 Hengjie Yuan1,2†
Hengjie Yuan1,2† Jianning Zhang
Jianning Zhang Jing-fei Dong
Jing-fei Dong Zilong Zhao
Zilong Zhao- 1Tianjin Institute of Neurology, Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin, China
- 2BloodWorks Research Institute, Seattle, WA, United States
- 3Division of Hematology, Department of Medicine, University of Washington, School of Medicine, Seattle, WA, United States
Transcription factors bind promoter or regulatory sequences of a gene to regulate its rate of transcription. However, they are also detected in anucleated platelets. The transcription factors RUNX1, GATA1, STAT3, NFκB, and PPAR have been widely reported to play key roles in the pathophysiology of platelet hyper-reactivity, thrombosis, and atherosclerosis. These non-transcriptional activities are independent of gene transcription or protein synthesis but their underlying mechanisms of action remain poorly defined. Genetic and acquired defects in these transcription factors are associated with the production of platelet microvesicles that are known to initiate and propagate coagulation and to promote thrombosis. In this review, we summarize recent developments in the study of transcription factors in platelet generation, reactivity, and production of microvesicles, with a focus on non-transcriptional activities of selected transcription factors.
1 Introduction
Transcription factors (TFs) are a group of mediators that bind the promoter or regulatory sequence of a gene to control its rate of transcribing genetic information from DNA to messenger RNA (1). This transcription control is key to ensuring an adequate level of expression of a given protein in targeted cells at a particular developmental stage. It not only directs the processes of proliferation, growth, and death of a cell, but also controls the rate of cell migration and organizational development during embryonic development, as well as regulating cellular response to the extracellular matrices. Thus far, more than 1600 transcription factors have so far been identified (2, 3), and they work in a coordinated fashion to down- as well as up-regulate target genes. The activation of a given gene can be regulated by multiple transcriptional factors and one transcription factor can regulate multiple genes. Such a multivalent activity is possible because of the modular structure of a transcriptional factor, which typically includes a DNA-binding domain, signal-sensing domain that contains binding sites for transcription co-regulators, and an optional transactivation domain, which senses external signals and transmits them to the rest of the transcription complex (4, 5). Because of their roles in regulating gene transcription, the activation and suppression of transcription factors is extensively reported in cancer development (6).
Paradoxically, multiple transcription factors have been reported to express and be active in platelets (Table 1), the anucleated offspring of megakaryocytes with a very limited capacity for protein synthesis (7). An obvious question is whether these transcription factors are merely leftover from parental megakaryocytes or have unique activities in platelets. Reports from studies on platelet transcription factors have been scarce in the literature, but increasing evidence suggests that transcription factors in platelets have unique activities of their own independent of their transcriptional activities (8–10). However, past research on transcription factors in platelets is often limited to reporting their presence and activation status, without further investigation of their activities in regulating platelet functions and, more importantly the underlying mechanism of their regulatory activities.
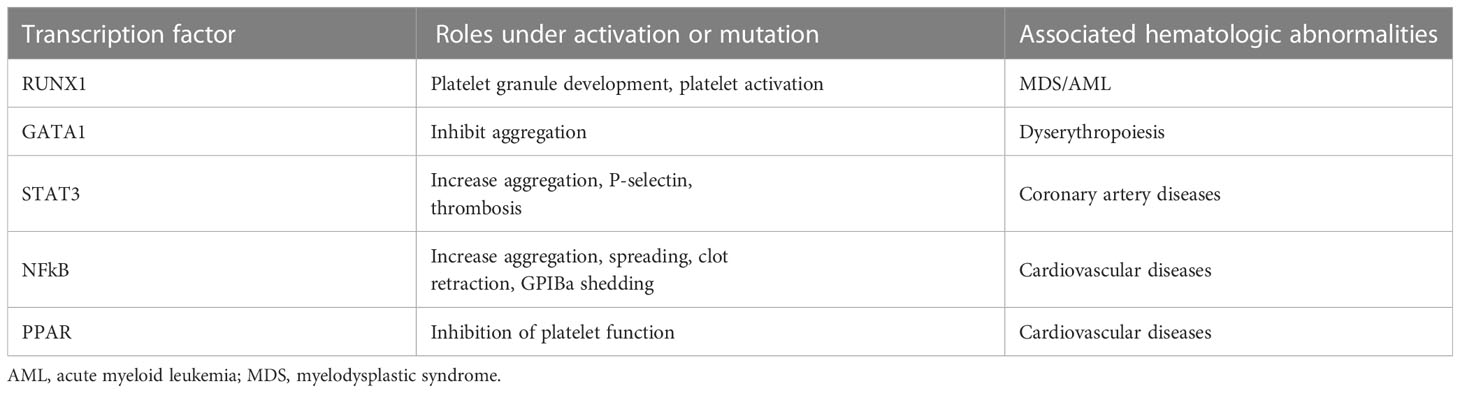
Table 1 Roles of transcription factors in platelets.
Platelets circulate along the vessel wall and act to stop bleeding at sites of vessel injury. This hemostatic process requires multiple ligand-receptor interactions to tether, activate, and aggregate platelets. The tightly controlled platelet activation and aggregation that occurs at the site of vascular injury during hemostasis can become dysregulated in pathological conditions, promoting thrombosis and inflammation. For example, platelets promote arterial thrombosis or thromboembolism when activated either on the surface of a ruptured atherosclerotic plaque or by pathological levels of high fluid shear stress in the area of arterial stenosis, leading to acute thrombotic events such as ischemic stroke and myocardial infarction (11). Emerging evidence further suggests that platelets also act as a cellular mediator in a variety of pathophysiological conditions such as cancer, rheumatoid arthritis, atherosclerosis, trauma, and immune response (12–14). How transcription factors regulate platelet production from megakaryocytes has been extensively reported, but their non-transcriptional activities (i.e., activity independent of gene regulations) have only begun to be recognized. Here, we discuss several transcription factors that have been reported to regulate platelet production and function.
