 Qijun Lu1
Qijun Lu1 Yukun Chen
Yukun Chen Jianwen Li
Jianwen Li Feng Zhu
Feng Zhu Zhan Zheng
Zhan Zheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1139595
This article is part of the Research Topic New insights into Innate Immune cell-based immunotherapies in cancer View all 19 articles
The cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway is critical in cancer immunity. Autophagy is a highly conserved process that is responsible for the degradation of cytoplasmic material and is involved in both innate and adaptive immunity. Recently, cGAS-STING and autophagy have been shown to be interconnected, which may influence the progression of cancer. Although cGAS-STING and autophagy have been shown to be interrelated in innate immunity, little has been reported about cancer immunity. As cancer immunity is key to treating tumors, it is essential to summarize the relationship and interactions between the two. Based on this, we systematically sorted out the recent findings of cGAS-STING and autophagy in cancer immunity and explored the interactions between cGAS-STING and autophagy, although these interactions have not been extensively studied. Lastly, we provide an outlook on how cGAS-STING and autophagy can be combined, with the hope that our research can help people better understand their potential roles in cancer immunity and bring light to the treatment of cancer.
Cancer is one of the world’s most serious threats to human health, with high morbidity and mortality rates, and according to the latest global data, 9.96 million patients will die from cancer in 2020 (1). Cancer is a genetic abnormal disease triggered by a long-term combination of multiple factors. When the human body is affected by chemical, physical, viral, and other carcinogenic substances in the environment or due to its own genetic, endocrine, gender, age, and other factors, a series of abnormal genetic changes will occur to form malignant tumors (2). Tumor cell growth is initiated by mutations that activate oncogenic drivers. This process is combined with the genetic or non-genetic activation or inactivation of genes that promote or inhibit tumor proliferation (3). In many cancers, oncogenesis is accompanied by the accumulation of mutations, which can provide a selective advantage to cancer cell populations by increasing the degree of genetic diversity and accelerating their evolutionary adaptation (4, 5). However, this diversity comes at a cost: the more the cancer cell differs from normal cells, the more likely it is to be recognized as a foreign agent by the immune system.
Current clinical treatment of malignancies is still dominated by radiotherapy, chemotherapy and surgery, but the 5-year survival rate of patients is still very low (6). Along with the advancement of human understanding of tumor immunity, immunotherapy has become increasingly sophisticated and offers new hope for cancer treatment (7–10). Immune checkpoint inhibitors, such as therapeutic monoclonal antibodies targeting the programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) pathway, have been approved as monotherapy or combination therapy for oncology treatment (11). One of the main targets of immune checkpoint inhibitors is the release of effector T cells. The positive correlation between T-cell infiltration in the tumor stroma and prognosis, as well as the clinical success of chimeric antigen receptor (CAR) T-cell infusion in certain hematologic malignancies, suggest a critical role for T cells in tumor immunity (12). These clinical successes have led to a T-cell-centric view of tumor immunity. There is a strong link between cancer and the immune system (13). Adaptive immunity, as well as innate immunity, make up the immune system. However, the effector function of T cells is not autonomous (14). The immune system promotes or suppresses tumor growth by recognizing and killing cancer cells. the initiation and maintenance of T cell responses and the development of durable protective memory T cells are dependent on the innate immune response (15). Innate immunity involves various types of myeloid cells, including dendritic cells (DCs), monocytes, macrophages, polymorphonuclear cells, mast cells, and innate lymphocytes (ILCs), such as natural killer (NK) cells (14). Innate immunity is the host’s first and fastest line of defense against invading pathogens. Different pattern recognition receptors (PRRs) are used to activate the innate immune response when host cells recognize conserved pathogen patterns known as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (16). In eukaryotic cells, DNA is usually present in the nucleus and mitochondria. The DNA present in the cytoplasm is usually due to microbial infection or DNA damage. Thus, cytoplasmic DNA is a red flag that triggers a strong innate immune response (17). Recognition of cytoplasmic DNA is an important host defense mechanism. Cyclic GMP-AMP synthase (cGAS) is thought to be a key sensor mediating cytoplasmic DNA recognition.
The STING pathway has emerged as a promising drug target for the treatment of cancer (18). By triggering the cGAS-STING pathway, the innate immune system can be activated, promoting acquired immunity to fight cancer and thus improving survival (19). In addition, autophagy, a tightly regulated mechanism of cellular self-degradation, is essential for maintaining intracellular homeostasis under stressful conditions (20). Autophagy is extensively involved in the survival, development, and maturation of immune cells (21). It can contribute to the initiation or inhibition of tumor growth by regulating the development of innate and adaptive immunity (22). cGAS-STING pathway can trigger autophagy in several ways, and autophagy can also regulate the cGAS-STING pathway (23). Therefore, in this review, we systematically discuss the interaction between the cGAS-STING signaling pathway and autophagy in cancer immunity, hoping to provide a direction for exploring new cancer immune mechanisms and therapeutic approaches (Figure 1).
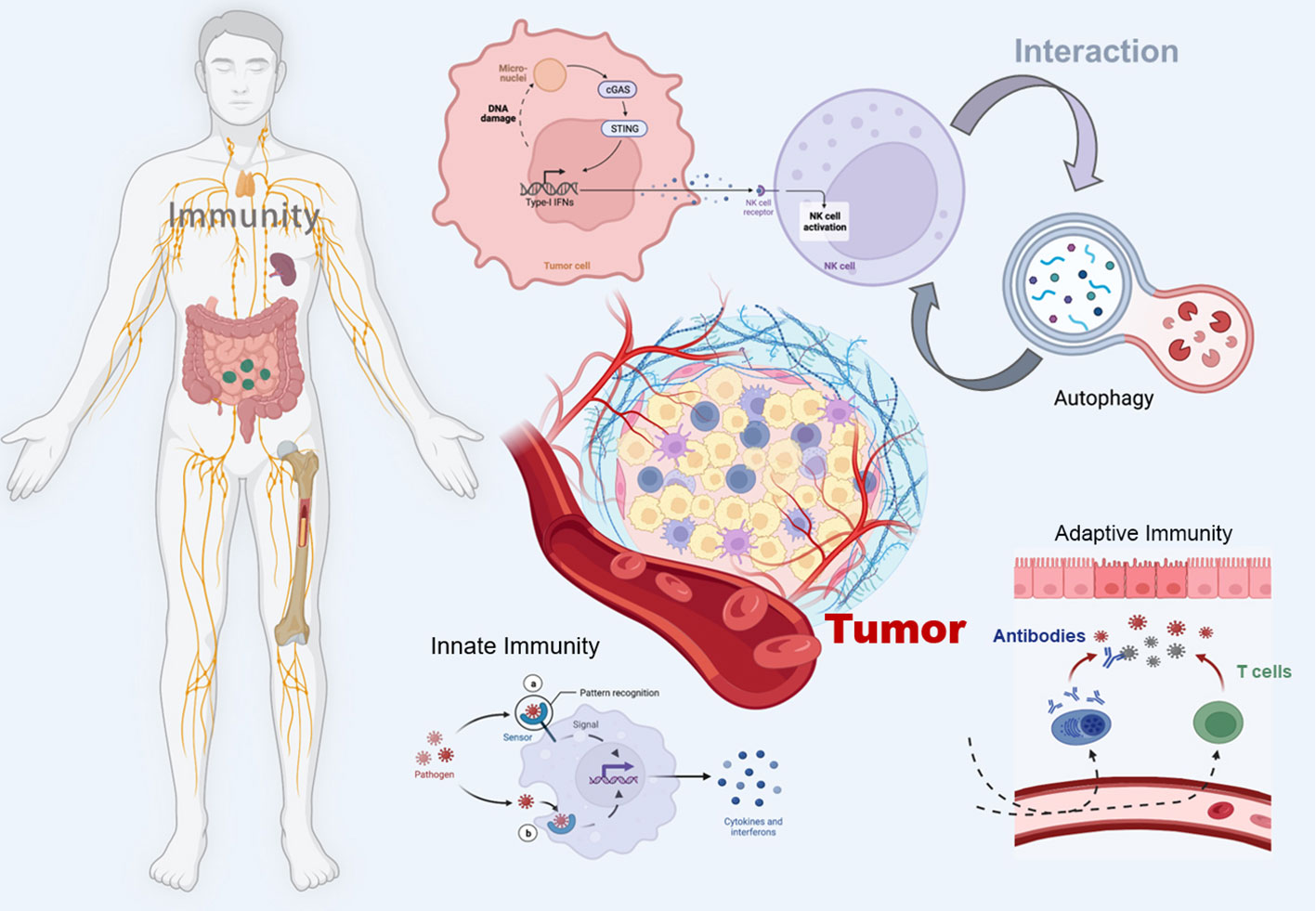
Figure 1 Schematic illustration of the crosstalk between the cGAS-STING pathway and autophagy in cancer immunity. The figure was created with BioRender (https://biorender.com/).
The immune system recognizes different pathogens to protect the body and maintain homeostasis. Innate immunity functions as the first line of defense against pathogenic microorganisms and as a basis for adaptive immune responses. The host cells become aware of a pathogen invasion through pattern recognition receptors, which will initiate a series of signaling events. Many pattern recognition receptors exist, such as Toll-like receptors, Nod-like receptors, and Scavenger receptors. A recently discovered pathogen recognition receptor, cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), can activate any sequence of double-stranded DNA (dsDNA) (24) and participate in various cellular processes, including proliferation, apoptosis, differentiation, and invasion of cancer cells.
