares JL, Oliveira EM, Pontillo A. Variants in NLRP3 and NLRc4 inflammasome associate with susceptibility and severity of multiple sclerosis. Mult Scler Relat Disord (2019) 29:26–34. doi: 10.1016/j.msard.2019.01.023
76. Gil-Varea E, Urcelay E, Vilariño-Güell C, Costa C, Midaglia L, Matesanz F, et al. Exome sequencing study in patients with multiple sclerosis reveals variants associated with disease course. J Neuroinflamm (2018) 15(1):265. doi: 10.1186/s12974-018-1307-1
77. Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med (2017) 214(5):1351–70. doi: 10.1084/jem.20150237
78. Gharagozloo M, Mahmoud S, Simard C, Yamamoto K, Bobbala D, Ilangumaran S, et al. NLRX1 inhibits the early stages of CNS inflammation and prevents the onset of spontaneous autoimmunity. PloS Biol (2019) 17(9):e3000451. doi: 10.1371/journal.pbio.3000451
79. Eitas TK, Chou WC, Wen H, Gris D, Robbins GR, Brickey J, et al. The nucleotide-binding leucine-rich repeat (NLR) family member NLRX1 mediates protection against experimental autoimmune encephalomyelitis and represses macrophage/microglia-induced inflammation. J Biol Chem (2014) 289(7):4173–9. doi: 10.1074/jbc.M113.533034
80. Tuladhar S, Kanneganti TD. NLRP12 in innate immunity and inflammation. Mol Aspects Med (2020) 76:100887. doi: 10.1016/j.mam.2020.100887
81. Gharagozloo M, Mahvelati TM, Imbeault E, Gris P, Zerif E, Bobbala D, et al. The nod-like receptor, NLRP12, plays an anti-inflammatory role in experimental autoimmune encephalomyelitis. J Neuroinflamm (2015) 12:198. doi: 10.1186/s12974-015-0414-5
82. Huang WX, Huang P, Hillert J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler (2004) 10(5):482–7. doi: 10.1191/1352458504ms1071oa
83. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA (2012) 109(26):10480–5. doi: 10.1073/pnas.1201836109
84. Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, et al. NLRc3 expression in dendritic cells attenuates CD4(+) T cell response and autoimmunity. EMBO J (2019) 38(16):e101397. doi: 10.15252/embj.2018101397
85. Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med (2019) 216(11):2562–81. doi: 10.1084/jem.20190377
86. Martin BN, Wang C, Zhang CJ, Kang Z, Gulen MF, Zepp JA, et al. T Cell-intrinsic ASC critically promotes Th17-mediated experimental autoimmune encephalomyelitis. Nat Immunol (2016) 17(5):583–92. doi: 10.1038/ni.3389
87. Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, et al. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity (2015) 42(4):654–64. doi: 10.1016/j.immuni.2015.03.006
88. Gharagozloo M, Mahmoud S, Simard C, Mahvelati TM, Amrani A, Gris D. The dual immunoregulatory function of NLRP12 in T cell-mediated immune response: Lessons from experimental autoimmune encephalomyelitis. Cells (2018) 7(9):119. doi: 10.3390/cells7090119
89. Ming X, Li W, Maeda Y, Blumberg B, Raval S, Cook SD, et al. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci (2002) 197(1-2):9–18. doi: 10.1016/s0022-510x(02)00030-8
90. Mahmoud S, Gharagozloo M, Simard C, Amrani A, Gris D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells (2019) 8(5). doi: 10.3390/cells8050400
91. Martinez-Martinez RE, Abud-Mendoza C, Patiño-Marin N, Rizo-Rodríguez JC, Little JW, Loyola-Rodríguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol (2009) 36(12):1004–10. doi: 10.1111/j.1600-051X.2009.01496.x
92. Moen K, Brun JG, Valen M, Skartveit L, Eribe EK, Olsen I, et al. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial dnas. Clin Exp Rheumatol (2006) 24(6):656–63.
93. Reichert S, Haffner M, Keyßer G, Schäfer C, Stein JM, Schaller HG, et al. Detection of oral bacterial DNA in synovial fluid. J Clin Periodontol (2013) 40(6):591–8. doi: 10.1111/jcpe.12102
94. Romero-Figueroa MDS, Ramírez-Durán N, Montiel-Jarquín AJ, Horta-Baas G. Gut-joint axis: Gut dysbiosis can contribute to the onset of rheumatoid arthritis via multiple pathways. Front Cell Infect Microbiol (2023) 13:1092118. doi: 10.3389/fcimb.2023.1092118
95. Wang Y, Wei J, Zhang W, Doherty M, Zhang Y, Xie H, et al. Gut dysbiosis in rheumatic diseases: A systematic review and meta-analysis of 92 observational studies. EBioMedicine (2022) 80:104055. doi: 10.1016/j.ebiom.2022.104055
96. Addobbati C, da Cruz HLA, Adelino JE, Melo Tavares Ramos AL, Fragoso TS, Domingues A, et al. Polymorphisms and expression of inflammasome genes are associated with the development and severity of rheumatoid arthritis in Brazilian patients. Inflammation Res (2018) 67(3):255–64. doi: 10.1007/s00011-017-1119-2
97. Cheng L, Liang X, Qian L, Luo C, Li D. NLRP3 gene polymorphisms and expression in rheumatoid arthritis. Exp Ther Med (2021) 22(4):1110. doi: 10.3892/etm.2021.10544
98. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Biologics in Rheumatoid Arthritis G, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis (2014) 73(6):1202–10. doi: 10.1136/annrheumdis-2013-203276
99. Guo C, Fu R, Wang S, Huang Y, Li X, Zhou M, et al. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin Exp Immunol (2018) 194(2):231–43. doi: 10.1111/cei.13167
100. Choulaki C, Papadaki G, Repa A, Kampouraki E, Kambas K, Ritis K, et al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther (2015) 17(1):257. doi: 10.1186/s13075-015-0775-2
101. Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, et al. Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (Il)-1β via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: A possible implication for therapeutic decision in these patients. Clin Exp Immunol (2015) 182(1):35–44. doi: 10.1111/cei.12667
102. Li Z, Guo J, Bi L. Role of the NLRP3 inflammasome in autoimmune diseases. BioMed Pharmacother (2020) 130:110542. doi: 10.1016/j.biopha.2020.110542
103. Ippagunta SK, Brand DD, Luo J, Boyd KL, Calabrese C, Stienstra R, et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a card (Asc) in T cell priming is critical for collagen-induced arthritis. J Biol Chem (2010) 285(16):12454–62. doi: 10.1074/jbc.M109.093252
104. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature (2014) 512(7512):69–73. doi: 10.1038/nature13322
