 Catarina Lopes1,2,3†
Catarina Lopes1,2,3† Tatiana C. Almeida1†
Tatiana C. Almeida1† Pedro Pimentel-Nunes4,5
Pedro Pimentel-Nunes4,5 Mário Dinis-Ribeiro1,6
Mário Dinis-Ribeiro1,6 Carina Pereira1,2*
Carina Pereira1,2*- 1Precancerous Lesions and Early Cancer Management Group, Research Center of IPO Porto (CI‐IPOP)/Rise@CI‐IPOP (Health Research Group), Portuguese Institute of Oncology of Porto (IPO Porto)/Porto Comprehensive Cancer Center (Porto.CCC), Porto, Portugal
- 2CINTESIS – Center for Health Technology and Services Research, University of Porto, Porto, Portugal
- 3ICBAS-UP – Institute of Biomedical Sciences Abel Salazar, University of Porto, Porto, Portugal
- 4Department of Surgery and Physiology, Faculty of Medicine, University of Porto (FMUP), Porto, Portugal
- 5Department of Gastroenterology, Unilabs, Porto, Portugal
- 6Department of Gastroenterology, Portuguese Institute of Oncology of Porto, Porto, Portugal
Upper gastrointestinal endoscopy is considered the gold standard for gastric lesions detection and surveillance, but it is still associated with a non-negligible rate of missing conditions. In the Era of Personalized Medicine, biomarkers could be the key to overcome missed lesions or to better predict recurrence, pushing the frontier of endoscopy to functional endoscopy. In the last decade, microbiota in gastric cancer has been extensively explored, with gastric carcinogenesis being associated with progressive dysbiosis. Helicobacter pylori infection has been considered the main causative agent of gastritis due to its interference in disrupting the acidic environment of the stomach through inflammatory mediators. Thus, does inflammation bridge the gap between gastric dysbiosis and the gastric carcinogenesis cascade and could the microbiota-inflammation axis-derived biomarkers be the answer to the unmet challenge of functional upper endoscopy? To address this question, in this review, the available evidence on the role of gastric dysbiosis and chronic inflammation in precancerous conditions of the stomach is summarized, particularly targeting the nuclear factor-κB (NF-κB), toll-like receptors (TLRs) and cyclooxygenase-2 (COX-2) pathways. Additionally, the potential of liquid biopsies as a non-invasive source and the clinical utility of studied biomarkers is also explored. Overall, and although most studies offer a mechanistic perspective linking a strong proinflammatory Th1 cell response associated with, but not limited to, chronic infection with Helicobacter pylori, promising data recently published highlights not only the diagnostic value of microbial biomarkers but also the potential of gastric juice as a liquid biopsy pushing forward the concept of functional endoscopy and personalized care in gastric cancer early diagnosis and surveillance.
1 An overview: Beyond and before imaging
Gastric cancer (GC) remains one of the deadliest cancers in the world. It became the 6th most diagnosed cancer type in 2020, with approximately 1.09 million new cases worldwide, and 769 thousand cancer-related deaths, data revealed by the International Agency for Research on Cancer (IARC) (1). Although GC rates have decreased over the last few decades, GC continues to be a major concern for public health, with an expected increase in the burden due to its prevalence and high mortality-to-incidence ratio associated with its detection at advanced stages (2).
GC develops through a series of steps caused by the exposure and interaction of several factors, often referred to as the “Correa Cascade” (3). Most of these factors have a cumulative effect on the gastric mucosal microenvironment, triggering degenerative changes, starting with chronic gastritis, most often caused by Helicobacter pylori (H. pylori) infection, and progressing via atrophy (AG), intestinal metaplasia (IM), dysplasia, and finally, the development of cancer (4). Genes involved in cell adhesion, signal transduction, differentiation, development, and DNA repair are all susceptible to being mutated by carcinogens throughout the carcinogenesis process (5). Somatic gene mutations or single nucleotide polymorphisms (SNPs) and epigenetic modifications involving DNA methylation are just a few examples of the wide range of important genetic effects that contribute to the molecular pathogenesis (6). These alterations are theorized to drive the progression of cells from stage to stage, culminating in the development of cancer (6).
