 Zongliang Huang1,2†
Zongliang Huang1,2† Wei Wang1,3†
Wei Wang1,3† Pengpeng Xu2†Shangyu Gong2Yingshan Hu2Yan Liu2Fang Su2Khalid Mahmood Anjum4Wu-Min Deng5
Pengpeng Xu2†Shangyu Gong2Yingshan Hu2Yan Liu2Fang Su2Khalid Mahmood Anjum4Wu-Min Deng5 Suping Yang3Jiyong Liu2*
Suping Yang3Jiyong Liu2* Renjie Jiao2*
Renjie Jiao2* Jianming Chen1,2*
Jianming Chen1,2*- 1Fujian Key Laboratory on Conservation and Sustainable Utilization of Marine Biodiversity, Fuzhou Institute of Oceanography, Minjiang University, Fuzhou, Fujian, China
- 2Sino-French Hoffmann Institute, School of Basic Sciences, Guangzhou Medical University, Guangzhou, Guangdong, China
- 3Department of Bioengineering and Biotechnology, College of Chemical Engineering, Huaqiao University, Xiamen, Fujian, China
- 4Department of Wildlife and Ecology, University of Veterinary and Animal Sciences, Lahore, Punjab, Pakistan
- 5Department of Biological Science, Florida State University, Tallahassee, FL, United States
Sterile alpha and HEAT/Armadillo motif-containing protein (SARM) is conserved in evolution and negatively regulates TRIF-dependent Toll signaling in mammals. The SARM protein from Litopenaeus vannamei and its Drosophila orthologue Ectoderm-expressed (Ect4) are also involved in immune defense against pathogen infection. However, the functional mechanism of the protective effect remains unclear. In this study, we show that Ect4 is essential for the viral load in flies after a Drosophila C virus (DCV) infection. Viral load is increased in Ect4 mutants resulting in higher mortality rates than wild-type. Overexpression of Ect4 leads to a suppression of virus replication and thus improves the survival rate of the animals. Ect4 is required for the viral induction of STAT-responsive genes, TotA and TotM. Furthermore, Ect4 interacts with Stat92E, affecting the tyrosine phosphorylation and nuclear translocation of Stat92E in S2 cells. Altogether, our study identifies the adaptor protein Ect4 of the Toll pathway contributes to resistance to viral infection and regulates JAK/STAT signaling pathway.
1 Introduction
Viral infections seriously threaten human health and majorly cause mortality worldwide. The fruit fly Drosophila melanogaster has been proven to be a powerful model for deciphering antiviral immune responses (1). To defend against viruses, Drosophila relies on antiviral immunity, including RNA interference (RNAi) and inducible responses (2). Studies have shown RNAi to play a major role in defense against viruses in Drosophila. After detecting viral RNAs, Dicer-2 processes them into small interfering RNAs (siRNAs), which are loaded onto the RISC (RNA-induced silencing complex) complex that contains Argonaute-2 (AGO2) to target the complementary viral sequences for silencing (3). Two cellular processes, autophagy and phagocytosis are involved in antiviral defense. Autophagy plays a relatively minor role in antiviral defenses, whereas phagocytosis only contributes to virus-specific immune responses (4). Genetic studies suggest an involvement of the evolutionarily conserved innate immune pathways in controlling viral infections. The cytosolic DNA sensor cyclic GMP-AMP (cGAMP) synthase (CGAS) catalyzes 2’ 3’-cGAMP and activates Sting-dependent antiviral responses in mammals. Recently a class of cGAS-like receptors (cGLRs) was identified in Drosophila playing key roles in defense against viral infections (5, 6). Inactivation of the Toll pathway results in increased susceptibility to Drosophila X virus (DXV) infection, and the Imd pathway is required for an effective antiviral immune response against Cricket Paralysis Virus (CrPV) (7, 8). Another pathway contributing to Drosophila antiviral immunity involves Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling (9). Deficiency in JAK/STAT pathway leads to increased DCV viral loads and higher mortality. In contrast to the Toll and Imd pathways, the JAK/STAT pathway is often activated by different types of stresses, such as mechanical pressure, heat shock, septic wounds, UV irradiation, and damage-associated molecular patterns (DAMPs) released from dead cells, instead of sensing microorganisms (10).
The evolutionarily conserved JAK/STAT pathway plays roles in various biological processes, including hematopoiesis, stress responses, and innate immunity (11–13). Dysregulation of the JAK/STAT pathway has been associated with several human diseases, such as autoimmune disease, allergy, and cancer (14–16). In Drosophila, JAK/STAT signaling is initiated by three cytokines of the Unpaired (Upd) family (Upd1, Upd2, and Upd3). The binding of Upd induces Domeless (Dome) dimerization and activation of the receptor-associated JAK molecules (termed Hopscotch). Activated Hopscotch then phosphorylates Dome, creating a docking site for the single Drosophila STAT family transcription factor, Stat92E. Phosphorylated Stat92E migrates into the nucleus in dimers, promoting target genes transcription (17). Infection with DCV has been shown to trigger the expression of JAK/STAT-dependent genes, including virus-induced RNA 1 (vir-1) and stress response genes Turandot A and M (TotA and TotM) (18). Although the function of these JAK/STAT-dependent genes in Drosophila remains unknown, JAK/STAT signaling has been proposed to be involved in host resistance and tolerance to viral or parasitoid challenges (10, 19).
The Ectoderm-expressed 4 (Ect4) protein is evolutionarily conserved from arthropods to mammals (20). The mammalian Ect4 orthologue, Sterile-alpha and Armadillo motif-containing protein (SARM) has been identified as a negative regulator of TLR-mediated NF-κB activation and to mediate axonal death (21, 22). In Drosophila and Pacific white shrimp (Litopenaeus vannamei), the production of antimicrobial peptides (AMPs) was downregulated by Ect4 and LvSarm (23, 24), suggesting the involvement of Ect4 homologs in Toll pathway suppression is conserved in crustaceans and mammals. Interestingly, in invertebrate species including C. elegans, Drosophila, and L. vannamei, Ect4 homologs were demonstrated to play a positive role in host defense against pathogen infections (24–26). The positive and negative contributions to innate immunity suggested that the invertebrate Ect4 homologs are also involved in immune defense independent of the Toll pathway. This study investigated the role of Ect4 in antiviral defense against DCV infection. As a result, Ect4 mutant flies exhibit increased susceptibility to infection by DCV, whereas overexpression of Ect4 confers resistance against DCV infection; Ect4 regulates the expression of JAK/STAT pathway target genes TotA and TotM; Ect4 interacts with Stat92E to alter the tyrosine phosphorylation status of Stat92E.
2 Materials and methods
2.1 Fly strains and mutant generation
w1118 flies were used as wild-type control. The wIR; dcr-2L811fsX mutant flies have been previously described (27). ubi-Gal4,tub-Gal80ts was a gift from Dr. D. Ferrandon. hopTum-l, ppl-Gal4, da-Gal4, hs-Gal4 were obtained from Bloomington Stock center. The generation of transgenic UAS-Ect4 and U6:3-gRNA-Ect4 lines was performed as previously described (28). Ect4-IR was obtained from NIG-FLY stocks (HMJ30091). For the generation of Ect4 mutant lines, transgenic U6:3-gRNA-Ect4 flies were crossed with the nos-Cas9 flies to get male F0 (nos-Cas9/+; U6:3-gRNA-Ect4/+) that were crossed with w1118; TM3, Sb/TM6B, Tb to obtain F1 progenies. Singular F1 flies were crossed with w1118; TM3, Sb/TM6B, Tb. PCR products amplified from F1 flies before being cloned into the pMD19-T vector according to the manufacturer’s instructions (TAKARA) for mutation identification.
2.2 Plasmid construction
pAC5.1-Ect4-Flag was made by cloning Ect4 cDNA into pAC5.1-Flag vectors. For pAC5.1-Ect4-GFP constructs, the EGFP fragment was amplified from pEGFP-C1 and assembled with the Ect4 fragment into pAC5.1-V5 vectors using ClonExpress MultiS One Step Cloning Kit (Vazyme). Stat92E cDNA was inserted in pAC5.1-HA to generate pAC5.1-Stat92E-HA. The hop (or Dome) cDNA was inserted in pAC5.1-V5 to generate pAC5.1-hop-V5 (or pAC5.1-Dome-V5). For the truncated Ect4 constructs, ARM domain (aa 318-701), SAM domain (aa 680-826), and TIR domain (aa 829-1360) were amplified from pAC5.1-Ect4-Flag before assembled into pAC5.1-Flag empty vector, respectively.
2.3 Cell transfection, co-immuno-precipitation, and Western blot
S2 cells were cultured at 25°C in Sf-900™ III SFM (Gibco). All S2 cell transfection experiments were carried out with the Effectene Transfection Reagent (QIAGEN). For a co-immunoprecipitation assay, S2 cells were transfected with different plasmids. After 48h, cells were collected and lysed in lysis buffer (150 mM NaCl, 25mM Tris-HCL, pH 7.4, 5% glycerol, 1% NP-40, 1mM EDTA, complete protease inhibitor cocktail tablets [Roche] and phosphatase inhibitor cocktail tablets [Roche]). Lysates were incubated overnight at 4°C with Anti-Flag M2 affinity gel (Sigma) or EZview Red Anti-HA Affinity Gel (Sigma). After centrifugation, pellets were washed with 1ml lysis buffer three times before resuspension in 2X Laemmli SDS-PAGE buffer and detection by Western blot. Western blot was performed according to standard procedures.
