 Ting Sun
Ting Sun Dengju Li
Dengju Li Liang Huang
Liang Huang Xiaojian Zhu
Xiaojian Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 May 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1141779
This article is part of the Research Topic Tumor Microenvironment and Hematological Malignancies: New Evidences and New Questions View all 7 articles
Chimeric antigen receptor T-cell (CAR-T) therapy has shown remarkable effects in treating various hematological malignancies. However, hematotoxicity, specifically neutropenia, thrombocytopenia, and anemia, poses a serious threat to patient prognosis and remains a less focused adverse effect of CAR-T therapy. The mechanism underlying lasting or recurring late-phase hematotoxicity, long after the influence of lymphodepletion therapy and cytokine release syndrome (CRS), remains elusive. In this review, we summarize the current clinical studies on CAR-T late hematotoxicity to clarify its definition, incidence, characteristics, risk factors, and interventions. Owing to the effectiveness of transfusing hematopoietic stem cells (HSCs) in rescuing severe CAR-T late hematotoxicity and the unignorable role of inflammation in CAR-T therapy, this review also discusses possible mechanisms of the harmful influence of inflammation on HSCs, including inflammatory abrasion of the number and the function of HSCs. We also discuss chronic and acute inflammation. Cytokines, cellular immunity, and niche factors likely to be disturbed in CAR-T therapy are highlighted factors with possible contributions to post-CAR-T hematotoxicity.
Chimeric antigen receptor T-cell (CAR-T) therapy is a type of adoptive T-cell immunotherapy (1), in which T cells are genetically modified to express a CAR consisting of a specific antigen recognition domain from a B-cell receptor and essential signaling elements for T cells (1). The antigen recognition domain is encoded by single-chain variable fragments (scFv) and can be substituted to target various kinds of cells. The signaling elements are composed of a co-stimulation domain (two co-stimulation domains for the third generation of CAR) and a signal transduction domain (1, 2). The fourth generation of CAR also contains a cytokine secretion domain (3). Such genetic modification capacitates CAR-T cells with vigorous non-major-histocompatibility-complex (MHC)-restricted cytotoxicity (1). Cytokines released during the eradication of target cells also activate neighboring immunocytes, which can exert a synergistic therapeutic effect (1). Subsequently, part of the CAR-T cells may enter the memory pool, circulating in the body for a long time and supervising the primary disease (1).
CAR-T therapy has achieved encouraging success in relapsed/refractory (R/R) hematological malignancies (4). In patients with R/R non-Hodgkin lymphoma (NHL), the long-term follow-up of Axicabtagene ciloleucel, an anti-CD19 CAR-T therapy, reported a median overall survival (OS) of 25.8 months [95% confidence interval (CI), 12.8–NE] (4), while the median OS was only 6.3 months (95% CI, 5.9–7.0) in R/R NHL patients (n = 636) treated with chemotherapies or autologous stem cell transplantation (5). The application of CAR-T has also extended to solid carcinomas, presenting a challenging and promising prospect (6). However, specific adverse events emerge simultaneously, such as cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), B-cell aplasia, hypogammaglobulinemia, and hematotoxicity (i.e., cytopenia) (7, 8). The most highlighted hematotoxic events are neutropenia, thrombocytopenia, and anemia (9, 10). Furthermore, leukopenia and lymphocytopenia are also the manifestations of cytopenia.
CAR-T late hematotoxicity refers to cytopenia of single or multiple lineages in recurrence, persistent myelosuppression (11–17), or occasionally reported delayed onset (12, 14). However, there is still a lack of consensus on the exact time to determine a late event in cytopenia. Cytopenia that occurs immediately after the infusion may recover gradually during residency. However, delayed or recurrent cytopenia may occur (12, 15) during the outpatient follow-up and might miss timely intervention. Moreover, persistent aplasia could prolong the time of hospitalization, adding to the medical expenses and the difficulty of clinical management (18). Severe cytopenia casts a shadow on the survival of the patients, raising the risk of severe infection, lethal bleeding, and extreme fatigue (10). Therefore, CAR-T hematotoxicity, specifically late events, should be studied thoroughly.
Hematopoietic stem cells (HSCs) lie at the top hierarchy of the hematopoietic output (19). Several clinical studies have reported the success of transfusing HSCs, also called hematopoietic stem cell boosting (HSCB), in rescuing severe and prolonged hematotoxicity (20–22). The importance of HSCs in the pathology of CAR-T hematotoxicity has been highlighted, although the role of differentiated and mature hematocytes could not be excluded. Moreover, patients are persistently challenged by inflammation owing to the previous multiple lines of chemotherapies or radiotherapies, CRS following CAR-T therapy, infections, and other pro-inflammatory events. Therefore, inflammation is likely a vital factor influencing hematopoietic recovery after CAR-T infusion.
In this review, we aim to discuss the possible mechanisms underlying CAR-T late hematotoxicity considering the negative impact of inflammation on the hematopoietic system, especially on HSCs. We aim to summarize current studies on the features of CAR-T late hematotoxicity, potential influencing factors, clinical management, and pathogenesis. Moreover, the crucial role of HSCs and inflammation as well as the negative influence of inflammation on HSCs will also be reviewed to provide information on pathogenic mechanisms.
Diagnosis and evaluation of CAR-T hematotoxicity are based on the Common Terminology Criteria for Adverse Events (CTCAE) (11–17). Despite this consensus, the definition of CAR-T late hematotoxicity, summarized in Table 1, varies in studies. One divergence was the severity of cytopenia. Most studies concentrated on grade 3–4 cytopenia, while less attention was given to grade 1–2 cytopenia, because more severe cytopenia might correlate with a higher incidence of infection (18, 23), lethal bleeding, and a worse prognosis (10, 18), thus requiring more intensive investigation. However, milder cytopenia should not be neglected because the main goal is normalizing hematopoietic recovery. Another divergence is the time point to define a late event, which went from 21 to 90 days post-infusion (11–17) (Table 1). Based on these existing reports, a standard time point for late events remains unclear.
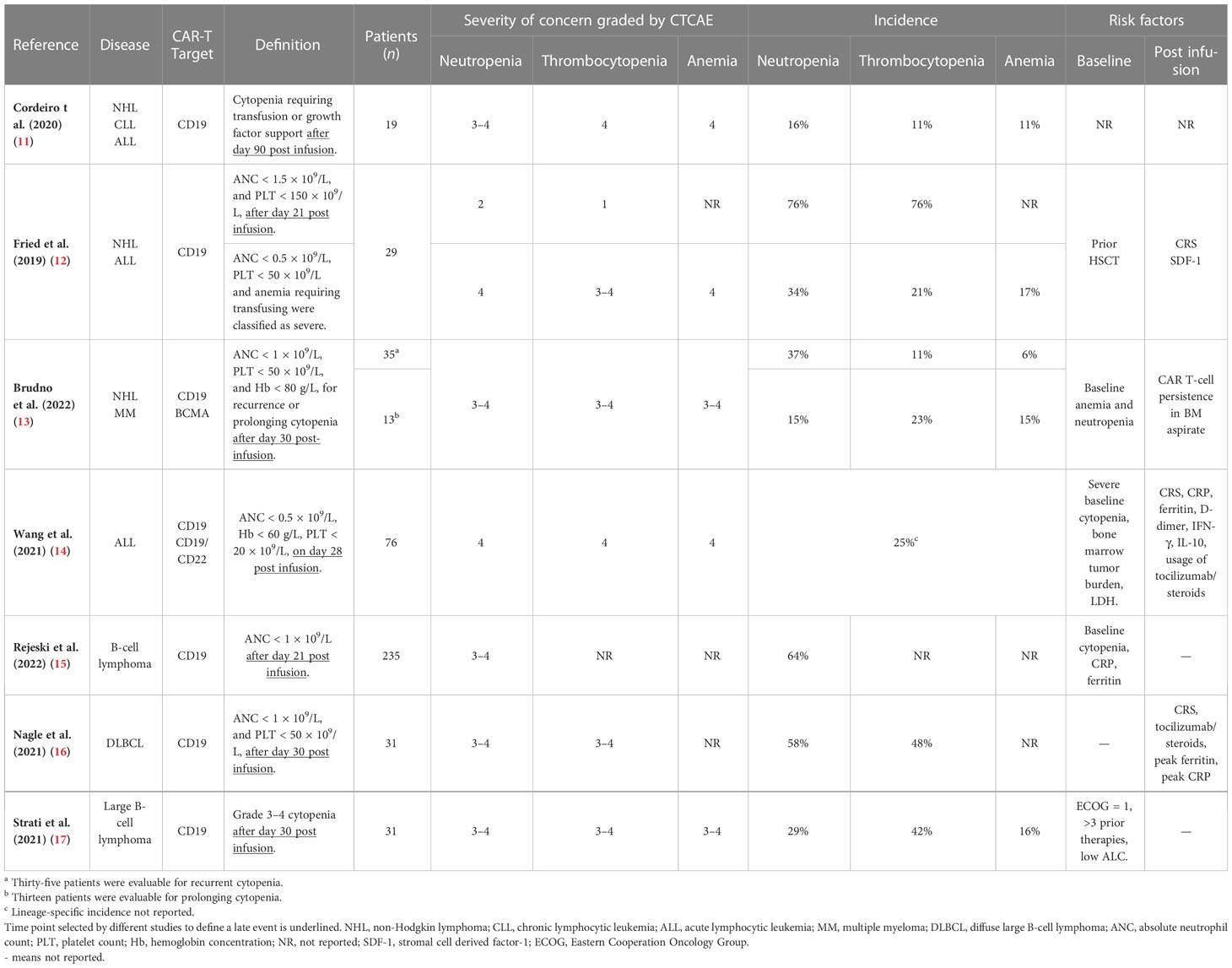
Table 1 Definition of CAR-T late hematotoxicity and severity of cytopenia of concern in different studies.
Among patients with B-cell lymphoma and treated with anti-CD19 CAR-T cells, the onset of cytopenia was observed from the day of infusion, or occasionally up to a month post-infusion (12). The median time to onset of neutropenia and thrombocytopenia was 3 days (range, 0–21) and 0 days (range, 0–38), respectively (12). For those patients who developed cytopenia a few days following infusion, previous studies reported a “biphasic” pattern of hematopoietic recovery (12, 15). The first trough of neutrophil count recovered within 3 weeks in 77% of the patients (n = 149) (15). However, 52% of the patients will experience a second trough of neutrophil count after a month of infusion (15). Moreover, some patients underwent severe myelosuppression, which could prolong for weeks, months, or even years without recovery and could be resistant to intensive clinical interventions (11, 13, 15, 17, 22, 24). Researchers held the opinion that the early phase of cytopenia might be related to lymphodepletion chemotherapies and CRS (12, 15). However, these factors should have subsided within 3 weeks of infusion (12, 15). Therefore, late-phase events, which are in the form of delayed-onset, recurrent, or persistent cytopenia, might be attributed to unknown mechanisms and require further investigation. Figure 1 depicts the different dynamics of hematopoietic reconstruction in CAR T-cell-treated patients.