STING is a receptor protein located on the endoplasmic reticulum (ER) that is critical for the response pathway in innate immunity. It is usually observed in the resting state as a dimer. By liquid-liquid phase separation, cGAS and dsDNA interact to form micrometer-sized drops that activate cGAS. As the reactants concentrate, these lipid droplets generate cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) which can be catalyzed from ATP and GTP (25, 26). STING is activated by cGAMP in the ER and becomes a tetramer by oligomerization (27). Sulfated glycosaminoglycans induce STING to translocate into the Golgi apparatus and perinuclear endosomes from the ER (28), during which STING is palmitoylated in the Golgi apparatus and caused to activate IRF3 by recruiting TBK1 kinase, which undergoes transautophosphorylation, thus enhancing affinity for interferon (IFN) regulatory factors. When IRF3 is activated, it enters the nucleus, which works synergistically with NF-κB to promote the transcription of type I IFN genes and related immunomodulatory factors (29–31). STING is rapidly degraded by sorting into lysosomes after signaling (32). In addition, STING can mediate the activation of the NF-κB pathway downstream of DNA damage signaling independently of cGAS, and the E3 ubiquitin ligase TRAF6, P53, DNA damage kinase ataxia telangiectasia mutated, enzyme poly(ADP-ribose) polymerase 1, and interferon-γ-inducible factor 16 combine to form a distinct STING signaling complex that induces TRAF6-dependent NF-κB activation (33–37). However, the exact mechanism of STING-dependent NF-κB pathway activation remains unknown (Figure 2).
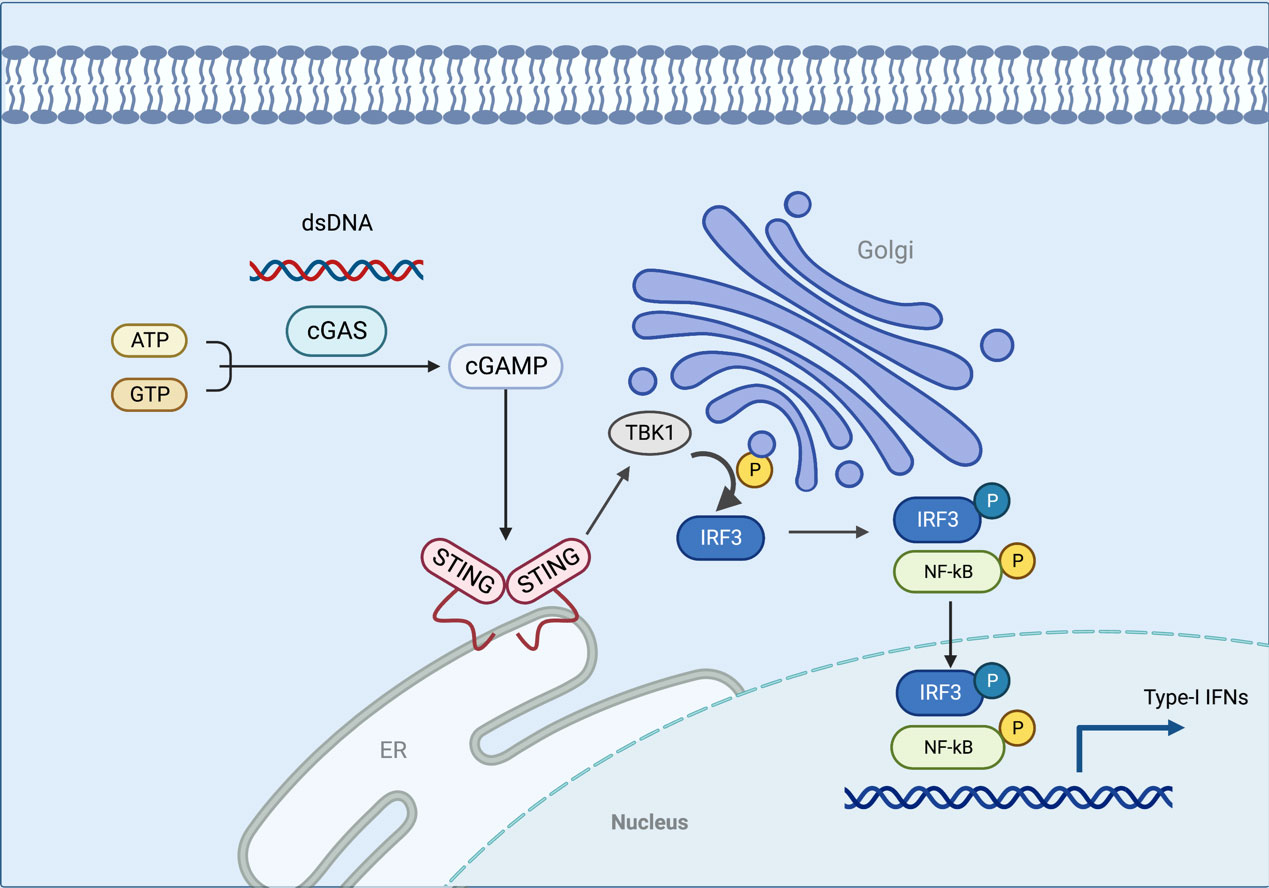
Figure 2 In this model, the cGAS interacts with the dsDNA via liquid-liquid phase separation, which activates the cGAS. STING is activated in the ER when cGAMP is generated in response to the concentration of the reactants. As STING is transferred to the Golgi apparatus, TBK1 is recruited to activate IRF3. When IRF3 is activated, it enters the nucleus and functions with NF-κB to produce type I IFN. The figure was created with BioRender (https://biorender.com/).
Many tissues have been found to express STING, such as the heart, spleen, lung, ovary, and various antigen-presenting cells (APCs). However, it was less expressed in tissues such as the brain, liver, kidney, small intestine, and colon. According to The Cancer Genome Atlas dataset, cGAS and STING gene expression was detected in all types of cancer, but the expression varied according to the stage and type of cancer (38). STING expression is significantly increased in murine pancreatic cancer models and human pancreatic tumors, as well as tongue squamous cell carcinoma, while down-regulated in malignant melanoma (39, 40). In addition, patients with lung adenocarcinoma had lower cGAS expression and longer survival (41). Based on the evidence presented above, cGAS-STING is inextricably linked to cancer.
In further studies, cGAS-STING was found to have a tumor-suppressive effect. By regulating the initiation of intestinal inflammation, STING may hinder the progression of colon cancer, and it may also regulate various signaling pathways such as signal transducer and activator of transcription-3 and NF-κB (42). However, tumors can develop when the cGAS-STING pathway is overactivated. By activating STING, the carcinogen 7,12-dimethyl-Benz[a]anthracene can cause DNA breaks in mice, resulting in skin tumors (43). In the same way, STING activation is associated with Lewis lung cancer growth (44).
Furthermore, the cGAS-STING pathway is involved in cancer metastasis. Cancer cells can transfer cGAMP to astrocytes via the cancer-astrocyte gap junction channel, which activates STING in astrocytes and subsequently produces inflammatory cytokines such as IFN-α and TNF-α, which in turn activate signal transducers and activator of transcription 1 (STAT1) and NF-κB signaling pathways in the cancer cell, leading to brain metastasis (45). In metastatic breast cancer, cGAS-STING signaling can activate atypical NF-κB pathways, which can promote metastasis due to epithelial-mesenchymal transition (EMT) (46). Meanwhile, the elimination of STING can inhibit breast cancer metastasis by reducing the expression of the EMT gene (46).
cGAS-STING participates in the remodeling of the tumor microenvironment (TME) (47), which induces the production of antitumor cytokines such as interleukin 10 and invariant surface glycoprotein (ISG) that inhibit tumor growth (48). Macrophages serve as powerful APCs by engulfing foreign pathogens and priming host defenses (49). The cGAS-STING pathway could significantly regulate macrophage polarization, which is considered an essential part of innate immunity and may be adopted as a target for immunotherapy-related diseases. Administration of liposome-derived cGAMP nanoparticles (cGAMP-NP) to tumor cells can activate STING in macrophages, repolarize M2-type macrophages into M1-type macrophages, improve MHC-like molecules or costimulatory molecules, and then induce differentiation of CD4+ and CD8+ T cells, thus producing a potent antitumor response (50).
In tumor cells, activating the cGAS-STING pass-through may inhibit the development of early tumors by upregulating type I IFN and other inflammatory genes. TME contains multiple proangiogenic factors that stimulate the formation of new blood vessels during tumor angiogenesis (51). Endothelial STING controls T-cell transendothelial migration in association with IFN-I (52). Activating STING increases the immune response to the TME and normalizes the tumor vasculature. In addition, the cGAS-STING pathway affects CD8+ T cell-mediated antitumor immunity by type I IFN. Downregulation of the cGAS-STING pathway leads to a reduction in tumor-infiltrating CD3+ CD8+ T cells by inhibiting type I IFN downstream genes, including chemokine ligands 9 and 10 (53).
In the immune system, DCs also play an essential antitumor immunity role. The STING protein in DCs amplifies signals from cytoplasmic DNA sensors, enhancing the adaptive immune system of the tumor. After being absorbed by tumor-infiltrating DCs, exosomal DNA activates STING signaling (54). DCs respond to NARK signaling by phagocytosing dead/damaged tumor cells, transferring exosomes, and forming cGAMP gap junctions. After injecting type I IFN, DCs drain lymph nodes and trigger tumor specific CD8+ T cells to migrate to the tumor. Finally, these CD8+ T cells proliferate in lymph nodes, killing the tumor cells (55). During TME, phagosomes degrade mtDNA from tumor cells, causing the production of type I IFN in the DC cytoplasm; inhibiting CD47 suppresses this degradation, enhancing adaptive immunity against tumors (56). If STING is deleted in DC, the ability to present antigens is abolished, and tumor infiltrating lymphocyte abundance is decreased (57). A similar effect was observed in colon tumors with MC38 after radiation exposure by mobilizing myeloid-derived suppressor cells (MDSCs) dependent on the host STING molecule (58).
