 Xin Li
Xin Li Jianbo Zhou
Jianbo Zhou Xue Wang
Xue Wang Chunxi Li
Chunxi Li Zifan Ma
Zifan Ma Qiaoling Wan
Qiaoling Wan Fu Peng
Fu Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1152312
This article is part of the Research Topic Fibrosis, inflammation, and cancers: dangerous liaisons to be depicted and targeted View all 10 articles
Pancreatic cancer is one of the most dangerous types of cancer today, notable for its low survival rate and fibrosis. Deciphering the cellular composition and intercellular interactions in the tumor microenvironment (TME) is a necessary prerequisite to combat pancreatic cancer with precision. Cancer-associated fibroblasts (CAFs), as major producers of extracellular matrix (ECM), play a key role in tumor progression. CAFs display significant heterogeneity and perform different roles in tumor progression. Tumor cells turn CAFs into their slaves by inducing their metabolic dysregulation, exacerbating fibrosis to acquire drug resistance and immune evasion. This article reviews the impact of metabolic reprogramming, effect of obesity and cellular crosstalk of CAFs and tumor cells on fibrosis and describes relevant therapies targeting the metabolic reprogramming.
Pancreatic cancer is one of the most aggressive types of cancer, being more common in developed countries and by low survival rates (1). As the main form of pancreatic cancer, pancreatic ductal adenocarcinoma (PDAC) has a discouraging prognosis, with a very low five-year survival rate (2). There is a correlation between lifestyle habits including smoking, alcohol consumption, and genetic and environmental factors and the onset of pancreatic cancer (1). Notably, the hormones, pro-angiogenic factors and pro-inflammatory cytokines secreted by obese tissues make obesity a risk factor for the occurrence of pancreatic cancer (3, 4). Diabetes associated with obesity and chronic pancreatitis also show a relevance to pancreatic cancer (5). Surgery is the treatment that has the potential to cure pancreatic cancer now, whilst chemotherapy, immunotherapy and targeted therapies have been demonstrated to help enhance the overall survival rate of patients (6–8).
Fibrosis driven by chronic inflammation occurs commonly in a variety of cancers, such as liver, pancreatic, and lung cancers (9–11). This formation of excessive intratumoral connective tissue is also referred to as desmoplasia by pathologists (12). Desmoplasia is one of the major pathological features and is intimately connected with its occurrence, progression and prognosis of pancreatic cancer. The desmoplastic reaction caused by inflammation gives pancreatic cancer an extraordinarily rich ECM (13). The fibrotic response in tumors is by the same mechanism as wound healing, being an excessive accumulation of ECM components and involving multiple cytokines and growth factors (14). ECM proteins are rich in composition, including fiber-forming proteins, glycoproteins, proteoglycans and matricellular proteins (15). The dense stroma leads to hypoxia in the tumor microenvironment and makes it difficult for chemotherapeutic agents to penetrate, thus imparting chemoresistance to pancreatic cancer (16).
TME of pancreatic cancer contains abundant stroma, blood vessels, and soluble proteins (17). Apart from cancer cells, three types of normal cells are found in the TME, namely stromal cells, fibroblasts, and immune cells (18). TME as a dynamic system has a changing composition and influences the progression of fibrosis in pancreatic cancer. Cancer-associated fibroblasts are extraordinarily abundant and secrete a range of extracellular matrix proteins, growth factors, and cytokines (19). CAFs crosstalk with tumor cells and immune cells and perform metabolic reprogramming to promote tumor development and fibrosis. In this review, we give a summary of current information about the heterogeneity of CAFs in pancreatic cancer cells, as well as updates on the metabolic reprogramming, crosstalk and therapies in the TME.
CAFs were initially thought to be homogeneous, but subsequent studies proved that CAFs varied in origin, expression, and function (20). Their typing remains incompletely elucidated, but existing work demonstrates that functionally distinct or even completely opposite subtypes exist. Öhlund et al. found pancreatic stellate cells (PSCs) were able to differentiate into two CAF subtypes, myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs) in mouse PDAC (21). They differed significantly in spatial distribution and cytokine expression. myCAFs were distributed in the periglandular region at a closer distance from tumor cells, with high expression of α-smooth muscle actin (α-SMA) and low expression of interleukin (IL)-6, whereas iCAFs were distributed more distantly throughout the tumor, with low expression of α-SMA but high expression of cytokines such as IL-6, IL-11, and leukemia inhibitory factor (LIF) (21). This classification still has not reached the end point, as three subgroups of iCAFs were identified (22). Antigen-presenting CAFs (apCAFs) was identified in PDAC, named for its ability to express MHC class II molecules (23). A new CAF subtype with a highly activated metabolic state (meCAFs) was found in loose-type PDAC (24). Complement-secreting CAFs (csCAFs) were found in PDAC featuring a specific expression of complement components such as C3, C7, C1R/S, CFD, CFH, CFI (25). In the same study, Chen et al. defined PSCs as a subtype of CAF and found that PSCs dominated in PDAC stages I, II and III (25). The state of differentiation is reversible as iCAFs and myCAFs are able to convert into each other and apCAFs can also differentiate into myCAFs (21, 23). Modulation of transforming growth factor-β (TGF-β), IL-1/JAK/STAT signaling and hedgehog signaling impact on the differentiation of myCAFs and iCAFs (26, 27). Hypoxia within the TME probably converted fibroblasts to iCAFs (28). Neuzillet et al. proved the presence of at least four CAF subtypes in PDAC, which were featured by distinct mRNA expression profiles, with POSTN, MYH11, and PDPN as markers for three of the subtypes (29). PDPN-positive CAFs are molecularly similar to an iCAF subset, while POSTN-positive CAFs are not associated with the classical myCAF/iCAF classification (30). And these two subsets cooperate in the TME to induce the recruitment of monocytes/macrophages (30). It is worth mentioning that the identified subtypes of CAFs are not only present in pancreatic cancer, but also can be found in breast, ovarian and lung cancer models (31).
The major CAF subtypes show significant heterogeneity not only in phenotypes but also in function (Figure 1). The pathways enriched by myCAFs included ECM organization, and collagen formation were significantly upregulated, and its high α-SMA expression indicated its possible involvement in ECM formation and fibrosis (23). iCAFs highly expressed inflammatory cytokines, and up-regulated IFN-γ response, TNF/NF-κB, IL-2/STAT5, IL-6/JAK/STAT3, and the complement pathway (23). PDAC iCAFs were classified into different subsets, and OGN was a unique marker for one of those linked to a good prognosis (22). are correlated with a poorer prognosis, whereas another study linked higher abundance of iCAFs to a better prognosis (28, 30). This may result from the presence of different subgroups in iCAFs, but it also demonstrates that iCAFs simultaneously have tumor-promoting and inhibiting properties. circCUL2 regulated miR-203a-5p/MyD88/NF-κB/IL-6 axis to induce the production of iCAFs, which increased the secretion of IL-6, thereby promoting PDAC progression and immunosuppression (32). Huang et al. found that mesothelial cells were induced to differentiate into apCAFs by IL-1/NF-κB and TGF-β signaling (33). apCAFs promoted the transition of naive CD4+ T cells into regulatory T cells (Tregs), which means that it may be related to immunosuppression (33).
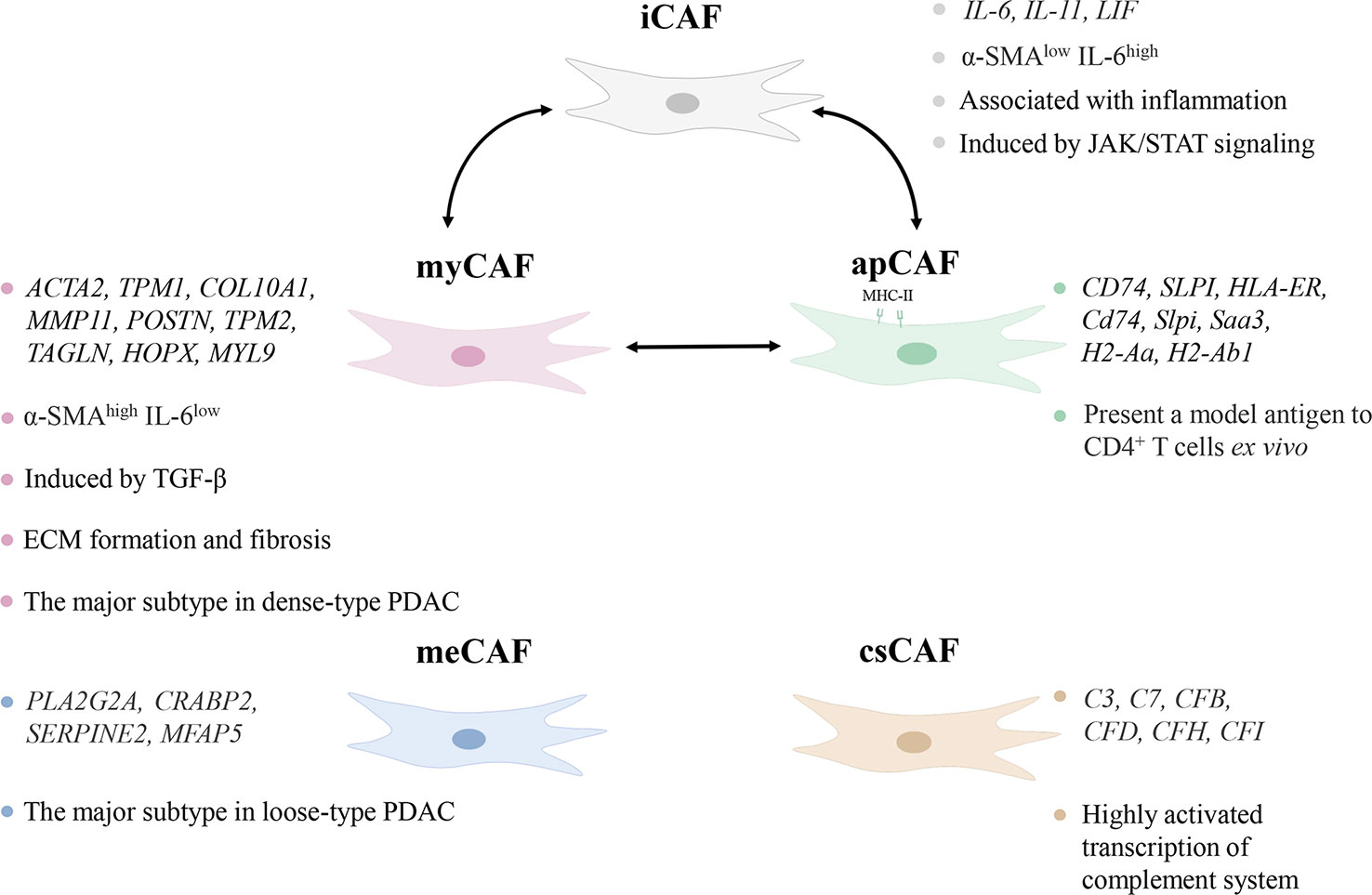
Figure 1 Characteristics of subtypes of cancer-associated fibroblasts (CAFs). The different subtypes of CAFs show heterogeneity in distribution, marker genes, and pathways in the tumor microenvironment. iCAF, inflammatory CAF; α-SMA, α-smooth muscle actin; IL-6, interleukin-6; myCAF, myofibroblastic CAF; TGF-β, transforming growth factor-β; ECM, extracellular matrix; PDAC, pancreatic ductal adenocarcinoma; apCAF, antigen-presenting CAF; meCAF, CAF with a highly activated metabolic state; csCAF complement-secreting CAF.