The detection of early stages in this multistep process, namely AG and IM, is of utmost importance, since it allows for less invasive and more effective treatments, leading to increased survival (76% in Korea, where 90% of cancers are diagnosed early vs 25% in Europe) and quality of life of patients, and reduces the economic burden associated with the treatment of more advanced stages (7–10). Several strategies are available for the early detection of GC, from non-invasive detection of circulating biomarkers to upper digestive endoscopy (10, 11). The latter is considered the gold standard, although proven not cost-effective for screening outside high-risk regions (12). Moreover, patients at higher risk of developing early GC can be stratified using endoscopic and histologic classifications like the operative link on gastritis assessment (OLGA), and the operative link on gastric intestinal metaplasia assessment (OLGIM), which rank the severity and topography of atrophic and IM alterations in gastritis, respectively, from stages 0 to IV, with stage III or IV considered to be high risk for GC development (13). In fact, in Europe and according to published guidelines the endoscopic surveillance of patients diagnosed with stomach precancerous conditions/lesions is endorsed as a strategy to detect GC when minimally invasive treatment is possible (14). However, the upper gastrointestinal (GI) endoscopy is still associated with a non-negligible rate of missed conditions (about 9.4 %), as reported in the systematic review and meta-analysis by Pimenta-Melo et al. (15), and the minimally invasive removal of early lesions is associated with a significant risk of metachronous lesions development (16). In the Era of Personalized Medicine, should other biomarkers besides age, sex, family history and morphology/histology be added to the equation? And when should we do it? When first performing the endoscopy to overcome missed lesions? Or during surveillance to better predict recurrence/secondary lesions? Could the microbiota-inflammation axis-derived biomarkers challenge the purely histological and imaging nature of current approaches towards the concept of functional endoscopy and individualization of care (Figure 1)?
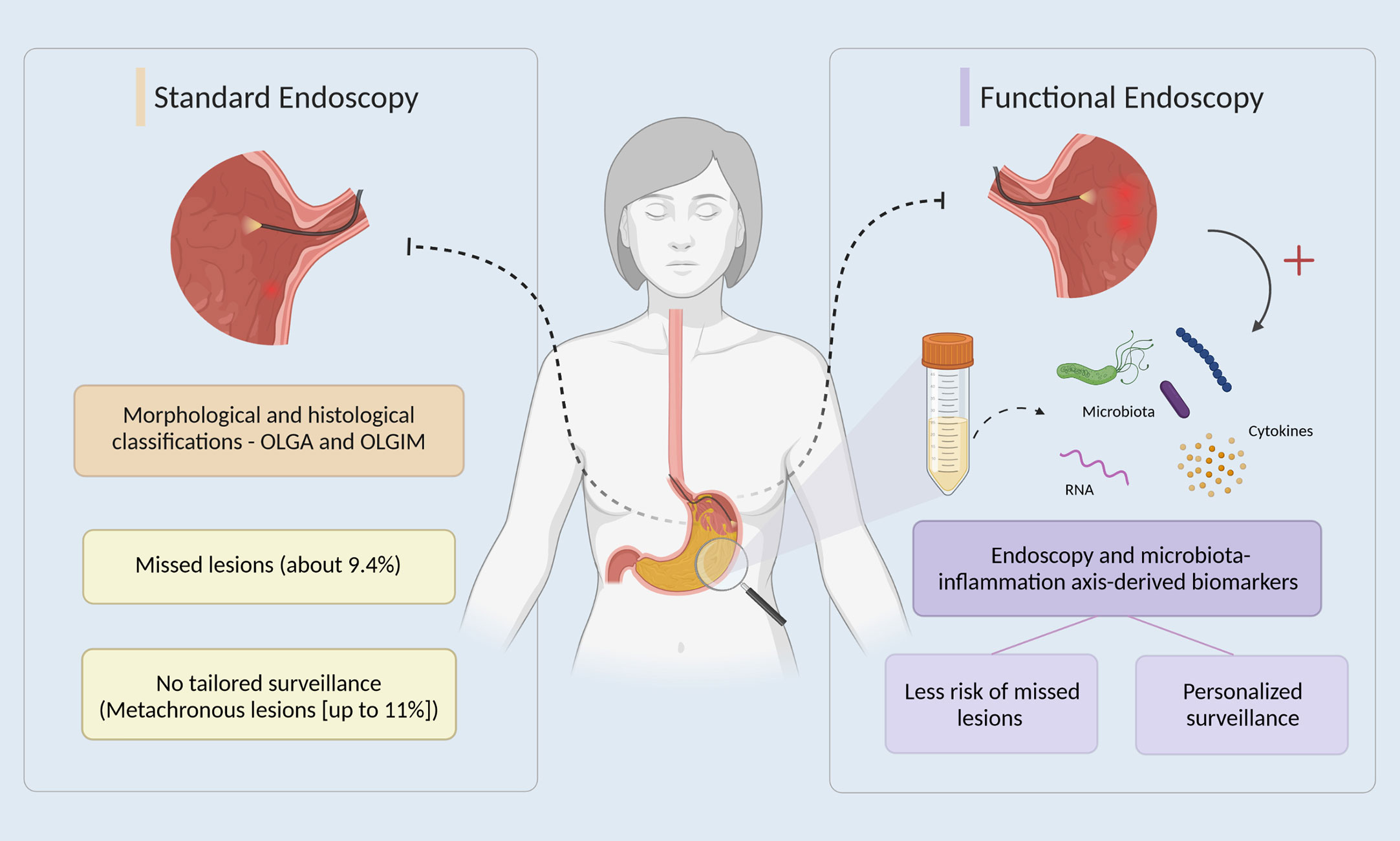
Figure 1 The potential of functional endoscopy in gastric cancer early detection and management. In standard endoscopy (left side), after an upper gastrointestinal endoscopy, patients are stratified using OLGA and OLGIM classifications, which rank the severity and topography of atrophic and IM alterations in gastritis (13). However, endoscopy alone is associated with a non-negligible rate of missed conditions and a significant risk of metachronous lesions development (15, 16). On the right side, the promise of functional endoscopy by also integrating, for example, dysbiosis-driven inflammatory biomarkers derived from liquid biopsies, namely gastric juice, towards the individualization of care by targeting a reduction of missed lesions and personalization of patients’ management in gastric cancer early detection.