105. Delgado-Arévalo C, Calvet-Mirabent M, Triguero-Martínez A, Vázquez de Luis E, Benguría-Filippini A, Largo R, et al. NLRC4-mediated activation of CD11c+ DC contributes to perpetuation of synovitis in rheumatoid arthritis. JCI Insight (2022) 7(22). doi: 10.1172/jci.insight.152886
106. Jakobs C, Perner S, Hornung V. AIM2 drives joint inflammation in a self-DNA triggered model of chronic polyarthritis. PloS One (2015) 10(6):e0131702. doi: 10.1371/journal.pone.0131702
107. Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, et al. Cutting edge: AIM2 and endosomal tlrs differentially regulate arthritis and autoantibody production in DNAse ii-deficient mice. J Immunol (2015) 194(3):873–7. doi: 10.4049/jimmunol.1402573
108. Yang Z, Cao J, Yu C, Yang Q, Zhang Y, Han L. Caspase-1 mediated interleukin-18 activation in neutrophils promotes the activity of rheumatoid arthritis in a NLRP3 inflammasome independent manner. Joint Bone Spine (2016) 83(3):282–9. doi: 10.1016/j.jbspin.2015.07.006
109. Zhao C, Gu Y, Zeng X, Wang J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin Immunol (2018) 197:154–60. doi: 10.1016/j.clim.2018.09.007
110. Prado DS, Veras FP, Ferreira RG, Damasceno LEA, Melo PH, Zamboni DS, et al. NLRP12 controls arthritis severity by acting as a checkpoint inhibitor of Th17 cell differentiation. FASEB J (2020) 34(8):10907–19. doi: 10.1096/fj.202000795R
111. Chen Y, Fujuan Q, Chen E, Yu B, Zuo F, Yuan Y, et al. Expression of AIM2 in rheumatoid arthritis and its role on fibroblast-like synoviocytes. Mediators Inflammation (2020) 2020:1693730. doi: 10.1155/2020/1693730
112. Lin Y, Luo Z. NLRP6 facilitates the interaction between Tab2/3 and Trim38 in rheumatoid arthritis fibroblast-like synoviocytes. FEBS Lett (2017) 591(8):1141–9. doi: 10.1002/1873-3468.12622
113. Li XF, Shen WW, Sun YY, Li WX, Sun ZH, Liu YH, et al. MicroRNA-20a negatively regulates expression of NLRP3-inflammasome by targeting txnip in adjuvant-induced arthritis fibroblast-like synoviocytes. Joint Bone Spine (2016) 83(6):695–700. doi: 10.1016/j.jbspin.2015.10.007
114. Tian J, Zhou D, Xiang L, Liu X, Zhang H, Wang B, et al. Mir-223-3p inhibits inflammation and pyroptosis in monosodium urate-induced rats and fibroblast-like synoviocytes by targeting NLRP3. Clin Exp Immunol (2021) 204(3):396–410. doi: 10.1111/cei.13587
115. Liu YR, Yan X, Yu HX, Yao Y, Wang JQ, Li XF, et al. NLRc5 promotes cell proliferation via regulating the NF-κB signaling pathway in rheumatoid arthritis. Mol Immunol (2017) 91:24–34. doi: 10.1016/j.molimm.2017.08.024
116. Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes: Pros and cons. Diabetes (2008) 57(11):2863–71. doi: 10.2337/db07-1023
117. Sechi LA, Rosu V, Pacifico A, Fadda G, Ahmed N, Zanetti S. Humoral immune responses of type 1 diabetes patients to Mycobacterium avium subsp. Paratuberculosis lend support to the infectious trigger hypothesis. Clin Vaccine Immunol (2008) 15(2):320–6. doi: 10.1128/cvi.00381-07
118. Magitta NF, Bøe Wolff AS, Johansson S, Skinningsrud B, Lie BA, Myhr KM, et al. A coding polymorphism in NALP1 confers risk for autoimmune addison's disease and type 1 diabetes. Genes Immun (2009) 10(2):120–4. doi: 10.1038/gene.2008.85
119. Sun X, Xia Y, Liu Y, Wang Y, Luo S, Lin J, et al. Polymorphisms in NLRP1 gene are associated with type 1 diabetes. J Diabetes Res (2019) 2019:7405120. doi: 10.1155/2019/7405120
120. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity (2010) 43(8):583–9. doi: 10.3109/08916930903540432
121. Sun X, Xu L, Xia Y, Luo S, Lin J, Xiao Y, et al. Rs3806265 and Rs4612666 of the NLRP3 gene are associated with the titer of glutamic acid decarboxylase antibody in type 1 diabetes. Front Endocrinol (Lausanne) (2022) 13:835054. doi: 10.3389/fendo.2022.835054
122. Xu L, Sun X, Xia Y, Luo S, Lin J, Xiao Y, et al. Polymorphisms of the NLRc4 gene are associated with the onset age, positive rate of gada and 2-h postprandial c-peptide in patients with type 1 diabetes. Diabetes Metab Syndr Obes (2020) 13:811–8. doi: 10.2147/dmso.S244882
123. Hu C, Ding H, Li Y, Pearson JA, Zhang X, Flavell RA, et al. NLRP3 deficiency protects from type 1 diabetes through the regulation of chemotaxis into the pancreatic islets. Proc Natl Acad Sci USA (2015) 112(36):11318–23. doi: 10.1073/pnas.1513509112
124. Leite JA, Pessenda G, Guerra-Gomes IC, de Santana AKM, André Pereira C, Ribeiro Campos Costa F, et al. The DNA sensor AIM2 protects against streptozotocin-induced type 1 diabetes by regulating intestinal homeostasis via the IL-18 pathway. Cells (2020) 9(4). doi: 10.3390/cells9040959
125. Wu C, Pan LL, Niu W, Fang X, Liang W, Li J, et al. Modulation of gut microbiota by low methoxyl pectin attenuates type 1 diabetes in non-obese diabetic mice. Front Immunol (2019) 10:1733. doi: 10.3389/fimmu.2019.01733
126. Jia LL, Zhang M, Liu H, Sun J, Pan LL. Early-life fingolimod treatment improves intestinal homeostasis and pancreatic immune tolerance in non-obese diabetic mice. Acta Pharmacol Sin (2021) 42(10):1620–9. doi: 10.1038/s41401-020-00590-4
127. Quaglia M, Merlotti G, De Andrea M, Borgogna C, Cantaluppi V. Viral infections and systemic lupus erythematosus: New players in an old story. Viruses (2021) 13(2). doi: 10.3390/v13020277
128. Chen C, Yan Q, Yao X, Li S, Lv Q, Wang G, et al. Alterations of the gut virome in patients with systemic lupus erythematosus. Front Immunol (2022) 13:1050895. doi: 10.3389/fimmu.2022.1050895
129. Li Y, Wang HF, Li X, Li HX, Zhang Q, Zhou HW, et al. Disordered intestinal microbes are associated with the activity of systemic lupus erythematosus. Clin Sci (Lond) (2019) 133(7):821–38. doi: 10.1042/cs20180841
130. Li BZ, Zhou HY, Guo B, Chen WJ, Tao JH, Cao NW, et al. Dysbiosis of oral microbiota is associated with systemic lupus erythematosus. Arch Oral Biol (2020) 113:104708. doi: 10.1016/j.archoralbio.2020.104708
131. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity (2012) 45(4):271–8. doi: 10.3109/08916934.2011.637532
132. Loras-Duclaux I, Descos B, Souillet G, Lachaux A, Badinand P, Montagnon N, et al. [Congenital intrinsic factor deficiency. apropos of 3 cases in a sibship]. Arch Fr Pediatr (1988) 45(3):197–9.
