 Dun Pan
Dun Pan Guo Li
Guo Li Chunlin Jiang1†
Chunlin Jiang1† Xiangming Hu
Xiangming Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 22 May 2023
Sec. Molecular Innate Immunity
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1149366
This article is part of the Research TopicPlasticity and Metabolic Switching in Adipose Tissue MacrophagesView all 11 articles
In adipose tissue, macrophages are the most abundant immune cells with high heterogeneity and plasticity. Depending on environmental cues and molecular mediators, adipose tissue macrophages (ATMs) can be polarized into pro- or anti-inflammatory cells. In the state of obesity, ATMs switch from the M2 polarized state to the M1 state, which contributes to chronic inflammation, thereby promoting the pathogenic progression of obesity and other metabolic diseases. Recent studies show that multiple ATM subpopulations cluster separately from the M1 or M2 polarized state. Various factors are related to ATM polarization, including cytokines, hormones, metabolites and transcription factors. Here, we discuss our current understanding of the potential regulatory mechanisms underlying ATM polarization induced by autocrine and paracrine factors. A better understanding of how ATMs polarize may provide new therapeutic strategies for obesity-related diseases.
Obesity has become a major public health problem because it increases the risk of many diseases (e.g., type 2 diabetes mellitus, hypertension, osteoarthritis and several cancers) (1). Obesity is characterized by accumulation of adipose tissue (AT), which leads to infiltration of immune cells and chronic low-grade inflammation (2, 3). Being a major portion of AT immune cells, adipose tissue macrophages (ATMs) are key for healthy adipose homeostasis but can also contribute to the pathogenic progression of obesity and other metabolic diseases (4). ATMs can be polarized into distinct phenotypes under different physiological or pathological conditions. In addition to the pro-inflammatory M1 phenotype and anti-inflammatory M2 phenotype (5), several novel ATM subpopulations (e.g., MMe, Mox and LAMs) have been discovered in recent years (6–8). Polarization of ATMs are regulated by organokines and related signaling pathways. In this article, we summarize the characteristics and functions of different ATM subpopulations. In addition, we also highlight the regulatory pathways and mechanisms of ATM polarization, providing novel insights for the treatment of obesity-diseases.
Many studies have classified macrophages according to the M1/M2 system, which is revealed by the secretory profile of cytokines and interleukins (9). Previous studies have described a ‘‘mixed’’ M1/M2 phenotype for ATMs that can be activated into a switching phenotype (10, 11). ATMs are mainly polarized towards the pro-inflammatory M1 phenotype in the obesity state, conversely, in the lean state, ATMs are polarized towards the anti-inflammatory M2 phenotype (5, 12). As technologies have advanced, accumulated evidence has suggested that ATM polarization is more variable than the M1 or M2 state. Distinct ATM subpopulations express specific markers and have unique transcriptional profiles and functions (as summarized in Table 1).
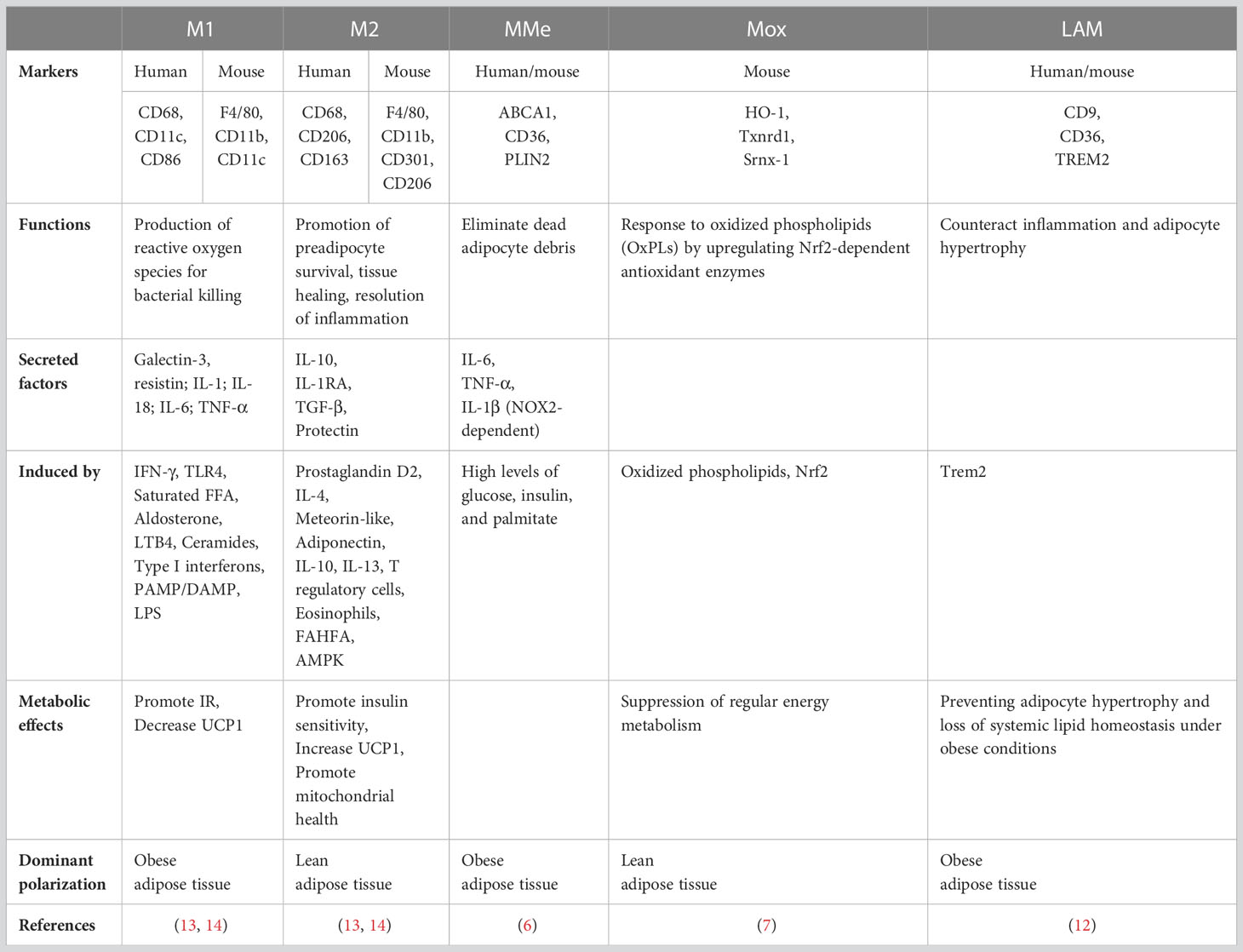
Table 1 Adipose tissue macrophages (ATMs) subpopulations.