Although PSCs are generally considered to be the major precursor cells for CAFs within pancreatic cancer, a recent study indicated that PSCs produced only a small fraction of CAFs in PDAC (34). However, the promotion of fibrosis by PSCs remains an important component of pancreatic cancer progression. While activated PSCs are considered to be CAFs, for a clearer representation of the source, PSCs are described separately from CAFs in this review. PSCs were first identified in the intralobular and interlobular connective tissues of normal pancreas with lipid droplets containing vitamin A in 1982 (35). A study showed that vitamin A deficiency contributed to the transition of PSCs from a quiescent to the activated state (36). When injury or inflammation activates the quiescent PSCs, this vitamin A droplet disappears while the expression of collagen, fibronectin, laminin and α-SMA increases, and EMT production rises. The activation of PSCs is influenced by a variety of factors, including alcohol, diabetes, oxidative stress, cytokines, growth factors, etc. TGF-β1 is considered to be the main regulator, while platelet derived growth factor (PDGF), IL-6, IL-11, c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) are also implicated (37–40).
Tumor cells still produce energy through less efficient aerobic glycolysis even under adequate oxygen, enhancing glucose transformation to pyruvate, termed the Warburg effect (41). However, this is not due to mitochondrial damage as originally envisioned by Warburg, but rather spontaneous metabolic reprogramming of tumor cells, where activation of a series of signaling factors and pathways leads to a switch from oxidative phosphorylation to glycolysis (42). Similar metabolic reprogramming exists in CAFs, and the Warburg effect is more obvious (43). Pancreatic cancer is one of the most severely hypoxic tumors as known, and hypoxia-inducible factors (HIFs) are the main regulators of hypoxia adaptation (44). Since the identification of HIF-1α in 1995, a wide range of roles of HIF-1 is continuously revealed in angiogenesis, cell metabolism, cell survival, and so forth (45, 46). In breast cancer, ROS production by cancer cells induces loss of Cav-1 in stromal cells, allowing CAFs to accumulate ROS and activate HIF-1α, consequently reprogramming CAFs and inducing autophagy (47, 48). The same alterations are shown in the PDAC model, where Cav-1 is lost in response to PSCs activation, correlating with stromal and cancer cells metabolic coupling (49). To conclude, HIF-1α connects oxidative stress and metabolic reprogramming of CAFs. Under such harsh conditions with hypoxia and low nutrition, there is metabolic crosstalk between CAFs with tumor cells and immune cells, all of which interact with each other to make TME a more habitable system (Figure 2).
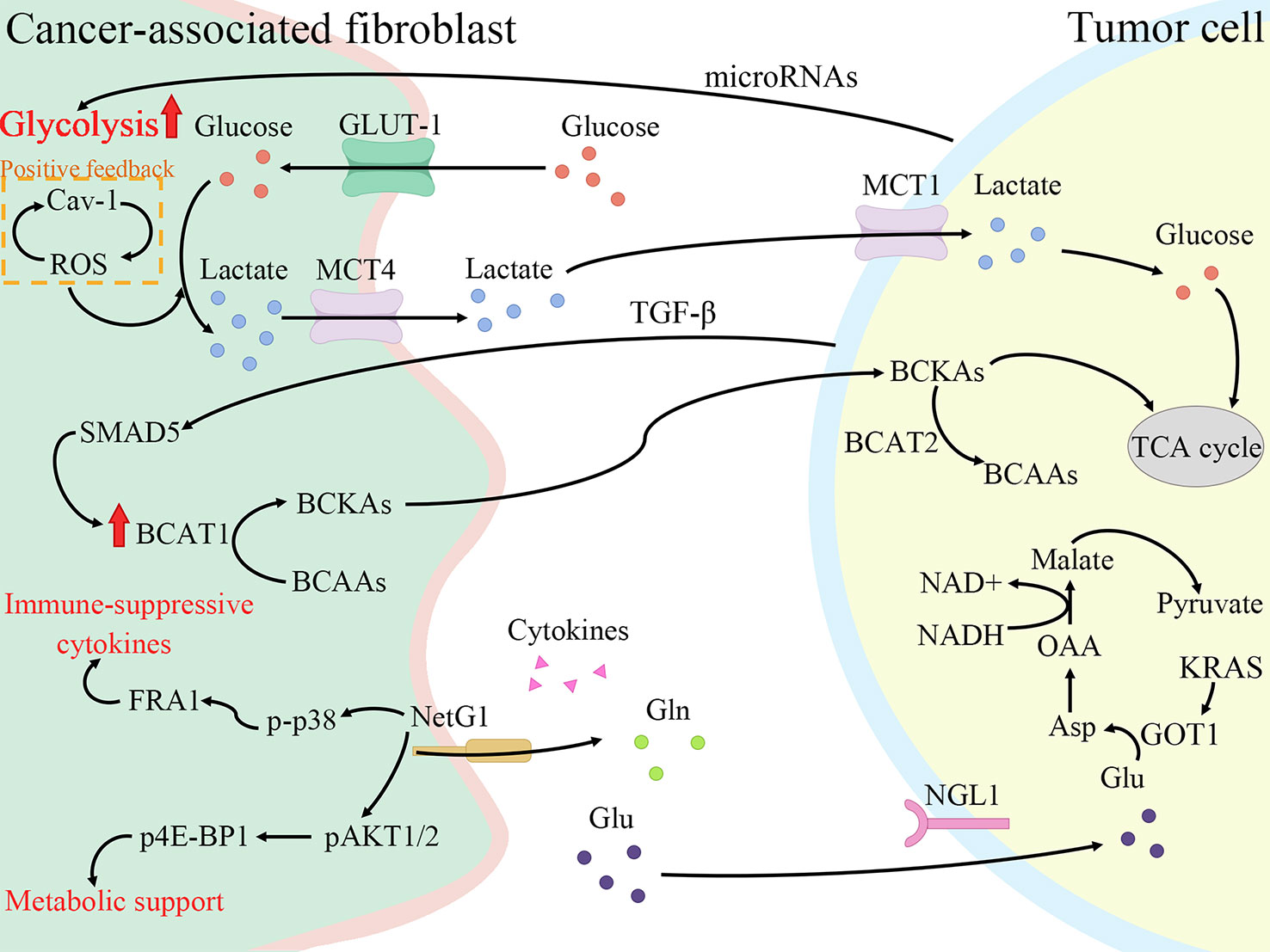
Figure 2 Cancer-associated fibroblasts (CAFs) promote fibrosis and tumor growth through metabolic reprogramming. CAFs increase glycolysis and glutamine secretion to supply lactate, branched-chain α-keto acids (BCKAs), glutamine, and cytokines to tumor cells. Meanwhile, tumor cells also secrete cytokines and microRNAs to regulate the metabolic reprogramming of CAFs to enable themselves to survive in a low-nutrient environment. Cav-1, Caveolin-1; ROS, reactive oxygen species; GLUT-1, glucose transporter-1; MCT4, monocarboxylate transporters 4; BCAT1, branched-chain amino acid transaminase 1; BCAAs, branched-chain amino acids; NetG1, Netrin G1; p-p38, phosphorylation of p38; FRA1, FOS-related antigen 1; pAKT1/2, phospho-AKT1/2; p4E-BP1, p4E-BP1; Gln, glutamine; Glu, glutamate; NGL1, Netrin-G ligand-1; TGF-β, transforming growth factor-β; MCT1, monocarboxylate transporters 1; BCAT2, branched-chain amino acid transaminase 2; TCA cycle, tricarboxylic acid cycle; GOT1, aspartate transaminase; Asp, aspartate; OAA, oxaloacetate.