Primary antibodies: Mouse anti-V5 (1:8000, Proteintech 66007-1-Ig); mouse anti-HA (1:8000, Milipore 05-904); mouse anti-α-Tubulin (1:20,000 Sigma T8203); goat anti-Stat (1:5000, Santa Cruz Biotechnology dN-17); mouse anti-FLAG (1:8000, Sigma F3165); rabbit anti-DCV (1:5000, Abcam ab92954); mouse anti-PY20 (1:2000, Abcam ab10321). Secondary antibodies: HRP-linked anti-mouse IgG (1:8000, Cell Signaling Technology 7076P2); HRP-linked anti-rabbit IgG (1:5000, Cell Signaling Technology 7074P2); HRP-linked anti-goat IgG (1:5000 Millipore AP106P); Alexa Fluor 555 goat anti-mouse IgG (1:500, life technologies A21422).
2.4 RNA analysis
According to the manufacturer’s instructions, total RNA was extracted from infected flies using RNAiso Plus (TAKARA), and cDNA was synthesized with the HiScript II Q RT SuperMix (Vazyme). The ChamQ SYBR qPCR Master Mix (Vazyme) was used for quantitative. Expression of the gene of interest was normalized to the Rpl32 RNA level. The following primers were used for qPCR: RpL32 (forward 5’-GACGCTTCAAGGGACAGTATCTG-3’; reverse 5’-AAACGCGGTTCTGCATGAG-3’), vir-1 (forward 5’-GATCCCAATTTTCCCATCAA-3’; reverse 5’-GATTACAGCTGGGTGCACAA-3’), DCV (forward 5’-TCATCGGTATGCACATTGCT-3’; reverse 5’-CGCATAACCATGCTCTTCTG-3’), TotA (forward 5’-CCCTGAGGAACGGGAGAGTA-3’; reverse 5’-CTTTCCAACGATCCTCGCCT-3’), TotM (forward 5’-ACCGGAACATCGACAGCC-3’; reverse 5’-CCAGAATCCGCCTTGTGC-3’), Ect4 (forward 5’-GCCTCCAGTATTACGGT-3’; reverse 5’-ATGTTTCTCCTGACTGATGA-3’), Vago (forward 5’-TGCAACTCTGGGAGGATAGC-3’; reverse 5’-AATTGCCCTGCGTCAGTTT-3’).
2.5 Virus infection
Virus stocks were prepared as described previously (29). All fly lines confirmed the absence of Wolbachia by PCR and were cured whenever necessary. For infection, 3-6 d old flies were anesthetized with CO2 and injected with PBS (Gibco) or virus suspension intra-thoracically using the Nanoject II injector (Drummond). Infected flies were monitored daily for survival rate or frozen for RNA analysis at the indicated time points.
2.6 Cell immunofluorescence and eye-pigmentation measurement
S2 cells were transfected with pAC5.1-Ect4-GFP and pAC5.1-Stat92E-HA plasmids, and approximately 1×106 cells were transferred to 24 well plates containing coverslips 48 h after transfection. Twelve h later, cells were washed in 0.5ml PBS and fixed with 4% formaldehyde in PBS for 15 min, then washed twice in 0.5 PBT (PBS containing 0.1% Tween-20) before blocking with 5% BSA in TBST for 1 h. Cells were then incubated with primary antibody (anti-HA 1:1000) overnight at 4°C before 2×5 min TBST washes. The secondary antibody was incubated for 4 h at room temperature. Nuclei were stained with PBS with 10 μg/ml DAPI for 5 min. Immunostaining samples were photographed with a Zeiss confocal microscope.
For eye pigment assay, the heads of 50 female flies (2-3 d old, raised at 25°C) of each indicated genotype were homogenized in methanol (1 ml, acidified with 0.1% HCl). After centrifugation, the supernatants were measured for absorbance at 480 nm.
2.7 RNAi knockdown in S2 cells and drug treatment
dsRNA targeting Ect4 and GFP were synthesized according to standard protocol. S2 cells were treated with a culture medium containing 10 μg/ml dsRNA for 3 d. After dsRNA treatment, a solution containing 2 mM H2O2 and 1 mM sodium vanadate (final concentrations; Sigma) pre-incubated for 15 min was added to S2 cells to induce tyrosine phosphorylated Stat92E for 30 min. Cell lysates were prepared with the lysis buffer before immune precipitation with an anti-Stat92E antibody at 4°C and incubated with Pierce Protein A/G Plus Agarose (Thermo Scientific) beads. Co-immuno-precipitated proteins were detected with an anti-Stat92E antibody or anti-PY20 antibody.
2.8 Statistical analysis
Survival data were analyzed by the Kaplan-Meier method using GraphPad Prism. Quantification of immunoblots was performed with ImageJ 1.51p. Altered protein levels were presented as normalized fold change compared to the control value. Statistical analysis was performed using the Student’s t-test. Grey value analysis was performed by the ZEN 2012 (blue edition) system. P-values below 0.05 were considered significantly different.
3 Results
3.1 Reduced resistance of Ect4 mutants to DCV infection
To investigate the role of Ect4 in Drosophila antiviral defense, an Ect4 mutant line was generated with the CRISPR/Cas9 system. The mutation, Ect417, covers a genomic deletion of 17 bp in the coding region of Ect4 (Figure 1A). Ect417 homozygous mutants are lethal at the second instar larval stage as judged by examining the development of both homo- and heterozygous animals distinguished by a GFP marker (Figure S1A), and heterozygous mutants were used for further experiments.
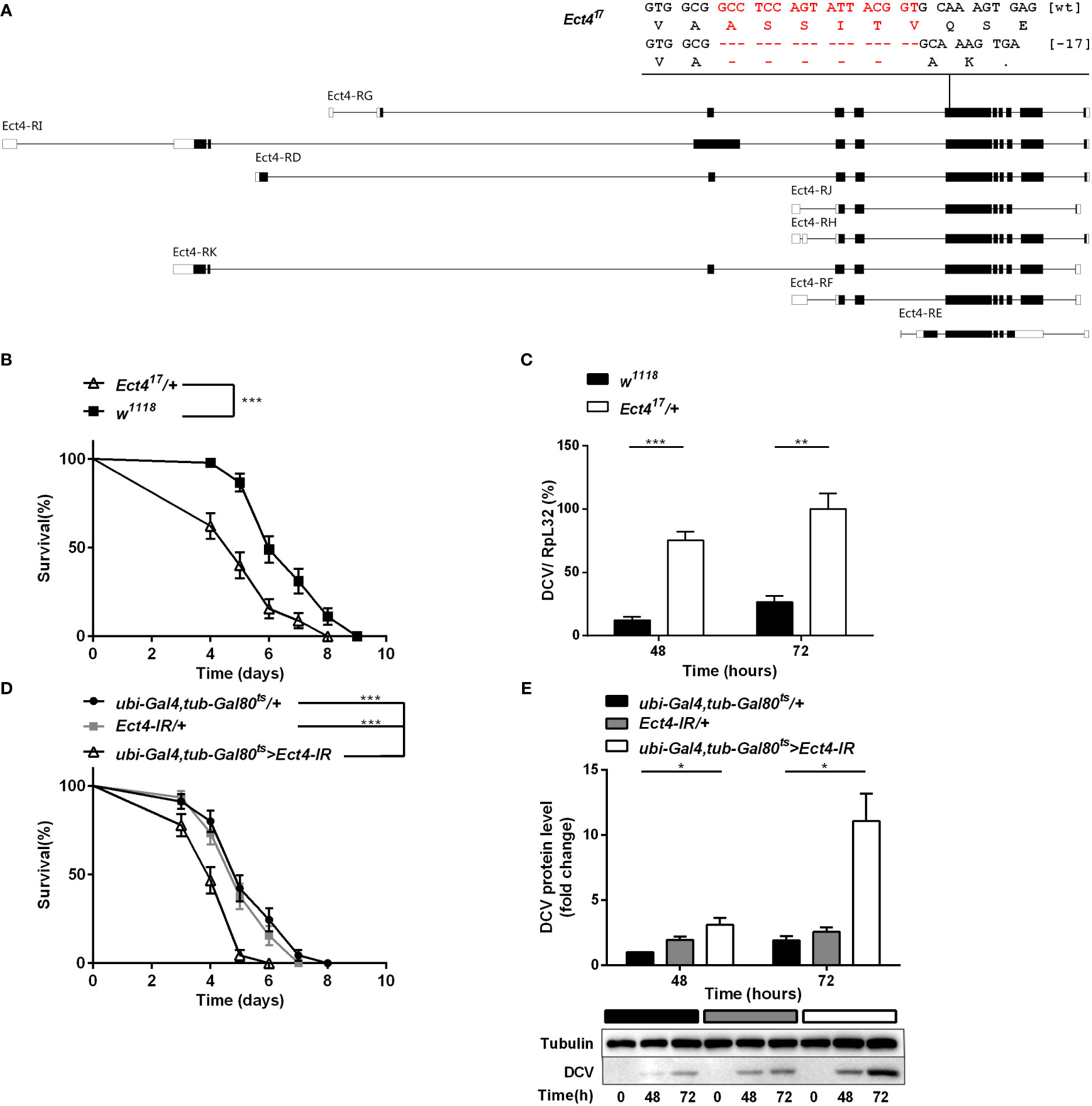
Figure 1 Depletion of Ect4 in adult flies leads to a reduction in viral resistance upon DCV infection. (A) Schematic representation of deletions at exon of Ect4 gene induced by CRISPR/Cas9. The deletion of 17 nucleotides (marked in red) caused a frameshift and created an early stop codons in the Ect417 mutant. Exons are represented by boxes, and introns by lines. UTRs are shown in white, and coding sequences are shown as black blocks. (B) Survival of Ect4 mutants and wild-type flies was monitored daily at 25°C. (C) Quantitative RT-PCR analysis of the accumulation of viral RNA at 48 and 72 h post-infection in wild-type and Ect4 mutant flies. (D) Survival of flies carrying the temperature-dependent Ect4 knockdown system and genetic control flies upon DCV infection at 29°C. (E) Immunoblot of the accumulation of DCV capsid polyprotein in Ect4-RNAi or control flies, color blocks represented the genotype as indicated. Data represent the means ± standard errors of 3 independent pools of 15 male flies (B, D) or 10 male flies (C, E) for each genotype. Log-rank test (B, D) and t-test (C, E): *P< 0.05, **P< 0.01, ***P< 0.001.