In contrast, cGAS-STING signaling may promote tumor growth and metastasis. Chronic activation may induce an immunosuppressive TME (17). STING was associated with poor prognosis in a subset of patients with colorectal cancer (38), suggesting that STING may contribute to tumor growth and immune evasion. Recent research found that STING agonists activate cell stress in T cells and trigger cell death (59). Another study found that constitutive activation of STING impaired T lymphocyte proliferation, a process dependent on NF-kB and triggered by STING relocalization to the Golgi apparatus (60). These findings suggest that cGAS/STING, as an innate sensor, also has the potential to impair the adaptive immune system.
Immune responses to DNA in the TME are influenced by tumor antigenicity, which is underappreciated. Through the induction of indoleamine 2,3-dioxygenase (IDO), the cGAS-STING pathway promotes tumor progression with low antigenicity (44). However, it remains unclear how cGAS-STING signaling stimulates cells to express PD-L1, which is known to mediate immune evasion of cancer cells (61). Mutations in the liver kinase B1 (LKB1) cause primary resistance to immunotherapy in non-small cell lung cancer (NSCLC). When LKB1 is lost, STING is inhibited, and cytoplasmic dsDNA is not sensitive to detection. Cancers resistant to immune checkpoint blockade may benefit from reactivating the LKB1 or STING pathways (62). In tongue squamous cell carcinoma samples, STING expression increased with tumor progression, with STING protein activation seen in papillomavirus positive specimens. In contrast, STING gene silencing does not affect cell viability or apoptosis but promotes IL-10, IDO, and CCL22, thus enhancing immunosuppressive cytokines and regulatory T-cell infiltration, suggesting that STING regulates the TME and influencing tumor progression (63).
Autophagy is a tightly regulated and stress-induced catabolic process that regulates cancer in eukaryotes (64). Macroautophagy, also known as canonical autophagy, can be divided into several stages including initiation, nucleation, or phagophore formation, elongation of the phagophore membrane to form the autophagosome, fusion of the autophagosome with the lysosomes, and degradation of the contents of the autophagosome (65). Macroautophagy was initially thought to be a massive degradation process activated by cellular starvation. Nevertheless, new findings suggest that autophagy also functions as a quality control mechanism for specific organelles and proteins (66). Through lysosomal or endosomal invagination, cytoplasmic cargo is engulfed during microautophagy (67).
During canonical autophagy, signaling pathways such as mTOR and AMPK sense metabolic stress and thus activate the Unc-51-like kinase 1 (ULK1 and ULK2) complex (68–70). In the initiation phase, ULK1/2 activates VPS34 and the complexes of VPS34, VPS15, autophagy-related gene (ATG)14, Beclin-1, and P150 catalyze the production of phosphoinositol-3-phosphate, recruiting a further boost to the autophagic pathway (70–72). The phosphoinositide 3-kinase (PI3K) complex is responsible for the expansion and maturation of autophagic vesicles (73). Furthermore, ATG5-ATG12-ATG16 and the LC3 ubiquitin-like system contribute to the extension of autophagosome membranes (74). In particular, ATG5-ATG12 non-covalently binds to interact with ATG16 to form the ATG5-ATG12/ATG16 complex (75). ATG4, ATG7, and ATG3 cleave the precursors of LC3-like proteins, maturing and conjugating them with phosphatidylethanolamine (PE) to form LC3-II, which drives the elongation and closure of cell membranes and, ultimately, the formation of autophagosomes (76, 77). P62 via a LIR motif (LC3 interacting region) interacts with LC3. P62 has also an ubiquitin binding domain (UBD) and can bind to autophagy cargo (the ubiquitinated proteins). thus, P62 is an adaptor protein, linking LC3 to its cargo (78). At the maturity stage, LC3-II is digested and autophagosome forms that fuse with a lysosome, causing cell cargo degradation (76) (Figure 3).
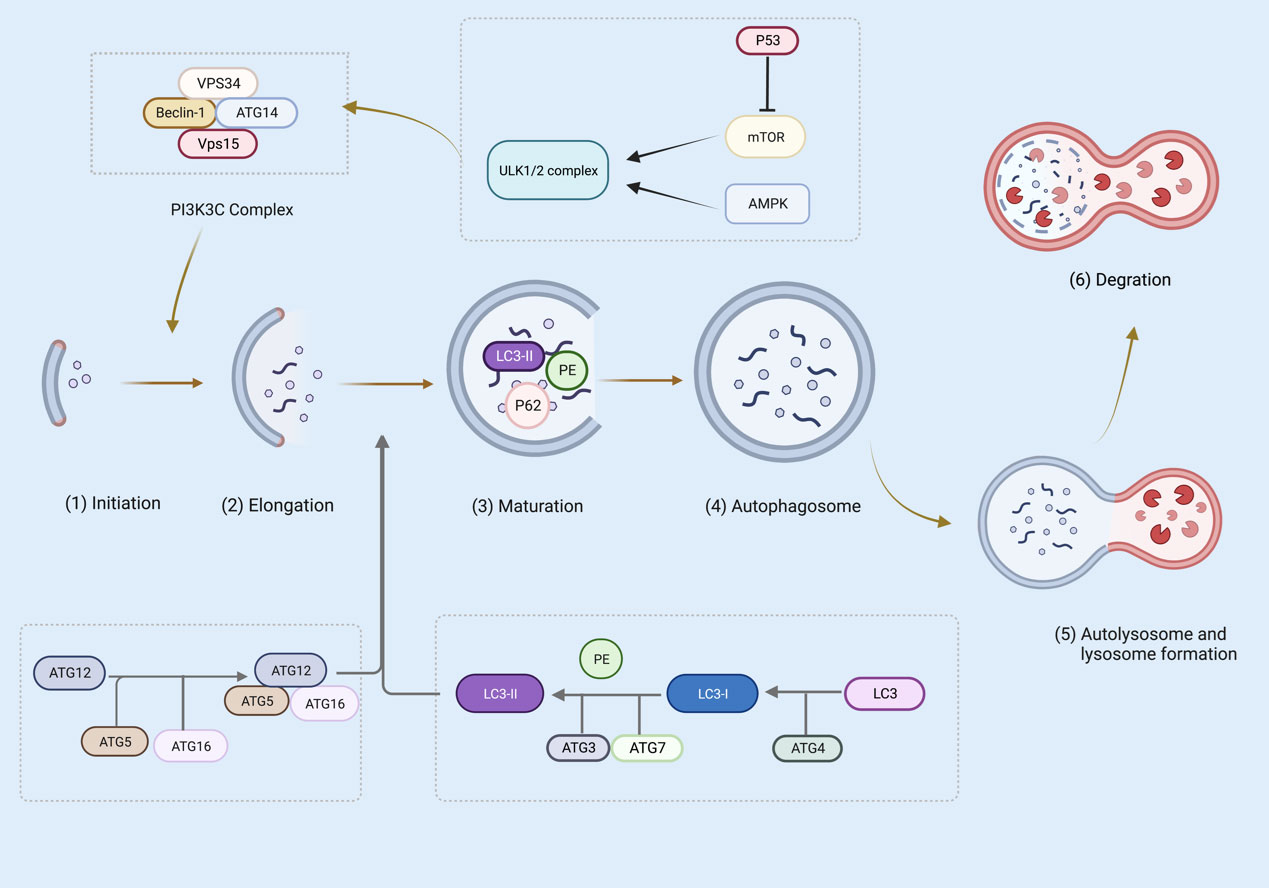
Figure 3 Several steps are involved in canonical autophagy: (1) initiation; (2) nucleation or phagosome extension; (3) maturation; (4) autophagosome formation; (5) autophagosome and lysosome formation; (6) degradation. The figure was created with BioRender (https://biorender.com/).
Although canonical and non-canonical autophagy pathways share overlapping machinery, they differ in several important ways (79, 80). Non-canonical autophagy processes include microautophagy, chaperone-mediated autophagy (CMA), and LC3-associated phagocytosis (LAP) (81). Microautophagy occurs when lysosomes or vesicular endosomes directly engulf intracellular material for degradation (82). CMA is the process of binding intracytoplasmic proteins to molecular chaperones and transferring them to the lysosomal lumen, where lysosomal enzymes digest them (83). However, CMA is selective in removing proteins and is a soluble protein (84). During phagocytosis, pathogens engage extracellular receptors, such as Toll-like receptors, to initiate LAP, a non-canonical form of autophagy (85). Also, immune complexes and dying cells can trigger LAP (86). Furthermore, LAP is an important mediator in the response to immune tolerance, in addition to participating in the degradation of engulfed pathogens (87). With increased research on non-canonical autophagy, the concept of autophagy has been better understood and appreciated.
Cells need to adapt to environmental disturbances to maintain homeostasis in the body. In this process, autophagy serves as a recycling pathway that participates in the turnover of cellular components (88). Also, autophagy is crucial for cancer cell survival in conditions of nutrient and oxygen deprivation by degrading protein and lipid bulks for nutrient recycling (89–91). Tumor types and tumor models affect autophagy in cancer progression (89, 90). Defects in autophagy in vivo have been linked to an increased risk of tumor initiation (92). However, it is still unclear how autophagy-deficient tumors sustain their growth. Hepatocellular tumors are more likely to progress in autophagy-deficient livers when the group box1 is released from autophagy-deficient hepatocytes, which increases proliferation capacity (93). A lack of autophagy inhibits the killing of triple-negative breast cancer cells both in vitro and in vivo (94). Phosphorylation of Beclin1 controls autophagy and promotes or inhibits it (95). It is reported to have increased Beclin1 expression in cancer tissues in 110 patients with prostatic carcinoma, suggesting that autophagy could promote tumorigenesis (96).