Obese AT is recognized as a low-grade, chronic inflammation condition accompanied by accumulation of ATMs and a phenotypic switch (2, 3). Obese adipocytes secrete various chemokines and adipokines (e.g., MCP-1, CXCL12 and Leptin), which recruit and switch macrophages from the M2 state to the M1 state (5). ATMs are recognized as cells that co-express F4/80 and CD11b in mice or CD68 and CD11b in human (2, 15). Moreover, CD11c is used as phenotypic markers of M1-ATMs, which produce pro-inflammatory mediators like IL-1β, TNF-α and nitric oxide (NO), acting as main effectors of inflammatory signals, impaired adipocyte function and insulin sensitivity (13, 14). Ablation of CD11c positive cells alleviated inflammation and improved insulin sensitivity in obese mice (16). The significance of M1-ATMs is also supported in human studies where CD11c positive ATMs has been associated with glucose intolerance and metabolic syndrome (15).
Unlike pro-inflammatory M1-ATMs, M2-ATMs attenuate inflammation to maintain adipose homeostasis (5). In the lean state, the dominant ATMs are considered as resident macrophages which express markers of M2 macrophages (e.g., CD206, CD301 and CD163) (2, 5). M2-ATMs are further divided into three major subtypes: M2a, M2b and M2c (17), which express specific markers and have unique functions. M2a-TAMs are characterized by high surface expression of IL-R and FIZZ1, and secrete TGF-𝛽, IGF and fibronectin to contribute to tissue repair (18, 19). M2b-TAMs express high levels of IL-10, CCL1 and TNFSF14, and low levels of IL-12, exhibiting anti-inflammatory and immune-regulated effects (20). M2c-TAMs expressing multiple markers like CD14 and TLR1, have high expression of IL-10, TGF-𝛽 and Mer receptor tyrosine kinase, and are considered as anti-inflammatory and phagocytic macrophages (17).
ATMs have a particular metabolically activated phenotype called “MMe”, which exhibit a mixture of M1 and M2 characteristics. The MMe phenotype, which can be identified by their surface markers CD36, ABCA1, and PLIN2, is stimulated by high levels of glucose, insulin, and palmitate (6). MMe macrophages not only promote insulin resistance (IR) via producing inflammatory cytokines, but also clear away dead adipocyte through lysosomal exocytosis, which protect AT from the deleterious effects of excess free fatty acids (FFAs) (21). Therefore, MMe macrophages perform both detrimental and beneficial functions during obesity.
Recently, a novel macrophage phenotype has been identified, known as Mox, mainly stimulated by oxidized lipids (7). High expression of Txnrd-1, Srnx-1 and HO-1 distinguishes Mox from the M1 or M2 phenotype. Compared with M1- and M2-TAMs, Mox macrophages exhibit restricted bioenergetics and more antioxidant production. Recent study has shown that Mox macrophages are the predominant ATMs in lean AT, while more energetic macrophages like M1- or M2-TAMs predominate during the development of obesity (7).
Recently, a novel ATM subpopulation defined as lipid-associated macrophages (LAMs) were discovered surrounding apoptotic adipocytes of obese AT (8). LAMs express highly conserved genes, including CD9, CD36, and the lipid receptor Trem2. LAMs utilize Trem2 as an extracellular lipid sensor and perform protective functions to combat adipocyte inflammation, hypertrophy, and metabolic dysfunction (22). In addition, LAMs express many immunosuppression-related genes such as Lgals1/3, suggesting that they may be involved in regulating inflammatory response induced by lipid accumulation (8).
Accumulating evidences suggest that AT, liver, skeletal muscle and gut function as endocrine organs, producing various organokines (adipokines, hepatokines, myokines and gut cytokines) that are capable of recruiting macrophages or switching ATM phenotypes.
AT secretes a variety of hormones/cytokines, which are called adipokines. Leptin, the first classical adipokine, is initially considered as a satiety signal that regulates body weight by reducing food intake and increasing energy expenditure (23, 24). Mice with leptin deficiency (ob/ob) or leptin receptor deficiency (db/db) develop morbid obesity due to hyperphagia (23, 25). However, due to leptin resistance, most forms of obesity of animals and human are associated with higher leptin levels rather than leptin deficiency (26).
In addition to its role in energy balance, leptin also functions as an immunomodulatory cytokine, inducing immunologic alterations in different cell types, including ATMs. The immunoregulatory effects of leptin were first assessed in ob/ob mice. Macrophages from ob/ob mice have impaired phagocytic activity and pro-inflammatory cytokine production, and exogenous leptin treatment improves the above defects (27). Leptin treatment in ob/ob mice also ameliorates IR while up-regulating the expression of M2 markers (e.g., Fizz-1, Arg-1 and Mgl-1) (28). Similarly, in vitro leptin treatment, CD14 human macrophages up-regulates typical M2 markers, while being able to increase the expression of M1 markers (e.g., IL-6, IL-1𝛽, and MCP-1) (29). These studies suggest that leptin could be a contributor to the distinct ATMs phenotype. Besides, leptin induces the expression of vascular cell adhesion molecules, which can increase macrophage infiltration into AT (30). Leptin also stimulates macrophage proliferation in a dose-dependent manner (31), meaning obesity-associated hyperleptinemia can increase the proliferation of ATMs. Hence, increased leptin levels in the obese AT are, in part, responsible for accumulation and activation of ATMs.