Pavlides et al. proposed the reverse Warburg effect, elucidating that CAFs were able to perform glycolysis, producing pyruvate and lactate and making them available to cancer cells for use in the mitochondrial tricarboxylic acid (TCA) cycle (50). In other words, CAFs are captured by engaging with cancer cells and reprogrammed to a glycolytic phenotype. thereby supplying metabolic intermediates that enable cancer cells to compensatively generate energy via mitochondrial OXPHOS (51, 52). Glycolysis is the main metabolic mode of CAFs due to the increased expression of HIF-1α and monocarboxylate transporter (MCT) 4 (53). HIF-1α is a key cytokine that enables cells to adjust to hypoxic environments and undergo metabolic changes by promoting glycolysis through genes which encode glucose transporter proteins and enzymes of the glycolytic pathway (54). MCTs are passive transporter proteins that transport monocarboxylic acid ions and are highly expressed in tumors (55). MCT1 and MCT4 exhibit proton-coupled symport, with MCT4 generally involved in the export of lactate and MCT1 generally involved in the import of lactate (56). The expression of two glycolytic enzymes, lactate dehydrogenase A and pyruvate kinase M2, was found to be increased in CAFs (57). Furthermore, when pancreatic cancer cells were co-cultured with CAFs, MCT1 protein, succinate dehydrogenase and fumarate hydratase expression increased, demonstrating the metabolic coupling existing between CAFs and cancer cells (57). Positive feedback of Caveolin-1-ROS signaling prompted activation of PSCs and upregulated the expression of glycolytic enzymes, and the transporter protein MCT4, and downregulated the expression of OXPHOS enzymes and the transporter protein MCT1, while the protein expression in cancer cells was completely opposite (49). Moreover, MiR-21 promotes glucose uptake and lactate secretion by CAFs, indirectly enhancing pancreatic cancer cell invasion (58). Interestingly, CAFs also show heterogeneity in metabolic pathways, for iCAF had the highest metabolic activity and was more biased to glycolysis, whereas myCAF scored higher in OXPHOS than iCAF and apCAF (28).
Glutamine, an amide of glutamate, is an essential origin of carbon and nitrogen in pancreatic cancer (59). Son et al. found that PDAC cells metabolized glutamine using a specific aspartate transaminase (glutamic-oxoacetic transaminase 1)-mediated pathway to produce biomass precursors and redox power (60). Glutamine also serves as an important energy source for CAFs and is metabolized and secreted into metabolites such as glutamate, α-ketoglutarate, aspartate and malate (53). Both Netrin G1 (NetG1) on NetG1+ CAFs and NetG1 ligand on tumor cells were highly expressed, resulting in the provision of glutamate/glutamine to tumor cells (61). NetG1 acts as a key regulator involved in ECM deposition, survival under low nutrient conditions and immunosuppression through the regulation of downstream pathways p38/FRA1 and AKT/4E-BP1 (61). PSCs increase glutamine synthetase expression by regulating the Wnt/β-catenin/TCF7 axis, thus promoting glutamine synthesis (62).
In addition to glutamine, alanine also acts as an important carbon source in the TCA of tumor cells. Tumor cells stimulate CAFs to catabolize metabolized proteins through autophagy to produce alanine and transaminate it to pyruvate (63). When glutamine is depleted, CAFs take up extracellular proteins through CaMKK2-AMPK-RAC1 signaling-dependent macropinocytosis and supply the produced amino acids to tumor cells (64). The macropinocytosis recovers CAFs to restore the production of collagen VI and fibronectin which is inhibited during glutamine depletion (64). In addition, the study also showed that protein-derived alanine was a secreted amino acid when serum albumin was cultured as a nutritional source for PSCs (64). It was demonstrated that pancreatic cancer cells and PSCs express SLC38A2 and SLC1A4 respectively to perform alanine exchange so as to meet the high alanine requirement of pancreatic cancer cells (65).
Furthermore, branched-chain amino acids (BCAAs), also known as leucine, isoleucine and valine, participate in metabolic reprogramming and crosstalk in CAFs and pancreatic cancer cells (66). Branched-chain amino acid transaminases (BCATs) can reversibly catalyze the transamination reaction of BCAAs to branched-chain α-keto acids (BCKAs) (67). TGF-β secreted by cancer cells upregulates BCAT1 activity by activating SMAD5 in CAFs, thereby increasing the secretion of BCKAs, which are supplied to cancer cells for BCAA synthesis (68).
Lipids form an important part of cellular biological membranes and building blocks, and are also involved in signaling and supplying energy (69). Multiple studies have demonstrated the existence of lipid metabolic reprogramming of CAFs in different cancer types, but regrettably there are not enough research in pancreatic cancer (70–72). PSCs undergo lipidomic remodeling upon activation, releasing lysophosphatidylcholine in large quantities to promote migration and proliferation of PDAC cells via the lysophosphatidylcholine-autotaxin-lysophosphatidic acid axis (73). Recently, a study found that activation of one PSC subpopulation is associated with elevated expression of lipoprotein-uptake very low-density lipoprotein receptor, which drives the expression of IL-33 (74). ROS-induced, endoplasmic reticulum stress-dependent increase in IL-33 expression mediates innate lymphoid type-2 cells activation, which induces proliferation and activation of PSCs, thereby stimulating pancreas fibrosis (74).
CAFs take part in multiple stages of tumor progression, enabling bidirectional communication with other cells in the TME through intercellular contacts, secreted proteins and extracellular vesicles (75). Tumor cells signal CAFs to activate or secrete cytokines and matrix proteins, while CAFs promote drug resistance, proliferation, and migration of tumor cells. Here, we mainly summarize the signals from tumor cells that are significant for fibrosis.
Extracellular vesicles are a form of intercellular communication that is currently of great interest. They are classified as prostasomes, apoptotic bodies, microvesicles and exosomes due to their size and origin (76). Exosomes contain a variety of nucleic acids(DNA, microRNA, lncRNA, circRNA), proteins, lipids and cytokines (77). We mention the ability of cancer cells to initiate metabolic reprogramming of CAFs, allowing them to provide nutrients to cancer cells. CD9, a specific exosome marker present on the surface of extracellular vesicles rich in annexin A6, enhances p38 mitogen-activated protein kinase signaling to induce PDAC cell migration (78, 79). Exosomes derived from PDAC cells expressing oncogenic KRAS mutants contain Survivin, imparting cell survival benefits to nearby CAFs (80). MiR-1246 and miR-1290 contained in pancreatic cancer cell-derived exosomes promote the expression of profibrogenic genes in PSCs (81).
Mutated KRAS induces upregulation of plasminogen activator inhibitor-1 (PAI-1) in pancreatic cancer cells which induces PSCs activation via LRP-1/ERK/c-JUN pathway to promote immunosuppression and fibrosis (82). Meanwhile, PAI-1 expression was regulated by acyl-CoA synthetase long-chain 3, which may be associated with the regulation of TGF-β (83). High expression of PAI-1 not only promoted PSCs activation but also was associated with a high tumor infiltration of M2 macrophages (83). TGF-β1 represents a critical factor in the activation of PSCs. The secretion of TGF-β1 in pancreatic cancer cells is modulated by proteasome activator subunit 3-mediated activation protein-1, thus regulating the proliferation of PSCs (84). The induction of CAFs by TGF-β1 can be indirect, mediated through extracellular matrix proteins and growth factors such as PDGF, vascular endothelial growth factor (VEGF) and IL-6 (85). PDGF activates the hippo pathway and adds phosphorylation of yes-associated protein 1 in PSCs, and yes-associated protein 1 regulates the transcription of genes triggered by the TGF-β1/SMAD pathway, such as connective tissue growth factor and IL-6 (86). It has been shown that overexpression of galectin-1 stimulates the TGF-β1/Smad signaling pathway, with tissue inhibitor of metalloproteinase-1 (TIMP-1) expression increasing more than matrix metallopeptidase (MMP) 2, resulting in inhibition of ECM degradation and increased expression of fibronectin, collagen I and α-SMA (87). In addition, the paracrine of galectin-1 enhances the tumorigenic capacity of pancreatic epithelial cells (88). CXCL12/CXCR4 participates in the fibrotic process and the conversion of fibroblasts to myofibroblasts in multiple organs (89). Tumor-produced lactate causes epigenomic reprogramming when mesenchymal stem cells differentiate into CAFs (90). The increase of α-ketoglutarate causes C-X-C motif chemokine receptor 4 (CXCR4) promoter demethylation, leading to CXCR4 upregulation (90). Increase of special AT-rich sequence-binding protein 1 (SATB-1) expression in pancreatic cancer cells by CAFs through the SDF-1/CXCR4 axis further promotes CAFs activation (91). Furthermore, it has been established that tumor cells and CAFs crosstalk through nuclear factor KB (NF-κB) activated by paracrine-IL-1β. NF-κB activation by tumor-secreted IL-1β induces the expression of ESE3 in PSCs, then epithelium-specific E-twenty six factor 3 (ESE3) binds to the promoters of α-SMA, collagen-I and IL-1β, activating PSCs and promoting PDAC fibrosis (92). PDAC cells secrete IL-1β to activate IL-1 receptor-associated kinase 4 (IRAK4) in CAFs, forming an IL1β-IRAK4 feedforward circuitry that initiates fibrotic function in CAFs (93).
Autophagy refers to a catabolic process to maintain intracellular homeostasis (94). But there is growing proof that autophagy takes part in the process of cellular secretion (95). Meanwhile, tumor cells are capable of secreting cytokines to induce autophagy in PSCs (63, 96). TGF-β1/Smad signaling-mediated autophagy promotes the conversion of fibroblasts to CAFs and facilitates their glycolysis (97). Activation of PSCs depends on autophagy, which is associated with the production of ECM and the secretion of IL-6 (96). CAFs conduct ribosomal RNA autophagy in a nuclear fragile X mental retardation-interacting protein 1 (NUFIP1)-dependent way, producing nucleosides available for PDAC cells under low nutrient conditions and initiating metabolic reprogramming (98). Collagen secretion can be facilitated by the mitophagy-regulated synthesis of proline in CAFs (99). In addition, a recent study found that PDAC cells generate lnc-FSD2-31:1 to promote the autophagy of CAFs via miR-4736, thereby inhibiting the activation of CAFs (100).
In TME, CAFs secrete large amounts of ECM proteins and remodeling enzymes to reorganize and stiffen the matrix (101). The main contribution of tumor cells to ECM deposition is the recruitment and activation of stromal cells. Multiple pathways of intercellular communication including protein secretion and extracellular vesicles enable pancreatic cancer cells to regulate the cellular activities of CAFs. Cancer cells are involved in the cross-linked sclerosis and degradation of ECM, aiding their invasion and migration from different aspects. Pancreatic cancer cells rely on multiple cytokines such as TGF-β, IL-1, sonic hedgehog (SHH), and microRNAs to activate CAFs and thus promote ECM stiffness (102). Meanwhile, pancreatic cancer cells also produce enzymes to promote matrix protein cross-linking in ECM such as lysyl oxidase-like protein 2 (LOXL2) (103). We summarize the cytokines and modes of action associated with fibrosis during the crosstalk between pancreatic cancer cells and CAFs (Table 1).
