 Jia-Tong Ding
Jia-Tong Ding Kang-Ping Yang
Kang-Ping Yang Hao-Nan Zhou
Hao-Nan Zhou Ying-Feng Huang1
Ying-Feng Huang1 Hui Li
Hui Li Zhen Zong
Zhen Zong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 April 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1149622
CD8+ T cells, a cytotoxic T lymphocyte, are a key component of the tumor immune system, but they enter a hyporeactive T cell state in long-term chronic inflammation, and how to rescue this depleted state is a key direction of research. Current studies on CD8+ T cell exhaustion have found that the mechanisms responsible for their heterogeneity and differential kinetics may be closely related to transcription factors and epigenetic regulation, which may serve as biomarkers and potential immunotherapeutic targets to guide treatment. Although the importance of T cell exhaustion in tumor immunotherapy cannot be overstated, studies have pointed out that gastric cancer tissues have a better anti-tumor T cell composition compared to other cancer tissues, which may indicate that gastrointestinal cancers have more promising prospects for the development of precision-targeted immunotherapy. Therefore, the present study will focus on the mechanisms involved in the development of CD8+ T cell exhaustion, and then review the landscapes and mechanisms of T cell exhaustion in gastrointestinal cancer as well as clinical applications, which will provide a clear vision for the development of future immunotherapies.
T cell exhaustion (TEX) is an effective low-response T cell state during chronic infection. This hyporeactive state is thought to be due to the inability of pathogens to be rapidly cleared in chronic inflammation and the continued stimulation of this specific T cell proliferation, resulting in the upregulation of many immune checkpoints on the surface of T cells, leading to a reduction in their proliferation and ability to capture pathogens (1, 2). Many studies have been devoted to exploring the mechanisms behind TEX, starting with attempts to overcome its problems. Researchers have tried to explore how TEX causes a reduction in T cell effector function and proliferation levels, as well as leading to the expression of suppressive immune checkpoint receptors and ultimately immune escape of the tumor (3, 4). There is evidence that CD8+ T cell infiltration below 2.2% predicts a fourfold higher risk of disease progression after cancer surgery (hazard ratio (HR) = 3.84, p < 0.01), but the proportion of CD8+ T cells does not correlate with other clinical data, suggesting that the mechanisms that trigger differences in T cell heterogeneity and kinetics are unclear (5, 6). Therefore, it is particularly important to identify the molecular determinants that regulate the number, spatial distribution, and heterogeneity of CD8+ T cells in tumors, which could help more patients to benefit from immunotherapy (7).
Current studies on TEX have found that the mechanisms behind it may be closely related to transcription factors and epigenetic regulation (8, 9). For example, in a study using a mouse model of chronic LCMV infection, the transcription factor TCF-1 was found to promote the function of depleted CD8+ T cells by promoting the expression of a series of key effector function-related transcriptional regulators, including Foxo1, Zeb2, Id3, and Eomes (10). A study identified progressive heritable methylation programs that limit T cell expansion and clonal diversity during PD-1 blockade therapy, and when these programs were reversed, TEX no longer inhibited immune checkpoint blocker (ICB) therapy (11). These findings suggest that transcription factors and epigenetic regulation may serve as biomarkers of CD8+ TEX and potential immunotherapeutic targets, which would provide precision therapy for tumor immunization patients (12).
The importance of TEX in tumor immunotherapy has been confirmed by many studies, however, what needs more attention is that gastrointestinal tumors are perhaps more suitable for deeper work in this direction than other tumors. For example, a pan-cancer analysis showed that the presence of a lower percentage of triminal CD8+ Tex and a higher percentage of CD8+ Trm cells in STAD tissues implies that the gastric cancer tumor microenvironment has a better composition of anti-tumor T cells, which would point to the development of more promising precision-targeted immunotherapies (3, 6, 9). There are many ongoing studies on CD8+ TEX in gastrointestinal cancers regarding immune checkpoint inhibition, CAR T cells, and synergistic therapy with chemotherapy, but many mechanisms are still unclear (13).
Therefore, this review will describe the origin, function, detection modalities, and mechanisms involved in CD8+ TEX, and review TEX in gastrointestinal cancers and related clinical applications, which will provide a clear and comprehensive vision for the development of future immunotherapies.
CD8+ T cells are cytotoxic T lymphocytes (CTL cells) that are produced by the body to fight against viruses, tumors, and other pathogens (14). They produce and express αβ-T cell receptors with CD8 in the thymus and destroy them by recognizing MHC class I on target cells (15). When the body’s CD8+ T cells or function is diminished, anti-tumor immune function will decrease and the risk of tumor growth and cancer metastasis will increase (16).
CD8+ T cells develop from CD34+ hematopoietic stem cells located in the bone marrow, which express CD2, CD5, and CD7 before leaving the bone marrow and entering the thymus to become CD3-expressing lymphoid progenitors, and subsequently undergo a double negative (DN) (CD8-CD4-) phase and a double positive (DP) phase (CD8+ CD4+) and finally became single positive (SP) CD8+ thymocytes. These cells are selected by positive and negative clones to become CD8+ T-αβ cells and are released into the circulation (5, 17). Antigen-presenting cells such as dendritic cells (DCs) usually present endogenous antigenic peptides in MHC class I molecules (18). CD8+ T cells are activated by recognition of antigenic peptides by CD8+ co-receptors on TCRs and CD8+ T cells and activated CD8+ T cells can lead to clonal expansion of antigen-specific CD8+ T cells, which then differentiate into effector or memory cells(Figure 1) (19).
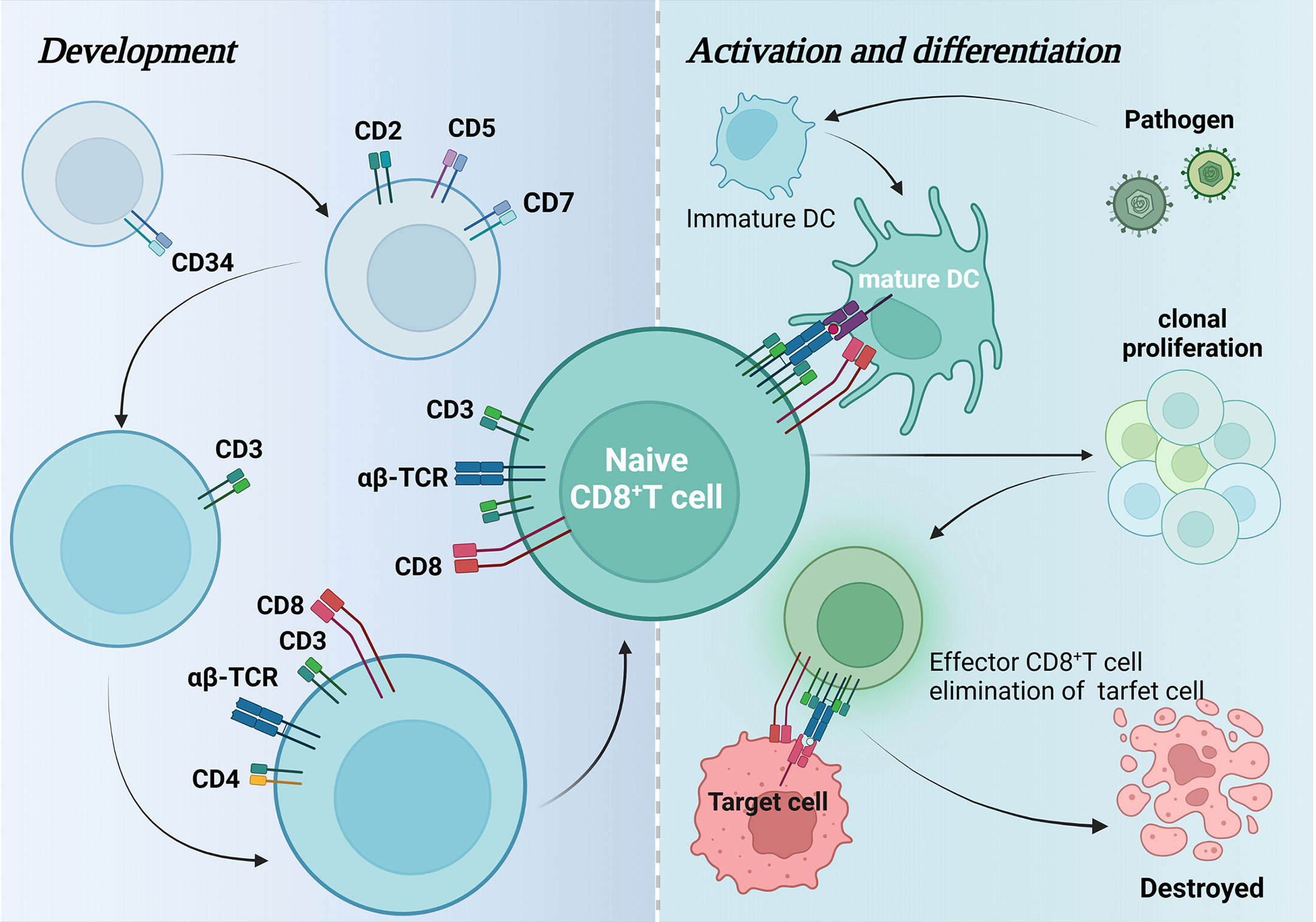
Figure 1 Development, activation, and differentiation of CD8+ T cells. This figure maps the changes in surface-specific antigens during CD8+ T cell development and how they are activated by dendritic cells to differentiate and generate effector CD8+ T cells that attack pathogenic target cells and eventually cause them to autophagy.
CD8+ T cells can exert antiviral or antitumor immune effects by mediating direct or indirect killing responses to target cells (20, 21). The main pathways of killing are: a) causing apoptosis of target cells through the release of lysozyme in intercellular contacts; b) acting on the target cell-expressed receptor Fas via Fas ligand (Fas-L), leading to apoptosis of target cells through a cysteine-dependent pathway; and c) indirectly inducing death of peripheral tumor cells through the secretion of cytokines (22, 23).
CD8+ TEX has been used to describe the unresponsive, low-effect state of T cells during tumor progression and is commonly seen in advanced progressive tumors that exhibit a host-cancer cell stalemate pattern (12, 24, 25). In the early stages of tumor development, i.e., before sufficient numbers of cancer cells and tumor antigens form and/or appear, tumor-reactive CD8+ T cells may remain in a state of ignorance (26, 27). As cancer progresses, CD8+ T cells are continuously stimulated by tumor antigens and enter a late dysfunctional state late (28).Exhausted-like T cells isolated in progressive tumors will exhibit tumor-infiltrating lymphocytes (TIL) that are responsive to tumor antigens expressing multiple inhibitory receptors and failing to produce effector cytokines or cytotoxic molecules (29–32). The mechanism that CD8 +T cells cannot eliminate cancer is attributed to factors such as cancer cells and TME, etc. (33–35).
Mechanisms mediated by cancer cells include loss of MHC expression, loss of antigen, loss or defect in antigen presentation, or expression of inhibitory receptor ligands. Loss of MHC class I expression of PD-L1 or other ICPs has been noted in many cancer types (36). expression of most components of the MHC I antigen presentation pathway is nonessential for cell survival and therefore is usually loss of function in cancer cells, leading to T cell depletion. transcription factors such as NLRC5, IRF1 and IRF2 are thought to play an important role in induced MHC I transcription expression and do not affect cell viability (37, 38). A new study indicates that post-translational protein modification, SUMOylation, also induces cancer cells to evade CD8+ T cell-mediated immune surveillance by inhibiting MHC-I antigen processing and presentation mechanisms (39).
Mechanisms mediated by TME include TGFβ, IL-10, and metabolic interaction. Tumor microenvironment (TME) is an ecological niche that can suppress CD8+ T cell immunometabolism and cytotoxicity. In a study of malignant breast cancer cells, tumor-derived extracellular vesicles could transport active TGF-β type II receptors (TβRII) to CD8+ T cells, thereby activating SMAD3 expression in CD8 T cells, ultimately leading to TEX and resulting in failure of immunotherapy (40). IL-10 release from HMOX1 myeloid cells induces TEX in the glioblastoma microenvironment (41). Cetuximab-based IL-10 fusion protein has also been shown to prevent dendritic cell-mediated TEX by regulating the IFN-γ pathway (42). Notably the role of lactate metabolism in the regulation of TEX is controversial. It has been suggested that lactate increases the stemness of CD8+ T cells and enhances antitumor immunity (43), while it has been suggested that tumor-derived lactate inhibits the cytotoxicity of CD8+ T cells (44, 45). Therefore, finding a level of lactate that balances cytotoxic CD8+ T cells with enhanced antitumor immunity is worth exploring. The mechanisms of T-cell loss are also influenced by tumor, treatment modality, and model, and research in this area is scarce yet necessary.
Single-cell transcriptomics is an important tool used to study the mechanisms of CD8+ TEX in cancer (46). For example, one study investigated the heterogeneity of high-grade serous ovarian cancer within its TME by scRNA-seq and found high expression of the immunosuppressive receptor TIGIT on CD8+ TEX cells. In a mouse model, the study blocked TIGIT, leading to f slowing of ovarian cancer tumor growth (47).These studies could provide new perspectives for further immunological studies and immunotherapy of relevant tumors (47, 48).
TEX as a sign-specific cell type needs to be distinguished from canonical functional T cell subpopulations phenotypically similar to it [e.g., naïve T cells (TN), effector T cells (TEFF), and memory T cells (TEME), often with great difficulty, despite the great functional, transcriptomic, and epigenomic differences in TEX (49, 50). This is because TEX expresses many markers that are equally expressed by TEFF and TEME (51). However, TEX has significant heterogeneity, such as TEX subpopulations with ancestral-progenitor relationships, or groups with different degrees of failure or homeostatic potential, which may be directly associated with disease progression. And TEX has a guiding role in the use of clinical immunosuppressants, which can be used to determine the suitability of immunosuppressant use and to predict the prognosis (32). Therefore, considering TEX heterogeneity, many studies have used mass spectrometry flow techniques (CyTOF) to perform high-dimensional number analysis for accurate assessment of TEX [47].
Although flow cytometry and single-cell transcriptomics have provided unprecedented insightful solutions to reveal the mechanisms controlling CD8+ T cell function in cancer, however, these techniques have failed to capture important spatial-spatial information, including intercellular interactions and tissue localization. Many new studies have now identified intra-tumor immune ecological sites, tertiary lymphoid structures (52), and tumor-draining lymph nodes as key sites of intercellular communication (53), and regulation of these sites plays an important role in the spatial determination of CD8+ T cell function.