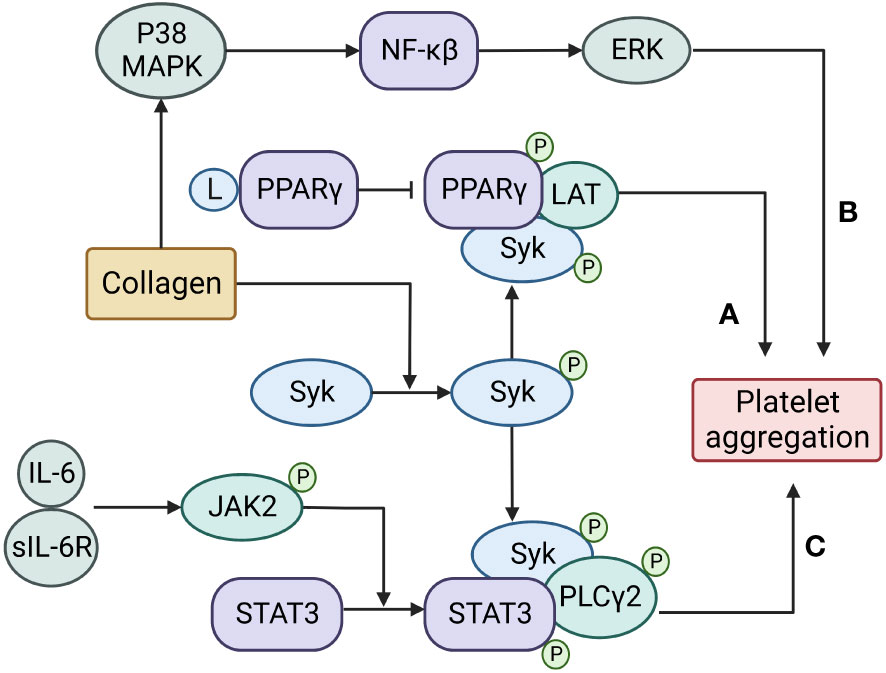
Figure 1 Transcription factors regulate platelet aggregation through non-transcriptional activities. (A) PPARγ is recruited and phosphorylated by Syk to promote the recruitment of LAT and enhance platelet aggregation;(B) NFκB is activated by upstream p38 mitogen-activated protein kinase (MAPK) and promotes platelet aggregation by regulating downstream extracellular signal-regulated kinase (ERK);(C) A complex of IL-6 with its soluble receptor IL-6R activates JAK2 to phosphorylate and dimerize STAT3, then the activated STAT3 serves as a protein scaffold to facilitate the catalytic interaction between the spleen tyrosine kinase (Syk) and its substrate PLCγ2 to promote platelet aggregation.
2 Transcription factors in platelet production
2.1 Runt-related transcription factor 1
In 1969, Weiss, et al. identified a family with an autosomal dominant inherited thrombocytopenia, caused primarily by decreased dense granule contents (15). A heterozygous Y260X mutation in the RUNX1 gene was subsequently shown to be the genetic basis of this inherited platelet defect (15, 16). To date, more than 200 families with RUNX1 variants have been reported (17). RUNX1/AML1 (also known as CBFA2 and PEBP2αB) is a member of the Runt family, which has three known transcription factors (RUNX1, RUNX2, and RUNX3), which share the Runt homology domain near the N-terminus. This domain interacts with CBFb to bind specific sequences of DNA to regulate its transcription (18).
RUNX1 regulates several genes that control platelet production, structure, function, and intracellular signaling. One report found that 22 patients in a family with autosomal dominant thrombocytopenia had mutations in the RUNX1 gene (19) and 6 of them developed hematologic malignancies (20). RUNX1-deficient mice die in uterus due to defective hematopoiesis and resultant severe bleeding (21, 22). Mice with the conditional knockout survive but have an impaired megakaryocyte maturation with a significant reduction in megakaryocyte polyploidization (23). Variations in the RUNX1 gene often result in bleeding diathesis, primarily because of defective platelet granules (15, 16), which reduce platelet activation and aggregation (24). For example, mice carrying the RUNX1 p.Leu43Ser variant (equivalent to human p.Leu56Ser) exhibit a prolonged bleeding time because of defective α-granule secretion and platelet spreading (25).RUNX1 deficiency can result in pallidin dysregulation and deficient dense granules in platelets (26) as well as the Ras-related protein RAB31-mediated early endosomal trafficking of von Willebrand factor (VWF) and epidermal growth factor receptor (EGFR) in megakaryocytes (27). RUNX1 regulates the development of platelet granules through interaction with genes involved in the biogenesis of platelet granules such as the nuclear factor erythroid 2 (NF-E2).
In addition, RUNX1 can also regulate genes related to platelet functions. For example, it regulates the transcription of the non-muscle myosin IIA (MYH9) and IIB (MYH10) genes, which encode non-muscle myosin II heavy chains; RUNX1 mutations are associated with dysregulated expression of MYH10 in platelets (28); and the expression level of non-muscle myosin is used as a marker for changes in transcriptional activity of RUNX1 as well as friend leukemia integration 1 transcription factor (FLI1) (29). RUNX1 also regulates the expression of the arachidonate 12-lipoxygenase gene (ALOX12) (30), which encodes the enzyme that acts on polyunsaturated fatty acid substrates to generate bioactive lipid mediators to regulate platelet function (30). PCTP (phosphatidylcholine transfer protein) regulates the intermembrane transfer of phosphatidylcholine and its upregulation by RUNX1 sensitizes platelet response to thrombin through protease-activated receptor 4 (31). RUNX1 also regulates the expression of platelet factor 4 through coordination with transcription factors in the ETS family that share a conserved winged helix-turn-helix DNA binding domain that recognizes unique DNA sequences containing GGAA/T (32). Platelet factor 4 belongs to the CXC chemokine family and is released from α-granules of activated platelets to promote coagulation and to participate in heparin-induced thrombocytopenia (33, 34). A recent report shows that RUNX-1 haploinsufficiency inhibits the differentiation of hematopoietic progenitor cells (HPCs) into megakaryocytes (35).
