 Chen Li1†
Chen Li1† Yan Zhang2†
Yan Zhang2† Qiulong Yan3†
Qiulong Yan3† Ruochun Guo4†
Ruochun Guo4† Changming Chen5†
Changming Chen5† Shenghui Li4
Shenghui Li4 Yue Zhang3
Yue Zhang3 Jinxin Meng4Jie Ma6Wei You7Zhisong Wu8
Jinxin Meng4Jie Ma6Wei You7Zhisong Wu8 Wen Sun6,9,10*
Wen Sun6,9,10*- 1Department of Rheumatology, Fangshan Hospital, Beijing University of Chinese Medicine, Beijing, China
- 2Department of Traditional Chinese Medicine, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 3Department of Microbiology, College of Basic Medical Sciences, Dalian Medical University, Dalian, China
- 4Puensum Genetech Institute, Wuhan, China
- 5Department of Rheumatology and Immunology, The Second Affiliated Hospital of Guizhou University of Traditional Chinese Medicine, Guiyang, China
- 6School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China
- 7Beijing Key Laboratory of Acupuncture Neuromodulation, Department of Acupuncture and Moxibustion, Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, Beijing, China
- 8Department of Intensive Care Medicine, Dongfang Hospital Beijing University of Chinese Medicine, Beijing, China
- 9Key Laboratory of Health Cultivation of the Ministry of Education, Beijing University of Chinese Medicine, Beijing, China
- 10Beijing Key Laboratory of Health Cultivation, Beijing University of Chinese Medicine, Beijing, China
Introduction: Ankylosing spondylitis (AS), a chronic autoimmune disease, has been linked to the gut bacteriome.
Methods: To investigate the characteristics of the gut virome in AS, we profiled the gut viral community of 193 AS patients and 59 healthy subjects based on a metagenome-wide analysis of fecal metagenomes from two publicly available datasets.
Results: AS patients revealed a significant decrease in gut viral richness and a considerable alteration of the overall viral structure. At the family level, AS patients had an increased abundance of Gratiaviridae and Quimbyviridae and a decreased abundance of Drexlerviridae and Schitoviridae. We identified 1,004 differentially abundant viral operational taxonomic units (vOTUs) between patients and controls, including a higher proportion of AS-enriched Myoviridae viruses and control-enriched Siphoviridae viruses. Moreover, the AS-enriched vOTUs were more likely to infect bacteria such as Flavonifractor, Achromobacter, and Eggerthellaceae, whereas the control-enriched vOTUs were more likely to be Blautia, Ruminococcus, Collinsella, Prevotella, and Faecalibacterium bacteriophages. Additionally, some viral functional orthologs differed significantly in frequency between the AS-enriched and control-enriched vOTUs, suggesting the functional role of these AS-associated viruses. Moreover, we trained classification models based on gut viral signatures to discriminate AS patients from healthy controls, with an optimal area under the receiver operator characteristic curve (AUC) up to 0.936, suggesting the clinical potential of the gut virome for diagnosing AS.
Discussion: This work provides novel insight into the AS gut virome, and the findings may guide future mechanistic and therapeutic studies for other autoimmune diseases.
Introduction
Ankylosing spondylitis (AS) is a chronic autoimmune disease that mainly affects the sacroiliac joints and the spine; ankylosis and spinal deformity can occur in severe cases (1). The prevalence rate of AS in mainland China is approximately 0.29%, and the ratio of males to females is approximately 2~3:1. The disease often occurs in people aged 20~30 years old and is rare in those over 40 years and under 8 years old (2). AS can lead to physiological injury and disability (3), resulting in loss of physical function, decreased quality of life, and heavy financial burden on patients, families, and society (4).
Current studies suggest that AS is a genetic disease that is closely associated with HLA-B27 (5). The pathogenesis of AS includes host genetic factors and environmental triggers, such as the gut microbiota (6). Increasing evidence also linked AS to some gastrointestinal disorders, such as irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) (7, 8). AS and IBD share similarities in terms of genetic risk factors and pathogenesis, and interaction with the gut microbiome is closely related to the pathogenesis of IBD (7). Therefore, AS may also be a gut microbiome-driven disease. The gut microbiome is considered to play an important role in the human defense system, with a wide range of physiological roles. Disturbance of interactions between the immune system and the gut microbiome may induce complex pathological changes (9). Clinical studies have shown that up to 70% of patients with AS have subclinical intestinal inflammation, and 5~10% of these patients will develop IBD, which is the common extra-articular manifestation of AS (10). In addition, a study of 211 Chinese subjects showed that AS was characterized by severe gut dysbiosis, increased relative abundances of Prevotella melaninogenica, Prevotella copri, and Prevotella sp. C561, and decreased Bacteroides spp (11). Another study showed that the microbiota characteristics of AS patients included increased Proteobacteria and decreased Bacteroidetes abundances, which was due to enrichment of Escherichia-Shigella, Veillonella, Lachnospiraceae NK4A136 group and reduction of Prevotella strain 9, Megamonas, and Fusobacterium (12). These studies suggest that the gut microbiota is associated with AS and may even be one of the critical inducing factors of AS. As the gut microbiota contains multiple components, including bacteria, fungi, and viruses, an obvious gap in knowledge is the role each component plays.
Although studies to date have focused on gut bacteria as one of the main components of the human gut, gut viruses also deserve attention. The human gut viral community is mainly composed of bacteriophages, most of which remain undiscovered, and very little is known about their role in the formation of the gut microbiome and its impact on human health. The virome is closely related to the gut microbiota, and recent studies have highlighted the association between the gut virome and many diseases, especially IBD (13), autoimmune diseases (14), and type 2 diabetes mellitus (15). Furthermore, evidence that the virome directly affects human health through the immune system has been found (16, 17). More importantly, our previous study has preliminary showed significant changes in the gut virome associated with several immune diseases, including AS (18). Given these findings, it makes sense to investigate the association between AS and the virome in more detail.
Overall, some viruses with low abundance or that are difficult to sequence might not be detected through metagenomic sequencing technology. However, gut virome studies can be performed by virus-like particle (VLP) metagenomic sequencing (19–21). By combining metagenomic and VLP metagenomic sequencing analyses, researchers can obtain a highly comprehensive map of the gut virus population. In this study, to identify changes in the gut virome in AS, we reanalyzed raw data from publicly available metagenomic data for two cohorts, which included a total of 252 human fecal samples (193 AS patients and 59 healthy subjects). Specifically, we compared the viral composition of AS patients with that of healthy individuals based on metagenome-based sequencing data and investigated the relationship between viruses and bacteria. In light of the exploration of the gut virome of AS patients, a better understanding of the etiology and pathogenesis of AS will contribute to developing prevention and treatment strategies for AS from new perspectives.
Methods
Sample information
This study involved 252 fecal metagenomic sequencing samples from two public cohorts (Table S1). The first cohort (cohort A) was recruited from Guangzhou, South China, and comprised 113 patients with AS and 37 healthy controls (HCs) (22). The other cohort (cohort B) included 80 AS patients and 22 HCs living in Beijing, North China, after removing 40 HC samples from another study to avoid batch effects (23). The sample metadata of cohort B is available in Zhou et al. study. For cohort B, AS patients were treatment-naive individuals, and had no significant difference in age (Student’s t test, p value = 0.87) and body mass index (Student’s t test, p value = 0.63) compared with HCs.