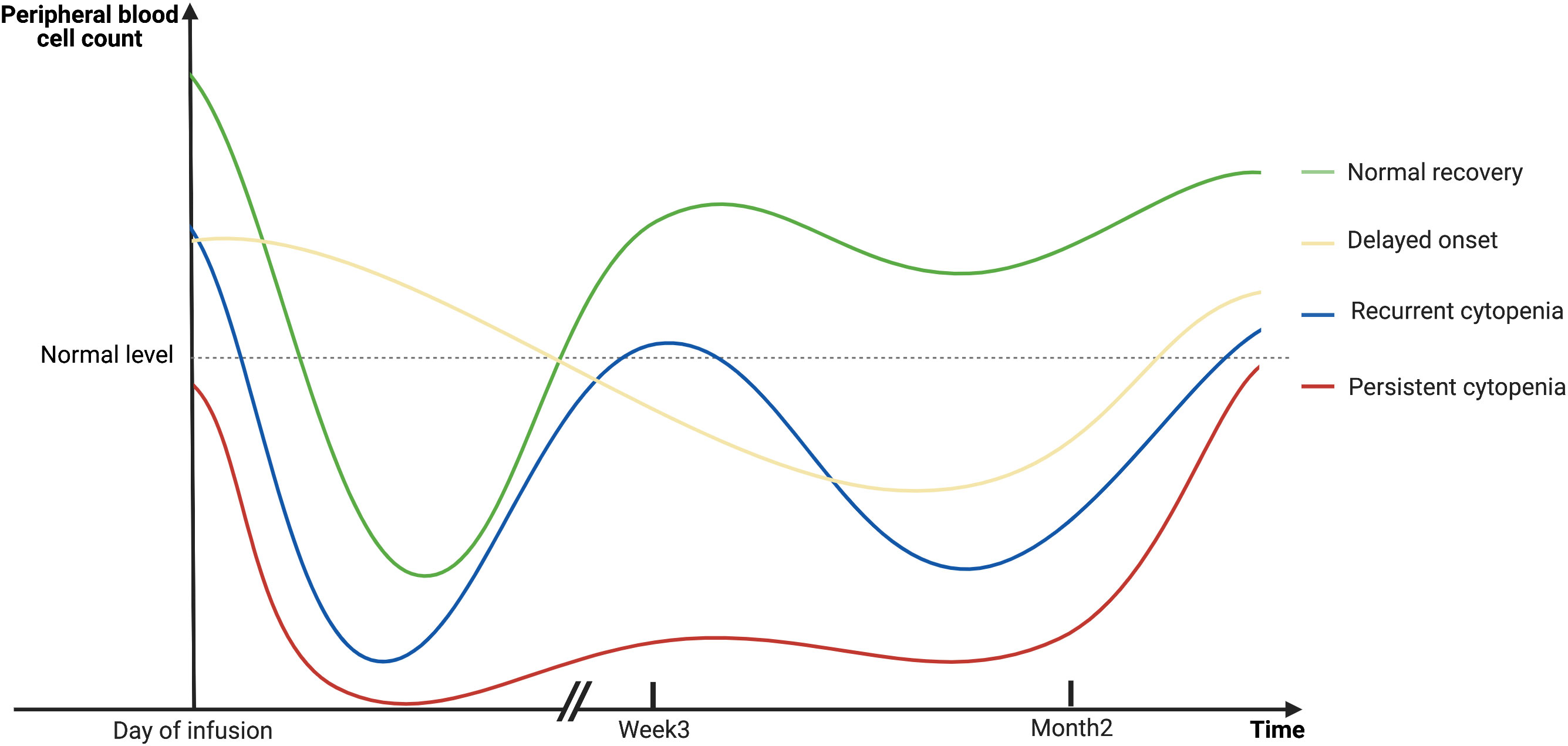
Figure 1 A schematic diagram showing the dynamics of hematopoietic reconstitution in CAR T-cell-treated patients. The early phase of cytopenia may be related to lymphodepletion chemotherapies and CRS, and will gradually recover in the majority of patients within 3 weeks after infusion. However, late-phase events, which are in the form of delayed-onset, recurrent, or persistent cytopenia in the second month or even later, cannot be explained for the same reasons. Created with BioRender.com.
The incidence of CAR-T hematotoxicity reported by different clinical trials has been thoroughly reviewed recently (6, 10). Briefly, the overall incidence of severe (grade ≥ 3) neutropenia, thrombocytopenia, and anemia ranges from 29% to 95%, from 15% to 65%, and from 14% to 77%, respectively (6, 10). However, a fewer number of studies distinguished late-phase from early-phase cytopenia. Those studies specifying CAR-T late-phase (>21 days post-infusion) hematotoxicity reported the highest incidence of grade 3–4 neutropenia, thrombocytopenia, and anemia to be 64%, 48%, and 17%, respectively (Table 1).
Retrospective studies have analyzed the clinical characteristics to identify associated factors related to CAR-T late hematotoxicity (Table 1). These studies were carried out among patients with R/R B-cell malignancies. CD19 was the most common therapeutic target, while other targets like B-cell maturation antigen (BCMA), CD22, and multi-target therapies were less reported. Juvenile patients were included in the analysis (12). However, no specific attention was given to the underage subgroup, and existing reports found no correlation between age and the incidence and severity of hematotoxicity (11–17). For the risk factors, different studies arrived at contradictory conclusions, specifically on the severity of CRS. Several studies reported that increased CRS severity was a predictor for slower recovery from cytopenia in the first month after CAR-T infusion (13, 25, 26). In a study of 83 patients with B-cell malignancies, grade ≥3 CRS was significantly associated with the absence of complete hematological recovery at a month (p = 0.002) (25). In another study (n = 164), the mean time to neutrophil recovery was 4.2 and 12.8 days in patients with grade 0 and ≥3 CRS, respectively (26). These findings indicated that CRS might be a predictor for the persistence of early-phase cytopenia. Moreover, some reports have stated that late hematotoxicity might be more common in patients with severe CRS grades (12, 14). Higher CRS grade showed a strong correlation with cytopenia > 21 days (p = 0.003, 0.018, and 0.04 for late anemia, thrombocytopenia, and anemia, respectively) (12). However, some reports stated otherwise (15, 17, 25, 27). The largest population (n = 235), and also the only multi-center analysis of CAR-T hematotoxicity carried out in R/R B-cell lymphoma patients revealed that CRS severity was not a risk factor (15). Aside from CRS, the cytokine profile was also analyzed. However, no specific cytokine was identified as a dependent influencing factor for hematotoxicity (12–14, 25, 26). Therefore, the specific contribution of cytokines to late hematotoxicity is difficult to determine. Other inflammatory indicators like C-reactive protein (CRP) and ferritin were correlated factors (14–16). Aside from inflammation-associated factors, baseline (before lymphodepleting chemotherapies) cytopenia was repeatedly reported as a strong indicator of persistent myelosuppression after CAR-T infusion (13–15, 17). For all 35 patients with NHL or MM available for evaluation, the hemoglobin level was lower among patients who developed delayed cytopenia (p = 0.0079) (13). For 76 patients with acute lymphocytic leukemia (ALL), researchers also identified that patients without baseline cytopenia exhibited easier hematologic restoration (p = 0.028) (14). These findings indicated that baseline cytopenia was strongly related to CAR-T hematotoxicity in different diseases.
CAR-T HEMATOTOX (HT), the only available predictive model for severe CAR-T hematotoxicity, was proposed based on the analysis of clinical characteristics in adult patients with R/R B-cell lymphomas (15). This evaluation system strengthens the importance of clinical features at baseline, including peripheral counts of neutrophils and platelets, hemoglobin concentration, CRP, and ferritin levels. Patients with lower baseline peripheral counts and higher CRP and ferritin levels will get a higher HT score. They are more likely to develop severe infections, specifically bacterial infections, and are more vulnerable to non-relapsed mortality (23). Although HT successfully predicts the occurrence of severe and prolonged neutropenia lasting longer than 14 days post-infusion (15), the recurrence of severe cytopenia cannot be indicated, nor can it reveal the prognosis of the patients with severe hematotoxicity (28). Therefore, future efforts must be dedicated to optimizing the evaluation model.
Clinical interventions for CAR-T late hematotoxicity are typically supportive and without specific targets. The most common strategies are blood transfusion, growth factor utilization, and HSCB (20–22, 29). Growth factors are widely used interventions for cytopenia for various reasons. However, only recently have their therapeutic effects on CAR-T hematotoxicity management been carefully investigated (29). A study of 197 patients revealed that, while prophylactic administration of granulocyte-colony stimulating factor (G-CSF) could shorten the duration of neutropenia after CAR-T infusion, reducing later recurrence is ineffective (29). Thrombopoietin receptor agonists (TPO-RA) have also been reported in treating prolonged myelosuppression after CAR-T infusion (30–32). In a retrospective study, 11 patients were treated with TPO-RA, a median of 17 days was needed to gain infusion independency, and a median of 46 days was needed to gain a plate recovery of ≥50 × 109/L after the initiation of TPO-RA (31). Another study has reported similar results in six patients with transfusion-dependent cytopenia (32). However, determining the exact benefit of TPO-RA in these retrospective studies is difficult because no comparison has been made between treated and non-treated patients.
Two clinical studies reported the effectiveness of HSCB in relieving CAR-T late hematotoxicity (20, 21). In a study with 12 patients, the median duration of severe neutropenia and thrombocytopenia was 42 days after CAR-T infusion and the cumulative response rate at day 30 after HSC infusion was 82% for neutropenia and 60% for thrombocytopenia (21). In another study with 31 patients, the response rate for neutropenia was 84%, and the responding patients showed higher survival than non-responding patients (20). HSCB was reported to be ineffective in rescuing patients of CAR-T late hematotoxicity during severe infection (20). However, previous research proposed that infusion of HSC could improve the survival rate of sepsis by 50%–60% (33), indicating that HSCB might still be considered an effective therapy for patients with severe infection, but HSCB is limited by its availability. The failure of mobilization is a challenge, specifically for patients with baseline cytopenia. Alternatively, allogenic sources of HSCs can be used occasionally (20, 21); however, allogenic sources may require careful evaluation for safety and efficiency. Hematotoxicity resistance to HSCB is also a challenging issue. In a case report, clinicians used rapamycin (Sirolimus), an mechanistic target of Rapamycin (mTOR) inhibitor, to suppress CAR-T amplification and successfully rescued a heavily treated patient with persistent myelosuppression who had failed HSCB (24).
Aside from clinical features, a few reports shed light on the pathogenesis of CAR-T late hematotoxicity. A deep sequencing approach to determine the prevalence of clonal hematopoiesis of indeterminate potential (CHIP), which is closely associated with chronic inflammation, revealed that CHIP is not related to the dynamics of hematopoietic recovery (34). In another study, single-cell analysis and serum cytokine analysis revealed a case of aplastic CAR-T hematotoxicity with bone marrow failure featuring oligoclonal T-cell expansion and altered cytokine-related features (35). Inflammation is the current focus of discussion, and further research is urgently needed for the pathogenetic investigations of CAR-T late hematotoxicity.