2.2 GATA-binding protein 1
GATA-binding protein 1 (GATA1) is a transcription factor that contains two zinc finger domains: a C-terminal zinc finger that binds the (T/A) GATA(A/G) motif of DNA and an N-terminal zinc finger that is required for stabilizing the C-terminal structure and also interacts with a nuclear co-factor protein called friend for GATA1 (FOG1), which stabilizes GATA1 binding (36, 37). GATA plays a pivotal role in hematopoietic development and is found in megakaryocytes (38). GATA1-deficient mice die before birth at approximately embryonic day 10, primarily because of severe anemia (39). However, mutations in the N-terminal zinc finger domain, which reduces the transcriptional activation of GATA1 (36, 40), are found in patients with myeloproliferative disorders and acute megakaryoblastic leukemia (41), suggesting that GATA1-FOG1 interaction is essential for the development and maturation of megakaryocytes, the parental cells of platelets. Decreased GATA-1 expression has also been reported in patients with myelodysplastic syndrome (42).
Embryonic stem cells from GATA1-deficient mice are smaller and show low expression of megakaryocytic markers, but have a high rate of proliferation (43). Complementation of these cells with a wild-type GATA1 gene allows megakaryocytes and erythrocytes to develop in response to a variety of cytokines. Additionally, cell division is attenuated in the megakaryocytic progenitor G1ME cells that overexpress GATA1. A recent report further shows that impaired MYH10 silencing causes GATA1-related polyploidization defect during megakaryocyte differentiation (44).
Furthermore, platelet aggregation induced by collagen is inhibited in GATA1-deficient mice (45), primarily due to reduced expression of the collagen receptor GPVI. Platelet adhesion and aggregation induced by shear stress are also reduced in GATA1-deficient mice (45). How a GATA1 deficiency causes these changes in platelet reactivity remains unknown, but these phenotypic changes in the mice provide the first indication that transcription factors could perform non-transcriptional activities in anucleated platelets.
3 Non-transcriptional activity in platelets
3.1 Signal transducer and activator of transcription 3
STAT includes a family of transcription factors critical for inflammatory and acute-phase reactions (46, 47). They also play vital roles in cancer development and hematopoiesis (48). The homologous STAT1, STAT3, and STAT5 are expressed in human platelets and are reported to regulate platelet reactivity through residual or mitochondrial transcriptional activity in platelets. For example, STAT3 affects mitochondrial transcription by binding to the regulatory D-loop region of mitochondrial DNA upon platelet activation (49).
However, STAT3 can also be activated (phosphorylated) and dimerized in platelets stimulated with thrombopoietin (49, 50), suggesting that STAT3 can also regulate platelet reactivity through non-transcriptional means. We have shown that STAT3 is activated and dimerized in collagen-stimulated platelets to serve as a protein scaffold that facilitates the catalytic interaction between spleen tyrosine kinase (Syk) and its substrate, PLCγ to enhance collagen-induced calcium mobilization and platelet activation (8). More importantly, STAT3 is activated to form dimers by a complex of IL-6 with its soluble receptor IL-6Rα, which activates JAK2 (51). The pharmacological inhibition of platelet STAT3 reduces collagen-induced platelet aggregation and thrombus formation on the collagen matrix (8, 52). Platelets from STAT3-deficient mice or mice infused with a STAT3 inhibitor have reduced collagen-induced aggregation. This non-transcriptional activity of STAT3 may be critical for the development of platelet hyper-reactivity, which has been widely associated with inflammation, especially that related to the activity of the proinflammatory cytokine IL-6 (8). We have also shown that the piper longum derivative piperlongumine (PL) blocks collagen-induced platelet reactivity in a dose-dependent manner by targeting STAT3 (53). Consistent with our observations, the small molecular STAT3 inhibitor SC99 has been shown to reduce platelet activation and aggregation induced by collagen and thrombin (54). These findings offer a new pathway for reducing platelet hyper-reactivity in conditions of inflammation and in prothrombotic states associated with trauma, cancer, autoimmune diseases, and severe infection.
3.2 Nuclear factor kappa β
Nuclear factor kappa β (NFκB) is a well-defined redox-sensitive transcription factor that regulates the immune response and inflammation by controlling the expression of multiple genes activated by inflammatory mediators (55–57). Blocking NFκB can therefore improve outcomes of inflammatory diseases (58). NFκB is composed of p50 and p65 subunits, normally as an inactive cytoplasmic complex. The inhibitory proteins of the IκB family tightly bind the subunits of NFκB (59). Upon activation, the IκK complex phosphorylates IκBα, thus activating NFκB by detaching it from IkBα (60–62). Three IκK family members, α, β, and γ, are expressed in platelets, with β being the most abundant, and are reported to regulate platelet reactivity through non-transcriptional activity (9, 10, 63). For example, the pharmacological inhibition of IκKβ leads to reduced agonist-induced platelet activation, increased bleeding time, and prolonged thrombus formation in a mouse model (64). NF-κB has also been reported to be partially involved in the regulation of SERCA activity to regulate calcium homeostasis in platelets (65). IκKβ-deficient platelets lose the ability to shed the ectodomain of GP Ibα in response to ADP or collagen stimulations (66) but preserve thrombin-induced GP Ibα shedding (67). Collagen-induced p65 and IκKβ phosphorylation is blocked by inhibition of MAP kinase, but not by inhibition of ERK in platelets (68). The thrombin-induced GP Ibα shedding requires p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) as its upstream and downstream molecules (68, 69).
