
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 31 March 2023
Sec. Primary Immunodeficiencies
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1135824
This article is part of the Research Topic Emerging Talents in Primary Immunodeficiencies: 2022 View all 7 articles
 Omaima Abdelmajeed1*
Omaima Abdelmajeed1* Muna Mohammed Dawoud Ali2
Muna Mohammed Dawoud Ali2 Nahla Hashim Erwa3
Nahla Hashim Erwa3 Alamin Mustafa4Yassin Abdelraheem Ahmed5Rogaia Hasap Alrasoul Ahmed5Hala Hamza Eltayeb Mohammed5Malaz Elsadeg Hassan5
Alamin Mustafa4Yassin Abdelraheem Ahmed5Rogaia Hasap Alrasoul Ahmed5Hala Hamza Eltayeb Mohammed5Malaz Elsadeg Hassan5 Monzir Ahmed5
Monzir Ahmed5 Shima Algam4
Shima Algam4Introduction: Mendelian susceptibility to mycobacterial disease (MSMD) is a rare inherited condition characterized by selective susceptibility to weakly virulent mycobacteria, such as substrains of the bacille Calmette–Guérin (BCG) vaccine and different environmental mycobacteria.
Case presentation: A 7-year-old Sudanese boy was referred to the immunology clinic with a suspected diagnosis of MSMD. This followed multiple presentations with disseminated tuberculosis and typhoid fever. Genetic testing surprisingly revealed pathogenic homozygous variants in IL12RB1 Exon 9, c.913A>T (p. Lys305*) in both the patient and his father, with a completely healthy asymptomatic carrier mother who is not blood related to the patient’s father.
Conclusion: It is challenging to diagnose MSMD, especially in developing countries where health systems are poor and have limited resources. Family history and genetic tests may help in early MSMD treatment and avoiding disease complications.
Inborn errors of immunity, also known as primary immunodeficiencies, are a group of rare genetic disorders characterized by a compromised immune system. These disorders often result in increased susceptibility to infections and other immunological problems (1). One of the key regulatory pathways in the immune system is the interferon-gamma (IFN-γ)-interleukin (IL)12/23 circuit, which is crucial for the development and function of T cells and the production of IL-12. IL-12 is an important cytokine that stimulates the immune response against intracellular pathogens, such as bacteria and viruses (2). Understanding the IFN-IL12/23 circuit and its role in primary immunodeficiencies is crucial for the diagnosis and management of disorders leading to increased susceptibility to mycobacterial disease.
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare inherited condition characterized by selective susceptibility to weakly virulent mycobacteria, such as substrains of the bacille Calmette–Guérin (BCG) vaccine and different environmental mycobacteria (3). Many patients diagnosed with MSMD have invasive infections caused by other intra-macrophagic microorganisms, such as Salmonella, or mucocutaneous infections caused by Candida species (3–5). Because of incomplete penetrance and genetic differences, the presentation of MSMD is variable. Severe forms of MSMD cause early-onset, disseminated, and persistent life-threatening mycobacterial infections, while the milder forms may have a late onset, improve with age, or even remain clinically silent (3, 5, 6). Almost 20 genes are known to cause 34 different forms of MSMD. Most of these defects affect the IL-12/IFN-γ pathway proteins with the exception of CYBB and ZNFX1 defects that affect the oxidative burst and the recruitment of stress granules, respectively (7). CYBB is a gene that regulates the respiratory burst in myeloid cells, including not only macrophages but also neutrophils. The Nicotinamide Adenine Dinucleotide Phosphate Oxidase, which plays a crucial role in the respiratory burst, is primarily located in neutrophils. This highlights the important role of CYBB in regulating cellular defense mechanisms in myeloid cells, particularly in response to pathogens and other foreign invaders (8, 9). The most frequent genetic cause of MSMD is IL12RB1 deficiency (10).