Preprocessing of metagenomic datasets
The raw metagenomic datasets were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with project accession IDs PRJEB28545 and PRJEB29373, respectively. The downloaded raw reads were filtered using fastp v0.20.1 (24) with the options ‘-l 90 -q 20 -u 30 -y –trim_poly_g’ to remove low-quality reads. The filtered reads were mapped to the GRCh38 reference genome using bowtie2 v2.4.1 (25) with default options to remove human reads. Finally, we obtained an average of 8.98 Gb clean data per sample (after quality control and human read removal).
Gut viral reference
To analyze the composition of the gut viral community in metagenomic samples, we used a gut virus catalog comprising over 67,000 nonredundant viral operational taxonomic units (vOTUs) as the reference. This Chinese gut virus catalog (cnGVC) was constructed from over 10,000 publicly available fecal metagenomes from the Chinese population (including all samples used in the current study) (18). Briefly, the clean reads of each metagenomic sample were assembled into contigs using Megahit v1.2.9 (26). Only contigs ≥5 kb in length were used to identify viral sequences in each sample. Identification, decontamination, and dereplication of viral sequences were performed according to our prior study (27). The quality of viral sequences was estimated using CheckV v0.7.0 (28). Taxonomic classification, host prediction, and functional annotation of viral sequences were also performed according to our prior study (18).
Taxonomic profiling of the gut virome
To generate taxonomic profiles of gut viral communities, the clean reads in each metagenomic sample were mapped to cnGVC using bowtie2 v2.4.1 with options ‘–end-to-end –fast –no-unal’. The total mapping reads of all samples were randomly subsampled to the same sequencing amount (1.2 million). The relative abundance of each vOTU was measured as the ratio of the number of reads mapped to the vOTU to the total number of reads mapped to any vOTU in each metagenome. In addition, the relative abundance of each viral family was generated by summing the relative abundances of vOTUs annotated with the corresponding family.
Statistical analyses
Statistical analysis and visualization were implemented in R v4.0.3 (29).
Alpha and beta diversities
Gut virome diversity was estimated based on the profiles at the vOTU level. The observed number of vOTUs was measured as the number of vOTUs with the relative abundance of >0. Shannon and Simpson diversity indices were measured by the diversity function in the vegan package. Bray-Curtis distances between samples were measured by the vegdist function in the vegan package. Principal coordinate analysis (PCoA) of Bray-Curtis distances was carried out via the pcoa function in the ape package. Permutational multivariate analysis of variance (PERMANOVA) was carried out using the adonis function in the vegan package.
Identification of viral markers
First, the identification of viral markers at the vOTU-level was performed between AS patients and healthy controls in each cohort using the wilcox.test function. We further performed Fisher’s method for combining p-values from independent tests in two cohorts by the sumlog function in the metap package. The combined p-values were adjusted via the p.adjust function with the option ‘method=BH’. A vOTU with adjusted p-value (FDR) of < 0.05 was recognized as a AS-associated viral marker.
Comparison analysis of functions between AS- and HC-enriched vOTUs
The occurrence rate of each KO in a group was measured as the number of vOTUs with this KO divided by the total number of vOTUs in the group. Fisher’s exact test was performed using the fisher.test function to determine whether the occurrence rate of each KO differed significantly between two groups. Then the obtained p-values were adjusted via the p.adjust function with the option ‘method=BH’. A KO with adjusted p-value (FDR) of < 0.05 was deemed to be significantly different between two groups.
Classification models
The intra-cohort models based on AS-associated viral markers were constructed using the randomForest function followed by 5 times of 5 fivefold cross-validations, and their performances were evaluated based on AUC scores calculated by the roc function in the pROC package. The cross-dataset model was evaluated by training it on one cohort and testing it on the other. The importance ranking of viral markers was obtained by the importance function.
Results
Study population and gut virome composition
To establish the composition of the gut viral community, we mapped clean reads of each sample to the cnGVC database, including 67,096 dereplicated vOTUs with high completeness (18). The rates of reads mapping to cnGVC ranged from 12.68% to 30.24% (Figure 1A), suggesting efficient capture of gut viral reads. Notably, 19,844 of 67,096 vOTUs accounted for 99% of mapping reads across all samples (Figure 1B) and were further used to characterize the gut viral composition of the two cohorts. Similar to previous studies (30, 31), the dominant families in the gut viral composition included Siphoviridae (mean relative abundance, 13.7%), Myoviridae (6.4%), Quimbyviridae (6.1%), and crAss-like viruses (1.4%), in addition to unclassified viruses (Figure 1C).
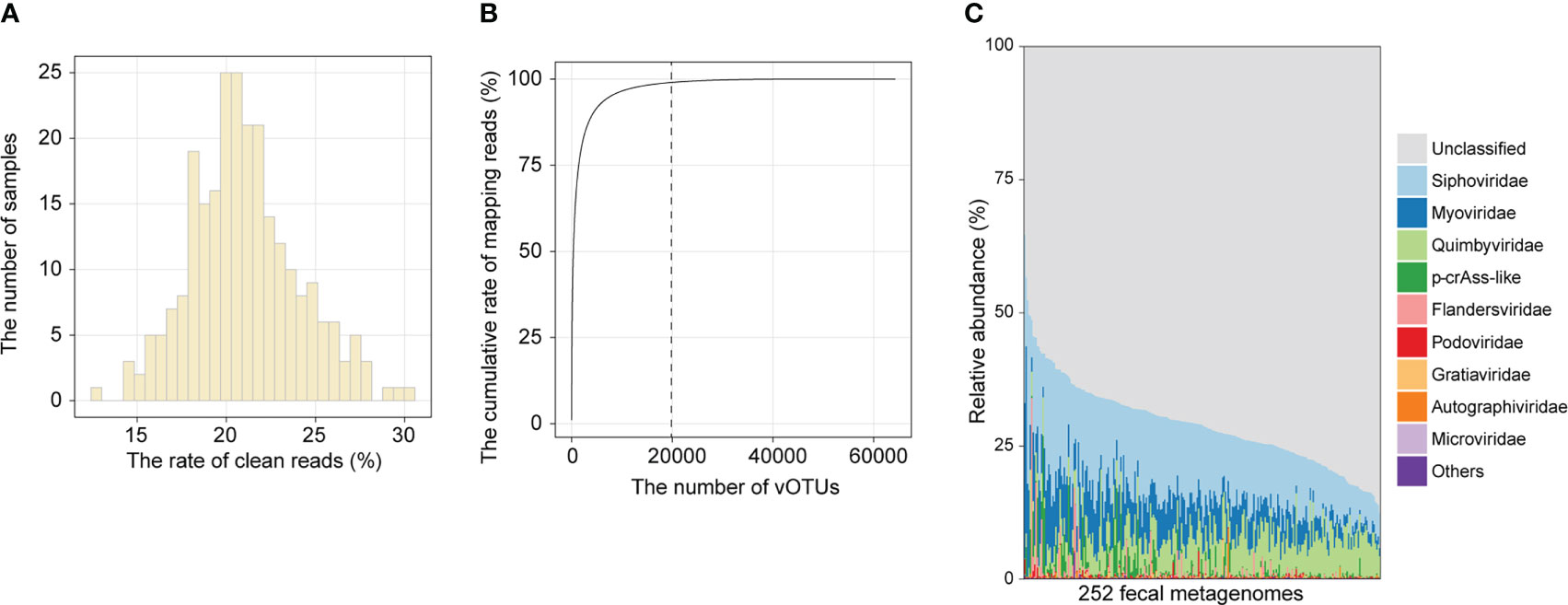
Figure 1 Overview of the gut viral community. (A) Histogram showing the proportion of metagenomic reads mapped to cnGVC. (B) The accumulative rate of mapping reads as a function of the number of vOTUs. A total of 67,096 vOTUs were sorted in descending order based on their average relative abundance across all metagenomic samples. (C) The gut viral composition of all samples at the family level.