3.3 Peroxisome proliferator-activated receptors
The peroxisome proliferator-activated receptors (PPARs) are ligand-activated receptors in the nuclear hormone receptor family. They contain three subtypes (PPARα, PPARβ/δ, and PPARγ), which are essential in the regulation of cell differentiation, development, and metabolism (70–72). All PPARs heterodimerize with retinoid X receptor (RXR) and subsequently bind to a specific region of target genes called a peroxisome proliferator response element (PPRE) (73). PPARγ plays a transcription factor role in regulating platelet production from megakaryocytes, but the PPARγ ligand thiazolidinedione inhibits platelet aggregation induced by ADP under hydrostatic pressure and in diabetic mice (74–76). Similarly, activating PPARβ/δ also reduces platelet reactivity to ADP, thrombin, and collagen (77, 78). However, PPARα is also required for platelet activation and thrombus formation, in which it regulates the dense granule secretion of platelets in hyperlipidemic mice (79). The reason for this apparent contradiction remains to be further investigated. PPARγ is recruited and phosphorylated by Syk to promote the recruitment of the protein called Linker for the Activation of T cells (LAT), which is necessary for collagen-induced platelet activation through glycoprotein VI (80).
While transcription factors are critically involved in megakaryocyte development and platelet production, they may also regulate platelet reactivity to conventional and specific platelet agonists (Figure 1). The latter is independent of transcriptional activity, for which it is present but at a residual level. This non-transcriptional activity remains poorly understood and requires further investigation because it helps understanding how platelets are activated either by conventional agonists for hemostasis or as complications found in patients treated with drugs that block transcriptional activity of cells (e.g., cancer treatments). Such research will also play an important role in developing new therapeutics targeting these transcription factors to enhance or reduce platelet reactivity.
4 Transcription factors in extracellular vesicles released from platelets
Extracellular vesicles (EVs) are shed membrane fragments, intracellular organelles, and nuclear components from cells undergoing active microvesiculation (81–84) or apoptosis (85–87). The former is triggered by the activation of the cysteine protease calpain, which disrupts the membrane-cytoskeleton association (88–91). Platelets are the primary source of EVs circulating in blood, accounting for approximately 80% of total EVs (92–94). The subcellular size of EVs allows them to travel to areas where parental cells are unable to go. In additional to inherent functions from their parental cells, EVs also perform unique activities of their own because of molecules expressed on their surface and carried by them, the latter of which include transcription factors such as STAT3, STAT5, and PPARγ (95) as well as regulators of transcription factors (96, 97). This EV-derived transcriptional activity has been scarcely reported but hold greats potential for influencing biological activities of target cells. For example, PPARγ in platelet EVs is taken up by monocytic THP-1 cells, where it induces the expression of fatty acid-binding protein-4 (FABP4). Monocytes receiving PPARγ-containing platelet EVs produce less inflammatory mediators and become more adherent through increased fibronectin production (95). Although reports on platelet-derived transcription factors remain very limited, a large body of evidence in the literature shows that platelet-derived EVs, especially EV-carried microRNAs, can change transcriptional activities, thus regulating the function of target cells. Platelet EV-carried NLR family pyrin domain containing 3 (NLRP3) stimulates endothelial cells to undergo pyroptosis through the NLRP3/nuclear factor (NF)-κB pathway (98). EVs from platelets stimulated with bacteria provoke proinflammatory activity of monocytes through the TRAF6/NFκB pathway (99). MicroRNA-142-3p carried by platelet-derived EVs promotes the proliferation of endothelial cells (100), whereas microRNA-126-3p-carrying platelet EVs can be internalized by macrophages to dose-dependently downregulate expression of target mRNA (101). These observations mostly pertain to phenotypic characterization with less information regarding the underlying pathways involved. Systemic studies of EV-carrying transcription factors and related mediators are therefore urgently needed.
5 Conclusion
Platelets lack a nucleus and de novo transcription, but a number of transcription factors are found in platelets and may have non-transcriptional activities that regulate platelet function. Transferring transcription factors between platelets and target cells through platelet EVs could also be a novel regulatory mechanism of cell-cell communications and a potential therapeutic target for a variety of pathologies.