Several reviews have discussed the possible mechanisms of CAR-T hematotoxicity (6, 10). Maintaining HSC homeostasis, the interaction between bone marrow niches, regulation by inflammatory cytokines, disorders of cellular immunity, and others could all play a part in the complex mechanisms (6, 10). However, no clinical disorders referred to by these detailed reviews (6, 10), such as acquired aplastic anemia (AA) and myelodysplastic syndromes (MDS), could fully concur with the characteristics of CAR-T late hematotoxicity. The biphasic or aplastic recovery feature indicates an intermittent or profound reduction of hematopoietic cells and a temporary or prolonged impairment of hematopoietic function. Considering the success of HSCB in relieving CAR-T late hematotoxicity and the crucial role of inflammation during CAR-T therapy, the negative role of inflammation on HSCs can be summarized.
Homeostatic hematopoietic output is a carefully regulated hierarchy process. In homeostasis, most HSCs are quiescent in the G0 phase (36). While long-term HSCs (LT-HSCs) are responsible for lifelong persistent hematopoiesis, multipotent progenitors (MPPs) dominate the homeostatic hematopoietic output. MPP1 is a cluster of metabolically active HSCs, also identified as short-term HSCs (ST-HSCs) (19); MPP2–4 are subsets of lineage-biased MPPs with reduced self-renewal potency, of which the most abundant MPP4 generates primarily myeloid and lymphoid output (37). MPPs may serve as the hematopoietic buffer, rapidly adapting to stimulations without uncontrolled activation of HSCs, which may be detrimental (37).
Maintaining the balance between quiescence and proliferation is essential for a long-term stabilized stem cell pool (38). However, this homeostasis can be disrupted when confronted with challenges such as inflammation. Intrinsic or extrinsic cell death mechanisms are also important factors contributing to diminishing the number of hematopoietic cells.
Quiescent HSCs harbor the most robust self-renewal capacity, and they are indispensable for a persistent stable hematopoietic output (39, 40). Quiescent HSCs exhibit better multi-lineage repopulation ability in long-term transplantation experiments (41). The conferring of dormant HSCs into an active cell cycle is not a fully reversible process, generating daughter phenotypic HSCs, which may not be as potent even after the return to dormancy (42, 43). Moreover, consistent activation and frequent division are detrimental to self-renewal and may eventually result in the depletion of the stem cell pool (44, 45).
The risk of chronic inflammation in HSCs has been well-established (45–48). In vivo chronic stimulation of lipopolysaccharide (LPS), pI:pC, or different cytokines, such as interferons (IFNs), transforming growth factors (TNFs), and IL-1 (48–51), induces HSC depletion via interruption of quiescence, promoting proliferation and differentiation at the expense of self-renewal. The effects can be either directly mediated by Toll-like receptors (TLRs) (52) or cytokine receptors on HSCs (53) or indirectly mediated via interfering with bone marrow niches (54). Chronic inflammation due to multiple lines of therapies and tumor-bearing status may already reduce the number and potency of HSCs before CAR-T treatment. Some patients certainly developed cytopenia before the routine lymphodepleting chemotherapy, which is a predictive factor of severe and prolonged cytopenia after CAR-T infusion (13–15, 17). However, chronic inflammation is constantly related to myeloid-biased hematopoietic output (55), which is commonly observed in MDS but seldom reported in CAR-T hematotoxicity. CHIP has not been proven to contribute to CAR-T hematotoxicity (34).
Acute inflammation also exerts influences on HSCs, although whether this effect is prolonged varies between investigations. A previous study showed that acute inflammation was not likely to considerably influence HSC potency in the long term (56). However, later investigations indicated that the impairment was prolonged or persistent after acute inflammation (41, 43). Peripheral virus infection activated HSCs by inflammatory cytokines and chemokines (43). The bone marrow of murine cytomegalovirus (MCMV)-infected mice was extracted at 4 days, 21 days, and 4 months post-infection. Competitive and secondary transplantation experiments revealed a significantly impaired function both in HSCs harvested 4 days post-infection during the acute phase and in HSCs harvested 21 days post-infection after returning to phenotypic quiescence (43). However, such impairment was not observed in the bone marrow harvested 4 months post-infection, indicating a long-term, but not infinite, impairment (43). In a more recent investigation (41), wild-type C57BL/6J mice were injected with blocks of pI:pC to mimic rounds of discrete acute inflammation in moderate intensity during virus infection. LT-HSCs demonstrated more rapid differentiation kinetics and faster exit form quiescence than their phosphate buffer saline (PBS)-treated counterparts, along with compromised overall in vitro proliferative potential (41). HSCs from mice treated with three blocks of pI:pC (i.e., eight individual injections spread over 8 weeks for each block) showed a significantly reduced functional potency than their age-matched counterparts even after a 12-month recovery (41). Further investigation of the Scl-tTA; H2BGFP mouse model revealed that H2B-GFP-retaining undivided HSCs, which remained quiescent throughout the challenges, maintained better functional potency than the divided subset, and the shrink of the dormant HSC pool was responsible for a persistently impaired hematopoietic output (41). Acute inflammation such as CRS or infection is common after CAR-T therapy. Therefore, acute inflammation may contribute at least partially to temporary or prolonged cytopenia in this scenario. However, clinical investigations have contradictory conclusions as to whether the grade of CRS has associated with CAR-T late hematotoxicity. Given the distinctive cytokine profile of CRS from viral infection (43, 57), CRS needs to be verified whether the profound effects of acute inflammation on LT-HSCs observed in viral infection are also applicable to CAR-T therapy.
Previous studies show that chronic and acute inflammation can activate dormant HSCs into proliferation at the expense of self-renewal, though activation of only a small clutch of HSCs is not likely to provide a grievous blow to the hematopoietic system (41, 56). A complicated network of intrinsic and extrinsic factors, such as cell cycle regulators, transcription factors, epigenetic factors, niche factors, and metabolism regulators, is involved in maintaining the quiescence of HSCs and has been thoroughly reviewed previously (Figure 2) (58, 59). Pathologic gene mutations are common in patients with hematological malignancies. For instance, P53, a commonly tested gene mutation in clinical practice, is an important transcription factor regulating HSC quiescence (58). Whether P53, along with other gene mutations, is associated with the incidence of CAR-T late hematotoxicity remains unknown. Moreover, epigenetic factors should be carefully considered. Inflammation has been proven to exert a long-lasting epigenetic effect on LT-HSCs, in a way designated as “trained immunity” (60) or leading to accelerated senescence (41). The contribution of epigenetic alternations to late hematotoxicity remains unknown. Niche factors, which are highly likely to be disrupted, should also be emphasized. For example, CD150 high bone marrow regulatory T cells (Tregs) maintain HSC quiescence via adenosine (61). However, Tregs are reduced during CAR-T therapy due to lymphodepleting chemotherapy, although whether niche-resident Tregs are also depleted remains unidentified. Hence, an essential factor in maintaining HSC dormancy would be diminished. Furthermore, inflammation is strongly associated with increased reactive oxygen species (ROS) and altered HSC metabolism, regulating glycolysis, oxidative phosphorylation (OXPHOS), and fatty acid oxidation (FAO), eventually influencing fate decisions (45, 62).
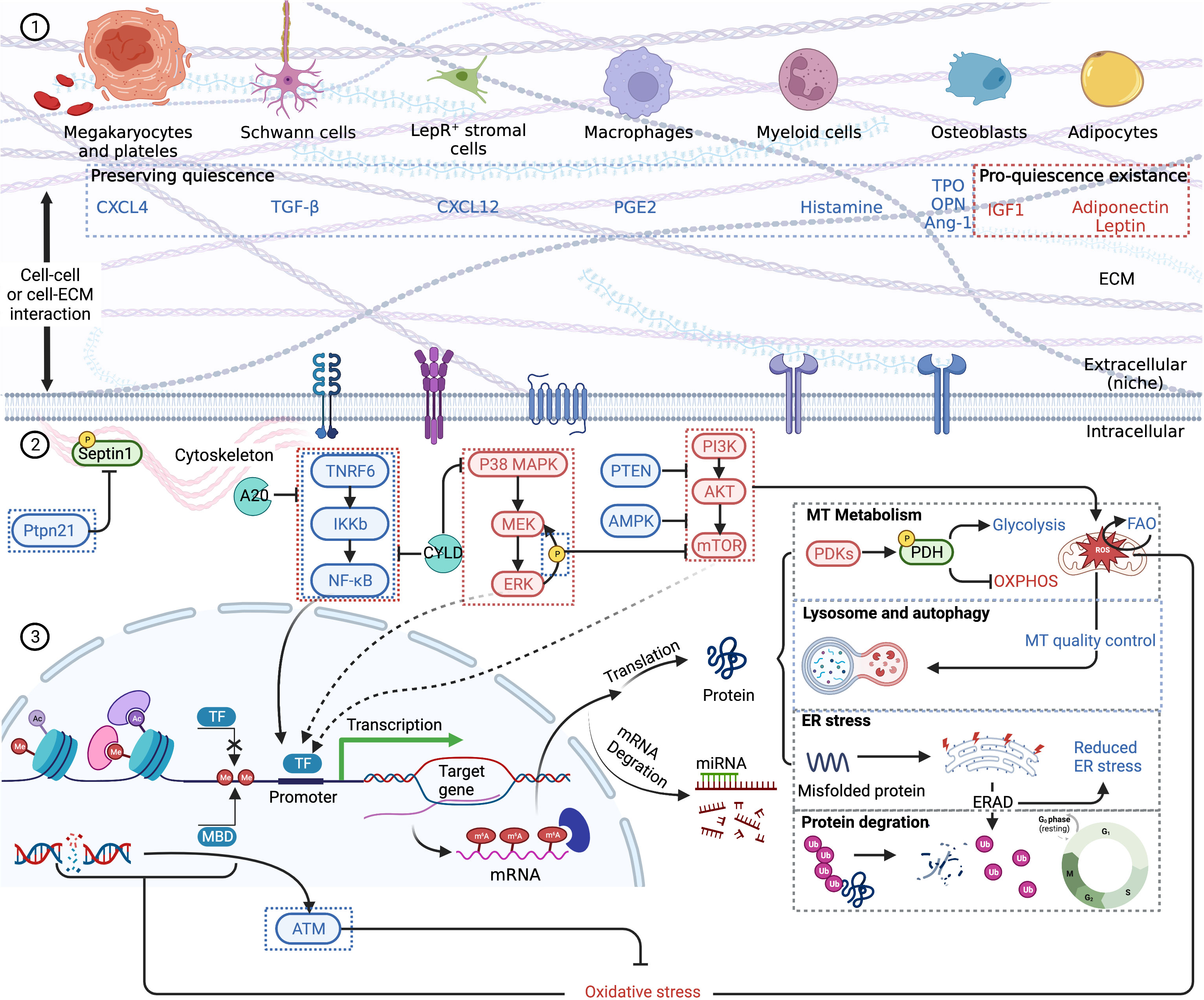
Figure 2 A schematic summary of HSC quiescence regulation based on previous reviews (58, 59). Blue font and boxes label the factors maintaining quiescence, while red font and boxes label the factors promoting quiescence existence. (1) HSC extrinsic regulation. Niche cells and respective cytokines are pictured. HSCs receive stimulation via membrane receptors. Cell–cell interaction and cell–ECM interaction are also vital to quiescence retention. (2) HSC intrinsic regulation. NF-kB pathway, MAPK pathway, and mTOR pathway are the main signal pathways regulating HSC quiescence. Quiescent HSCs mainly depend on glycolysis for energy, and the self-renewal and maintenance of stem-cell pool rely on FAO. Lysosome and autophagy are important to clear excessively active mitochondria. Epigenetic changes, including histone and DNA modification, which influence gene transcription, are also the consequences of oxidative stress. After transcription, m6A modifications are significant epigenetic factors regulating HSC quiescence. Misfolded or unfolded protein depends on ERAD to degrade via ubiquitination. Other proteins including cell cycle proteins are also eliminated by ubiquitination, thus regulating cell dormancy and cycle activation. CXCL, CXC motif chemokine ligand; TGF, transforming growth factor; PGE, prostaglandin E; TPO, thrombopoietin; OPN, osteopontin; IGF, insulin-like growth factor; ECM, extracellular matrix; MT, mitochondrion; PDK, PDH kinases; PDH, pyruvate dehydrogenase; OXPHOS, oxidative phosphorylation; ER, endoplasmic reticulum; TF, transcription factor; MBD, methyl binding domain protein; ATM, ataxia telangiectasia mutated kinase. Created with BioRender.com.