Variation in the pastrel gene is associated with natural resistance to DCV infection in D. melanogaster. The non-synonymous single nucleotide polymorphism (SNP) position 598, located in the last exon, has the strongest effect on DCV susceptibility (30). Sequencing of the pastrel locus revealed that all the strains of D. melanogaster tested contained the susceptible allele (data not shown), thus limiting the effect of discordance in the SNP profile between different fly lines to the difference in DCV resistance.
Ect4 transcription in Ect417/+ heterozygotes was reduced by 45% compared with the wild-type flies (Figure S1B). Wild-type and Ect4 mutant files were challenged with DCV by intra-thoraxic injection. Ect4 mutants were more sensitive to infection than wild-type flies, with a significantly different mean survival of 5 and 6 d for Ect417/+ and w1118 male flies, respectively (Figure 1B). Notably, a significant increase in the DCV viral loading was observed in Ect417/+ flies at 48 and 72 h post-infection (Figure 1C), indicating that Ect4 mutants are more sensitive to DCV infection.
To consolidate the DCV sensitivity phenotype observed with heterozygous Ect4 individuals, the temperature-sensitive Gal80ts allele (31) was used to knockdown Ect4 expression in adult flies by shifting the culture temperature from 18-20°C to 29°C before and during the infection of DCV. RT-qPCR shows that Ect4 expression decreased after the temperature shift to 29°C (Figure S3A). As expected, flies with knockdown of Ect4 succumbed earlier to DCV infection than the control flies (Figure 1D). Consistently, the down-regulation of Ect4 increased viral proteins (Figure 1E). Since DCV replicates mainly in fat bodies (32), we employed a fat body-specific driver, ppl-Gal4, to knock down Ect4 expression in the fat body. Specific depletion of Ect4 in the fat body under the control of ppl-Gal4 also affected the survival rate and viral load upon DCV infection (Figures S2A, B). The decreased survival rate was correlated with the increased viral burden in Ect4-RNAi flies.
3.2 Ect4 protects flies from DCV infection
To verify the specificity of the function of Ect4 in DCV infection, UAS-Ect4 transgenic flies were crossed with a ubiquitous Gal4 driver, da-Gal4, to express the Ect4 transgene ectopically. Remarkably, ubiquitous overexpression of Ect4 promoted survival after the viral challenge (Figure 2A). Further, the increased dose of Ect4 led to decreased viral burden in infected flies (Figure 2B). Interestingly, as shown in Figures 2C, D, flies overexpressing Ect4, specifically in the fat body using the ppl-Gal4 driver, showed significantly more resistance to DCV infection than control flies and significantly decreased DCV replication levels. More importantly, rescue experiments by the expression of Ect4 in Ect417/+ flies under the control of da-Gal4 were performed to prove the specific role of Ect4 in protecting flies from viral infections. The decreased survival rate of Ect4 mutants after DCV infection, as well as increased viral load, was rescued to similar levels of the control flies following transgenic expression of Ect4 in heterozygous Ect4 mutants (Figures 2E, F). Similar results were obtained when a hs-Gal4 driver was used for the rescue experiment. Ect4 heterozygous mutant flies expressing Ect4 under the control of hs-Gal4 exhibited a decreased viral replication at 48 h post-infection. (Figure S2C). These results indicate that Ect4 confers resistance against DCV infection and is required to control the accumulation of viruses.
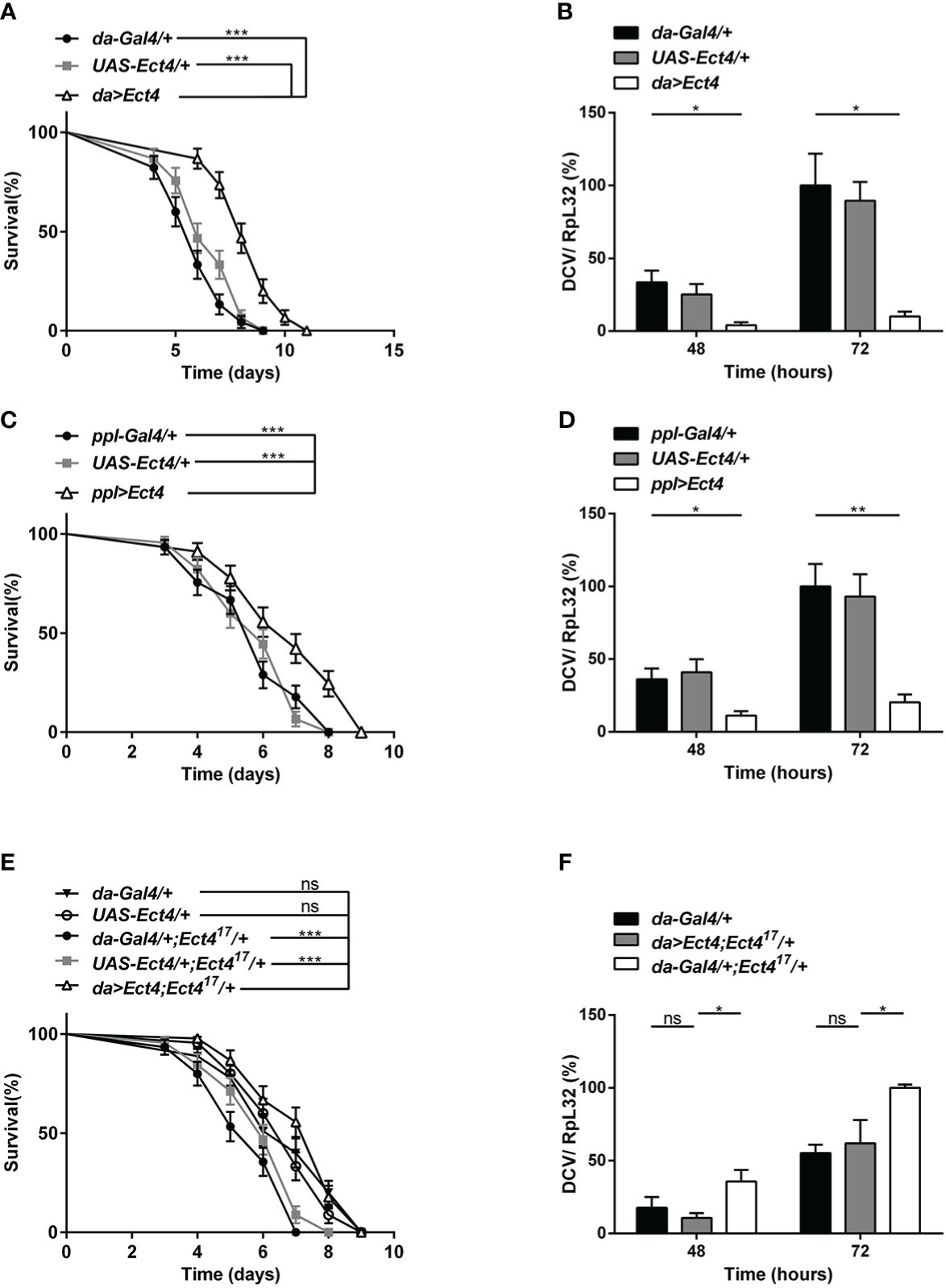
Figure 2 Overexpression of Ect4 provides strong protection against DCV. (A) Survival of flies expressing Ect4 transgene in whole flies by da-Gal4 driver and control flies following DCV infection. (B) Quantitative RT-PCR analysis of the accumulation of viral RNA at 48 and 72 h post-infection in Ect4 overexpression and control flies. (C) Survival of flies expressing Ect4 transgene specifically in the fat body by the ppl-Gal4 driver and control flies following DCV infection. (D) Quantitative RT-PCR analysis of the accumulation of viral RNA at 48 and 72 h post-infection in Ect4 overexpression and control flies, specifically in the fat body. (E) Survival of Ect4 mutant flies expressing Ect4 transgene under the control of da-Gal4 and control flies post-DCV infection. (F) Quantitative RT-PCR analysis of the accumulation of viral RNA at 48 and 72 h post-infection in control or Ect4 mutant flies expressing Ect4 transgene. Data represent the means ± standard errors of 3 independent pools of 15 male flies (A, C, E) or 10 male flies (B, D, F) for each genotype. Log-rank test (A, C, E) and t-test (B, D, F): *P< 0.05, **P< 0.01, ***P< 0.001, ns, not significant.