Gut viral structure and diversity in ankylosing spondylitis
We performed PCoA based on the Bray-Curtis distance of the vOTU-level profiles to assess the difference in gut virome composition between AS patients and healthy controls and visualized the first two principal coordinate axes explaining 29% of the total variation (Figure 2A). The first principal coordinates of AS samples were larger than those of HC samples, and the direction of the effect of disease status was consistent across the two cohorts. Similarly, the gut viral composition of the two cohorts showed a relationship with disease status in the same direction along the second principal coordinate. In addition, PERMANOVA showed a significant difference in the overall virome composition between AS patients and HCs within each cohort (Figure 2A; adonis, p value <0.05).
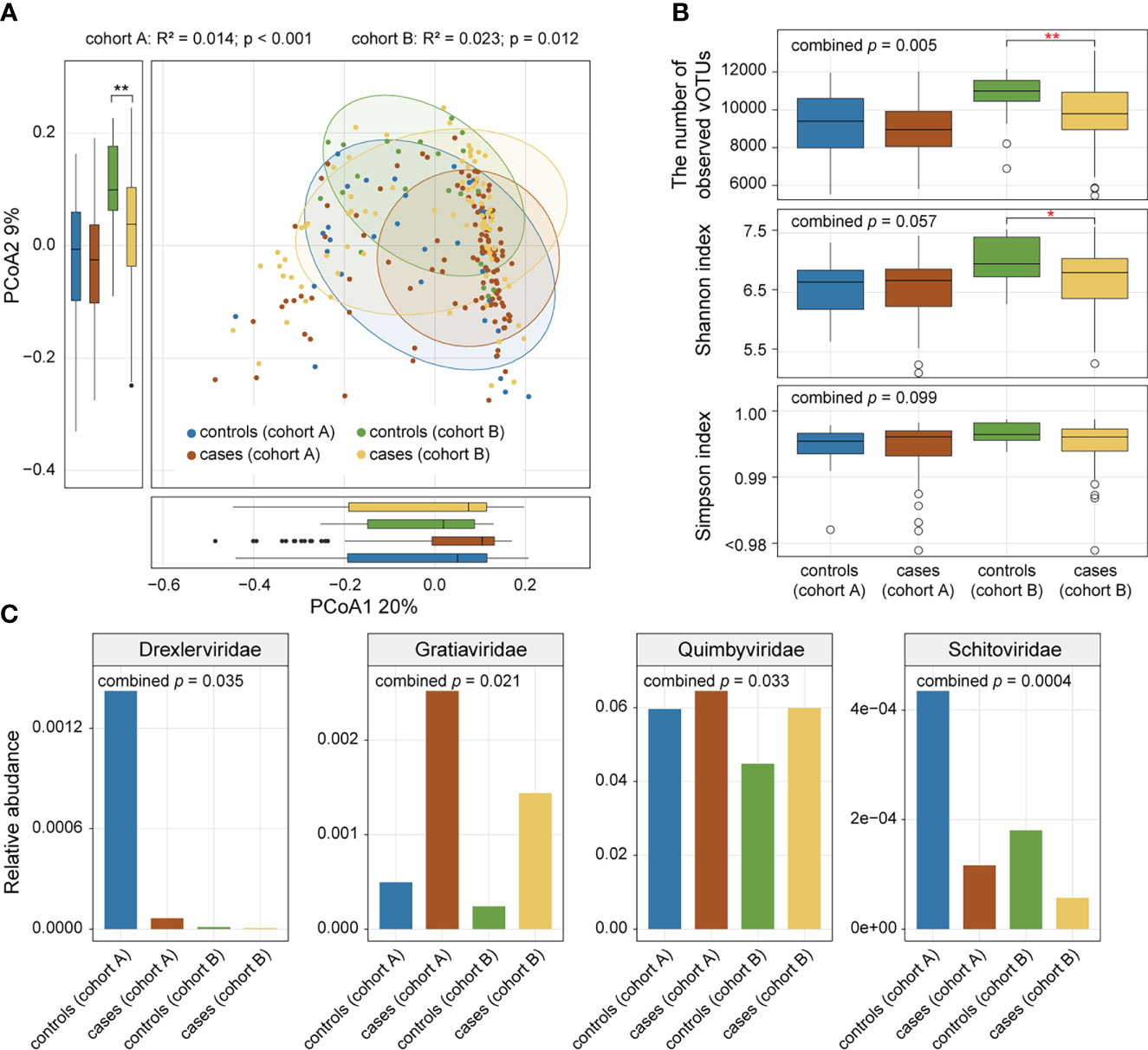
Figure 2 Comparison of gut viral diversity and composition between AS patients and healthy controls. (A) PCoA of Bray-Curtis distances of the gut virome at the vOTU level. Samples are shown at the first and second principal coordinates (PCoA1 and PCoA2), and the ratio of variance contributed by these two PCoAs is shown. Ellipsoids represent a 95% confidence interval surrounding each group. The bottom and left boxplots show the sample scores in PCoA1 and PCoA2 (boxes show medians/quartiles; error bars extend to the most extreme values within 1.5 interquartile ranges). (B) Boxplot showing the number of observed vOTUs (upper panel), Shannon diversity index (middle panel), and Simpson diversity index (bottom panel) of the gut virome of all samples. Wilcoxon rank-sum test: *p<0.05; **p<0.01. (C) Bar plot showing relative abundances of differentially abundant viral families between patients and controls.
To estimate the effect of disease status on gut viral richness and evenness, alpha diversities of each sample were calculated using three indices (i.e., number of observed vOTUs, Shannon diversity index, and Simpson index). Comparison analysis was conducted based on Fisher’s method of combining p values from Wilcoxon rank-sum tests. AS patients showed a significant decrease in viral richness (number of observed vOTUs) compared with controls (combined p value = 0.005), particularly in cohort A (Wilcoxon rank-sum test, p value = 0.002; Figure 2B). However, there was no significant difference in viral evenness (Shannon index and Simpson index) between the two groups (combined p value > 0.05; Figure 2B).
Comparison of the family-level composition revealed that AS patients had an increased abundance of Gratiaviridae and Quimbyviridae but that HCs had a higher abundance of Drexlerviridae and Schitoviridae (combined p value < 0.05; Figure 2C) in their viromes.
Identification of viral signatures associated with ankylosing spondylitis
We obtained 1,004 vOTUs with a significant difference between AS patients and HCs using the combined probability values from independent tests in two cohorts (BH-adjusted combined p value < 0.05), with 260 of these enriched in AS patients and 744 enriched in HCs (Table S2). For both the AS- and HC-enriched vOTUs, differential vOTUs were dominated by viruses belonging to Siphoviridae, Myoviridae, and unclassified taxa (Figure 3A). However, AS-enriched vOTUs presented a clearly higher proportion of Myoviridae viruses (19.2% vs. 10.9%); HC-enriched vOTUs contained more Siphoviridae viruses (43.5% vs. 25.4%). Moreover, AS-enriched vOTUs included more viruses belonging to Microviridae (9 vs. 3), p-crAss-like (8 vs. 4), and Gratiaviridae (8 vs. 2), whereas 18 Drexlerviridae and 9 Podoviridae viruses were observed among HC-enriched vOTUs, but no member of these two families was regarded as AS-enriched vOTUs.