Malignant tumors are closely linked to autophagy, especially the processes of recurrence, metastasis, and drug resistance (97). Cancer progression has been characterized by metastasis. Autophagy in metastasis is quite complex as a survival pathway and quality control mechanism. During the early stages of metastasis, autophagy serves primarily as a suppressor by restricting necrosis and mediating autophagic cell death (98). On the contrary, in the advanced stages of metastasis, autophagy as a dynamic degradation and recycling system can help to cope with intracellular and environmental stresses, such as hypoxia, nutrient shortage, or cancer therapy, thus favoring tumor progression. Moreover, Autophagy is upregulated in primary human glioblastoma, melanoma, esophageal cancer, and hepatocellular carcinoma upon progression to advanced metastatic disease, and autophagy markers in these cancers are associated with poor prognosis (99–101), indicating its importance throughout the metastatic cascade. Also, profilin 1 participates in cell proliferation and enhances autophagy-induced drug resistance by interacting with the Beclin1 complex in multiple myeloma (102).
Autophagy influences tumorigenesis by modulating the formation of TME, and this microenvironment causes changes in autophagy signaling pathways in tumors, stroma, and innate immune cells (103). Depending on the characteristics of the tumor, autophagy can promote or suppress the immune response of the TME. Autophagy of these cells can enhance antitumor immune responses and immunotherapy. As a major innate effector component of early immunity, NKs play a crucial role. When NK cells develop, autophagy protects them by removing damaged mitochondria and reactive oxygen species (ROS) (104). As a result of its interaction with ATG7, phosphorylated Forkhead box O (FoxO) 1 induces autophagy in iNKs (104). NK cell maturation may be affected by autophagy when ATG7 and FoxO1 are disrupted in the cytosol of immature NK cells (105). CCL5 overexpression was associated with significantly improved long-term survival in patients with melanoma. Targeting autophagy in a CCL5-dependent manner improves NK cell infiltration and inhibits melanoma growth (106). Therefore, autophagy can act as an inhibitor of the expression of protumor and antitumor chemokines, thus differentially influencing tumor progression.
Autophagy is involved in the processing and presentation of major histocompatibility complex (MHC) molecular antigens and T cell-mediated immune responses, which contribute to tumorigenesis or antitumor immune responses. Pancreatic ductal adenocarcinoma (PDAC) cells are targeted for selective degradation by the autophagy cargo receptor neighbor of BRCA1, inhibiting antigen presentation and killing T cells. On the other hand, inhibition of autophagy restored MHC I surface levels, improved antigen presentation, enhanced antitumor T cell responses, and reduced tumor growth in syngeneic host mice (107). Unlike LAP and LANDO14, canonical autophagy is required for the degradation of MHC I. This suggests that tumor cells can evade immune surveillance through autophagy-mediated degradation of MHC I. A significant component of tumor-induced immunosuppression is MDSCs, which produce DCs, macrophages, and neutrophils. Autophagy deficiency enhances the immunogenic properties of tumor-derived tumor-infiltrating autophagy-deficient monocytic MDSCs through impaired lysosomal degradation of MHC II molecules (108). Consequently, inhibition of autophagy in MDSCs may be beneficial in the treatment of cancer; however, it remains challenging to target specific myeloid subpopulations in TME. The ubiquitination of MHC II in DC affects homeostasis, phenotype, cytokine production, and Ag proteolysis by DC, affecting Ag presentation and T-cell and Ab-mediated immunity (109). By interacting with antigen-processing pathways in DCs, autophagy can effectively modulate adaptive immunity. Through autophagy, organelles and apoptotic proteins are degraded, promoting T-cell development and survival. Furthermore, autophagy in DCs was shown to process tumors intracellularly for the presentation of MHC II to CD4+ T cells (110). Fusion of viral and tumor antigens into the LC3-II protein of ATG8, which is located in autophagosomal membranes, increases the presentation to CD4+ T cells (111). A CD4+ T helper cell activates CD8+ T cells primed by DCs. An effector CD8+ T cell lacking autophagy cannot establish long-term memory for effective antiviral immunity (112). Mice lacking the autophagy genes Atg5, Atg14, or Atg16L1 suffer from synthetic tumor growth impairment (113). Also, Atg5-/- CD8+ T cells show enhanced glucose metabolism which results in altered histone methylation and higher transcription levels (113). In contrast, limiting glucose could inhibit the Atg5-dependent enhancement effector, therefore directly enhancing antitumor immunity via autophagy (113). In addition, DC activity can be inhibited by autophagy and antigen degradation. Through autophagy induction, the immune response is activated, inhibiting T cell activation after EMT and ROS (114, 115), affecting tumor killing. Inhibition of LAP in myeloid cells induces tumor-associated macrophages (TAMs) to develop a proinflammatory phenotype and increases phagocytosis of dying tumor cells, suggesting that LAP can increase immunity (116).
Furthermore, TME galectin-1 (Gal-1) improves tumor cell adhesion, invasiveness, angiogenesis, and immune evasion and contributes to tumor progression (117, 118). Through TLR2-activated secretory autophagy and MVB/Rab11/VAMP7-mediated vesicle trafficking, Hepatocellular carcinoma (HCC) cells stimulate TAMs to actively secrete Gal-1 (119). Autophagy-secreted Gal-1 promotes the growth of HCC in mice and is associated with a poor prognosis in patients with HCC (120). HCC cells can inhibit macrophage autophagy flux in vitro and stimulate the expression of PD-L1 (121). Another report shows that autophagy blockade drives PDAC to up-regulate and utilize the NRF2-induced alternative macrophagocytosis nutrient procurement pathway, which allows tumor cells to extract nutrients from extracellular sources and use them for energy production (122). As a result, combined autophagy and macropinocytosis inhibition may enhance cancer treatment.
As part of autophagy induction, the core complex Beclin-1-PI3KC3 generates a PtdIns-3-P-rich membrane that recruits autophagy proteins and forms autophagosomes (123). Rubicon interacts with the Beclin-1-PI3KC3 core complex, negatively regulating autophagy and PI3KC3 lipid kinase activity (124). Rubicon competes with cGAS in conjunction with Beclin1. Binding of the central NTase domain of cGAS to the central CCD of Beclin 1 inhibits cGAMP synthesis and subsequent IFN production, as well as stimulates Rubicon release from the Beclin 1 complex, which induces autophagy by activating PI3KC3, clearing cytoplasmic dsDNA, inhibiting cGAS activation and sustained immune stimulation (125). In conclusion, cGAS and Beclin-1 interact to coordinate the IFN and autophagic pathways and thereby regulate the innate immune response.
cGAS contains five LC3-interacting regions (LIRs) that bind to LC3 and induce noncanonical autophagy (126). In a recent study, ATG7 and ATG14 were found to depend on the involvement of cGAS to contribute constitutively to nucleus clearance, suggesting that this pathway occurs through typical autophagy, in contrast to STING1-mediated autophagy of the non-dependent ULK1 and BECN1 pathways (127). cGAS has also been shown to bind to dsDNA to form liquid-phase condensates (25). Interestingly, liquid-like condensates can recruit autophagy-related molecules like ATG, LC3, as well as P62 to form cytosomes and participate in the mTOR-mediated autophagic pathway to facilitate cargo degradation (128, 129).
In the immune system, cGAS may be a versatile sensor. Triplet motif containing 14 (TRIM14), a mitochondrial articulator that promotes innate immune signaling, is involved in various tumorigenesis processes. Through the PRYSPRY domain and the C terminus of cGAS, TRIM14 and cGAS interact (130). Researchers demonstrated that TRIM14 inhibits autophagic degradation of cGAS by preventing its entry into the autophagosome, which promotes immune responses (130).
In the drosophila model, previous research revealed that inflammation-induced STING-dependent autophagy limits Zika virus infection (131). In further experiments, it was found that STING may evolve to destroy intracellular pathogens, suggesting that cGAS/STING induces autophagy in an ancient and highly conserved way (132). Nuclear warhead protease B has been found to mediate genomic DNA damage and cell membrane DNA release, activating STING-dependent autophagy and leading to ferrotoxic death in human pancreatic cancer cells (133). This implies that STING-mediated autophagy is potentially promising for the treatment of cancer.
When STING binds to cGAMP, it changes conformation. As the oligomerized STING migrates from the ER to the Golgi apparatus, it passes through the ER-Golgi intermediate compartment (ERGIC). In ERGIC, STING plays an essential role in the induction of autophagy. The STING translocation requires both the COP-II complex and ARF GTPases. The STING-containing ERGIC is capable of lipidating LC3 membranes and thereby triggering the formation of autophagosomes (134). In STING-induced autophagy, the transport of STING from ERGIC to Golgi is unknown. After sensing c-di-AMP, STING disrupts ER homeostasis, leading to the stress of the ER, mTOR inactivation, and ER phagocytosis to coordinate autophagy, thus rescuing dead cells. A recent study has demonstrated that activated STING can undergo intercellular transfer and stimulate RAB22A-mediated non-canonical autophagy derived from the ER, thereby propagating antitumor immunity (135).
Additionally, STING activated the unfolded protein response (UPR) (136). ER stress is induced by unfolded or misfolded proteins, which trigger the UPR to relieve it and restore ER homeostasis. The UPR signaling network activates transcription factor 6, PKR-like ER kinase (PERK), and Inositol-Requiring Protein-1 (137). UPR activation may affect autophagy (138). A lack of PERK has been implicated in converting MDSCs into antitumor CD8+ T cells and myeloid immune cells, leading to STING-dependent production of type I IFN and antitumor immunity (115).
By separating ULK1 from AMP-activated proteins, cGAMP generated by cGAS promotes autophagy independent of STING. Upon activation of ULK1, STING is phosphorylated at serine 366, which is then degraded by autophagy and inhibits IRF3 activity (139). In this regard, it is essential to note that, although cGAMP stimulates STING function, it is followed by negative feedback that inhibits the expression of pro-inflammatory molecules, emphasizing the complexity of STING trafficking.