Under inflammatory challenges, different forms of cell death take part in reducing the number of hematopoietic cells, such as apoptosis, necroptosis, and pyroptosis (47). Additionally, direct attack by immunity cells also leads to cell death. While dormant HSCs tend to be more resistant to harmful factors than differentiated hematocytes (41, 63, 64), HSCs can be conferred to death under certain circumstances.
In inflammation, some cytokines like IFNs and TNFs are proapoptotic (54). HSCs were primed to apoptosis under chronic exposure to type I IFNs. Once forced into the cell cycle, such as cell culture, transplantation, and myeloid-ablative treatment, the p53-dependent proapoptotic program was quickly activated, leading to HSC depletion and bone marrow failure (64). An earlier investigation revealed that IFN-γ treatment upregulated FAS along with other proapoptotic genes, sensitizing Lin-Sca-1+c-Kit- cells (LSKs) to apoptosis both in vitro and in vivo (65). In addition, LSKs consist of different cell subsets with heterogeneous vulnerability to the same treatment (66, 67); hence, earlier reports on these poorly purified populations should be judged carefully. For example, HSCs and MPP2/3 were resistant to the cytotoxicity of TNFα, while MPP4, granulocyte/macrophage progenitors (GMPs), and other committed and mature cells were susceptible to apoptosis in a dose-dependent manner, indicating that the neutrophil lineage may be more vulnerable (67). Moreover, TNFα initiates necroptosis in a receptor-interacting serine/threonine kinase 1 (RIPK1)-dependent manner (68).
Necroptosis and apoptosis share a common molecular pathway. Apoptosis can transform into necroptosis when suppressed (69, 70). Apoptosis-defect mice with Bax, Bak, and Bid triple knockout showed massive necroptosis in myeloid progenitors, mediated by abnormally raised RIPK1 expression (68). Increased cytokine levels and excessive stem cell proliferation were observed and eventually led to stem cell exhaustion and bone marrow failure (BMF) (68). Knockdown of one RIPK1 allele to normalize RIPK1 expression to wild-type level or inhibition of TNFα signal effectively increased myeloid progenitors and ameliorated cytopenia (68). However, a deficiency of RIPK1 resulted in the overactivation of RIPK3 and MLKL, leading to necroptosis and hematopoietic stem/progenitor cell (HSPC) depletion, indicating that a normal level of RIPK1 is necessary for HSPC survival from necroptosis (71). TNFα is a commonly focused cytokine in necroptosis, inducing apoptosis or necroptosis to lineage-committed progenitor and mature cells in a dose-dependent manner (67). However, TNFα mediated the upregulation of the p65/NF-κB-cIAP2 pro-survival pathway in HSCs, protecting them from necroptosis (67). Nevertheless, with the attenuation of the TNFα signal, this pro-survival pathway would quickly get inactivated, creating a vulnerable time window to necroptosis in activated HSCs, which leads to the contraction of the HSC pool and a transient but significant hematopoietic impairment (67). Only after the return to dormancy is this vulnerability removed (67).
Pyroptosis is another vital cell death mechanism. Similar to necroptosis, pyroptosis leads to the production of pro-inflammatory substrates (72). Viral infection and chemotherapy induce massive hematopoietic progenitor cell death via NACHT leucine-rich-repeat protein 1 (NLRP1)-mediated pyroptosis (73). Chronic TGF-β exposure followed by pI:pC acute inflammatory stimulation induced prolonged cytopenia. Consistent upregulation of Caspase1, a key molecule in pyroptosis, was observed in HSCs; however, this research did not further confirm pyroptosis to be the exact reason for BMF (64).
Necroptosis and pyroptosis lead to the production of damage-associated molecular patterns (DAMPs) (74, 75). As reported in sepsis, DAMPs like high mobility group protein B1 (HMGB1) and mitochondrial DNA (mtDNA) are potent inducers of type I interferons, which negatively regulate emergency hematopoiesis (72). HMGB1 is a late-phase inflammatory substrate secreted by activated megakaryocytes in sepsis (76). While targeting early-phase cytokines gained only limited therapeutic effects, the administration of HMGB1 antagonists significantly improved the survival of systemic inflammatory response syndrome (SIRS) in rodents (77–79). HMGB1 has also been reported to be the mechanism of various cytopenia diseases, even though not directed on HSCs, such as chronic idiopathic neutropenia (CIP) (80), immune thrombocytopenia (ITP) (81, 82), anemia of inflammation (AI) (83, 84), and BMF (85) (Figure 3). Pyroptosis is critical for CAR-T assassination of target cells and is proven to be the mechanism of CRS (86). Because one of the dominant HMGB1 receptors, TLR4 (79), is expressed in HSCs (42), HMGB1 may exert a direct impact on HSCs. Nevertheless, whether this impact contributes to CAR-T late hematotoxicity remains unclear.
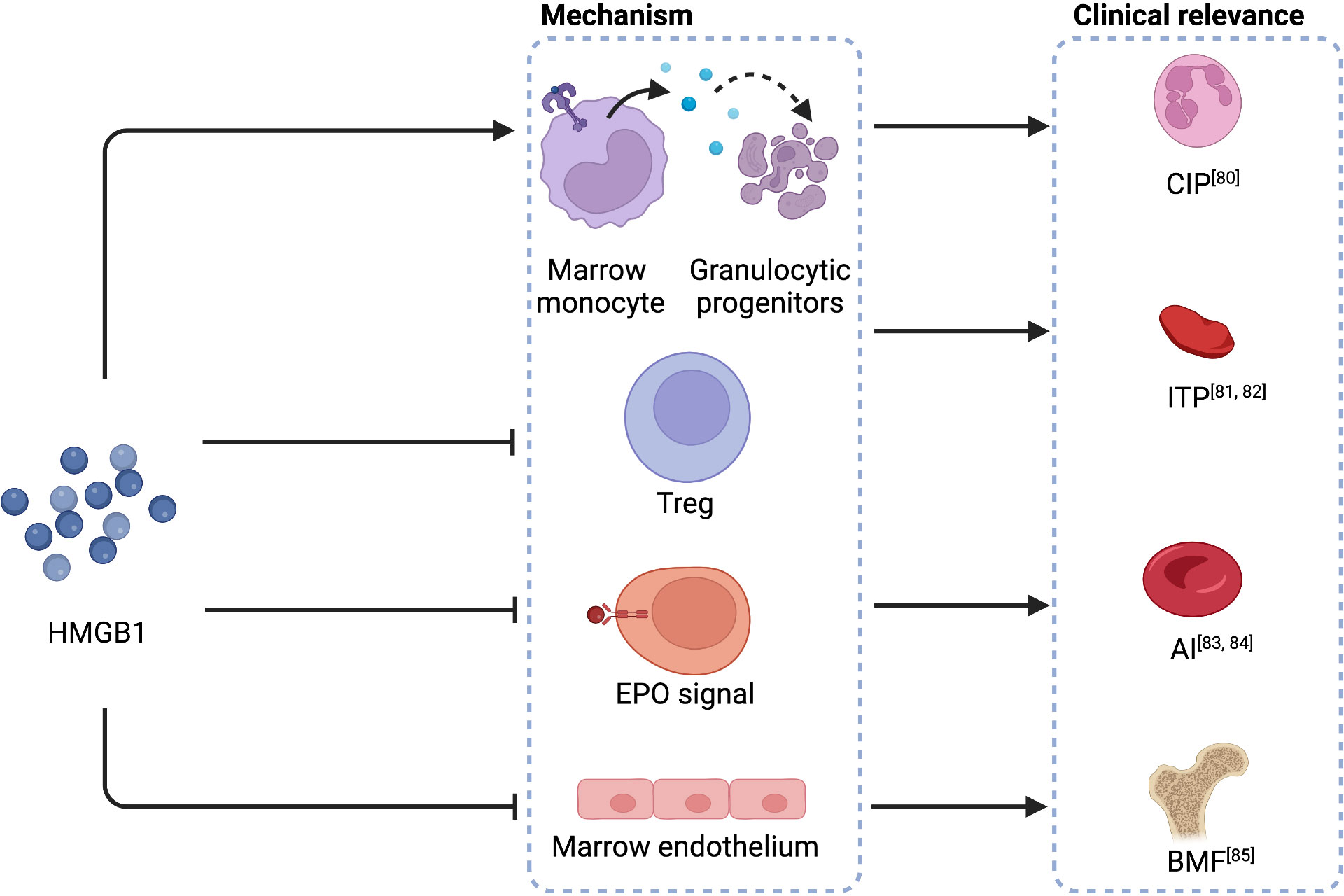
Figure 3 HMGB1 in the mechanisms of cytopenia diseases. HMGB1 causes the apoptosis of granulocytic progenitors, interferes with Tregs, disrupts the regular EPO signal, and impairs bone marrow endothelium, leading to CIP, ITP, AI, and BMF, respectively. HMGB1, high mobility group protein B1; CIP, chronic idiopathic neutropenia; Treg, regulatory T cells; ITP, immune thrombocytopenia; anemia of inflammation, AI; anemia of inflammation; EPO, erythropoietin; BMF, bone marrow failure. Created with BioRender.com.