3.3 Deficiency in Ect4 does not alter the activity of the siRNA pathway
RNA interference (RNAi) acts as the first line of defense against viruses in Drosophila (33). There was strong genetic evidence that one RNAi-related pathway, the siRNA pathway, plays a major role in antiviral immunity in Drosophila (7, 34). Since heterozygous Ect4 mutant flies are hypersensitive to DCV infection, we asked whether the down-regulation of Ect4 affects the function of the siRNA pathway. To address this question, siRNA pathway activity was monitored using an in vivo sensor assay, wherein the endogenous white gene is silenced by the expression of a hairpin dsRNA corresponding to an exon of white. Expression of UAS-wIR using the eye-specific driver GMR-Gal4 alters eye pigmentation to a white color or pale orange if the silencing is incomplete. Studies have shown that the siRNA pathway is inactivated without Dicer-2 (Dcr-2). Therefore eye pigmentation of a Dcr-2 null mutant (dcr-2L811fsX/dcr-2L811fsX) in GMR>UAS-wIR background is red, whereas Dcr-2 heterozygous mutants (dcr-2L811fsX/+) display pale orange (27). Our results show that mutation in Ect4 did not lead to any changes in the eye pigmentation in wIR; dcr-2L811fsX/+ (Figures 3A, B). Moreover, the expression of Vago, induced in DCV infection dependent on Dicer-2 (33), did not differ between wild-type and Ect4 mutant flies at 48 and 72 hpi (Figure 3C). These results suggest that Ect4 does not directly affect the antiviral siRNA pathway.
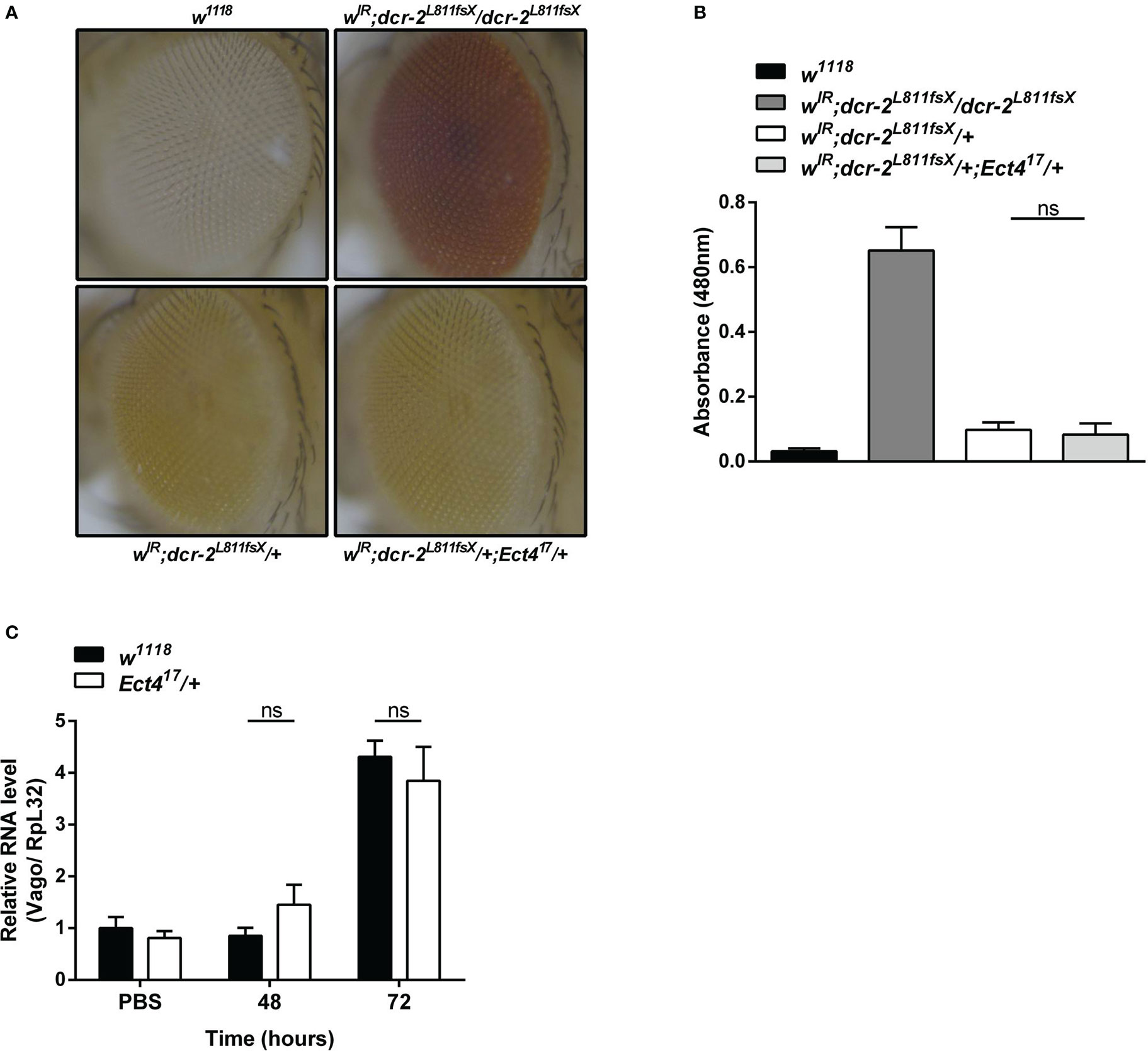
Figure 3 Ect4 deficiency did not affect siRNA-mediated gene silencing. (A) Effects of altering dosages of Ect4 in wIR; dcr-2L811fsX background. The eye color of a white null mutant fly (upper left panel) and a white+ fly carrying GMR>UAS-wIR transgene and homozygous mutant for dcr-2L811fsX (upper right panel). The eye color of a fly carrying heterozygous dcr-2L811fsX mutation with (bottom right panel) or without (bottom left panel) one Ect4 mutant allele. (B) Red-eye pigment levels of the indicated phenotype were determined by measuring absorbance at a wavelength of 480 nm (n = 50 for each group). (C) Expression levels of Vago at 48 and 72 h post-infection in wild-type or Ect4 mutant flies challenged with DCV. Data represent the means ± standard errors of 3 independent pools of 50 female flies (B) or 10 male flies (C) for each genotype. The t-test (B, C): ns, not significant.
3.4 Ect4 regulates the expression of JAK/STAT-dependent genes, TotA, and TotM
The JAK/STAT pathway was shown to contribute to the antiviral response in Drosophila (9), where several genes are induced following viral infection via the JAK/STAT pathway including virus-induced RNA-1 (vir-1), the stress-induced genes Turandot A and M (TotA and TotM) (18). To examine whether the downregulation of Ect4 affected JAK/STAT pathway activation, we examined the expression of vir-1, TotA, and TotM by RT-qPCR at 48 and 72 h after DCV infection (hpi). As previously reported, DCV infection induced a strong up-regulation of vir-1, TotA, and TotM in wild-type flies (18). However, vir-1 induction in response to DCV infection in Ect4 mutant flies was indistinguishable from the control (Figure 4C). Similar results were observed using a ubiquitous temperature-sensitive Gal4 driver, ubi-Gal4 (Figures S3B, C). A genetic interaction experiment was performed to assess further the relationship between Ect4 and the JAK/STAT pathway. TotA and TotM were expressed in flies carrying a JAK gain-of-function allele Tum-l (hopTum-l), which encodes a hyperactive JAK kinase due to a G341E substitution (35). Reducing the dosage of Ect4 by half resulted in a large reduction of the RNA levels of TotA and TotM in hopTum-l flies (Figure 4D). The TotA and TotM response was also attenuated in the fat body of flies where Ect4 was downregulated by expressing the Ect4-IR transgene using a ppl-Gal4 driver (Figures S3D, E). This TotA and TotM expression reduction was rescued by ubiquitously expressed Ect4 (Figures 4E, F). Together, these results suggest that Ect4 genetically interacts with the JAK/STAT pathway to regulate the expression of TotA and TotM in response to DCV infection.