133. Huang T, Yin H, Ning W, Wang X, Chen C, Lin W, et al. Expression of inflammasomes NLRP1, NLRP3 and AIM2 in different pathologic classification of lupus nephritis. Clin Exp Rheumatol (2020) 38(4):680–90.
134. da Cruz HLA, Cavalcanti CAJ, de Azevedo Silva J, de Lima CAD, Fragoso TS, Barbosa AD, et al. Differential expression of the inflammasome complex genes in systemic lupus erythematosus. Immunogenetics (2020) 72(4):217–24. doi: 10.1007/s00251-020-01158-6
135. Inoue K, Ishizawa M, Kubota T. Monoclonal anti-dsDNA antibody 2c10 escorts DNA to intracellular DNA sensors in normal mononuclear cells and stimulates secretion of multiple cytokines implicated in lupus pathogenesis. Clin Exp Immunol (2020) 199(2):150–62. doi: 10.1111/cei.13382
136. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol (2013) 190(3):1217–26. doi: 10.4049/jimmunol.1202388
137. Bonomini F, Dos Santos M, Veronese FV, Rezzani R. NLRP3 inflammasome modulation by melatonin supplementation in chronic pristane-induced lupus nephritis. Int J Mol Sci (2019) 20(14). doi: 10.3390/ijms20143466
138. Liu J, Berthier CC, Kahlenberg JM. Enhanced inflammasome activity in systemic lupus erythematosus is mediated via type I interferon-induced up-regulation of interferon regulatory factor 1. Arthritis Rheumatol (2017) 69(9):1840–9. doi: 10.1002/art.40166
139. Liu Y, Tao X, Tao J. Strategies of targeting inflammasome in the treatment of systemic lupus erythematosus. Front Immunol (2022) 13:894847. doi: 10.3389/fimmu.2022.894847
140. Faliti CE, Gualtierotti R, Rottoli E, Gerosa M, Perruzza L, Romagnani A, et al. P2X7 receptor restrains pathogenic tfh cell generation in systemic lupus erythematosus. J Exp Med (2019) 216(2):317–36. doi: 10.1084/jem.20171976
141. Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol (2017) 69(8):1636–46. doi: 10.1002/art.40155
142. Fu R, Xia Y, Li M, Mao R, Guo C, Zhou M, et al. Pim-1 as a therapeutic target in lupus nephritis. Arthritis Rheumatol (2019) 71(8):1308–18. doi: 10.1002/art.40863
143. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med (2000) 343(13):938–52. doi: 10.1056/nejm200009283431307
144. Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem (2016) 136 Suppl 1:29–38. doi: 10.1111/jnc.13217
145. Ramagopalan SV, Sadovnick AD. Epidemiology of multiple sclerosis. Neurol Clin (2011) 29(2):207–17. doi: 10.1016/j.ncl.2010.12.010
146. Steinman L. No quiet surrender: Molecular guardians in multiple sclerosis brain. J Clin Invest (2015) 125(4):1371–8. doi: 10.1172/jci74255
147. Attfield KE, Jensen LT, Kaufmann M, Friese MA, Fugger L. The immunology of multiple sclerosis. Nat Rev Immunol (2022) 22(12):734–50. doi: 10.1038/s41577-022-00718-z
148. Bar-Or A. The immunology of multiple sclerosis. Semin Neurol (2008) 28(1):29–45. doi: 10.1055/s-2007-1019124
149. Morales Y, Parisi JE, Lucchinetti CF. The pathology of multiple sclerosis: Evidence for heterogeneity. Adv Neurol (2006) 98:27–45.
150. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun (2018) 9(1):2036. doi: 10.1038/s41467-018-04376-5
151. Ma C, Li S, Hu Y, Ma Y, Wu Y, Wu C, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med (2021) 218(5). doi: 10.1084/jem.20201796
152. Barclay WE, Aggarwal N, Deerhake ME, Inoue M, Nonaka T, Nozaki K, et al. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight (2022) 7(8). doi: 10.1172/jci.insight.155563
153. Gharagozloo M, Gris KV, Mahvelati T, Amrani A, Lukens JR, Gris D. NLR-dependent regulation of inflammation in multiple sclerosis. Front Immunol (2017) 8:2012. doi: 10.3389/fimmu.2017.02012
154. Geng YM, Wu BX. [One case of haemolysis after transcatheter closure of atrial septal defect]. Zhonghua Xin Xue Guan Bing Za Zhi (2012) 40(12):1062–3.