To tackle these questions, we reviewed and will, in the next sections, summarize the available evidence on the role of gastric dysbiosis and chronic inflammation, particularly targeting the nuclear factor-κB (NF-κB), toll-like receptors (TLRs) and cyclooxygenase-2 (COX-2) pathways, in the early phases of gastric carcinogenesis. The potential of liquid biopsies as a non-invasive source, as well as the clinical utility of studied biomarkers, will be explored in the last sections.
2 Gastric dysbiosis as a main player in early gastric carcinogenesis
The predominant role of H. pylori in GC development has triggered an increasing interest in the study of gastric dysbiosis in last decade (17). In fact, gastric microbiota composition is highly variable between individuals (18). However, current research has found five main phyla in the normal stomach, including Firmicutes, Bacteroidites, Actinobacteria, Fusobacteria, and Proteobacteria, with Prevotella, Streptococcus, Veillonella, Rothia, and Haemophilus as the most common taxa (19–23). Human health depends on maintaining a balanced microbiome (18). Dysbiosis, characterized by alterations in microbiome content, has been linked to a broad set of disorders, including cancer (24). Studies that examined the gastric microbiome in the context of GC identified several bacterial communities characterized by different species (25, 26). Although several studies have documented stomach microbiota in GC patients, research on precancerous lesions is far more limited (26–32). An overview of the most common bacteria found throughout the Correa cascade can be found in Table 1.
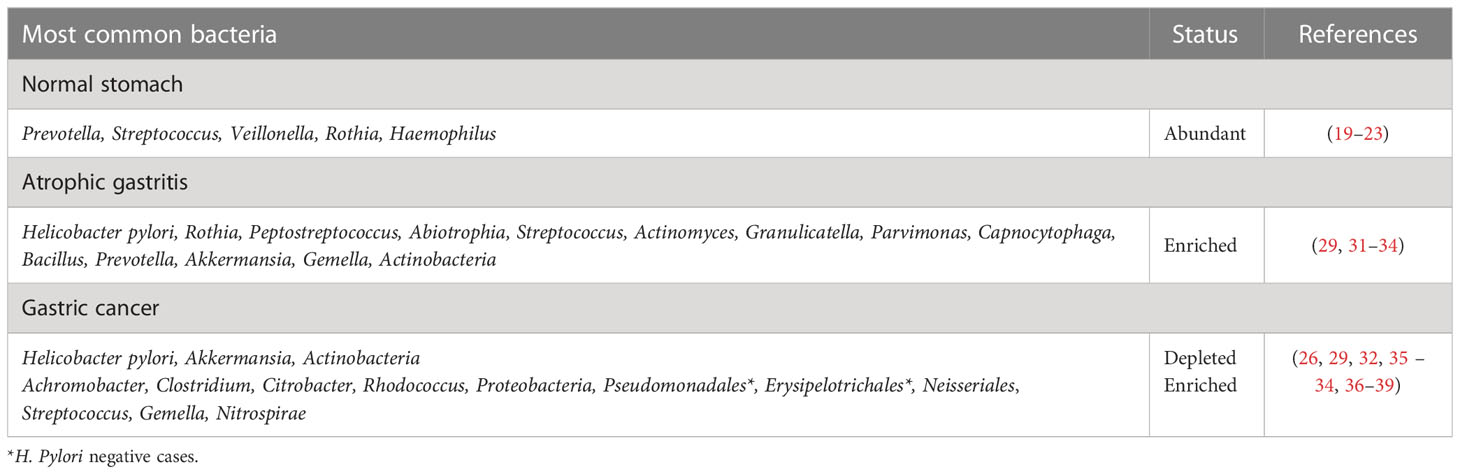
Table 1 Gastric dysbiosis throughout the Correa Cascade.