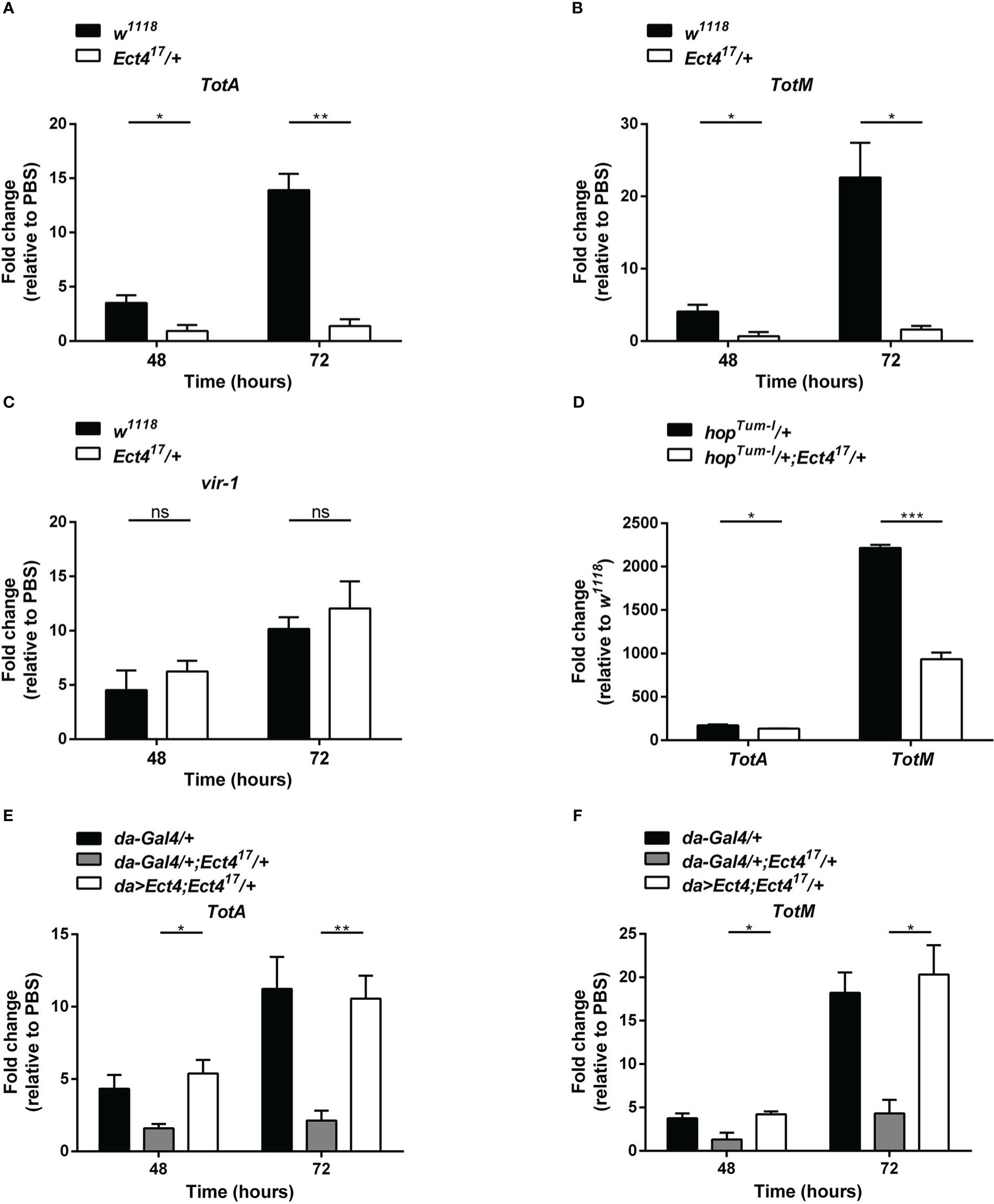
Figure 4 Ect4 is required for TotA and TotM induction in response to DCV infection. (A–C) Expression of JAK/STAT-dependent gene TotA, TotM, and vir-1 at 48 and 72 h after DCV infection determined by RT-qPCR in whole flies. Expression of the gene of interest was normalized to transcript levels of the housekeeping gene Rpl32 and expressed as fold change relative to mock infection (PBS). (D) Expression levels of TotA and TotM on 3-5 d-old unchallenged flies carrying one copy of hopTum-l allele with or without the Ect4 mutant allele. Expression of TotA and TotM was normalized to transcript levels of the housekeeping gene Rpl32 and expressed as fold change relative to wild-type (w1118) flies. (E, F) Expression levels of TotA and TotM at 48 and 72 h in control flies or Ect4 mutant flies overexpressing Ect4 transgene under a ubiquitous da-Gal4 driver upon DCV infection. Data represent the means ± standard errors of 3 independent pools of 10 male flies (A–F) for each genotype. T-test (A–F): *P< 0.05, **P< 0.01, ***P< 0.001, ns, not significant.
3.5 Ect4 physically interacts with Stat92E
To unravel the molecular mechanism underlying the relationship between Ect4 and JAK/STAT pathway, we examined whether Ect4 interacted with any known components of the JAK/STAT pathway. Differentially tagged forms of JAK/STAT pathway components and Ect4 were expressed in S2 cells, and co-immunoprecipitation studies were performed. As shown in Figure 5A, Ect4 is associated with the transcription factor Stat92E but not other key components (Hop or Dome) of the JAK/STAT pathway. Consistent with previous findings (36), Stat92E protein was located both in the cytoplasm and nucleus as visualized by immunofluorescence staining. Since the green fluorescent protein (GFP)-Ect4 fusion protein was localized in the cytoplasm, the interaction between the two proteins occurs in the cytoplasm (Figure 5B).
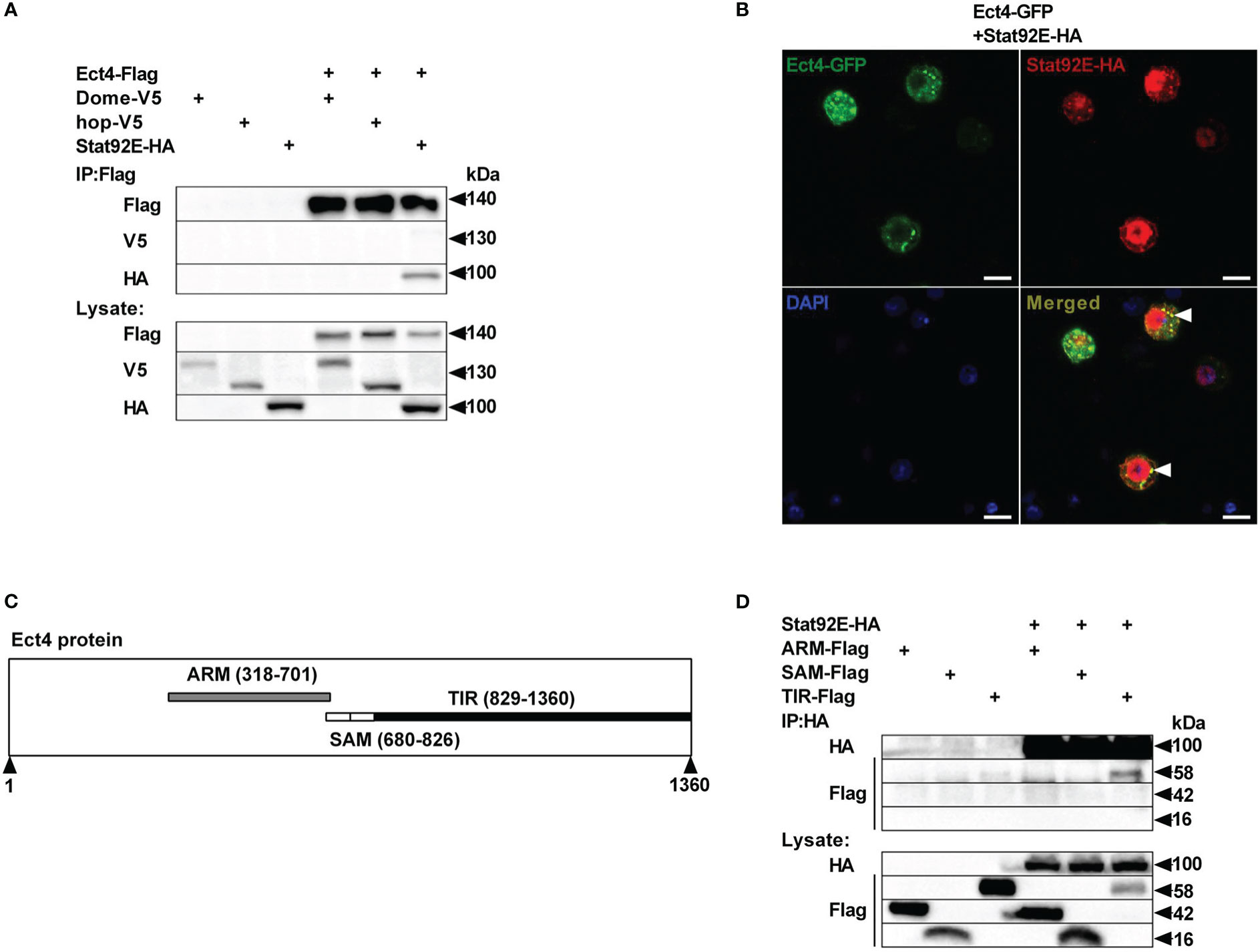
Figure 5 Ect4 co-localizes and physically associates with Stat92E. (A) S2 cells were transfected with combinations of expression plasmids as indicated. Cell lysates were immunoprecipitated with anti-Flag beads, followed by immunoblot analysis with the indicated antibodies. (B) S2 cells were transfected with Ect4-GFP in combination with Stat92E-HA, stained with DAPI (blue) and anti-HA antibody (Red), and imaged by confocal microscopy. Ect4 was co-localized with Stat92E in the cytoplasm (white arrow). (C) Ect4 protein includes ARM (gray), SAM (white), and TIR (black) domain. (D) S2 cells were cotransfected with Stat92E-HA and domains of Ect4. Cell lysates were immunoprecipitated with anti-HA beads and immunoblot analysis with the indicated antibodies. Scale bar: 10 μm.