155. Cui Y, Yu H, Bu Z, Wen L, Yan L, Feng J. Focus on the role of the NLRP3 inflammasome in multiple sclerosis: Pathogenesis, diagnosis, and therapeutics. Front Mol Neurosci (2022) 15:894298. doi: 10.3389/fnmol.2022.894298
156. Keane RW, Dietrich WD, de Rivero Vaccari JP. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol (2018) 9:135. doi: 10.3389/fneur.2018.00135
157. Voo VTF, O'Brien T, Butzkueven H, Monif M. The role of vitamin d and P2X7r in multiple sclerosis. J Neuroimmunol (2019) 330:159–69. doi: 10.1016/j.jneuroim.2019.03.004
158. Tavazzi B, Batocchi AP, Amorini AM, Nociti V, D'Urso S, Longo S, et al. Serum metabolic profile in multiple sclerosis patients. Mult Scler Int (2011) 2011:167156. doi: 10.1155/2011/167156
159. Lazzarino G, Amorini AM, Eikelenboom MJ, Killestein J, Belli A, Di Pietro V, et al. Cerebrospinal fluid atp metabolites in multiple sclerosis. Mult Scler (2010) 16(5):549–54. doi: 10.1177/1352458510364196
160. Burger D, Molnarfi N, Weber MS, Brandt KJ, Benkhoucha M, Gruaz L, et al. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc Natl Acad Sci USA (2009) 106(11):4355–9. doi: 10.1073/pnas.0812183106
161. Mellergård J, Edström M, Vrethem M, Ernerudh J, Dahle C. Natalizumab treatment in multiple sclerosis: Marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler (2010) 16(2):208–17. doi: 10.1177/1352458509355068
162. Noroozi S, Meimand HAE, Arababadi MK, Nakhaee N, Asadikaram G. The effects of IFN-beta1 a on the expression of inflammasomes and apoptosis-associated speck-like proteins in multiple sclerosis patients. Mol Neurobiol (2017) 54(4):3031–7. doi: 10.1007/s12035-016-9864-8
163. Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol (2010) 185(2):974–81. doi: 10.4049/jimmunol.0904145
164. Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, et al. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PloS Pathog (2014) 10(1):e1003885. doi: 10.1371/journal.ppat.1003885
165. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37(1):96–107. doi: 10.1016/j.immuni.2012.07.006
166. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The caterpiller protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem (2005) 280(48):39914–24. doi: 10.1074/jbc.M502820200
167. Sun SC. Non-canonical NF-κB signaling pathway. Cell Res (2011) 21(1):71–85. doi: 10.1038/cr.2010.177
168. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/s0140-6736(16)30173-8
169. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: A review. JAMA (2018) 320(13):1360–72. doi: 10.1001/jama.2018.13103
170. Weyand CM, Wu B, Goronzy JJ. The metabolic signature of T cells in rheumatoid arthritis. Curr Opin Rheumatol (2020) 32(2):159–67. doi: 10.1097/bor.0000000000000683
171. Edilova MI, Akram A, Abdul-Sater AA. Innate immunity drives pathogenesis of rheumatoid arthritis. BioMed J (2021) 44(2):172–82. doi: 10.1016/j.bj.2020.06.010
172. Kotake S, Nanke Y, Yago T, Kawamoto M, Kobashigawa T, Yamanaka H. Elevated ratio of Th17 cell-derived Th1 cells (CD161(+)Th1 cells) to CD161(+)Th17 cells in peripheral blood of early-onset rheumatoid arthritis patients. BioMed Res Int (2016) 2016:4186027. doi: 10.1155/2016/4186027
173. Chen J, Li S, Shi J, Zhang L, Li J, Chen S, et al. Serum progranulin irrelated with Breg cell levels, but elevated in RA patients, reflecting high disease activity. Rheumatol Int (2016) 36(3):359–64. doi: 10.1007/s00296-015-3372-4
174. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers (2018) 4:18001. doi: 10.1038/nrdp.2018.1
175. Pap T, Dankbar B, Wehmeyer C, Korb-Pap A, Sherwood J. Synovial fibroblasts and articular tissue remodelling: Role and mechanisms. Semin Cell Dev Biol (2020) 101:140–5. doi: 10.1016/j.semCDb.2019.12.006
176. Deane KD, Holers VM. The natural history of rheumatoid arthritis. Clin Ther (2019) 41(7):1256–69. doi: 10.1016/j.clinthera.2019.04.028
177. Carubbi F, Alunno A, Gerli R, Giacomelli R. Post-translational modifications of proteins: Novel insights in the autoimmune response in rheumatoid arthritis. Cells (2019) 8(7). doi: 10.3390/cells8070657
178. Wu XY, Li KT, Yang HX, Yang B, Lu X, Zhao LD, et al. Complement C1q synergizes with Ptx3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J Autoimmun (2020) 106:102336. doi: 10.1016/j.jaut.2019.102336
179. Alippe Y, Mbalaviele G. Omnipresence of inflammasome activities in inflammatory bone diseases. Semin Immunopathol (2019) 41(5):607–18. doi: 10.1007/s00281-019-00753-4
180. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflammation (2017) 2017:3908061. doi: 10.1155/2017/3908061
181. Wahamaa H, Schierbeck H, Hreggvidsdottir HS, Palmblad K, Aveberger AC, Andersson U, et al. High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther (2011) 13(4):R136. doi: 10.1186/ar3450
182. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature (2019) 570(7760):246–51. doi: 10.1038/s41586-019-1263-7
183. Lochhead RB, Ordoñez D, Arvikar SL, Aversa JM, Oh LS, Heyworth B, et al. Interferon-gamma production in Lyme arthritis synovial tissue promotes differentiation of fibroblast-like synoviocytes into immune effector cells. Cell Microbiol (2019) 21(2):e12992. doi: 10.1111/cmi.12992
184. Mousavi MJ, Karami J, Aslani S, Tahmasebi MN, Vaziri AS, Jamshidi A, et al. Transformation of fibroblast-like synoviocytes in rheumatoid arthritis; from a friend to foe. Auto Immun Highlights (2021) 12(1):3. doi: 10.1186/s13317-020-00145-x
185. Morgan R, Endres J, Behbahani-Nejad N, Phillips K, Ruth JH, Friday SC, et al. Expression and function of aminopeptidase N/CD13 produced by fibroblast-like synoviocytes in rheumatoid arthritis: Role of CD13 in chemotaxis of cytokine-activated T cells independent of enzymatic activity. Arthritis Rheumatol (2015) 67(1):74–85. doi: 10.1002/art.38878
186. Harada S, Yamamura M, Okamoto H, Morita Y, Kawashima M, Aita T, et al. Production of interleukin-7 and interleukin-15 by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum (1999) 42(7):1508–16. doi: 10.1002/1529-0131(199907)42:7<1508::Aid-anr26>3.0.Co;2-l
187. Liu R, Zhao P, Zhang Q, Che N, Xu L, Qian J, et al. Adiponectin promotes fibroblast-like synoviocytes producing IL-6 to enhance T follicular helper cells response in rheumatoid arthritis. Clin Exp Rheumatol (2020) 38(1):11–8.