Author contributions
HY and YL performed the literature search and compiled all the information from the researched articles and wrote the manuscript. ZZ, J-FD and JZ formulated, proposed, guided and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by Young Scientists Award 82022020 from the National Natural Science Foundation of China (ZZ), National Natural Science Foundation of China 81971176 (ZZ), 81271361, 81271359 (JZ), 81102447 (HY), National Natural Science Foundation of China State Key Program Grant 81330029, National Natural Science Foundation of China Major International Joint Research Project 81720108015 (JZ), and Postdoctoral Science Foundation of China Grants 2013M541190 (HY).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tippens ND, Vihervaara A, Lis JT. Enhancer transcription: what, where, when, and why? Genes Dev (2018) 32:1–3. doi: 10.1101/gad.311605.118
2. Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol (2004) 14:283–91. doi: 10.1016/j.sbi.2004.05.004
3. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell (2018) 172:650–65. doi: 10.1016/j.cell.2018.01.029
4. Warnmark A, Treuter E, Wright AP, Gustafsson JA. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol Endocrinol (2003) 17:1901–9. doi: 10.1210/me.2002-0384
5. Latchman DS. Transcription factors: an overview. Int J Biochem Cell Biol (1997) 29:1305–12. doi: 10.1016/S1357-2725(97)00085-X
6. Chen KS, Lim JWC, Richards LJ, Bunt J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett (2017) 410:124–38. doi: 10.1016/j.canlet.2017.09.015
7. Ezumi Y, Takayama H, Okuma M. Thrombopoietin, c-mpl ligand, induces tyrosine phosphorylation of Tyk2, JAK2, and STAT3, and enhances agonists-induced aggregation in platelets in vitro. FEBS Lett (1995) 374:48–52. doi: 10.1016/0014-5793(95)01072-M
8. Zhou Z, Gushiken FC, Bolgiano D, Salsbery BJ, Aghakasiri N, Jing N, et al. Signal transducer and activator of transcription 3 (STAT3) regulates collagen-induced platelet aggregation independently of its transcription factor activity. Circulation (2013) 127:476–85. doi: 10.1161/CIRCULATIONAHA.112.132126
9. Liu F, Morris S, Epps J, Carroll R. Demonstration of an activation regulated NF-kappaB/I-kappaBalpha complex in human platelets. Thromb Res (2002) 106:199–203. doi: 10.1016/S0049-3848(02)00130-5
10. Spinelli SL, Casey AE, Pollock SJ, Gertz JM, McMillan DH, Narasipura SD, et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler Thromb Vasc Biol (2010) 30:591–8. doi: 10.1161/ATVBAHA.109.197343
11. Badimon L, Padró T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease, European heart journal. Acute Cardiovasc Care (2012) 1:60–74. doi: 10.1177/2048872612441582
12. Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res (2013) 112:1506–19. doi: 10.1161/CIRCRESAHA.113.300512
13. Garraud O, Hamzeh-Cognasse H, Pozzetto B, Cavaillon JM, Cognasse F. Bench-to-bedside review: Platelets and active immune functions - new clues for immunopathology? Crit Care (2013) 17:236. doi: 10.1186/cc12716
14. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
15. Weiss HJ, Chervenick PA, Zalusky R, Factor A. A familialdefect in platelet function associated with imapired release of adenosine diphosphate. New Engl J Med (1969) 281:1264–70. doi: 10.1056/NEJM196912042812303
16. Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood (2002) 99:1364–72. doi: 10.1182/blood.V99.4.1364
17. Homan CC, King-Smith SL, Lawrence DM, Arts P, Feng J, Andrews J, et al. The RUNX1 database (RUNX1db): establishment of an expert curated RUNX1 registry and genomics database as a public resource for familial platelet disorder with myeloid malignancy. Haematologica (2021) 106:3004–7. doi: 10.3324/haematol.2021.278762
18. Coffman JA. Runx transcription factors and the developmental balance between cell proliferation and differentiation. Cell Biol Int (2003) 27:315–24. doi: 10.1016/S1065-6995(03)00018-0
19. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet (1999) 23:166–75. doi: 10.1038/13793
20. Dowton SB, Beardsley D, Jamison D, Blattner S, Li FP. Studies of a familial platelet disorder. Blood (1985) 65:557–63. doi: 10.1182/blood.V65.3.557.557
21. Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. (1996). Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis, in: Proceedings of the National Academy of Sciences of the United States of America, Vol. 93. pp. 3444–9.
22. Hayashi K, Natsume W, Watanabe T, Abe N, Iwai N, Okada H, et al. Diminution of the AML1 transcription factor function causes differential effects on the fates of CD4 and CD8 single-positive T cells. J Immunol (Baltimore Md. 1950) (2000) 165:6816–24. doi: 10.4049/jimmunol.165.12.6816
23. Ichikawa M, Asai T, Chiba S, Kurokawa M, Ogawa S. Runx1/AML-1 ranks as a master regulator of adult hematopoiesis. Cell Cycle (Georgetown Tex.) (2004) 3:722–4.
24. Stockley J, Morgan NV, Bem D, Lowe GC, Lordkipanidze M, Dawood B, et al. Phenotyping of platelets study, enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood (2013) 122:4090–3. doi: 10.1182/blood-2013-06-506873
25. Marín-Quílez A, García-Tuñón I, Fernández-Infante C, Hernández-Cano L, Palma-Barqueros V, Vuelta E, et al. Characterization of the platelet phenotype caused by a germline RUNX1 variant in a CRISPR/Cas9-generated murine model. Thromb Haemostasis (2021) 121:1193–205. doi: 10.1055/s-0041-1723987