The name ‘resistin’ is coined from the original observation that it induces IR and has been proposed to link obesity and diabetes (32). Resistin is a 12.5-kDa peptide hormone, which is predominantly expressed in white adipocytes in rodents whereas in humans its main source is peripheral blood mononuclear cells (PBMCs) and macrophages (33). In mice, circulating resistin levels are positively correlated to obesity and IR, and resistin-treated mice or resistin-overexpressing transgenic mice exhibit glucose intolerance and IR (34). Conversely, resistin knockout or resistin neutralization with antibodies improves insulin sensitivity in diet-induced obese mice (32). However, in humans, the link between increased resistin levels and obesity/IR remains under debate and needs more epidemiological studies (34).
In obese conditions, there is an increase in adipocytes and ATMs leading to increased resistin expression. Resistin induces the expression of chemokines (e.g., CCL2 and CXCL1) as well as adhesion molecules (e.g., ICAM-1 and VCAM-1) to promote monocytes infiltration into a variety of tissues and organs including AT (35). M0 macrophages originated from monocytes can further differentiate into distinct ATM subsets, including LAMs (8), M1 and M2 macrophages (36), under varying circumstances. Resistin originated from adipose resident macrophages stimulates the expression of pro-inflammatory cytokines like IL-6, IL-12 and TNF-α (37), indicating that resistin might promote an M1-like phenotype in ATMs. Thus, resistin works by autocrine, paracrine and endocrine modes, and affects the accumulation and polarization of macrophages in AT.
Wingless‐type (Wnt)-inducible signaling pathway protein-1 (WISP1), a matricellular protein, is a novel adipokine associated with inflammation in obesity (38–40). WISP1 levels in plasma and subcutaneous AT are elevated in obese subjects and are positively correlated with systemic inflammation and IR (38). WISP1 plays a pro‐inflammatory role in AT inflammation. Stimulation of macrophages with WISP1 induces the secretion of pro-inflammatory cytokines (e.g., TNF-a and IL-6) and promotes M1 macrophage polarization (38). However, in adipocytes, WISP1 neither induces cytokine expression nor affects insulin signaling (38). This suggests that WISP1 contributes to AT inflammation and IR by regulating macrophages rather than adipocytes.
As the best-known and most abundant adipokine in circulation, adiponectin is exclusively secreted from adipocytes (41). Unlike pro-inflammatory adipokines, which have a positive trend in conditions of obesity, adiponectin levels are reduced in obesity and are up-regulated after weight loss (42, 43). Many evidences proved that adiponectin shows protective activity in obesity and IR (44, 45).
Adiponectin acts as anti-inflammatory factor and plays important roles in the accumulation and polarization of ATMs. Adiponectin has been reported to promote calreticulin receptor-dependent clearance of apoptotic adipocytes (46). Since apoptosis of adipocytes is a key initial event that contributes to macrophage infiltration into AT (47), the inhibition of adipocyte apoptosis using adiponectin can suppress the infiltration of ATMs. Furthermore, adiponectin induces anti-inflammatory M2 macrophage proliferation in AT by the activation of AKT signaling (48), while suppresses pro-inflammatory M1 macrophage proliferation via inhibiting NF-κB signaling (49). Additionally, adiponectin induces the M1 to M2 macrophage polarization switch in AT. Macrophages from adiponectin deficient mice display increased M1 markers and decreased M2 markers, while systemic administration of adenovirus expressing adiponectin results in an increased expression of M2 markers in AT (50). Overall, adiponectin regulates the infiltration, proliferation, and polarization of ATMs, which accounts for its anti-inflammatory properties.
Fetuin-A is a fatty acid-binding glycoprotein that is primarily expressed in the liver (51). Circulating Fetuin-A levels are higher in human subjects with obesity, type 2 diabetes mellitus (T2DM) and nonalcoholic fatty liver disease (NAFLD) (52). In obese AT, fatty acid-binding Fetuin-A acts as an endogenous ligand of TLR4 to promote inflammation and IR (53). By binding to the extracellular domain of TLR4, Fetuin-A activates NF-κB signaling to induce pro-inflammatory cytokines release and promote M1 macrophages polarization (53, 54). Moreover, Fetuin-A also serves as a chemoattractant, inducing the infiltration of macrophages into AT (55).
Growth differentiation factor 15 (GDF-15), a distant member of the TGF-β superfamily, is highly expressed in the liver (56) and AT (57). GDF-15 has been shown to be a stress responsive cytokine associated with obesity and diabetes (58). GDF-15 exerts its known anti-inflammatory properties against obesity by regulating at least in part the activation of ATMs. GDF-15 transgenic mice fed a HFD exhibit reduced NLRP3 inflammasome activity and lower levels of macrophage infiltration into AT (59).Recently, a study suggests that GDF-15 plays a role in the polarization of ATMs. GDF-15 expression in macrophages is induced by IL-4, which promotes M2 polarization of ATMs via the upregulation of oxidative metabolism (60).
Irisin, a 12 kDa peptide, is a novel myokine that is cleaved from fibronectin type III domain protein 5 (FNDC5) (61). In obese subjects, circulating irisin levels are reduced and are related with insulin sensitivity (62). Irisin has been reported to ameliorate adipose inflammation via up-regulating anti-inflammatory cytokine (e.g., adiponectin) and down-regulating pro-inflammatory cytokine (e.g., leptin and IL-6) (63). FNDC5 overexpression attenuates adipose tissue inflammation in HFD-induced obese mice by inhibiting macrophage recruitment and M1 phenotype polarization (64).Therefore, irisin’s anti-inflammatory effect in AT includes reducing production of pro-inflammatory cytokines, suppressing macrophage proliferation and infiltration, and inducing M2 macrophage polarization.