During acute infection, naïve CD8+ T cells are activated and differentiate into effector CD8+ T cells, including short-lived effector cells (SLEC) and memory precursor effector cells (MPEC) (54–56). Effector CD8+ T cells are characterized by extensive reprogramming of transcriptional network, epigenetic modulation, and metabolism, and the acquisition of cardinal effector function, including changed tissue homing, and dramatic numerical expansion (54, 55). After the clearance of antigens, the majority of effector T cells die, and the subset-MPEC would then convert into memory CD8+ T cells. Memory CD8+ T cells could be reactivated easily and rapidly when encountering the secondary infection, and their maintenance is independent of antigen stimulation but a stem cell-like self-renewal dependent on interleukin-7(IL-7) and IL-15 (54). Whereas during chronic infection or cancers, an altered differentiated state of T cells appears and is termed TEX due to persistent antigen stimulation. Exhausted CD8+ T cells have various cardinal features, including progressive loss of effector function, sustained upregulation of multiple co-inhibitory receptors such as PD-1, CTLA4, LAG3, and TIM3, downregulation of co-stimulatory receptors, and altered transcriptional network, epigenetic modulation, and metabolism compared to functional effector cells and memory T cells (57). Contrary to memory T cells, exhausted CD8+ T cells respond poorly to IL-7, and IL-15, and their persistence are via continuous stimulation of antigens (58–60), although a minority of them might be persistent without the continuous antigen stimulation in some contexts (61–63). Of note, while exhausted T cells are relatively hypofunctional, they still provide a crucial killing effect against pathogens, which exerts inseparable roles for host-pathogen balance, hence, another thought toward TEX has been popular recently that the generation of TEX is a protection mechanism for hosts to avoid the inflammation damage due to immune system hyper-activated by persistent antigen stimulation. Whereas the generation of TEX involves multiple factors, herein, we manage to review the correlated studies to describe the mechanism of TEX from the perspective of transcription factors and epigenetic modulation.
It has been known that transcription factor T-bet and Eomes are both essential for the differentiation of effector CD8+ T cells and memory CD8+ T cells during acute infection, where their roles are partially overlapping but the expression discrepancies also exist in effector and memory CD8+ T cells, indicating that they respectively have unique function in the development process of effector CD8+ T cells and memory CD8+ T cells (55). In brief, early effector CD8+ T cells (T-bet+Eomes+) increase the expression of T-bet to turn into terminally differentiated effectors (T-bet++Eomes+), while memory CD8+ T cells express a higher level of Eomes (T-bet+Eomes++) (64). However, if the acute infection progresses into a chronic infection, terminally differentiated effectors would reduce the expression level of T-bet but express more Eomes (T-bet+Eomes+++) to execute the exhaustion program, instead of differentiating into memory CD8+ T cells. T-bet and Eomes influenced the fate decision between memory T cells and exhausted T cells. Exhausted CD8+ cells expressed less T-bet and more Eomes versus CD8+effectors, and higher Eomes expression seemed to predispose CD8+ effectors to become exhausted T cells (T-bet+Eomes+++) rather than memory T cells (T-bet+Eomes++) when encountering persistent infection. During chronic LCMV infection, the expression pattern of T-bet and Eomes is also closely associated with the differentiation within the heterogenous Exhausted CD8+ T cells, and it seems that T-bet and Eomes have antagonistic roles for Tex development (65). Researchers found that two Tex subsets identified according to the expression levels of T-bet and Eomes, in conjunction with PD-1, have different residual effector functions although both subsets were effector impaired versus memory CD8+ T cells. T-betHiEomesLoPD-1int Tex cells could still proliferate and release medium amounts of TNFα and IFNγ, and they were potential to be reinvigorated with anti-PD-1/PD-L1 therapy (66). Whereas T-betLoEomesHiPD-1Hi Tex cells were less potential to proliferate, produced lower quantities of effector cytokines, and simultaneously expressed higher levels of other inhibitory receptors except for PD-1 and CTLA4, including CD160, Lag-3, and Tim-3. It suggested that a high expression level of T-bet was correlated to a progenitor-like Tex subset with mild exhaustion performance, high expression of Eomes was associated with a Tex subset with severe exhaustion manifestation, and these two transcription factors have an opposite regulatory effect on the expression of Tex markers including PD-1. Although exactly how they influenced the fate decision between memory T cells and exhausted T cells and the development process of Tex remains poorly understood, more comprehensive events that appear between T-bet and Eomes and exhaustion program have recently been revealed by Mclane et al., which exhibited that T-bet and Eomes competed for the same binding DNA sequences, including the Pdcd1 (encoding for PD1) (67). A high level of nuclear T-bet vigorously inhibited Pdcd1 transcription, while a low level of nuclear T-bet gave rise to Eomes-mediated weaker repression on Pdcd1 in Tex, and blocking PD-1 signaling in Tex could upregulate T-bet level, which restored potent inhibition for Pdcd1 transcription and T-bet associated gene expression of activation, chemotaxis and homing (67).
In 2016, He et al. found that during chronic infection, a subset of exhausted CD8+ T cells expressing the chemokine receptor CXCR5 could migrate into B-cell follicles, which was similar to follicular helper T cells, and the CXCR5+ subset expressed lower amounts of inhibitory receptors and retained more powerful cytotoxicity compared with CXCR5- subset (68). This specific CXCR+ subset was also observed in HIV patients, and its amount was inversely associated with viral load. Besides, when adoptively transferred into chronically infected mice, the CXCR+ subset exhibited a synergistic effect with the anti-PD-L1 treatment to reduce viral load versus CXCR- subset, which showed a greater therapeutic potential (64). In succession, another study also reported that in the mice model with chronic infection of LCMV, the CXCR5+PD-1+ subset expressed more co-stimulatory molecules including ICOS and CD28 versus the CXCR5-PD-1+ subset, and the specific subset was able to proliferate, almost accounting for the whole proliferative burst, in response to PD-1 signaling blockade (64). Besides, the transcription profiling demonstrated that the CXCR5+PD-1+ subset was highly enriched for the transcription factor TCF-1, indicating an important role of TCF-1 in the progenitor-like Tex subset (64). Later, multiple studies demonstrated that the TCF-1+ subset represents the progenitors of exhausted T cells, which proliferates more TCF-1+ progenitors, or loses the expression of TCF-1 and differentiates into the terminally effector subset (32, 69–71). On the one hand, TCF-1 itself functions as a direct downstream effector of the WNT/β-catenin signaling pathway that exerts essential roles for stem cell self-renewal, on the other hand, the depletion of TCF1 indeed could deprive the proliferative potential and aggravate the exhausted manifestation of Tex cells in chronic infection or tumors (72–74). Hence, people gradually recognized that TCF-1 seems to take up a core position to regulate various transcription factors and further edit the exhaustion program. Transcriptional profiling suggested that TCF-1 cKO CD8+ T cells exhibited increased TimhighBlimphigh gene expression signatures but reduced TimlowBlimplow gene expression signatures, indicating that TCF1 promoted the downregulation of Tim and Blimp, and expressed lower amounts of mRNA for transcription factors, including Bcl6, Ikzf2, and Aff3, and surface receptors such as Ccr7, CXCR5, Il23r, Tnfsf8 (encoding CD30L) and Sell (encoding CD62L) compared to WT CD8+ T cells (68). Microassays showed that TCF1 down-regulated Prdm1(Blimp1) and Havcr2 (Tim3), which might implicate the repression mechanism for Blimp1 and Tim3 (71). On the contrary, Bcl6, a crucial regulator of follicular helper T cell differentiation that counteracts Blimp1 activity (75, 76), was found to be induced by overexpression of TCF1 (68). Additionally, in the early phase of Tex development, TCF1 repressed the expression of T-bet and ID2 and promoted the expression of Eomes, c-Myb, and Bcl-2, mediating a transcription factor transition from Tex precursors to terminally differentiated Tex (69).
Persistent antigen stimulation is considered necessary for the execution of the exhaustion program, in other words, TCR activation is indispensable for the initial maintenance of exhausted CD8+ T cells. Whereas acute infection also requires TCR activation, hence, regarding the outcome discrepancies, a different pattern should exist in TCR activation between acute infection and persistent infection and tumors. In acute infection, activation of TCR and CD28 on the surface of T cells generates signaling cascades, which further activates transcription factor NFAT as well as its partner AP1 to form the NFAT/AP1 complex, and the complex is crucial for the activation of CD8+ T cells and the acquisition of various effector functions. Hence, NFAT and AP1 are both elevated during the development of effector cells. In contrast, in exhausted CD8+ T cells, NFAT (especially NFAT2) is separately upregulated, while the expression level of AP1 is lower (77), indicating that NFAT might exert its role independent of AP1 in exhausted T cells (Figure 2). Consistently, Martinez et al. confirmed that NFAT proteins triggered the alteration of the transcriptional network to execute the exhaustion program, and NFAT that was engineered as sequestered from AP1 protein could strongly induce exhaustion (78), demonstrating that the individual NFAT exerted essential roles in the execution of exhaustion program. Engineered NFAT proteins were also proved to bind to the regulatory sites of inhibitory receptors, including LAG3, PD-1, and TIM3, thus enforcing the exhaustion program (74). Of note, in the study of Martinez et al. (74), the NFAT proteins were designed as sequestered from AP1 protein, and it was uncertain that individual NFAT induced the above outcomes by itself or cooperating with other transcription factors, maybe the modulating axis also existed between NFAT and other molecules. A recent study showed that, in response to persistent TCR signaling, NFAT proteins promoted the expression of IRF4 (interferon regulatory factor 4) and BATF (basic leucine zipper ATF-like transcription factor), and NFAT proteins could cooperate with IRF4 and BATF (NFAT/IRF4/BATF) to induce the expression of genes involved in multiple pathways mediating exhaustion, for example, Pdcd1(PD1), Havcr2(TIM-3), and Ctla4(CTLA-4), whereas the NFAT/IRF4/BATF complex repressed the expression of Tcf7(TCF-1) which was necessary for the maintenance of progenitor-like T cells in Tex, suggesting that NFAT/IRF4/BATF complex promote the development of terminally differentiated Tex cells through repressing TCF-1 (79). Additionally, chronic tumor antigen stimulation as well as induced NFAT activation could give rise to the expression of transcription factor Tox that is highly expressed in dysfunctional tumor-specific CD8+ T cells and exhausted CD8+ T cells during chronic infection (80, 81). Tox is a crucial center regulator of exhaustion. Ectopic expression of Tox in effector T cells induced a transcriptional program associated with TEX in vitro, and deletion of Tox could rescind the exhaustion program in tumor-specific CD8+ T cells, for example, Tox-deleted exhausted CD8+ T cells could not upregulate genes encoding for inhibitory receptors, including Entpd1, Pdcd1, Tigit, Havcr2 and Cd244 and reserved high expression level of TCF-1 (37). Similarly, transcription factor NR4A was also confirmed to be induced by NFAT in exhausted T cells, and NR4A-deficient CD8+CAR+PD-1highTIM3high tumor-infiltration T cells exhibited upregulated cytokine expression and reduced expression of inhibitory receptors (82). The authors also proved the existence of positive-feedback regulation in the expression between Tox and NR4A, thus cooperating to promote CD8+ TEX (77). Therefore, it suggests that Tox and NR4A are the downstream targets of NFAT and contribute to TEX to a great extent, which might be potential to serve as therapeutic targets for cancer control.
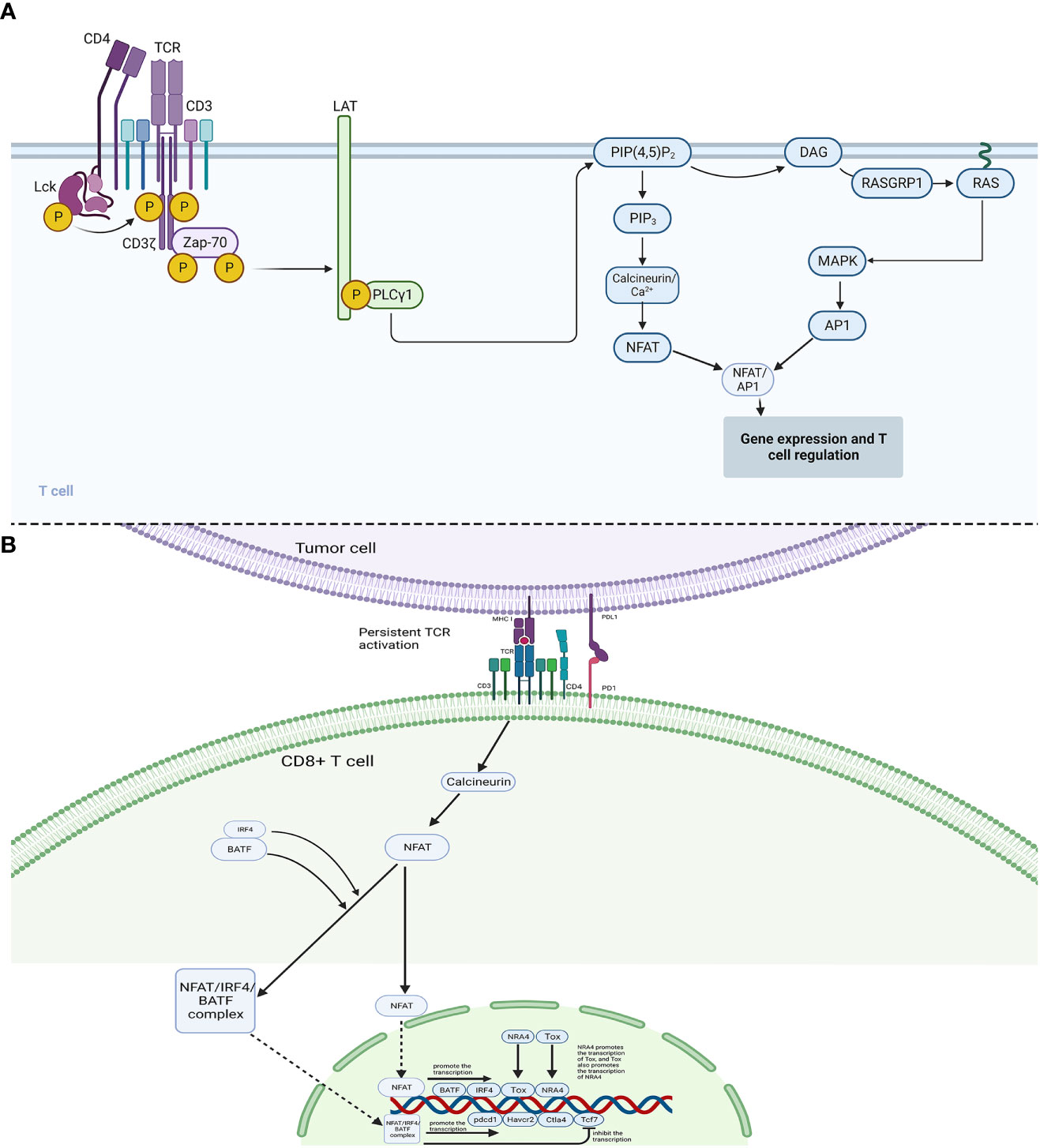
Figure 2 Mechanisms of CD8+ T cell regulation and exhaustion. (A): In acute infection, phosphorylated Lck phosphorylates CD3 and ζ chains, which then link ZAP-70. ZAP-70 could phosphorylate LAT, and then LAT would bind PLCγ1, which activates PIP(4,5)P2. Subsequently, in one pathway, PIP(4,5)P2 activates DAG/RAS/MAPK and AP1, in another pathway, PIP(4,5)P2 could induce the activation of Calcineurin/Ca+ and NFAT. (B): In the case between tumor cells and CD8+ T cells, the interaction between PD1 and PDL1 interferes the original powerful immune-activated pathways, which could induce abnormal NFAT signaling to promote Tex. Continuous TCR activation leads to the activation of calcineurin pathways, which dephosphorylates the cytoplasmic nuclear factor of activated T cells (NFAT). The cytoplasmic NFAT would then translocate into the nucleus and promote transcription of BATF, IRF4, Tox, and NRA4. Cytoplasmic BATF and IRF4 proteins could also cooperate with cytoplasmic NFAT protein (NFAT/IRF4/BATF complex), which then translocates into the nucleus. The nuclear NFAT/IRF4/BATF complex could promote the transcription of inhibitory receptor genes, including pdcd1(PD-1), Havcr2(TIM-3), and Ctla4(CTLA-4), and repress the gene Tcf7(TCF-1).