2.1 Helicobacter pylori-triggered gastric carcinogenesis
One of the most clinically significant bacteria in the context of carcinogenesis is H. pylori, a mobile, microaerophilic, rod-shaped bacterium that is able to survive in the stomach environment (40). H. pylori its enclosed in a protective capsule, make it more resistant to adverse conditions and antibiotics, and so, its classified as a Gram-negative bacterium (41). Like other Gram-negative bacteria, H. pylori cell wall comprises of lipopolysaccharide (LPS), which has been linked to pathogenicity by contributing to stomach H. pylori colonization and persistence (42). LPS is composed of lipid A, a core oligosaccharide, and an O-specific chain (43). H. pylori LPS O-specific surface chain has a structural similarity to Lewis blood type antigens, which are commonly present in the human gastric mucosa (42–44). Molecular mimicry of Lewis antigens gives H. pylori the ability to evade host immune detection, allowing its survival and prevalence in the stomach (44). However, compared to other bacterial LPS, H. pylori LPS exhibits lower toxic activity (45).
H. pylori infects around 50% of global population and is associated with 79% of newly diagnosed cases of GC each year (46). Since there is enough evidence to conclude that it can cause cancer in humans, IARC categorizes H. pylori as a class I carcinogen (47). Different strains around the world are shown to have different effects on cancer risk (48). East Asia has high incidences of H. pylori infection and high incidences of GC, while some other highly infected populations in South Asia and Africa do not, possibly explained by genotype differences of H. pylori (48). Falush et al. (49) sequenced the strains from 27 geographical ethnic groups, and defined four major groups: hpEurope, hpEastAsia, hpAfrica1 and hpAfrica2, names associated with their geographic distribution. Later, hpEastAsia was subdivided into hspEAsia (East Asia), hspMaori (Polynesians) and hspAmerind (native Americans), while hpAfrica1 was divided into hspWAfrica (South Africa, Americas and West Africa) and hspSAfrica (South Africa) (49). From these clusters, population with hpEastAsia strain showed the highest rates of GC, in contrast with hpAfrica1 and hpAfrica2 (49). Virulent strains vary in the expression of cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA), two of the most prominent virulence factors of H. pylori that contribute to inflammation, with these particularly aggressive strains (VacA and CagA positive [+]) linked to higher risk of GC and peptic ulcer disease (50–52) . East Asian type CagA is present in over 70% of patients with GC, and statistical studies have shown that East Asian type is more dangerous than CagA of the Western, explaining the high incidence of GC in East Asia (53, 54). In the hpAfrica2 strain, genomic structures are distinct, lacking the CagA gene, and hence explaining the high prevalence, but low rate, of GC (49, 55). On the other hand, the VacA gene is present in all strains, varying in its vacuolating activity (56). S1/m1 and s1/m2 VacA strains have high or moderate toxin activity, a stronger inflammatory response, more DNA damage to epithelial cells, and higher carcinogenic potential (more associated with hspEAsia), while s2/m2 strains do not vacuolize (57). H. pylori adhesins, especially blood group antigen-binding adhesin gene A (BabA), play an important role in bacteria colonization of gastric epithelial cells and contribute to the development of GC (58).
H. pylori produces the enzyme urease, which temporarily buffers the acidic environment by hydrolyzing urea into ammonia and carbon dioxide, forming a protection around the bacterium that allows it to survive the stomach acid (59). As mentioned above, H. pylori infection is considered the initial trigger for AG. Initiation of a chronic inflammatory process and direct toxic action of virulence factors are recognized as the key processes by which H. pylori leads to GC (60). However, the precise method by which this happens is still understood (61). Once established in the gastric mucosa, H. pylori induces inflammation, increasing the risk of gastric epithelium precancerous lesions (Figure 2) (27). After translocation, CagA activates NF-κB and promotes interleukin (IL)-8 synthesis, neutrophils are heavily recruited in response to infection and infiltrated into the stomach mucosa, which increases the severity of the inflammatory response (66). VacA protein destroys the tight junctions of epithelial cells, causing cell death by apoptosis and changes in cell signaling (50, 75–77). It also suppresses interleukin 2 (IL-2) synthesis, which is important for T-cell proliferation and survival, and decreased expression of surface IL-2 receptor (50, 75–77). Such outcomes are the result of VacA ability to suppress the activation of nuclear factor of activated T-cells (NFAT), a transcription factor that works as a central regulator of immune response genes and is crucial for efficient T-cell activation (78, 79). Ammonia produced by urea degradation causes damage at the cellular level and also induces the production and activation of inflammatory cytokines and phagocytes (60). Eradication of H. pylori may have a long-term positive effect on AG, as a meta-analysis indicated a regression of this condition after H. pylori treatment, although this effect was not detected for IM (80).