A 7-year-old male patient (the proband, IV-4; Figure 1A) had a history of two episodes of mycobacterial lymphadenitis and recurrent typhoid fever. He was referred to the pediatric immunology clinic due to a newly diagnosed disseminated tuberculosis (TB) affecting the lungs and multiple lymph nodes that included the mesenteric lymph nodes. The patient had a history of fever and left axillary lymphadenitis at the age of 6 months, for which he received oral and injectable antibiotics without improvement. Excisional biopsy and histopathology showed caseating granuloma diagnosed as BCGitis, and he was treated with four anti-TB drugs (rifampicin, isoniazid, pyrazinamide, and ethambutol) for only 6 months. At 2 years of age, he developed fever, weight loss, and cervical lymphadenopathy. Fine-needle aspiration cytology showed caseating granuloma. He received the same combination of anti-TB drugs as before for 6 months, and his symptoms improved (Figure 1B).
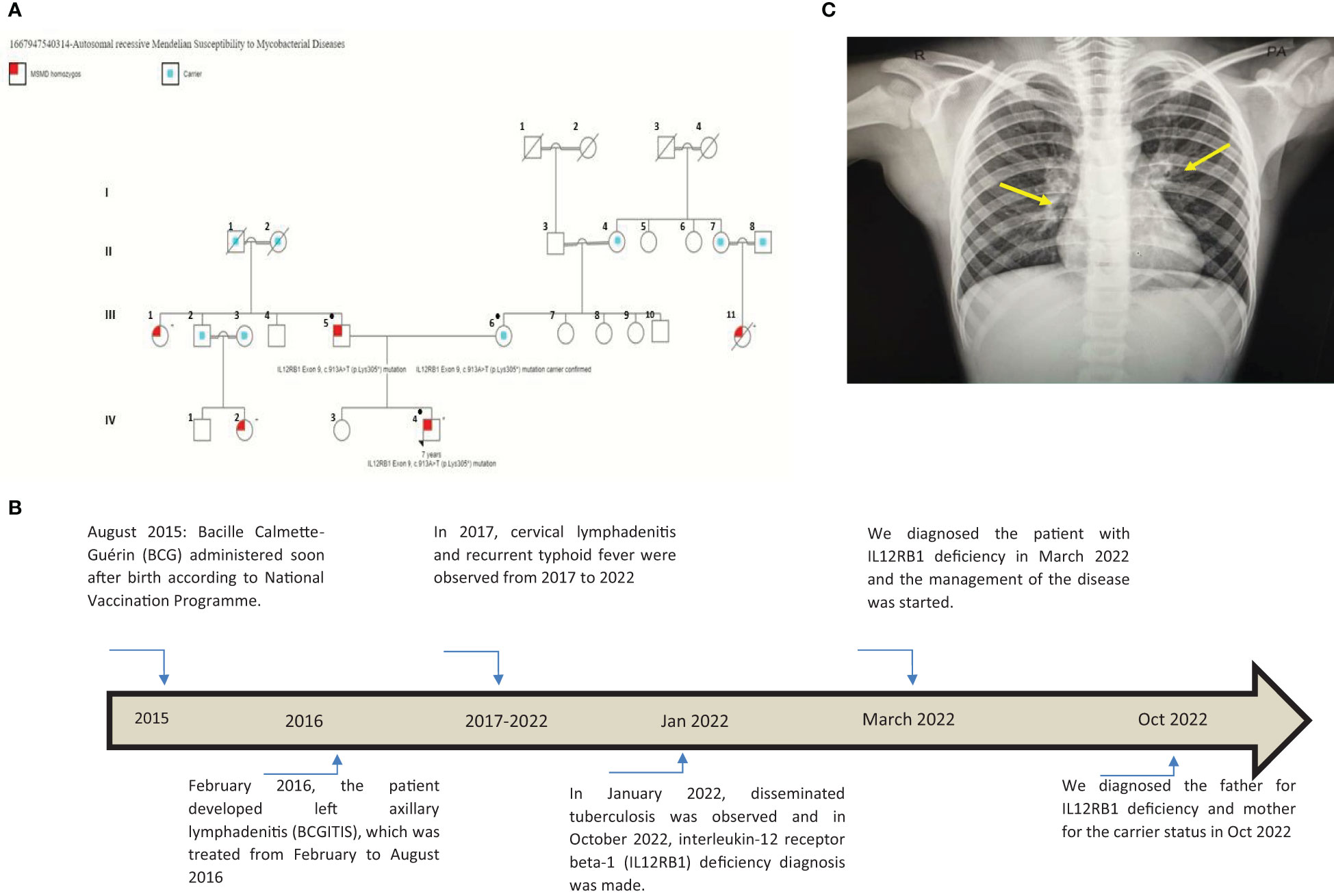
Figure 1 (A) This pedigree shows four generations of relations to the proband who is indicated with the black arrowhead IV-4. Red color in III-1, III-5, III-11, IV-2, and IV-4 indicates individuals affected by symptoms. Only the father (III-5), the mother (III-6), and the patient (IV-4) received genetic testing (indicated by ● in the pedigree); other affected individuals had clinical presentations highly suggestive of Mendelian susceptibility to mycobacterial disease (MSMD) and a history of multiple recurrent tuberculosis infections in childhood. III-7, III-8, III-9, III-10, and their offspring (not shown) have no MSMD symptoms or complaints. This pedigree was constructed using https://www.progenygenetics.com. (B) This figure illustrates the timeline of care for the patient (IV-4), with relevant date points highlighted, and identifies the major point of care. (C) This photographic image shows the chest x-ray for the patient, which is showing bilateral prominent hilar lymph nodes and bilateral diffuse reticulation with multiple hyperlucencies involving bilateral upper, middle, and left lower lobes (yellow arrows). These chest x-ray findings indicate early bronchiectasis.
He also had recurrent febrile illnesses associated with abdominal pain and mesenteric lymphadenitis that were diagnosed as typhoid fever on many occasions based on a very high antibody titer on Widal test (culture not done) (Figure 1B). On each occasion, his condition improved remarkably after treatment. There was no history of other infections (pneumonia, otitis media, nor skin, bone, or joint infections). The patient’s new TB condition started 6 weeks before referral to the Pediatric Tropical and Infectious Diseases unit. He appeared to have normal growth with a weight of 20 kg (10th) and a height of 118 cm (25th). He presented with high-grade fever, mainly at night with sweating, which improved with oral paracetamol. He also had severe abdominal pain disturbing his