Ect4 protein harbor three different domains: ARM (Armadillo motif) domains followed by two SAM (Sterile Alpha motif) domains and TIR (Toll -Interleukin-1 receptor) domain (Figure 5C). To further investigate the molecular basis of the interaction between Ect4 and Stat92E, a series of truncated forms of Ect4 were generated. Co-immuno-precipitation studies showed that ARM and SAM domains were likely not required for Ect4 to interact with Stat92E, whereas the TIR domain was essential since only the TIR domain co-immuno-precipitated with Stat92E (Figure 5D). Together, these results suggest that Ect4 may regulate the JAK/STAT signaling activity by interacting with Stat92E.
3.6 Ect4 is required for phosphorylation and nuclear translocation of Stat92E
As described thus far, we show that Ect4 regulates the expression of JAK/STAT-dependent genes TotA and TotM and is associated with Stat92E. It is intriguing to predict that Ect4 may affect Stat92E phosphorylation. To test this hypothesis, we employed an RNAi approach to knock down Ect4 in S2 cells (Figure 6A). Previous studies have shown that tyrosine residues of Stat92E are phosphorylated after treatment of S2 cells with pervanadate, which activates Stat92E in a ligand-independent manner, while activation is not present in untreated cells (36, 37). As shown in Figure 6, treatment with dsRNA targeting Ect4 mRNA resulted in a significant reduction of tyrosine phosphorylated Stat92E upon pervanadate treatment, as compared with the control using dsRNA targeting GFP. It was noted that upon DCV infection of S2 cells, phosphorylated Stat92E (p-Stat92E) was not detected by immunostaining, likely due to the transient activity of p-Stat92E dimers. Therefore, is Ect4 required for the nuclear translocation of Stat92E? As expected, reduced nuclear translocation of Stat92E in response to the pervanadate stimulus was detected in cells treated with Ect4 RNAi (Figures 6C, D).
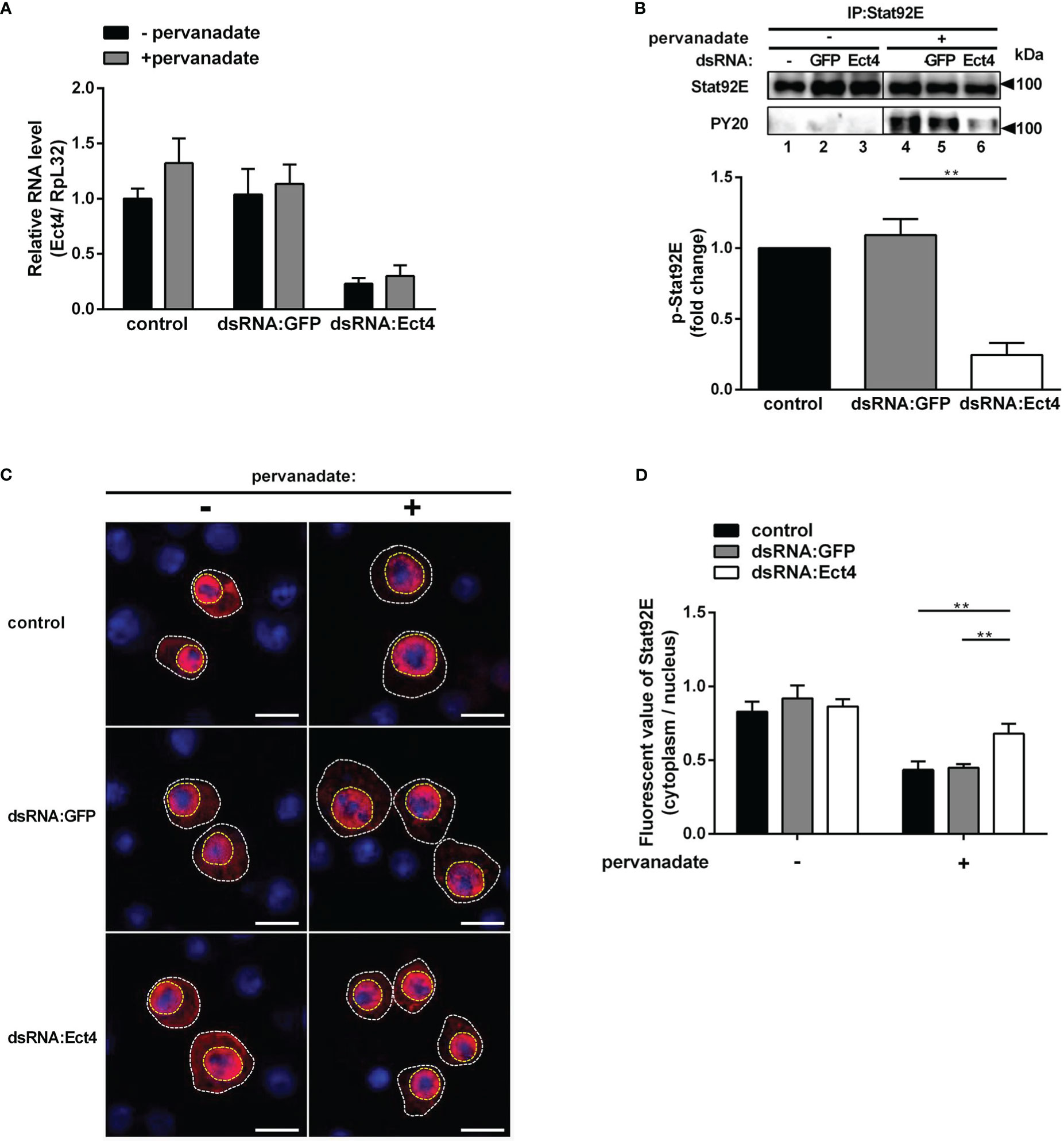
Figure 6 Knockdown of Ect4 mRNA decreased the phosphorylation of Stat92E. (A) S2 cells were pretreated with or without dsRNA targeting GFP or Ect4 transcripts for 72 h, and measurement of Ect4 mRNA by RT-qPCR to confirm RNAi efficiency. (B) After dsRNA treatment, S2 cells were either unstimulated or treated with pervanadate. Stat92E proteins were immunoprecipitated with Stat92E antibody, followed by immunoblot analysis with the Stat92E or PY20 antibodies. In the absence of pervanadate, phosphorylated-tyrosine Stat92E (p-Stat92E) was undetectable (lane 1-3). In contrast, after pervanadate treatment, p-Stat92E increased to levels detectable by western blot (lanes 4-6). The phosphorylated Stat92E was quantified from n = 3 independent experiments. (C) After transfection with Stat92E-HA, S2 cells were treated with or without dsRNA targeting GFP or Ect4 for 72 h and then left unstimulated or treated with pervanadate. Cells were stained with anti-HA antibody (red) and DAPI (blue) and imaged by confocal microscopy. (D) Quantification assays of the ratio between the fluorescent signal intensity of Stat92E in the nucleus (surrounded by a yellow dashed line) and in the cytoplasm (the area in the white dotted line subtracts the area from the yellow dashed line). Cell samples collected from (C), n=15. Scale bar: 10 μm. Data represent the means ± standard errors. The t-test (B, D): **P< 0.01.
4 Discussion
The innate immune system processes pathogen-induced pathways that detect the pathogen and induce the expression of antiviral effectors that control its proliferation (38). Consequently, it is expected that insufficient resistance mechanisms will lead to an increase in viral load, increased morbidity, and reduced survival. An in-depth understanding of antiviral resistance is important for developing novel methods for treating viral infections and other diseases. Nevertheless, mechanisms of resistance still need to be clearly understood.
Invertebrate Ect4 orthologues play a positive role in innate immunity. The Ect4 orthologue in C. elegans (TIR-1) and L. vannamei (LvSarm) were required to express antimicrobial peptides. Depletion of both led to decreased survival of the animals upon bacterial infections (24, 26, 39). Kemp et al. (40) reported that Ect4 is responsive to DCV infection. And about a 2-fold increase of Ect4 transcription at 72 hpi was observed in this study (data not shown), indicating that Ect4 participated in the immune response upon DCV infection. Furthermore, our study showed that Ect4 contributes to resistance and regulates JAK/STAT signaling in Drosophila. Flies with down-regulated Ect4 showed significantly elevated viral replication and earlier mortality after the DCV challenge, and a high level of Ect4 expression was associated with increased resistance to DCV.
Compared with invertebrate Ect4 orthologues, the mammalian orthologue SARM acts as a negative regulator of TLR signaling and is not directly antiviral, as mice lacking SARM show enhanced survival after Bunyavirus infection (41) because SARM family members have acquired diverse biological functions during evolution. For example, while Ect4 is essential for development in Drosophila, SARM1 is redundant for viability in mice (21). A previous study revealed that SARM is expressed mainly in the mouse brain, whereas its expression in other tissues, such as the spleen and the lymph node, was low (42). However, due to the lack of suitable anti-Ect4 antibodies, detecting Ect4 protein expression in Drosophila tissue sections was unsuccessful in the present study. Our results show that ectopic overexpression or knockdown of Ect4 in the fat body has a positive or negative effect on immune resistance upon DCV infection, suggesting that Ect4 may regulate antiviral immune system function mainly in the fat body.