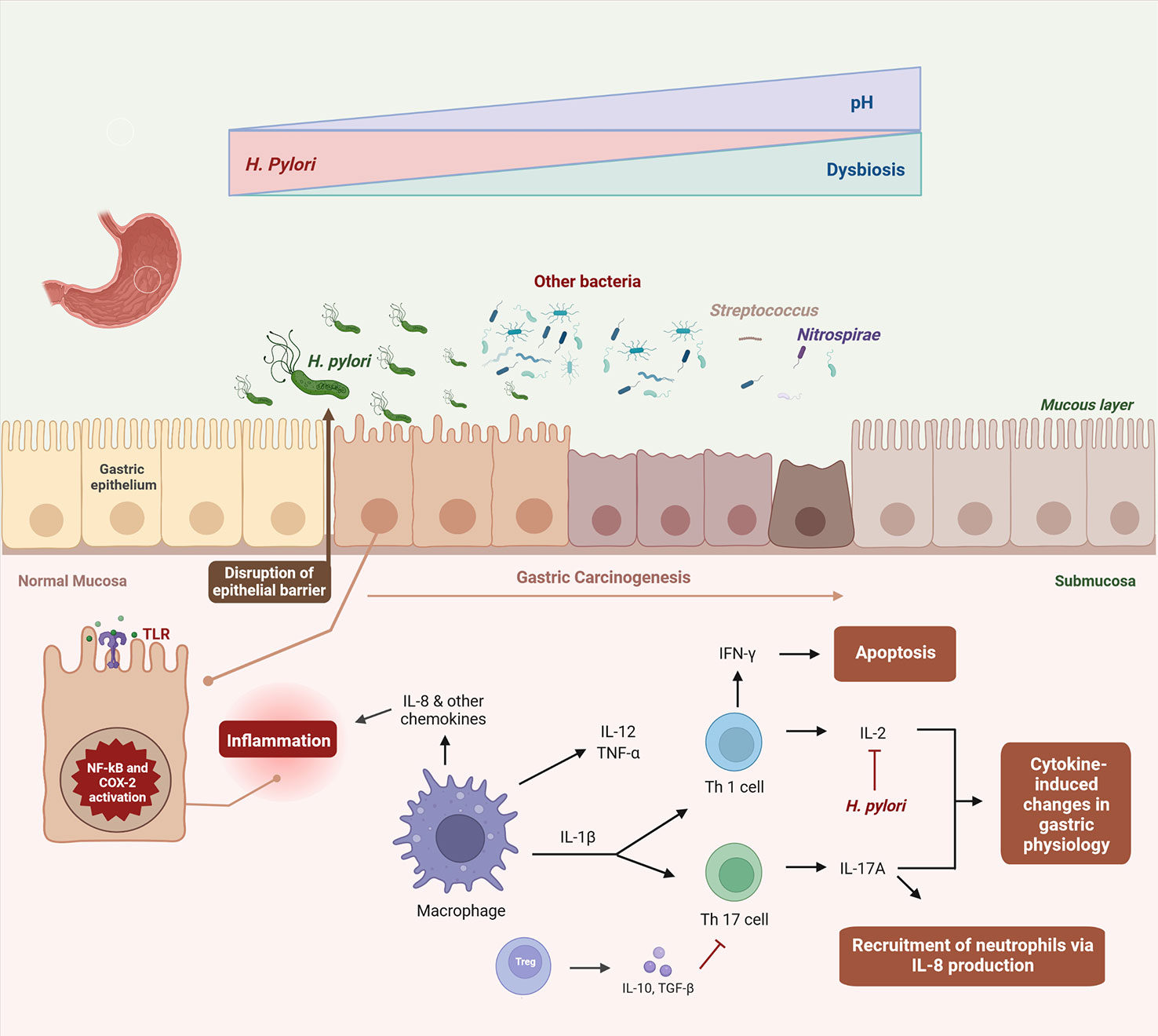
Figure 2 Dysbiosis-driven inflammation in gastric carcinogenesis. Once established in the gastric mucosa, H. pylori induces and maintains an inflammatory response, disrupting the epithelial barrier and altering the stomach acidity (27). In this altered environment, H. pylori colonization declines in the gastric carcinogenesis and other bacteria invade the gastric niche, culminating in dysbiosis (22, 32, 62–65). Virulence factors of H. pylori activate NF-κB and COX-2 pathways, promoting interleukin (IL)-8 synthesis and other immune cells, which are heavily recruited in response to infection, leading to cytokine-induced changes in gastric physiology (66–74). It also suppresses IL-2 synthesis, which is important for T-cell proliferation and survival (50, 75–77).