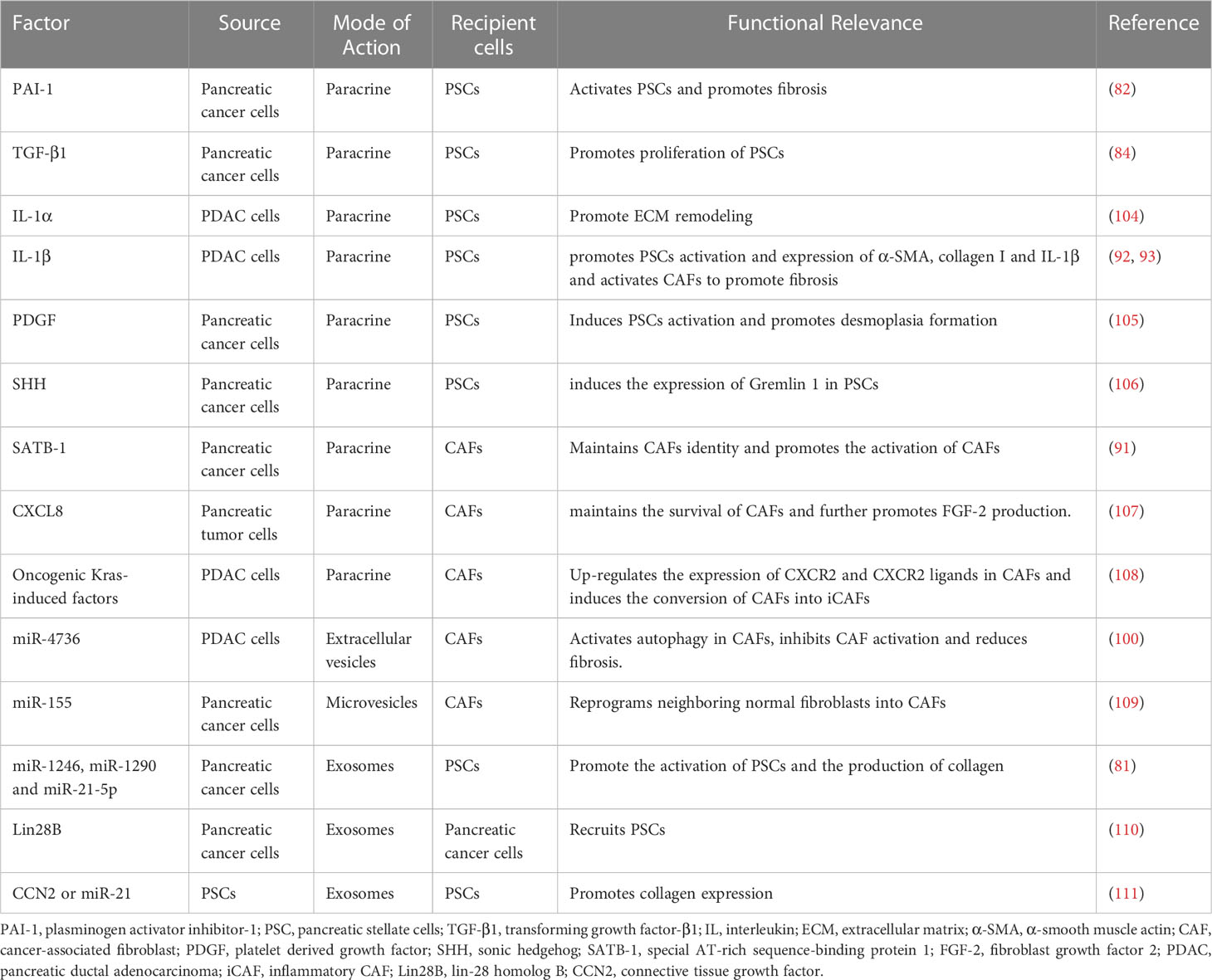
Table 1 Overview of the impact of crosstalk between CAFs and tumor cells on fibrosis.
Obesity is a critical independent risk factor for pancreatic cancer and is consistently associated with the development of pancreatic cancer. Obesity leads to hypertrophy and hyperplasia of adipocytes and causes chronic inflammation of the adipose tissue around or within the pancreas, which promotes tumor progression (112). And along with the advancement of pancreatic cancer stages, patients with pancreatic cancer experience adipose tissue loss as one of the manifestations of cachexia (113). Adipose tissue is divided into white, brown, and beige adipose tissue, while white adipose tissue is further classified into subcutaneous white adipose tissue and visceral white adipose tissue, with the latter playing a more pivotal role in the progression of pancreatic cancer (114). The cellular composition of white adipose tissue includes adipocytes, preadipocytes, immune cells, pericytes, endothelial cells, and multipotent stem cells (115). Some researches demonstrated the correlation of adipose tissue with fibroblast transformation and the formation and remodeling of ECM.
On the one hand, cells in adipose tissue have the ability to be reprogrammed into CAFs by pancreatic cancer cells. Adrenomedullin in the exosomes of pancreatic cancer cells promotes lipolysis in adipocytes (116). The lipolysis may explain the weight loss of the patients and represents a phenomenon of adipocytes dedifferentiation. Consequently, the dedifferentiation possibly connects the cachexia with fibroplasia in pancreatic cancer. When co-cultured with pancreatic cancer cells, 3T3-L1 adipocytes dedifferentiated to fibroblast-like cells, losing lipid droplets and expressing S100A4, MMP11, collagen I and α-SMA (117, 118). The reprogramming is closely correlated with WNT5a signaling (119). Adipose tissue-derived stromal cells can also be recruited to extrapancreatic invasive lesions and differentiate into CAFs, producing a more rigid ECM (120). Mucin 5AC secreted by pancreatic cancer cells recruits mesenchymal stem cells (MSCs) to a-SMA+ CAFs (121). Activin A produced by PDAC cells was found to be associated with the loss of adipose tissue and the promotion of fibrosis, with an induction of trans-differentiation of white adipocytes into fibrotic cells (122). On the other hand, adipocytes mediate fibrosis by crosstalk with neighboring cells via paracrine secretion. Adipocytes secrete IL-1β to recruit neutrophils, thereby enhancing the activation of PSCs (123).
Ideas for targeting CAFs as therapeutic targets in pancreatic cancer for clinical benefit are diverse including depletion of CAFs, reprogramming CAFs to make them normal, and blocking signals from CAFs (Figure 3) (124). However, studies concerning the depletion of CAFs demonstrated that this treatment could lead to the exact opposite of what was expected, a facilitation of tumor progression (125, 126). Reprogramming CAFs to the stationary case is currently considered a feasible approach. It has been preliminarily demonstrated to be viable to normalize CAFs through endogenous substances, gene regulation, agents and intercellular interactions. Lipoxin A4 reversed the activation of PSCs to CAFs for matrix reprogramming, with decreased expression of α-SMA and collagen I (127). The increase of retinoic acid was able to inhibit CAFs and reduce the expression of α-SMA and FAP (128). Zhao et al. constructed a targeted drug delivery system based on red blood cells vesicles partial protection to deliver retinoic acid to CAFs to disrupt the Golgi apparatus and thereby inhibit the secretion of proteins such as MMP2, MMP9 and CCL2 (129). In addition, all-transretinoic acid inactivated PSCs by inhibiting Yes-associated protein 1 (YAP1) (130). Vitamin D and its receptor were involved in stromal reprogramming as well by inactivating CAF/PSC (131, 132). The activation of p53 could directly induce the accumulation of cytoplasmic lipid droplets in PSCs, thus effectively reprogramming PSCs to a quiescent state (133). Integrin subtype α 11 was also considered as a viable target for controlling the phenotype and activation of PSCs (134). Several studies have shown that metformin can reprogram PSCs to improve desmoplasia (135–137). Metformin inhibited TGF-β1 secretion by activating AMPK in pancreatic cancer cells, leading to blocking the activation of PSCs (136). In addition, eribulin also showed potential for normalizing CAFs due to its simulation of TGF-β downregulation (138). Mechanical regulation of intercellular interactions such as N-cadherin and N-cadherin ligand linkages could reprogram PSCs to a stationary state, however not in all cases, as this reprogramming was associated with mechanical dosing (139). Unfortunately, studies on the regulation of the metabolism of CAFs are scarce, because the mechanism of metabolic reprogramming of CAFs is still not entirely clarified. Chen et al. designed a liposome carrying hydroxychloroquine and paclitaxel to target autophagy in CAFs, with advantages for crosstalk and fibrosis inhibition (140). A biomimetic nanocarrier was devised to disrupt metabolic crosstalk by blocking lactate production in both CAFs and cancer cells (141).