Although terminally differentiated exhausted CD8+ T cells express higher amounts of PD-1 receptor compared to initial progenitor exhausted CD8+ T cells, anti-PD-1: PD-L1 treatment almost could not reinvigorate the terminally differentiated CD8+ subset, suggesting that the exhaustion program is also fixed by other factors. Focused on the limitation of anti-PD-1:PD-L1 treatment, with genome-wide epigenetic profiling technologies, researchers found that Tex acquired a distinct epigenetic landscape compared to effector T cells and memory T cells, and only ~10% of the epigenetic landscape was remodeled after PD-1 blockade, indicating that Tex was a distinct lineage of CD8+ T cells and the stable epigenetic landscape of Tex might limit the current immunotherapies (83). Additionally, even in the absence of antigens, the results remained similar (84–86). A recent study revealed that using mice models, after transferring the Tex cells into the infection-free mice, although partial Tex cells that were TCF-1+ Tex progenitors acquired the transcriptional features of memory T cells, their recall ability remained compromised versus memory T cells and their chromatin landscape largely conserved as the exhausted state (79). Therefore, it suggests that exhaustion could cause persistent and stubborn epigenetic landscape alteration which is minimally reinvigorated by PD-1 blockade or removal of antigen.
Although the precise molecular mechanism maintaining the exhausted chromatin state remains unclear, multiple types of research showed that in addition to serving as a transcription factor, TOX also participates in the epigenetic remodeling in both tumors and chronic viral infection (80, 81, 87). Transcription factor NR4a has also been reported to drive Tex via orchestrating epigenetic changes in CD8+ tumor-infiltrating lymphocytes (TIL) and chimeric antigen receptor (CAR) T cells (82, 88, 89). In addition, the de novo methyltransferase DNMT3A was also confirmed to maintain the Tex epigenetic state (11). Ghneim et al. found that during chronic infection, activated CD8+ T cells whose de novo methylation was blocked could still retain effector functions, and even though during PD-1 blockade, the exhaustion linked de novo DNA methylation remained preserved. Corresponding to the differentiated degree of Tex, only the terminally differentiated Tex TILs with high PD-1 receptors acquired the exhaustion-linked DNA methylation (85) which might explain the reason that the PD-1 blockade only revives those progenitor Tex cells to some degree.
Gastrointestinal cancer occurs in gastrointestinal tract and accessory organs of digestive system, including esophagus cancer, gastric cancer, colorectal cancer, and others. As gastric cancer and colorectal cancer are leading causes of cancer death globally, present review mainly focuses on these two kinds of cancer types (90, 91).
Gastric cancer (GC) is one of the most common malignancies and the leading cause of cancer-related mortality worldwide (92). Unfortunately, though proven novel effective anticancer therapies in several cancers, programmed cell death protein 1 (PD-1) inhibitor (nivolumab) and anti-PD-L1 antibody (avelumab) showed limited benefits in improving survival outcomes for GC patients (93, 94). Hence, a clear understanding of the landscape of TEX in GC is pivotal to identifying novel targets for improving clinical management and decision-making. Figure 3 shows the major mechanisms of T-cell exhaustion in GC.
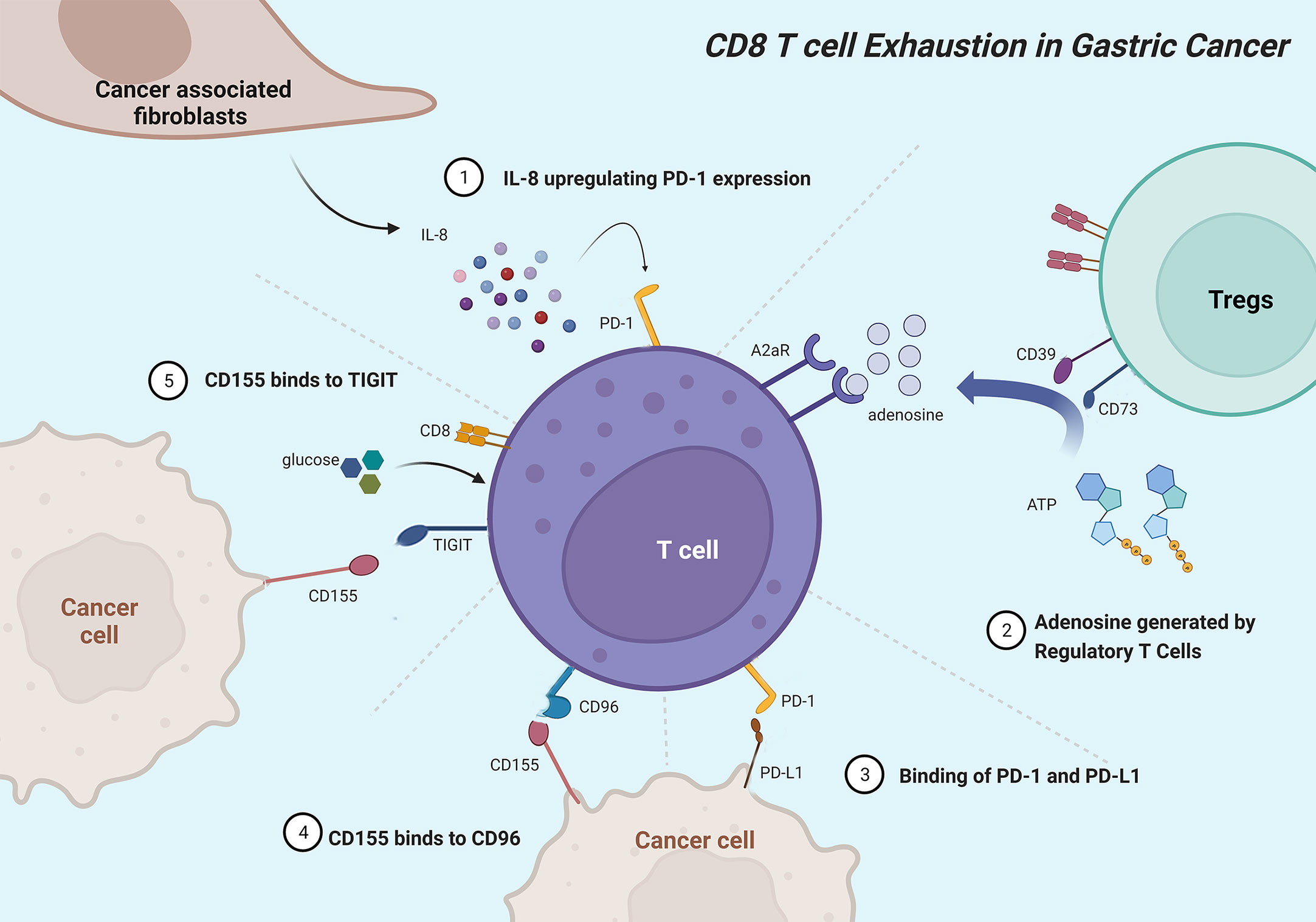
Figure 3 Mainly mechanisms of CD8+ T cell exhaustion in gastric cancer. Cancer-associated fibroblasts originate IL-8 to upregulate PD-1 expression in CD8+ T cells. Ectonucleotidases CD39 and CD73 catalyze adenosine that interactives with A2aR to inhibit CD8+ T cells. PD-L1 on tumor cells interacts with PD-1 to induce T-cell exhaustion. CD96 mainly binds to CD155 and associates with CD8+ T cell exhaustion phenotype. Gastric cancer cells expressing CD155 deprive CD8+ T cells of glucose and impair CD8+ T cell effector functions, which can be rescued by additional glucose.
The advancement and proliferation of single-cell RNA sequencing (scRNA-seq) technology make it possible to exactly map the status and differentiation trajectories of exhausted T cells in the tumor. Recent scRNA-seq research on tumor samples and adjacent non-tumor samples from nine untreated non-metastatic GC patients revealed that CD8+ T cells exhibited no significant increase of exhaustion levels and expressed low levels of exhaustion markers, including PDCD1, cytotoxic T-lymphocyte antigen 4 (CTLA-4), HAVCR2, LAG-3, and TIGIT (T-cell immunoreceptor with immunoglobulin and ITIM domains), which may partly explain the limited benefit of immunotherapy among GC patients (95). The other scRNA-seq analysis with tumor tissues, adjacent normal tissue, and matched peripheral blood of ten primary GC patients revealed that tumor-infiltrating CD8+ T cells reached exhausted state via two exhaustion trajectories. Tissue-resident memory T cells differentiate into IL-17CD8+ T cells (Tc17) and subsequently exhausted T cells with increasing cytolytic scores, while the blood-derived cytolytic CD8+ T cells turn to the exhausted population with lower exhaustion scores and decreasing cytolytic score, which are attributed to distinct transcription programs. Tc17-derived exhausted T cells highly express keratin KRT86, while cytolytic-cell-derived exhausted T cells express high levels of GZMK. As for transcription factors, PRDM1, TOX2, NR3C1, CEBPD, ATF3, and EOMES were expressed in cytolytic-cell-derived exhausted T cells, while BHLHE40, CREM, and RUNX2 were expressed in Tc17-derived exhausted T cells (96). The high heterogeneity between the two studies may associate with differences in patient populations. The degree of confidence in the results is also limited by the limited sample sizes, which requires further validation in large-scale scRNA-seq cohorts.
Immune checkpoint molecules and regulators exert key roles in maintaining tolerance, preventing T cell hyperactivation, and regulating TEX. TIGIT, an immune checkpoint expressed on the T-cell surface, belongs to the CD28 family and binds to CD155 to suppress T-cell activation (97). In GC, TIGIT+CD8+ T cells with functional exhaustion occupy a higher proportion. GC tissue and cell lines expressing CD155 deprive CD8+ T cells of glucose, and impair CD8+ T-cell effector functions and IFNγ production, which can be rescued by additional glucose or TIGIT blockade, especially combined with PD-1 blockade (98). As a novel immune checkpoint, CD96 mainly binds to CD155 and impact CD8+ TEX phenotype and immunosuppressive microenvironments, leading to poor prognosis in GC (99, 100). PD-1 acts as a negative co-stimulatory receptor and induces TEX via interacting with programmed death ligand 1 (PD-L1) or PD-L2, which is recognized as the main mechanism of tumor cells to escape anti-tumor immune reaction (101). Elevated serum interleukin-8 (sIL-8) prominently originated from GC-associated fibroblasts upregulates PD-1 expression in CD8+ T cells via activating JAK2/STAT3 signaling and inhibiting PD-1 ubiquitination through Fbxo38 down-regulation, leading to immunosuppressive environments, lymph node metastasis, and poor prognosis (102). FoxP3+ Tregs in GC tissues can decompose ATP to adenosine with CD39 and CD73, and then adenosine interactives with A2aR to induce apoptosis and inhibit immune response and proliferation of CD8+ T cells (103, 104). Transcriptional repressor cAMP response element modulator (CREM) is induced by cAMP signaling pathway and positively correlated with exhausted marker genes in gastric adenocarcinoma (105). Dexamethasone suppress the transcription of PD-L1 and IDO1 via nuclear translocation of GR/STAT3 complex to inhibit TEX and immune evasion in coculture system, 3D organoid model and humanized mouse model (106).
There is growing evidence showing the pivotal roles of naïve, effector, memory, and exhausted T cells in the progression of colorectal cancer (CRC) (107). The infiltration level and exhaustion state of CD8+ T cells in tumor microenvironments have been recognized as two well-recognized immune escape mechanisms reflecting the resistance levels to anti-PD1 treatment (108, 109). Although MSI (microsatellite instable)/MSS (microsatellite stable) status, TMB (tumor mutational burden), POLE/POLD1 mutation, and MSI-like gene signature are widely used as indications of whether CRC a patient should receive immunotherapy, these features cannot fully explain anti-PD1 resistance and exhibit limited accuracy in predicting the response to immune checkpoint inhibitor treatment, due to they only directly or indirectly indicate the potential of high quantity of tumor-infiltrating CD8+ T cells in tumor samples while ignoring the exhaustion state of CD8+ T cells (110–112). Therefore, dissecting the TEX status in the tumor microenvironment seems to occupy a more important position in predicting the response to immunotherapy in CRC. Figure 4 shows the major mechanisms of T cell exhaustion in CRC.