Myostatin (MSTN), also termed as growth differentiation factor 8, is primarily produced by skeletal muscle, and negatively regulates skeletal muscle mass. A recent study has reported that serum myostatin levels are positively correlated with adipose inflammation, obesity and IR (65). Inhibition of myostatin in mice suppresses HFD-induced infiltration of macrophages and reduces the expression of pro-inflammatory cytokines in AT (66). In addition, myostatin inhibition increases irisin production and induces M2 macrophage polarization in AT, thus suppressing inflammation (66).
GLP-1 is a hormone secreted by intestinal L-cells and is associated with obesity-related inflammation. GLP-1 alleviates macrophage infiltration in AT of ob/ob mice and reduces M1-polarized specific mRNA expression (67). Another study has shown that GLP-1/GLP-1R signaling in macrophages suppresses M1 polarization and triggers M2 polarization (68).In light of these findings, we speculate that GLP-1 alleviates obesity-related inflammation via inhibiting macrophage recruitment and promoting M2 macrophage polarization in AT.
Ghrelin is a gastrointestinal cytokine that increases appetite and promotes obesity (69). A number of studies have described that ghrelin has strong anti-inflammatory properties, however, the effects of ghrelin in macrophages are complex. In vitro model, ghrelin inhibits LPS-induced production of pro-inflammatory cytokines in macrophages (70). Ghrelin’s function is mediated by its receptors GHSR. In vivo work has demonstrated that GHSR knockout or administration of Des-acyl ghrelin (functions as ghrelin antagonist), in obese mice, reduces macrophage infiltration, promotes macrophage polarization to M2 in AT, thus suppressing adipose inflammation (71, 72). These data suggest that Ghrelin/GHSR axis can act as a pro-inflammatory mediator in AT.
Studies have demonstrated that several other organokines, including adipokines (pro-inflammatory: Chemerin, PGRN and LCN2; anti-inflammatory: Omentin, Spexin, and Sfrp5), Hepatokines (pro-inflammatory: RBP4 and DPP4; anti-inflammatory: FGF21), Myokines (pro-inflammatory: IL-6; anti-inflammatory: DEL-1) and insulin are important players in regulation of the recruitment or polarization of macrophages in AT, and thus promote inflammation associated with obesity and metabolic disease (73, 74) (Figure 1).
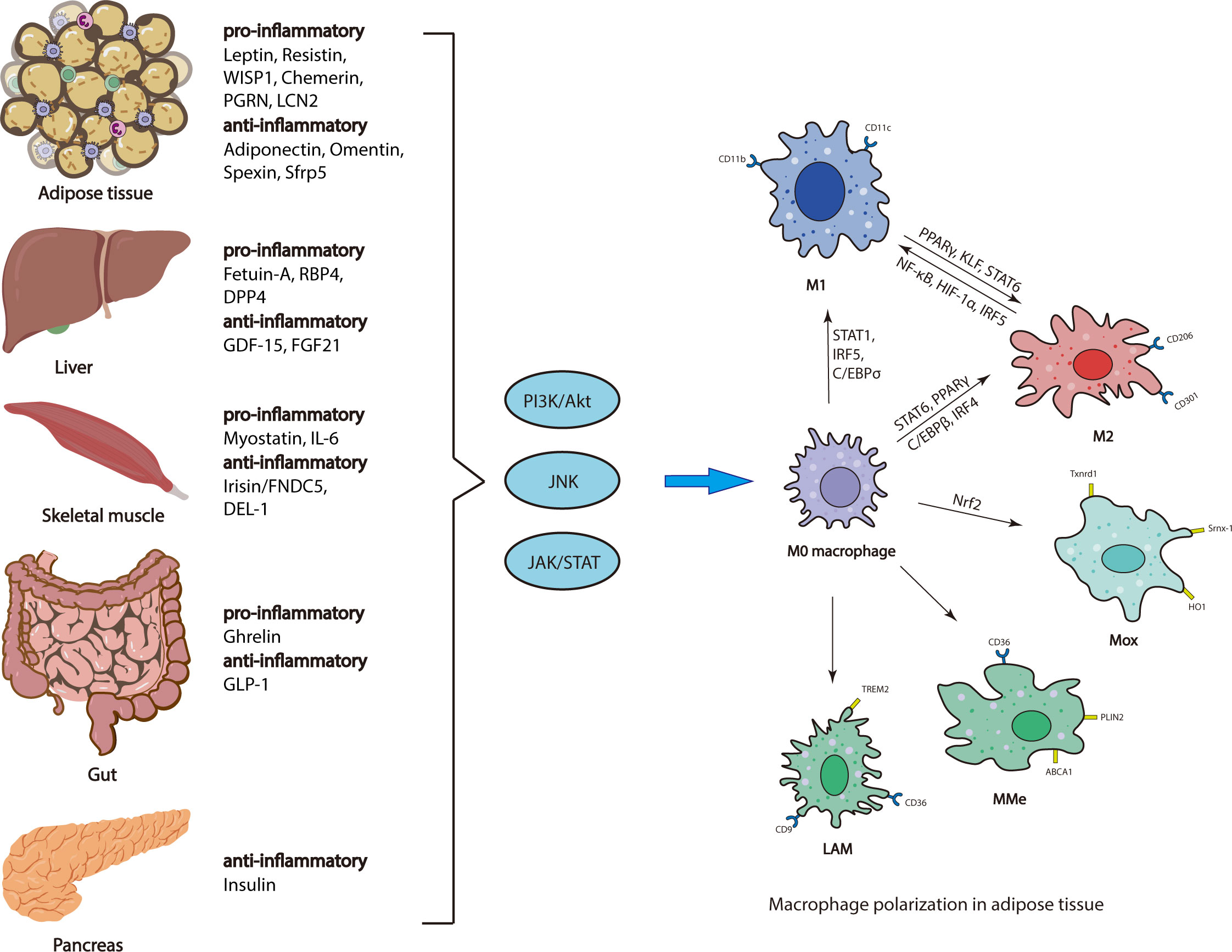
Figure 1 Regulatory mechanisms of ATM polarization. Adipose tissue, liver, skeletal muscle, gut and pancreas secrete various organokines, which are increased or decreased, initiating multiple downstream signaling pathways, thus affecting ATM polarization.