sleep and daily living activity, but there was no abdominal distension, diarrhea, constipation, or vomiting. The mother noted that the patient had significant weight loss and fatigability. Two weeks later, the patient developed a cough, which started dry and then became productive with thick mucoid sputum without blood. He did not suffer from chest pain or shortness of breath. In this occasion (January 2022), his diagnosis was disseminated TB (Figure 1B). His family history was remarkable for the affection of four other members (Figure 1A: III-1, III-5, III-11, IV-2, and IV-4) across two generations including his father (Figure 1A: III-5), with a similar condition. The patient’s father (Figure 1A: III-5), who was 40 years old at the time of presentation of the proband, had recurrent TB adenitis since early childhood, which was diagnosed based on histopathology and acid-fast Bacillus (AFB) stain. Furthermore, the proband’s parents reported that the father had three episodes of meningitis. One of these was cerebrospinal fluid (CSF)-confirmed tuberculous meningitis. In two occasions, failure of therapy was followed by the detection of Aspergillus fungal hyphae on the CSF. As a result of complicated TB and fungal meningitis, the father is now deaf, blind, and with shunted hydrocephalus. The patient’s mother (III-6) aged 35 years is phenotypically healthy, is not blood related to the father, and is not from the same region. After taking a detailed family history, the mother mentioned a similar presentation of disseminated BCG infection leading to early infantile death in one of her own cousins (Figure 1A: III-11). There is a positive family history of recurrent adenitis among members of the patient’s paternal family including his paternal aunt (III-1) and paternal cousin (IV-2) (Figure 1A).
To investigate the patient’s abdominal complaints, an abdominal ultrasound scan was requested for the patient. This showed minimal intra-abdominal fluid and mesenteric and para-aortic lymph node adenitis, which was reported as highly suggestive of abdominal TB based on his previous history. The chest x-ray showed bilateral prominent hilar lymph nodes (Figure 1C). The diagnosis of TB infection was further confirmed with PCRs using IS6110 and mtp40 primers on blood and sputum, respectively. Immunological investigation revealed elevated immunoglobulin G (IgG) for his age, 2,117 mg/dl (normal range: 380–1,400 mg/dl). IgG subclasses and specific antibody titers for protein and polysaccharide vaccines were not done, as they are unavailable in Sudan. The patient’s IgM and IgE levels were normal at 69 mg/dl (normal range: 37–225 mg/dl) and 53 IU/ml (normal range up to 155 IU/ml), respectively. His IgA level was low, 19 mg/dl (normal range: 30–250 mg/dl), dihydrorhodamine reduction (DHR) test by flow cytometer showed normal oxidative burst, and the patient’s complete blood count and lymphocyte subset are shown in Tables 1 and Table 2, respectively.