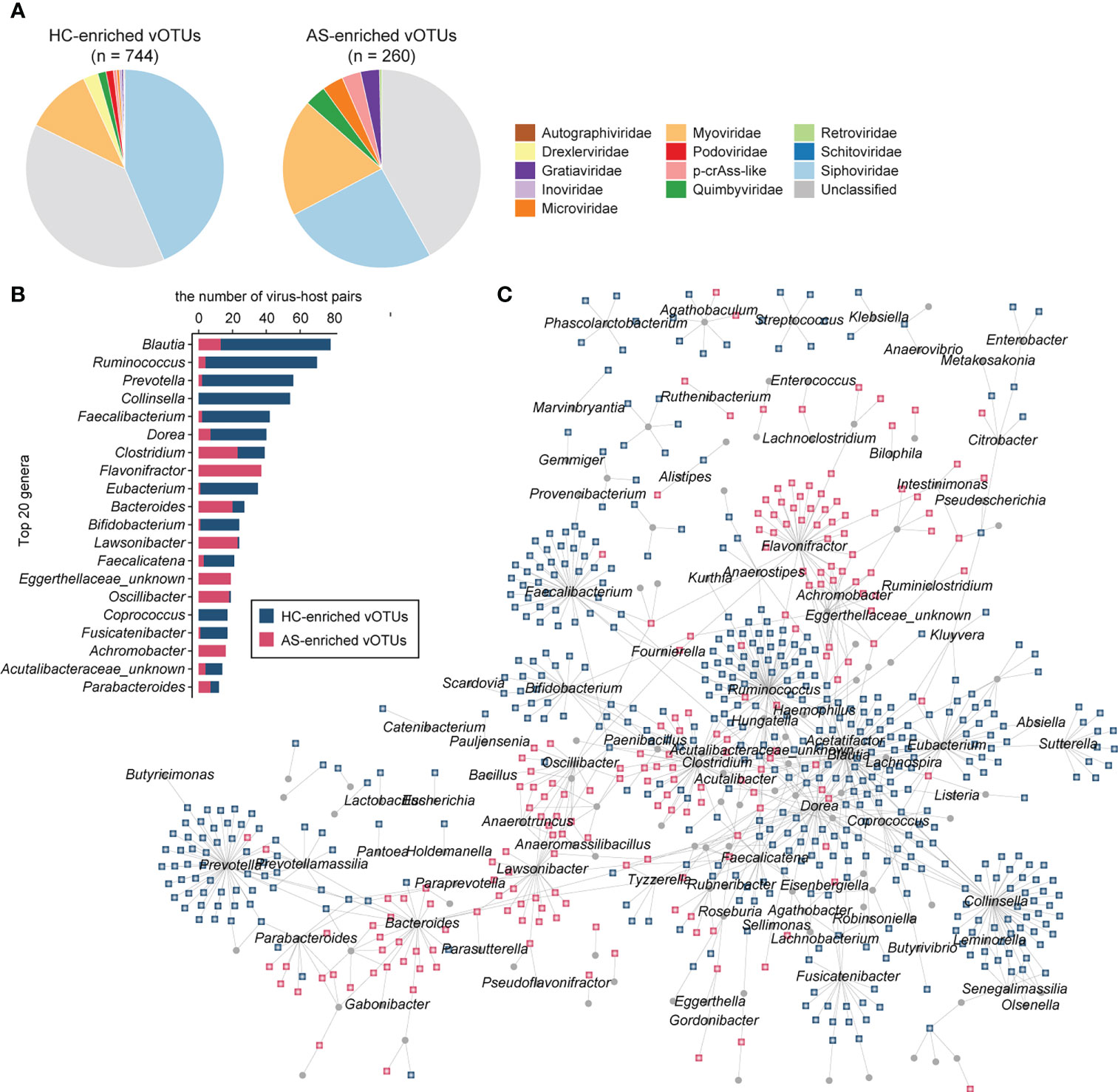
Figure 3 AS-associated gut viral signatures. (A) Taxonomic distribution of AS-enriched and HC-enriched vOTUs. (B) Distribution of prokaryotic hosts of AS-associated vOTUs. (C) Network showing vOTUs and corresponding prokaryotic hosts.
Then, we shaped the virus-host infection networks based on the alignment of genomic homology and the matching of CRISPR spacer sequences according to a previous method (27, 32) and observed a remarkable difference in host preference between AS-enriched and HC-enriched vOTUs (Figure 3B). Most AS-associated vOTUs (66.2%, n = 665) could be assigned to at least one prokaryotic host, including 474 HC-enriched vOTUs and 191 AS-enriched vOTUs. The HC-enriched vOTUs are predicted to more frequently infect members of Blautia (such as B. wexlerae and B. obeum), Ruminococcus (such as R. callidus and R. bicirculans), Collinsella (such as C. aerofaciens), Prevotella, Faecalibacterium (such as F. prausnitzii), Dorea (such as D. longicatena and D. formicigenerans), Eubacterium (such as E. hallii and E. ventriosum), and Bifidobacterium (such as B. adolescentis, B. breve, and B. catenulatum) (Figure 3C). In contrast to the HC-enriched vOTUs, the dominant hosts of the AS-enriched vOTUs included members of Flavonifractor, Lawsonibacter, Clostridium, Bacteroides, Oscillibacter, Achromobacter and an unknown genus of Eggerthellaceae. Strikingly, no species belonging to Flavonifractor, Achromobacter, or Eggerthellaceae was linked to the HC-enriched vOTUs, and several members of other genera, such as Lawsonibacter asaccharolyticus and Oscillibacter sp., were rarely connected to HC-enriched vOTUs. These findings indicate distinct virus-host relationships between AS-enriched and HC-enriched vOTUs.
Functions of viral signatures associated with ankylosing spondylitis
To explore the functional potential of gut viral signatures associated with AS, we identified 73,576 putative protein-coding genes from 1,004 AS-associated vOTUs and annotated them against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, leading to 11,478 genes annotated with KEGG functional orthologs (KOs). These annotated genes frequently participate in replication and repair mechanisms, protein family functions, transcription, prokaryotic defense systems, cell growth and death, and amino acid metabolism, among others (Figure 4A). Comparison analysis revealed that the occurrence rates of 27 KOs were significantly different between HC-enriched and AS-enriched vOTUs (Fisher’s exact test, adjusted p value < 0.05; Figure 4B; Table S3). Among these KOs, 7 are encoded by HC-enriched vOTUs, including 4 enzymes involved in genetic information processing and expression (K01160, K03111, K07474, and K07496), 2 lysozymes (K01185 and K07273) and 1 caseinolytic protease (K01358). The other 20 KOs were more frequently detected in AS-enriched vOTUs, including 11 functions involving genetic information processing and expression, 5 metabolic functions, and 4 potential structure and signaling proteins. Notably, 5 metabolic functions contain K01448 (N-acetylmuramoyl-L-alanine amidase), K00969 (nicotinate-nucleotide adenylyltransferase), K02217 (ferritin), K01768 (adenylate cyclase), and K01144 (exodeoxyribonuclease V).