The past decades have revealed several key regulatory pathways of ATM polarization.
The PI3K singling cascade is a central metabolic regulator, which is activated by metabolic stimuli including insulin, glucose, FFAs and various organokines. ATMs are exposed to increased levels of these stimuli in the obese state and adopts metabolism-dependent phenotypes. Numerous studies have implicated that PI3K/AKT singling plays an inhibitory role in TLRs-mediated inflammation and contributes to M2 macrophage polarization (75). Obesity is associated with increased circulating LPS, which initiates adipose inflammation and macrophage activation by activating TLR4 (76), suggesting PI3K/AKT singling can regulate ATM polarization. Indeed, a recent study has demonstrated that macrophage-intrinsic PI3K signaling promotes a beneficial ATM population characterized by lipid uptake (77). It is worth noting that Akt isoforms differentially contributes to macrophage polarization, with AKT1 ablation induing M1 activation and AKT2 ablation resulting in M2 phenotype activation (78). However, how individual Akt isoforms are activated by PI3Ks in the context of macrophages polarization remains to be elucidated.
Mixed-lineage kinase 3 (MLK3) deficiency attenuates JNK activation, reduces ATM accumulation and M1 activation in HFD-fed mice (79). In macrophages, PPARγ is efficiently phosphorylated by JNK (80). PPARγ knockout in murine myeloid cells induces M1 activation of ATMs, obesity and IR (81). Acute exercise improves insulin signaling in the White AT, at least in part by inducing macrophage polarization to M2 via down-regulating phospho-JNK (82). These studies have demonstrated that JNK signaling plays a pivotal role in both ATM accumulation and polarization.
IFNγ is the best known inducer of JAK-STAT signaling (83). In recent studies, deletion of either TRIM29 or TRIM18 increases the release of IFNγ that enhances inflammatory cytokine production and induces M1 macrophage polarization through activating STAT1 (84–86). SOCS proteins act as feedback inhibitors of the JAK/STAT signaling. Myeloid SOCS3 knockout mice exhibit prolonged activation of JAK/STAT signaling and increased expression of pro-inflammatory cytokines in macrophages (87). In contrast to STAT1, STAT6 promotes M2 macrophage polarization. Activation of STAT6 by IL-4 attenuates adipose inflammation by inducing proliferation of local ATMs and polarization of M2 phenotype (88).
The infiltration and polarization of macrophages in adipose tissue are beginning to be recognized as pivotal instigators of metabolic dysfunction and obesity. In response to over-nutrition, endocrine organs change metabolic phenotypes, release distinct secretome profiles, and shift adipose homeostasis into one that promotes macrophage invasion and polarization, and supports downstream chronic inflammation and IR.
Several endocrine organs are involved in regulation of ATM polarization, including adipose tissue, gut, liver, skeletal muscle and pancreas. Various organokines derived from these endocrine organs bind to their respective receptors to initiate multiple downstream signaling pathways (e.g., PI3K/AKT, JNK, and JAK/STAT), which regulate ATM polarization via different transcriptional regulators (e.g., PPARγ, STAT, C/EBP and IRFs) (Figure 1).
It’s worth noting that the increase in the M1/M2 ratio of ATMs during obesity cannot be explained simply by the transformation from M2- to M1-phenotype macrophages but rather by the infiltration of circulating monocytes to AT followed by differentiating into M1 and M2 macrophages (36, 89). Mouse ATMs can be generated from circulating monocytes classified as Ly6C+ and Ly6C− that are generally thought to preferentially differentiate into M1 and M2 macrophages, respectively (17). Human CD14+ CD16− and CD14+ CD16+ monocytes are considered to resemble mouse Ly6C+ inflammatory monocytes, while CD14dim CD16+ monocytes resemble Ly6C− anti‐inflammatory monocytes (17, 90). In human obese AT, pro-inflammatory macrophages have been described as CD14+CD16+ cells with high levels of M1 markers (91). Polarization of inflammatory monocytes has been implicated in the pathogenesis of obesity-related diseases including T2DM and atherosclerosis (92, 93), thus underlying mechanisms and approaches for resolving monocyte polarization conducive to disease regression need to be established.
Despite the great achievements made over the past few decades, a lot of questions on regulatory mechanisms of ATM polarization and its physiological and pathological functions are yet to be answered. Another limitation in the field is that current ATM-markers are ubiquitously expressed in macrophages in different tissues but not specifically in ATMs. More specific markers need to be identified, which will greatly facilitate our understanding on ATMs in health and metabolic diseases. Our understanding of the complexity of ATM subpopulations is inadequate since macrophages are highly plastic and heterogeneous cell populations. New technologies, including single-cell analysis, computational biology and bioinformatics, are being incorporated in this field and are expected to hopefully help address those challenges. With increasing understanding of regulatory mechanisms of ATM polarization, novel insights and treatment strategies should emerge in the prevention of obesity-related diseases.
DP, GL and CJ drafted the original manuscript. DP and GL designed and created the figures and tables. JH and XH structured and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81902842 to XH, 81801974 to JH), the Natural Science Foundation of Fujian Province of China (2020J01615 to JH, 2021J01669 to XH), Joint Funds for the innovation of science and Technology, Fujian province (2020Y9006 to JH, 2019Y9014 to DP).