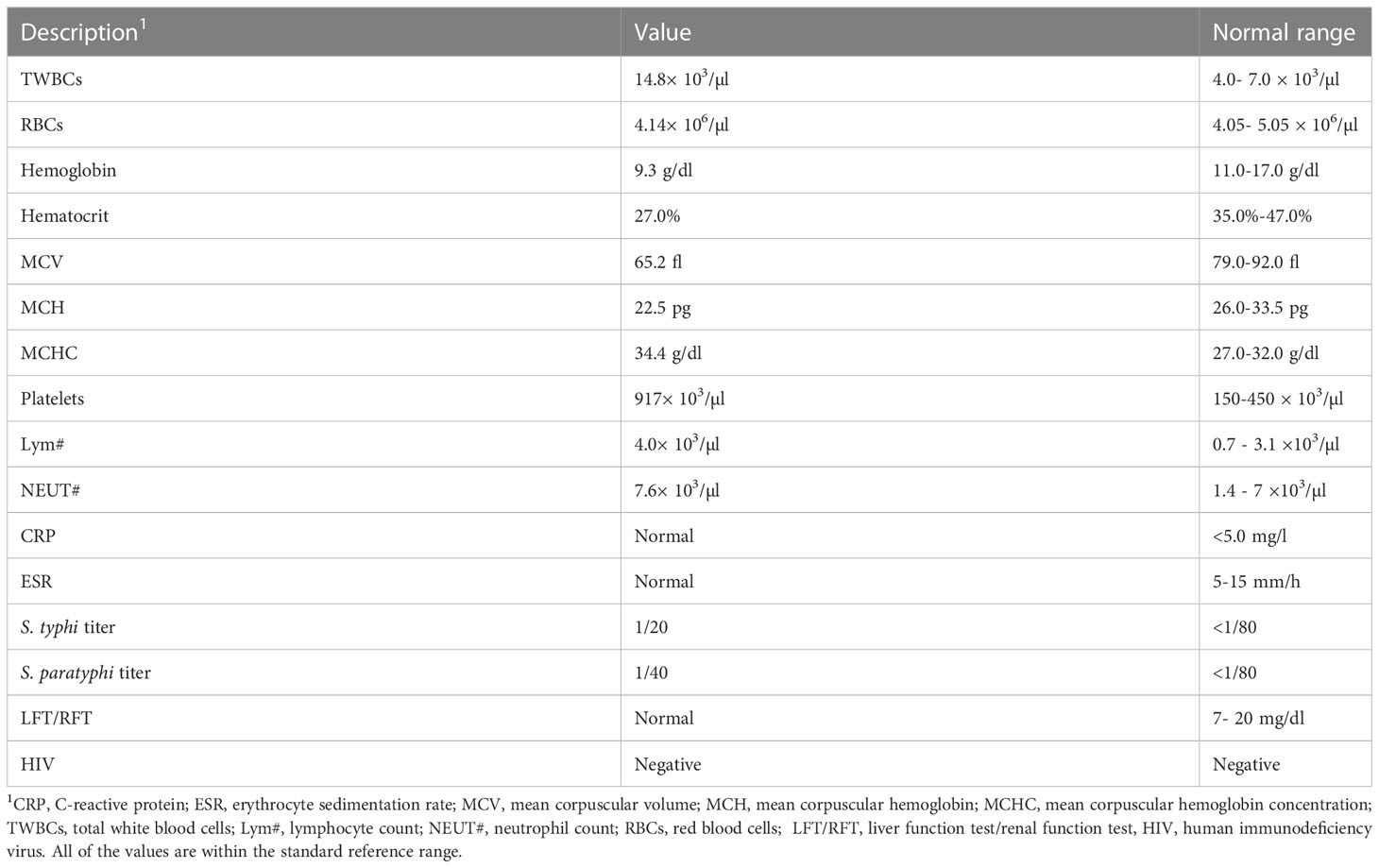
Table 1 Hematological, LFT, RFT, and HIV findings in the patient’s blood sample.