2.2 Non-H. pylori bacteria-mediated gastric carcinogenesis
A few studies found an association between certain gastric bacteria and precancerous lesions in the stomach (28–30). Due to the decreased production of gastric acid, various microorganisms are able to colonize the stomach in a patient with AG (35). Sung et al. (31) reported that Rothia, Peptostreptococcus, Abiotrophia, Streptococcus, Actinomyces, Granulicatella and Parvimonas were linked to AG and found in patients with IM after H. pylori eradication. Additionally, it has been postulated that the relative abundance of Capnocytophaga, Bacillus, and Prevotella bacteria rises as precancerous lesions evolve to dysplasia and GC, whereas the relative abundance of Helicobacter species declined (29). In a study by Ferreira et al. (26), Achromobacter, Clostridium, Citrobacter and Rhodococcus were found to be more common in Portuguese patients with GC than in those with chronic gastritis, in contrast with the notable decline of H. pylori in more advanced stages. Deng et al. (33) recently identified a substantial but non-statistically significant increase from 38% to 60% of Proteobacteria in biopsies from antral GC compared to biopsies from antrum gastritis, followed by a substantial reduction in Actinobacteria. In addition, they found an increase of Pseudomonadales and Erysipelotrichales in people with H. pylori-negative antral GC, compared to the infected individuals, who were enriched with Neisseriales (33). A recent study by Park et al. (34) reported a depletion of Akkermansia in GC stage, compared with his overabundance in the AG group, results consistent with other studies. This bacterium contributes to the maintenance of mucin and short-chain fatty acid balance by synthesizing a protein that degrades mucin, therefore promoting the barrier function (34). Hence, Akkermansia might play a preventative role in gastric carcinogenesis by impacting the integrity of the stomach mucosa (34). The authors also reported a significant increase of Lactobacillus and Veillonella in GC compared to other stages (34). Lactobacillus species are part of the human commensal gut bacteria (81). However, their overproliferation has been associated with IM or GC in humans and mice (26, 34). One possible explanation is that cancer cells are fueled by exogenous lactate supplied by lactic acid bacteria, which in turn promotes inflammation, angiogenesis, metastasis, and immune evasion (81).
In a study by Pimentel-Nunes et al. (32), Streptococcus and Gemella were present in patients with advanced AG, persist during the carcinogenesis cascade. Other studies support these findings, with Streptococcus being abundant in tumor tissue compared to normal (26, 35–38). Nitrospirae was also found in tumor tissue but absent in patients with chronic gastritis (39). Several Nitrospirae phylum members have been implicated in nitrate and nitrite metabolism (82). Consumption of nitrates is associated with an increased risk of developing GC, and it is possible that nitrosating bacteria, such as Nitrospirae, Staphylococcus, Veillonella, Clostridium, Haemophilus, Streptococcus, and Neisseria, can contribute to the formation of carcinogenic N-nitroso compounds (35, 83, 84).
2.3 Microbiota interplay in gastric carcinogenesis
The presence of H. pylori can alter the normal gastric microbiota, leading to dysbiosis and potentially the progression to GC (22, 62). Constant stimulation of the host immune system due to an imbalance in the gastric microbiota may lead to chronic inflammation of the gastric mucosa (62). It has been reported a decrease in the abundance of common phyla in the stomach when H. pylori is present (85). Animal studies have shown that the probiotic bacteria Lactobacillus, Bifidobacterium, and Saccharomyces have antagonistic effects against H. pylori, preventing from adhering, colonizing, and proliferating within the stomach, while Eubacterium limosum was shown to have the opposite effect (86–88). Three ways in which H. pylori interacts and disturbs the stomach microbiota have been proposed: 1) altering the stomach acidity, 2) providing substrates suitable for the proliferation of certain bacterial strains, and 3) modulating the host lifestyle and diet (89–92).
Overall, current research reports a greater diversity of gastric microbiota in healthy stomachs followed by a constant decrease throughout the cascade stages (63–65). The decline in mucosal microbial variety and richness may indicate that the disease is progressing (63–65) . H. pylori assumes its dominancy in initial atrophy stages, but not in IM (32).
Understanding how H. pylori and the microbiota affect the development of gastritis and cancer has come a long way. However, there have been discrepancies across studies, which could be related to the method used or differences in the host environment, such as diet, age, ethnicity, or genetics (93–95). In fact, some studies report considerable changes in stomach microbiota according to H. pylori status, while others fail to achieve statistical significance (96–98).
More detailed studies are still needed in well-defined human populations, to compare the topographic heterogeneity in microbial composition of the stomach of people with H. pylori infection with and without neoplastic lesions.
3 Dysbiosis-driven inflammation in gastric carcinogenesis: The role of NF-κB, TLR, and COX-2 pathways
In 2011, tumor-promoting inflammation was added to the hallmarks of cancer and the infiltration of tumors with both innate and adaptive immune system cells has been long recognized (99). Most major risk factors for common types of cancer, including chronic infections, tobacco, alcohol, or obesity, are triggers of chronic inflammation (100). Chronic inflammation driven by immune cells is a rather complex process requiring multiple cellular players (e.g., macrophages, lymphocytes) and pivotal regulators, including signaling pathways (e.g., NF-κB, JAK-STAT, TLR, MAPK), growth factors (e.g., VEGF, TGF-ß), as well as inflammatory factors (e.g., cytokines, chemokines), inflammasome, and inflammatory metabolites (e.g., prostaglandins, leukotrienes) , as reviewed by Zhao et al. (100). Tumor growth is promoted by an inflammatory response, which in turn is induced by inflammatory mediators that can attract immune cells to the lesion (101). That promotes the perfect conditions for the growth, invasion and metastization of tumor cells, known as the tumor microenvironment (101).