Our study demonstrated that down-regulation in Ect4 does not directly interfere with the siRNA pathway. Instead, Ect4 regulates JAK/STAT dependent gene expression, TotA, and TotM, in response to DCV infection. A previous study revealed that the proper level of JAK/STAT signaling activation is required for normal immune response: hyper-activation of JAK/STAT triggered early mortality and loss of function mutations of hop in flies causing reduced JAK/STAT activation in flies, also decreasing resistance upon a challenge with DCV (18, 43). Despite being elicited by DCV and commonly used as a read-out of JAK/STAT activation, the function of TotA and TotM in Drosophila remains unclear. The protein products encoded by the Turandot gene family are protein chaperones or signaling molecules, which are produced in the fat body and secreted into the hemolymph (44, 45). This inflammatory response is reminiscent of the acute phase response in mammals, which can be activated by infection and produce acute phase protein. These proteins are involved in the immune responses, including host defense, vascular permeability, and coagulation.
Furthermore, TotM enhanced tolerance against fungal sexually transmitted infections (STIs), and TotA confers resistance to heat stress (45, 46). The Tot gene family regulates diverse fly physiology aspects that coordinate resistance or tolerance to immune challenges. Indeed, the present study showed that Ect4 is required for virus-induced expression of TotA and TotM genes. However, Ect4 is dispensable for vir-1 induction in response to DCV infection. It is likely that different factors are involved in the regulation of JAK/STAT downstream genes and that less p-Stat92E in Ect4 RNAi flies sufficient for inducing vir-1 expression, which requires a lower threshold of STAT activity.
Ect4 interacts with Stat92E in S2 cells through the highly conserved TIR domain of SARM family origin, which have roles in cell death and neuronal destruction in mammals (47). As an adaptor, SARM has been reported that interact with the mitochondrial antiviral-signaling protein MAVS in the mitochondria to mediate cell death during virus infection (41). Mitochondrial localization of tyrosine-phosphorylated STAT5, a homolog of Stat92E in mammalian, has been supposed to modulate cellular metabolism in cytokine-stimulated cells (48, 49). We show that down-regulation of Ect4 reduced phosphorylated Stat92E upon pervanadate treatment in S2 cells, which suggests a role of Ect4 in regulating cell death through the modulation of JAK/STAT via the interaction with Stat92E.
In addition to antiviral immune defense, apoptosis is a conserved mechanism of programmed cell death that can prevent the infection before viral replication is completed (50, 51). Our in vivo study revealed that Ect4 mutants showed enhanced mortality and increased viral load upon DCV challenge. We seek to further elucidate the unknown mechanisms of antiviral response in Drosophila by assessing whether Ect4 affects host resistance to viral infection by regulating cell death.
Our results demonstrate the novel roles for Drosophila Ect4 in regulation of JAK/STAT signaling pathway and protection against DCV infection. It is still unclear if Ect4 also participated in the control of other virus infection. The contribution of JAK/STAT signaling to Drosophila antiviral protection is virus-specific. Although JAK/STAT pathway can be activated by RNA viruses, including DCV, CrPV, FHV, and DXV, it is only required for resistance against two Dicistroviridae family members, DCV and CrPV (18). Our data suggest that Ect4 is required for phosphorylation and nuclear translocation of Stat92E. Future studies should investigate if the involvement of Ect4 in activating the JAK/STAT pathway impart resistance in Drosophila to other virus infection.
The tight regulation of immune-related signal transduction cascades is essential for the defense against a wide range of pathogens. However, although the key components of the JAK/STAT pathway have been identified, the ‘non-core’ pathway activity regulators are less known. In mammals, poly (ADP-ribose) polymerase PARP9 was recently reported as a noncanonical sensor for RNA viruses that depends on the PI3K/AKT3 pathway to produce antiviral type I interferon (52). PARP9 interacted with the E3 ubiquitin ligase DTX3L and STAT1 functioned as a chaperone to enhance levels of the PARP9-DTX3L protein complex and STAT1-mediated interferon-stimulated gene expression (53). Another E3 ubiquitin ligase TRIM18 recruited protein phosphatase 1A (PPM1A), a negative regulator of STAT1, to dampen type I interferon-mediated antiviral innate immunity for promoting virus infection (54, 55). Given the conserved nature of the JAK/STAT pathway, Drosophila homologs of PARP9 and TRIM18 are potential candidates for JAK/STAT pathway regulators. It will be intriguing to investigate whether other factors or pathways are involved in Ect4-mediated JAK/STAT pathway modulation and defense against viral infection. Further exploration will yield more insights into the current understanding of the JAK/STAT pathway immune regulatory mechanism and contributes to establishing an immune signaling network.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
ZH and PX carried out experiments. SG contributed to data collection. YH analyzed confocal images. YL, SY, and FS conducted statistical analysis. KA and W-MD revised the manuscript. JL and RJ contributed to the conception and design of the experiments. WW wrote the first draft of the manuscript. JC directed the studies. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Fujian Province of China (2020J01856), Fuzhou Science and Technology Planning Project of (2022-P-008), Research Program of Minjiang University (MYK21004), and Special Projects for Local Science and Technology Development Guided by the Central Government of Fujian Province (2021L3020022).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1135625/full#supplementary-material
References
1. Younes S, Al-Sulaiti A, Nasser EAA, Najjar H, Kamareddine L. Drosophila as a model organism in host–pathogen interaction studies. Front Cell Infect Mi (2020) 10:214. doi: 10.3389/fcimb.2020.00214
2. Swevers L, Liu J, Smagghe G. Defense mechanisms against viral infection in Drosophila: RNAi and non-RNAi. Viruses-Basel (2018) 10(5):230. doi: 10.3390/v10050230
3. Leggewie M, Schnettler E. RNAi-mediated antiviral immunity in insects and their possible application. Curr Opin Virol (2018) 32:108–14. doi: 10.1016/j.coviro.2018.10.004
4. Lamiable O, Arnold J, de Faria IJ, Olmo RP, Bergami F, Meignin C, et al. Analysis of the contribution of hemocytes and autophagy to Drosophila antiviral immunity. J Virol (2016) 90:5415–26. doi: 10.1128/JVI.00238-16
5. Holleufer A, Winther KG, Gad HH, Ai X, Chen Y, Li L, et al. Two cGAS-like receptors induce antiviral immunity in Drosophila. Nat (2021) 597:114–8. doi: 10.1038/s41586-021-03800-z
6. Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, et al. cGAS-like receptors sense RNA and control 3’2’-cGAMP signalling in drosophila. Nature (2021) 597:109–13. doi: 10.1038/s41586-021-03743-5
7. Machado SR, van der Most T, Miesen P. Genetic determinants of antiviral immunity in dipteran insects–compiling the experimental evidence. Dev Comp Immunol (2021) 119:104010. doi: 10.1016/j.dci.2021.104010
8. Schneider J, Imler JL. Sensing and signalling viral infection in Drosophila. Dev Comp Immunol (2021) 117:103985. doi: 10.1016/j.dci.2020.103985
9. Tafesh-Edwards G, Eleftherianos I. Drosophila immunity against natural and nonnatural viral pathogens. Virol (2020) 540:165–71. doi: 10.1016/j.virol.2019.12.001
10. Myllymäki H, Rämet M. JAK/STAT pathway in Drosophila immunity. Scand J Immunol (2014) 79(6):377–85. doi: 10.1111/sji.12170
11. Tokusumi Y, Tokusumi T, Schulz RA. Mechanical stress to Drosophila larvae stimulates a cellular immune response through the JAK/STAT signaling pathway. Biochem Biophys Res Co (2018) 502(3):415–21. doi: 10.1016/j.bbrc.2018.05.192
12. Zhang Z, Zhang Y, Gao M, Cui X, Yang Y, Van Duijn B, et al. Steamed Panax notoginseng attenuates anemia in mice with blood deficiency syndrome via regulating hematopoietic factors and JAK-STAT pathway. Front Pharmacol (2020) 10:1578. doi: 10.3389/fphar.2019.01578
13. Bang IS. JAK/STAT signaling in insect innate immunity. Entomol Res (2019) 49(8):339–53. doi: 10.1111/1748-5967.12384
14. Su Y, Tao T, Liu X, Su W. JAK-STAT signaling pathway in non-infectious uveitis. Biochem Pharmacol (2022) 204:115236. doi: 10.1016/j.bcp.2022.115236
15. Hu X, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct Tar (2021) 6(1):1–33. doi: 10.1038/s41392-021-00791-1