26. Mao GF, Goldfinger LE, Fan DC, Lambert MP, Jalagadugula G, Freishtat R, et al. Dysregulation of PLDN (pallidin) is a mechanism for platelet dense granule deficiency in RUNX1 haplodeficiency. J Thromb Haemost (2017) 15:792–801. doi: 10.1111/jth.13619
27. Jalagadugula G, Mao G, Goldfinger LE, Wurtzel JG, Del Carpio-Cano F, Lambert MP, et al. Defective RAB31-mediated megakaryocytic early endosomal trafficking of VWF, EGFR, and M6PR in RUNX1 deficiency, blood advances. Blood Adv (2022) 6(17):5100–12. doi: 10.1182/bloodadvances.2021006945
28. Bluteau D, Glembotsky AC, Raimbault A, Balayn N, Gilles L, Rameau P, et al. Dysmegakaryopoiesis of FPD/AML pedigrees with constitutional RUNX1 mutations is linked to myosin II deregulated expression. Blood (2012) 120:2708–18. doi: 10.1182/blood-2012-04-422337
29. Antony-Debre I, Bluteau D, Itzykson R, Baccini V, Renneville A, Boehlen F, et al. MYH10 protein expression in platelets as a biomarker of RUNX1 and FLI1 alterations. Blood (2012) 120:2719–22. doi: 10.1182/blood-2012-04-422352
30. Mitsui T, Makino S, Tamiya G, Sato H, Kawakami Y, Takahashi Y, et al. ALOX12 mutation in a family with dominantly inherited bleeding diathesis. J Hum Genet (2021) 66:753–9. doi: 10.1038/s10038-020-00887-6
31. Mao G, Songdej N, Voora D, Goldfinger LE, Del Carpio-Cano FE, Myers RA, et al. Transcription factor RUNX1 regulates platelet PCTP (Phosphatidylcholine transfer protein): Implications for cardiovascular events: Differential effects of RUNX1 variants. Circulation (2017) 136:927–39. doi: 10.1161/CIRCULATIONAHA.116.023711
32. Okada Y, Watanabe M, Nakai T, Kamikawa Y, Shimizu M, Fukuhara Y, et al. RUNX1, but not its familial platelet disorder mutants, synergistically activates PF4 gene expression in combination with ETS family proteins. J Thromb Haemost (2013) 11:1742–50. doi: 10.1111/jth.12355
33. Kowalska MA, Rauova L, Poncz M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res (2010) 125:292–6. doi: 10.1016/j.thromres.2009.11.023
34. Johnston I, Sarkar A, Hayes V, Koma GT, Arepally GM, Chen J, et al. Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood (2020) 135:1270–80. doi: 10.1182/blood.2018881607
35. Estevez B, Borst S, Jarocha D, Sudunagunta V, Gonzalez M, Garifallou J, et al. RUNX-1 haploinsufficiency causes a marked deficiency of megakaryocyte-biased hematopoietic progenitor cells. Blood (2021) 137:2662–75. doi: 10.1182/blood.2020006389
36. Yu C, Niakan KK, Matsushita M, Stamatoyannopoulos G, Orkin SH, Raskind WH. X-Linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood (2002) 100:2040–5. doi: 10.1182/blood-2002-02-0387
37. Freson K, Matthijs G, Thys C, Marien P, Hoylaerts MF, Vermylen J, et al. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum Mol Genet (2002) 11:147–52. doi: 10.1093/hmg/11.2.147
38. Nei Y, Obata-Ninomiya K, Tsutsui H, Ishiwata K, Miyasaka M, Matsumoto K, et al. (2013). GATA-1 regulates the generation and function of basophils, in: Proceedings of the National Academy of Sciences of the United States of America, Vol. 110. pp. 18620–5.
39. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. (1996). Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1, in: Proceedings of the National Academy of Sciences of the United States of America, Vol. 93. pp. 12355–8.
40. Balduini CL, Pecci A, Loffredo G, Izzo P, Noris P, Grosso M, et al. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb Haemostasis (2004) 91:129–40. doi: 10.1160/TH03-05-0290
41. Xu G, Nagano M, Kanezaki R, Toki T, Hayashi Y, Taketani T, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of down syndrome. Blood (2003) 102:2960–8. doi: 10.1182/blood-2003-02-0390
42. Kim H, Lee MK, Kim HR. Difference in megakaryocyte expression of GATA-1, IL-6, and IL-8 associated with maintenance of platelet counts in patients with plasma cell neoplasm with dysmegakaryopoiesis. Exp Hematol (2019) 73:13–17.e12. doi: 10.1016/j.exphem.2019.02.005
43. Stachura DL, Chou ST, Weiss MJ. Early block to erythromegakaryocytic development conferred by loss of transcription factor GATA-1. Blood (2006) 107:87–97. doi: 10.1182/blood-2005-07-2740
44. Saultier P, Cabantous S, Puceat M, Peiretti F, Bigot T, Saut N, et al. GATA1 pathogenic variants disrupt MYH10 silencing during megakaryopoiesis. J Thromb Haemost (2021) 19:2287–301. doi: 10.1111/jth.15412
45. Hughan SC, Senis Y, Best D, Thomas A, Frampton J, Vyas P, et al. Selective impairment of platelet activation to collagen in the absence of GATA1. Blood (2005) 105:4369–76. doi: 10.1182/blood-2004-10-4098
46. Drachman JG, Sabath DF, Fox NE, Kaushansky K. Thrombopoietin signal transduction in purified murine megakaryocytes. Blood (1997) 89:483–92. doi: 10.1182/blood.V89.2.483
47. Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood (2007) 109:353–8. doi: 10.1182/blood-2006-07-033969
48. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi: 10.1038/nrc2734
49. Vassilev AO, Lorenz DR, Tibbles HE, Uckun FM. Role of the leukemia-associated transcription factor STAT3 in platelet physiology. Leuk Lymphoma (2002) 43:1461–7. doi: 10.1080/1042819022386716
50. Majka M, Ratajczak J, Villaire G, Kubiczek K, Marquez LA, Janowska-Wieczorek A, et al. Thrombopoietin, but not cytokines binding to gp130 protein-coupled receptors, activates MAPKp42/44, AKT, and STAT proteins in normal human CD34+ cells, megakaryocytes, and platelets. Exp Hematol (2002) 30:751–60. doi: 10.1016/S0301-472X(02)00810-X