Emerging technologies such as single-cell RNA-sequencing (scRNA-seq) are delivering unprecedent insights of elusive immune-mediated mechanisms of gastric carcinogenesis by providing a comprehensive view of the cellular landscape and interactions within a tissue at a single-cell resolution, with the potential to contribute towards the development of more effective diagnostics and therapeutic targets (102–104).
In the stomach, the presence of inflammatory cells constitutes gastritis, which can evolve to atrophy and later to adenocarcinoma in a multistep process as aforementioned (105). This progression has been linked to gastric microbiota, since sterile mice colonized with three strains of Altered Schaedler’s flora (ASF) presented increased levels of pro-inflammatory mediators, as well as cancer-related genes, compared to aseptic mice (106).
In this section, the role of H. pylori in gastric inflammation will be explored, as well as the association between microbiota and major inflammatory players (NF-κB, TLRs, and COX-2).
3.1 Helicobacter pylori-triggered chronic gastritis
The main cause of chronic AG is the persistence of H. pylori infection (107). That requires a broad spectrum of adhesion factors, and H. pylori genomes from various strains contain over 30 genes encoding outer membrane proteins divided into Hop, Helicobacter outer membrane porins, and Hor, hop-related (108). BabA-mediated adherence of the bacterium, a member of the Hop family of proteins and the first adhesin discovered, is pH sensitive and allows the infection to adapt to shifts in stomach acidity resulting from chronic inflammation through mutations and other events in BabA-related genes (108, 109). Upon adhesion, the type IV secretion system (T4SS) components within the cagPAI pathogenicity island assemble a structure that mediates the transfer of CagA into the host cells in the epithelium, promoting the increased release of IL-1β and IL-8 (110, 111). The former allows a more effective inflammatory response, whereas the latter is a proinflammatory chemokine involved in neutrophils chemotaxis, quickly responding to stimulus from infectious agents and initializing the reaction (110). On the other hand, the same cagPAI strains that induce IL-1β also induce IL-10, an anti-inflammatory cytokine known to inhibit NF-κB activation and IL-8 expression, promoting the growth of bacteria (110). H. pylori response to neutrophils appears to vary from pro- to anti-inflammatory, depending on the integrity of T4SS and cagPAI (110). Additionally, loss of matrix metalloproteinase 7 (MMP-7), secreted mainly by epithelial cells, has been associated with significantly more severe gastric inflammation in mice infected with H. pylori (112). MMP-7 affects the extracellular matrix, potentially altering the microenvironment, and its loss leads to an increase in M1 macrophage activation and proinflammatory cytokines secretion by those cells (112).
Overall, the inflammatory microenvironment promoted by H. pylori can increase mutation rates, DNA damage through double strand breaks and aberrant DNA methylation, as well as other epigenetic changes (113). These alterations may lead to the progression of the gastric carcinogenesis cascade from chronic gastritis to atrophic gastritis, and residual DNA methylation after eradication may be involved in GC development (113). In its turn, H. pylori infection leads to the activation of anti-microbial mechanisms through the infiltration of macrophages and neutrophils into the gastric mucosa (114). These cells release the aforementioned chemokines and cytokines, reactive oxygen species (ROS) and reactive nitrogen species (RNS), increasing oxidative stress, MMPs, and PGs, such as PGE2, maintaining the tumor-promoting inflammatory microenvironment and enhancing genomic instability (114).
3.2 Microbiota-mediated NF-κB pathway in gastric carcinogenesis
The NF-κB family comprises five genes, NF-κB1, NF-κB2, RelA, c-Rel, and RelB, that encode seven proteins with a common Rel homology domain (RHD) able to mediate DNA binding, interaction with inhibitors and dimerization (115). These proteins remain predominantly in the cell cytoplasm due to their interaction with inhibitors, IκBs, which bind to the RHDs of NF-κB (115). A variety of stimulus can trigger NF-κB activation, including growth factors, cytokines, and signaling pathways, such as PI3K/Akt and Ras/MAPK (115). Upon activation, the NF-κB signaling pathway affects cellular proliferation and apoptosis, by targeting molecules such as Bcl2 and cyclins, as well as immune response and inflammation, by upregulating chemokines and cytokines as the ones mentioned in the previous section (116).
Induction of NF-κB pathway by H. pylori has been known for over 20 years (117). During infection, this pathway is readily employed to activate antibacterial immunity, playing an important role in microbiota-associated gastric tumorigenesis (116). In fact, in H. pylori-infected individuals, NF-κB activity was markedly increased compared to controls, suggesting an association between gastritis due to neutrophil infiltration and the activation of this pathway (118). On the other hand, NF-κB has been shown to be implicated in genomic instability through R-loop induction, associated with DNA damage and replication stress, and through DNA double strand breaks as a result of H. pylori infection mediated by T4SS (119, 120).