Autophagy proteins have alternative functions, such as LAP, which is involved in phagosome maturation and subsequent signaling mechanisms. Through its direct interaction with LC3, STING mediates autophagy through its classical LIRs. However, STING does not require TBK1 or IRF3 for autophagy to be induced (140). Similarly, autophagy proteins of myeloid cells in the TME are involved in the immunosuppression of T lymphocytes by affecting LAP-induced oncogene expression and triggering the STING-mediated TAM type I IFN response (116).
There is a potential connection between DNA sensing and autophagy: cytosolic DNA inhibits STING-dependent delivery of microbes to autophagosomes that destroy intracellular pathogens (141). The ATG5-dependent autophagy machinery in the ER, which is a key membrane source for autophagosome formation, may regulate innate immune signaling through STING (140). Cytosolic DNA accumulates in cells depleted of ATG5 and ATG7, induced by the expression of STING, STAT1, and ISG15. Activation of the STAT1-ISG15 axis leads to cell migration, invasion, and proliferation, suggesting that inhibition of autophagy can promote tumor-associated phenotypes by activating STING (142). Atg9a is the only multitransmembrane protein identified as an ATG protein in mammals (143) that delivers membranes to the trans-Golgi network (TGN) to form autophagosomes between the plasma membrane and the TGN (144, 145). After dsDNA stimulation, STING colocalizes with the autophagy-associated protein Atg9a and the microtubule-associated protein LC3. When Atg9a is disrupted, the assembly of STING and TBK1 dsDNA is promoted, leading to aberrant activation of innate immunity (146, 147). Interestingly, STING can activate autophagy without Beclin1, Ulk1, or Atg9a (140). A lack of Atg9a led to enhanced STING signaling, suggesting that Atg9a is independent of autophagy in the regulation of STING signaling [118]. Furthermore, activated STING has been reported to recruit ATG16L1 to lipidated LC3 for single membrane perinuclear vesicles through its structural domain WD40, a process that bypasses the requirement for canonical upstream autophagy (148). STING-induced ERGIC or Golgi membrane damage induces the V-ATPase (vacuolar-type H+-ATPase) to lapidate LC3 on the Golgi membrane and participates in non-canonical autophagy (85, 149). These findings suggest that STING can interact with LC3 and participate in noncanonical autophagy.
Activating the cGAS-STING pathway can regulate intrinsic cellular programs, such as inducing autophagy in tumor cells (150). Increasing evidence suggests that cargo receptors provide substrates for selective autophagy (151, 152). As a chaperone-like protein, ubiquitously expressed prefoldin like chaperone was vital for suppressing excessive activation of STING1-mediated type I IFN signaling through autophagic degradation of STING1 through sequestosome 1 (153). The Unc-93 homolog B1 attenuates the cGAS-STING signaling pathway by targeting STING for degradation in autophagy lysosomes (154). This provides new insight into the function of STING in innate antiviral immunity, which functions as a checker to prevent hyperactivation.
P62 has been implicated in tumor development as an autophagy selective substrate (155, 156). In cancer cells, increased expression of p62 is associated with defective autophagy, which promotes tumor growth (157). Autophagy can be induced even in the absence of p62 in the presence of ectopic expression of STING (140), indicating that p62 is not necessary for STING-dependent autophagy. The ubiquitination of STING promotes both activation and negative regulation of STING during autophagosome degradation (158). Microtubule-associated protein one LC3 promotes the recruitment of ubiquitinated carriers to the autophagosome membrane through its ubiquitin-associated structural domain. The interaction of LC3-p62 interaction and autophagic degradation is regulated by the structural domain of LIRs (78). By connecting to K63, STING is ubiquitinated and recruited into p62 positive compartments. This results in TBK1 phosphorylating p62 in a manner that depends on IRF3 but not on transcription, thus increasing the affinity of ubiquitin for it. Therefore, p62-deficient cells do not degrade STING, resulting in elevated levels of type I IFN and ISG (159). STING and p62 interact in autophagy and immune regulation, which requires further research.
The mediator of IRF3 activation, a regulator of innate immunity, regulates autophagy flux to promote cell death in breast cancer cells (160). Autophagy may also regulate the stability of IRF3. PSMD14/POH1 deubiquitinase prevents IRF3 autophagy by cleaving its K27-linked polyubiquitin chain in lysine 313 to promote IRF3-mediated type I IFN activation (161). STING also triggers non-canonical autophagy in response to dsDNA, which is crucial for the activation of both IRF3/7 and NF-κB (139). Consequently, selective autophagy-mediated degradation of IRF3 causes immunosuppression by preventing excessive IFN signaling. Nevertheless, IRF3 does not appear to understand the molecular mechanisms that lead to STING degradation. The future of precise immunosuppression may involve activation of the IRF3 pathway, although autophagy may be an important contributor to IRF3-dependent type I IFN signaling (Figure 4).
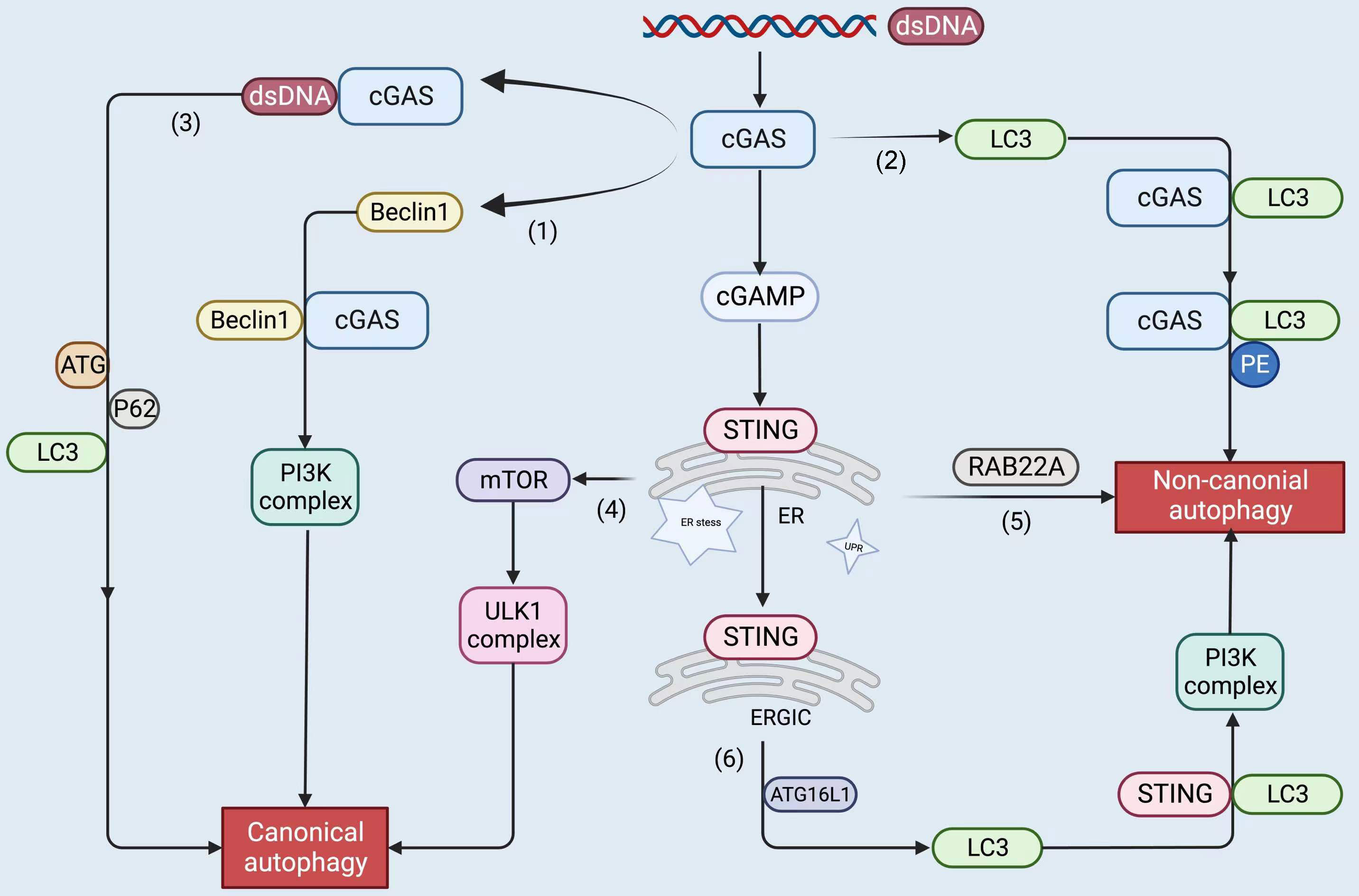
Figure 4 The upstream and downstream of the cGAS-STING pathway, including STING proteins, trigger autophagy by the following roughly divided mechanisms: cGAS binds to dsDNA to form liquid-phase condensates. (1) cGAS interacts with Beclin1 and triggers canonical autophagy; (2) cGAS binds to LC3 to induce non-canonical autophagy; (3) cGAS binds to dsDNA and recruits ATG, LC3, and P62 to participate in canonical autophagy; (4) STING leads to ER stress, mTOR inactivation, and coordinates autophagy; (5) STING stimulates RAB22A-mediated non-canonical autophagy derived from the ER; (6) STING recruits ATG16L1 to lipidated LC3, induces non-canonical autophagy. The figure was created with BioRender (https://biorender.com/).