We apologize to all our colleagues whose studies we could not include into this review. We thank members in XH Lab for discussion.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127(1):1–4. doi: 10.1172/JCI92035
2. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
3. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/JCI200319451
4. Yao J, Wu D, Qiu Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front Immunol (2022) 13:977485. doi: 10.3389/fimmu.2022.977485
5. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
6. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab (2014) 20(4):614–25. doi: 10.1016/j.cmet.2014.08.010
7. Serbulea V, Upchurch CM, Schappe MS, Voigt P, DeWeese DE, Desai BN, et al. Macrophage phenotype and bioenergetics are controlled by oxidized phospholipids identified in lean and obese adipose tissue. Proc Natl Acad Sci USA (2018) 115(27):E6254–E6263. doi: 10.1073/pnas.1800544115
8. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. (2019) 178(3):686–98.e14. doi: 10.1016/j.cell.2019.05.054
9. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. (2010) 32(5):593–604. doi: 10.1016/j.immuni.2010.05.007
10. Nakajima S, Koh V, Kua LF, So J, Davide L, Lim KS, et al. Accumulation of CD11c+CD163+ adipose tissue macrophages through upregulation of intracellular 11beta-HSD1 in human obesity. J Immunol (2016) 197(9):3735–45. doi: 10.4049/jimmunol.1600895
11. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
12. Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: unique polarization and bioenergetics in obesity. Immunol Rev (2020) 295(1):101–13. doi: 10.1111/imr.12853
13. Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care (2011) 14(4):341–6. doi: 10.1097/MCO.0b013e328347970b
14. Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism: Clin experimental. (2017) 72:120–43. doi: 10.1016/j.metabol.2017.04.005
15. Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. (2010) 59(7):1648–56. doi: 10.2337/db09-0287
16. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab (2008) 8(4):301–9. doi: 10.1016/j.cmet.2008.08.015
17. Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology. (2018) 155(4):407–17. doi: 10.1111/imm.13002
18. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
19. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. (2008) 57(12):3239–46. doi: 10.2337/db08-0872
20. Lefevre L, Gales A, Olagnier D, Bernad J, Perez L, Burcelin R, et al. PPARgamma ligands switched high fat diet-induced macrophage M2b polarization toward M2a thereby improving intestinal candida elimination. PLoS One (2010) 5(9):e12828. doi: 10.1371/journal.pone.0012828
21. Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep (2017) 20(13):3149–61. doi: 10.1016/j.celrep.2017.08.096
22. Kim K, Park SE, Park JS, Choi JH. Characteristics of plaque lipid-associated macrophages and their possible roles in the pathogenesis of atherosclerosis. Curr Opin lipidology. (2022) 33(5):283–8. doi: 10.1097/MOL.0000000000000842
23. Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered (1950) 41(12):317–8. doi: 10.1093/oxfordjournals.jhered.a106073
24. Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Sci (New York NY). (1995) 269(5223):546–9. doi: 10.1126/science.7624778
25. Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Sci (New York NY). (1966) 153(3740):1127–8. doi: 10.1126/science.153.3740.1127
26. Friedman JM. Leptin and the endocrine control of energy balance. Nat Metab (2019) 1(8):754–64. doi: 10.1038/s42255-019-0095-y
27. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, et al. Leptin regulates proinflammatory immune responses. FASEB J Off Publ Fed Am Societies Exp Biol (1998) 12(1):57–65. doi: 10.1096/fsb2fasebj.12.1.57
28. Hoffmann A, Kralisch S, Duhring S, Ebert T, Jeromin F, Kloting N, et al. Effects of leptin on macrophages in vivo. Atherosclerosis. (2014) 235(2):E194–E. doi: 10.1016/j.atherosclerosis.2014.05.572
29. Acedo SC, Gambero S, Cunha FG, Lorand-Metze I, Gambero A. Participation of leptin in the determination of the macrophage phenotype: an additional role in adipocyte and macrophage crosstalk. In Vitro Cell Dev Biol Anim (2013) 49(6):473–8. doi: 10.1007/s11626-013-9629-x
30. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112(12):1785–8. doi: 10.1172/JCI20514
31. Monteiro L, Pereira J, Palhinha L, Moraes-Vieira PMM. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J leukocyte Biol (2019) 106(3):703–16. doi: 10.1002/JLB.MR1218-478R
32. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. (2001) 409(6818):307–12. doi: 10.1038/35053000
33. Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, et al. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun (2003) 300(2):472–6. doi: 10.1016/S0006-291X(02)02841-3
34. Taouis M, Benomar Y. Is resistin the master link between inflammation and inflammation-related chronic diseases? Mol Cell Endocrinol (2021) 533:111341. doi: 10.1016/j.mce.2021.111341
35. Acquarone E, Monacelli F, Borghi R, Nencioni A, Odetti P. Resistin: a reappraisal. Mech Ageing Dev (2019) 178:46–63. doi: 10.1016/j.mad.2019.01.004
36. Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. (2009) 58(11):2574–82. doi: 10.2337/db08-1475