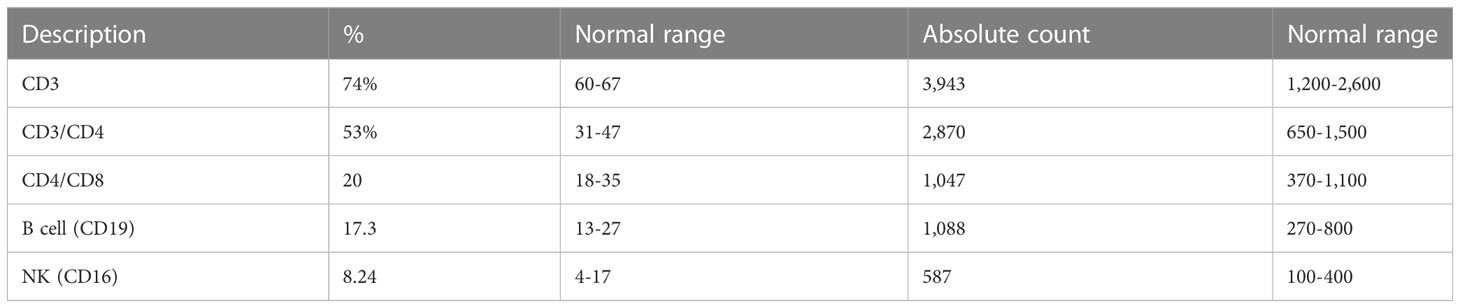
Table 2 Patient lymphocyte subset flow cytometry results.
Primary immunodeficiency genetic testing performed on the NextSeq Illumina platform revealed two pathogenic homozygous variants in IL12RB1 gene of our patient at Exon 9, c.913A>T (p. Lys305*). This IL12RB1 mutation is associated with autosomal recessive MSMD, which is reported by Altare et al. (11). This sequence change creates a premature translational stop signal (p.Lys305*) in the IL12RB1 gene, which is expected to result in an absent or disrupted protein product (11). Such loss-of-function mutations in IL12RB1 are pathogenic (PMID: 9603733, 12591909). The patient’s variant was not present in population databases (gnomAD no frequency), but this premature translational stop signal was observed in individuals with clinical features of MSMD (PMID: 9603732; Invitae). ClinVar also contained an entry for this variant (Variation ID: 488832). The father’s genotype showed the same homozygous pathogenic variants in IL12RB1 Exon 9, c.913A>T (p. Lys305*), and interestingly, the mother, who is not blood related to the father, is a carrier for the same mutation. The patient is now on anti-TB drugs and planned to continue treatment for 2 years and then for bone marrow transplant.
Disseminated BCG infection is the most serious complication of BCG vaccination. Fatal infection has occurred at a rate of 0.06–1.56 cases per million doses; these deaths occurred primarily among immunocompromised people (12, 13). The management of disseminated BCG infection should be prolonged for up to 24–36 months with a pyrazinamide-free 4-drug regimen (rifampicin, isoniazid, ethambutol, and levofloxacin ± clarithromycin). Then, a prophylactic anti-TB regimen with two drugs should be continued until complete immunological reconstitution after hematopoietic stem cell transplantation is achieved, which is found curative in MSMD cases (14).
The patient has IL12RB1 Exon 9, c.913A>T (p.Lys305*) mutation, which results in the replacement of a lysine (K) by a stop codon (X) at amino acid position 305 and is referred to as K305X. This defect is associated with autosomal recessive MSMD, and it disrupts the coding region upstream of the transmembrane segment, preventing the receptor from being expressed on the surface of T lymphocytes and natural killer (NK) cells (11). The proband acquired a homozygous mutation from his parents. This is interesting, as the father is homozygous and affected by the disease, while the mother is a carrier of the same mutation despite the fact that there is no direct blood relation between the parents (Figure 1A).