188. Eljaafari A, Tartelin ML, Aissaoui H, Chevrel G, Osta B, Lavocat F, et al. Bone marrow-derived and synovium-derived mesenchymal cells promote Th17 cell expansion and activation through caspase 1 activation: Contribution to the chronicity of rheumatoid arthritis. Arthritis Rheum (2012) 64(7):2147–57. doi: 10.1002/art.34391
189. Lin J, Zhou Z, Huo R, Xiao L, Ouyang G, Wang L, et al. Cyr61 induces IL-6 production by fibroblast-like synoviocytes promoting Th17 differentiation in rheumatoid arthritis. J Immunol (2012) 188(11):5776–84. doi: 10.4049/jimmunol.1103201
190. Lee DG, Woo JW, Kwok SK, Cho ML, Park SH. Mrp8 promotes Th17 differentiation via upregulation of il-6 production by fibroblast-like synoviocytes in rheumatoid arthritis. Exp Mol Med (2013) 45(4):e20. doi: 10.1038/emm.2013.39
191. Wang B, Ma Z, Wang M, Sun X, Tang Y, Li M, et al. IL-34 upregulated Th17 production through increased IL-6 expression by rheumatoid fibroblast-like synoviocytes. Mediators Inflammation (2017) 2017:1567120. doi: 10.1155/2017/1567120
192. Gao S, Qi X, Li J, Sang L. Upregulated Kat7 in synovial fibroblasts promotes Th17 cell differentiation and infiltration in rheumatoid arthritis. Biochem Biophys Res Commun (2017) 489(2):235–41. doi: 10.1016/j.bbrc.2017.05.143
193. Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum (2007) 56(5):1497–506. doi: 10.1002/art.22573
194. Jäger E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun (2020) 11(1):4243. doi: 10.1038/s41467-020-17749-6
195. Mukherjee S, Kumar R, Tsakem Lenou E, Basrur V, Kontoyiannis DL, Ioakeimidis F, et al. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat Immunol (2020) 21(6):626–35. doi: 10.1038/s41590-020-0681-x
196. Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, et al. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum (2009) 60(12):3651–62. doi: 10.1002/art.25006
197. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430(7000):694–9. doi: 10.1038/nature02794
198. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/jci94549
199. Clark M, Kroger CJ, Tisch RM. Type 1 diabetes: A chronic anti-self inflammatory response. Front Immunol (2017) 8:1898. doi: 10.3389/fimmu.2017.01898
200. Tisch R, MCDevitt H. Insulin-dependent diabetes mellitus. Cell (1996) 85(3):291–7. doi: 10.1016/s0092-8674(00)81106-x
201. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91(1):79–118. doi: 10.1152/physrev.00003.2010
202. Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(11). doi: 10.1101/cshperspect.a007641
203. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers (2017) 3:17016. doi: 10.1038/nrdp.2017.16
204. Pearson JA, Agriantonis A, Wong FS, Wen L. Modulation of the immune system by the gut microbiota in the development of type 1 diabetes. Hum Vaccin Immunother (2018) 14(11):2580–96. doi: 10.1080/21645515.2018.1514354
205. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455(7216):1109–13. doi: 10.1038/nature07336
206. Lebreton F, Berishvili E, Parnaud G, Rouget C, Bosco D, Berney T, et al. NLRP3 inflammasome is expressed and regulated in human islets. Cell Death Dis (2018) 9(7):726. doi: 10.1038/s41419-018-0764-x
207. Carlos D, Costa FR, Pereira CA, Rocha FA, Yaochite JN, Oliveira GG, et al. Mitochondrial DNA activates the NLRP3 inflammasome and predisposes to type 1 diabetes in murine model. Front Immunol (2017) 8:164. doi: 10.3389/fimmu.2017.00164
208. Pereira CA, Carlos D, Ferreira NS, Silva JF, Zanotto CZ, Zamboni DS, et al. Mitochondrial DNA promotes NLRP3 inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes. Front Physiol (2019) 10:1557. doi: 10.3389/fphys.2019.01557
209. Schott WH, Haskell BD, Tse HM, Milton MJ, Piganelli JD, Choisy-Rossi CM, et al. Caspase-1 is not required for type 1 diabetes in the nod mouse. Diabetes (2004) 53(1):99–104. doi: 10.2337/diabetes.53.1.99
210. Wen L, Green EA, Stratmann T, Panosa A, Gomis R, Eynon EE, et al. In vivo diabetogenic action of CD4+ T lymphocytes requires fas expression and is independent of IL-1 and IL-18. Eur J Immunol (2011) 41(5):1344–51. doi: 10.1002/eji.201041216
211. Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, et al. IL-1 receptor deficiency slows progression to diabetes in the nod mouse. Diabetes (2004) 53(1):113–21. doi: 10.2337/diabetes.53.1.113
212. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through toll-like receptors. Proc Natl Acad Sci USA (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
213. Pearson JA, Kakabadse D, Davies J, Peng J, Warden-Smith J, Cuff S, et al. Altered gut microbiota activate and expand insulin B15-23-reactive CD8+ T cells. Diabetes (2019) 68(5):1002–13. doi: 10.2337/db18-0487
214. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol (2017) 18(5):552–62. doi: 10.1038/ni.3713
215. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi: 10.1126/science.1233521
216. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi: 10.1016/j.cell.2011.04.022
217. Fattinger SA, Geiser P, Samperio Ventayol P, Di Martino ML, Furter M, Felmy B, et al. Epithelium-autonomous Naip/NLRc4 prevents TNF-driven inflammatory destruction of the gut epithelial barrier in Salmonella-infected mice. Mucosal Immunol (2021) 14(3):615–29. doi: 10.1038/s41385-021-00381-y
218. Leber A, Hontecillas R, Zoccoli-Rodriguez V, Bienert C, Chauhan J, Bassaganya-Riera J. Activation of NLRX1 by NX-13 alleviates inflammatory bowel disease through immunometabolic mechanisms in CD4+ T cells. J Immunol (2019) 203(12):3407–15. doi: 10.4049/jimmunol.1900364