The cGAS-STING pathway has been identified as a significant immune pathway to recognize cytosolic DNA. It has now made great progress in multiple immune pathways. To support antitumor effects, the host can activate the cGAS-STING pathway, but excessive activation can also contribute to tumor progression. STING activity can be precisely modulated to affect the immune response, including terminating STING-mediated excessive immune activation, which could lead to further investigation. Autophagy exhibits similar dichotomous effects on tumor development. With the advancement of research, autophagy is becoming a more prominent part of tumor immunity. Most of them are focused on the field of canonical autophagy, and non-canonical autophagy remains an area that needs to continue to be explored in depth, which appears to be more comprehensive for better control of mechanistic studies of autophagy in cancer immunity (162).
This review explores the interactions between the upstream and downstream regulators of cGAS-STING and autophagy-related proteins and their relevant effects on cancer immunity. Future research could focus on finding herbal medicine and ingredients that can promote immune cells with antitumor effects. Herbal medicine can be used in combination with chemotherapy or targeted drugs, or immunotherapy represented by PD-1 and PD-L1 inhibitors to have a selective synergistic effect, improving the killing effect of cancer cells, while reducing the side effects of these therapies on healthy ones. In clinical practice, this expectation is consistent with what we have observed. The combination of herbal medicine and various therapies can enhance tumor inhibition more effectively than single drugs (163). Meanwhile, we found that herbal medicine can enhance the cytotoxic effect of chemotherapy on NSCLC by inhibiting cisplatin-induced protective autophagy (164). This way, the application of synergistic treatment of tumors with herbal medicine combined with chemotherapy or targeted drugs, or immunotherapy will be appropriate. This fundamental study can better facilitate the design and development of future antitumor-targeting drugs. Based on the function of cGAS-STING, we will take this pathway as the main means to test the anticancer effect of herbal medicine.
Many interesting questions remain for future investigation and interpretation, although cGAS-STING can trigger both canonical and non-canonical autophagy through multiple pathways. First, in different types of cancer, cGAS-STING inhibits the cell growth cycle through cellular senescence, necrosis, and apoptosis (165–168). What determines cell fate after cGAS-STING mediation? Does the presence of cGAS without transmembrane domains and its localization have an impact on this, including the onset of autophagy? Furthermore, the degree of STING activity and the intrinsic changes in the cancer cells themselves are also taken into account. Second, we need to find other pathways to connect cGAS-STING to autophagy more directly. At present, there is only a preliminary linkage between the two, but there is no more comprehensive systematic evidence to combine them and coordinate a series of downstream pathways to improve tumor immune efficiency in response to various foreign stimuli. Third, it is worthwhile to think about how to more fully elucidate the specific structures and modes of interaction between STING and some of the factors associated with autophagy along with drug trials and applications concerning each of them. Overall, combining cGAS-STING with autophagy can help to deepen the understanding of the intersection of innate and acquired immunity, which provides a new avenue for studying antitumor immunity.
QL wrote the first version of the manuscript, and YC finalized the manuscript. JL downloaded the references and processed the figures in the manuscript. FZ collected the data. ZZ (corresponding author) conceived and coordinated the study and critically evaluated the data. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Shanghai, China (No.20ZR1459200).
The authors would like to thank the reviewers and also the authors of all references. The reviewer’s advice really makes a great improvement to this paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Wang J-J, Lei K-F, Han F. Tumor microenvironment: recent advances in various cancer treatments. Eur Rev Med Pharmacol Sci (2018) 22:3855–64. doi: 10.26355/eurrev_201806_15270
3. Rosemurgy A. Special issue editorial-cancer genetics. Cancer Genet (2016) 209:535–6. doi: 10.1016/j.cancergen.2016.11.003
4. Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: Treating cancer with specificity. Eur J Pharmacol (2018) 834:188–96. doi: 10.1016/j.ejphar.2018.07.034
5. Naser R, Dilabazian H, Bahr H, Barakat A, El-Sibai M. A guide through conventional and modern cancer treatment modalities: A specific focus on glioblastoma cancer therapy (Review). Oncol Rep (2022) 48:190. doi: 10.3892/or.2022.8405
6. Gregory S, Kelley M, Lalani T. Novel therapies in oncology: An individualized approach. AACN Adv Crit Care (2021) 32:315–23. doi: 10.4037/aacnacc2021102
7. Callahan MK, Wolchok JD. At The bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol (2013) 94:41–53. doi: 10.1189/jlb.1212631
8. Sznol M, Melero I. Revisiting anti-CTLA-4 antibodies in combination with PD-1 blockade for cancer immunotherapy. Ann Oncol (2021) 32:295–7. doi: 10.1016/j.annonc.2020.11.018
9. Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol (2016) 17:976–83. doi: 10.1016/S1470-2045(16)30053-5
10. De Giglio A, Di Federico A, Nuvola G, Deiana C, Gelsomino F. The landscape of immunotherapy in advanced NSCLC: Driving beyond PD-1/PD-L1 inhibitors (CTLA-4, LAG3, IDO, OX40, TIGIT, vaccines). Curr Oncol Rep (2021) 23:126. doi: 10.1007/s11912-021-01124-9
11. Nouri Rouzbahani F, Shirkhoda M, Memari F, Dana H, Mahmoodi Chalbatani G, Mahmoodzadeh H, et al. Immunotherapy a new hope for cancer treatment: A review. Pak J Biol Sci (2018) 21:135–50. doi: 10.3923/pjbs.2018.135.150
12. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci (2019) 15:2548–60. doi: 10.7150/ijbs.34213
13. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
14. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
15. Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in universal CAR-T cell therapy. Front Immunol (2021) 12:744823. doi: 10.3389/fimmu.2021.744823
16. Ding C, Song Z, Shen A, Chen T, Zhang A. Small molecules targeting the innate immune cGAS−STING−TBK1 signaling pathway. Acta Pharm Sin B (2020) 10:2272–98. doi: 10.1016/j.apsb.2020.03.001
17. Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS–STING pathway in cancer. Cancer Discovery (2020) 10:26–39. doi: 10.1158/2159-8290.CD-19-0761
18. Zhang X, Bai X, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity (2020) 53:43–53. doi: 10.1016/j.immuni.2020.05.013
19. Du H, Xu T, Cui M. cGAS-STING signaling in cancer immunity and immunotherapy. BioMed Pharmacother (2021) 133:110972. doi: 10.1016/j.biopha.2020.110972
20. Jang YJ, Kim JH, Byun S. Modulation of autophagy for controlling immunity. Cells (2019) 8:138. doi: 10.3390/cells8020138
21. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol (2017) 176:55–62. doi: 10.1016/j.clim.2017.01.007
22. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13:722–37. doi: 10.1038/nri3532
23. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
24. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science (2019) 363:eaat8657. doi: 10.1126/science.aat8657
25. Du M, Chen ZJ. DNA-Induced liquid phase condensation of cGAS activates innate immune signaling. Science (2018) 361:704–9. doi: 10.1126/science.aat1022
26. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi: 10.1126/science.1229963
27. Shang G, Zhang C, Chen ZJ, Bai X, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
28. Fang R, Jiang Q, Guan Y, Gao P, Zhang R, Zhao Z, et al. Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization and activation of the cGAMP sensor STING. Immunity (2021) 54:962–975.e8. doi: 10.1016/j.immuni.2021.03.011
29. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
30. Zhang C, Shang G, Gui X, Zhang X, Bai X-C, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
31. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
32. Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N, et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep (2017) 21:3234–42. doi: 10.1016/j.celrep.2017.11.061
33. Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun (2017) 8:14392. doi: 10.1038/ncomms14392
34. Hinz M, Stilmann M, Arslan SÇ, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol Cell (2010) 40:63–74. doi: 10.1016/j.molcel.2010.09.008
35. Piret B, Schoonbroodt S, Piette J. The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene (1999) 18:2261–71. doi: 10.1038/sj.onc.1202541
36. Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell (2009) 36:365–78. doi: 10.1016/j.molcel.2009.09.032
37. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell (2018) 71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034
38. An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol Ther - Nucleic Acids (2019) 14:80–9. doi: 10.1016/j.omtn.2018.11.003
39. Baird JR, Friedman D, Cottam B, Dubensky TW, Kanne DB, Bambina S, et al. Radiotherapy combined with novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res (2016) 76:50–61. doi: 10.1158/0008-5472.CAN-14-3619
40. Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, et al. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PloS One (2014) 9:e91473. doi: 10.1371/journal.pone.0091473
41. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA (2017) 114:E4612–20. doi: 10.1073/pnas.1705499114
42. Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, et al. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. JI (2014) 193:4779–82. doi: 10.4049/jimmunol.1402051
43. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun (2014) 5:5166. doi: 10.1038/ncomms6166
44. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res (2016) 76:2076–81. doi: 10.1158/0008-5472.CAN-15-1456
45. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature (2016) 533:493–8. doi: 10.1038/nature18268
46. Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature (2018) 553:467–72. doi: 10.1038/nature25432
47. Shen R, Liu D, Wang X, Guo Z, Sun H, Song Y, et al. And activation of cGAS/STING pathway induce tumor microenvironment remodeling. Front Cell Dev Biol (2022) 9:828657. doi: 10.3389/fcell.2021.828657
48. Ng KW, Marshall EA, Bell JC, Lam WL. cGAS–STING and cancer: Dichotomous roles in tumor immunity and development. Trends Immunol (2018) 39:44–54. doi: 10.1016/j.it.2017.07.013
49. Guerriero JL. Macrophages: Their untold story in T cell activation and function. Int Rev Cell Mol Biol (2019) 342:73–93. doi: 10.1016/bs.ircmb.2018.07.001
50. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer. JCI Insight (2018) 3:e120638. doi: 10.1172/jci.insight.120638
51. Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res (2020) 39:204. doi: 10.1186/s13046-020-01709-5
52. Anastasiou M, Newton GA, Kaur K, Carrillo-Salinas FJ, Smolgovsky SA, Bayer AL, et al. Endothelial STING controls tcell transmigration in an IFN-I dependent manner. JCI Insight (2021) 6:e149346. doi: 10.1172/jci.insight.149346
53. Tan YS, Sansanaphongpricha K, Xie Y, Donnelly CR, Luo X, Heath BR, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res (2018) 24:4242–55. doi: 10.1158/1078-0432.CCR-17-2807
54. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist N-P, Chapman JR, Ueberheide BM, et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res (2018) 6:910–20. doi: 10.1158/2326-6066.CIR-17-0581
55. Ritchie C, Cordova AF, Hess GT, Bassik MC, Li L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol Cell (2019) 75:372–381.e5. doi: 10.1016/j.molcel.2019.05.006
56. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity (2017) 47:363–373.e5. doi: 10.1016/j.immuni.2017.07.016
57. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
58. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun (2017) 8:1736. doi: 10.1038/s41467-017-01566-5
59. Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A. Cutting edge: Activation of STING in T cells induces type I IFN responses and cell death. JI (2017) 199:397–402. doi: 10.4049/jimmunol.1601999
60. Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg M-C, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med (2017) 214:1769–85. doi: 10.1084/jem.20161674
61. He L, Xiao X, Yang X, Zhang Z, Wu L, Liu Z. STING signaling in tumorigenesis and cancer therapy: A friend or foe? Cancer Lett (2017) 402:203–12. doi: 10.1016/j.canlet.2017.05.026
62. Della Corte CM, Byers LA. Evading the STING: LKB1 loss leads to STING silencing and immune escape in KRAS-mutant lung cancers. Cancer Discovery (2019) 9:16–8. doi: 10.1158/2159-8290.CD-18-1286
63. Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, et al. Activated STING enhances tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim Biophys Acta (2015) 1852:2494–503. doi: 10.1016/j.bbadis.2015.08.011
64. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
65. Murthy AMV, Robinson N, Kumar S. Crosstalk between cGAS–STING signaling and cell death. Cell Death Differ (2020) 27:2989–3003. doi: 10.1038/s41418-020-00624-8
66. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discovery (2019) 9:1167–81. doi: 10.1158/2159-8290.CD-19-0292
67. Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J Biol Chem (2018) 293:5414–24. doi: 10.1074/jbc.R117.818237
68. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol (2020) 21:183–203. doi: 10.1038/s41580-019-0199-y
69. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi: 10.1038/ncb2152
70. Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol (2013) 15:741–50. doi: 10.1038/ncb2757
71. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy (2018) 14:207–15. doi: 10.1080/15548627.2017.1378838
72. Wang J, Davis S, Zhu M, Miller EA, Ferro-Novick S. Autophagosome formation: Where the secretory and autophagy pathways meet. Autophagy (2017) 13:973–4. doi: 10.1080/15548627.2017.1287657
73. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
74. Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D. The double-edge sword of autophagy in cancer: From tumor suppression to pro-tumor activity. Front Oncol (2020) 10:578418. doi: 10.3389/fonc.2020.578418
75. Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J (2012) 31:4304–17. doi: 10.1038/emboj.2012.278
76. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40:280–93. doi: 10.1016/j.molcel.2010.09.023
77. Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science (2016) 354:1036–41. doi: 10.1126/science.aaf6136
78. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282:24131–45. doi: 10.1074/jbc.M702824200
79. Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol (2010) 298:C776–785. doi: 10.1152/ajpcell.00507.2009
80. Sprenkeler EGG, Gresnigt MS, van de Veerdonk FL. LC3-associated phagocytosis: a crucial mechanism for antifungal host defence against aspergillus fumigatus. Cell Microbiol (2016) 18:1208–16. doi: 10.1111/cmi.12616
81. Sil P, Muse G, Martinez J. A ravenous defense: canonical and non-canonical autophagy in immunity. Curr Opin Immunol (2018) 50:21–31. doi: 10.1016/j.coi.2017.10.004
82. Petroni G, Galluzzi L. “Canonical versus noncanonical autophagy.,”. In: Non-canonical autophagy. Elsevier (2021). p. 1–8. doi: 10.1016/B978-0-12-820538-9.00008-9
83. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity (2021) 54:437–53. doi: 10.1016/j.immuni.2021.01.018
84. Dupont N, Codogno P. Non-canonical autophagy: Facts and prospects. Curr Pathobiol Rep (2013) 1:263–71. doi: 10.1007/s40139-013-0030-y
85. Hooper KM, Jacquin E, Li T, Goodwin JM, Brumell JH, Durgan J, et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J Cell Biol (2022) 221:e202105112. doi: 10.1083/jcb.202105112
86. Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol (2012) 13:7–12. doi: 10.1038/nrm3249
87. Fracchiolla D, Martens S. Sorting out “non-canonical” autophagy. EMBO J (2018) 37:e988. doi: 10.15252/embj.201798895
88. Gerada C, Ryan KM. Autophagy, the innate immune response and cancer. Mol Oncol (2020) 14:1913–29. doi: 10.1002/1878-0261.12774
89. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer (2017) 17:528–42. doi: 10.1038/nrc.2017.53
90. Singh SS, Vats S, Chia AY-Q, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene (2018) 37:1142–58. doi: 10.1038/s41388-017-0046-6
91. Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J Cell Biochem (2017) 118:3237–48. doi: 10.1002/jcb.25972
92. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25:795–800. doi: 10.1101/gad.2016211
93. Khambu B, Hong H, Liu S, Liu G, Chen X, Dong Z, et al. The HMGB1-RAGE axis modulates the growth of autophagy-deficient hepatic tumors. Cell Death Dis (2020) 11:333. doi: 10.1038/s41419-020-2536-7
94. Li Z-L, Zhang H-L, Huang Y, Huang J-H, Sun P, Zhou N-N, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-c degradation. Nat Commun (2020) 11:3806. doi: 10.1038/s41467-020-17395-y
95. Nam RK, Benatar T, Amemiya Y, Sherman C, Seth A. Mir-139 regulates autophagy in prostate cancer cells through beclin-1 and mTOR signaling proteins. Anticancer Res (2020) 40:6649–63. doi: 10.21873/anticanres.14689
96. Holah NS, El-Dien MMS, Mahmoud SF. Expression of autophagy markers Beclin1 and LC3B in prostatic carcinoma: An immunohistochemical case-control study. Iran J Pathol (2022) 17:75–84. doi: 10.30699/IJP.2021.530887.2649
97. Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: A potential link between chemoresistance, recurrence, and metastasis. Biores Open Access (2015) 4:97–108. doi: 10.1089/biores.2014.0035
98. Yao D, Wang P, Zhang J, Fu L, Ouyang L, Wang J. Deconvoluting the relationships between autophagy and metastasis for potential cancer therapy. Apoptosis (2016) 21:683–98. doi: 10.1007/s10495-016-1237-2
99. Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene (2013) 32:699–712. doi: 10.1038/onc.2012.111
100. Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis (2013) 34:1343–51. doi: 10.1093/carcin/bgt063
101. Whelan KA, Chandramouleeswaran PM, Tanaka K, Natsuizaka M, Guha M, Srinivasan S, et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and parkin-mediated mitochondrial clearance. Oncogene (2017) 36:4843–58. doi: 10.1038/onc.2017.102
102. Lu Y, Wang Y, Xu H, Shi C, Jin F, Li W. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci (2018) 109:2706–16. doi: 10.1111/cas.13711
103. Mowers EE, Sharifi MN, Macleod KF. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J (2018) 285:1751–66. doi: 10.1111/febs.14388
104. Wang S, Xia P, Huang G, Zhu P, Liu J, Ye B, et al. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun (2016) 7:11023. doi: 10.1038/ncomms11023
105. López-Soto A, Bravo-San Pedro JM, Kroemer G, Galluzzi L, Gonzalez S. Involvement of autophagy in NK cell development and function. Autophagy (2017) 13:633–6. doi: 10.1080/15548627.2016.1274486
106. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U.S.A. (2017) 114:E9271–9. doi: 10.1073/pnas.1703921114
107. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
108. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest (2018) 128:3840–52. doi: 10.1172/JCI120888
109. Wilson KR, Jenika D, Blum AB, Macri C, Xu B, Liu H, et al. MHC class II ubiquitination regulates dendritic cell function and immunity. J Immunol (2021) 207:2255–64. doi: 10.4049/jimmunol.2001426
110. Ghislat G, Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol (2018) 15:944–52. doi: 10.1038/cmi.2018.2
111. Jin Y, Sun C, Feng L, Li P, Xiao L, Ren Y, et al. Regulation of SIV antigen-specific CD4+ T cellular immunity via autophagosome-mediated MHC II molecule-targeting antigen presentation in mice. PloS One (2014) 9:e93143. doi: 10.1371/journal.pone.0093143