The patient’s homozygous mutation and MSMD phenotype could be explained by the high incidence of consanguinity and intermarriages in Sudan (15). The mother’s mutation could be due to an uncovered founder mutation in the Sudanese population (16). MSMD is considered a rare condition, and the population frequency of mutations leading to MSMD and other primary immunodeficiencies is not known in the Sudanese population. These mutations could be as frequent as cystic fibrosis (CF) pathogenic alleles in Caucasian populations where the CF phenotype penetrance occurs only when homozygous or compound heterozygous inheritance is exhibited. In such cases, consanguinity would enhance the occurrence of homozygosity and phenotype penetrance (16). Performing Sanger’s sequencing Whole‐Exome Sequencing on IL12RB1 deficiency population would help in confirming gene variants in the Sudanese population. On the other hand, a haplotype search using Single Nucleotide Polymorphism array or WES on next-generation sequencing (NGS) followed by homozygosity mapping would reveal shared regions of homozygosity in the parents (17, 18).
Due to the unavailability of genetic testing in Sudan, a definitive diagnosis of MSMD remains challenging, and there are no data on the prevalence of MSMD in Sudan. The challenge to identify this phenotype increases in countries with prevalent endemic TB and the need for early BCG vaccination coupled with poor health systems and limited resources. Over the past 2 years, the Pediatric Immunology and Pediatric Tropical and Infectious Diseases units at the Tropical Diseases Hospital, Omdurman, Sudan, have encountered 44 patients with disseminated BCGosis (unpublished data). In this group of patients, a diagnosis of severe combined immunodeficiency (SCID) was made in 21 patients, chronic granulomatous disease (CGD) in eight patients, and MSMD in 15. These diagnoses were reached using the combination of clinical picture and immunological lab parameters. Only two out of the 15 patients suspected with MSMD had a confirmed diagnosis through genetic testing. Poor awareness regarding these conditions delayed the diagnosis of the father for many years (40 years old), which led to him being adversely affected with several TB infections throughout his life. The case of the father reflects the natural history of delayed diagnosis and treatment of patients with IL12RB1 mutations.
Although no genetic testing was performed in other members of the family, it was found that many members were affected by recurrent or disseminated TB including the mother’s maternal cousin (III-11) who died at age 2 because of recurrent TB infections (Figure 1A). There was a similar clinical presentation with recurrent TB infections in the patient’s paternal aunt (III-1) and the patient’s paternal cousin (IV-2). This family pedigree also shows six first-degree consanguineous marriages that increase the possibility of occurrence of autosomal recessive conditions such as autosomal recessive MSMD. Unfortunately, we were only able to perform MSMD genetic testing on the patient and his parents. Other affected MSMD individuals were suspected by virtue of the history of recurrent TB infections.
Sudan is an East African country with complex genetic and population structures. This complexity stemmed from the linguistic and cultural differences between its ethnic groups acting in parallel with other, sometimes opposing, population genetic forces, e.g., consanguinity, admixture, and migration. For instance, 67% of marriages in some parts of the country are consanguineous (15, 19).
The frequency of the carrier state for MSMD causing mutations in the Sudanese population needs to be investigated to prevent their deleterious effects. This family’s offspring had a 50% chance of being diseased. Patients with MSMD typically present with BCG infections or environmental non-tuberculous mycobacteria (NTM) (3). Patients with MSMD are also susceptible to other intracellular pathogens, such as extraintestinal infection with typhoid, non-typhoid Salmonella, and mucocutaneous infections (5, 20). Here, the patient had a typical presentation with BCGitis after he received the BCG vaccine, but he was not investigated properly for disseminated BCGosis, and he received a short treatment regimen including pyrazinamide against which Mycobacterium bovis is naturally resistant (3).
Also, the patient had oral thrush that was due to candidiasis. This is consistent with studies that found that mucocutaneous candidiasis is present in approximately 25% of patients with IL12RB1 mutation (10, 20, 21). This is likely due to impaired IL-23-dependent IL-17 immunity. The IL12RB1 gene encodes the IL-12 receptor beta 1 subunit, which forms a heterodimeric receptor with the IL-12 receptor alpha 1 subunit. This receptor is responsible for the transduction of signals from the cytokines IL-12 and IL-23 (22). Impairment of the IL12RB1 gene thus results in decreased signaling through the IL-12/IL-23 receptor complex, potentially impacting the immune function.