219. Leber A, Hontecillas R, Tubau-Juni N, Zoccoli-Rodriguez V, Abedi V, Bassaganya-Riera J. NLRX1 modulates immunometabolic mechanisms controlling the host-gut microbiota interactions during inflammatory bowel disease. Front Immunol (2018) 9:363. doi: 10.3389/fimmu.2018.00363
220. Truax AD, Chen L, Tam JW, Cheng N, Guo H, Koblansky AA, et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe (2018) 24(3):364–78.e6. doi: 10.1016/j.chom.2018.08.009
221. Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol (2017) 18(5):541–51. doi: 10.1038/ni.3690
222. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12(12):716–30. doi: 10.1038/nrrheum.2016.186
223. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: A cellular perspective. Trends Mol Med (2017) 23(7):615–35. doi: 10.1016/j.molmed.2017.05.006
224. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med (2016) 14(1):156. doi: 10.1186/s12967-016-0911-z
225. Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self double-stranded (ds)DNA induces IL-1beta production from human monocytes by activating NLRP3 inflammasome in the presence of anti-(ds)DNA antibodies. J Immunol (2013) 190(4):1407–15. doi: 10.4049/jimmunol.1201195
226. Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, et al. U1-small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol (2012) 188(10):4769–75. doi: 10.4049/jimmunol.1103355
227. Shin MS, Kang Y, Wahl ER, Park HJ, Lazova R, Leng L, et al. Macrophage migration inhibitory factor regulates U1 small nuclear rnp immune complex-mediated activation of the NLRP3 inflammasome. Arthritis Rheumatol (2019) 71(1):109–20. doi: 10.1002/art.40672
228. Miao EA, Rajan JV, Aderem A. Caspase-1-Induced pyroptotic cell death. Immunol Rev (2011) 243(1):206–14. doi: 10.1111/j.1600-065X.2011.01044.x
229. Pisetsky DS, Lipsky PE. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat Rev Rheumatol (2020) 16(10):565–79. doi: 10.1038/s41584-020-0480-7
230. Brinkmann V, Zychlinsky A. Beneficial suicide: Why neutrophils die to make nets. Nat Rev Microbiol (2007) 5(8):577–82. doi: 10.1038/nrmicro1710
231. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
232. Eloranta ML, Ronnblom L. Cause and consequences of the activated type I interferon system in SLE. J Mol Med (Berl) (2016) 94(10):1103–10. doi: 10.1007/s00109-016-1421-4
233. Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal (2012) 5(225):ra38. doi: 10.1126/scisignal.2002767
234. Zhao J, Wang H, Dai C, Wang H, Zhang H, Huang Y, et al. P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/Caspase 1 pathway. Arthritis Rheum (2013) 65(12):3176–85. doi: 10.1002/art.38174
235. Zhu FG, Jiang W, Bhagat L, Wang D, Yu D, Tang JX, et al. A novel antagonist of toll-like receptors 7, 8 and 9 suppresses lupus disease-associated parameters in NZBW/F1 mice. Autoimmunity (2013) 46(7):419–28. doi: 10.3109/08916934.2013.798651
236. Zhao J, Wang J, Zhou M, Li M, Li M, Tan H. Curcumin attenuates murine lupus via inhibiting NLRP3 inflammasome. Int Immunopharmacol (2019) 69:213–6. doi: 10.1016/j.intimp.2019.01.046
237. Li D, Shi G, Wang J, Zhang D, Pan Y, Dou H, et al. Baicalein ameliorates pristane-induced lupus nephritis via activating Nrf2/Ho-1 in myeloid-derived suppressor cells. Arthritis Res Ther (2019) 21(1):105. doi: 10.1186/s13075-019-1876-0
238. Lu A, Li H, Niu J, Wu S, Xue G, Yao X, et al. Hyperactivation of the NLRP3 inflammasome in myeloid cells leads to severe organ damage in experimental lupus. J Immunol (2017) 198(3):1119–29. doi: 10.4049/jimmunol.1600659
239. Nawata A, Hisano S, Shimajiri S, Wang KY, Tanaka Y, Nakayama T. Podocyte and endothelial cell injury lead to nephrotic syndrome in proliferative lupus nephritis. Histopathology (2018) 72(7):1084–92. doi: 10.1111/his.13454
240. Lech M, Lorenz G, Kulkarni OP, Grosser MO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-beta receptor signalling. Ann Rheum Dis (2015) 74(12):2224–35. doi: 10.1136/annrheumdis-2014-205496
241. Panchanathan R, Xin H, Choubey D. Disruption of mutually negative regulatory feedback loop between interferon-inducible p202 protein and the E2f family of transcription factors in lupus-prone mice. J Immunol (2008) 180(9):5927–34. doi: 10.4049/jimmunol.180.9.5927
242. Haywood ME, Rose SJ, Horswell S, Lees MJ, Fu G, Walport MJ, et al. Overlapping BXSB congenic intervals, in combination with microarray gene expression, reveal novel lupus candidate genes. Genes Immun (2006) 7(3):250–63. doi: 10.1038/sj.gene.6364294
243. Yin Q, Sester DP, Tian Y, Hsiao YS, Lu A, Cridland JA, et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep (2013) 4(2):327–39. doi: 10.1016/j.celrep.2013.06.024
244. Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozören N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host IPAF. J Biol Chem (2006) 281(46):35217–23. doi: 10.1074/jbc.M604933200
245. Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT. Coordinated host responses during pyroptosis: Caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J Immunol (2011) 187(5):2748–54. doi: 10.4049/jimmunol.1100477
246. Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity (2012) 36(3):388–400. doi: 10.1016/j.immuni.2012.01.018
247. Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell (2008) 132(5):818–31. doi: 10.1016/j.cell.2007.12.040
248. Luzio JP, Pryor PR, Bright NA. Lysosomes: Fusion and function. Nat Rev Mol Cell Biol (2007) 8(8):622–32. doi: 10.1038/nrm2217
249. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA (2011) 108(49):19725–30. doi: 10.1073/pnas.1108586108
250. Syed FM, Hahn HS, Odley A, Guo Y, Vallejo JG, Lynch RA, et al. Proapoptotic effects of caspase-1/Interleukin-Converting enzyme dominate in myocardial ischemia. Circ Res (2005) 96(10):1103–9. doi: 10.1161/01.RES.0000166925.45995.ed
251. Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, et al. Fundamental role of the Rip2/Caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci USA (2003) 100(26):16012–7. doi: 10.1073/pnas.2534856100
252. Javanmard Khameneh H, Leong KWK, Mencarelli A, Vacca M, Mambwe B, Neo K, et al. The inflammasome adaptor ASC intrinsically limits CD4(+) T-cell proliferation to help maintain intestinal homeostasis. Front Immunol (2019) 10:1566. doi: 10.3389/fimmu.2019.01566
253. Bruchard M, Rebe C, Derangere V, Togbe D, Ryffel B, Boidot R, et al. The receptor NLRP3 is a transcriptional regulator of Th2 differentiation. Nat Immunol (2015) 16(8):859–70. doi: 10.1038/ni.3202
254. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T Cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74(1):5–17. doi: 10.1016/j.cyto.2014.09.011
255. Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2ra variation lowers il-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188(9):4644–53. doi: 10.4049/jimmunol.1100272
256. Chou WC, Guo Z, Guo H, Chen L, Zhang G, Liang K, et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature (2021) 591(7849):300–5. doi: 10.1038/s41586-021-03231-w
257. Ziegler SF, Buckner JH. Foxp3 and the regulation of Treg/Th17 differentiation. Microbes Infect (2009) 11(5):594–8. doi: 10.1016/j.micinf.2009.04.002
258. Wu H, Deng Y, Long D, Yang M, Li Q, Feng Y, et al. The IL-21-Tet2-AIM2-C-Maf pathway drives the T follicular helper cell response in lupus-like disease. Clin Transl Med (2022) 12(3):e781. doi: 10.1002/ctm2.781
259. Yang M, Long D, Hu L, Zhao Z, Li Q, Guo Y, et al. AIM2 deficiency in b cells ameliorates systemic lupus erythematosus by regulating Blimp-1-Bcl-6 axis-mediated B-cell differentiation. Signal Transduct Target Ther (2021) 6(1):341. doi: 10.1038/s41392-021-00725-x
260. Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and AKT. Nat Med (2015) 21(8):906–13. doi: 10.1038/nm.3908
261. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
262. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
263. Pan RY, Kong XX, Cheng Y, Du L, Wang ZC, Yuan C, et al. 1,2,4-trimethoxybenzene selectively inhibits NLRP3 inflammasome activation and attenuates experimental autoimmune encephalomyelitis. Acta Pharmacol Sin (2021) 42(11):1769–79. doi: 10.1038/s41401-021-00613-8
264. Sanchez-Fernandez A, Skouras DB, Dinarello CA, Lopez-Vales R. Olt1177 (Dapansutrile), a selective NLRP3 inflammasome inhibitor, ameliorates experimental autoimmune encephalomyelitis pathogenesis. Front Immunol (2019) 10:2578. doi: 10.3389/fimmu.2019.02578
265. Marchetti C, Swartzwelter B, Koenders MI, Azam T, Tengesdal IW, Powers N, et al. NLRP3 inflammasome inhibitor Olt1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res Ther (2018) 20(1):169. doi: 10.1186/s13075-018-1664-2
266. Chen Y, He H, Lin B, Chen Y, Deng X, Jiang W, et al. Rrx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell Mol Immunol (2021) 18(6):1425–36. doi: 10.1038/s41423-021-00683-y
267. Guo C, Fulp JW, Jiang Y, Li X, Chojnacki JE, Wu J, et al. Development and characterization of a hydroxyl-sulfonamide analogue, 5-Chloro-N-[2-(4-Hydroxysulfamoyl-Phenyl)-Ethyl]-2-Methoxy-Benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci (2017) 8(10):2194–201. doi: 10.1021/acschemneuro.7b00124
268. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10(4). doi: 10.15252/emmm.201708689
269. Peng X, Yang T, Liu G, Liu H, Peng Y, He L. Piperine ameliorated lupus nephritis by targeting Ampk-mediated activation of NLRP3 inflammasome. Int Immunopharmacol (2018) 65:448–57. doi: 10.1016/j.intimp.2018.10.025
270. Ka SM, Lin JC, Lin TJ, Liu FC, Chao LK, Ho CL, et al. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arthritis Res Ther (2015) 17:331. doi: 10.1186/s13075-015-0844-6
271. Shen C, Xu M, Xu S, Zhang S, Lin W, Li H, et al. Myricitrin inhibits fibroblast-like synoviocyte-mediated rheumatoid synovial inflammation and joint destruction by targeting AIM2. Front Pharmacol (2022) 13:905376. doi: 10.3389/fphar.2022.905376
272. Desu HL, Plastini M, Illiano P, Bramlett HM, Dietrich WD, de Rivero Vaccari JP, et al. Ic100: A novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J Neuroinflamm (2020) 17(1):143. doi: 10.1186/s12974-020-01826-0
273. Chen C, Zhou Y, Ning X, Li S, Xue D, Wei C, et al. Directly targeting ASC by lonidamine alleviates inflammasome-driven diseases. J Neuroinflamm (2022) 19(1):315. doi: 10.1186/s12974-022-02682-w
274. Liu W, Yang J, Fang S, Jiao C, Gao J, Zhang A, et al. Spirodalesol analog 8a inhibits NLRP3 inflammasome activation and attenuates inflammatory disease by directly targeting adaptor protein ASC. J Biol Chem (2022) 298(12):102696. doi: 10.1016/j.jbc.2022.102696
275. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci USA (2018) 115(26):E6065–e74. doi: 10.1073/pnas.1722041115
276. Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage (2003) 11(10):738–46. doi: 10.1016/s1063-4584(03)00153-5
277. Zhang X, Zhang Y, Wang F, Liu Y, Yong VW, Xue M. Necrosulfonamide alleviates acute brain injury of intracerebral hemorrhage via inhibiting inflammation and necroptosis. Front Mol Neurosci (2022) 15:916249. doi: 10.3389/fnmol.2022.916249
278. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science (2020) 369(6511):1633–7. doi: 10.1126/science.abb9818
279. Lang Y, Chu F, Liu L, Zheng C, Li C, Shen D, et al. Potential role of Bay11-7082, a NF-kappaB blocker inhibiting experimental autoimmune encephalomyelitis in C57BL/6j mice via declining NLRP3 inflammasomes. Clin Exp Immunol (2021) 207(3):378–86. doi: 10.1093/cei/uxab022
280. Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-κB activation. Int Immunopharmacol (2013) 17(1):116–22. doi: 10.1016/j.intimp.2013.05.027
281. Pang Z, Wang G, Ran N, Lin H, Wang Z, Guan X, et al. Inhibitory effect of methotrexate on rheumatoid arthritis inflammation and comprehensive metabolomics analysis using ultra-performance liquid chromatography-quadrupole time of flight-mass spectrometry (Uplc-Q/Tof-Ms). Int J Mol Sci (2018) 19(10). doi: 10.3390/ijms19102894
282. Su B, Ye H, You X, Ni H, Chen X, Li L. Icariin alleviates murine lupus nephritis via inhibiting NF-κB activation pathway and NLRP3 inflammasome. Life Sci (2018) 208:26–32. doi: 10.1016/j.lfs.2018.07.009
283. Nikfar S, Saiyarsarai P, Tigabu BM, Abdollahi M. Efficacy and safety of interleukin-1 antagonists in rheumatoid arthritis: A systematic review and meta-analysis. Rheumatol Int (2018) 38(8):1363–83. doi: 10.1007/s00296-018-4041-1
284. Alten R, Gomez-Reino J, Durez P, Beaulieu A, Sebba A, Krammer G, et al. Efficacy and safety of the human anti-IL-1β monoclonal antibody canakinumab in rheumatoid arthritis: Results of a 12-week, phase ii, dose-finding study. BMC Musculoskelet Disord (2011) 12:153. doi: 10.1186/1471-2474-12-153
285. Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet (2013) 381(9881):1905–15. doi: 10.1016/s0140-6736(13)60023-9
286. Seelig E, Timper K, Falconnier C, Stoeckli R, Bilz S, Oram R, et al. Interleukin-1 antagonism in type 1 diabetes of long duration. Diabetes Metab (2016) 42(6):453–6. doi: 10.1016/j.diabet.2016.08.005
287. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. Mcc950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15(6):556–9. doi: 10.1038/s41589-019-0277-7
288. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, et al. NLRP3 inflammasome inhibition by Mcc950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb VASC Biol (2017) 37(8):1457–61. doi: 10.1161/atvbaha.117.309575
289. Zhang C, Zhu X, Li L, Ma T, Shi M, Yang Y, et al. A small molecule inhibitor Mcc950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab Syndr Obes (2019) 12:1297–309. doi: 10.2147/dmso.s199802
290. Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, et al. Mcc950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep (2018) 8(1):8618. doi: 10.1038/s41598-018-26775-w
291. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental Nash in mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
292. Wang L, Lei W, Zhang S, Yao L. Mcc950, a NLRP3 inhibitor, ameliorates lipopolysaccharide-induced lung inflammation in mice. Bioorg Med Chem (2021) 30:115954. doi: 10.1016/j.bmc.2020.115954
293. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17(8):588–606. doi: 10.1038/nrd.2018.97
294. Ltd IU. Safety and tolerability, pharmacokinetic and pharmacodynamic study with inzomelid. (2019).
295. Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ (2005) 12 Suppl 1:942–61. doi: 10.1038/sj.CDd.4401556
296. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
297. Imani D, Azimi A, Salehi Z, Rezaei N, Emamnejad R, Sadr M, et al. Association of NOD-like receptor protein-3 single nucleotide gene polymorphisms and expression with the susceptibility to relapsing-remitting multiple sclerosis. Int J Immunogenet (2018) 45(6):329–36. doi: 10.1111/iji.12401
298. Jiang Q, Wang X, Huang E, Wang Q, Wen C, Yang G, et al. Inflammasome and its therapeutic targeting in rheumatoid arthritis. Front Immunol (2021) 12:816839. doi: 10.3389/fimmu.2021.816839
299. Sode J, Vogel U, Bank S, Andersen PS, Thomsen MK, Hetland ML, et al. Anti-TNF treatment response in rheumatoid arthritis patients is associated with genetic variation in the NLRP3-inflammasome. PloS One (2014) 9(6):e100361. doi: 10.1371/journal.pone.0100361
300. Kastbom A, Verma D, Eriksson P, Skogh T, Wingren G, Söderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish tira project). Rheumatol (Oxford) (2008) 47(4):415–7. doi: 10.1093/rheumatology/kem372
301. Shaw PJ, MCDermott MF, Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med (2011) 17(2):57–64. doi: 10.1016/j.molmed.2010.11.001
302. Wu Z, Wu S, Liang T. Association of NLRP3 Rs35829419 and Rs10754558 polymorphisms with risks of autoimmune diseases: A systematic review and meta-analysis. Front Genet (2021) 12:690860. doi: 10.3389/fgene.2021.690860
303. Smigoc Schweiger D, Goricar K, Hovnik T, Mendez A, Bratina N, Brecelj J, et al. Dual role of Ptpn22 but not NLRP3 inflammasome polymorphisms in type 1 diabetes and celiac disease in children. Front Pediatr (2019) 7:63. doi: 10.3389/fped.2019.00063
304. Griess K, Rieck M, Müller N, Karsai G, Hartwig S, Pelligra A, et al. Sphingolipid subtypes differentially control proinsulin processing and systemic glucose homeostasis. Nat Cell Biol (2023) 25(1):20–9. doi: 10.1038/s41556-022-01027-2
Keywords: autoimmunity, self-tolerance, inflammasomes, immunoregulation, inflammation
Citation: Ke Q, Greenawalt AN, Manukonda V, Ji X and Tisch RM (2023) The regulation of self-tolerance and the role of inflammasome molecules. Front. Immunol. 14:1154552. doi: 10.3389/fimmu.2023.1154552
Received: 30 January 2023; Accepted: 17 March 2023;
Published: 04 April 2023.
Edited by:
Joanna Davies, San Diego Biomedical Research Institute, United StatesReviewed by:
Andreas B. Wild, Universtätsklinikum Erlangen, GermanyJianan Zhao, Shanghai University of Traditional Chinese Medicine, China
Copyright © 2023 Ke, Greenawalt, Manukonda, Ji and Tisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland Michael Tisch, rmtisch@med.unc.edu