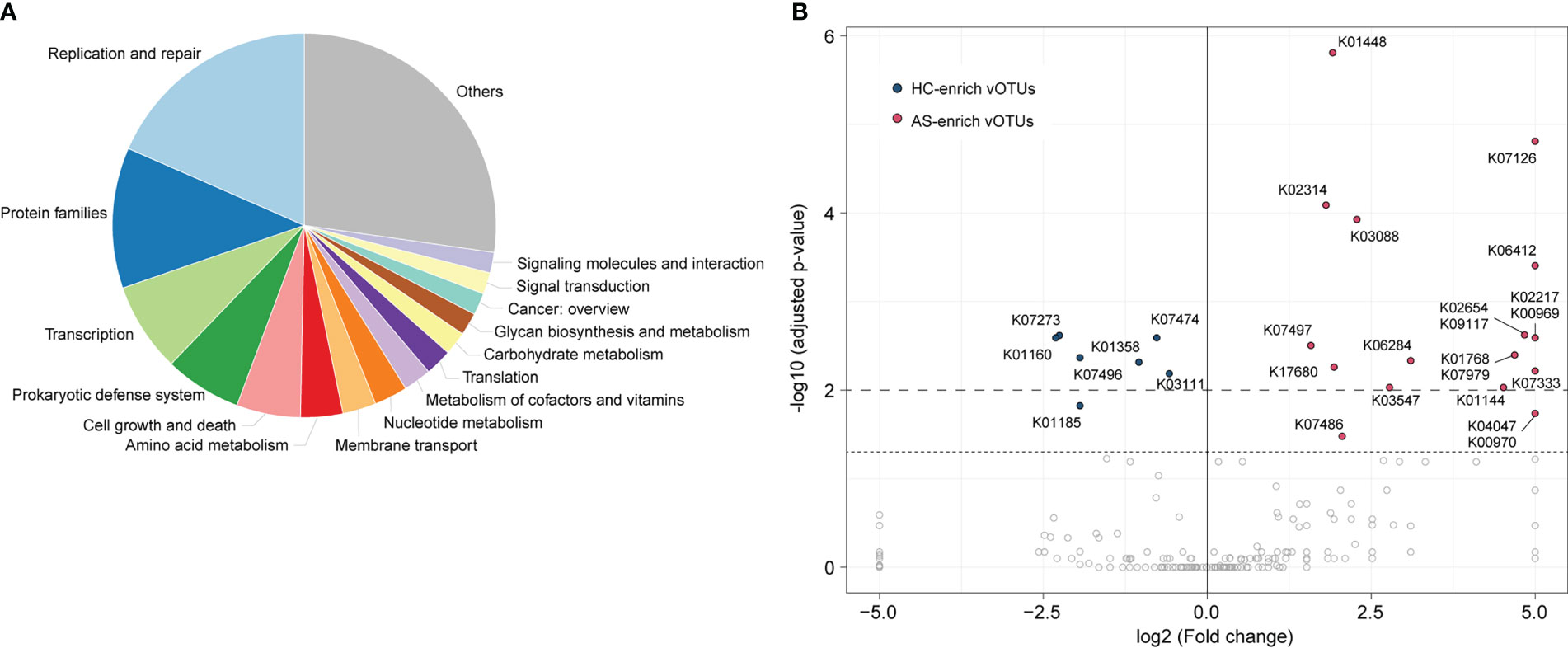
Figure 4 Comparison of functions between AS-enriched and HC-enriched vOTUs. (A) Composition of the functions of AS-associated vOTUs, as categorized at KEGG pathway level A. (B) Comparison of the occurrence rate of functions between AS-enriched and HC-enriched vOTUs.
Performance of the classification model based on the gut viral community
We built classification models for the AS patients and HCs based on the relative abundances of 1,004 AS-associated vOTUs using the random forest algorithm with 5 fivefold cross-validations, and we assessed the model performance based on the AUC score. AUC scores were 86.8% (95% confidence interval [CI] = 80.1%-93.6%), 89.5% (95% CI = 84.4%-94.6%), and 89.3% (95% CI = 83.7%-94.9%) in all samples, within cohort A, and within cohort B, respectively (Figure 5A). Furthermore, to optimize the model, we built random forest models based on a small set of vOTUs. All AS-associated vOTUs were first ranked by the importance level (i.e., mean decrease accuracy) obtained from the model for all samples. Then, we compared the performances of the models using various numbers of top-ranked vOTUs and observed approaching the optimal performance (AUC: 93.6% to 95.1%) in the models for each dataset based on 30 top-ranked vOTUs (Figure 5B). Notably, the 30 top-ranked vOTUs also showed excellent potential for classifying controls vs. patients in cross-dataset prediction (AUC > 80%; Figure 5C). These findings suggest that gut viral signatures have high effectiveness for AS prediction.
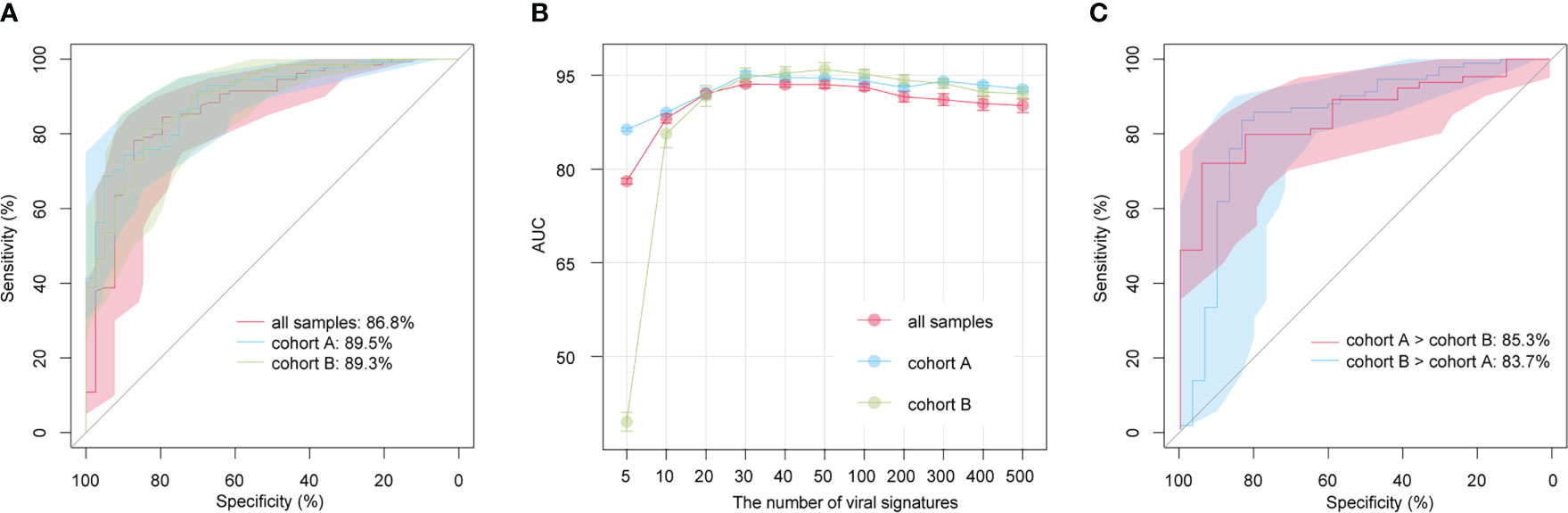
Figure 5 Prediction models of AS status based on gut viral signatures. (A) Receiver operating characteristic (ROC) analysis of the classification of disease/control status using the random forest algorithm. (B) Average AUC values for different numbers of AS-associated vOTUs. (C) ROC analysis of the classification of disease/control status in cross-cohort models.