112. Xu X, Araki K, Li S, Han J-H, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15:1152–61. doi: 10.1038/ni.3025
113. DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8+ T cell anti-tumor immunity. Cell Rep (2019) 27:502–513.e5. doi: 10.1016/j.celrep.2019.03.037
114. Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res (2013) 73:2418–27. doi: 10.1158/0008-5472.CAN-12-2432
115. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy (2013) 9:1292–307. doi: 10.4161/auto.25399
116. Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell (2018) 175:429–441.e16. doi: 10.1016/j.cell.2018.08.061
117. Goud NS, Soukya PSL, Ghouse M, Komal D, Alvala R, Alvala M. Human galectin-1 and its inhibitors: Privileged target for cancer and HIV. Mini Rev Med Chem (2019) 19:1369–78. doi: 10.2174/1389557519666190304120821
118. Martínez-Bosch N, Navarro P. Galectins in the tumor microenvironment: Focus on galectin-1. Adv Exp Med Biol (2020) 1259:17–38. doi: 10.1007/978-3-030-43093-1_2
119. Davuluri GVN, Chen C-C, Chiu Y-C, Tsai H-W, Chiu H-C, Chen Y-L, et al. Autophagy drives galectin-1 secretion from tumor-associated macrophages facilitating hepatocellular carcinoma progression. Front Cell Dev Biol (2021) 9:741820. doi: 10.3389/fcell.2021.741820
120. Su H, Yang F, Fu R, Li X, French R, Mose E, et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell (2021) 39:678–693.e11. doi: 10.1016/j.ccell.2021.02.016
121. Deust A, Chobert M-N, Demontant V, Gricourt G, Denaës T, Thiolat A, et al. Macrophage autophagy protects against hepatocellular carcinogenesis in mice. Sci Rep (2021) 11:18809. doi: 10.1038/s41598-021-98203-5
122. Mondal G, Debnath J. NRF2 activates macropinocytosis upon autophagy inhibition. Cancer Cell (2021) 39:596–8. doi: 10.1016/j.ccell.2021.03.011
123. Cheng S, Wu Y, Lu Q, Yan J, Zhang H, Wang X. Autophagy genes coordinate with the class II PI/PtdIns 3-kinase PIKI-1 to regulate apoptotic cell clearance in c. elegans. Autophagy (2013) 9:2022–32. doi: 10.4161/auto.26323
124. Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, et al. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem (2011) 286:185–91. doi: 10.1074/jbc.M110.126425
125. Liang Q, Seo GJ, Choi YJ, Kwak M-J, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe (2014) 15:228–38. doi: 10.1016/j.chom.2014.01.009
126. Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, et al. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy (2014) 10:913–25. doi: 10.4161/auto.28260
127. Zhao M, Wang F, Wu J, Cheng Y, Cao Y, Wu X, et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy (2021) 17:3976–91. doi: 10.1080/15548627.2021.1899440
128. Sun D, Wu R, Li P, Yu L. Phase separation in regulation of aggrephagy. J Mol Biol (2020) 432:160–9. doi: 10.1016/j.jmb.2019.06.026
129. Fujioka Y, Noda NN. Biomolecular condensates in autophagy regulation. Curr Opin Cell Biol (2021) 69:23–9. doi: 10.1016/j.ceb.2020.12.011
130. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell (2016) 64:105–19. doi: 10.1016/j.molcel.2016.08.025
131. Liu Y, Gordesky-Gold B, Leney-Greene M, Weinbren NL, Tudor M, Cherry S. Inflammation-induced, STING-dependent autophagy restricts zika virus infection in the drosophila brain. Cell Host Microbe (2018) 24:57–68.e3. doi: 10.1016/j.chom.2018.05.022
132. Liu Y, Cherry S. Zika virus infection activates sting-dependent antiviral autophagy in the drosophila brain. Autophagy (2019) 15:174–5. doi: 10.1080/15548627.2018.1528813
133. Kuang F, Liu J, Li C, Kang R, Tang D. Cathepsin b is a mediator of organelle-specific initiation of ferroptosis. Biochem Biophys Res Commun (2020) 533:1464–9. doi: 10.1016/j.bbrc.2020.10.035
134. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
135. Gao Y, Zheng X, Chang B, Lin Y, Huang X, Wang W, et al. Intercellular transfer of activated STING triggered by RAB22A-mediated non-canonical autophagy promotes antitumor immunity. Cell Res (2022) 32:1086–104. doi: 10.1038/s41422-022-00731-w
136. Wu J, Chen Y-J, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med (2019) 216:867–83. doi: 10.1084/jem.20182192
137. Zhang L, Wang A. Virus-induced ER stress and the unfolded protein response. Front Plant Sci (2012) 3:293. doi: 10.3389/fpls.2012.00293
138. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci (2015) 40:141–8. doi: 10.1016/j.tibs.2015.01.002
139. Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell (2013) 155:688–98. doi: 10.1016/j.cell.2013.09.049
140. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ (2019) 26:1735–49. doi: 10.1038/s41418-018-0251-z
141. Watson RO, Manzanillo PS, Cox JS. Extracellular m. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150:803–15. doi: 10.1016/j.cell.2012.06.040
142. Kong E, Kim HD, Kim J. Deleting key autophagy elongation proteins induces acquirement of tumor-associated phenotypes via ISG15. Cell Death Differ (2020) 27:2517–30. doi: 10.1038/s41418-020-0519-y
143. Webber JL, Young ARJ, Tooze SA. Atg9 trafficking in mammalian cells. Autophagy (2007) 3:54–6. doi: 10.4161/auto.3419
144. Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, et al. Regulation of mATG9 trafficking by src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res (2017) 27:184–201. doi: 10.1038/cr.2016.146
145. Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci (2006) 119:3888–900. doi: 10.1242/jcs.03172
146. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U.S.A. (2009) 106:20842–6. doi: 10.1073/pnas.0911267106
147. Saitoh T, Fujita N, Yoshimori T, Akira S. Regulation of dsDNA-induced innate immune responses by membrane trafficking. Autophagy (2010) 6:430–2. doi: 10.4161/auto.6.3.11611
148. Fischer TD, Wang C, Padman BS, Lazarou M, Youle RJ. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Biol (2020) 219:e202009128. doi: 10.1083/jcb.202009128
149. Gao Y, Liu Y, Hong L, Yang Z, Cai X, Chen X, et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis (2016) 7:e2330–0. doi: 10.1038/cddis.2016.236
150. Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell (2018) 34:361–78. doi: 10.1016/j.ccell.2018.05.013
151. Green DR, Levine B. To be or not to be? how selective autophagy and cell death govern cell fate. Cell (2014) 157:65–75. doi: 10.1016/j.cell.2014.02.049
152. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J (2021) 40:e108863. doi: 10.15252/embj.2021108863
153. Pan M, Yin Y, Hu T, Wang X, Jia T, Sun J, et al. UXT attenuates the CGAS-STING1 signaling by targeting STING1 for autophagic degradation. Autophagy (2022) 19:440–56. doi: 10.1080/15548627.2022.2076192
154. Zhu H, Zhang R, Yi L, Tang Y-D, Zheng C. UNC93B1 attenuates the cGAS-STING signaling pathway by targeting STING for autophagy-lysosome degradation. J Med Virol (2022) 94:4490–501. doi: 10.1002/jmv.27860
155. Tao M, Liu T, You Q, Jiang Z. p62 as a therapeutic target for tumor. Eur J Med Chem (2020) 193:112231. doi: 10.1016/j.ejmech.2020.112231
156. Zhang J, Yang Z, Dong J. P62: An emerging oncotarget for osteolytic metastasis. J Bone Oncol (2016) 5:30–7. doi: 10.1016/j.jbo.2016.01.003
157. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen H-Y, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell (2009) 137:1062–75. doi: 10.1016/j.cell.2009.03.048
158. Ni G, Konno H, Barber GN. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci Immunol (2017) 2:eaah7119. doi: 10.1126/sciimmunol.aah7119
159. Prabakaran T, Bodda C, Krapp C, Zhang B, Christensen MH, Sun C, et al. Attenuation of c GAS - STING signaling is mediated by a p62/SQSTM 1-dependent autophagy pathway activated by TBK1. EMBO J (2018) 37:e97858. doi: 10.15252/embj.201797858
160. Bhatelia K, Singh K, Prajapati P, Sripada L, Roy M, Singh R. MITA modulated autophagy flux promotes cell death in breast cancer cells. Cell Signal (2017) 35:73–83. doi: 10.1016/j.cellsig.2017.03.024
161. Wu Y, Jin S, Liu Q, Zhang Y, Ma L, Zhao Z, et al. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy (2021) 17:1379–92. doi: 10.1080/15548627.2020.1761653
162. Chen Y, Scarcelli V, Legouis R. Approaches for studying autophagy in caenorhabditis elegans. Cells (2017) 6:E27. doi: 10.3390/cells6030027
163. Wang S, Long S, Deng Z, Wu W. Positive role of Chinese herbal medicine in cancer immune regulation. Am J Chin Med (2020) 48:1577–92. doi: 10.1142/S0192415X20500780
164. Zheng Z, Ma Y, Wang L, Deng H, Wang Z, Li J, et al. Chinese Herbal medicine feiyanning cooperates with cisplatin to enhance cytotoxicity to non-small-cell lung cancer by inhibiting protective autophagy. J Ethnopharmacol (2021) 276:114196. doi: 10.1016/j.jep.2021.114196
165. Wu S, Zhang Q, Zhang F, Meng F, Liu S, Zhou R, et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat Cell Biol (2019) 21:1027–40. doi: 10.1038/s41556-019-0352-z
166. Zeng P-H, Yin W-J. The cGAS/STING signaling pathway: a cross-talk of infection, senescence and tumors. Cell Cycle (2022) 22:38–56. doi: 10.1080/15384101.2022.2109899
167. An M, Yu C, Xi J, Reyes J, Mao G, Wei W-Z, et al. Induction of necrotic cell death and activation of STING in the tumor microenvironment via cationic silica nanoparticles leading to enhanced antitumor immunity. Nanoscale (2018) 10:9311–9. doi: 10.1039/c8nr01376d
Keywords: antitumor, autophagy, cancer, cGAS-STING, immunity
Citation: Lu Q, Chen Y, Li J, Zhu F and Zheng Z (2023) Crosstalk between cGAS-STING pathway and autophagy in cancer immunity. Front. Immunol. 14:1139595. doi: 10.3389/fimmu.2023.1139595
Received: 07 January 2023; Accepted: 20 February 2023;
Published: 01 March 2023.
Edited by:
Mary Poupot-Marsan, INSERM U1037 Centre de Recherche en Cancérologie de Toulouse, FranceReviewed by:
Mariana Pavel-Tanasa, Grigore T. Popa University of Medicine and Pharmacy, RomaniaCopyright © 2023 Lu, Chen, Li, Zhu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhan Zheng, emhlbmd6aGFuMjY5NkAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.