51. Lu WJ, Lin KC, Huang SY, Thomas PA, Wu YH, Wu HC, et al. Role of a janus kinase 2-dependent signaling pathway in platelet activation. Thromb Res (2014) 133:1088–96. doi: 10.1016/j.thromres.2014.03.042
52. Latorre AM, Santos MT, Vallés J, Bonanad S, Moscardó A. Signal transducer and activator of transcription 3 (STAT3) phosphorylation regulates thromboxane A(2) receptor activity in human platelets. Br J Haematology (2020) 188:e39–42. doi: 10.1111/bjh.16309
53. Yuan H, Houck KL, Tian Y, Bharadwaj U, Hull K, Zhou Z, et al. Piperlongumine blocks JAK2-STAT3 to inhibit collagen-induced platelet reactivity independent of reactive oxygen species. PloS One (2015) 10:e0143964. doi: 10.1371/journal.pone.0143964
54. Xu Z, Xu YJ, Hao YN, Ren LJ, Zhang ZB, Xu X, et al. A novel STAT3 inhibitor negatively modulates platelet activation and aggregation. Acta Pharmacol Sin (2017) 38:651–9. doi: 10.1038/aps.2016.155
55. Ghisletti S, Meda C, Maggi A, Vegeto E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol (2005) 25:2957–68. doi: 10.1128/MCB.25.8.2957-2968.2005
56. Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest (2001) 107:255–64. doi: 10.1172/JCI10373
57. Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, et al. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med (2013) 5:1017–34. doi: 10.1002/emmm.201202318
58. Chang CC, Lu WJ, Ong ET, Chiang CW, Lin SC, Huang SY, et al. A novel role of sesamol in inhibiting NF-kappaB-mediated signaling in platelet activation. J BioMed Sci (2011) 18:93. doi: 10.1186/1423-0127-18-93
59. Fuentes E, Rojas A, Palomo I. NF-kappaB signaling pathway as target for antiplatelet activity. Blood Rev (2016) 30:309–15. doi: 10.1016/j.blre.2016.03.002
60. Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol (2008) 8:837–48. doi: 10.1038/nri2423
61. Baeuerle PA, Baltimore D. I Kappa b: a specific inhibitor of the NF-kappa b transcription factor. Science (1988) 242:540–6. doi: 10.1126/science.3140380
62. Baeuerle PA, Baltimore D. A 65-kappaD subunit of active NF-kappaB is required for inhibition of NF-kappaB by I kappaB. Genes Dev (1989) 3:1689–98. doi: 10.1101/gad.3.11.1689
63. Malaver E, Romaniuk MA, D'Atri LP, Pozner RG, Negrotto S, Benzadon R, et al. NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost (2009) 7:1333–43. doi: 10.1111/j.1538-7836.2009.03492.x
64. Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, et al. IkappaB kinase phosphorylation of SNAP-23 controls platelet secretion. Blood (2013) 121:4567–74. doi: 10.1182/blood-2012-11-470468
65. Paul M, Kemparaju K, Girish KS. Inhibition of constitutive NF-kappaB activity induces platelet apoptosis via ER stress. Biochem Biophys Res Commun (2017) 493:1471–7. doi: 10.1016/j.bbrc.2017.10.011
66. Wei S, Wang H, Zhang G, Lu Y, An X, Ren S, et al. Platelet IkappaB kinase-beta deficiency increases mouse arterial neointima formation via delayed glycoprotein ibalpha shedding. Arterioscler Thromb Vasc Biol (2013) 33:241–8. doi: 10.1161/ATVBAHA.112.300781
67. Brill A, Chauhan AK, Canault M, Walsh MT, Bergmeier W, Wagner DD. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc Res (2009) 84:137–44. doi: 10.1093/cvr/cvp176
68. Lu WJ, Lin KH, Hsu MJ, Chou DS, Hsiao G, Sheu JR. Suppression of NF-kappaB signaling by andrographolide with a novel mechanism in human platelets: regulatory roles of the p38 MAPK-hydroxyl radical-ERK2 cascade. Biochem Pharmacol (2012) 84:914–24. doi: 10.1016/j.bcp.2012.06.030
69. Lu WJ, Lee JJ, Chou DS, Jayakumar T, Fong TH, Hsiao G, et al. A novel role of andrographolide, an NF-kappa b inhibitor, on inhibition of platelet activation: the pivotal mechanisms of endothelial nitric oxide synthase/cyclic GMP. J Mol Med (Berl) (2011) 89:1261–73. doi: 10.1007/s00109-011-0800-0
70. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International union of pharmacology. LXI. peroxisome proliferator-activated receptors. Pharmacol Rev (2006) 58:726–41. doi: 10.1124/pr.58.4.5
71. Laudet V, Auwerx J, Gustafsson JA, Wahli W. A unified nomenclature system for the nuclear receptor superfamily. Cell (1999) 97:161–3. doi: 10.1016/S0092-8674(00)80726-6
72. Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell (1992) 68:879–87. doi: 10.1016/0092-8674(92)90031-7
73. Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature (1992) 358:771–4. doi: 10.1038/358771a0
74. Rao F, Yang RQ, Chen XS, Xu JS, Fu HM, Su H, et al. PPARγ ligands decrease hydrostatic pressure-induced platelet aggregation and proinflammatory activity. PloS One (2014) 9:e89654. doi: 10.1371/journal.pone.0089654
75. Bodary PF, Vargas FB, King SA, Jongeward KL, Wickenheiser KJ, Eitzman DT. Pioglitazone protects against thrombosis in a mouse model of obesity and insulin resistance. J Thromb Haemost (2005) 3:2149–53. doi: 10.1111/j.1538-7836.2005.01551.x
76. Sahler J, Woeller C, Spinelli S, Blumberg N, Phipps R. A novel method for overexpression of peroxisome proliferator-activated receptor-γ in megakaryocyte and platelet microparticles achieves transcellular signaling. J Thromb Haemost (2012) 10:2563–72. doi: 10.1111/jth.12017
77. Ali FY, Davidson SJ, Moraes LA, Traves SL, Paul-Clark M, Bishop-Bailey D, et al. Role of nuclear receptor signaling in platelets: antithrombotic effects of PPARbeta. FASEB J (2006) 20:326–8. doi: 10.1096/fj.05-4395fje
78. Ali FY, Hall MG, Desvergne B, Warner TD, Mitchell JA. PPARbeta/delta agonists modulate platelet function via a mechanism involving PPAR receptors and specific association/repression of PKCalpha–brief report. Arterioscler Thromb Vasc Biol (2009) 29:1871–3. doi: 10.1161/ATVBAHA.109.193367