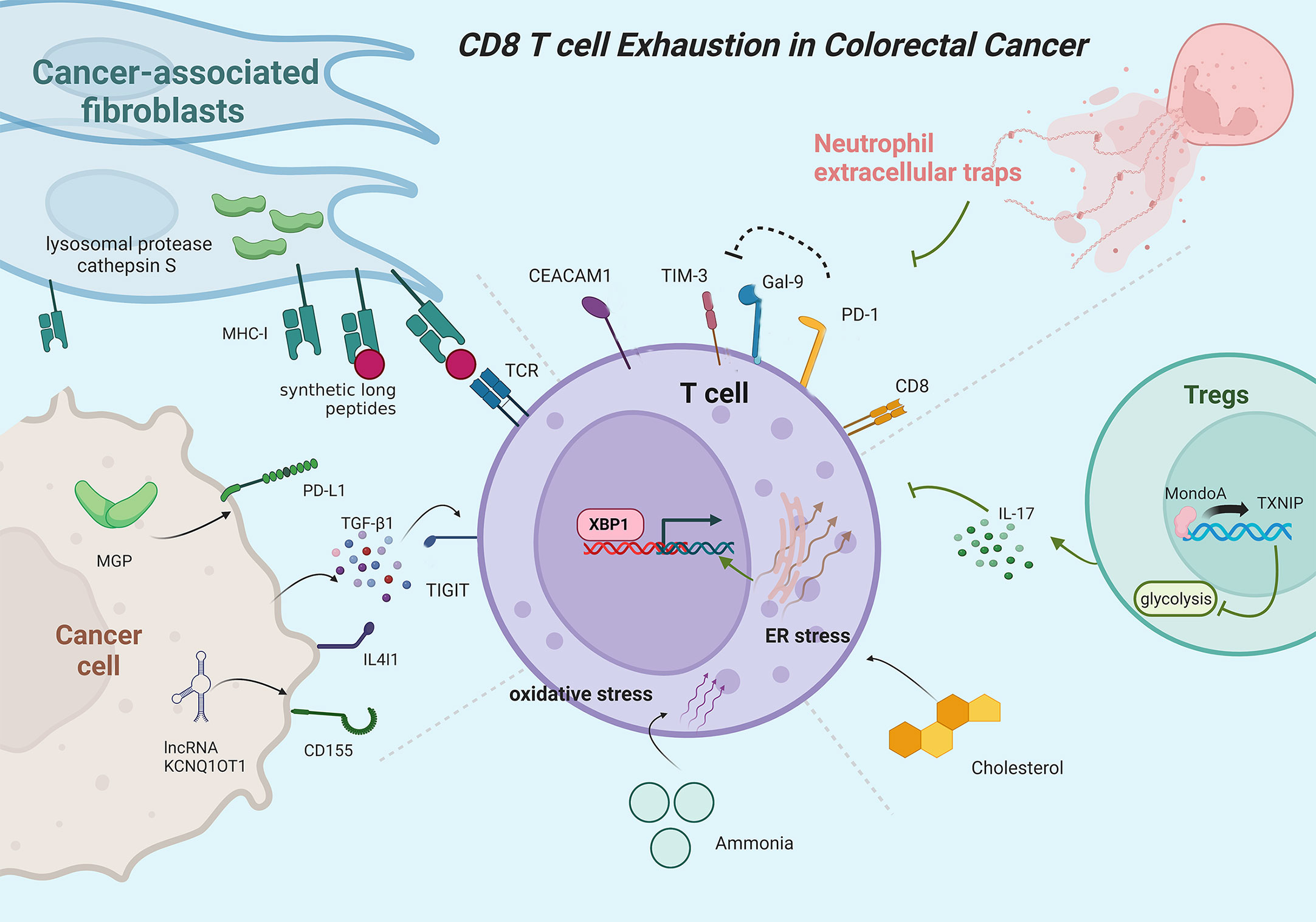
Figure 4 Mainly mechanisms of CD8+ T cell exhaustion in colorectal cancer. PD-1 protects exhausted T cells from Gal-9-induced cell death. CEACAM1 co-expresses with TIM-3 to promote T-cell exhaustion. Neutrophil extracellular traps can promote T cell exhaustion. Inhibition of the MondoA–TXNIP axis induces hyperglycolytic regulatory T cells to produce IL-17A and promotes T cell exhaustion. Cholesterol increases endoplasmic reticulum stress and activates XBP1 to induce transcription of immune checkpoints. Ammonia induces oxidative stress and T-cell exhaustion. MGP activates PD-L1 expression in cancer cells. Cancer cells can secrete TGF-β1 to inhibit T cell function. CD155 expression positively correlates with lncRNA KCNQ1OT1. Cancer-associated fibroblasts upregulate lysosomal protease cathepsin S expression and boost the capacity to cross-present synthetic long peptides, promoting T cell exhaustion.
Single-cell analyses observe exhausted T cells in CRC samples but low or undetectable PD1 expression in eight polyps with CODEX imaging, suggesting that TEX only appears in CRC instead of precancerous adenomas (113). The mRNA levels of several inhibitory immune checkpoints, transcription factors, and other TEX-related markers are elevated in CRC tumor tissues, including PD-1, CTLA-4, TIGIT, T cell immunoglobulin, and mucin domain-containing protein 3 (TIM-3), CD160, CD244, killer cell lectin-like receptor subfamily G member 1 (KLRG1), Thymocyte selection-associated high-mobility-group box 2 (TOX2) and TOX3, gene name for Blimp1 (PRDM1), and Ki-67, while PD-1 and CD160 are downregulated in tumor tissue of advanced stages (114, 115). Similarly, Yassin et al. have reported that PD-1 expression increases on mucosal T cell subsets of colon and ileum with the progression of inflammation-induced CRC in the azoxymethane (AOM)/dextran sulphate sodium (DSS)-treated mice. Dysplastic aberrant crypt foci in the ascending colon increases the expression of immune checkpoint and TEX gene PDCD1 but decreases cytotoxic T cell effector gene expression and inhibits interferon signaling (116). Yang et al. also reported a significant positive correlation between PDCD1 and several transcription factors, including NR4A1, BATF, and VDR. Due to the persistent stimuli of tumor antigens, tumor-reactive CD103+CD39+CD8+ T cells in CRC tumor microenvironments display exhausted phenotypes with high expression of CTLA4, HAVCR2, LAYN, and TOX (117). In metastatic sites of CRC, the expression levels of PD-1 and TIM-3 are abundantly upregulated compared to primary sites along with the elevated number of CD8+ T cells (118). The research combining single-cell mass cytometry with transcriptome sequencing in 18 patients with MSS CRC revealed that immunosenescent CD28−CD8+ T cells with decreasing proliferation dominate the T cell compartment at the immunosuppressive tumor microenvironment which takes shape early during CRC development (119). Dynamic network biomarker applied in a scRNA-seq data set identifies CCT6A as a biomarker for pre-exhausted T cell subpopulation, which drives TUBA1B expression to promote CD8+ TEX in CRC. Exhaustion status is also induced by the receptor-ligand interactions between terminally differentiated exhausted T cells and pre-exhausted T cells (120).
Similar to GC, higher infiltration levels of exhausted TIGIT+CD8+ T cells are observed in CRC tumor tissues with low levels of killer cytokines, including IFN-γ, IL-2, and TNF-α, but higher inhibitory receptors expression, such as PD-1, LAG-3, and TIM-3. Interestingly, CRC cells can secrete TGF-β1 to promote the expression of TIGIT, induce TIGIT+CD8+ T cell expansion, and inhibit CD8+ T cell function (121). The expression level of CD155 is also positively correlated with lncRNA KCNQ1OT1 in CRC cancer cells, and knockdown of lncRNA KCNQ1OT1 downregulates CD155 expression in HCT116 and SW620 cells, thereby enhancing the immune response of CD8+ T cells (122). Considered a novel immune checkpoint, the Natural Killer group protein 2A (NKG2A) interacts with the CD94 chain to form inhibitory receptors on NK cells and CD8+ T cells (123). Enriching in CRC tumor tissues, NKG2A CD8+ T cells characterize as tissue-resident T cells mainly in terminal exhaustion status with high functional avidity but impaired proliferative capacity (124). Galectin-9 (Gal-9) can be upregulated by interferons and interacts with TIM-3 to induce cell death of terminally exhausted T cells (PD-1TIM-3), but this process can be interfered by PD-1, which contributes to the persistence of PD-1TIM-3 T cells (125, 126). Carcinoembryonic antigen cell adhesion molecule 1 (CEACAM1) co-expresses with TIM-3 on CD8+ T cells to promote TEX, with TIM-3CEACAM1 CD8+ T cells exhibiting the most dysfunctional status and least IFN-γ production capability in CRC (127).
Besides, multiple complex mechanisms are also involved in the exhausted status of CD8+ T cells in CRC. MondoA, a member of Mondo family transcription factors, and thioredoxin-interacting protein (TXNIP) participate in regulating glucose metabolism and redox state (128–130). Inhibition of MondoA–TXNIP axis induces hyperglycolytic Th17-like regulatory T cells with reduced immunosuppressive functions, which formulates interleukin 17A prominent microenvironment, promotes TEX, and drives the advancement of CRC (131). Cholesterol in the tumor microenvironment increases endoplasmic reticulum stress and subsequently activates XBP1 to induce transcription of immune checkpoint in CD8+ T cells, contributing to TEX status with upregulated expression of PD-1, 2B4, TIM-3, and LAG-3 (132). Ammonia accumulated in the CRC microenvironment inhibits transsulfuration pathway and induce oxidative stress, paving the way to impaired T cell proliferation capacity and increased TEX. Thus, clearing tumor-associated ammonia reboots T cells and improves anti-PD-L1 efficacy (133). Elevated in CRC cancer cells, Matrix Gla protein (MGP) increases intracellular free Ca2+ levels, promotes NF-κB phosphorylation, and activates PD-L1 expression, leading to CD8+ TEX (134). PTPN2 mediates the generation of the Tim-3 subpopulation, and loss of Ptpn2 promotes the differentiation and formation of the Tim-3+CD8+ T cells and increases Tim-3+CD8+ T cell responses in MC38 cancer models (135). In MSS CRC, vascular endothelial growth factor-A (VEGF-A) induces the expression of transcription factor TOX in T cells to enhance the exhaustion-specific transcription program and drive TEX (136). Interleukin-4-induced gene 1 (IL4I1) enhances the translocation of Aryl hydrocarbon receptor (AHR) from the cytoplasm to nuclear in CD8+ T cells to regulate gene expression and thereby promote TEX (137). Kynurenine upregulate immune checkpoint expression and promote TEX in CRC via upregulating TOX expression, while IDO1 inhibitors and TOX knockdown can restore the anti-tumor activity of CD8+ T cells (138). Cbl-b, an E3 ubiquitin ligase, is upregulated in PD1+Tim3+ exhausted T cells, while Cbl-b deficiency leads to reduced endogenous CD8+ TEX (139). Gut microbiome dysbiosis leads to elevated colon tumor susceptibility with increased CD8+ IFNγ T cells in precancerous colon lamina propria but decreased CD8+ IFNγ T cells in tumor tissues, suggesting that microbial perturbations hyper stimulate CD8+ T cells to promote early TEX (140). CD39+CD8+ T cells display higher PD-1 expression and partially impaired functions, which correlates with the initial progression stages of the tumor (141). Epigenetic mechanisms also participate in the regulation of T-cell exhaustion. For example, downregulated histone 3 lysine 9 trimethylation and upregulated histone 3 lysine 4 trimethylation are observed in the promoters of PD-L1 and TOX2 in CRC tumor tissues (142). High DNA methylation levels in PDCD1 promoter are related to upregulated NR4A1 and VDR expression in several CD8+ T cells (117). Other components in the tumor microenvironment can also influence the exhaustion status of T cells. Neutrophil extracellular traps promote CD4+ and CD8+ T cells displaying functional and metabolic exhaustion with multiple inhibitory receptors (143). CRC-derived cross-presenting cancer-associated fibroblasts cognately interact with CD8+ T cells to suppress T cell activation, decrease cytotoxicity, and increase exhaustion marker expression, which is associated with upregulated lysosomal protease cathepsin S expression and boosted capacity to cross-present neoantigen-derived synthetic long peptides of cancer-associated fibroblasts (144).
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UD), causes intestinal inflammation and mucosal dysplasia, which triggers the development of IBD-related CRC (145, 146). CD39+ and CD39+PD-1+ CD8+ T cells accumulate in the intestinal tissue during inflammation in CD, and the exhaustion of CD39-expressing CD8+ T cells is related to attenuation and remission of disease activity, which agrees with the report that TEX informs the clinical differences in adult onset IBD (147, 148). Detailed roles of TEX in IBD deserve further exploration of the in-depth mechanisms, which may explain the likely promoting role of TEX in the course of IBD-related CRC.
With bioinformatics tools, researchers have identified multiple dysregulated molecules related to TEX and prognosis in GC or CRC tissues, such as CLDN10, C3AR1, ANTXR1, ANXA1, and PDPN (149–153). However, their exact regulatory mechanisms in T cell exhaustion remain for further validation and investigation.