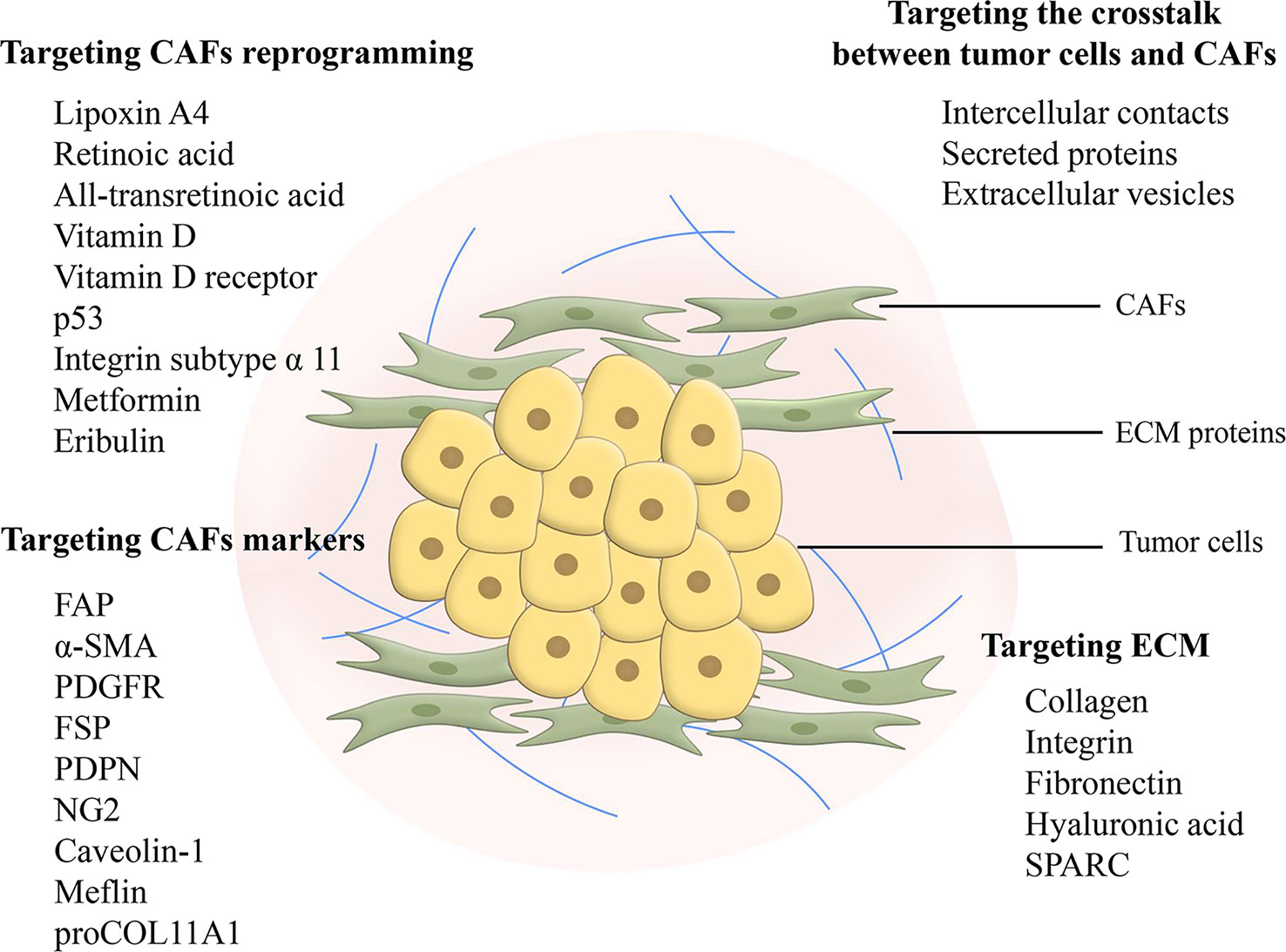
Figure 3 Feasible design ideas for targeting fibrosis. Targeting CAFs in metabolic reprogramming and signaling communication with cancer cells is considered as a promising therapeutic modality. Targeting markers of CAFs can inhibit CAFs, but there is a problem of non-specificity and further search and research is still needed. Targeting ECM elimination requires limitations, as sustained defibrosis implies enhanced invasion. In addition, ECM protein interactions may become an emerging therapeutic target. CAF, cancer-associated fibroblast; FAP, fibroblast activation protein; α-SMA, α-smooth muscle actin; PDGFR, platelet-derived growth factor receptor; FSP, fibroblast-specific protein; PDPN, podoplanin; NG2, nerve/glial antigen 2; ECM, extracellular matrix; SPARC, secreted protein acidic and rich in cysteine.
CAF synthesizes, remodels and crosslinks ECM to increase stiffness leading to the generation of a dense fibrotic tumor stroma (101). CAFs act in pancreatic cancer progression as an essential component of the stroma. Five subtypes of CAFs have been identified so far, namely myCAFs, iCAFs, apCAFs, meCAFs and csCAFs, showing differences in expression and function in pancreatic cancer. This is still not the endpoint of the classification of CAFs, and the subtypes may contain subpopulations. As we mentioned before, different subpopulations of iCAFs may have opposite effects on tumor development. It implies that therapies targeting CAFs need more specific biomarkers. Different subtypes of CAFs relate to the discrepant prognosis of pancreatic cancer patients (24, 28).
Cells in the TME interact with each other to co-construct a microenvironment suitable for tumor survival. CAFs conduct metabolic reprogramming to provide available metabolites to tumor cells (Table 2). Oxidative stressed-driven metabolic changes in CAFs are known as the reverse Warburg effect, manifested by glycolysis as the main mode of metabolism and increased utilization of glutamine. Multiple forms of crosstalk including direct contact, extracellular vesicles, paracrine and autophagy-dependent secretion between tumor cells and CAFs activate CAFs for fibrosis on the one hand and enhance tumor cells proliferation and migration on the other. Cellular communication also exists between adipocytes and other cells in the TME. Lipolysis occurs when adipocytes dedifferentiate into CAFs, which perhaps partially explains both the cachexia and desmoplasia.
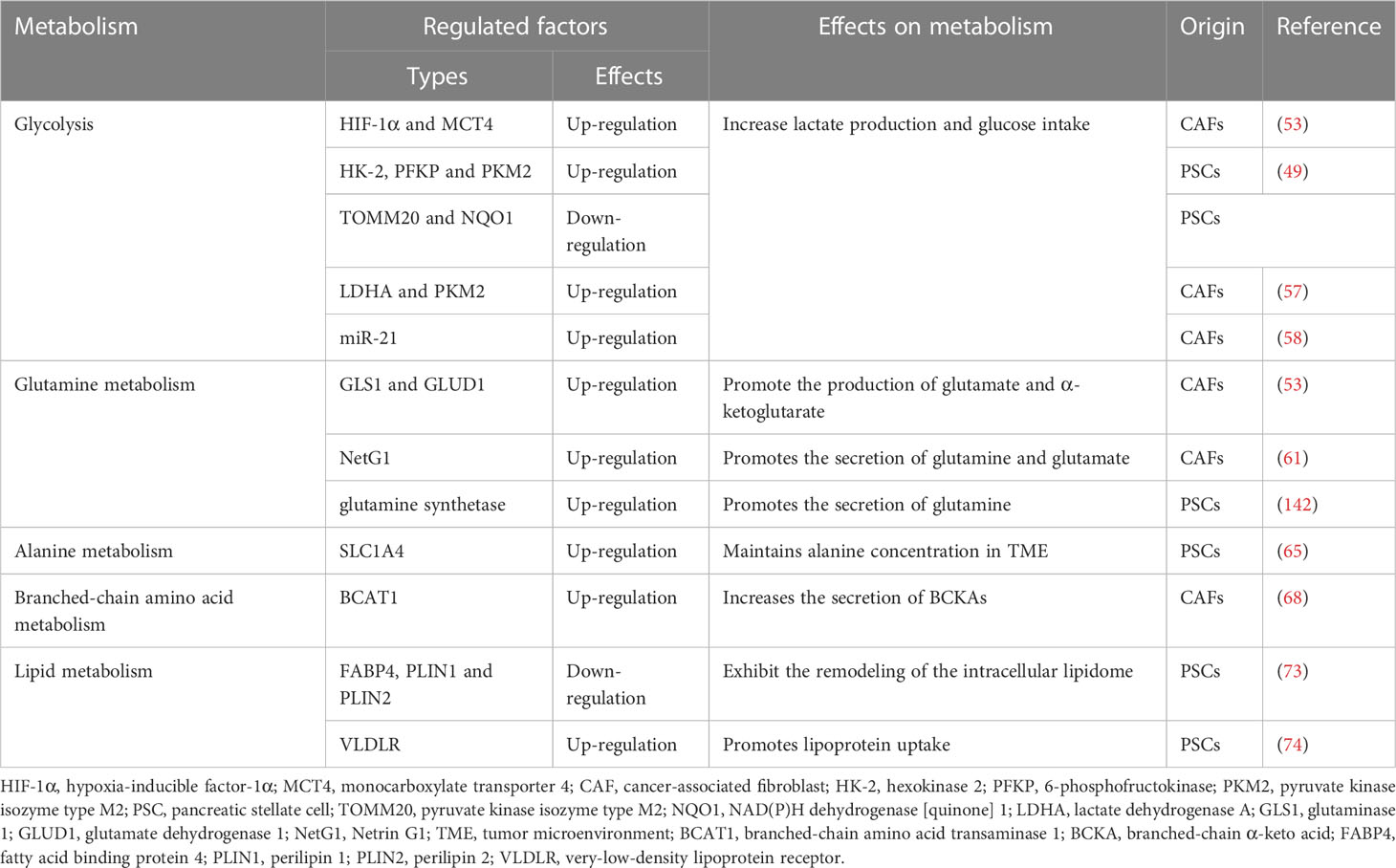
Table 2 Summary of CAFs metabolic reprogramming.
Experimentally, depletion of CAFs proved to be an infeasible treatment. Reprogramming CAFs to a normal state or blocking signaling may be promising ways to target pancreatic cancer fibrosis. In conclusion, CAFs are important targets to explain fibrosis and drug resistance in pancreatic cancer, but further studies on the heterogeneity of CAFs and the mechanisms of crosstalk are still needed to provide more basis for targeting CAFs for therapy.
The manuscript was written by XL, JZ, XW, ZM, CL and QW. XL and XW designed and made the figures. FP and JZ revised the manuscript. FP supported the study. All authors contributed to the article and approved the submitted version.
The study was supported by National Natural Science Foundation of China (no.82003879 and U19A2010), the Key Project of Science and Technology Department of Sichuan Province (no. 2020YFS0053; 2021YFS0044), and Youth Talent Promotion Project of China Association for Science and Technology (CACM-2020-QNRC1-01) and Project of State Administration of Traditional Chinese Medicine of China (ZYYCXTD-D-202209).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
TME, tumor microenvironment; ECM, extracellular matrix; CAF, cancer-associated fibroblast; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cell; myCAF, myofibroblastic CAF; iCAF, inflammatory CAF; α-SMA, α-smooth muscle actin; IL, interleukin; apCAF, antigen-presenting CAF; meCAF, CAF with a highly activated metabolic state; csCAF, complement-secreting CAF; LIF, leukemia inhibitory factor; TGF-β, transforming growth factor-β; Tregs, regulatory T cells; PDGF, platelet derived growth factor; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; TCA, tricarboxylic acid; HIF, hypoxia-inducible factor; MCT, monocarboxylate transporter; NetG1, Netrin G1; BCAA, branched-chain amino acid; BCAT, branched-chain amino acid transaminase; BCKA, branched-chain α-keto acid; PAI-1, plasminogen activator inhibitor-1; VEGF, vascular endothelial growth factor; TIMP-1, tissue inhibitor of metalloproteinase-1; MMP, matrix metallopeptidase; CECR4, C-X-C motif chemokine receptor 4; SATB-1, special AT-rich sequence-binding protein 1; NF-κB, nuclear factor KB; ESE3, epithelium-specific E-twenty six factor 3; IRAK4, IL-1 receptor-associated kinase 4; NUFIP1, nuclear fragile X mental retardation-interacting protein 1; SHH, sonic hedgehog; LOXL2, lysyl oxidase-like protein 2; MSC, mesenchymal stem cell; YAP1, Yes-associated protein 1.
1. Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol (2021) 18(7):493–502. doi: 10.1038/s41575-021-00457-x
2. Jungmann F, Kaissis GA, Ziegelmayer S, Harder F, Schilling C, Yen HY, et al. Prediction of tumor cellularity in resectable PDAC from preoperative computed tomography imaging. Cancers (Basel) (2021) 13(9):2069. doi: 10.3390/cancers13092069
3. Quoc Lam B, Shrivastava SK, Shrivastava A, Shankar S, Srivastava RK. The impact of obesity and diabetes mellitus on pancreatic cancer: Molecular mechanisms and clinical perspectives. J Cell Mol Med (2020) 24(14):7706–16. doi: 10.1111/jcmm.15413
4. Mukherjee AG, Wanjari UR, Gopalakrishnan AV, Bradu P, Sukumar A, Patil M, et al. Implications of cancer stem cells in diabetes and pancreatic cancer. Life Sci (2023) 312:121211. doi: 10.1016/j.lfs.2022.121211
5. Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, et al. Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes (2017) 66(5):1103–10. doi: 10.2337/db16-1477
6. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet (2020) 395(10242):2008–20. doi: 10.1016/S0140-6736(20)30974-0
7. Jiang S, Fagman JB, Ma Y, Liu J, Vihav C, Engstrom C, et al. A comprehensive review of pancreatic cancer and its therapeutic challenges. Aging (Albany NY) (2022) 14(18):7635–49. doi: 10.18632/aging.204310
8. Wood LD, Canto MI, Jaffee EM, Simeone DM. Pancreatic cancer: Pathogenesis, screening, diagnosis, and treatment. Gastroenterology (2022) 163(2):386–402.e381. doi: 10.1053/j.gastro.2022.03.056
9. Affo S, Yu LX, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol (2017) 12:153–86. doi: 10.1146/annurev-pathol-052016-100322
10. Kinoshita T, Goto T. Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: A review. Int J Mol Sci (2019) 20(6):1461. doi: 10.3390/ijms20061461
11. Huang C, Iovanna J, Santofimia-Castaño P. Targeting fibrosis: The bridge that connects pancreatitis and pancreatic cancer. Int J Mol Sci (2021) 22(9):4970. doi: 10.3390/ijms22094970
12. Abbas O, Mahalingam M. Desmoplasia: not always a bad thing. Histopathology (2011) 58(5):643–59. doi: 10.1111/j.1365-2559.2010.03617.x
13. Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: A complex relationship. Cancers (Basel) (2018) 10(9):316. doi: 10.3390/cancers10090316
14. Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res (2015) 3(1):1–11. doi: 10.1158/2326-6066.CIR-14-0209
15. Tracy LE, Minasian RA, Caterson EJ. Extracellular matrix and dermal fibroblast function in the healing wound. Adv Wound Care (New Rochelle) (2016) 5(3):119–36. doi: 10.1089/wound.2014.0561
16. McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol (2014) 5:141. doi: 10.3389/fphys.2014.00141
17. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res (2012) 18(16):4266–76. doi: 10.1158/1078-0432.CCR-11-3114
18. Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer (2019) 18(1):14. doi: 10.1186/s12943-018-0927-5
19. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - accomplices in tumor malignancy. Cell Immunol (2019) 343:103729. doi: 10.1016/j.cellimm.2017.12.003
20. Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev (2021) 302(1):259–72. doi: 10.1111/imr.12978
21. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
22. Dings MPG, Manoukian P, Waasdorp C, Quik JSE, Strijker M, Lodestijn SC, et al. Serum levels of iCAF-derived osteoglycin predict favorable outcome in pancreatic cancer. Int J Cancer (2023) 152(3):511–23. doi: 10.1002/ijc.34276
23. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
24. Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discovery (2021) 7(1):36. doi: 10.1038/s41421-021-00271-4
25. Chen K, Wang Q, Li M, Guo H, Liu W, Wang F, et al. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine (2021) 66:103315. doi: 10.1016/j.ebiom.2021.103315
26. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
27. Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin Cancer Res (2021) 27(7):2023–37. doi: 10.1158/1078-0432.CCR-20-3715
28. Hu B, Wu C, Mao H, Gu H, Dong H, Yan J, et al. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann Transl Med (2022) 10(5):262. doi: 10.21037/atm-22-407
29. Neuzillet C, Tijeras-Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol (2019) 248(1):51–65. doi: 10.1002/path.5224
30. Neuzillet C, Nicolle R, Raffenne J, Tijeras-Raballand A, Brunel A, Astorgues-Xerri L, et al. Periostin- and podoplanin-positive cancer-associated fibroblast subtypes cooperate to shape the inflamed tumor microenvironment in aggressive pancreatic adenocarcinoma. J Pathol (2022) 258(4):408–25. doi: 10.1002/path.6011
31. Desbois M, Wang Y. Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol Rev (2021) 302(1):241–58. doi: 10.1111/imr.12982
32. Zheng S, Hu C, Lin H, Li G, Xia R, Zhang X, et al. circCUL2 induces an inflammatory CAF phenotype in pancreatic ductal adenocarcinoma via the activation of the MyD88-dependent NF-κB signaling pathway. J Exp Clin Cancer Res (2022) 41(1):71. doi: 10.1186/s13046-021-02237-6
33. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell (2022) 40(6):656–673.e657. doi: 10.1016/j.ccell.2022.04.011
34. Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal lineage heterogeneity underlies nonredundant functions of pancreatic cancer-associated fibroblasts. Cancer Discovery (2022) 12(2):484–501. doi: 10.1158/2159-8290.CD-21-0601
35. Watari N, Hotta Y, Mabuchi Y. Morphological studies on a vitamin a-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin a administration. Okajimas Folia Anat Jpn (1982) 58(4-6):837–58. doi: 10.2535/ofaj1936.58.4-6_837
36. Zhou Y, Zhou J, Sun B, Xu W, Zhong M, Li Y, et al. Vitamin a deficiency causes islet dysfunction by inducing islet stellate cell activation via cellular retinol binding protein 1. Int J Biol Sci (2020) 16(6):947–56. doi: 10.7150/ijbs.37861
37. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut (1999) 44(4):534–41. doi: 10.1136/gut.44.4.534
38. Xu XF, Liu F, Xin JQ, Fan JW, Wu N, Zhu LJ, et al. Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-β1 (TGF-β1). Biochem Biophys Res Commun (2018) 501(2):365–73. doi: 10.1016/j.bbrc.2018.04.176
39. Zheng M, Li H, Sun L, Brigstock DR, Gao R. Interleukin-6 participates in human pancreatic stellate cell activation and collagen I production via TGF-β1/Smad pathway. Cytokine (2021) 143:155536. doi: 10.1016/j.cyto.2021.155536
40. Ng B, Viswanathan S, Widjaja AA, Lim WW, Shekeran SG, Goh JWT, et al. IL11 activates pancreatic stellate cells and causes pancreatic inflammation, fibrosis and atrophy in a mouse model of pancreatitis. Int J Mol Sci (2022) 23(7):3549. doi: 10.3390/ijms23073549
41. Vaupel P, Schmidberger H, Mayer A. The warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
42. Vaupel P, Multhoff G. Revisiting the warburg effect: historical dogma versus current understanding. J Physiol (2021) 599(6):1745–57. doi: 10.1113/JP278810
43. Zhu Y, Li X, Wang L, Hong X, Yang J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front Endocrinol (Lausanne) (2022) 13:988295. doi: 10.3389/fendo.2022.988295
44. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci (2019) 20(2:238). doi: 10.3390/ijms20020238
45. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U.S.A. (1995) 92(12):5510–4. doi: 10.1073/pnas.92.12.5510
46. Ratcliffe PJ, O'Rourke JF, Maxwell PH, Pugh CW. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol (1998) 201(Pt 8):1153–62. doi: 10.1242/jeb.201.8.1153
47. Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle (2012) 11(12):2272–84. doi: 10.4161/cc.20717