Besides the influence of cytokines, cellular immunity is also involved in reducing the HSC pool. Cytotoxic T cells attack HSPCs and result in aplastic aplasia (87). In this process, IFN-γ is involved in the recruitment of T cells and the enhancement of the vulnerability of HSPCs to CD8+-T-cell cytotoxicity and mediates the cytotoxic effect (65). Natural killer (NK) cells also participate in the direct attack of HSPCs. NK cells are reported to reduce the number of HSPCs with genotoxic stress by exerting cytotoxicity through the interaction between natural killer group 2 member D (NKG2D) and NKG2D ligand. Inhibition of this interaction could improve cytopenia in vivo (88). In normal conditions, HSCs are protected from the attack of cellular immunity. Bone marrow-resident Tregs participate in maintaining an inhibitory immune microenvironment (89, 90). If these Tregs are affected in CAR-T therapy, immune-inhibitive protection would be disrupted. Additionally, CD47 is a “do not eat me” signal expressed on HSCs, while the mobilization of HSCs into the periphery will downregulate its expression (89). DAMPs, which are abundantly generated in CRS (75), were reported to be a powerful mobilization factor (91). SDF-1 level, a cytokine essential for HSC retention in the bone marrow and B-cell development, has been reported to be correlated with late neutropenia, though another report later challenged this conclusion (12, 13). Whether these factors contribute to the reduction of the HSC pool by increasing the vulnerability to cellular immunity attacks is unknown.
Cytokines are fundamental in regulating hematopoietic output (92), and serum cytokine level has been repeatedly analyzed in clinical reports on CAR-T late hematotoxicity (12–14, 25, 26). Among the diversity of cytokines, including the IL-1 family, hematopoietin (class I cytokine) family, IFN (class II cytokine) family, growth factors, TNF family, IL-17 family, and chemokines (25), some have been identified as relevant to hematopoietic recovery after CAR-T infusion, even though contradictions exist between studies. These cytokines include IL-6, IL-10, IFN-γ, TGF-β1, and SDF-1 (12–14, 25, 26). Higher serum concentrations of IL-6 (26), IL-10, and IFN-γ (14) were associated with lower blood cell counts, while a higher concentration of TGF-β1 (26) and SDF-1 was related to higher blood cell counts (12). IL-6 is a cytokine promoting neutrophil production in emergency hematopoiesis (Table 2) (53, 92, 105–107). IL-6 was not included in Table 2 because it mainly regulates progenitor cells, although the peak level of IL-6 is negatively related to hematopoietic recovery. Moreover, the IL-6 receptor is lowly expressed in HSCs (108). Therefore, even chronically exposing LT-HSCs to IL-6 will not bring apparent functional deficiency (105).
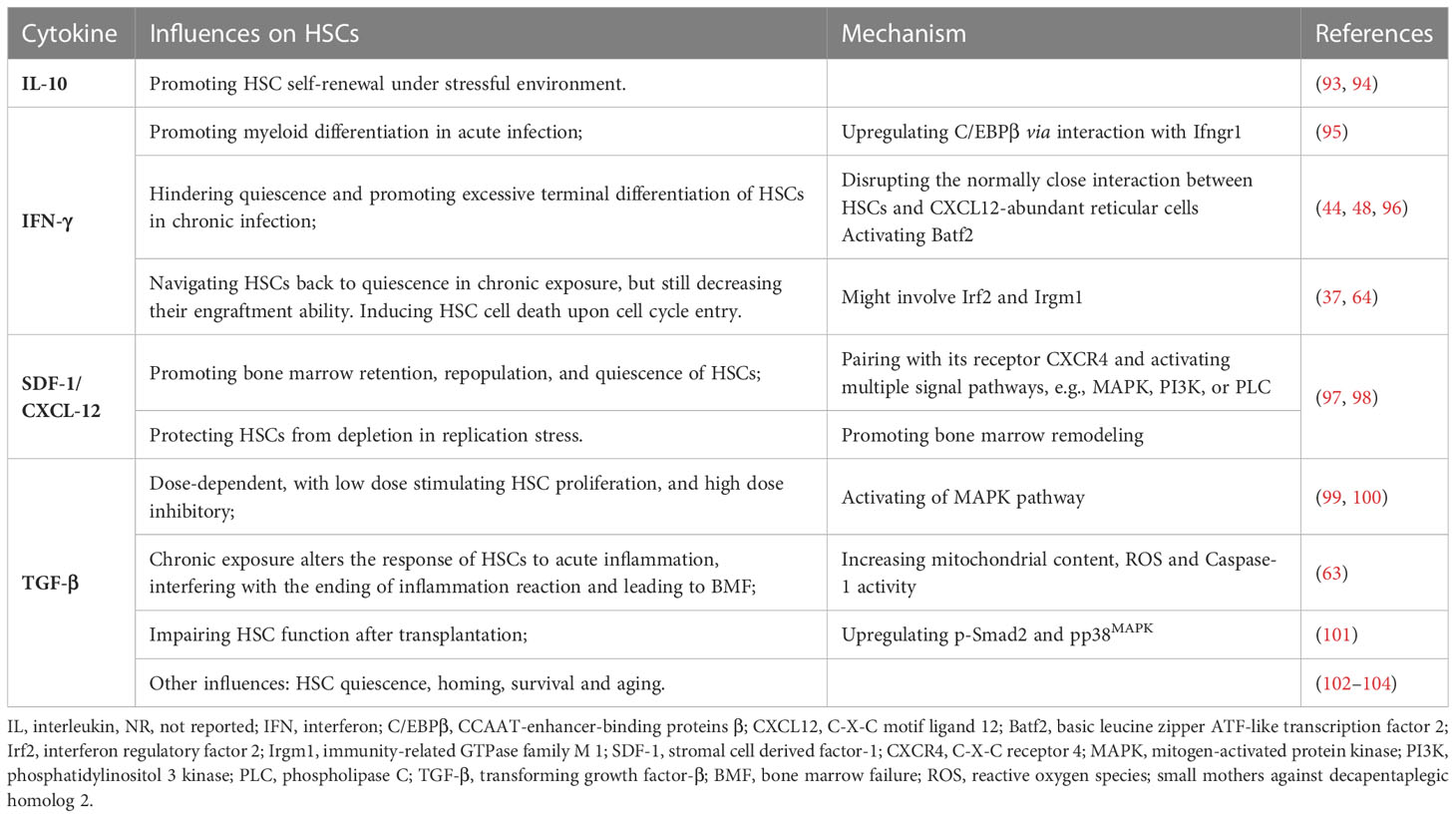
Table 2 Cytokines reported to be associated with CAR-T late hematotoxicity and their direct impact on HSCs.
Cytokines exert a complicated influence on hematopoietic output; however, the actual effects of cytokines tend to be dose-dependent and time-dependent. The dose and acute or chronic exposure may bring opposite influences (53, 54, 109), which has been observed in IL-1 (45, 110–112), TNFα (109), IFNs, and TGF-β (Table 2). Besides the direct influence on HSCs, specific cytokines can also interfere with the normal signals controlling hematopoietic output downstream of stem cells. IFN-γ binds to TPO and inhibits hematopoiesis by interfering with the interaction between TPO and its receptor c-MPL as proven in AA (113, 114). In AI, HMGB1 disrupts the signal transduction of erythropoietin (EPO), causing prolonged anemia after sepsis recovery (84, 115). However, as previously shown, the bone marrow cytokine profile might be different from the peripheral (89). Therefore, identifying the bone marrow cytokine profile might be the most accurate and helpful method to determine the critical influencing factors on HSCs after CAR-T therapy.
Some cytokines such as IL-1 (116), IL-6 (54), and TNFα (117, 118) induce inflammation of HSCs (119). The function of HSC is also significantly altered with senescence, even though they still harbor the same stem cell phenotype (117). For one thing, senescent HSCs show diminished hematopoietic potency, myeloid-biased differentiation, and clonal hematopoiesis (117). For another thing, the inflammation-associated signal, like the NF-kB pathway, is upregulated in senescent HSCs, raising their sensitivity to inflammatory challenges (116, 117, 120). However, clinical investigations have shown that older age is not associated with CAR-T late hematotoxicity (11–17). Nevertheless, the biological age of the patient may not reflect the actual age of HSCs. The methyl clock analysis has proved that repeated inflammatory challenges will cause premature aging of LT-HSCs (121). Multiple lines of therapies before CAR-T, which raise inflammatory challenges in bone marrow (122), may be risk factors for the accelerated aging of HSCs. Epigenetic analysis to determine the actual age of HSCs may help clarify the role of senescence in CAR-T late hematotoxicity. mTOR is reported to be vital in regulating the function of senescent HSCs (123). The mTOR signaling pathway is essential for the function of HSCs, by regulating important activities like autophagy, metabolism, and the transformation between quiescence and expansion (124, 125). Rapamycin, an mTOR inhibitor reported to be effective in relieving prolonged CAR-T late hematotoxicity, is an intervention to improve the function of senescent HSCs (123, 125). This mTOR inhibitor has also been proven effective in preserving HSPCs and relieving BMF in mouse models (47). Moreover, apart from cell-intrinsic regulation, the mTOR pathway also regulates the function of bone marrow niches, which provide vital support for normal hematopoiesis. mTORC1 activation enhances the production of IL-19 by osteocytes, and IL-19 has been proven to be a robust stimulation of granulopoieses, relieving chemotherapy- and irradiation-induced neutropenia even more effectively than G-CSF (126).
Niche dysfunction is also harmful to HSCs. Endothelial cells with chronically activated mitogen-activated protein kinase (MAPK) signals are reported to cause HSCs to lose their engraftment ability (127). The impairment of endothelial cells by IFN-γ-induced HMGB1 drives myelosuppression (85). Stromal cells with oxidative stress lead to DNA damage in HSCs (128). Among niche factors promoting hematopoiesis, bone marrow T cells are essential for hematopoietic recovery after stressful challenges. Aside from the function of Tregs, which has already been mentioned earlier in this review, bone marrow-resident group 2 innate lymphoid cells (ILC2) have also been proven vital for hematopoietic recovery as an abundant source of granulocyte-macrophage colony-stimulating factor (GM-CSF) (122). B-cell progenitors produce IL-33, inducing ICL2s to secrete GM-CSF via the activation of MyD88, thus promoting neutrophil recovery and bone marrow engraftment (122). ICL2s are also activated by mesenchymal-derived prostaglandin D2 (PGD2) through the interaction with the receptor CRTH2 (129). Activated ICL2s secrete IL-5 and IL-13, and IL-5 further promotes the expansion of HSPCs via the stimulation of CD4+CD25+IL5Rα+ Tregs (129). These observations strengthen the importance of niche factors, especially niche-resident T cells in hematopoiesis after stressful challenges. Other niche-resident cells also provide significant cytokines influencing HSC function, such as IL-1β-secreting dendritic cells (DCs) (130) and myeloid cells (116), IL-6-secreting CXCL12-abundant reticular cells (105), IL-19-secreting osteocytes (126), and CXC motif chemokine ligand (CXCL)-12 secreting stromal cells (97, 131).
This review summarizes CAR-T late hematotoxicity, including definition, incidence, clinical characteristics, risk factors, and interventions. Current clinical investigations on CAR-T late hematotoxicity highlight the following: (1) The chosen time point for late events is arbitrary and inconsistent between studies. Determining the exact time point for the late event immediately after CAR-T infusion, based on the duration of earlier cytopenia, may help answer this issue. (2) Research attention is being paid to severe cytopenia of grade 3–4, while less is devoted to cytopenia in grade 1–2. If the normalization of hematopoietic recovery is considered a standard, the challenge of CAR-T late hematotoxicity should be more severe. (3) Baseline cytopenia is a relatively definite indicator of severe late hematotoxicity. Therefore, to eliminate the influence of CAR-T-associated factors, analyses should focus on patients without baseline cytopenia. (4) While HSCB exhibits positive therapeutic effects, the availability of this treatment is limited. Interventions are mainly supportive without definite targets. Moreover, HSCB is mainly for aplastic CAR-T late hematotoxicity, and whether recurrent cytopenia requires positive interventions is unknown.