Discussion
AS is a chronic, progressive inflammatory disease. In addition to involving the spine, patients often have some extrajoint manifestations, such as gastrointestinal problems. Studies have found that AS is closely related to gut inflammation, and approximately 70% of AS patients have subclinical gut inflammation (33), which suggests that gut dysbiosis may contribute to this phenomenon. In recent years, many studies have shown that the occurrence of AS is influenced by the gut microbiota (1, 34). The gut microbiota mainly consists of bacteria, archaea, viruses, fungi and parasites. In a study of the terminal ileum, researchers found a higher abundance of five bacterial families (Lachnospiraceae, Prevotellaceae, Rikenellaceae, Porphyromonadaceae, and Bacteriaceae) in AS patients and a lower abundance of Rumincoccaceae and Rikenellaceae. Several bacteria play key roles in the pathogenesis of AS, such as Klebsiella pneumoniae and Bacteroides vulgatus (35). In addition, several mechanisms to explain the role of the microbiome in the etiology of AS have been proposed, such as altered intestinal permeability (36), stimulation of the immune response (37), and molecular simulation (38). However, none of these fully explain the etiology of AS, and researchers continue to search for possible etiologies.
The gut microbiota is closely related to AS and may even be one of the key inducers; however, enteroviruses are often overlooked. In this study, we explored changes in the gut viral population of 193 AS patients compared with 59 healthy controls. Our study enhances the previous metagenomic datasets based on the gut virome of AS by adding more information. Metagenome-based virome sequencing improves the observed diversity of viruses, as it can capture more information about previously overlooked viruses. Compared with the HCs, the AS patients in our study exhibited a significant decrease in viral richness (number of observed vOTUs), particularly those in cohort A, though there was no significant difference in viral evenness (Shannon index and Simpson index) between the two groups. Specifically, comparison of the family-level composition revealed that the virome AS patients had an increased abundance of Gratiaviridae and Quimbyviridae but that HCs showed a higher abundance of Drexlerviridae and Schitoviridae. Gratiaviridae viruses are a putative novel family of viruses that is able to infect Bacteroides, and it consists of less abundant viruses that are remotely related to the Autographiviridae, Drexlerviridae, and Chaseviridae families (39). Quimbyviridae members are abundant, hypervariable viruses that often infect Bacteroides and are suspected to be obligate lytic viruses; they are a recently described novel family of viruses that are highly abundant and widespread in the human gut (39). Enrichment of these two novel viruses in AS suggests that they may play an important role in its pathogenesis. Interestingly, many viral viruses of the Drexlerviridae family, such as KM18 and IME268, have cleavage activity against Klebsiella pneumoniae or even its biofilms, which may be due to their putative endosialidase (depolymerase) enzyme (40, 41). Schitoviridae is a novel viral family of Escherichia N4-like phages, including eight subfamilies and many new genera, and there is a poorly reported association between Schitoviridae and human disease (42).
We used the combined probability values from independent tests in two cohorts to obtain 1004 vOTUs with significant differences between AS patients and HCs, of which 260 were enriched in AS patients and 744 in HCs. The AS-enriched vOTUs presented a higher proportion of Myoviridae viruses, Microviridae, p-crAss-like, and Gratiaviridae; the HC-enriched vOTUs contained more viruses of Siphoviridae, Drexlerviridae, and Podoviridae.
From the aspect of viral host, we found that most AS-associated vOTUs could be assigned to at least one prokaryotic host, including 474 HC-enriched vOTUs and 191 AS-enriched vOTUs. The dominant hosts of AS-enriched vOTUs included members of Flavonifractor, Lawsonibacter, Clostridium, Bacteroides, Oscillibacter, Achromobacter and an unknown genus from Eggerthellaceae. HC-enriched vOTUs are predicted to frequently infect members of Blautia, Ruminococcus, Collinsella, Prevotella, Faecalibacterium, Dorea, Eubacterium, and Bifidobacterium. Flavonifractor plautii is a flavonoid-degrading bacterium that was reported to affect antigen-induced T helper 2 cell (Th2) immune responses in mice, and it is also increased in young-onset colorectal cancer patients (43, 44). There are few articles on the relationship between Lawsonibacter and human diseases, though one species of Lawsonibacter was identified as a butyrate-producing bacterium (45). Interestingly, the relative abundance of Clostridium was found to be decreased in the gut microbiota of AS patients in many previous reports (46–48), but we found that Clostridium was the dominant host of AS-enriched vOTUs. Differentially abundant taxa have been identified in patients with juvenile idiopathic arthritis with the HLA-B27 allele, including Bilophila, Clostridium cluster XIVb, Oscillibacter, and Parvimonas (49), and Oscillibacter was shown to be involved in the integrity of the mouse intestinal barrier (50). Achromobacter may be involved in the development of systemic-onset juvenile idiopathic arthritis (51). Eggobacteriaceae is considered a bacterial biomarker of alopecia universalis and radiation enteritis (52, 53). These studies suggest the close association between the host bacteria of these AS-enriched vOTUs and AS. On the other hand, the relative abundance of Blautia is reportedly lower in HLA-B27+ offspring (54), in accordance with our results. In addition, Ruminococcus gnavus group, Faecalibacterium prausnitzii and Eubacterium ruminantium group are present at a lower abundance in AS patients (48, 55). However, in contrast to our results, the relative abundances of Collinsella, Prevotella, Dorea, and Bifidobacterium are increased in AS patients (47, 48, 55). These findings indicate distinct virus-host relationships between AS-enriched and HC-enriched vOTUs.
We also focused on the functions of viral signatures associated with AS and found that 4 enzymes involved in genetic information processing and expression (K01160, K03111, K07474, and K07496), 2 lysozymes (K01185 and K07273) and 1 caseinolytic protease (K01358) to be encoded by HC-enriched vOTUs. Eleven functions involve genetic information processing and expression, 5 metabolic functions, and 4 potential structure and signaling proteins. Notably, 5 metabolic functions containing K01448 (N-acetylmuramoyl-L-alanine amidase), K00969 (nicotinate-nucleotide adenylyltransferase), K02217 (ferritin), K01768 (adenylate cyclase), and K01144 (exodeoxyribonuclease V) were more frequently detected in AS-enriched vOTUs. To date, many attempts to screen biomarkers of AS have been made. Based on miRNA-451a and miRNA-125a expression levels (56) and collagen protein of serum samples (57), the models generally achieved the performance of AUCs between 0.60 to 0.80. Herein, we obtained a more accurate and higher AUC of 0.868, 0.895, and 0.893 using only 1004 gut vOTUs. This analysis showed highest AUC for the model (0.936 to 0.951) when using a subset of 30 top-ranked vOTUs. These findings are encouraging developments that suggest the high diagnostic potential of the gut virome in AS discrimination.