PD-1, the key coinhibitory receptor on activated T cells, interacts with overexpressed PD-L1 on cancer cells, tumor-infiltrating lymphocytes, and stroma cells to inhibit T cell proliferation and function (154). CTLA-4, an inhibitory receptor primarily expressed by T cells, binds to CD80/CD86 on antigen-presenting cells resulting in impaired T cell functions (155). Since ipilimumab became the world-first approved immune checkpoint inhibitor (ICI) by the Food and Drug Administration, ICIs, mainly including monoclonal antibodies targeting PD-1, PD-L1, and CTLA-4, have been widely used and exhibited significant clinical activity in a spectrum of malignancies, including GC and CRC (156, 157). Tables 1, 2 list the latest representative clinical trials of ICIs in GC and CRC, respectively. A meta-analysis revealed that ICIs improve overall survival (OS) and progression free survival (PFS) for gastric/gastroesophageal junction cancer patients regardless to combined positive score (187). ICIs also exhibit high durable response rates and improved survival outcomes metastatic CRC patients with deficient mismatch repair or high microsatellite instability (188). However, although ICI is a promising method for GC and CRC, restricted effectiveness rate and common drug resistance limit its clinical application (157, 188, 189). What is worse, hyperactivated T cells induced by ICI can lead to multiple kinds of immune-related adverse events (irAEs), such as cutaneous toxicities, neurologic complications, ICI-associated myocarditis, and ICI-induced type 1 diabetes (190–195). Even so, treatment-related adverse events with ICI are less frequent and severe than chemotherapy (196). Xu et al. have summarized several biomarkers such as circulating blood cells, serum pro-inflammatory cytokines, co-expression antigens, and tumor burden as risk factors for ICIs-associated toxicities, which may help perform a timely process for ICIs-related toxicities in clinical practice (197). Moreover, ICIs combined with other interventions that are cytotoxic to tumor cells, chemotherapy for example, might enhance ICIs activity and improve clinical outcomes in GC and CRC. In a phase III clinical trial, patients with advanced GC, GEJ, or esophageal adenocarcinoma received nivolumab in combination with chemotherapy (capecitabine and oxaliplatin or leucovorin, fluorouracil, and oxaliplatin) had a longer OS, PFS, and an acceptable safety profile than did patients receiving chemotherapy alone (198). This synergy seems to be more pronounced in CRC. The efficacy of PD-1/PD-L1 inhibitors is unsatisfactory in mismatch repair proficient CRC, but patients receiving pembrolizumab combined with maraviroc exhibited prolonged disease stabilizations, limited clinical activity, improved treatment efficacy, and longer overall survival than expected (173, 199, 200). Nonetheless, the improved clinical benefits of combination therapy with ICIs and chemotherapy appear to lie in the multiple chances of response to a single agent, rather than the additive or synergistic effects of the drugs (201). Huang et al. have reported the correlation between clinical response of PD-1 blockade and the scale of reinvigorated circulating exhausted CD8+ T cells that are associated with the pretreatment tumor burden (202). These facts may suggest that chemotherapy reduces tumor burden to decrease antigenic load, thereby remitting TEX and improving the activity of ICIs (Figure 5). In a nutshell, ICI has an excellent clinical application prospect, especially combining with radiotherapy, chemotherapy, molecular target agents or other ICIs. However, considering the current limitations of ICI, novel treatment strategy and productive biomarkers for anti-tumor efficacy or toxicities of ICI need to be developed to maximize its efficacy, and deciphering the molecular mechanisms of TEX is likely to provide a rational avenue to reverse TEX and improve anti-tumor T cell activity. As described in the previous section, a variety of immune checkpoints, cytokines, and small molecule compounds are involved in the induction and persistence of TEX in GC and CRC. This suggests that targeting these molecules might alter the immunosuppressive microenvironment and activate exhausted tumor-reactive T cells, therefore, have therapeutic potential.
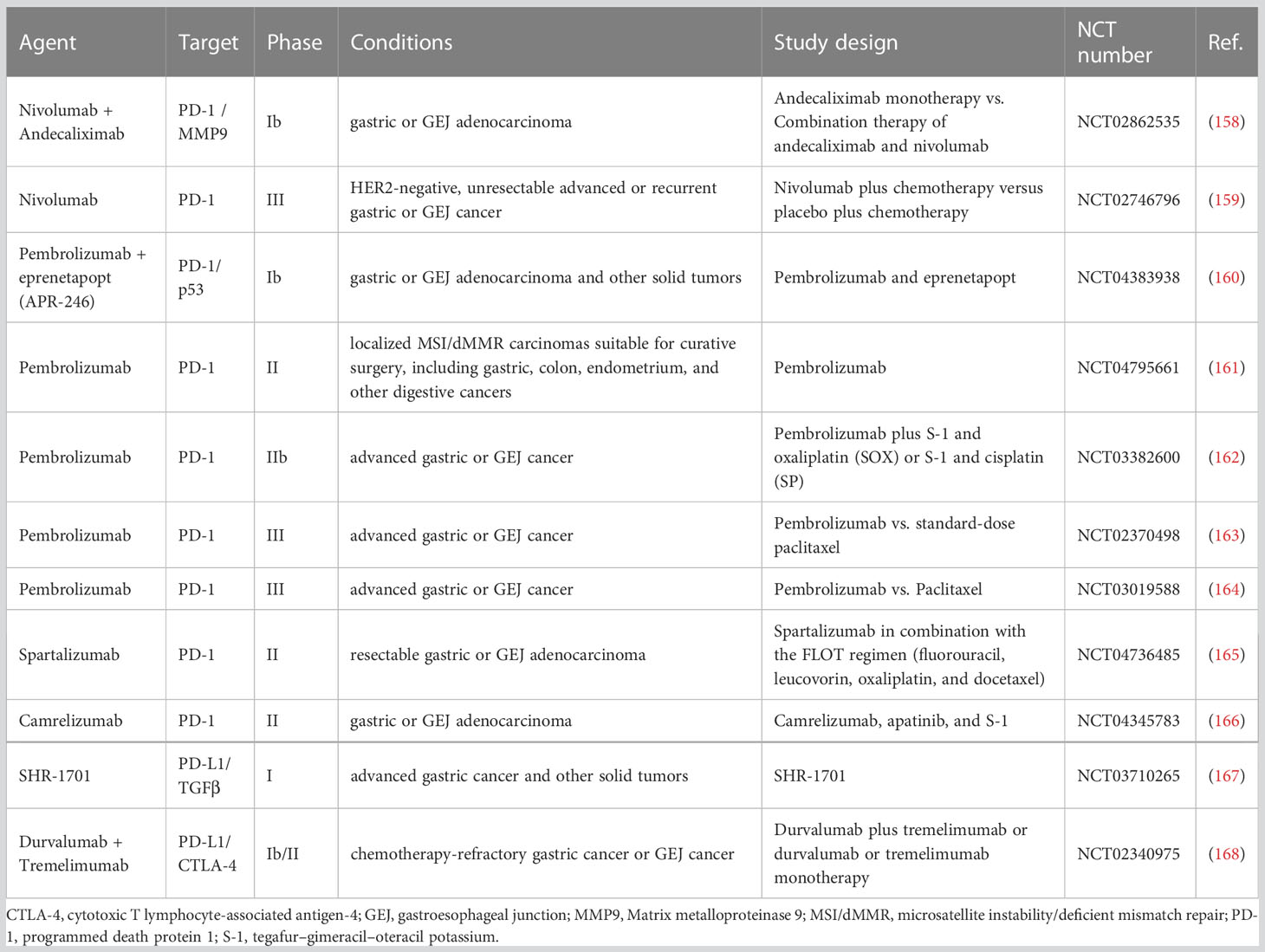
Table 1 Overview of the latest representative clinical trials of ICIs in gastric cancer.
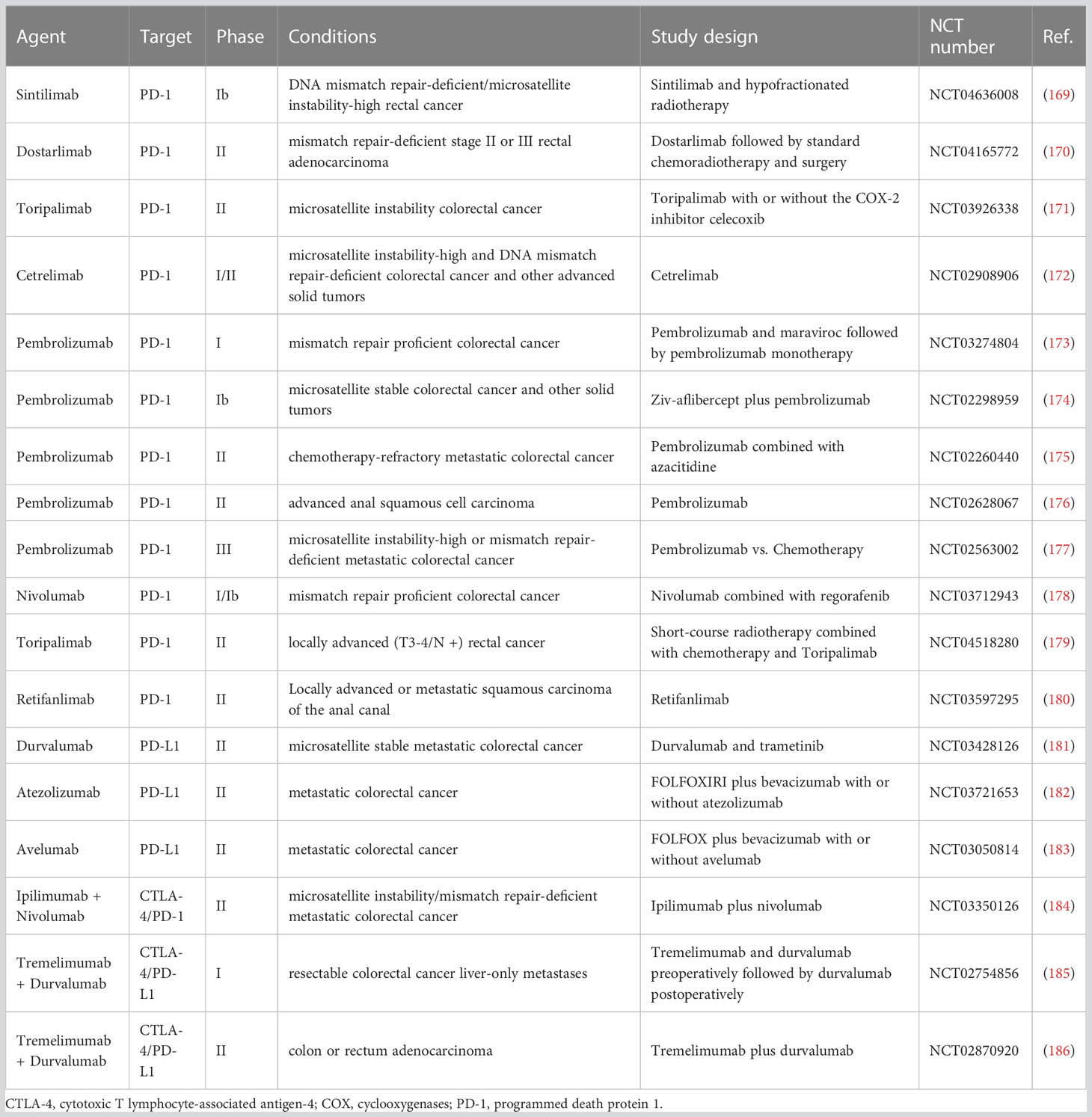
Table 2 Overview of the latest representative clinical trials of ICIs in colorectal cancer.
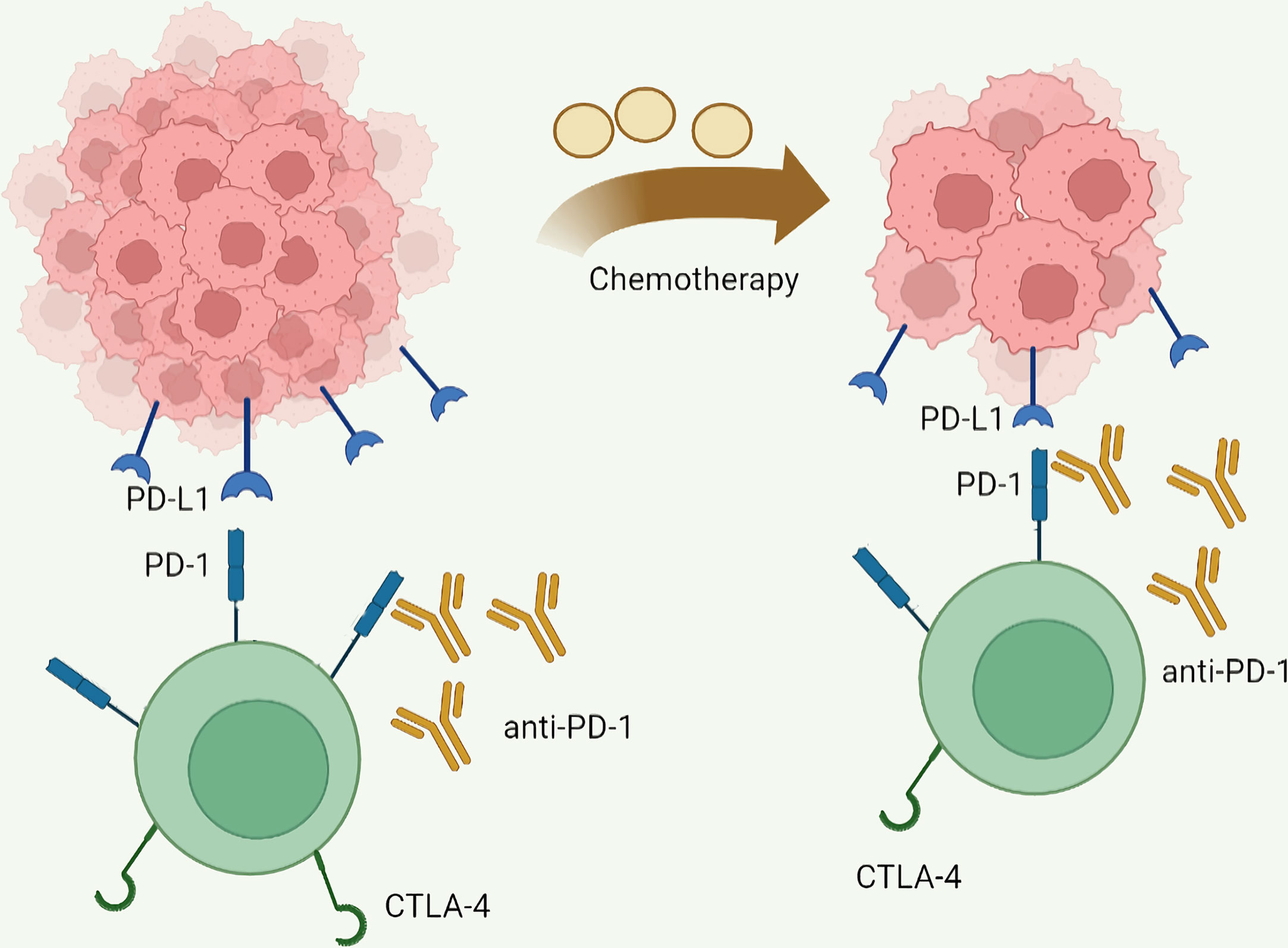
Figure 5 Mainly mechanisms of chemotherapy enhanced immune checkpoint inhibitor activity. Chemotherapy reduces antigen load by reducing tumor burden, thereby reducing T cell exhaustion and increasing the activity of immune checkpoint inhibitors.