79. Li L, Zhou J, Wang S, Jiang L, Chen X, Zhou Y, et al. Critical role of peroxisome proliferator-activated receptor α in promoting platelet hyperreactivity and thrombosis under hyperlipidemia. Haematologica (2022) 107:1358–73. doi: 10.3324/haematol.2021.279770
80. Moraes LA, Spyridon M, Kaiser WJ, Jones CI, Sage T, Atherton RE, et al. Non-genomic effects of PPARgamma ligands: inhibition of GPVI-stimulated platelet activation. J Thromb Haemost (2010) 8:577–87. doi: 10.1111/j.1538-7836.2009.03732.x
81. Siljander P, Farndale RW, Feijge MA, Comfurius P, Kos S, Bevers EM, et al. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: Involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol (2001) 21:618–27. doi: 10.1161/01.ATV.21.4.618
82. Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood (1997) 90:2615–25. doi: 10.1182/blood.V90.7.2615
83. Alkhamis TM, Beissinger RL, Chediak JR. Artificial surface effect on red blood cells and platelets in laminar shear flow. Blood (1990) 75:1568–75. doi: 10.1182/blood.V75.7.1568.1568
84. Owens AP 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res (2011) 108:1284–97. doi: 10.1161/CIRCRESAHA.110.233056
85. Shcherbina A, Remold-O'Donnell E. Role of caspase in a subset of human platelet activation responses. Blood (1999) 93:4222–31. doi: 10.1182/blood.V93.12.4222
86. Dale GL, Friese P. Bax activators potentiate coated-platelet formation. J Thromb Haemost (2006) 4:2664–9. doi: 10.1111/j.1538-7836.2006.02211.x
87. Brown SB, Clarke MC, Magowan L, Sanderson H, Savill J. Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. a caspase-independent cell clearance program. J Biol Chem (2000) 275:5987–96. doi: 10.1074/jbc.275.8.5987
88. Fox JE, Austin CD, Reynolds CC, Steffen PK. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem (1991) 266:13289–95. doi: 10.1016/S0021-9258(18)98837-X
89. Fox JE, Austin CD, Boyles JK, Steffen PK. Role of the membrane skeleton in preventing the shedding of procoagulant-rich microvesicles from the platelet plasma membrane. J Cell Biol (1990) 111:483–93. doi: 10.1083/jcb.111.2.483
90. Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics (2010) 7:31–42. doi: 10.1016/j.nurt.2009.11.002
91. Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol (2013) 9:201–10. doi: 10.1038/nrneurol.2013.9
92. Flaumenhaft R, Mairuhu AT, Italiano JE. Platelet- and megakaryocyte-derived microparticles. Semin Thromb hemostasis (2010) 36:881–7. doi: 10.1055/s-0030-1267042
93. Flaumenhaft R, Dilks JR, Richardson J, Alden E, Patel-Hett SR, Battinelli E, et al. Megakaryocyte-derived microparticles: direct visualization and distinction from platelet-derived microparticles. Blood (2009) 113:1112–21. doi: 10.1182/blood-2008-06-163832
94. Aatonen MT, Ohman T, Nyman TA, Laitinen S, Grönholm M, Siljander PR. Isolation and characterization of platelet-derived extracellular vesicles. J extracellular vesicles (2014) 3. doi: 10.3402/jev.v3.24692
95. Ray DM, Spinelli SL, Pollock SJ, Murant TI, O'Brien JJ, Blumberg N, et al. Peroxisome proliferator-activated receptor gamma and retinoid X receptor transcription factors are released from activated human platelets and shed in microparticles. Thromb haemostasis (2008) 99:86–95. doi: 10.1160/TH07-05-0328
96. Duchez AC, Boudreau LH, Naika GS, Bollinger J, Belleannee C, Cloutier N, et al. (2015). Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA, in: Proceedings of the National Academy of Sciences of the United States of America, Vol. 112. pp. E3564–3573.
97. Dinkla S, van Cranenbroek B, van der Heijden WA, He X, Wallbrecher R, Dumitriu IE, et al. Platelet microparticles inhibit IL-17 production by regulatory T cells through p-selectin. Blood (2016) 127:1976–86. doi: 10.1182/blood-2015-04-640300
98. Di L, Zha C, Liu Y. Platelet-derived microparticles stimulated by anti-beta(2)GPI/beta(2)GPI complexes induce pyroptosis of endothelial cells in antiphospholipid syndrome. Platelets (2023) 34:2156492. doi: 10.1080/09537104.2022.2156492
99. Bei JJ, Liu C, Peng S, Liu CH, Zhao WB, Qu XL, et al. Staphylococcal SSL5-induced platelet microparticles provoke proinflammatory responses via the CD40/TRAF6/NFkappaB signalling pathway in monocytes. Thromb haemostasis (2016) 115:632–45. doi: 10.1160/th15-04-0322
100. Bao H, Chen YX, Huang K, Zhuang F, Bao M, Han Y, et al. Platelet-derived microparticles promote endothelial cell proliferation in hypertension via miR-142-3p. FASEB J (2018) 32:3912–23. doi: 10.1096/fj.201701073R
Keywords: transcription factor, platelet reactivity, platelet-microparticles, thrombosis, non-transcriptional manner
Citation: Yuan H, Liu Y, Zhang J, Dong J-f and Zhao Z (2023) Transcription factors in megakaryocytes and platelets. Front. Immunol. 14:1140501. doi: 10.3389/fimmu.2023.1140501
Received: 09 January 2023; Accepted: 01 March 2023;
Published: 09 March 2023.
Edited by:
Fabrice Cognasse, INSERM U1059 SAnté INgéniérie BIOlogie, FranceReviewed by:
Ananda L. Roy, National Institutes of Health (NIH), United StatesLucy Kornblith, University of California, San Francisco, United States
Copyright © 2023 Yuan, Liu, Zhang, Dong and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zilong Zhao, emlsb25nemhhb3RpYW5qaW5AaG90bWFpbC5jb20=; Jing-fei Dong, amZkb25nQEJsb29kV29ya3NOVy5vcmc=
†These authors have contributed equally to this work