16. Tzeng HT, Chyuan IT, Lai JH. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: a focus on molecular mechanisms and therapeutic potential. Biochem Pharmacol (2021) 193:114760. doi: 10.1016/j.bcp.2021.114760
17. Trivedi S, Starz-Gaiano M. Drosophila Jak/STAT signaling: regulation and relevance in human cancer and metastasis. Int J Mol Sci (2018) 19(12):4056. doi: 10.3390/ijms19124056
18. Merkling SH, Bronkhorst AW, Kramer JM, Overheul GJ, Schenck A, Van Rij RP. The epigenetic regulator G9a mediates tolerance to RNA virus infection in Drosophila. PloS Pathog (2015) 11:e1004692. doi: 10.1371/journal.ppat.1004692
19. Yang L, Qiu LM, Fang Q, Stanley DW, Ye GY. Cellular and humoral immune interactions between Drosophila and its parasitoids. Insect Sci (2021) 28(5):1208–27. doi: 10.1111/1744-7917.12863
20. Carty M, Bowie AG. SARM: From immune regulator to cell executioner. Biochem Pharmacol (2019) 161:52–62. doi: 10.1016/j.bcp.2019.01.005
21. Panneerselvam P, Ding JL. Beyond TLR signaling–the role of SARM in antiviral immune defense, apoptosis development. Int Rev Immunol (2015) 34(5):432–44. doi: 10.3109/08830185.2015.1065826
22. Balaji SP, Chand CV, Justin A, Ramanathan M. Telmisartan mediates anti-inflammatory and not cognitive function through PPAR-γ agonism via SARM and MyD88 signaling. Pharmacol Biochem Be (2015) 137:60–8. doi: 10.1016/j.pbb.2015.08.007
23. Akhouayri I, Turc C, Royet J, Charroux B. Toll-8/Tollo negatively regulates antimicrobial response in the Drosophila respiratory epithelium. PloS Pathog (2011) 7(10):e1002319. doi: 10.1371/journal.ppat.1002319
24. Wang PH, Gu ZH, Wan DH, Zhu WB, Qiu W, Weng SP, et al. Litopenaeus vannamei sterile-alpha and armadillo motif containing protein (LvSARM) is involved in regulation of penaeidins and antilipopolysaccharide factors. PloS One (2013) 8(2):e52088. doi: 10.1371/journal.pone.0052088
25. Monsanto-Hearne V, Tham AL, Wong ZS, Asgari S, Johnson KN. Drosophila miR-956 suppression modulates ectoderm-expressed 4 and inhibits viral replication. Virol (2017) 502:20–7. doi: 10.1016/j.virol.2016.12.009
26. Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, et al. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol (2004) 5(5):488–94. doi: 10.1038/ni1060
27. Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, et al. Distinct roles for drosophila dicer-1 and dicer-2 in the siRNA/miRNA silencing pathways. Cell (2004) 117:69–81. doi: 10.1016/S0092-8674(04)00261-2
28. Bischof J, Sheils EM, Bjorklund M, Basler K. Generation of a transgenic ORFeome library in Drosophila. nat. Protoc (2014) 9:1607–20. doi: 10.1038/nprot.2014.105
29. Mukherjee P, Winkler CW, Taylor KG, Woods TA, Nair V, Khan BA, et al. SARM1, not MyD88, mediates TLR7/TLR9-induced apoptosis in neurons. J Immunol (2015) 195:4913–21. doi: 10.4049/jimmunol.1500953
30. Magwire MM, Fabian DK, Schweyen H, Cao C, Longdon B, Bayer F, et al. Genome-wide association studies reveal a simple genetic basis of resistance to naturally coevolving viruses in Drosophila melanogaster. PloS Genet (2012) 8(11):e1003057. doi: 10.1371/journal.pgen.1003057
31. Mondal K, Dastidar AG, Singh G, Madhusudhanan S, Gande SL, VijayRaghavan K, et al. Design and isolation of temperature-sensitive mutants of Gal4 in yeast and Drosophila. J. Mol Biol (2007) 370(5):939–50. doi: 10.1016/j.jmb.2007.05.035
32. Kemp C, Imler JL. Antiviral immunity in drosophila. Curr Opin Immunol (2009) 21:3–9. doi: 10.1016/j.coi.2009.01.007
33. Deddouche S, Matt N, Budd A, Mueller S, Kemp C, Galiana-Arnoux D, et al. The DExD/H-box helicase dicer-2 mediates the induction of antiviral activity in drosophila. Nat Immunol (2008) 9:1425–32. doi: 10.1038/ni.1664
34. Bronkhorst AW, van Rij RP. The long and short of antiviral defense: small RNA-based immunity in insects. Curr Opin Virol (2014) 7:19–28. doi: 10.1016/j.coviro.2014.03.010
35. Luo H, Hanratty WP, Dearolf CR. An amino acid substitution in the Drosophila hopTum-l jak kinase causes leukemia-like hematopoietic defects. EMBO J (1995) 14:1412–20. doi: 10.1002/j.1460-2075.1995.tb07127.x
36. Ekas LA, Cardozo TJ, Flaherty MS, McMillan EA, Gonsalves FC, Bach EA. Characterization of a dominant-active STAT that promotes tumorigenesis in Drosophila. Dev Biol (2010) 344:621–36. doi: 10.1016/j.ydbio.2010.05.497
37. Sweitzer SM, Calvo S, Kraus MH, Finbloom DS, Larner AC. Characterization of a stat-like DNA binding activity in Drosophila melanogaster. J Biol Chem (1995) 270:16510–3. doi: 10.1074/jbc.270.28.16510
38. Mohammadi Pour P, Fakhri S, Asgary S, Farzaei MH, Echeverria J. The signaling pathways, and therapeutic targets of antiviral agents: focusing on the antiviral approaches and clinical perspectives of anthocyanins in the management of viral diseases. Front Pharmacol (2019) 10:1207. doi: 10.3389/fphar.2019.01207
39. Kurz CL, Shapira M, Chen K, Baillie DL, Tan MW. Caenorhabditis elegans pgp-5 is involved in resistance to bacterial infection and heavy metal and its regulation requires TIR-1 and a p38 map kinase cascade. Biochem Biophys Res Co (2007) 363(2):438–43. doi: 10.1016/j.bbrc.2007.08.190
40. Kemp C, Mueller S, Goto A, Barbier V, Paro S, Bonnay F, et al. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J Immunol (2013) 190:650–8. doi: 10.4049/jimmunol.1102486
41. Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity (2013) 38(4):705–16. doi: 10.1016/j.immuni.2013.02.013
42. Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile virus pathogenesis. J Virol (2009) 83:9329–38. doi: 10.1128/JVI.00836-09
43. Dostert C, Jouanguy E, Irving P, Troxler L, Galiana-Arnoux D, Hetru C, et al. The jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol (2005) 6:946–53. doi: 10.1038/ni1237
44. Ekengren S, Hultmark D. A family of turandot-related genes in the humoral stress response of Drosophila. Biochem Biophys Res Commun (2001) 284:998–1003. doi: 10.1006/bbrc.2001.5067
45. Ekengren S, Tryselius Y, Dushay MS, Liu G, Steiner H, Hultmark D. A humoral stress response in Drosophila. Curr Biol (2001) 11:714–8. doi: 10.1016/S0960-9822(01)00203-2
46. Zhong W, McClure CD, Evans CR, Mlynski DT, Immonen E, Ritchie MG, et al. Immune anticipation of mating in Drosophila: Turandot m promotes immunity against sexually transmitted fungal infections. Proc Biol Sci (2013) 280:2013–8. doi: 10.1098/rspb.2013.2018
47. Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc Natl Acad Sci USA (2016) 113:E6271–e80. doi: 10.1073/pnas.1601506113
48. Chueh FY, Leong KF, Yu CL. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem Biophys Res Commun (2010) 402:778–83. doi: 10.1016/j.bbrc.2010.10.112
49. Summers DW, DiAntonio A, Milbrandt J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci (2014) 34:9338–50. doi: 10.1523/JNEUROSCI.0877-14.2014
50. Settles EW, Friesen PD. Flock house virus induces apoptosis by depletion of Drosophila inhibitor-of-apoptosis protein DIAP1. J Virol (2008) 82:1378–88. doi: 10.1128/JVI.01941-07
51. Liu B, Behura SK, Clem RJ, Schneemann A, Becnel J, Severson DW, et al. P53-mediated rapid induction of apoptosis conveys resistance to viral infection in Drosophila melanogaster. PloS Pathog (2013) 9:e1003137. doi: 10.1371/journal.ppat.1003137
52. Xing J, Zhang A, Du Y, Fang M, Minze LJ, Liu YJ, et al. Identification of poly (ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat Commun (2021) 12(1):1–17. doi: 10.1038/s41467-021-23003-4
53. Zhang Y, Mao D, Roswit WT, Jin X, Patel AC, Patel DA, et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat Commun (2015) 16(12):1215–27. doi: 10.1038/ni.3279
54. Fang M, Zhang A, Du Y, Lu W, Wang J, Minze LJ, et al. TRIM18 is a critical regulator of viral myocarditis and organ inflammation. J Biomed Sci (2022) 29(1):1–20. doi: 10.1186/s12929-022-00840-z
Keywords: Ect4, Drosophila C virus, tyrosine phosphorylation, JAK/STAT pathway, innate immunity
Citation: Huang Z, Wang W, Xu P, Gong S, Hu Y, Liu Y, Su F, Anjum KM, Deng W-M, Yang S, Liu J, Jiao R and Chen J (2023) Drosophila Ectoderm-expressed 4 modulates JAK/STAT pathway and protects flies against Drosophila C virus infection. Front. Immunol. 14:1135625. doi: 10.3389/fimmu.2023.1135625
Received: 01 January 2023; Accepted: 23 January 2023;
Published: 03 February 2023.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Jules Hoffmann, Université de Strasbourg, FranceXiaochuan Liu, University of California, Riverside, United States
Copyright © 2023 Huang, Wang, Xu, Gong, Hu, Liu, Su, Anjum, Deng, Yang, Liu, Jiao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiyong Liu, bWFuZ3JpdmVyQGhvdG1haWwuY29t; Renjie Jiao, cmppYW9AZ3pobXUuZWR1LmNu; Jianming Chen, Y2hlbmptQG1qdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship