 Brian J. Laight1,2,3
Brian J. Laight1,2,3 Natasha A. Jawa2,4
Natasha A. Jawa2,4 Kathrin Tyryshkin5
Kathrin Tyryshkin5 David M. Maslove6,7J. Gordon Boyd4,6,7Peter A. Greer1,3*
David M. Maslove6,7J. Gordon Boyd4,6,7Peter A. Greer1,3*- 1Department of Pathology and Molecular Medicine, Faculty of Health Sciences, Queen’s University, Kingston, Ontario, ON, Canada
- 2School of Medicine, Faculty of Health Sciences, Queen’s University, Kingston, Ontario, ON, Canada
- 3Queen’s Cancer Research Institute, Queen’s University, Kingston, Ontario, ON, Canada
- 4Centre for Neuroscience Studies, Faculty of Health Sciences, Queen’s University, Kingston, Ontario, ON, Canada
- 5School of Computing, Queen’s University, Kingston, Ontario, ON, Canada
- 6Division of Medicine and Critical Care Medicine, Department of Medicine, Faculty of Health Sciences, Queen’s University, Kingston, Ontario, ON, Canada
- 7Departments of Medicine and Critical Care Medicine, Kingston General Hospital, Kingston, Ontario, ON, Canada
Introduction: Sepsis is a result of initial over-activation of the immune system in response to an infection or trauma that results in reduced blood flow and life-threatening end-organ damage, followed by suppression of the immune system that prevents proper clearance of the infection or trauma. Because of this, therapies that not only limit the activation of the immune system early on, but also improve blood flow to crucial organs and reactivate the immune system in late-stage sepsis, may be effective treatments. The tyrosine kinase FES may fulfill this role. FES is present in immune cells and serves to limit immune system activation. We hypothesize that by enhancing FES in early sepsis and inhibiting its effects in late sepsis, the severity and outcome of septic illness can be improved.
Methods and analysis: In vitro and in vivo modeling will be performed to determine the degree of inflammatory signaling, cytokine production, and neutrophil extracellular trap (NET) formation that occurs in wild-type (WT) and FES knockout (FES-/-) mice. Clinically available treatments known to enhance or inhibit FES expression (lorlatinib and decitabine, respectively), will be used to explore the impact of early vs. late FES modulation on outcomes in WT mice. Bioinformatic analysis will be performed to examine FES expression levels in RNA transcriptomic data from sepsis patient cohorts, and correlate FES expression data with clinical outcomes (diagnosis of sepsis, illness severity, hospital length-of-stay).
Ethics and dissemination: Ethics approval pending from the Queen’s University Health Sciences & Affiliated Teaching Hospitals Research Ethics Board. Results will be disseminated through scientific publications and through lay summaries to patients and families.
Highlights
•This will be the first study to explore the utility of FES modulation in the treatment of sepsis.
•This study will illustrate the importance of differential regulation of sepsis at both early and late timepoints.
•This study will employ in vivo, in vitro, and clinical research methodologies to elucidate the role of FES in sepsis.
•There exists the potential for off-target effects of the drugs used in this study that may complicate the interpretation of results or outcomes, as they are not specific enhancers or inhibitors of FES.
Introduction
Sepsis is the leading cause of death among hospitalized patients in North America, and is present in over 50% of adult hospitalizations resulting in death or discharge to hospice care (1). Globally, sepsis accounts for 19.7% of all deaths (2), and is the most expensive healthcare problem requiring in-hospital treatment (3), costing the healthcare system approximately $1 billion per year in Ontario alone ($670 million for severe cases, $420 million for non-severe cases) (4). Importantly, the consequences of sepsis extend beyond the duration of the patient’s hospitalization, with lasting impairments being increasingly recognized as comprising “post-sepsis syndrome” (5); resulting in additional costs to the healthcare system for auxiliary care after discharge. Targeted treatments for sepsis are therefore urgently needed to simultaneously improve patient outcomes and alleviate the burden of sepsis on global healthcare systems.
Sepsis is characterized by widespread inflammation and life-threatening organ dysfunction, resulting from dysregulation of the patient’s response to infection and coagulation cascades (6, 7). Specifically, sepsis occurs upon disruption of the tightly regulated balance of pro- and anti-inflammatory mediators activated in response to pathogen- (e.g., virus, bacteria) or damage-associated molecular patterns (e.g., trauma; PAMPs and DAMPs, respectively) (8). Binding of PAMPs and DAMPs to toll-like receptors (TLRs) on the surface of antigen presenting cells (APCs) and monocytes causes nuclear translocation of nuclear factor kappa light chain enhancer of activated B cells (NF-κB), resulting in expression of genes encoding pro-inflammatory cytokines, tumor necrosis factor (TNF)-α, interferons, and components of complement and coagulation pathways (2). Clinically, activation of these pathways results in hypercoagulation, endothelial dysfunction, and ultimately to end organ damage (2).
While sepsis pathophysiology remains to be fully elucidated, we have previously shown that the FES kinase, which is highly expressed in APCs, plays a role in limiting over-stimulation of the innate immune system (9). FES knockout mice are hypersensitive to endotoxin lipopolysaccharide (LPS), a TLR-4 agonist, as a result of overactive TLR-4 signaling-induced innate immune responses (10) including increased transcription of genes encoding pro-inflammatory mediators, such as TNF-α (9). Our group and others have implicated FES as a regulator of TLR-4 signaling (9, 11). TLR-4 is also a critical inducer of both platelet-dependent and -independent NET formation, where neutrophils and platelets express both FES and TLR-4. Hyperactive TLR-4 signaling may therefore exacerbate the immune response and coagulation in severe sepsis (12, 13). Furthermore, FES knockout (FES-/-) mice demonstrate dysregulation of signal transducer and activator of transcription 3 (STAT3) (13), which plays a vital role in the pathophysiology of sepsis by initiating endothelial dysfunction, vasoplegia, coagulopathy, and multi-organ failure (14, 15). FES may also contribute to thrombin formation (16), which is known to contribute to hypercoagulation during the early inflammatory stage of sepsis (17).
There are currently no approved targeted molecular therapies available for the treatment of sepsis (6). The mainstay of treatment for sepsis involves management of infection, optimizing the patient’s fluid balance, improving hemodynamic stability, and sedative management; however, there is controversy over whether these interventions contribute to reduced mortality and morbidity in this cohort (3, 6, 18). Furthermore, early pre-clinical trials employing anti-TNF antibodies or corticosteroids to block proinflammatory cytokine cascades have since been shown to be ineffective in larger and later-stage clinical trials (3, 6). Identifying and targeting the key events and their regulators that precede these dysregulated immune responses characteristic of sepsis is therefore critical to optimize patients’ responses to systemic infection (6).
Due to its expression in innate immune cells and its involvement in regulating TLR-4 signaling (9, 11), we expect that increased FES expression will limit hyper-inflammatory phenotypes of immune cells (9, 11), reduce platelet aggregation (19), and we hypothesize it will also reduce NET formation (12, 20, 21). Consequently, targeting FES may allow for specific inhibition of two key aspects underlying the pathophysiology of sepsis: the coagulation cascade and the innate immune system. This study aims to elucidate the role of FES in sepsis pathophysiology using both an established mouse model of sepsis and a repository of data from patients with sepsis, as a first step toward being able to develop targeted immune-based therapies to enhance patient care and improve outcomes.
Overarching hypothesis
We hypothesize that FES expression is related to outcomes in in vivo and in vitro models of sepsis, as well as in clinical populations of patients with sepsis. We further hypothesize that modulating FES can be used as a method of improving sepsis outcomes. Figure 1 illustrates the hypothesized role of FES in the pathophysiology of sepsis. Figure 2 illustrates how FES regulation could be used therapeutically to treat sepsis.
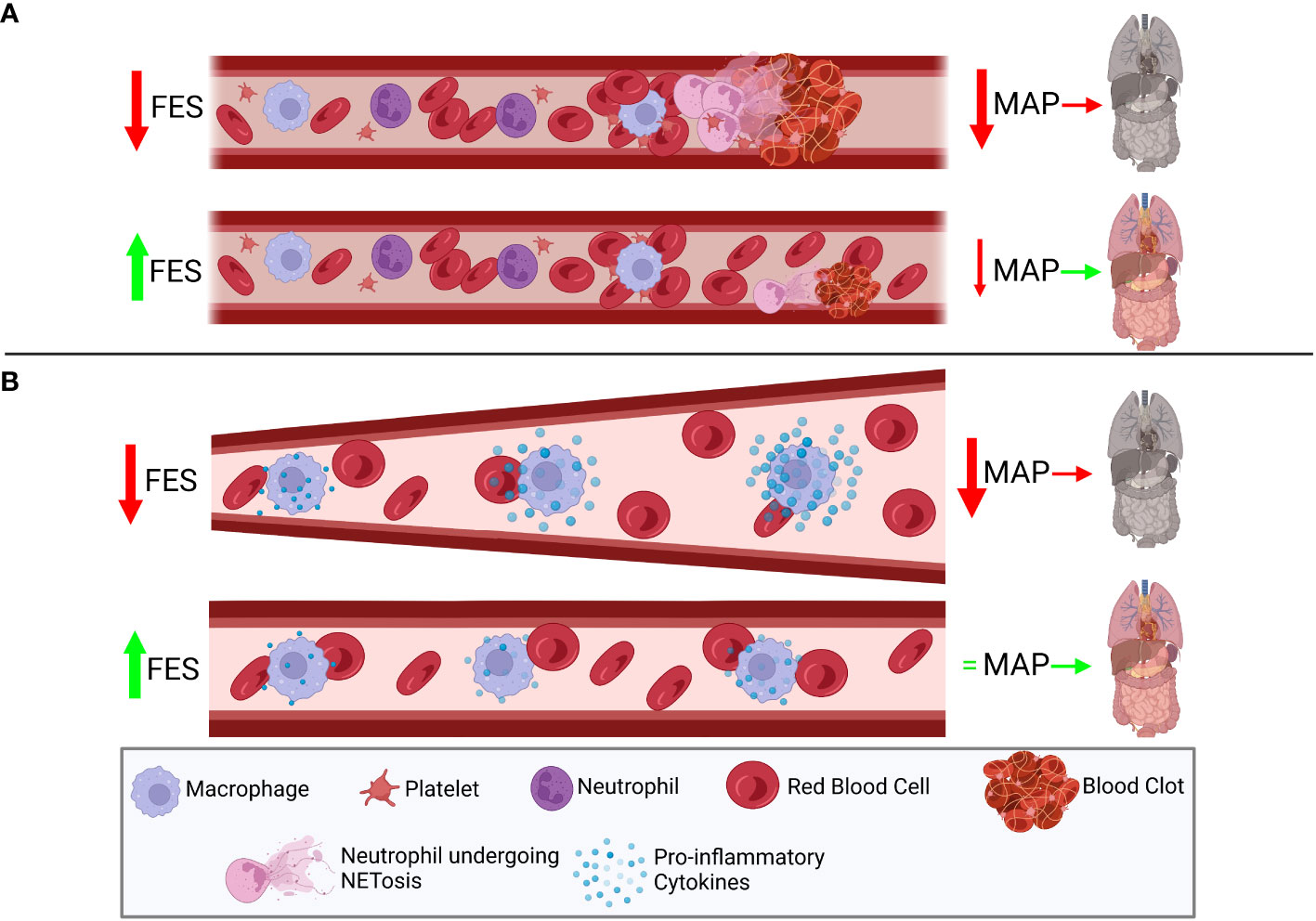
Figure 1 Decreased FES levels may correlate with worse septic outcomes by modulating host immune and coagulation pathways. (A) FES has been shown to play a role in limiting coagulation cascades through modulation of platelets and we hypothesize a role for FES in decreasing NETosis from neutrophils. Therefore, low FES expression is hypothesized to correlate with reduced lysis of blood clots, and an increase in NETosis in both a platelet-dependent and –independent manner, thus leading to reduced mean arterial pressure (MAP), leading to end organ damage. However, in patients with high FES expression, there is hypothesized to be reduced NETosis, and increased disaggregation of platelets, leading to a less dramatic impact on MAP, and improved perfusion of end organs. (B) FES has been shown to suppress the toll-like receptor (TLR) 4 signaling cascade, which leads to the production of pro-inflammatory cytokines regulated by NF-κB and IRF3. In patients with low FES expression, it is hypothesized that this signaling cascade will be poorly regulated, leading to the over-activation of the TLR4 cascade, stimulated by components of bacterial cell walls (lipopolysaccharide) or in response to tissue damage (high mobility group box 1), and ultimately leading to the over-production of inflammatory cytokines. This production of excess cytokines can lead to vasodilation, leading to reduced MAP, and reducing perfusion to end organs. However, with high FES expression, this cascade is tightly regulated, and the over-production of pro-inflammatory cytokines is limited, reducing changes in vasodilation, and preventing drastic changes in MAP, thus protecting end organs. Created with BioRender.com.
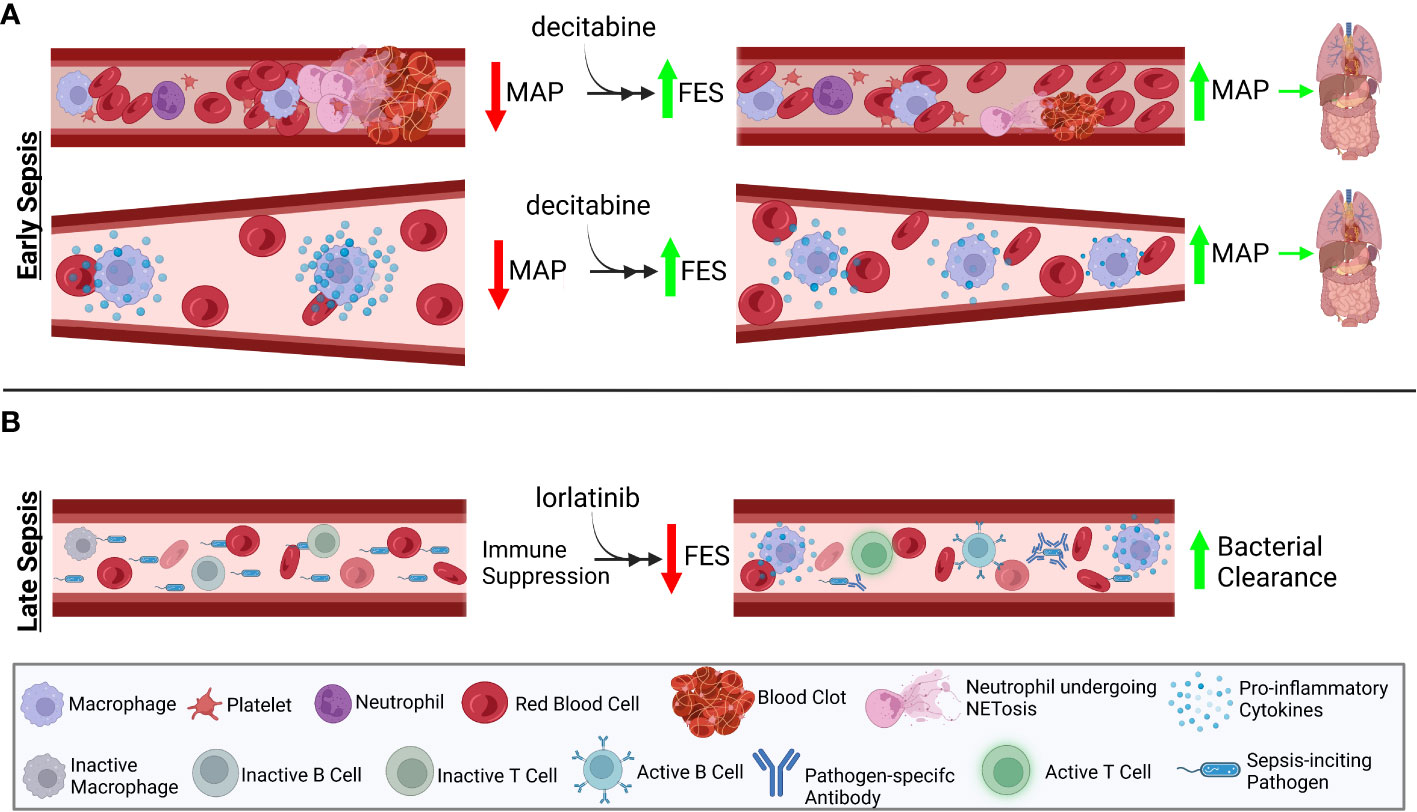
Figure 2 Therapeutic efficacy of FES expression level changes is stage-dependent. Sepsis can be separated into two stages: early stage, characterized by overactive immune responses leading to decreased end organ perfusion; and late stage, characterized by immune suppression, preventing the efficient clearance of the sepsis-inciting event. (A) Due to the over-stimulation of the immune system in early sepsis, treatment with decitabine, known to induce increased FES expression, will prevent overactivation of the immune system. (B) In contrast, overcoming the characteristic late-stage immunosuppression by treatment with lorlatinib, an inhibitor of FES activity, will promote greater pro-inflammatory cytokine production from immune cells. This increase in immune activation will aid in resolving the inciting event of sepsis. Created with BioRender.com.
Aims and specific hypotheses
Cell types responsible for sepsis severity and their effect on sepsis outcomes
This study aims to determine the cell types responsible for increased sepsis severity and their effects on outcomes in WT or FES-/- mice. We hypothesize that FES limits over-activation of innate immune cells and suppresses vascular hypercoagulation. We expect that FES-/- immune cells will take on a hyper-inflammatory phenotype when stimulated with infectious or damaging agents and will produce increased levels of pro-inflammatory cytokines (such as Interleukin (IL) -1, IL-6, Tumor Necrosis Factor (TNF)-α) and NETs relative to WT mice. In vivo, we expect to find an increase in both immune cell numbers and their degree of activation, as well as increased coagulation and end-organ damage in septic FES-/- mice compared to WT.
Timing of FES regulation on sepsis outcomes
This study will aim to evaluate whether FES regulation, and the timing of FES regulation, can affect sepsis outcomes. Early sepsis pathogenesis is characterized by hyperinflammation and coagulation, while late sepsis pathogenesis is characterized by immune suppression and organ dysfunction. It is hypothesized that the observed late-stage immunosuppression is as a commensal response to the initial overactive immune response. The early hyper-inflammatory stage of sepsis is characterized by increased immune cell activation and production proinflammatory cytokine secretion such as IL-1, IL-6, and TNF-α (22), a dangerous condition known as cytokine storm (23). This late stage is characterized by production of anti-inflammatory cytokines, such as IL-10 and Transforming Growth Factor-β (TGF-β), which serve to repress the overstimulated immune cells, reduced antigen presenting cells functions, lymphocyte anergy and apoptosis, and reduced responsiveness to activating stimuli, such as LPS (24). Simultaneous inflammation and immunosuppression is now being described in late-stage sepsis (22), suggesting that while there may be immunostimulatory molecules circulating, immune cells may be unable to appropriately respond to these stimuli. This provides additional rational for regulating FES, as it plays a role in limiting innate immune cell activation in response to stimulatory molecules such as LPS. We therefore hypothesize that artificially enhancing FES activation will improve early-stage sepsis, and that inhibiting FES will improve outcomes in late-stage sepsis. Figure 2 illustrates the timeline of sepsis and the expected impact of FES regulation at each stage of illness.
Association between FES expression and clinical sepsis presentation and outcomes
This study will seek to determine whether the RNA expression levels of FES are associated with the incidence, severity, and outcome of sepsis using bioinformatic analysis. We expect that FES will be upregulated in early-stage and downregulated in late-stage sepsis. Furthermore, we hypothesize that decreased FES RNA levels will be associated with receiving a clinical diagnosis of sepsis, increased illness severity, and poorer clinical outcomes including greater risk for ICU mortality and increased length of hospitalization.
Furthermore, we aim to determine whether clinical sepsis outcomes are correlated with single nucleotide polymorphisms (SNPs) associated with differential FES expression. Recently, two SNPs have been associated with reduced FES expression: rs17514846, located within in the 5’ proximal region of the neighbouring gene FURIN, and rs1894401, located within the FES gene. The presence or absence of these two SNPs will be correlated with clinical outcomes in a cohort of sepsis patients (25).
Methods and analysis
Patient and public involvement
Patient partners who have recovered from sepsis will be recruited from the ICU Follow-Up Clinic at Kingston Health Sciences Centre (Kingston, Ontario). Patient partners will help to guide decision-making around the clinical outcomes that are most important to study, such that our analysis can be focused on patient-centred outcomes.
Study design and setting
To expand our understanding of FES involvement in mediating sepsis, we will perform in vitro and in vivo modeling to elucidate the degree of inflammatory signaling, cytokine production, and NET formation that occurs in wild-type (WT) and FES knockout (FES-/-) mice. Using clinically available treatments that either enhance FES expression or inhibit FES activity, we will explore the utility of early vs. late FES modulation on outcomes in WT mice. Finally, we will explore the clinical relevance of FES in the pathogenesis of sepsis by using bioinformatic analyses to examine FES expression levels in RNA transcriptomic data from publicly available cohorts of patients with sepsis and healthy controls and correlating this with clinical outcomes. All analyses will take place in the Queen’s Cancer Research Institute facility at Queen’s University (for experimental laboratory-based aims), or the Centre for Advanced Computing (for the clinical bioinformatics aims).
In vitro methodology
Bone marrow derived macrophages (BMDMs) (26) or neutrophils (27) isolated from WT or FES-/- mice will be stimulated with infectious surrogates (LPS or PolyI:C), or a surrogate for tissue damage-mediated sepsis (High Mobility Group Box 1; HMGB1). The degree of inflammatory signaling (e.g., NF-κB and IRF-3 phosphorylation) will be assessed via immunoblot analysis. Supernatants will be analyzed by cytokine multiplex (28) methods to determine differential cytokine production in WT or FES-/- immune cells, and the type of cytokines (e.g., proinflammatory, such as IL-1, IL-6, IL-12, TNF-α, and Interferon-α/β (23); or anti-inflammatory, such as, IL-10 and TGF-β (29)) (Figure 3A). Finally, WT and FES-/- neutrophils will be evaluated for their ability to produce NETs using flow cytometry (30) (Figure 3B).
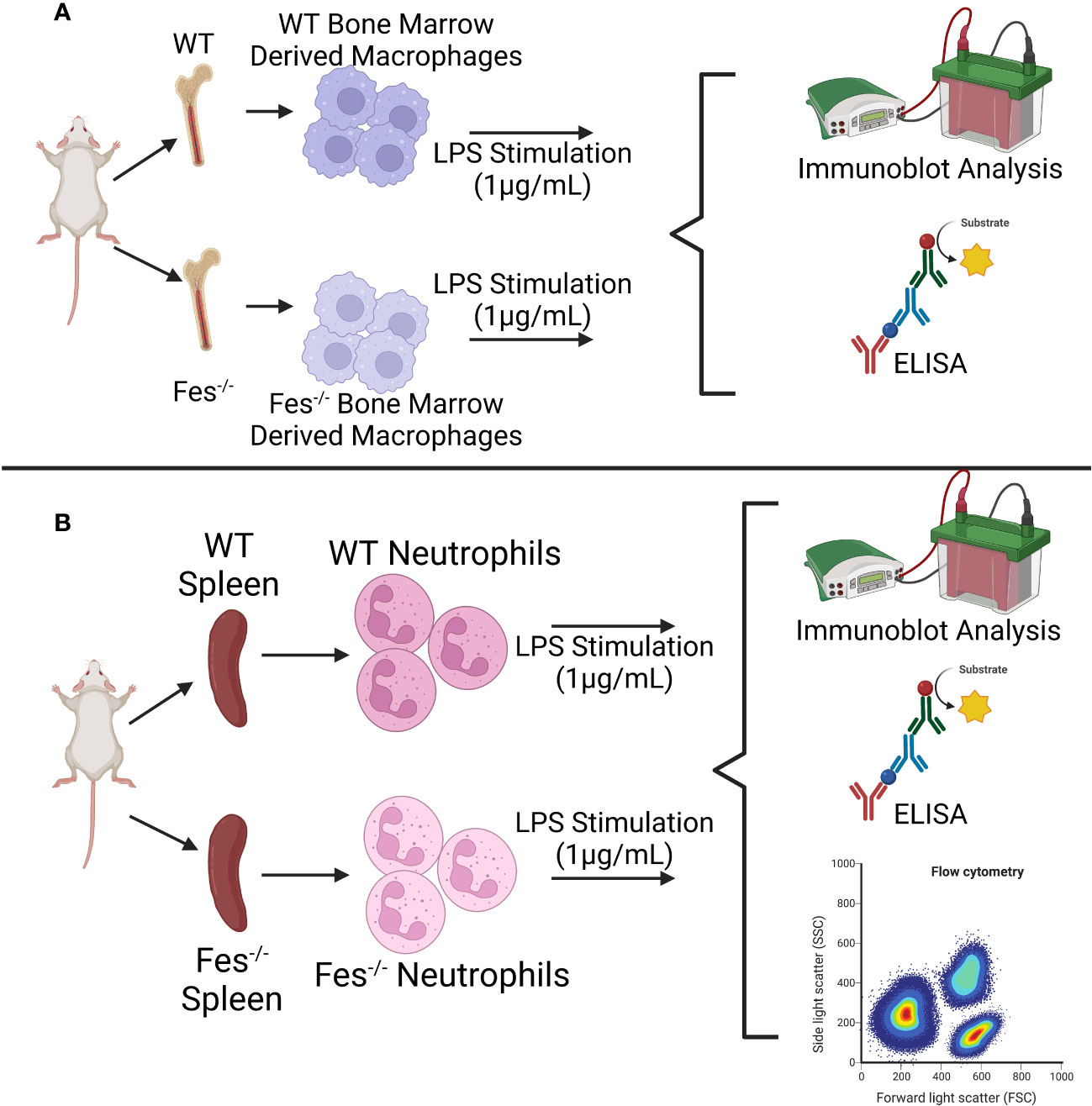
Figure 3 Graphical depiction of in vitro methodology. (A) Wildtype (WT) or FES-/- bone marrow derived macrophages will be stimulated with LPS or other stimulating agents and analyzed for differential signaling cascade activation (immunoblot analysis) and cytokine production (enzyme-linked immunosorbent assay (ELISA)). (B) WT or FES-/- neutrophils isolated from the spleen will be stimulated with LPS or other stimulating agents and analyzed for differential signaling cascade activation (immunoblot analysis), cytokine production (ELISA), and neutrophil extracellular trap formation (flow cytometry). The same methodology will be followed with the addition of decitabine and lorlatinib. Created with BioRender.com.
BMDMs and neutrophils isolated from WT or FES-/- mice will be pre-treated with either a drug that inhibits FES activity (lorlatinib (31)), or one that increases FES transcription (decitabine (32)). These cells will then be stimulated with LPS, PolyI:C, or HMGB1 to simulate the immune cell activation associated with sepsis, and the same methodology as in Aim 1 will be used to assess increased (in the case of lorlatinib treatment) or decreased (in the case of decitabine treatment) hyper-inflammatory signaling, cytokine production, and NET production.
In vivo methodology
WT or FES-/- mice will be treated in one of two ways, either simulating different insults resulting in sepsis: 1) cecal ligation and puncture (CLP), the gold-standard sepsis model (33) or 2) a cecal slurry (CS) of 0.5mL of a solution at a fecal concentration of 45mg/mL (33–35). Following induction of sepsis, mice will be closely monitored and scored on the murine sepsis score (MSS), which assesses sepsis severity based on appearance, level of consciousness, activity, response to stimulus, eyes, respiration rate, and respiration quality (35). The MSS has been shown to have a specificity of 57%, and a sensitivity of 100% for predicting onset of severe sepsis and death following CS. Blood will be collected by cardiac puncture at the 24h time point and analyzed by cytokine multiplex to assess differential cytokine production, which will demonstrate differences in cytokine production from either WT or FES-/- genotypes and can also be correlated with sepsis severity (35). Additionally, the following biomarkers will be assayed from the blood, as these have been associated with sepsis in murine models: C-reactive protein (CRP), soluble triggering receptor expressed on myeloid cells-1 (sTERM-1), CD163, procalcitonin (PCT) and hypoxia-inducible factor (HIF-1α) by ELISA, where CD163 and procalcitonin can stratify mice further into a state of severe sepsis (36) (Figure 4). Spleens will be isolated, dissociated and analyzed by flow cytometry for the presence and activation of immune cells (i.e., macrophages, neutrophils, T cells, B cells), as well as NET formation (30). Finally, differences in clotting ability and end-organ damage between WT and FES-/- mice will be assessed by histopathological analysis of hematoxylin & eosin-stained necropsy tissues. Briefly, livers, kidneys, small intestines, lungs, and brains will be assessed for necrosis, edema, accumulation of immune cells within tissues (e.g. macrophages, neutrophils, T-cells) and signs of damage to the tissues (e.g., loss of goblet cells and villi in small intestines, debris, and loss of organization and structure of epithelium) (35). As CS with a fecal concentration of 45mg/mL is associated with a 40% survival in C57BL/6 wildtype mice, mice will be assessed for neurocognitive impairments: a significant sequalae of post-sepsis syndrome (37, 38) A recent systematic review listed several common forms of cognitive impairment found in pre-clinical models of sepsis (e.g., aversive memory, learning, locomotor and exploratory activities, short-term and long-term memories), which may mimic what happens in patient populations (37). Therefore, any surviving mice will be subjected to the Y-maze novel arm preference test (a measure of testing spatial memory and attention), buried food test (a measure of attention and organized thinking), attentional set-shifting task (a measure of attention and cognitive flexibility), and finally the open field test (a measure of motor performance) (39) (Figure 4). Following neurocognitive assessments, mice will be euthanized and assessed for hyper-inflammation, coagulation, and histopathology as described above.
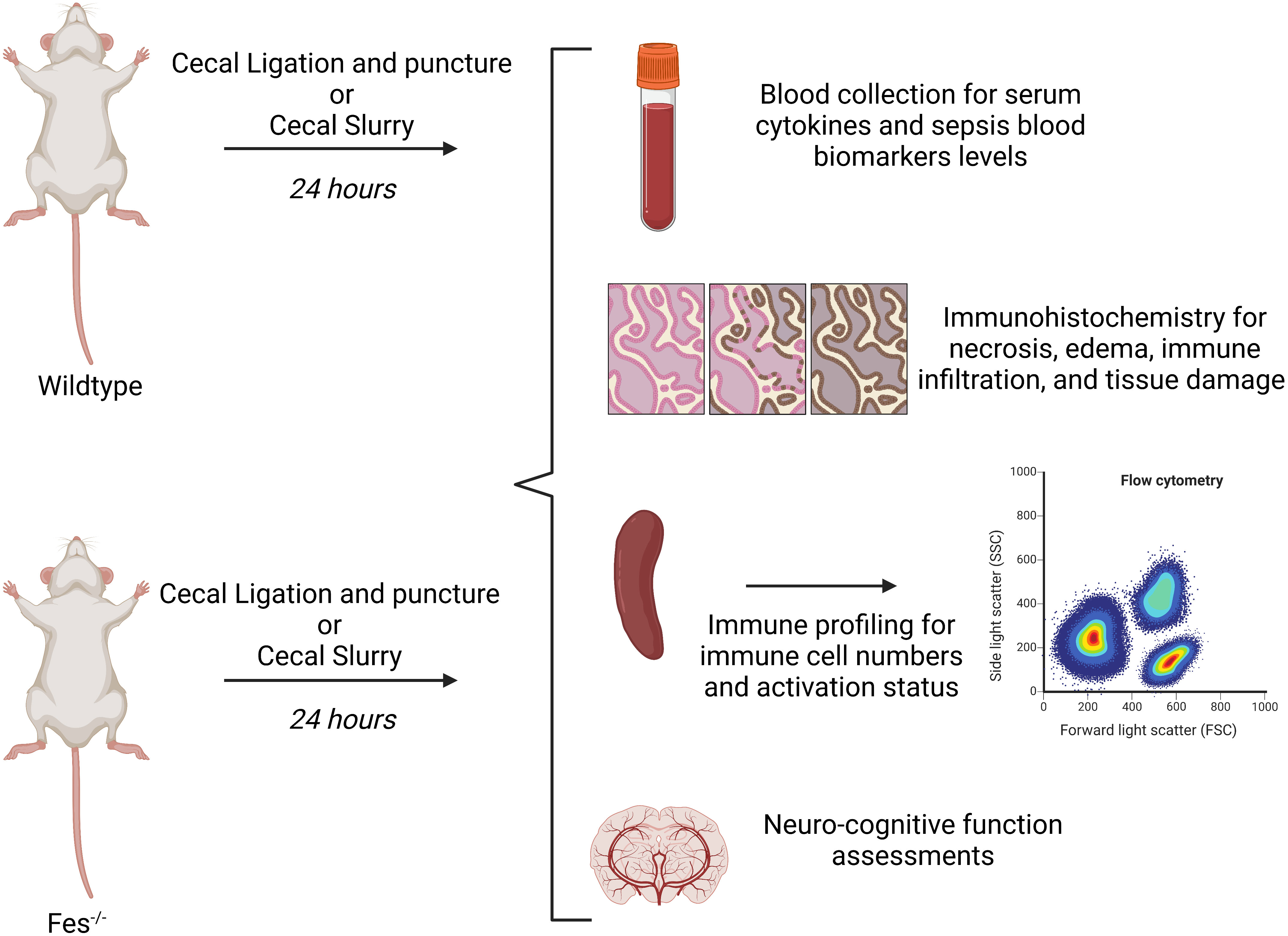
Figure 4 Graphical depiction of in vivo methodology. Wildtype (WT) or FES-/- mice will be undergo a cecal ligation and puncture (CLP) or receive a cecal slurry injected intraperitoneally. 24 hours later, mice will be assessed for the following: neurologic function assessment, serum cytokine and biomarker analysis, immunohistochemistry, and spleen immune profiling. The same methodology will be used with the addition of decitabine or lorlatinib. Created with BioRender.com.