37. Jiang CY, Wang W, Tang JX, Yuan ZR. The adipocytokine resistin stimulates the production of proinflammatory cytokines TNF-alpha and IL-6 in pancreatic acinar cells via NF-kappaB activation. J Endocrinol Invest. (2013) 36(11):986–92. doi: 10.3275/9002
38. Murahovschi V, Pivovarova O, Ilkavets I, Dmitrieva RM, Docke S, Keyhani-Nejad F, et al. WISP1 is a novel adipokine linked to inflammation in obesity. Diabetes. (2015) 64(3):856–66. doi: 10.2337/db14-0444
39. Tacke C, Aleksandrova K, Rehfeldt M, Murahovschi V, Markova M, Kemper M, et al. Assessment of circulating Wnt1 inducible signalling pathway protein 1 (WISP-1)/CCN4 as a novel biomarker of obesity. J Cell Commun Signal (2018) 12(3):539–48. doi: 10.1007/s12079-017-0427-1
40. Barchetta I, Cimini FA, Capoccia D, De Gioannis R, Porzia A, Mainiero F, et al. WISP1 is a marker of systemic and adipose tissue inflammation in dysmetabolic subjects with or without type 2 diabetes. J Endocr Soc (2017) 1(6):660–70. doi: 10.1210/js.2017-00108
41. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270(45):26746–9. doi: 10.1074/jbc.270.45.26746
42. Hoffstedt J, Arvidsson E, Sjolin E, Wahlen K, Arner P. Adipose tissue adiponectin production and adiponectin serum concentration in human obesity and insulin resistance. J Clin Endocrinol Metab (2004) 89(3):1391–6. doi: 10.1210/jc.2003-031458
43. Yang WS, Lee WJ, Funahashi T, Tanaka S, Matsuzawa Y, Chao CL, et al. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J Clin Endocrinol Metab (2001) 86(8):3815–9. doi: 10.1210/jcem.86.8.7741
44. Adiyaman SC, Ozer M, Saydam BO, Akinci B. The role of adiponectin in maintaining metabolic homeostasis. Curr Diabetes Rev (2020) 16(2):95–103. doi: 10.2174/1573399815666190702155733
45. Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest (2019) 129(10):4041–9. doi: 10.1172/JCI129193
46. Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest (2007) 117(2):375–86. doi: 10.1172/JCI29709
47. Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem (2010) 285(5):3428–38. doi: 10.1074/jbc.M109.074252
48. Hui X, Gu P, Zhang J, Nie T, Pan Y, Wu D, et al. Adiponectin enhances cold-induced browning of subcutaneous adipose tissue via promoting M2 macrophage proliferation. Cell Metab (2015) 22(2):279–90. doi: 10.1016/j.cmet.2015.06.004
49. Luo Y, Liu M. Adiponectin: a versatile player of innate immunity. J Mol Cell Biol (2016) 8(2):120–8. doi: 10.1093/jmcb/mjw012
50. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem (2010) 285(9):6153–60. doi: 10.1074/jbc.M109.088708
51. Denecke B, Gräber S, Schäfer C, Heiss A, Wöltje M, Jahnen-Dechent W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-b and fetuin-a. Biochem J (2003) 376(Pt 1):135–45. doi: 10.1042/bj20030676
52. Chung HS, Choi KM. Organokines in disease. Adv Clin Chem (2020) 94:261–321. doi: 10.1016/bs.acc.2019.07.012
53. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-a acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med (2012) 18(8):1279–85. doi: 10.1038/nm.2851
54. Chattopadhyay D, Das S, Guria S, Basu S, Mukherjee S. Fetuin-a regulates adipose tissue macrophage content and activation in insulin resistant mice through MCP-1 and iNOS: involvement of IFNgamma-JAK2-STAT1 pathway. Biochem J (2021) 478(22):4027–43. doi: 10.1042/BCJ20210442
55. Chatterjee P, Seal S, Mukherjee S, Kundu R, Mukherjee S, Ray S, et al. Adipocyte fetuin-a contributes to macrophage migration into adipose tissue and polarization of macrophages. J Biol Chem (2013) 288(39):28324–30. doi: 10.1074/jbc.C113.495473
56. Adolph TE, Grabherr F, Mayr L, Grander C, Enrich B, Moschen AR, et al. Weight loss induced by bariatric surgery restricts hepatic GDF15 expression. J Obes (2018) 2018:7108075. doi: 10.1155/2018/7108075
57. Imbert A, Vialaneix N, Marquis J, Vion J, Charpagne A, Metairon S, et al. Network analyses reveal negative link between changes in adipose tissue GDF15 and BMI during dietary-induced weight loss. J Clin Endocrinol Metab (2022) 107(1):e130–e42. doi: 10.1210/clinem/dgab621
58. Keipert S, Ost M. Stress-induced FGF21 and GDF15 in obesity and obesity resistance. Trends Endocrinol Metab (2021) 32(11):904–15. doi: 10.1016/j.tem.2021.08.008
59. Wang X, Chrysovergis K, Kosak J, Eling TE. Lower NLRP3 inflammasome activity in NAG-1 transgenic mice is linked to a resistance to obesity and increased insulin sensitivity. Obesity. (2014) 22(5):1256–63. doi: 10.1002/oby.20638
60. Jung SB, Choi MJ, Ryu D, Yi HS, Lee SE, Chang JY, et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun (2018) 9(1):1551. doi: 10.1038/s41467-018-03998-z
61. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. (2012) 481(7382):463–8. doi: 10.1038/nature10777
62. Moreno-Navarrete JM, Ortega F, Serrano M, Guerra E, Pardo G, Tinahones F, et al. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J Clin Endocrinol Metab (2013) 98(4):E769–78. doi: 10.1210/jc.2012-2749
63. Mazur-Bialy AI, Bilski J, Pochec E, Brzozowski T. New insight into the direct anti-inflammatory activity of a myokine irisin against proinflammatory activation of adipocytes. implication for exercise in obesity. J Physiol Pharmacol (2017) 68(2):243–51.