Limited resources prevent identifying whether the disseminated MSMD disease is due to Mycobacterium tuberculosis infection or reactivated latent infection with M. bovis infection acquired from the BCG vaccine, as the patient received a non-sufficient regimen for BCG infection in his early life. This is consistent with several studies that mentioned that before a diagnosis of MSMD, acquired and inherited immunodeficiencies that confer a predisposition to mycobacterial diseases must be excluded first (3, 23, 24).
Finally, it is important to note the importance of early diagnosis of such patients. Our patient was not investigated for possible immunodeficiency despite TB recurrences and a highly suggestive family history. This reflects the lack of awareness regarding conditions that result from genetic susceptibility to infection such as MSMD and other inborn errors of immunity. To ensure the best possible outcomes for this patient and the family, he will be under the care and close follow-up of the Pediatric Immunology and the Pediatric Tropical and Infectious Diseases units.
Diagnosing IL12RB1 mutations should be considered in patients with recurrent, severe, or persistent TB infection especially after obtaining the BCG vaccination. BCGosis is another indicator of MSMD. Taking a detailed infection, immunization, and family history together with conducting immunological and genetic tests will identify MSMD early enough to avoid complications from the disease. Awareness programs regarding the identification of MSMD and other inborn errors of immunity are needed to improve patient outcomes and to enhance better care for such patients.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Tropical Diseases Teaching Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
OA, MuA, and NE cared the patient, performed medical examinations, and approved the study. AM, OA, and YA took the lead in writing the manuscript and family pedigree constriction. OS, AM, RH, HM, MH, MoA, and SA revised and wrote the manuscript. All authors reviewed the manuscript and provided critical feedback and agreed on the final manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol (2018) 14:1–12. doi: 10.1186/s13223-018-0290-5
2. Fleisher TA. Immunological tests–from the microscope to whole genome analysis. Primary Immunodeficiency Disord Elsevier (2014), 51–63. doi: 10.1016/B978-0-12-407179-7.00005-9
3. Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of ifn-Γ immunity. Semin Immunol (2014). doi: 10.1016/j.smim.2014.09.008
4. Glanzmann B, Möller M, Moncada-Velez M, Peter J, Urban M, van Helden PD, et al. Autosomal dominant ifn-Γr1 deficiency presenting with both atypical mycobacteriosis and tuberculosis in a bcg-vaccinated south African patient. J Clin Immunol (2018) 38(4):460–3. doi: 10.1007/s10875-018-0509-8
5. Hatipoglu N, Güvenç BH, Deswarte C, Koksalan K, Boisson-Dupuis S, Casanova J-L, et al. Inherited il-12rβ1 deficiency in a child with bcg adenitis and oral candidiasis: A case report. Pediatrics (2017) 140(5). doi: 10.1542/peds.2016-1668
6. Muriel-Vizcaino R, Yamazaki-Nakashimada M, López-Herrera G, Santos-Argumedo L, Ramírez-Alejo N. Hemophagocytic lymphohistiocytosis as a complication in patients with msmd. J Clin Immunol (2016) 36(5):420–2. doi: 10.1007/s10875-016-0292-3
7. Xia L, Liu X-H, Yuan Y, Lowrie DB, Fan X-Y, Li T, et al. An updated review on msmd research globally and a literature review on the molecular findings, clinical manifestations, and treatment approaches in China. Front Immunol (2022), 3672. doi: 10.3389/fimmu.2022.926781
8. de Oliveira-Junior E, Bustamante J, Newburger PE, Condino-Neto A. The human nadph oxidase: Primary and secondary defects impairing the respiratory burst function and the microbicidal ability of phagocytes. Scandinavian J Immunol (2011) 73(5):420–7. doi: 10.1111/j.1365-3083.2010.02501.x