To determine the effect of the timing of FES regulation on outcomes, WT or FES-/- mice will be subjected to either CLP or CS and treated with lorlatinib or decitabine at either 1h (mimicking regulation in early-stage sepsis) or 12h (mimicking regulation in late-stage sepsis). Hyper-inflammation, coagulation, histopathology, and neurocognition will be assessed at 24h, as previously mentioned. Finally, to determine differential susceptibility of WT mice to sepsis, we will perform an LD50 analysis of both early- and late-stage treatment regimens with either drug.
Clinical and bioinformatics methodology
We have previously extracted RNA from whole blood samples in critically ill patients at various time points during their admission, as well as from healthy controls, as part of the “Prevention of nosocomial infections in critically ill patients with lactoferrin (PREVAIL)” study (NCT01996579). Briefly, blood was collected into PAXgene tubes, frozen, RNA extracted, and gene expression was profiled using the Affymetrix PrimeView microarray (40). Data on 69 patients and 21 healthy controls from this analysis is publicly available (41) and will be analyzed for the current study. Machine-learning approaches will be used to identify key variables associated with FES RNA expression levels, including a) clinical diagnosis of sepsis; b) illness severity, as measured by Acute Physiology and Chronic Health Evaluation II (APACHE II) and Sequential Organ Failure Assessment (SOFA) scores; and c) clinical outcomes including hospital length of stay and ICU mortality.
A publicly available dataset comprised of clinical outcome data as well as SNP analysis from genome wide association studies (GWAS) will be sought through collaborations with local and international experts in the field of sepsis genetics. Multivariable analysis will be used to determine the association between the two SNPs (rs17514846 and rs1894401) related to differential FES expression, and clinical, patient-centred outcomes. Additionally, we will apply for access to the United Kingdom BioBank (https://www.ukbiobank.ac.uk/), a large-scale database with in-depth genetic and clinical outcomes for a wide variety of diseases (including sepsis) in over half a million patients. This database currently has SNP and transcriptomic data available to be paired with clinical outcomes and will soon be releasing proteomic data which can be analyzed further to pair the two SNPs (rs17514846 and rs1894401) related to differential FES expression with protein expression, and clinical outcomes.
Expected impact
These results will determine if FES modulates sepsis severity by preventing hyper-inflammation and suppressing coagulation cascades, providing a rationale for using FES regulation as a therapeutic strategy to reduce sepsis severity. The results of these experiments will further improve our understanding of the process by which NETs are produced, which has implications not only for sepsis but also for many other diseases.
In lorlatinib treated WT immune cells, we expect to see a phenocopy of the FES-/- immune cells, and prevention of inflammatory signaling with decitabine. Irrespective of the timing of treatment, we do not expect either lorlatinib or decitabine to affect sepsis severity in FES-/- mice (as there is no FES present to be regulated by either of these drugs). By contrast, in WT mice we expect to see increased septic severity following early lorlatinib treatment, and decreased septic severity following early decitabine treatment. We further expect to see decreased septic severity following late lorlatinib treatment (protective effect) and increased septic severity with late decitabine treatment (detrimental effect). These findings would support the notion that FES is a critical regulator of sepsis; and would further demonstrate that sepsis severity can be attenuated using currently available treatments by differentially modulating FES depending on the stage of a patient’s illness progression.
While it is our expectation that high FES expression levels in early sepsis and low FES expression levels in late sepsis will result in improved murine and patient outcomes, it is possible that this will not be the case. However, irrespective of the direction of the association, we will capitalize on the FES expression results found in order to improve in vitro, in vivo outcomes. For example, if we find that low FES expression is associated with improved outcomes in early sepsis (contrary to our hypothesis), we will expand upon this finding by seeking to reduce FES expression in our models of early sepsis. Similarly, if–contrary to our expectations–we find that high FES expression is associated with improved outcomes in late sepsis, we will aim to increase FES expression in our models of late sepsis and evaluate changes in outcomes seen as a result of this shift in expression. Additionally, if FES expression plays no role in sepsis severity, it will allow us to rule out a potential regulator heavily involved in many aspects of sepsis, which will help to narrow in on true regulators of sepsis severity and outcomes. We expect that FES expression levels on microarray analysis can be used alone or in combination with other genetic features as a biomarker to determine the presence, stage, severity, and outcome of sepsis. The heterogeneity of sepsis presentation and progression has long posed a problem for determining which patients will benefit from specific therapies, and the appropriate timing of these interventions. If we find that FES expression differs between time points in the progression of septic illness (i.e., between early- vs. late-stage sepsis), these results may then be used in future studies to help with targeting treatments toward specific subsets of patients who may differentially benefit from therapeutic interventions applied at different time points.
Finally, we expect that two SNPs associated with differential FES expression will be useful as prognostic biomarkers to stratify patients with sepsis based on risk for adverse outcomes that are relevant to patients, with the potential predictive value to inform FES-targeted treatments based on risk.