64. Xiong XQ, Geng Z, Zhou B, Zhang F, Han Y, Zhou YB, et al. FNDC5 attenuates adipose tissue inflammation and insulin resistance via AMPK-mediated macrophage polarization in obesity. Metabolism: Clin experimental. (2018) 83:31–41. doi: 10.1016/j.metabol.2018.01.013
65. Amor M, Itariu BK, Moreno-Viedma V, Keindl M, Jurets A, Prager G, et al. Serum myostatin is upregulated in obesity and correlates with insulin resistance in humans. Exp Clin Endocrinol Diabetes. (2019) 127(8):550–6. doi: 10.1055/a-0641-5546
66. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes (2005). (2016) 40(3):434–42. doi: 10.1038/ijo.2015.200
67. Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, et al. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia. (2012) 55(9):2456–68. doi: 10.1007/s00125-012-2592-3
68. Wan S, Sun H. Glucagon-like peptide-1 modulates RAW264.7 macrophage polarization by interfering with the JNK/STAT3 signaling pathway. Exp Ther Med (2019) 17(5):3573–9. doi: 10.3892/etm.2019.7347
69. Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. (2000) 407(6806):908–13. doi: 10.1038/35038090
70. Waseem T, Duxbury M, Ito H, Ashley SW, Robinson MK. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery. (2008) 143(3):334–42. doi: 10.1016/j.surg.2007.09.039
71. Yuan F, Ma J, Xiang X, Lan H, Xu Y, Zhao J, et al. Improvement of adipose macrophage polarization in high fat diet-induced obese GHSR knockout mice. BioMed Res Int (2018) 2018:4924325. doi: 10.1155/2018/4924325
72. Yuan F, Zhang Q, Dong H, Xiang X, Zhang W, Zhang Y, et al. Effects of des-acyl ghrelin on insulin sensitivity and macrophage polarization in adipose tissue. J Transl Int Med (2021) 9(2):84–97. doi: 10.2478/jtim-2021-0025
73. Mancuso P. The role of adipokines in chronic inflammation. Immunotargets Ther (2016) 5:47–56. doi: 10.2147/ITT.S73223
74. Ren Y, Zhao H, Yin C, Lan X, Wu L, Du X, et al. Adipokines, hepatokines and myokines: focus on their role and molecular mechanisms in adipose tissue inflammation. Front endocrinology. (2022) 13:873699. doi: 10.3389/fendo.2022.873699
75. Stark AK, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol (2015) 23:82–91. doi: 10.1016/j.coph.2015.05.017
76. Caesar R, Tremaroli V, Kovatcheva-Datchary P, Cani PD, Backhed F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab (2015) 22(4):658–68. doi: 10.1016/j.cmet.2015.07.026
77. Brunner JS, Vogel A, Lercher A, Caldera M, Korosec A, Puhringer M, et al. The PI3K pathway preserves metabolic health through MARCO-dependent lipid uptake by adipose tissue macrophages. Nat Metab (2020) 2(12):1427–42. doi: 10.1038/s42255-020-00311-5
78. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198(3):1006–14. doi: 10.4049/jimmunol.1601515
79. Gadang V, Kohli R, Myronovych A, Hui DY, Perez-Tilve D, Jaeschke A. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am J Physiol Endocrinol Metab (2013) 305(4):E549–56. doi: 10.1152/ajpendo.00197.2013
80. Yin R, Dong YG, Li HL. PPARgamma phosphorylation mediated by JNK MAPK: a potential role in macrophage-derived foam cell formation. Acta Pharmacol Sin (2006) 27(9):1146–52. doi: 10.1111/j.1745-7254.2006.00359.x
81. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447(7148):1116–20. doi: 10.1038/nature05894
82. Oliveira AG, Araujo TG, Carvalho BM, Guadagnini D, Rocha GZ, Bagarolli RA, et al. Acute exercise induces a phenotypic switch in adipose tissue macrophage polarization in diet-induced obese rats. Obesity. (2013) 21(12):2545–56. doi: 10.1002/oby.20402
83. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity. (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
84. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi: 10.1038/ni.3580
85. Fang M, Zhang A, Du Y, Lu W, Wang J, Minze LJ, et al. TRIM18 is a critical regulator of viral myocarditis and organ inflammation. J BioMed Sci (2022) 29(1):55. doi: 10.1186/s12929-022-00840-z
86. Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among jak-STAT, toll-like receptor, and ITAM-dependent pathways in macrophage activation. J leukocyte Biol (2007) 82(2):237–43. doi: 10.1189/jlb.1206763
87. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol (2012) 189(7):3439–48. doi: 10.4049/jimmunol.1201168
88. Zheng C, Yang Q, Xu C, Shou P, Cao J, Jiang M, et al. CD11b regulates obesity-induced insulin resistance via limiting alternative activation and proliferation of adipose tissue macrophages. Proc Natl Acad Sci USA (2015) 112(52):E7239–48. doi: 10.1073/pnas.1500396113
89. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116(6):1494–505. doi: 10.1172/JCI26498
90. Geng S, Zhang Y, Yi Z, Lu R, Li L. Resolving monocytes generated through TRAM deletion attenuate atherosclerosis. JCI Insight (2021) 6(20). doi: 10.1172/jci.insight.149651
91. Poledne R, Malinska H, Kubatova H, Fronek J, Thieme F, Kauerova S, et al. Polarization of macrophages in human adipose tissue is related to the fatty acid spectrum in membrane phospholipids. Nutrients (2019) 12(1). doi: 10.3390/nu12010008
92. Liang W, Qi Y, Yi H, Mao C, Meng Q, Wang H, et al. The roles of adipose tissue macrophages in human disease. Front Immunol (2022) 13:908749. doi: 10.3389/fimmu.2022.908749
Keywords: adipose tissue macrophages, polarization, organokines, signaling pathway, obesity
Citation: Pan D, Li G, Jiang C, Hu J and Hu X (2023) Regulatory mechanisms of macrophage polarization in adipose tissue. Front. Immunol. 14:1149366. doi: 10.3389/fimmu.2023.1149366
Received: 21 January 2023; Accepted: 04 May 2023;
Published: 22 May 2023.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Mariana Renovato-Martins, Federal Fluminense University, BrazilCopyright © 2023 Pan, Li, Jiang, Hu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangming Hu, eG1odTIwMDNAZmptdS5lZHUuY24=; Jinfeng Hu, aHVqaW5mZW5nQGZqbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.