Owing to the limited research available, the exact pathogenesis is difficult to discuss. Based on the therapeutic effect of HSCB and the influence of inflammation in CAR-T therapy (20–22), we reviewed the negative impact of inflammation on HSCs. HSCB directly supplements the number of HSCs and improves hematopoiesis in the majority of patients, indicating that the loss of HSCs due to undetermined factors contributes to the incidence of CAR-T late hematotoxicity. However, a small proportion of patients were unresponsive to HSCB and showed poor prognosis, indicating that the function of HSCs is crucial. Therefore, we divided the discussion of the negative influence of inflammation on HSCs into the depletion of stem cell number and the impairment of hematopoietic function. Nevertheless, one limitation is that we have not emphasized the more differentiated and mature hematocytes as they have been summarized previously (10) and no clinical practice suggests that the interventions on these subgroups are effective. Additionally, the various incidences of neutropenia, thrombocytopenia, and anemia cannot be explained at the HSC level. The highest incidence of neutropenia may be explained by the shortest life of neutrophils, making ANC the most sensitive reflection of hematotoxicity (132). Nevertheless, lineage-specific hematotoxic factors and lineage-differentiated vulnerability cannot be excluded as well.
Inflammation exerts a complex influence on HSCs including the direct interference and indirect impact via the regulation of niche factors. Chronic inflammation not only impairs the self-renewal and function of HSCs but also may increase their vulnerability or alter the response (59) to later inflammatory challenges. Whether acute inflammation will impair hematopoiesis in the long term during CAR-T therapy is an open question. Identifying the significant factors from all the candidates will be challenging. However, considering the clinical manifestations of CAR-T late hematotoxicity, the high incidence suggests that such factors may exist in most patients. The late event suggests that such factors may persist for a long time or exhibit late-phase peak levels long after the ablation of CRS or other earlier factors. Among these factors, cytokines, niche factors, and cellular immunity attacks, which are dominant to the pathogenesis of CAR-T late hematotoxicity, are crucial and need consideration. Identifying the most critical elements from all these factors will be challenging; however, the mechanisms are fundamental in understanding CAR-T late hematotoxicity.
TS and XZ finished the conceptualization. TS performed document investigation, wrote the original draft, and prepared the table and figures. This work was supervised, revised, and edited by DL, LH, and XZ. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 812270183, to XZ).
We would like to thank Editage (www.editage.com) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Davila ML, Bouhassira DCG, Park JH, Curran KJ, Smith EL, Pegram HJ, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int J Hematology (2014) 99(4):361–71. doi: 10.1007/s12185-013-1479-5
2. Manier S, Ingegnere T, Escure G, Prodhomme C, Nudel M, Mitra S, et al. Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev (2022) 54:100929. doi: 10.1016/j.blre.2022.100929
3. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T cells targeting GD(2) and releasing inducible IL-18. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.839783
4. Jacobson C, Locke FL, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term (>= 4 year and >= 5 year) overall survival (OS) by 12-and 24-month event-free survival (EFS): an updated analysis of ZUMA-1, the pivotal study of axicabtagene ciloleucel (Axi- cel) in patients (Pts) with refractory Large b-cell lymphoma (LBCL). Blood (2021) 138:1764–+. doi: 10.1182/blood-2021-148078
5. Crump M, Neelapu SS, Farooq U, Van den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large b-cell lymphoma: results from the international SCHOLAR. Blood (2017) 130(16):1800–8.
6. Chen Q, Lu L, Ma W. Efficacy, safety, and challenges of CAR T-cells in the treatment of solid tumors. Cancers (2022) 14(23):5983. doi: 10.3390/cancers14235983
7. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Mueller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol (2021) 32(1):34–48. doi: 10.1016/j.annonc.2020.10.478
8. Hernani R, Benzaquen A, Solano C. Toxicities following CAR-T therapy for hematological malignancies. Cancer Treat Rev (2022) 111:102479. doi: 10.1016/j.ctrv.2022.102479
9. Sharma N, Reagan PM, Liesveld JL. Cytopenia after CAR-T cell therapy-a brief review of a complex problem. Cancers (2022) 14(6):1501. doi: 10.3390/cancers14061501
10. Si X, Gu T, Liu L, Huang Y, Han Y, Qian P, et al. Hematologic cytopenia post CAR T cell therapy: etiology, potential mechanisms and perspective. Cancer Lett (2022) 550:215920. doi: 10.1016/j.canlet.2022.215920
11. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol Blood Marrow Transplantation (2020) 26(1):26–33. doi: 10.1016/j.bbmt.2019.08.003
12. Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplantation (2019) 54(10):1643–50. doi: 10.1038/s41409-019-0487-3
13. Brudno JN, Natrakul D, Lam N, Dulau-Florea A, Yuan CM, Kochenderfer JN. Acute and delayed cytopenias following CAR T-cell therapy: an investigation of risk factors and mechanisms. Leukemia Lymphoma (2022) 63(8):1849–60. doi: 10.1080/10428194.2022.2056172
14. Wang L, Hong R, Zhou L, Ni F, Zhang M, Zhao H, et al. New-onset severe cytopenia after CAR-T cell therapy: analysis of 76 patients with relapsed or refractory acute lymphoblastic leukemia. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.702644
15. Rejeski K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, et al. CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large b-cell lymphoma. Blood (2021) 138(24):2499–513. doi: 10.1182/blood.2020010543
16. Nagle SJ, Murphree C, Raess PW, Schachter L, Chen A, Hayes-Lattin B, et al. Prolonged hematologic toxicity following treatment with chimeric antigen receptor T cells in patients with hematologic malignancies. Am J Hematology (2021) 96(4):455–61. doi: 10.1002/ajh.26113
17. Strati P, Varma A, Adkins S, Nastoupil LJ, Westin JR, Hagemeister FB, et al. Hematopoietic recovery and immune reconstitution after axicabtagene ciloleucel in patients with large b-cell lymphoma. Haematologica (2021) 106(10):2667–72. doi: 10.3324/haematol.2020.254045
18. Rejeski K, Perez A, Iacoboni G, Penack O, Bucklein V, Jentzsch L, et al. The CAR-HEMATOTOX risk-stratifies patients for severe infections and disease progression after CD19 CAR-T in R/R LBCL. J Immunotherapy Cancer (2022) 10(5):e004475. doi: 10.1136/jitc-2021-004475
19. Overbeeke C, Tak T, Koenderman L. The journey of neutropoiesis: how complex landscapes in bone marrow guide continuous neutrophil lineage determination. Blood (2022) 139(15):2285–93. doi: 10.1182/blood.2021012835
20. Gagelmann N, Wulf GG, Duell J, Glass B, Heteren PV, von Tresckow B, et al. Hematopoietic stem cell boost for persistent neutropenia after CAR-T cell therapy: a GLA/DRST study. Blood advances (2022) 7(4):555–9. doi: 10.1182/bloodadvances.2022008042
21. Rejeski K, Burchert A, Iacoboni G, Sesques P, Fransecky L, Buecklein V, et al. Safety and feasibility of stem cell boost as a salvage therapy for severe hematotoxicity after CD19 CAR T-cell therapy. Blood Advances (2022) 6(16):4719–25. doi: 10.1182/bloodadvances.2022007776
22. Rejeski K, Kunz WG, Rudelius M, Buecklein V, Blumenberg V, Schmidt C, et al. Severe candida glabrata pancolitis and fatal aspergillus fumigatus pulmonary infection in the setting of bone marrow aplasia after CD19-directed CAR T-cell therapy - a case report. BMC Infect Dis (2021) 21(1):121. doi: 10.1186/s12879-020-05755-4
23. Pradhan K, Lombardo A, Peeke SZ, Shah N, Pradhan K, Lombardo A, et al. Patterns of leukocyte recovery predict infectious complications after CD19 CAR-T cell therapy in a real-world setting. Stem Cell Invest (2021) 8:18–. doi: 10.21037/sci-2021-008
24. Xing L, Wang Y, Liu H, Gao S, Shao Q, Yue L, et al. Case report: sirolimus alleviates persistent cytopenia after CD19 CAR-T-Cell therapy. Front Oncol (2021) 11. doi: 10.3389/fonc.2021.798352
25. Jain T, Knezevic A, Pennisi M, Chen Y, Ruiz JD, Purdon TJ, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Advances (2020) 4(15):3776–87. doi: 10.1182/bloodadvances.2020002509
26. Juluri KR, Wu QV, Voutsinas J, Hou J, Hirayama AV, Mullane E, et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Advances (2022) 6(7):2055–68. doi: 10.1182/bloodadvances.2020004142
27. Schaefer A, Huang Y, Kittai A, Maakaron JE, Saygin C, Brammer J, et al. Cytopenias after CD19 chimeric antigen receptor T-cells (CAR-T) therapy for diffuse Large b-cell lymphomas or transformed follicular lymphoma: a single institution experience. Cancer Manage Res (2021) 13:8901–6. doi: 10.2147/CMAR.S321202
28. Faram RG, Davila ML. CAR T-cell hematotoxicity is inflammation the key? Blood (2021) 138(24):2447–8. doi: 10.1182/blood.2021012876
29. Miller KC, Johnson PC, Abramson JS, Soumerai JD, Yee AJ, Branagan AR, et al. Effect of granulocyte colony-stimulating factor on toxicities after CAR T cell therapy for lymphoma and myeloma. Blood Cancer J (2022) 12(10):146. doi: 10.1038/s41408-022-00741-2
30. Baur R, Jitschin R, Kharboutli S, Stoll A, Voelkl S, Buettner-Herold M, et al. Thrombopoietin receptor agonists for acquired thrombocytopenia following anti-CD19 CAR-t-cell therapy: a case report. J Immunotherapy Cancer (2021) 9(7):e002721. doi: 10.1136/jitc-2021-002721
31. Drillet G, Lhomme F, De Guibert S, Manson G, Houot R. Prolonged thrombocytopenia after CAR T-cell therapy: the role of thrombopoietin receptor agonists. Blood Adv (2022) 7(4):537–40. doi: 10.1182/bloodadvances.2022008066
32. Beyar-Katz O, Perry C, On YB, Amit O, Gutwein O, Wolach O, et al. Thrombopoietin receptor agonist for treating bone marrow aplasia following anti-CD19 CAR-T cells-single-center experience. Ann Hematology (2022) 101(8):1769–76. doi: 10.1007/s00277-022-04889-6