The functional states of CD8+ T cells in tumor microenvironments have received much attention due to their vital roles in the efficacy of immunotherapies. With the applications of scRNA-seq, exhausted CD8+ T cells are identified with multiple interconnected subpopulations with discernible heterogeneity (115). Nonetheless, the present results of scRNA-seq research might not be representative of the general population due to limited sample sizes and the high heterogeneity of patients, which requires further validation in large-scale cohorts. Researchers have also revealed numerous alters in tumor microenvironments promote the exhaustion status of T cells in GC and CRC, such as metabolite changes and cell communications. However, available research on the potential roles of TEX in IBD and IBD-related CRC is quite limited. As an inflammation-induced carcinogenesis, IBD-related CRC have different molecular pathogenesis compared with sporadic CRC that arises from adenoma (203). Thus, further exploration is needed to explore the detailed roles and mechanisms of TEX in the progression from IBD to CRC. As for anti-cancer immunotherapy, though often seen as a barrier to effective therapy, TEX is also an inevitable process that protects T cells from overstimulation-related cell death in the persistence of tumor antigens (13). Clinical trials have reported better effects of the combination of chemotherapy and inhibitory receptors blockade than monotherapy (198). Therefore, attenuating the progression of TEX seems a more promising cancer treatment modality than restoring exhausted T cells. As the current immunotherapies with ICIs are not fully satisfactory, novel treatment strategies are needed. In addition to PD-1/PD-L1 and CTLA-4, other immune checkpoints have been discovered and their functions are under investigation, which raises a fundamental question of what targets are most suited for individualizing immunotherapy among different patients. Multiple factors and cells regulating TEX in the tumor microenvironment provides a broader perspective on how to inhibit and reverse TEX and may serve as a promising avenue of synergy with anti-cancer immunotherapy.
All authors have discussed the proposed scope and content of the article before drafting. J-TD, K-PY, and H-NZ wrote and revised the paper. Y-FH collected literatures. HL and ZZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Number: 81860433, 82103645 and 82260596); Training Plan for Academic and Technical Young Leaders of Major Disciplines in Jiangxi Province (Grant Number: 20204BCJ23021); the Natural Science Youth Foundation of Jiangxi Province (Grant Numbers: 20192BAB215036); the Key Technology Research and Development Program of Jiangxi Province (Grant Number: 20202BBG73024).
The author J-TD thanks Ms. Yang for her support over the years. The authors acknowledge Biorender that is used to create schematic figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Philip M, Schietinger A. CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol (2022) 22(4):209–23. doi: 10.1038/s41577-021-00574-3
3. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol (2019) 19(11):665–74. doi: 10.1038/s41577-019-0221-9
4. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
5. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. (2019) 576(7787):465–70. doi: 10.1038/s41586-019-1836-5
6. Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science (2021) 374(6574):abe6474. doi: 10.1126/science.abe6474
7. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. (2017) 169(7):1342–56.e16. doi: 10.1016/j.cell.2017.05.035
8. Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol (2022) 23(6):848–60. doi: 10.1038/s41590-022-01224-z
9. Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q, et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell (2022) 40(7):768–86.e7. doi: 10.1016/j.ccell.2022.06.001
10. Wang Y, Hu J, Li Y, Xiao M, Wang H, Tian Q, et al. The transcription factor TCF1 preserves the effector function of exhausted CD8 T cells during chronic viral infection. Front Immunol (2019) 10:169. doi: 10.3389/fimmu.2019.00169
11. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. (2017) 170(1):142–57.e19. doi: 10.1016/j.cell.2017.06.007
12. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
13. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol (2022) 19(12):775–90. doi: 10.1038/s41571-022-00689-z
14. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
15. Reiser J, Banerjee A. Effector, memory, and dysfunctional CD8(+) T cell fates in the antitumor immune response. J Immunol Res (2016) 2016:8941260. doi: 10.1155/2016/8941260
16. Gonzalez SM, Taborda NA, Rugeles MT. Role of different subpopulations of CD8(+) T cells during HIV exposure and infection. Front Immunol (2017) 8:936. doi: 10.3389/fimmu.2017.00936
17. Pu X, Zhu P, Zhou X, He Y, Wu H, Du L, et al. CD34(+) cell atlas of main organs implicates its impact on fibrosis. Cell Mol Life Sci (2022) 79(11):576. doi: 10.1007/s00018-022-04606-6
18. Wang X, Waschke BC, Woolaver RA, Chen SMY, Chen Z, Wang JH. MHC class I-independent activation of virtual memory CD8 T cells induced by chemotherapeutic agent-treated cancer cells. Cell Mol Immunol (2021) 18(3):723–34. doi: 10.1038/s41423-020-0463-2
19. Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. (2021) 596(7870):119–25. doi: 10.1038/s41586-021-03704-y
20. Lisci M, Barton PR, Randzavola LO, Ma CY, Marchingo JM, Cantrell DA, et al. Mitochondrial translation is required for sustained killing by cytotoxic T cells. Science (2021) 374(6565):eabe9977. doi: 10.1126/science.abe9977
21. Wiedemann A, Depoil D, Faroudi M, Valitutti S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc Natl Acad Sci USA (2006) 103(29):10985–90. doi: 10.1073/pnas.0600651103
22. Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, et al. FAS-l, IL-10, and double-negative CD4- CD8- TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. (2009) 113(13):3027–30. doi: 10.1182/blood-2008-09-179630
23. Caron B, Patin E, Rotival M, Charbit B, Albert ML, Quintana-Murci L, et al. Integrative genetic and immune cell analysis of plasma proteins in healthy donors identifies novel associations involving primary immune deficiency genes. Genome Med (2022) 14(1):28. doi: 10.1186/s13073-022-01032-y
24. Hellström I, Hellström KE, Pierce GE, Yang JP. Cellular and humoral immunity to different types of human neoplasms. Nature. (1968) 220(5174):1352–4. doi: 10.1038/2201352a0
25. Hellstrom KE, Hellstrom I. From the hellstrom paradox toward cancer cure. Prog Mol Biol Transl Sci (2019) 164:1–24. doi: 10.1016/bs.pmbts.2018.11.002
26. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. (2001) 411(6841):1058–64. doi: 10.1038/35082583
27. Mascaux C, Angelova M, Vasaturo A, Beane J, Hijazi K, Anthoine G, et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature. (2019) 571(7766):570–5. doi: 10.1038/s41586-019-1330-0
28. Zinkernagel RM. Immunity against solid tumors? Int J Cancer (2001) 93(1):1–5. doi: 10.1002/ijc.1305
29. Alavi S, Emran AA, Tseng HY, Tiffen JC, McGuire HM, Hersey P. Nicotinamide inhibits T cell exhaustion and increases differentiation of CD8 effector T cells. Cancers (Basel). (2022) 14(2). doi: 10.3390/cancers14020323
30. Mitchell KG, Parra ER, Nelson DB, Zhang J, Wistuba II, Fujimoto J, et al. Tumor cellular proliferation is associated with enhanced immune checkpoint expression in stage I non-small cell lung cancer. J Thorac Cardiovasc Surg (2019) 158(3):911–9.e6. doi: 10.1016/j.jtcvs.2019.04.084
31. Yang M, Lin C, Wang Y, Chen K, Zhang H, Li W. Identification of a cytokine-dominated immunosuppressive class in squamous cell lung carcinoma with implications for immunotherapy resistance. Genome Med (2022) 14(1):72. doi: 10.1186/s13073-022-01079-x
32. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. (2020) 52(5):825–41.e8. doi: 10.1016/j.immuni.2020.04.014
33. Kersten K, Hu KH, Combes AJ, Samad B, Harwin T, Ray A, et al. Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer Cell (2022) 40(6):624–38.e9. doi: 10.1016/j.ccell.2022.05.004
34. Ludwig N, Whiteside TL. Potential roles of tumor-derived exosomes in angiogenesis. Expert Opin Ther Targets. (2018) 22(5):409–17. doi: 10.1080/14728222.2018.1464141
35. Whiteside TL. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin Immunol (2018) 35:69–79. doi: 10.1016/j.smim.2017.12.003
36. Lin SY, Hang JF, Lai CR, Chan IS, Shih YC, Jiang LY, et al. Loss of major histocompatibility complex class I, CD8 + tumor-infiltrating lymphocytes, and PD-L1 expression in ovarian clear cell carcinoma. Am J Surg Pathol (2023) 47(1):124–30. doi: 10.1097/PAS.0000000000001975
37. Yoshihama S, Vijayan S, Sidiq T, Kobayashi KS. NLRC5/CITA: a key player in cancer immune surveillance. Trends Cancer. (2017) 3(1):28–38. doi: 10.1016/j.trecan.2016.12.003
38. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
39. Demel UM, Böger M, Yousefian S, Grunert C, Zhang L, Hotz PW, et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J Clin Invest (2022) 132(9). doi: 10.1172/JCI152383
40. Xie F, Zhou X, Su P, Li H, Tu Y, Du J, et al. Breast cancer cell-derived extracellular vesicles promote CD8(+) T cell exhaustion via TGF-β type II receptor signaling. Nat Commun (2022) 13(1):4461. doi: 10.1038/s41467-022-31250-2
41. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, Kückelhaus J, et al. T-Cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun (2022) 13(1):925. doi: 10.1038/s41467-022-28523-1
42. Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis. Cancer Cell (2019) 35(6):901–15.e4. doi: 10.1016/j.ccell.2019.05.005
43. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun (2022) 13(1):4981. doi: 10.1038/s41467-022-32521-8
44. Elia I, Rowe JH, Johnson S, Joshi S, Notarangelo G, Kurmi K, et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab (2022) 34(8):1137–50.e6. doi: 10.1016/j.cmet.2022.06.008
45. Wang K, Zhang Y, Chen ZN. Metabolic interaction: tumor-derived lactate inhibiting CD8(+) T cell cytotoxicity in a novel route. Signal Transduct Target Ther (2023) 8(1):52. doi: 10.1038/s41392-023-01320-y
46. Giles JR, Ngiow SF, Manne S, Baxter AE, Khan O, Wang P, et al. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat Immunol (2022) 23(11):1600–13. doi: 10.1038/s41590-022-01338-4
47. Xu J, Fang Y, Chen K, Li S, Tang S, Ren Y, et al. Single-cell RNA sequencing reveals the tissue architecture in human high-grade serous ovarian cancer. Clin Cancer Res (2022) 28(16):3590–602. doi: 10.1158/1078-0432.CCR-22-0296
48. Xu L, Lu Y, Deng Z, Li X, Shi Y, Zhao K, et al. Single-cell landscape of immunocytes in patients with extrahepatic cholangiocarcinoma. J Transl Med (2022) 20(1):210. doi: 10.1186/s12967-022-03424-5
49. Winkler F, Bengsch B. Use of mass cytometry to profile human T cell exhaustion. Front Immunol (2020) 10. doi: 10.3389/fimmu.2019.03039
50. Schuyler RP, Jackson C, Garcia-Perez JE, Baxter RM, Ogolla S, Rochford R, et al. Minimizing batch effects in mass cytometry data. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.02367
51. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O'Brien S, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity. (2018) 48(5):1029–45.e5. doi: 10.1016/j.immuni.2018.04.026
52. Ding GY, Ma JQ, Yun JP, Chen X, Ling Y, Zhang S, et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J Hepatol (2022) 76(3):608–18. doi: 10.1016/j.jhep.2021.10.030
53. du Bois H, Heim TA, Lund AW. Tumor-draining lymph nodes: At the crossroads of metastasis and immunity. Sci Immunol (2021) 6(63):eabg3551. doi: 10.1126/sciimmunol.abg3551
54. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol (2013) 13(5):309–20. doi: 10.1038/nri3442
55. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
56. Obar JJ, Lefrancois L. Memory CD8+ T cell differentiation. Ann N Y Acad Sci (2010) 1183:251–66. doi: 10.1111/j.1749-6632.2009.05126.x
57. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
58. Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci USA (2004) 101(45):16004–9. doi: 10.1073/pnas.0407192101
59. Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med (2007) 204(4):941–9. doi: 10.1084/jem.20061937
60. Obar JJ, Fuse S, Leung EK, Bellfy SC, Usherwood EJ. Gammaherpesvirus persistence alters key CD8 T-cell memory characteristics and enhances antiviral protection. J Virol (2006) 80(17):8303–15. doi: 10.1128/JVI.00237-06
61. Alter G, Hatzakis G, Tsoukas CM, Pelley K, Rouleau D, LeBlanc R, et al. Longitudinal assessment of changes in HIV-specific effector activity in HIV-infected patients starting highly active antiretroviral therapy in primary infection. J Immunol (2003) 171(1):477–88. doi: 10.4049/jimmunol.171.1.477