9. Thomas DC. The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol Lett (2017) 192:88–96. doi: 10.1016/j.imlet.2017.08.016
10. de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, et al. Mutations in Stat3 and IL12RB1 impair the development of human Il-17–producing T cells. J Exp Med (2008) 205(7):1543–50. doi: 10.1084/jem.20080321
11. Altare F, Durandy A, Lammas D, Emile J-F, Lamhamedi S, Le Deist F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science (1998) 280(5368):1432–5. doi: 10.1126/science.280.5368.1432
12. Shahmohammadi S, Saffar MJ, Rezai MS. Bcg-osis after bcg vaccination in immunocompromised children: Case series and review. J Pediatr Rev (2014) 2(1):62–74. doi: 10.7508/JPR-V2-N1-62-74
13. Al Jumaah S, Al Hajjar S, Al Mousa H. Bacille calmette-guérin vaccination in Saudi Arabia: Benefits versus risks. King Faisal Specialist Hosp Res Centre (2012), 1–3. doi: 10.5144/0256-4947.2012.1
14. Cuello-García CA, Pérez-Gaxiola G, Gutiérrez-Castrellón P, Gutiérrez CJ, Menjívar-Rubio AH. Treatments for bcg-induced disease in children. Cochrane Database Systematic Rev (2010) 1). doi: 10.1002/14651858.CD008300
15. Temaj G, Nuhii N, Sayer J. The impact of consanguinity on human health and disease with an emphasis on rare diseases. J Rare Dis (2022) 1(1):2. doi: 10.1007/s44162-022-00004-5
16. Barbouche M-R, Mekki N, Ben-Ali M, Ben-Mustapha I. Lessons from genetic studies of primary immunodeficiencies in a highly consanguineous population. Front Immunol (2017) 8:737. doi: 10.3389/fimmu.2017.00737
17. Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet (2013) 132(11):1197–211. doi: 10.1007/s00439-013-1344-x
18. Evans JA. Old meets new: Identifying founder mutations in genetic disease. Cmaj (2015) 187(2):93–4. doi: 10.1503/cmaj.141509
19. Stevanin G, Hamed A, Mohamed I, Elseed M, Salih M, Elsadig S, et al. Clinical phenotyping and genetic diagnosis of a Large cohort of Sudanese families with hereditary spinocerebellar degeneration. (2022). doi: 10.21203/rs.3.rs-2219015/v1
20. Ouederni M, Sanal O, Ikincioğullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis (2014) 58(2):204–13. doi: 10.1093/cid/cit722
21. Conti HR, Gaffen SL. Host responses to candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect (2010) 12(7):518–27. doi: 10.1016/j.micinf.2010.03.013
22. Wojno EDT, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: Room for discovery. Immunity (2019) 50(4):851–70. doi: 10.1016/j.immuni.2019.03.011
23. Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev (2015) 264(1):103–20. doi: 10.1111/imr.12272
24. Esteve-Solé A, Sologuren I, Martínez-Saavedra MT, Deyà-Martínez À, Oleaga-Quintas C, Martinez-Barricarte R, et al. Laboratory evaluation of the ifn-Γ circuit for the molecular diagnosis of mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci (2018) 55(3):184–204. doi: 10.1080/10408363.2018.1444580
Keywords: immunology, bacille Calmette–Guérin (BCG) vaccine, Mendelian susceptibility to mycobacterial diseases, tuberculosis, IL12RB1 mutation
Citation: Abdelmajeed O, Ali MMD, Erwa NH, Mustafa A, Ahmed YA, Ahmed RHA, Mohammed HHE, Hassan ME, Ahmed M and Algam S (2023) Autosomal recessive IL12RB1 mutation: A case report of a Sudanese child and his father. Front. Immunol. 14:1135824. doi: 10.3389/fimmu.2023.1135824
Received: 01 January 2023; Accepted: 13 March 2023;
Published: 31 March 2023.
Edited by:
Yu Lung Lau, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Amy P. Hsu, National Institute of Allergy and Infectious Diseases (NIH), United StatesCopyright © 2023 Abdelmajeed, Ali, Erwa, Mustafa, Ahmed, Ahmed, Mohammed, Hassan, Ahmed and Algam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omaima Abdelmajeed, b21haW1hbmFpbEB5YWhvby5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.