Conclusion
Overall, based on the cross-cohort metagenome shotgun sequencing data, we identified some viral signatures with a significant difference between AS patients and healthy controls. These viral signatures may be potential targets for treating AS, contributing to developing prevention and treatment strategies for AS from new perspectives. In addition, the excellent predictive model (AUC >0.936) using only a small number of viruses indicate its excellent potential for clinical application.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
CL, QY, SL, and WS conceived of the study. YanZ, SL, RG, YueZ and JMen performed data interpretation. JMa and WY performed partial data analysis. CL, YanZ, QY, CC, SL, ZW, and WS drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the funding from the Beijing University of Chinese Medicine (NO. 2180072120049) and the National Natural Science Foundation of China (NO. 82074246).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1154380/full#supplementary-material
References
1. Liu B, Ding Z, Xiong J, Heng X, Wang H, Chu W. Gut microbiota and inflammatory cytokine changes in patients with ankylosing spondylitis. BioMed Res Int (2022) 2022:1005111. doi: 10.1155/2022/1005111
2. Zhao J, Huang C, Huang H, Pan JK, Zeng LF, Luo MH, et al. Prevalence of ankylosing spondylitis in a Chinese population: a systematic review and meta-analysis. Rheumatol Int (2020) 40:859–72. doi: 10.1007/s00296-020-04537-0
3. Sieper J, Poddubnyy D. Axial spondyloarthritis. Lancet (2017) 390:73–84. doi: 10.1016/S0140-6736(16)31591-4
4. Zhao SS, Radner H, Siebert S, Duffield SJ, Thong D, Hughes DM, et al. Comorbidity burden in axial spondyloarthritis: a cluster analysis. Rheumatol (Oxford) (2019) 58:1746–54. doi: 10.1093/rheumatology/kez119
5. Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis–insights into pathogenesis. Nat Rev Rheumatol (2016) 12:81–91. doi: 10.1038/nrrheum.2015.133
6. Yang L, Wang L, Wang X, Xian CJ, Lu H. A possible role of intestinal microbiota in the pathogenesis of ankylosing spondylitis. Int J Mol Sci (2016) 17. doi: 10.3390/ijms17122126
7. Thjodleifsson B, Geirsson AJ, Björnsson S, Bjarnason I. A common genetic background for inflammatory bowel disease and ankylosing spondylitis: a genealogic study in Iceland. Arthritis Rheum (2007) 56:2633–9. doi: 10.1002/art.22812
8. Feng H-Y, Chan C-H, Chu Y-C, Qu X-M, Wang Y-H, Wei JC-C. Patients with ankylosing spondylitis have high risk of irritable bowel syndrome: a long-term nationwide population-based cohort study. Postgraduate Med (2022) 134:290–6. doi: 10.1080/00325481.2022.2041338
9. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature (2016) 535:65–74. doi: 10.1038/nature18847
10. Robinson PC, Leo PJ, Pointon JJ, Harris J, Cremin K, Bradbury LA, et al. Exome-wide study of ankylosing spondylitis demonstrates additional shared genetic background with inflammatory bowel disease. NPJ Genom Med (2016) 1:16008. doi: 10.1038/npjgenmed.2016.8
11. Wen C, Zheng Z, Shao T, Liu L, Xie Z, Le Chatelier E, et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol (2017) 18:142. doi: 10.1186/s13059-017-1271-6
12. Li M, Dai B, Tang Y, Lei L, Li N, Liu C, et al. Altered bacterial-fungal interkingdom networks in the guts of ankylosing spondylitis patients. mSystems (2019) 4(2):e00176-18. doi: 10.1128/mSystems.00176-18
13. Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe (2019) 26:764–778 e5. doi: 10.1016/j.chom.2019.10.009
14. Tomofuji Y, Kishikawa T, Maeda Y, Ogawa K, Nii T, Okuno T, et al. Whole gut virome analysis of 476 Japanese revealed a link between phage and autoimmune disease. Ann Rheum Dis (2022) 81:278–88. doi: 10.1136/annrheumdis-2021-221267
15. Ma Y, You X, Mai G, Tokuyasu T, Liu C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome (2018) 6:24. doi: 10.1186/s40168-018-0410-y
16. Li Y, Handley SA, Baldridge MT. The dark side of the gut: Virome-host interactions in intestinal homeostasis and disease. J Exp Med (2021) 218. doi: 10.1084/jem.20201044
17. Keen EC, Dantas G. Close encounters of three kinds: Bacteriophages, commensal bacteria, and host immunity. Trends Microbiol (2018) 26:943–54. doi: 10.1016/j.tim.2018.05.009
18. Li S, Yan Q, Wang G, Zhang Y, Guo R, Zhang P, et al. Cataloguing and profiling of the gut virome in Chinese populations uncover extensive viral signatures across common diseases. bioRxiv (2022). doi: 10.1101/2022.12.27.522048
19. Coughlan S, Das A, O’Herlihy E, Shanahan F, O’Toole PW, Jeffery IB. The gut virome in irritable bowel syndrome differs from that of controls. Gut Microbes (2021) 13:1–15. doi: 10.1080/19490976.2021.1887719
20. Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe (2019) 26:527–541 e5. doi: 10.1016/j.chom.2019.09.009
21. Garmaeva S, Gulyaeva A, Sinha T, Shkoporov AN, Clooney AG, Stockdale SR, et al. Stability of the human gut virome and effect of gluten-free diet. Cell Rep (2021) 35:109132. doi: 10.1016/j.celrep.2021.109132
22. Huang R, Li F, Zhou Y, Zeng Z, He X, Fang L, et al. Metagenome-wide association study of the alterations in the intestinal microbiome composition of ankylosing spondylitis patients and the effect of traditional and herbal treatment. J Med Microbiol (2020) 69:797. doi: 10.1099/jmm.0.001107
23. Zhou C, Zhao H, Xiao X-Y, Guo R-J, Wang Q, Chen H, et al. Metagenomic profiling of the pro-inflammatory gut microbiota in ankylosing spondylitis. J Autoimmun (2020) 107:102360. doi: 10.1016/j.jaut.2019.102360
24. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
25. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods (2012) 9:357–9. doi: 10.1038/nmeth.1923
26. Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
27. Li S, Guo R, Zhang Y, Li P, Chen F, Wang X, et al. A catalogue of 48,425 nonredundant viruses from oral metagenomes expands the horizon of the human oral virome. iScience (2022) 25(6):104418. doi: 10.1016/j.isci.2022.104418
28. Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, Kyrpides NC. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol (2020) 39(5):578–85. doi: 10.1038/s41587-020-00774-7