62. Oxenius A, Price DA, Gunthard HF, Dawson SJ, Fagard C, Perrin L, et al. Stimulation of HIV-specific cellular immunity by structured treatment interruption fails to enhance viral control in chronic HIV infection. Proc Natl Acad Sci U S A. (2002) 99(21):13747–52. doi: 10.1073/pnas.202372199
63. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, et al. T Cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol (2013) 14(6):603–10. doi: 10.1038/ni.2606
64. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
65. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. (2012) 338(6111):1220–5. doi: 10.1126/science.1229620
66. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. (2008) 105(39):15016–21. doi: 10.1073/pnas.0801497105
67. McLane LM, Ngiow SF, Chen Z, Attanasio J, Manne S, Ruthel G, et al. Role of nuclear localization in the regulation and function of T-bet and eomes in exhausted CD8 T cells. Cell Rep (2021) 35(6):109120. doi: 10.1016/j.celrep.2021.109120
68. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature. (2016) 537(7620):412–28. doi: 10.1038/nature19317
69. Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-Centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. (2019) 51(5):840–55.e5. doi: 10.1016/j.immuni.2019.09.013
70. Zheng W, Wei J, Zebley CC, Jones LL, Dhungana Y, Wang YD, et al. Regnase-1 suppresses TCF-1+ precursor exhausted T-cell formation to limit CAR-t-cell responses against ALL. Blood. (2021) 138(2):122–35. doi: 10.1182/blood.2020009309
71. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1(6). doi: 10.1126/sciimmunol.aai8593
72. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. (2016) 537(7620):417–21. doi: 10.1038/nature19330
73. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5(+) follicular cytotoxic T cells control viral infection in b cell follicles. Nat Immunol (2016) 17(10):1187–96. doi: 10.1038/ni.3543
74. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
75. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
76. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. (2009) 325(5943):1001–5. doi: 10.1126/science.1176676
77. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27(4):670–84. doi: 10.1016/j.immuni.2007.09.006
78. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
79. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity. (2017) 47(6):1129–41.e5. doi: 10.1016/j.immuni.2017.11.021
80. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. (2019) 571(7764):270–4. doi: 10.1038/s41586-019-1324-y
81. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. (2019) 571(7764):211–8. doi: 10.1038/s41586-019-1325-x
82. Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
83. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
84. Abdel-Hakeem MS, Manne S, Beltra JC, Stelekati E, Chen Z, Nzingha K, et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol (2021) 22(8):1008–19. doi: 10.1038/s41590-021-00975-5
85. Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE, et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol (2021) 22(8):1020–9. doi: 10.1038/s41590-021-00979-1
86. Tonnerre P, Wolski D, Subudhi S, Aljabban J, Hoogeveen RC, Damasio M, et al. Differentiation of exhausted CD8(+) T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nat Immunol (2021) 22(8):1030–41. doi: 10.1038/s41590-021-00982-6
87. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. (2019) 571(7764):265–9. doi: 10.1038/s41586-019-1326-9
88. Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
89. Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. (2019) 567(7749):525–9. doi: 10.1038/s41586-019-0979-8
90. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. (2020) 396(10251):635–48. doi: 10.1016/S0140-6736(20)31288-5
91. Baidoun F, Elshiwy K, Elkeraie Y, Merjaneh Z, Khoudari G, Sarmini MT, et al. Colorectal cancer epidemiology: recent trends and impact on outcomes. Curr Drug targets. (2021) 22(9):998–1009. doi: 10.2174/1389450121999201117115717
92. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
93. Bang YJ, Ruiz EY, Van Cutsem E, Lee KW, Wyrwicz L, Schenker M, et al. Phase III, randomised trial of avelumab versus physician's choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN gastric 300. Ann Oncol (2018) 29(10):2052–60. doi: 10.1093/annonc/mdy264
94. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390(10111):2461–71. doi: 10.1016/S0140-6736(17)31827-5
95. Li Y, Hu X, Lin R, Zhou G, Zhao L, Zhao D, et al. Single-cell landscape reveals active cell subtypes and their interaction in the tumor microenvironment of gastric cancer. Theranostics. (2022) 12(8):3818–33. doi: 10.7150/thno.71833
96. Sun K, Xu R, Ma F, Yang N, Li Y, Sun X, et al. scRNA-seq of gastric tumor shows complex intercellular interaction with an alternative T cell exhaustion trajectory. Nat Commun (2022) 13(1):4943. doi: 10.1038/s41467-022-32627-z
97. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol (2011) 186(3):1338–42. doi: 10.4049/jimmunol.1003081
98. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res (2017) 77(22):6375–88. doi: 10.1158/0008-5472.CAN-17-0381
99. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res (2016) 22(21):5183–8. doi: 10.1158/1078-0432.CCR-16-0933
100. Xu C, Fang H, Gu Y, Yu K, Wang J, Lin C, et al. Impact of intratumoural CD96 expression on clinical outcome and therapeutic benefit in gastric cancer. Cancer Sci (2022) 113(12):4070–81. doi: 10.1111/cas.15537
101. Srinivasan P, Wu X, Basu M, Rossi C, Sandler AD. PD-L1 checkpoint inhibition and anti-CTLA-4 whole tumor cell vaccination counter adaptive immune resistance: a mouse neuroblastoma model that mimics human disease. PLoS Med (2018) 15(1):e1002497. doi: 10.1371/journal.pmed.1002497
102. Li X, Zhai J, Shen Y, Zhang T, Wang Y, He Y, et al. Tumor-derived IL-8 facilitates lymph node metastasis of gastric cancer via PD-1 up-regulation in CD8(+) T cells. Cancer Immunol Immunother. (2022) 71(12):3057–70. doi: 10.1007/s00262-022-03223-3
103. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
104. Shi L, Feng M, Du S, Wei X, Song H, Yixin X, et al. Adenosine generated by regulatory T cells induces CD8(+) T cell exhaustion in gastric cancer through A2aR pathway. BioMed Res Int (2019) 2019:4093214. doi: 10.1155/2019/4093214
105. Yu K, Kuang L, Fu T, Zhang C, Zhou Y, Zhu C, et al. CREM is correlated with immune-suppressive microenvironment and predicts poor prognosis in gastric adenocarcinoma. Front Cell Dev Biol (2021) 9:697748. doi: 10.3389/fcell.2021.697748
106. Xiang Z, Zhou Z, Song S, Li J, Ji J, Yan R, et al. Dexamethasone suppresses immune evasion by inducing GR/STAT3 mediated downregulation of PD-L1 and IDO1 pathways. Oncogene. (2021) 40(31):5002–12. doi: 10.1038/s41388-021-01897-0
107. Emambux S, Tachon G, Junca A, Tougeron D. Results and challenges of immune checkpoint inhibitors in colorectal cancer. Expert Opin Biol Ther (2018) 18(5):561–73. doi: 10.1080/14712598.2018.1445222
108. Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. (2020) 579(7798):274–8. doi: 10.1038/s41586-020-2056-8
109. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2019) 176(1-2):404. doi: 10.1016/j.cell.2018.12.034
110. Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 inhibitors in metastatic dMMR/MSI-h colorectal cancer. Front Oncol (2019) 9:396. doi: 10.3389/fonc.2019.00396
111. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. (2019) 19(3):133–50. doi: 10.1038/s41568-019-0116-x
112. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. (2019) 176(4):775–89.e18. doi: 10.1016/j.cell.2018.11.043
113. Becker WR, Nevins SA, Chen DC, Chiu R, Horning AM, Guha TK, et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet (2022) 54(7):985–95. doi: 10.1038/s41588-022-01088-x
114. Saleh R, Taha RZ, Toor SM, Sasidharan Nair V, Murshed K, Khawar M, et al. Expression of immune checkpoints and T cell exhaustion markers in early and advanced stages of colorectal cancer. Cancer Immunol Immunother. (2020) 69(10):1989–99. doi: 10.1007/s00262-020-02593-w
115. Thommen DS, Schumacher TN. T Cell dysfunction in cancer. Cancer Cell (2018) 33(4):547–62. doi: 10.1016/j.ccell.2018.03.012
116. Ideta T, Li B, Flynn C, Igarashi Y, Lowman G, Looney T, et al. The epithelial-stromal microenvironment in early colonic neoplasia. Mol Cancer Res (2022) 20(1):56–61. doi: 10.1158/1541-7786.MCR-21-0202
117. Yang R, Cheng S, Luo N, Gao R, Yu K, Kang B, et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol (2019) 21(1):2. doi: 10.1186/s13059-019-1921-y
118. Sakimura S, Nagayama S, Fukunaga M, Hu Q, Kitagawa A, Kobayashi Y, et al. Impaired tumor immune response in metastatic tumors is a selective pressure for neutral evolution in CRC cases. PLoS Genet (2021) 17(1):e1009113. doi: 10.1371/journal.pgen.1009113
119. Di J, Liu M, Fan Y, Gao P, Wang Z, Jiang B, et al. Phenotype molding of T cells in colorectal cancer by single-cell analysis. Int J Cancer. (2020) 146(8):2281–95. doi: 10.1002/ijc.32856
120. Hu J, Han C, Zhong J, Liu H, Liu R, Luo W, et al. Dynamic network biomarker of pre-exhausted CD8(+) T cells contributed to T cell exhaustion in colorectal cancer. Front Immunol (2021) 12:691142. doi: 10.3389/fimmu.2021.691142
121. Liang R, Zhu X, Lan T, Ding D, Zheng Z, Chen T, et al. TIGIT promotes CD8(+)T cells exhaustion and predicts poor prognosis of colorectal cancer. Cancer Immunol Immunother. (2021) 70(10):2781–93. doi: 10.1007/s00262-021-02886-8
122. Lin ZB, Long P, Zhao Z, Zhang YR, Chu XD, Zhao XX, et al. Long noncoding RNA KCNQ1OT1 is a prognostic biomarker and mediates CD8(+) T cell exhaustion by regulating CD155 expression in colorectal cancer. Int J Biol Sci (2021) 17(7):1757–68. doi: 10.7150/ijbs.59001
123. Carretero M, Cantoni C, Bellon T, Bottino C, Biassoni R, Rodriguez A, et al. The CD94 and NKG2-a c-type lectins covalently assemble to form a natural killer cell inhibitory receptor for HLA class I molecules. Eur J Immunol (1997) 27(2):563–7. doi: 10.1002/eji.1830270230
124. Ducoin K, Oger R, Bilonda Mutala L, Deleine C, Jouand N, Desfrancois J, et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. Oncoimmunology. (2022) 11(1):2046931. doi: 10.1080/2162402X.2022.2046931
125. Yang RY, Rabinovich GA, Liu FT. Galectins: structure, function and therapeutic potential. Expert Rev Mol Med (2008) 10:e17. doi: 10.1017/S1462399408000719
126. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun (2021) 12(1):832. doi: 10.1038/s41467-021-21099-2
127. Zhang Y, Cai P, Li L, Shi L, Chang P, Liang T, et al. Co-Expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int Immunopharmacol. (2017) 43:210–8. doi: 10.1016/j.intimp.2016.12.024
128. Richards P, Ourabah S, Montagne J, Burnol AF, Postic C, Guilmeau S. MondoA/ChREBP: the usual suspects of transcriptional glucose sensing; implication in pathophysiology. Metabolism. (2017) 70:133–51. doi: 10.1016/j.metabol.2017.01.033
129. Richards P, Rachdi L, Oshima M, Marchetti P, Bugliani M, Armanet M, et al. MondoA is an essential glucose-responsive transcription factor in human pancreatic beta-cells. Diabetes. (2018) 67(3):461–72. doi: 10.2337/db17-0595
130. Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell (2013) 49(6):1167–75. doi: 10.1016/j.molcel.2013.01.035
131. Lu Y, Li Y, Liu Q, Tian N, Du P, Zhu F, et al. MondoA-Thioredoxin-Interacting protein axis maintains regulatory T-cell identity and function in colorectal cancer microenvironment. Gastroenterology. (2021) 161(2):575–91.e16. doi: 10.1053/j.gastro.2021.04.041
132. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30(1):143–56.e5. doi: 10.1016/j.cmet.2019.04.002
133. Bell HN, Huber AK, Singhal R, Korimerla N, Rebernick RJ, Kumar R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab (2023) 35(1):134–49.e6. doi: 10.1016/j.cmet.2022.11.013
134. Rong D, Sun G, Zheng Z, Liu L, Chen X, Wu F, et al. MGP promotes CD8(+) T cell exhaustion by activating the NF-kappaB pathway leading to liver metastasis of colorectal cancer. Int J Biol Sci (2022) 18(6):2345–61. doi: 10.7150/ijbs.70137
135. LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. PTPN2 regulates the generation of exhausted CD8(+) T cell subpopulations and restrains tumor immunity. Nat Immunol (2019) 20(10):1335–47. doi: 10.1038/s41590-019-0480-4
136. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-a drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol (2019) 4(41). doi: 10.1126/sciimmunol.aay0555
137. Sun H, Han W, Wen J, Ma X. IL4I1 and tryptophan metabolites enhance AHR signals to facilitate colorectal cancer progression and immunosuppression. Am J Transl Res (2022) 14(11):7758–70.
138. Wu D, Zhu Y. Role of kynurenine in promoting the generation of exhausted CD8(+) T cells in colorectal cancer. Am J Transl Res (2021) 13(3):1535–47.
139. Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of cbl-b inhibits CD8(+) T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer (2021) 9(1). doi: 10.1136/jitc-2020-001688
140. Yu AI, Zhao L, Eaton KA, Ho S, Chen J, Poe S, et al. Gut microbiota modulate CD8 T cell responses to influence colitis-associated tumorigenesis. Cell Rep (2020) 31(1):107471. doi: 10.1016/j.celrep.2020.03.035
141. Gallerano D, Ciminati S, Grimaldi A, Piconese S, Cammarata I, Focaccetti C, et al. Genetically driven CD39 expression shapes human tumor-infiltrating CD8(+) T-cell functions. Int J Cancer. (2020) 147(9):2597–610. doi: 10.1002/ijc.33131
142. Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Epigenetic regulation of immune checkpoints and T cell exhaustion markers in tumor-infiltrating T cells of colorectal cancer patients. Epigenomics. (2020) 12(21):1871–82. doi: 10.2217/epi-2020-0267
143. Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil extracellular traps promote T cell exhaustion in the tumor microenvironment. Front Immunol (2021) 12:785222. doi: 10.3389/fimmu.2021.785222
144. Harryvan TJ, Visser M, de Bruin L, Plug L, Griffioen L, Mulder A, et al. Enhanced antigen cross-presentation in human colorectal cancer-associated fibroblasts through upregulation of the lysosomal protease cathepsin s. J Immunother Cancer (2022) 10(3). doi: 10.1136/jitc-2021-003591
145. Majumder S, Shivaji UN, Kasturi R, Sigamani A, Ghosh S, Iacucci M. Inflammatory bowel disease-related colorectal cancer: past, present and future perspectives. World J Gastrointest Oncol (2022) 14(3):547–67. doi: 10.4251/wjgo.v14.i3.547
146. Ephraim R, Feehan J, Fraser S, Nurgali K, Apostolopoulos V. Cancer immunotherapy: the checkpoint between chronic colitis and colorectal cancer. Cancers (Basel). (2022) 14(24). doi: 10.3390/cancers14246131
147. Globig AM, Mayer LS, Heeg M, Andrieux G, Ku M, Otto-Mora P, et al. Exhaustion of CD39-expressing CD8(+) T cells in crohn's disease is linked to clinical outcome. Gastroenterology. (2022) 163(4):965–81.e31. doi: 10.1053/j.gastro.2022.06.045
148. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-Cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. (2015) 523(7562):612–6. doi: 10.1038/nature14468
149. Rao X, Jiang J, Liang Z, Zhang J, Zhuang Z, Qiu H, et al. Down-regulated CLDN10 predicts favorable prognosis and correlates with immune infiltration in gastric cancer. Front Genet (2021) 12:747581. doi: 10.3389/fgene.2021.747581
150. Yang H, Li L, Liu X, Zhao Y. High expression of the component 3a receptor 1 (C3AR1) gene in stomach adenocarcinomas infers a poor prognosis and high immune-infiltration levels. Med Sci Monit (2021) 27:e927977. doi: 10.12659/MSM.927977
151. Huang X, Zhang J, Zheng Y. ANTXR1 is a prognostic biomarker and correlates with stromal and immune cell infiltration in gastric cancer. Front Mol Biosci (2020) 7:598221. doi: 10.3389/fmolb.2020.598221
152. Liang Z, Li X. Identification of ANXA1 as a potential prognostic biomarker and correlating with immune infiltrates in colorectal cancer. Autoimmunity. (2021) 54(2):76–87. doi: 10.1080/08916934.2021.1887148
153. Hu L, Zhang P, Sun W, Zhou L, Chu Q, Chen Y. PDPN is a prognostic biomarker and correlated with immune infiltrating in gastric cancer. Med (Baltimore). (2020) 99(19):e19957. doi: 10.1097/MD.0000000000019957
154. Tang W, Pan X, Han D, Rong D, Zhang M, Yang L, et al. Clinical significance of CD8(+) T cell immunoreceptor with ig and ITIM domains(+) in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology. (2019) 8(6):e1593807. doi: 10.1080/2162402X.2019.1593807
155. Van Coillie S, Wiernicki B, Xu J. Molecular and cellular functions of CTLA-4. Adv Exp Med Biol (2020) 1248:7–32. doi: 10.1007/978-981-15-3266-5_2
156. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
157. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
158. Yoshikawa AK, Yamaguchi K, Muro K, Takashima A, Ichimura T, Sakai D, et al. Safety and tolerability of andecaliximab as monotherapy and in combination with an anti-PD-1 antibody in Japanese patients with gastric or gastroesophageal junction adenocarcinoma: a phase 1b study. J Immunother Cancer (2022) 10(1). doi: 10.1136/jitc-2021-003518
159. Kang YK, Chen LT, Ryu MH, Oh DY, Oh SC, Chung HC, et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): a randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2022) 23(2):234–47. doi: 10.1016/S1470-2045(21)00692-6
160. Park H, Shapiro GI, Gao X, Mahipal A, Starr J, Furqan M, et al. Phase ib study of eprenetapopt (APR-246) in combination with pembrolizumab in patients with advanced or metastatic solid tumors. ESMO Open (2022) 7(5):100573. doi: 10.1016/j.esmoop.2022.100573
161. Coutzac C, Bibeau F, Ben Abdelghani M, Aparicio T, Cohen R, Coquan E, et al. Immunotherapy in MSI/dMMR tumors in the perioperative setting: the IMHOTEP trial. Dig Liver Dis (2022) 54(10):1335–41. doi: 10.1016/j.dld.2022.07.008
162. Yamaguchi K, Minashi K, Sakai D, Nishina T, Omuro Y, Tsuda M, et al. Phase IIb study of pembrolizumab combined with s-1 + oxaliplatin or s-1 + cisplatin as first-line chemotherapy for gastric cancer. Cancer Sci (2022) 113(8):2814–27. doi: 10.1111/cas.15462
163. Fuchs CS, Ozguroglu M, Bang YJ, Di Bartolomeo M, Mandala M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated PD-L1-positive advanced gastric or gastroesophageal junction cancer: 2-year update of the randomized phase 3 KEYNOTE-061 trial. Gastric Cancer. (2022) 25(1):197–206. doi: 10.1007/s10120-021-01227-z
164. Chung HC, Kang YK, Chen Z, Bai Y, Wan Ishak WZ, Shim BY, et al. Pembrolizumab versus paclitaxel for previously treated advanced gastric or gastroesophageal junction cancer (KEYNOTE-063): a randomized, open-label, phase 3 trial in Asian patients. Cancer. (2022) 128(5):995–1003. doi: 10.1002/cncr.34019
165. Dos Santos M, Lequesne J, Leconte A, Corbinais S, Parzy A, Guilloit JM, et al. Perioperative treatment in resectable gastric cancer with spartalizumab in combination with fluorouracil, leucovorin, oxaliplatin and docetaxel (FLOT): a phase II study (GASPAR). BMC Cancer. (2022) 22(1):537. doi: 10.1186/s12885-022-09623-z
166. Jing C, Wang J, Zhu M, Bai Z, Zhao B, Zhang J, et al. Camrelizumab combined with apatinib and s-1 as second-line treatment for patients with advanced gastric or gastroesophageal junction adenocarcinoma: a phase 2, single-arm, prospective study. Cancer Immunol Immunother. (2022) 71(11):2597–608. doi: 10.1007/s00262-022-03174-9
167. Liu D, Zhou J, Wang Y, Li M, Jiang H, Liu Y, et al. Bifunctional anti-PD-L1/TGF-betaRII agent SHR-1701 in advanced solid tumors: a dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med (2022) 20(1):408. doi: 10.1186/s12916-022-02605-9
168. Kelly RJ, Lee J, Bang YJ, Almhanna K, Blum-Murphy M, Catenacci DVT, et al. Safety and efficacy of durvalumab and tremelimumab alone or in combination in patients with advanced gastric and gastroesophageal junction adenocarcinoma. Clin Cancer Res (2020) 26(4):846–54. doi: 10.1158/1078-0432.CCR-19-2443
169. Li X, Fang C, Wang X, Yu Y, Wang Z, Qiu M. Neoadjuvant treatment of sintilimab plus hypofractionated radiotherapy for MSI-H/dMMR rectal cancer: a prospective, multicenter, phase ib study. Cancer Med (2022) 11(23):4405–10. doi: 10.1002/cam4.4720
170. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med (2022) 386(25):2363–76. doi: 10.1056/NEJMoa2201445
171. Hu H, Kang L, Zhang J, Wu Z, Wang H, Huang M, et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): a single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol Hepatol (2022) 7(1):38–48. doi: 10.1016/S2468-1253(21)00348-4
172. Felip E, Moreno V, Morgensztern D, Curigliano G, Rutkowski P, Trigo JM, et al. First-in-human, open-label, phase 1/2 study of the monoclonal antibody programmed cell death protein-1 (PD-1) inhibitor cetrelimab (JNJ-63723283) in patients with advanced cancers. Cancer Chemother Pharmacol (2022) 89(4):499–514. doi: 10.1007/s00280-022-04414-6
173. Haag GM, Springfeld C, Grun B, Apostolidis L, Zschabitz S, Dietrich M, et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer - the PICCASSO phase I trial. Eur J Cancer. (2022) 167:112–22. doi: 10.1016/j.ejca.2022.03.017
174. Rahma OE, Tyan K, Giobbie-Hurder A, Brohl AS, Bedard PL, Renouf DJ, et al. Phase IB study of ziv-aflibercept plus pembrolizumab in patients with advanced solid tumors. J Immunother Cancer (2022) 10(3). doi: 10.1136/jitc-2021-003569
175. Kuang C, Park Y, Augustin RC, Lin Y, Hartman DJ, Seigh L, et al. Pembrolizumab plus azacitidine in patients with chemotherapy refractory metastatic colorectal cancer: a single-arm phase 2 trial and correlative biomarker analysis. Clin Epigenetics. (2022) 14(1):3. doi: 10.1186/s13148-021-01226-y
176. Marabelle A, Cassier PA, Fakih M, Kao S, Nielsen D, Italiano A, et al. Pembrolizumab for previously treated advanced anal squamous cell carcinoma: results from the non-randomised, multicohort, multicentre, phase 2 KEYNOTE-158 study. Lancet Gastroenterol Hepatol (2022) 7(5):446–54. doi: 10.1016/S2468-1253(21)00382-4
177. Diaz LA Jr., Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol (2022) 23(5):659–70. doi: 10.1016/S1470-2045(22)00197-8
178. Kim RD, Kovari BP, Martinez M, Xie H, Sahin IH, Mehta R, et al. A phase I/Ib study of regorafenib and nivolumab in mismatch repair proficient advanced refractory colorectal cancer. Eur J Cancer. (2022) 169:93–102. doi: 10.1016/j.ejca.2022.03.026
179. Wang Y, Shen L, Wan J, Zhang H, Wu R, Wang J, et al. Short-course radiotherapy combined with CAPOX and toripalimab for the total neoadjuvant therapy of locally advanced rectal cancer: a randomized, prospective, multicentre, double-arm, phase II trial (TORCH). BMC Cancer. (2022) 22(1):274. doi: 10.1186/s12885-022-09348-z
180. Rao S, Anandappa G, Capdevila J, Dahan L, Evesque L, Kim S, et al. A phase II study of retifanlimab (INCMGA00012) in patients with squamous carcinoma of the anal canal who have progressed following platinum-based chemotherapy (POD1UM-202). ESMO Open (2022) 7(4):100529. doi: 10.1016/j.esmoop.2022.100529
181. Johnson B, Haymaker CL, Parra ER, Soto LMS, Wang X, Thomas JV, et al. Phase II study of durvalumab (anti-PD-L1) and trametinib (MEKi) in microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J Immunother Cancer (2022) 10(8). doi: 10.1136/jitc-2022-005332
182. Antoniotti C, Rossini D, Pietrantonio F, Catteau A, Salvatore L, Lonardi S, et al. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol (2022) 23(7):876–87. doi: 10.1016/S1470-2045(22)00274-1
183. Redman JM, Tsai YT, Weinberg BA, Donahue RN, Gandhy S, Gatti-Mays ME, et al. A randomized phase II trial of mFOLFOX6 + bevacizumab alone or with AdCEA vaccine + avelumab immunotherapy for untreated metastatic colorectal cancer. Oncologist. (2022) 27(3):198–209. doi: 10.1093/oncolo/oyab046
184. Cohen R, Bennouna J, Meurisse A, Tournigand C, de la Fouchardiere C, Tougeron D, et al. RECIST and iRECIST criteria for the evaluation of nivolumab plus ipilimumab in patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the GERCOR NIPICOL phase II study. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-001499
185. Kanikarla Marie P, Haymaker C, Parra ER, Kim YU, Lazcano R, Gite S, et al. Pilot clinical trial of perioperative durvalumab and tremelimumab in the treatment of resectable colorectal cancer liver metastases. Clin Cancer Res (2021) 27(11):3039–49. doi: 10.1158/1078-0432.CCR-21-0163
186. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: the Canadian cancer trials group CO.26 study. JAMA Oncol (2020) 6(6):831–8. doi: 10.1001/jamaoncol.2020.0910
187. Dubois M, Liscia N, Brunetti O, Ziranu P, Lai E, Argentiero A, et al. The role of immune checkpoint inhibitors in the treatment sequence of advanced gastric or gastro-esophageal junction cancer: a systematic review and meta-analysis of randomized trials. Crit Rev oncology/hematology. (2022) 173:103674. doi: 10.1016/j.critrevonc.2022.103674
188. Borelli B, Antoniotti C, Carullo M, Germani MM, Conca V, Masi G. Immune-checkpoint inhibitors (ICIs) in metastatic colorectal cancer (mCRC) patients beyond microsatellite instability. Cancers (Basel). (2022) 14(20). doi: 10.3390/cancers14204974
189. Kono K, Nakajima S, Mimura K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer. (2020) 23(4):565–78. doi: 10.1007/s10120-020-01090-4
190. Wang M, Zhai X, Li J, Guan J, Xu S, Li Y, et al. The role of cytokines in predicting the response and adverse events related to immune checkpoint inhibitors. Front Immunol (2021) 12:670391. doi: 10.3389/fimmu.2021.670391
191. Yoshikawa Y, Imamura M, Yamauchi M, Hayes CN, Aikata H, Okamoto W, et al. Prevalence of immune-related adverse events and anti-tumor efficacy following immune checkpoint inhibitor therapy in Japanese patients with various solid tumors. BMC Cancer. (2022) 22(1):1232. doi: 10.1186/s12885-022-10327-7
192. Ding JT, Yang KP, Lin KL, Cao YK, Zou F. Mechanisms and therapeutic strategies of immune checkpoint molecules and regulators in type 1 diabetes. Front Endocrinol (Lausanne). (2022) 13:1090842. doi: 10.3389/fendo.2022.1090842
193. Haugh AM, Probasco JC, Johnson DB. Neurologic complications of immune checkpoint inhibitors. Expert Opin Drug Saf. (2020) 19(4):479–88. doi: 10.1080/14740338.2020.1738382
194. Quach HT, Johnson DB, LeBoeuf NR, Zwerner JP, Dewan AK. Cutaneous adverse events caused by immune checkpoint inhibitors. J Am Acad Dermatol (2021) 85(4):956–66. doi: 10.1016/j.jaad.2020.09.054
195. Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor-associated myocarditis: manifestations and mechanisms. J Clin Invest. (2021) 131(5). doi: 10.1172/JCI145186
196. Chen C, Zhang F, Zhou N, Gu YM, Zhang YT, He YD, et al. Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. Oncoimmunology. (2019) 8(5):e1581547. doi: 10.1080/2162402X.2019.1581547
197. Xu Y, Fu Y, Zhu B, Wang J, Zhang B. Predictive biomarkers of immune checkpoint inhibitors-related toxicities. Front Immunol (2020) 11:2023. doi: 10.3389/fimmu.2020.02023
198. Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. (2021) 398(10294):27–40. doi: 10.1016/S0140-6736(21)00797-2
199. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
200. Parikh AR, Szabolcs A, Allen JN, Clark JW, Wo JY, Raabe M, et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat Cancer. (2021) 2(11):1124–35. doi: 10.1038/s43018-021-00269-7
201. Palmer AC, Izar B, Hwangbo H, Sorger PK. Predictable clinical benefits without evidence of synergy in trials of combination therapies with immune-checkpoint inhibitors. Clin Cancer Res (2022) 28(2):368–77. doi: 10.1158/1078-0432.CCR-21-2275
202. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-Cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. (2017) 545(7652):60–5. doi: 10.1038/nature22079
Keywords: T cell exhaustion, gastric cancer, colorectal cancer, immune checkpoints, immunotherapy
Citation: Ding J-T, Yang K-P, Zhou H-N, Huang Y-F, Li H and Zong Z (2023) Landscapes and mechanisms of CD8+ T cell exhaustion in gastrointestinal cancer. Front. Immunol. 14:1149622. doi: 10.3389/fimmu.2023.1149622
Received: 22 January 2023; Accepted: 13 April 2023;
Published: 25 April 2023.
Edited by:
Mohamed S. Abdel-Hakeem, Emory University, United StatesReviewed by:
Billur Akkaya, The Ohio State University, United StatesCopyright © 2023 Ding, Yang, Zhou, Huang, Li and Zong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Li, bmR5ZnkwNDQwOEBuY3UuZWR1LmNu; Zhen Zong, bmRlZnkxNjEzM0BuY3UuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.