An important note about the involvement of FES in the pathophysiology is the association of SNP rs4957796, located within an intron of the FER gene (Fps/Fes related tyrosine kinase), with reduced mortality from sepsis (42), and reduced incidence of bloodstream infections, but increased mortality in those who do develop a bloodstream infection (43). FES and FER share similar biological functions such as platelet aggregation (19), mast cell activation (13, 44) and signaling of the immunosuppressive cytokine IL-10 (13), an important signalling pathway known to limit inflammation, whose function may have differing important roles during early- (important to limit the overactivation of the immune system) and late-stage (contribution to characteristic late-stage immunosuppression) sepsis. These observations may suggest a partial explanation to the improved outcomes in patients containing the SNP, rs4957796 (42) as well as the observations surrounding bloodstream infections (43). This hints that the impact of FER and potentially FES is double-edged sword where it is crucial to have the right amount of expression at the right time to have protective effects and not exacerbate the illness. If true, this would suggest a problem that the application of precision medicine might be able to overcome.
Sepsis is a disease that is constantly under investigation to find an effective treatment because of the major consequences on human life and healthcare burden. Unfortunately, there has not been much success towards this aim; over 100 randomized clinical trials which identify a target, and directly modulate that target, have failed (45), suggesting a need for novel approaches to treating sepsis. Not only do we believe we have identified a novel target capable of regulating multiple aspects of sepsis pathophysiology, but also the timing of intervention. This lends itself to identifying FES as a treatable trait (46), where, depending on the timing of intervention and the levels of FES expression or activity, may result in a different approach to treating a patient’s sepsis (upregulating FES expression or inhibiting FES activity).
Significance
Sepsis is one of the greatest healthcare problems worldwide, causing significant mortality and costing the Canadian healthcare system billions of dollars annually (4). Even among those who survive, sepsis is associated with long-term morbidities and reduced quality of life (4, 47). This project will provide crucial insight into the pathophysiology underlying sepsis, laying the foundation for future work to manage the dangerous side effects of immunotherapy-based treatments, and study the effect of systemic FES inhibition on this vulnerable cohort.
Author contributions
BL, DM, and PG designed the study. BL and NJ wrote the study protocol. BL, NJ, KT, DM, JB, and PG reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Physician’ Services Incorporated Foundation: Research Trainee Award- (GRANT # 2022-2744) providing funding in support of the first author stipend and additional funding to be used on reagents and publication costs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rhee C, Jones TM, Hamad Y, Pande A, Varon J, O’Brien C, et al. Prevalence, underlying causes, and preventability of sepsis-associated mortality in us acute care hospitals. JAMA Network Open (2019) 2(2):e187571. doi: 10.1001/jamanetworkopen.2018.7571
2. Jarczak D, Kluge S, Nierhaus A. Sepsis-pathophysiology and therapeutic concepts. Front Med (Lausanne) (2021) 8:628302. doi: 10.3389/fmed.2021.628302
3. Gyawali B, Ramakrishna K, Dhamoon AS. Sepsis: The evolution in definition, pathophysiology, and management. SAGE Open Med (2019) 7:1–13. doi: 10.1177/2050312119835043
4. Farrah K, McIntyre L, Doig CJ, Talarico R, Taljaard M, Krahn M, et al. Sepsis-associated mortality, resource use, and healthcare costs: A propensity-matched cohort study. Crit Care Med (2021) 49(2):215–27. doi: 10.1097/CCM.0000000000004777
5. Mostel Z, Perl A, Marck M, Mehdi SF, Lowell B, Bathija S, et al. Post-sepsis syndrome - an evolving entity that afflicts survivors of sepsis. Mol Med (2019) 26(1):6. doi: 10.1186/s10020-019-0132-z
6. Gotts JE, Matthay MA. Sepsis: Pathophysiology and clinical management. BMJ (2016) 353:i1585. doi: 10.1136/bmj.i1585
7. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
8. Nedeva C, Menassa J, Puthalakath H. Sepsis: Inflammation is a necessary evil. Front Cell Dev Biol (2019) 7:108. doi: 10.3389/fcell.2019.00108
9. Parsons SA, Greer PA. The Fps/Fes kinase regulates the inflammatory response to endotoxin through down-regulation of Tlr4, nf-κb activation, and tnf-α secretion in macrophages. J Leukocyte Biol (2006) 80(6):1522–8. doi: 10.1189/jlb.0506350
10. Zirngibl RA, Senis Y, Greer PA. Enhanced endotoxin sensitivity in Fps/Fes-null mice with minimal defects in hematopoietic homeostasis. Mol Cell Biol (2002) 22(8):2472–86. doi: 10.1128/mcb.22.8.2472-2486.2002
11. Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, et al. Constitutive mhc class I molecules negatively regulate tlr-triggered inflammatory responses Via the fps-Shp-2 pathway. Nat Immunol (2012) 13(6):551–9. doi: 10.1038/ni.2283
12. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet Tlr4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
13. Greer P. Closing in on the biological functions of Fps/Fes and fer. Nat Rev Mol Cell Biol (2002) 3(4):278–89. doi: 10.1038/nrm783
14. Lei W, Liu D, Sun M, Lu C, Yang W, Wang C, et al. Targeting Stat3: A crucial modulator of sepsis. J Cell Physiol (2021) 236(11):7814–31. doi: 10.1002/jcp.30394
15. Clere-Jehl R, Mariotte A, Meziani F, Bahram S, Georgel P, Helms J. Jak-stat targeting offers novel therapeutic opportunities in sepsis. Trends Mol Med (2020) 26(11):987–1002. doi: 10.1016/j.molmed.2020.06.007
16. Sangrar W, Senis Y, Samis JA, Gao Y, Richardson M, Lee DH, et al. Hemostatic and hematological abnormalities in gain-of-Function Fps/Fes transgenic mice are associated with the angiogenic phenotype. J Thromb Haemost (2004) 2(11):2009–19. doi: 10.1111/j.1538-7836.2004.00956.x
17. Simmons J, Pittet JF. The coagulopathy of acute sepsis. Curr Opin Anaesthesiol (2015) 28(2):227–36. doi: 10.1097/ACO.0000000000000163