29. Team RC. R: A language and environment for statistical computing (2020). Available at: https://www.R-project.org/.
30. Gregory AC, Zablocki O, Zayed AA, Howell A, Bolduc B, Sullivan MB. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe (2020) 28:724–740 e8. doi: 10.1016/j.chom.2020.08.003
31. Guo R, Li S, Zhang Y, Zhang Y, Wang G, Ullah H, et al. Dysbiotic oral and gut viromes in untreated and treated rheumatoid arthritis patients. Microbiol Spectr (2022) 10(5):e00348–22. doi: 10.1128/spectrum.00348-22
32. Yan Q, Wang Y, Chen X, Jin H, Wang G, Guan K, et al. Characterization of the gut DNA and RNA viromes in a cohort of Chinese residents and visiting pakistanis. Virus Evol (2021) 7:veab022. doi: 10.1093/ve/veab022
33. Ciccia F, Rizzo A, Triolo G. Subclinical gut inflammation in ankylosing spondylitis. Curr Opin Rheumatol (2016) 28:89–96. doi: 10.1097/BOR.0000000000000239
34. Song ZY, Yuan D, Zhang SX. Role of the microbiome and its metabolites in ankylosing spondylitis. Front Immunol (2022) 13:1010572. doi: 10.3389/fimmu.2022.1010572
35. Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with bacteroides vulgatus or escherichia coli. Infect Immun (1999) 67:2969–74. doi: 10.1128/IAI.67.6.2969-2974.1999
36. Ciccia F, Ferrante A, Triolo G. Intestinal dysbiosis and innate immune responses in axial spondyloarthritis. Curr Opin Rheumatol (2016) 28:352–8. doi: 10.1097/BOR.0000000000000296
37. Fragoulis GE, Siebert S, McInnes IB. Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu Rev Med (2016) 67:337–53. doi: 10.1146/annurev-med-051914-021944
38. Fielder M, Pirt SJ, Tarpey I, Wilson C, Cunningham P, Ettelaie C, et al. Molecular mimicry and ankylosing spondylitis: possible role of a novel sequence in pullulanase of klebsiella pneumoniae. FEBS Lett (1995) 369:243–8. doi: 10.1016/0014-5793(95)00760-7
39. Benler S, Yutin N, Antipov D, Rayko M, Shmakov S, Gussow AB, et al. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome (2021) 9:78. doi: 10.1186/s40168-021-01017-w
40. Ku H, Kabwe M, Chan HT, Stanton C, Petrovski S, Batinovic S, et al. Novel drexlerviridae bacteriophage KMI8 with specific lytic activity against klebsiella michiganensis and its biofilms. PloS One (2021) 16:e0257102. doi: 10.1371/journal.pone.0257102
41. Nazir A, Qi C, Shi N, Gao X, Feng Q, Qing H, et al. Characterization and genomic analysis of a novel drexlervirial bacteriophage IME268 with lytic activity against klebsiella pneumoniae. Infect Drug Resist (2022) 15:1533–46. doi: 10.2147/IDR.S347110
42. Wittmann J, Turner D, Millard AD, Mahadevan P, Kropinski AM, Adriaenssens EM. From orphan phage to a proposed new family-the diversity of N4-like viruses. Antibiotics (Basel) (2020) 9. doi: 10.3390/antibiotics9100663
43. Yang Y, Du L, Shi D, Kong C, Liu J, Liu G, et al. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat Commun (2021) 12:6757. doi: 10.1038/s41467-021-27112-y
44. Ogita T, Yamamoto Y, Mikami A, Shigemori S, Sato T, Shimosato T. Oral administration of flavonifractor plautii strongly suppresses Th2 immune responses in mice. Front Immunol (2020) 11:379. doi: 10.3389/fimmu.2020.00379
45. Sakamoto M, Iino T, Yuki M, Ohkuma M. Lawsonibacter asaccharolyticus gen. nov., sp. nov., a butyrate-producing bacterium isolated from human faeces. Int J Syst Evol Microbiol (2018) 68:2074–81. doi: 10.1099/ijsem.0.002800
46. Cardoneanu A, Cozma S, Rezus C, Petrariu F, Burlui AM, Rezus E. Characteristics of the intestinal microbiome in ankylosing spondylitis. Exp Ther Med (2021) 22:676. doi: 10.3892/etm.2021.10108
47. Zhang L, Han R, Zhang X, Fang G, Chen J, Li J, et al. Fecal microbiota in patients with ankylosing spondylitis: Correlation with dietary factors and disease activity. Clin Chim Acta (2019) 497:189–96. doi: 10.1016/j.cca.2019.07.038
48. Cardoneanu A, Mihai C, Rezus E, Burlui A, Popa I, Cijevschi Prelipcean C. Gut microbiota changes in inflammatory bowel diseases and ankylosing spondilytis. J Gastrointestin Liver Dis (2021) 30:46–54. doi: 10.15403/jgld-2823
49. Di Paola M, Cavalieri D, Albanese D, Sordo M, Pindo M, Donati C, et al. Alteration of fecal microbiota profiles in juvenile idiopathic arthritis. associations with HLA-B27 allele and disease status. Front Microbiol (2016) 7:1703. doi: 10.3389/fmicb.2016.01703
50. Lam YY, Ha CW, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PloS One (2012) 7:e34233. doi: 10.1371/journal.pone.0034233
51. Janarthanan M, Gollapalli S, Sankaranarayanan S. Achromobacter xylosoxidans sepsis unveiling X-linked agammaglobulinemia masquerading as systemic-onset juvenile idiopathic arthritis. Indian Pediatr (2019) 56:423–5. doi: 10.1007/s13312-019-1541-3
52. Moreno-Arrones OM, Serrano-Villar S, Perez-Brocal V, Saceda-Corralo D, Morales-Raya C, Rodrigues-Barata R, et al. Analysis of the gut microbiota in alopecia areata: identification of bacterial biomarkers. J Eur Acad Dermatol Venereol (2020) 34:400–5. doi: 10.1111/jdv.15885
53. Li Y, Yan H, Zhang Y, Li Q, Yu L, Li Q, et al. Alterations of the gut microbiome composition and lipid metabolic profile in radiation enteritis. Front Cell Infect Microbiol (2020) 10:541178. doi: 10.3389/fcimb.2020.541178
54. Stoll ML, DeQuattro K, Li Z, Sawhney H, Weiss PF, Nigrovic PA, et al. Impact of HLA-B27 and disease status on the gut microbiome of the offspring of ankylosing spondylitis patients. Children (Basel) (2022) 9. doi: 10.3390/children9040569
55. Chen Z, Qi J, Wei Q, Zheng X, Wu X, Li X, et al. Variations in gut microbial profiles in ankylosing spondylitis: disease phenotype-related dysbiosis. Ann Transl Med (2019) 7:571. doi: 10.21037/atm.2019.09.41
56. Fotoh DS, Noreldin RI, Rizk MS, Elsabaawy MM, Esaily HA. miRNA-451a and miRNA-125a expression levels in ankylosing spondylitis: Impact on disease diagnosis, prognosis, and outcomes. J Immunol Res (2020) 2020:2180913. doi: 10.1155/2020/2180913
57. Bay-Jensen AC, Wichuk S, Byrjalsen I, Leeming DJ, Morency N, Christiansen C, et al. Circulating protein fragments of cartilage and connective tissue degradation are diagnostic and prognostic markers of rheumatoid arthritis and ankylosing spondylitis. PloS One (2013) 8:e54504. doi: 10.1371/journal.pone.0054504
Keywords: ankylosing spondylitis, gut virome, viral dysbiosis, viral operational taxonomic units, viral function, metagenome sequencing
Citation: Li C, Zhang Y, Yan Q, Guo R, Chen C, Li S, Zhang Y, Meng J, Ma J, You W, Wu Z and Sun W (2023) Alterations in the gut virome in patients with ankylosing spondylitis. Front. Immunol. 14:1154380. doi: 10.3389/fimmu.2023.1154380
Received: 30 January 2023; Accepted: 20 March 2023;
Published: 30 March 2023.
Edited by:
James Cheng-Chung Wei, Chung Shan Medical University Hospital, TaiwanReviewed by:
An-Ping Huo, Chung Shan Medical University Hospital, TaiwanLuminiţa-Smaranda Iancu, Grigore T. Popa University of Medicine and Pharmacy, Romania
Copyright © 2023 Li, Zhang, Yan, Guo, Chen, Li, Zhang, Meng, Ma, You, Wu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Sun, c3Vud2VuQGJ1Y20uZWR1LmNu
†These authors have contributed equally to this work