48. Arcucci A, Ruocco MR, Granato G, Sacco AM, Montagnani S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. BioMed Res Int (2016) 2016:4502846. doi: 10.1155/2016/4502846
49. Shao S, Qin T, Qian W, Yue Y, Xiao Y, Li X, et al. Positive feedback in cav-1-ROS signalling in PSCs mediates metabolic coupling between PSCs and tumour cells. J Cell Mol Med (2020) 24(16):9397–408. doi: 10.1111/jcmm.15596
50. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
51. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett (2016) 380(1):272–80. doi: 10.1016/j.canlet.2015.10.027
52. Avagliano A, Granato G, Ruocco MR, Romano V, Belviso I, Carfora A, et al. Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. BioMed Res Int (2018) 2018:6075403. doi: 10.1155/2018/6075403
53. Knudsen ES, Balaji U, Freinkman E, McCue P, Witkiewicz AK. Unique metabolic features of pancreatic cancer stroma: relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget (2016) 7(48):78396–411. doi: 10.18632/oncotarget.11893
54. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol (2021) 599(1):23–37. doi: 10.1113/JP280572
55. Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab (2020) 33:48–66. doi: 10.1016/j.molmet.2019.07.006
56. Halestrap AP. The SLC16 gene family - structure, role and regulation in health and disease. Mol Aspects Med (2013) 34(2-3):337–49. doi: 10.1016/j.mam.2012.05.003
57. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J, et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol Rep (2017) 37(4):1971–9. doi: 10.3892/or.2017.5479
58. Chen S, Chen X, Shan T, Ma J, Lin W, Li W, et al. MiR-21-mediated metabolic alteration of cancer-associated fibroblasts and its effect on pancreatic cancer cell behavior. Int J Biol Sci (2018) 14(1):100–10. doi: 10.7150/ijbs.22555
59. Qin C, Yang G, Yang J, Ren B, Wang H, Chen G, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer (2020) 19(1):50. doi: 10.1186/s12943-020-01169-7
60. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496(7443):101–5. doi: 10.1038/nature12040
61. Francescone R, Barbosa Vendramini-Costa D, Franco-Barraza J, Wagner J, Muir A, Lau AN, et al. Netrin G1 promotes pancreatic tumorigenesis through cancer-associated fibroblast-driven nutritional support and immunosuppression. Cancer Discovery (2021) 11(2):446–79. doi: 10.1158/2159-8290.CD-20-0775
62. Liu H, Zhang H, Liu X, Guo W, Liu Q, Chen L, et al. Pancreatic stellate cells exploit wnt/β-catenin/TCF7-mediated glutamine metabolism to promote pancreatic cancer cells growth. Cancer Lett (2022) 555:216040. doi: 10.1016/j.canlet.2022.216040
63. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
64. Zhang Y, Recouvreux MV, Jung M, Galenkamp KMO, Li Y, Zagnitko O, et al. Macropinocytosis in cancer-associated fibroblasts is dependent on CaMKK2/ARHGEF2 signaling and functions to support tumor and stromal cell fitness. Cancer Discovery (2021) 11(7):1808–25. doi: 10.1158/2159-8290.CD-20-0119
65. Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnación-Rosado J, et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discovery (2020) 10(7):1018–37. doi: 10.1158/2159-8290.CD-19-0959
66. Sivanand S, Vander Heiden MG. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell (2020) 37(2):147–56. doi: 10.1016/j.ccell.2019.12.011
67. Peng H, Wang Y, Luo W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene (2020) 39(44):6747–56. doi: 10.1038/s41388-020-01480-z
68. Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, et al. Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab (2020) 2(8):775–92. doi: 10.1038/s42255-020-0226-5
69. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med (2021) 218(1):e20201606. doi: 10.1084/jem.20201606
70. Nardi F, Fitchev P, Franco OE, Ivanisevic J, Scheibler A, Hayward SW, et al. PEDF regulates plasticity of a novel lipid-MTOC axis in prostate cancer-associated fibroblasts. J Cell Sci (2018) 131(13):jcs213579. doi: 10.1242/jcs.213579
71. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis (2020) 11(4):267. doi: 10.1038/s41419-020-2434-z
72. Hwang SH, Yang Y, Jung JH, Kim Y. Oleic acid from cancer-associated fibroblast promotes cancer cell stemness by stearoyl-CoA desaturase under glucose-deficient condition. Cancer Cell Int (2022) 22(1):404. doi: 10.1186/s12935-022-02824-3
73. Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discovery (2019) 9(5):617–27. doi: 10.1158/2159-8290.CD-18-1212
74. Yang X, Chen J, Wang J, Ma S, Feng W, Wu Z, et al. Very-low-density lipoprotein receptor-enhanced lipid metabolism in pancreatic stellate cells promotes pancreatic fibrosis. Immunity (2022) 55(7):1185–1199.e1188. doi: 10.1016/j.immuni.2022.06.001
75. Chang CH, Pauklin S. Extracellular vesicles in pancreatic cancer progression and therapies. Cell Death Dis (2021) 12(11):973. doi: 10.1038/s41419-021-04258-7
76. Urabe F, Kosaka N, Ito K, Kimura T, Egawa S, Ochiya T. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am J Physiol Cell Physiol (2020) 318(1):C29–c39. doi: 10.1152/ajpcell.00280.2019
77. Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther (2020) 5(1):145. doi: 10.1038/s41392-020-00261-0
78. Brosseau C, Colas L, Magnan A, Brouard S. CD9 tetraspanin: A new pathway for the regulation of inflammation? Front Immunol (2018) 9:2316. doi: 10.3389/fimmu.2018.02316
79. Nigri J, Leca J, Tubiana SS, Finetti P, Guillaumond F, Martinez S, et al. CD9 mediates the uptake of extracellular vesicles from cancer-associated fibroblasts that promote pancreatic cancer cell aggressiveness. Sci Signal (2022) 15(745):eabg8191. doi: 10.1126/scisignal.abg8191
80. Chang WH, Nguyen TT, Hsu CH, Bryant KL, Kim HJ, Ying H, et al. KRAS-dependent cancer cells promote survival by producing exosomes enriched in survivin. Cancer Lett (2021) 517:66–77. doi: 10.1016/j.canlet.2021.05.031
81. Masamune A, Yoshida N, Hamada S, Takikawa T, Nabeshima T, Shimosegawa T. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem Biophys Res Commun (2018) 495(1):71–7. doi: 10.1016/j.bbrc.2017.10.141
82. Wang HC, Lin YL, Hsu CC, Chao YJ, Hou YC, Chiu TJ, et al. Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics (2019) 9(24):7168–83. doi: 10.7150/thno.36830
83. Rossi Sebastiano M, Pozzato C, Saliakoura M, Yang Z, Peng RW, Galiè M, et al. ACSL3-PAI-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci Adv (2020) 6(44):eabb9200. doi: 10.1126/sciadv.abb9200
84. Yu L, Li JJ, Liang XL, Wu H, Liang Z. PSME3 promotes TGFB1 secretion by pancreatic cancer cells to induce pancreatic stellate cell proliferation. J Cancer (2019) 10(9):2128–38. doi: 10.7150/jca.30235
85. Heneberg P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit Rev Oncol Hematol (2016) 97:303–11. doi: 10.1016/j.critrevonc.2015.09.008
86. Hu C, Yang J, Su HY, Waldron RT, Zhi M, Li L, et al. Yes-associated protein 1 plays major roles in pancreatic stellate cell activation and fibroinflammatory responses. Front Physiol (2019) 10:1467. doi: 10.3389/fphys.2019.01467
87. Tang D, Wu Q, Zhang J, Zhang H, Yuan Z, Xu J, et al. Galectin-1 expression in activated pancreatic satellite cells promotes fibrosis in chronic pancreatitis/pancreatic cancer via the TGF-β1/Smad pathway. Oncol Rep (2018) 39(3):1347–55. doi: 10.3892/or.2018.6202
88. Orozco CA, Martinez-Bosch N, Guerrero PE, Vinaixa J, Dalotto-Moreno T, Iglesias M, et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc Natl Acad Sci U.S.A. (2018) 115(16):E3769–e3778. doi: 10.1073/pnas.1722434115
89. Wu X, Qian L, Zhao H, Lei W, Liu Y, Xu X, et al. CXCL12/CXCR4: An amazing challenge and opportunity in the fight against fibrosis. Ageing Res Rev (2023) 83:101809. doi: 10.1016/j.arr.2022.101809
90. Bhagat TD, Von Ahrens D, Dawlaty M, Zou Y, Baddour J, Achreja A, et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife (2019) 8:e50663. doi: 10.7554/eLife.50663
91. Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, et al. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis (2018) 9(11):1065. doi: 10.1038/s41419-018-1104-x
92. Zhao T, Xiao D, Jin F, Sun X, Yu J, Wang H, et al. ESE3-positive PSCs drive pancreatic cancer fibrosis, chemoresistance and poor prognosis via tumour-stromal IL-1β/NF-κB/ESE3 signalling axis. Br J Cancer (2022) 127(8):1461–72. doi: 10.1038/s41416-022-01927-y
93. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, et al. Tumor-stroma IL1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res (2018) 78(7):1700–12. doi: 10.1158/0008-5472.CAN-17-1366
94. Cao W, Li J, Yang K, Cao D. An overview of autophagy: Mechanism, regulation and research progress. Bull Cancer (2021) 108(3):304–22. doi: 10.1016/j.bulcan.2020.11.004
95. New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy (2019) 15(10):1682–93. doi: 10.1080/15548627.2019.1596479
96. Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology (2017) 152(6):1492–1506.e1424. doi: 10.1053/j.gastro.2017.01.010
97. Jena BC, Das CK, Banerjee I, Bharadwaj D, Majumder R, Das S, et al. TGF-β1 induced autophagy in cancer associated fibroblasts during hypoxia contributes EMT and glycolysis via MCT4 upregulation. Exp Cell Res (2022) 417(1):113195. doi: 10.1016/j.yexcr.2022.113195
98. Yuan M, Tu B, Li H, Pang H, Zhang N, Fan M, et al. Cancer-associated fibroblasts employ NUFIP1-dependent autophagy to secrete nucleosides and support pancreatic tumor growth. Nat Cancer (2022) 3(8):945–60. doi: 10.1038/s43018-022-00426-6