33. Morales-Mantilla DE, Kain B, Le D, Flores AR, Paust S, King KY. Hematopoietic stem and progenitor cells improve survival from sepsis by boosting immunomodulatory cells. Elife (2022) 11:e74561. doi: 10.7554/eLife.7456
34. Teipel R, Kroschinsky F, Kramer M, Kretschmann T, Egger-Heidrich K, Kruger T, et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Advances (2022) 6(6):1941–6. doi: 10.1182/bloodadvances.2021005747
35. Rejeski K, Wu Z, Blumenberg V, Kunz WG, Muller S, Kajigaya S, et al. Oligoclonal T-cell expansion in a patient with bone marrow failure after CD19 CAR-T therapy for Richter-transformed DLBCL. Blood (2022) 140(20):2175–9. doi: 10.1182/blood.2022017015
36. Sommerkamp P, Altamura S, Renders S, Narr A, Ladel L, Zeisberger P, et al. Differential alternative polyadenylation landscapes mediate hematopoietic stem cell activation and regulate glutamine metabolism. Cell Stem Cell (2020) 26(5):722–+. doi: 10.1016/j.stem.2020.03.003
37. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood (2017) 130(15):1693–8. doi: 10.1182/blood-2017-06-780882
38. Yamada T, Park CS, Lacorazza HD. Genetic control of quiescence in hematopoietic stem cells. Cell Cycle (2013) 12(15):2376–83. doi: 10.4161/cc.25416
39. Mann Z, Sengar M, Verma YK, Rajalingam R, Raghav PK. Hematopoietic stem cell factors: their functional role in self-renewal and clinical aspects. Front Cell Dev Biol (2022) 10. doi: 10.3389/fcell.2022.664261
40. Post Y, Clevers H. Defining adult stem cell function at its simplest: the ability to replace lost cells through mitosis. Cell Stem Cell (2019) 25(2):174–83. doi: 10.1016/j.stem.2019.07.002
41. Bogeska R, Mikecin A-M, Kaschutnig P, Fawaz M, Buechler-Schaeff M, Le D, et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell (2022) 29(8):1273–+. doi: 10.1016/j.stem.2022.06.012
42. Demel UM, Lutz R, Sujer S, Demerdash Y, Sood S, Gruenschlaeger F, et al. A complex proinflammatory cascade mediates the activation of HSCs upon LPS exposure in vivo. Blood Adv (2022) 6(11):3513–28. doi: 10.1182/bloodadvances.2021006088
43. Hirche C, Frenz T, Haas SF, Doering M, Borst K, Tegtmeyer P-K, et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep (2017) 19(11):2345–56. doi: 10.1016/j.celrep.2017.05.063
44. Florez MA, Matatall KA, Jeong Y, Ortinau L, Shafer PW, Lynch AM, et al. Interferon gamma mediates hematopoietic stem cell activation and niche relocalization through BST2. Cell Rep (2020) 33(12):108530. doi: 10.1016/j.celrep.2020.108530
45. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol (2017) 38(5):345–57. doi: 10.1016/j.it.2017.01.004
46. Nakagawa MM, Chen H, Rathinam CV. Constitutive activation of NF-kappa b pathway in hematopoietic stem cells causes loss of quiescence and deregulated transcription factor networks. Front Cell Dev Biol (2018) 6. doi: 10.3389/fcell.2018.00143
47. Wang J, Erlacher M, Fernandez-Orth J. The role of inflammation in hematopoiesis and bone marrow failure: what can we learn from mouse models? Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.951937
48. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465(7299):793–U9. doi: 10.1038/nature09135
49. Takizawa H, Manz MG. Impact of inflammation on early hematopoiesis and the microenvironment. Int J Hematology (2017) 106(1):27–33. doi: 10.1007/s12185-017-2266-5
50. Yan Y, Dong L, Chen C, Bunting KD, Li Q, Stieglitz E, et al. JMML tumor cells disrupt normal hematopoietic stem cells by imposing inflammatory stress through overproduction of IL-1 beta. Blood Advances (2022) 6(1):200–6. doi: 10.1182/bloodadvances.2021005089
51. Fang J, Muto T, Kleppe M, Bolanos LC, Hueneman KM, Walker CS, et al. TRAF6 mediates basal activation of NF-kappa b necessary for hematopoietic stem cell homeostasis. Cell Rep (2018) 22(5):1250–62. doi: 10.1016/j.celrep.2018.01.013
52. Nguyen TH, Abidin BM, Heinonen KM. Frizzled-6 promotes hematopoietic stem/progenitor cell mobilization and survival during LPS-induced emergency myelopoiesis. Stem Cell Rep (2022) 17(10):2303–17. doi: 10.1016/j.stemcr.2022.08.004
53. Chen Z, Ju Z. Inflamm-aging of hematopoietic stem cells. Blood Sci (Baltimore Md) (2019) 1(2):141–3. doi: 10.1097/BS9.0000000000000029
54. Caiado F, Pietras EM, Manz MG. Inflammation as a regulator of hematopoietic stem cell function in disease, and clonal selection. J Exp Med (2021) 218(7):e20201541. doi: 10.1084/jem.20201541
55. Asada S, Kitamura T. Clonal hematopoiesis and associated diseases: a review of recent findings. Cancer Science (2021) 112(10):3962–71. doi: 10.1111/cas.15094
56. Zhang X, Karatepe K, Chiewchengchol D, Zhu H, Guo R, Liu P, et al. Bacteria-induced acute inflammation does not reduce the long-term reconstitution capacity of bone marrow hematopoietic stem cells. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00626
57. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24(6):739–+. doi: 10.1038/s41591-018-0036-4
58. Chen Z, Guo Q, Song G, Hou Y. Molecular regulation of hematopoietic stem cell quiescence. Cell Mol Life Sci (2022) 79(4):218. doi: 10.1007/s00018-022-04200-w
59. Budgude P, Vaidya A, Kale V. Cell-intrinsic factors governing quiescence vis-a-vis activation of adult hematopoietic stem cells. Mol Cell Biochem (2022). doi: 10.1007/s11010-022-04594-y
60. Geiger SS, Essers MAG. Inflammation's epigenetic footprint in hematopoietic stem cells. Cell Stem Cell (2020) 26(5):611–2. doi: 10.1016/j.stem.2020.04.015
61. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150(high) bone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell (2018) 22(3):445–+. doi: 10.1016/j.stem.2018.01.017
62. Ito K, Bonora M, Ito K. Metabolism as master of hematopoietic stem cell fate. Int J Hematology (2019) 109(1):18–27. doi: 10.1007/s12185-018-2534-z
63. Javier J, Hinge A, Bartram J, Xu J, Filippi M-D. Transforming growth factor-beta signaling modifies the hematopoietic acute inflammatory response to drive bone marrow failure. Haematologica (2022) 107(6):1323–34. doi: 10.3324/haematol.2020.273292
64. Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211(2):245–62. doi: 10.1084/jem.20131043
65. Chen J, Feng X, Desierto MJ, Keyvanfar K, Young NS. IFN-gamma-mediated hematopoietic cell destruction in murine models of immune-mediated bone marrow failure. Blood (2015) 126(24):2621–31. doi: 10.1182/blood-2015-06-652453
66. Termini CM, Chute JP. Hematopoietic stem cell stress and regeneration. Curr Stem Cell Rep (2020) 6(4):134–43. doi: 10.1007/s40778-020-00181-3
67. Yamashita M, Passegue E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell (2019) 25(3):357–+. doi: 10.1016/j.stem.2019.05.019
68. Wagner PN, Shi Q, Salisbury-Ruf CT, Zou J, Savona MR, Fedoriw Y, et al. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood (2019) 133(2):107–20. doi: 10.1182/blood-2018-05-847335
69. Xiao Y, Li H, Zhang J, Volk A, Zhang S, Wei W, et al. TNF-alpha/Fas-RIP-1-induced cell death signaling separates murine hematopoietic stem cells/progenitors into 2 distinct populations. Blood (2011) 118(23):6057–67. doi: 10.1182/blood-2011-06-359448
70. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell (2008) 135(7):1311–23. doi: 10.1016/j.cell.2008.10.044
71. Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, et al. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci United States America (2014) 111(40):14436–41. doi: 10.1073/pnas.1409389111
72. Croker BA, Silke J, Gerlic M. Fight or flight: regulation of emergency hematopoiesis by pyroptosis and necroptosis. Curr Opin Hematology (2015) 22(4):293–301. doi: 10.1097/MOH.0000000000000148
73. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O'Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity (2012) 37(6):1009–23. doi: 10.1016/j.immuni.2012.08.027
74. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
75. Deng T, Tang C, Zhang G, Wan X. DAMPs released by pyroptotic cells as major contributors and therapeutic targets for CAR-t-related toxicities. Cell Death Disease (2021) 12(1):129. doi: 10.1038/s41419-021-03428-x
76. Wang HC, Bloom O, Zhang MH, Vishnubhakat JM, Ombrellino M, Che JT, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi: 10.1126/science.285.5425.248
77. Croker BA, O'Donnell JA, Gerlic M. Pyroptotic death storms and cytopenia. Curr Opin Immunol (2014) 26:128–37. doi: 10.1016/j.coi.2013.12.002
78. Andersson U, Yang H, Harris H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets (2018) 22(3):263–77. doi: 10.1080/14728222.2018.1439924
79. Yang H, Ochani M, Li JH, Qiang XL, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci United States America (2004) 101(1):296–301. doi: 10.1073/pnas.2434651100
80. Velegraki M, Koutala H, Tsatsanis C, Papadaki HA. Increased levels of the high mobility group box 1 protein sustain the inflammatory bone marrow microenvironment in patients with chronic idiopathic neutropenia via activation of toll-like receptor 4. J Clin Immunol (2012) 32(2):312–22. doi: 10.1007/s10875-011-9620-9
81. Wang H, Yu T, An N, Sun Y, Xu P, Han P, et al. Enhancing regulatory T cell function via inhibition of high mobility group box 1 protein signaling in immune thrombocytopenia. Haematologica (2022) 108(3):843–58. doi: 10.3324/haematol.2022.281557
82. Zhang G, Yang P, Liu X, Liu H, Wang J, Wang J, et al. HMGB1 is increased in patients with immune thrombocytopenia and negatively associates with tregs. Thromb Res (2022) 213:128–36. doi: 10.1016/j.thromres.2022.02.021
83. Valdes-Ferrer SI, Papoin J, Dancho ME, Olofsson PS, Li J, Lipton JM, et al. HMGB1 mediates anemia of inflammation in murine sepsis survivors. Mol Med (2015) 21:951–8. doi: 10.2119/molmed.2015.00243
84. Dulmovits BM, Tang Y, Papoin J, He M, Li J, Yang H, et al. HMGB1-mediated restriction of EPO signaling contributes to anemia of inflammation. Blood (2022) 139(21):3181–93. doi: 10.1182/blood.2021012048
85. Gopal A, Ibrahim R, Fuller M, Umlandt P, Parker J, Tran J, et al. TIRAP drives myelosuppression through an ifn gamma-Hmgb1 axis that disrupts the endothelial niche in mice. J Exp Med (2022) 219(3):e20200731. doi: 10.1084/jem.20200731
86. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin e-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
87. Zhu C, Lian Y, Wang C, Wu P, Li X, Gao Y, et al. Single-cell transcriptomics dissects hematopoietic cell destruction and T-cell engagement in aplastic anemia. Blood (2021) 138(1):23–33. doi: 10.1182/blood.2020008966
88. Dulmovits BM, Olson TS. Does immune destruction drive all forms of bone marrow failure? J Clin Invest (2022) 132(15):e161288. doi: 10.1172/JCI161288
89. Fidyk W, Mitrus I, Ciomber A, Smagur A, Chwieduk A, Glowala-Kosinska M, et al. Evaluation of proinflammatory and immunosuppressive cytokines in blood and bone marrow of healthy hematopoietic stem cell donors. Cytokine (2018) 102:181–6. doi: 10.1016/j.cyto.2017.09.001
90. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of T-reg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474(7350):216–U56. doi: 10.1038/nature10160
91. Thapa A, Adamiak M, Bujko K, Ratajczak J, Abdel-Latif AK, Kucia M, et al. Danger-associated molecular pattern molecules take unexpectedly a central stage in Nlrp3 inflammasome-caspase-1-mediated trafficking of hematopoietic stem/progenitor cells. Leukemia (2021) 35(9):2658–71. doi: 10.1038/s41375-021-01158-9
92. Jahandideh B, Derakhshani M, Abbaszadeh H, Movassaghpour AA, Mehdizadeh A, Talebi M, et al. The pro-inflammatory cytokines effects on mobilization, self-renewal and differentiation of hematopoietic stem cells. Hum Immunol (2020) 81(5):206–17. doi: 10.1016/j.humimm.2020.01.004
93. Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematology (2008) 15(4):307–11. doi: 10.1097/MOH.0b013e3283007db5
94. Kang Y-J, Yang S-J, Park G, Cho B, Min C-K, Kim T-Y, et al. A novel function of interleukin-10 promoting self-renewal of hematopoietic stem cells. Stem Cells (2007) 25(7):1814–22. doi: 10.1634/stemcells.2007-0002
95. Matatall KA, Shen C-C, Challen GA, King KY. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells (2014) 32(11):3023–30. doi: 10.1002/stem.1799
96. Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, et al. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep (2016) 17(10):2584–95. doi: 10.1016/j.celrep.2016.11.031
97. Herd OJ, Rani GF, Hewitson JP, Hogg K, Stone AP, Cooper N, et al. Bone marrow remodeling supports hematopoiesis in response to immune thrombocytopenia progression in mice. Blood Advances (2021) 5(23):4877–89. doi: 10.1182/bloodadvances.2020003887
98. Moll NM, Ransohoff RM. CXCL12 and CXCR4 in bone marrow physiology. Expert Rev Hematology (2010) 3(3):315–22. doi: 10.1586/ehm.10.16
99. Kale VP. Differential activation of MAPK signaling pathways by TGF-beta 1 forms the molecular mechanism behind its dose-dependent bidirectional effects on hematopoiesis. Stem Cells Dev (2004) 13(1):27–38. doi: 10.1089/154732804773099236
100. Kale VP, Vaidya AA. Molecular mechanisms behind the dose-dependent differential activation of MAPK pathways induced by transforming growth factor-beta 1 in hematopoietic cells. Stem Cells Dev (2004) 13(5):536–47. doi: 10.1089/scd.2004.13.536
101. Hinge A, Xu J, Javier J, Mose E, Kumar S, Kapur R, et al. p190-b RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat Commun (2017) 8:14382. doi: 10.1038/ncomms14382
102. Jiang L, Han X, Wang J, Wang C, Sun X, Xie J, et al. SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-beta signaling. J Exp Med (2018) 215(5):1337–47. doi: 10.1084/jem.20171477
103. Capron C, Lacout C, Lecluse Y, Jalbert V, Chagraoui H, Charrier S, et al. A major role of TGF-beta 1 in the homing capacities of murine hematopoietic stem cell/progenitors. Blood (2010) 116(8):1244–53. doi: 10.1182/blood-2009-05-221093
104. Montazersaheb S, Ehsani A, Fathi E, Farahzadi R. Cellular and molecular mechanisms involved in hematopoietic stem cell aging as a clinical prospect. Oxid Med Cell Longevity (2022) 2022:2713483. doi: 10.1155/2022/2713483
105. Gerosa RC, Boettcher S, Kovtonyuk LV, Hausmann A, Hardt W-D, Hidalgo J, et al. CXCL12-abundant reticular cells are the major source of IL-6 upon LPS stimulation and thereby regulate hematopoiesis. Blood Advances (2021) 5(23):5002–15. doi: 10.1182/bloodadvances.2021005531
106. Zhao JL, Ma C, O'Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell (2014) 14(4):445–59. doi: 10.1016/j.stem.2014.01.007
107. Hormaechea-Agulla D, Le DT, King KY. Common sources of inflammation and their impact on hematopoietic stem cell biology. Curr Stem Cell Rep (2020) 6(3):96–107. doi: 10.1007/s40778-020-00177-z
108. Mirantes C, Pctssegue E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res (2014) 329(2):248–54. doi: 10.1016/j.yexcr.2014.08.017
109. Chu T, Hu S, Qi J, Li X, Zhang X, Tang Y, et al. Bifunctional effect of the inflammatory cytokine tumor necrosis factor alpha on megakaryopoiesis and platelet production. J Thromb Haemostasis (2022) 20(12):2998–3010. doi: 10.1111/jth.15891
110. Chavez JS, Rabe JL, Hernandez G, Mills TS, Nino KE, Davizon-Castillo P, et al. PU.1 expression defines distinct functional activities in the phenotypic HSC compartment of a murine inflammatory stress model. Cells (2022) 11(4):680. doi: 10.3390/cells11040680
111. Chavez JS, Rabe JL, Loeffler D, Higa KC, Hernandez G, Mills TS, et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J Exp Med (2021) 218(6):e20201169. doi: 10.1084/jem.20201169
112. Wei Z, Li C, Zhang Y, Lin C, Zhang Y, Shu L, et al. Macrophage-derived IL-1 beta regulates emergency myelopoiesis via the NF-kappa b and c/ebp beta in zebrafish. J Immunol (2020) 205(10):2694–706. doi: 10.4049/jimmunol.2000473
113. Krause DS. IFN-gamma binds TPO to inhibit hematopoiesis. Blood (2019) 133(19):2004–5. doi: 10.1182/blood-2019-03-900977
114. Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-gamma. Blood (2019) 133(19):2043–55. doi: 10.1182/blood-2018-11-884486
115. Bodine DM. Anemia of inflammation is all the RAGE comment. Blood (2022) 139(21):3106–7. doi: 10.1182/blood.2021015337
116. Kovtonyuk LV, Caiado F, Garcia-Martin S, Manz E-M, Helbling P, Takizawa H, et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood (2022) 139(1):44–58. doi: 10.1182/blood.2021011570
117. He H, Xu P, Zhang X, Liao M, Dong Q, Cong T, et al. Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood (2020) 136(2):183–98. doi: 10.1182/blood.2019003910
118. Grants JM, Wegrzyn J, Hui T, O'Neill K, Shadbolt M, Knapp DJHF, et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood (2020) 135(25):2235–51. doi: 10.1182/blood.2019003105
119. Jose SS, Bendickova K, Kepak T, Krenova Z, Fric J. Chronic inflammation in immune aging: role of pattern recognition receptor crosstalk with the telomere complex? Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.01078
120. Zeng X, Li X, Shao M, Xu Y, Shan W, Wei C, et al. Integrated single-cell bioinformatics analysis reveals intrinsic and extrinsic biological characteristics of hematopoietic stem cell aging. Front Genet (2021) 12. doi: 10.3389/fgene.2021.745786
121. Burns SS, Kapur R. Turning the clock forward: inflammation accelerates the aging of hematopoietic stem cells. Cell Stem Cell (2022) 29(8):1156–8. doi: 10.1016/j.stem.2022.07.002
122. Sudo T, Motomura Y, Okuzaki D, Hasegawa T, Yokota T, Kikuta J, et al. Group 2 innate lymphoid cells support hematopoietic recovery under stress conditions. J Exp Med (2021) 218(5):e20200817. doi: 10.1084/jem.20200817
123. Hambright WS, Philippon MJ, Huard J. Rapamycin for aging stem cells. Aging-Us (2020) 12(15):15184–5. doi: 10.18632/aging.103816
124. Fernandes H, Moura J, Carvalho E. mTOR signaling as a regulator of hematopoietic stem cell fate. Stem Cell Rev Rep (2021) 17(4):1312–22. doi: 10.1007/s12015-021-10131-z
125. Wu F, Chen Z, Liu J, Hou Y. The akt-mTOR network at the interface of hematopoietic stem cell homeostasis. Exp Hematology (2021) 103:15–23. doi: 10.1016/j.exphem.2021.08.009
126. Xiao M, Zhang W, Liu W, Mao L, Yang J, Hu L, et al. Osteocytes regulate neutrophil development through IL-19: a potent cytokine for neutropenia treatment. Blood (2021) 137(25):3533–47. doi: 10.1182/blood.2020007731
127. Ramalingam P, Poulos MG, Lazzari E, Gutkin MC, Lopez D, Kloss CC, et al. Chronic activation of endothelial MAPK disrupts hematopoiesis via NFKB dependent inflammatory stress reversible by SCGF. Nat Commun (2020) 11(1):666. doi: 10.1038/s41467-020-14478-8
128. Li Y, Xue Z, Dong X, Liu Q, Liu Z, Li H, et al. Mitochondrial dysfunction and oxidative stress in bone marrow stromal cells induced by daunorubicin leads to DNA damage in hematopoietic cells. Free Radical Biol Med (2020) 146:211–21. doi: 10.1016/j.freeradbiomed.2019.11.007
129. Wu L, Lin Q, Ma Z, Chowdhury FA, Mazumder MHH, Du W. Mesenchymal PGD(2) activates an ILC2-treg axis to promote proliferation of normal and malignant HSPCs. Leukemia (2020) 34(11):3028–41. doi: 10.1038/s41375-020-0843-8
130. Li S, Yao J-C, Oetjen KA, Krambs JR, Xia J, Zhang J, et al. IL-1beta expression in bone marrow dendritic cells is induced by TLR2 agonists and regulates HSC function. Blood (2022) 140(14):1607–20. doi: 10.1182/blood.2022016084
131. Schajnovitz A, Itkin T, D'Uva G, Kalinkovich A, Golan K, Ludin A, et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat Immunol (2011) 12(5):391–U130. doi: 10.1038/ni.2017
Keywords: cytopenia, hematotoxicity, inflammation, CAR-T, hematopoietic stem cell
Citation: Sun T, Li D, Huang L and Zhu X (2023) Inflammatory abrasion of hematopoietic stem cells: a candidate clue for the post-CAR-T hematotoxicity? Front. Immunol. 14:1141779. doi: 10.3389/fimmu.2023.1141779
Received: 10 January 2023; Accepted: 21 April 2023;
Published: 08 May 2023.
Edited by:
James Kurnick, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Ming Jiang, the First Affiliated Hospital of Xinjiang Medical University, ChinaCopyright © 2023 Sun, Li, Huang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojian Zhu, emh1eGlhb2ppYW5AaHVzdC5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.