18. Gattinoni L, Brazzi L, Pelosi P, Latini R, Tognoni G, Pesenti A, et al. A trial of goal-oriented hemodynamic therapy in critically ill patients. Svo2 collaborative group. N Engl J Med (1995) 333(16):1025–32. doi: 10.1056/NEJM199510193331601
19. Senis YA, Sangrar W, Zirngibl RA, Craig AW, Lee DH, Greer PA. Fps/Fes and fer non-receptor protein-tyrosine kinases regulate collagen- and adp-induced platelet aggregation. J Thromb Haemost (2003) 1(5):1062–70. doi: 10.1046/j.1538-7836.2003.t01-1-00124.x
20. McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood (2017) 129(10):1357–67. doi: 10.1182/blood-2016-09-741298
21. Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat Commun (2021) 12(1):2776. doi: 10.1038/s41467-021-23086-z
22. Koutroulis I, Batabyal R, McNamara B, Ledda M, Hoptay C, Freishtat RJ. Sepsis immunometabolism: From defining sepsis to understanding how energy production affects immune response. Crit Care Explor (2019) 1(11):e0061. doi: 10.1097/cce.0000000000000061
23. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med (2020) 383(23):2255–73. doi: 10.1056/nejmra2026131
24. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306(23):2594. doi: 10.1001/jama.2011.1829
25. Karamanavi E, McVey DG, Laan SW, Stanczyk PJ, Morris GE, Wang Y, et al. The fes gene at the 15q26 coronary-Artery-Disease locus inhibits atherosclerosis. Circ Res (2022) 131(12):1004–17. doi: 10.1161/CIRCRESAHA.122.321146
26. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol (2008) 14:14.1.1–14.1.14. doi: 10.1002/0471142735.im1401s83
27. Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp (2013) 77):e50586. doi: 10.3791/50586
28. Leng SX, McElhaney JE, Walston JD, Xie D, Fedarko NS, Kuchel GA. Elisa And multiplex technologies for cytokine measurement in inflammation and aging research. Biol Sci Med Sci (2008) 63(8):879–84. doi: 10.1093/gerona/63.8.879
29. Bergmann CB, Beckmann N, Salyer CE, Hanschen M, Crisologo PA, Caldwell CC. Potential targets to mitigate trauma- or sepsis-induced immune suppression. Front Immunol (2021) 12:622601. doi: 10.3389/fimmu.2021.622601
30. Masuda S, Shimizu S, Matsuo J, Nishibata Y, Kusunoki Y, Hattanda F, et al. Measurement of net formation in vitro and in vivo by flow cytometry. Cytometry A (2017) 91(8):822–9. doi: 10.1002/cyto.a.23169
31. Nielsen SR, Strobech JE, Horton ER, Jackstadt R, Laitala A, Bravo MC, et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat Commun (2021) 12(1):3414. doi: 10.1038/s41467-021-23731-7
32. Shaffer JM, Smithgall TE. Promoter methylation blocks fes protein-tyrosine kinase gene expression in colorectal cancer. Genes Chromosomes Cancer (2009) 48(3):272–84. doi: 10.1002/gcc.20638
33. Stortz JA, Raymond SL, Mira JC, Moldawer LL, Mohr AM, Efron PA. Murine models of sepsis and trauma: Can we bridge the gap? ILAR J (2017) 58(1):90–105. doi: 10.1093/ilar/ilx007
34. Starr ME, Steele AM, Saito M, Hacker BJ, Evers BM, Saito H. A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PloS One (2014) 9(12):e115705. doi: 10.1371/journal.pone.0115705
35. Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, et al. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes (2014) 7(1):233. doi: 10.1186/1756-0500-7-233
36. Li J-L, Li G, Jing X-Z, Li Y-F, Ye Q-Y, Jia H-H, et al. Assessment of clinical sepsis-associated biomarkers in a septic mouse model. J Int Med Res (2018) 46(6):2410–22. doi: 10.1177/0300060518764717
37. Barichello T, Sayana P, Giridharan VV, Arumanayagam AS, Narendran B, Della Giustina A, et al. Long-term cognitive outcomes after sepsis: A translational systematic review. Mol Neurobiol (2019) 56(1):186–251. doi: 10.1007/s12035-018-1048-2
38. Mostel Z, Perl A, Marck M, Mehdi SF, Lowell B, Bathija S, et al. Post-sepsis syndrome – an evolving entity that afflicts survivors of sepsis. Mol Med (2020) 26(1):1–14. doi: 10.1186/s10020-019-0132-z
39. Illendula M, Osuru HP, Ferrarese B, Atluri N, Dulko E, Zuo Z, et al. Surgery, anesthesia and intensive care environment induce delirium-like behaviors and impairment of synaptic function-related gene expression in aged mice. Front Aging Neurosci (2020) 12:542421. doi: 10.3389/fnagi.2020.542421
40. Muscedere J, Maslove D, Boyd JG, O’Callaghan N, Lamontagne F, Reynolds S, et al. Prevention of nosocomial infections in critically ill patients with lactoferrin (Prevail study): Study protocol for a randomized controlled trial. Trials (2016) 17(1):474. doi: 10.1186/s13063-016-1590-z
41. [Dataset]. Available at: https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE118657.
42. Rautanen A, Mills TC, Gordon AC, Hutton P, Steffens M, Nuamah R, et al. Genome-wide association study of survival from sepsis due to pneumonia: An observational cohort study. Lancet Respir Med (2015) 3(1):53–60. doi: 10.1016/s2213-2600(14)70290-5
43. Rogne T, Damås JK, Flatby HM, Åsvold BO, DeWan AT, Solligård E. The role of fer Rs4957796 in the risk of developing and dying from a bloodstream infection: A 23-year follow-up of the population-based nord-trøndelag health study. Clin Infect Dis (2021) 73(2):e297–303. doi: 10.1093/cid/ciaa786
44. Udell CM, Samayawardhena LA, Kawakami Y, Kawakami T, Craig AWB. Fer and Fps/Fes participate in a Lyn-dependent pathway from fcϵri to platelet-endothelial cell adhesion molecule 1 to limit mast cell activation. J Biol Chem (2006) 281(30):20949–57. doi: 10.1074/jbc.m604252200
45. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med (2014) 20(4):195–203. doi: 10.1016/j.molmed.2014.01.007
46. Maslove DM, Tang B, Shankar-Hari M, Lawler PR, Angus DC, Baillie JK, et al. Redefining critical illness. Nat Med (2022) 28(6):1141–8. doi: 10.1038/s41591-022-01843-x
Keywords: sepsis, innate immunity, dysregulated inflammation, coagulation, personalized medicine
Citation: Laight BJ, Jawa NA, Tyryshkin K, Maslove DM, Boyd JG and Greer PA (2023) Establishing the role of the FES tyrosine kinase in the pathogenesis, pathophysiology, and severity of sepsis and its outcomes. Front. Immunol. 14:1145826. doi: 10.3389/fimmu.2023.1145826
Received: 16 January 2023; Accepted: 03 April 2023;
Published: 14 April 2023.
Edited by:
Yufeng Zhou, Fudan University, ChinaReviewed by:
Marisa Mariel Fernandez, Institute of Studies on Humoral Immunity (IDEHU), ArgentinaAntonio Ferrante, South Australia Pathology, Australia
Copyright © 2023 Laight, Jawa, Tyryshkin, Maslove, Boyd and Greer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter A. Greer, Z3JlZXJwQHF1ZWVuc3UuY2E=