99. Bai J, Liu T, Tu B, Yuan M, Shu Z, Fan M, et al. Autophagy loss impedes cancer-associated fibroblast activation via downregulating proline biosynthesis. Autophagy (2023) 19(2):632–43. doi: 10.1080/15548627.2022.2093026
100. Geng X, Li L, Luo Y, Yang W, Hu J, Zhao Z, et al. Tumor cell derived lnc-FSD2-31:1 contributes to cancer-associated fibroblasts activation in pancreatic ductal adenocarcinoma progression through extracellular vesicles cargo MiR-4736. Adv Sci (Weinh) (2023)2023:e2203324. doi: 10.1002/advs.202203324
101. Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer (2020) 1873(2):188356. doi: 10.1016/j.bbcan.2020.188356
102. Zhang Z, Zhang H, Shi L, Wang D, Tang D. Heterogeneous cancer-associated fibroblasts: A new perspective for understanding immunosuppression in pancreatic cancer. Immunology (2022) 167(1):1–14. doi: 10.1111/imm.13496
103. Tanaka N, Yamada S, Sonohara F, Suenaga M, Hayashi M, Takami H, et al. Clinical implications of lysyl oxidase-like protein 2 expression in pancreatic cancer. Sci Rep (2018) 8(1):9846. doi: 10.1038/s41598-018-28253-9
104. Tjomsland V, Pomianowska E, Aasrum M, Sandnes D, Verbeke CS, Gladhaug IP. Profile of MMP and TIMP expression in human pancreatic stellate cells: Regulation by IL-1α and TGFβ and implications for migration of pancreatic cancer cells. Neoplasia (2016) 18(7):447–56. doi: 10.1016/j.neo.2016.06.003
105. Kuo TL, Cheng KH, Shan YS, Chen LT, Hung WC. β-catenin-activated autocrine PDGF/Src signaling is a therapeutic target in pancreatic cancer. Theranostics (2019) 9(2):324–36. doi: 10.7150/thno.28201
106. Yu Y, Cheng L, Yan B, Zhou C, Qian W, Xiao Y, et al. Overexpression of gremlin 1 by sonic hedgehog signaling promotes pancreatic cancer progression. Int J Oncol (2018) 53(6):2445–57. doi: 10.3892/ijo.2018.4573
107. Awaji M, Futakuchi M, Heavican T, Iqbal J, Singh RK. Cancer-associated fibroblasts enhance survival and progression of the aggressive pancreatic tumor Via FGF-2 and CXCL8. Cancer Microenviron (2019) 12(1):37–46. doi: 10.1007/s12307-019-00223-3
108. Awaji M, Saxena S, Wu L, Prajapati DR, Purohit A, Varney ML, et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J (2020) 34(7):9405–18. doi: 10.1096/fj.201902990R
109. Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci (2015) 106(10):1362–9. doi: 10.1111/cas.12747
110. Zhang YF, Zhou YZ, Zhang B, Huang SF, Li PP, He XM, et al. Pancreatic cancer-derived exosomes promoted pancreatic stellate cells recruitment by pancreatic cancer. J Cancer (2019) 10(18):4397–407. doi: 10.7150/jca.27590
111. Charrier A, Chen R, Chen L, Kemper S, Hattori T, Takigawa M, et al. Connective tissue growth factor (CCN2) and microRNA-21 are components of a positive feedback loop in pancreatic stellate cells (PSC) during chronic pancreatitis and are exported in PSC-derived exosomes. J Cell Commun Signal (2014) 8(2):147–56. doi: 10.1007/s12079-014-0220-3
112. Rebours V, Gaujoux S, d'Assignies G, Sauvanet A, Ruszniewski P, Lévy P, et al. Obesity and fatty pancreatic infiltration are risk factors for pancreatic precancerous lesions (PanIN). Clin Cancer Res (2015) 21(15):3522–8. doi: 10.1158/1078-0432.CCR-14-2385
113. Yu SY, Luan Y, Dong R, Abazarikia A, Kim SY. Adipose tissue wasting as a determinant of pancreatic cancer-related cachexia. Cancers (Basel) (2022) 14(19):4754. doi: 10.3390/cancers14194754
114. Himbert C, Delphan M, Scherer D, Bowers LW, Hursting S, Ulrich CM. Signals from the adipose microenvironment and the obesity-cancer link-a systematic review. Cancer Prev Res (Phila) (2017) 10(9):494–506. doi: 10.1158/1940-6207.CAPR-16-0322
115. Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med (2013) 34(1):1–11. doi: 10.1016/j.mam.2012.10.001
116. Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS, et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut (2016) 65(7):1165–74. doi: 10.1136/gutjnl-2014-308350
117. Cai Z, Liang Y, Xing C, Wang H, Hu P, Li J, et al. Cancer−associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol Rep (2019) 42(6):2537–49. doi: 10.3892/or.2019.7365
118. Takehara M, Sato Y, Kimura T, Noda K, Miyamoto H, Fujino Y, et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci (2020) 111(8):2883–94. doi: 10.1111/cas.14527
119. Zoico E, Darra E, Rizzatti V, Budui S, Franceschetti G, Mazzali G, et al. Adipocytes WNT5a mediated dedifferentiation: a possible target in pancreatic cancer microenvironment. Oncotarget (2016) 7(15):20223–35. doi: 10.18632/oncotarget.7936
120. Okumura T, Ohuchida K, Kibe S, Iwamoto C, Ando Y, Takesue S, et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int J Cancer (2019) 144(6):1401–13. doi: 10.1002/ijc.31775
121. Ganguly K, Cox JL, Ghersi D, Grandgenett PM, Hollingsworth MA, Jain M, et al. Mucin 5AC-mediated CD44/ITGB1 clustering mobilizes adipose-derived mesenchymal stem cells to modulate pancreatic cancer stromal heterogeneity. Gastroenterology (2022) 162(7):2032–2046.e2012. doi: 10.1053/j.gastro.2022.02.032
122. Xu PC, You M, Yu SY, Luan Y, Eldani M, Caffrey TC, et al. Visceral adipose tissue remodeling in pancreatic ductal adenocarcinoma cachexia: the role of activin a signaling. Sci Rep (2022) 12(1):1659. doi: 10.1038/s41598-022-05660-7
123. Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discovery (2016) 6(8):852–69. doi: 10.1158/2159-8290.CD-15-1177
124. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
125. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
126. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
127. Schnittert J, Heinrich MA, Kuninty PR, Storm G, Prakash J. Reprogramming tumor stroma using an endogenous lipid lipoxin A4 to treat pancreatic cancer. Cancer Lett (2018) 420:247–58. doi: 10.1016/j.canlet.2018.01.072
128. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett (2014) 345(1):132–9. doi: 10.1016/j.canlet.2013.12.006
129. Zhao T, Zhang R, He Q, Zhou H, Song X, Gong T, et al. Partial ligand shielding nanoparticles improve pancreatic ductal adenocarcinoma treatment via a multifunctional paradigm for tumor stroma reprogramming. Acta Biomater (2022) 145:122–34. doi: 10.1016/j.actbio.2022.03.050
130. Xiao Y, Zhang H, Ma Q, Huang R, Lu J, Liang X, et al. YAP1-mediated pancreatic stellate cell activation inhibits pancreatic cancer cell proliferation. Cancer Lett (2019) 462:51–60. doi: 10.1016/j.canlet.2019.07.015
131. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell (2014) 159(1):80–93. doi: 10.1016/j.cell.2014.08.007
132. Mukai Y, Yamada D, Eguchi H, Iwagami Y, Asaoka T, Noda T, et al. Vitamin d supplementation is a promising therapy for pancreatic ductal adenocarcinoma in conjunction with current chemoradiation therapy. Ann Surg Oncol (2018) 25(7):1868–79. doi: 10.1245/s10434-018-6431-8
133. Saison-Ridinger M, DelGiorno KE, Zhang T, Kraus A, French R, Jaquish D, et al. Reprogramming pancreatic stellate cells via p53 activation: A putative target for pancreatic cancer therapy. PloS One (2017) 12(12):e0189051. doi: 10.1371/journal.pone.0189051
134. Schnittert J, Bansal R, Mardhian DF, van Baarlen J, Östman A, Prakash J. Integrin α11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J (2019) 33(5):6609–21. doi: 10.1096/fj.201802336R
135. Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PloS One (2015) 10(12):e0141392. doi: 10.1371/journal.pone.0141392
136. Duan W, Chen K, Jiang Z, Chen X, Sun L, Li J, et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett (2017) 385:225–33. doi: 10.1016/j.canlet.2016.10.019
137. Wu C, Qiu S, Zhu X, Lin H, Li L. OCT1-mediated metformin uptake regulates pancreatic stellate cell activity. Cell Physiol Biochem (2018) 47(4):1711–20. doi: 10.1159/000491003
138. Luong T, Cukierman E. Eribulin normalizes pancreatic cancer-associated fibroblasts by simulating selected features of TGFβ inhibition. BMC Cancer (2022) 22(1):1255. doi: 10.1186/s12885-022-10330-y
139. Zhang H, Zhu H, Feng J, Zhang Z, Zhang S, Wang Z, et al. Reprogramming of activated pancreatic stellate cells via mechanical modulation of transmembrane force-sensitive n-cadherin receptor. J Mol Biol (2023) 435(1):167819. doi: 10.1016/j.jmb.2022.167819
140. Chen X, Yu Q, Liu Y, Sheng Q, Shi K, Wang Y, et al. Synergistic cytotoxicity and co-autophagy inhibition in pancreatic tumor cells and cancer-associated fibroblasts by dual functional peptide-modified liposomes. Acta Biomater (2019) 99:339–49. doi: 10.1016/j.actbio.2019.09.003
141. Zang S, Huang K, Li J, Ren K, Li T, He X, et al. Metabolic reprogramming by dual-targeting biomimetic nanoparticles for enhanced tumor chemo-immunotherapy. Acta Biomater (2022) 148:181–93. doi: 10.1016/j.actbio.2022.05.045
Keywords: pancreatic cancer, cancer-associated fibroblasts, fibrosis, metabolic reprogramming, crosstalk, heterogeneity
Citation: Li X, Zhou J, Wang X, Li C, Ma Z, Wan Q and Peng F (2023) Pancreatic cancer and fibrosis: Targeting metabolic reprogramming and crosstalk of cancer-associated fibroblasts in the tumor microenvironment. Front. Immunol. 14:1152312. doi: 10.3389/fimmu.2023.1152312
Received: 27 January 2023; Accepted: 09 March 2023;
Published: 22 March 2023.
Edited by:
Denisa Baci, University of Insubria, ItalyReviewed by:
Luo Yuhao, Southwest Medical University, ChinaCopyright © 2023 Li, Zhou, Wang, Li, Ma, Wan and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fu Peng, cGVuZ2ZAc2N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.