Countless molecules have been shown to interact with the NF-κB pathway in response to H. pylori, either by inhibiting it or enhancing its effects and playing a role in gastritis. For example, bacterial infection in the early development stage leads to microRNA (miR)-204 downregulation, resulting in decreased baculoviral IAP repeat-containing 2 (BIRC2) expression, a protein that promotes IκBα degradation (121). Consequently, this leads to NF-κB signaling enhancement by allowing its entrance in the nucleus (121). Similarly, loss of trefoil factor 1 (TFF1) expression, a tumor suppressor gene, leads to an increase of NF-κB transcription factors expression, modulating the inflammatory response, through the activation of the tumor necrosis factor (TNF) receptor 1 (TNFR1)/IκB kinase (IKK) pathway (122). Reconstitution of TFF1 expression in cells led to a significantly suppression of the increase of NF-κB nuclear staining mediated by H. pylori (123). Accordingly, Tff1-knockout (KO) mice infected with the bacterium showed higher chronic inflammation compared to uninfected mice (123). In the same mouse model, increased levels of aurora kinase A (AURKA) were also associated with stronger chronic inflammation, resulting from the consequent increase of NF-κB phosphorylation and activation (124). Moreover, tumor necrosis factor receptor-associated factor-interacting protein with forkhead-associated domain (TIFA) was reported to bind to TRAF6 and TRAF2, forming TIFAsomes, which trigger NF-κB upon H. pylori infection of cells (125). On the other hand, tumor necrosis factor‐α‐induced protein‐8 like-2 (TIPE2) was identified as a negative regulator of this pathway, being able to maintain immune homeostasis in an independent manner (126). Furthermore, it has been shown to modulate HOX transcript antisense RNA (HOTAIR) levels, a long non-coding RNA enhanced in early stages of gastric tumorigenesis, particularly gastritis (126).
In contrast, there are also numerous molecules influenced by the NF-κB pathway upon bacterial infection, mainly with increased expressions, namely the oncogenes β-catenin and c-Myc (127), RASAL2 (128), Rev-erbα (127), dopamine- and cAMP-regulated phosphoprotein Mr 32 kDa (DARPP-32), whose overexpression occurs early in IM (129), miR233-3p, which decreases AT-rich interactive domain-containing protein A1 (ARID1A) expression and promoting proliferation and migration of cells directly associated with CagA (130). This H. pylori virulent factor induces the NF-κB-mediation of let-7a downregulation, resulting in increased human telomerase reverse transcriptase (hTERT) expression in AG and IM, indicating a role of this marker in cancer initiation (131).
Regarding other bacterial species, bacterial abundance and colonization rates were increased in Nfkb2-KO mice compared to control mice infected with Helicobacter felis (H. felis) (132). In fact, NF- κB2 signaling disruption appeared to be crucial for gastric atrophy development in response to that infection (132). In another study by Shibata et al. (133), a mice model with IKK depletion in gastric cells presented faster H. felis-dependent progression to dysplasia, which was associated with increased apoptosis and necrosis due to cellular stress induced by the bacterium. The latter process involved upregulation of IL-1 and C-X-C motif ligand 2 (CXCL2), a chemokine involved in leukocytes chemotaxis, consequently enhancing chronic inflammation (133). Furthermore, as a probiotic, Lactobacillus casei was shown to inhibit cell proliferation and promote apoptosis through NF-κB signaling modulation (134).
3.3 Microbial activation of toll-like receptors
TLRs constitute a group of type I transmembrane glycosylated proteins able to detect microbes or products from microorganisms either outside (TLR1, TLR2, TLR4, TLR5, and TLR10) or inside the cell (TLR3, TLR7, TLR8, TLR9) (135). These proteins are able to sense pathogen-associated molecular patterns (PAMPs) or microbe-associated molecular patterns (MAMPs), leading to the activation of downstream signaling pathways in response to bacteria, fungi, protozoa or viruses (135, 136). TLR signaling can be either MyD88- or Tikk/IL-1 receptor (TIR) domain-containing adaptor inducing interferon-β (TRIF)-dependent, ultimately reaching the same endpoint: NF-κB activation and production of type I interferons, chemokines, and cytokines to fight infection (135). TLR expression, particularly TLR2, TLR4, and TLR5 has been found upregulated throughout gastric carcinogenesis cascade, including IM, dysplasia, and adenocarcinoma, showing an association between these receptors and the development of lesions in the stomach (137–139).
The relationship between TLRs and H. pylori has been extensively explored and reviewed throughout the years (140–144). After colonization, bacteria interact with TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 in the cell surface, as well as TLR9 in the intracellular environment, whose expression is dynamically regulated (145, 146). In fact, H, pylori infection has been associated with increased expression of some of these transmembrane receptors (147, 148). Protein CagL, one of T4SS components, acts as a TLR5 activator, independent of flagellin (149). On the other hand, TLR2 and TLR4 are ligands for H. pylori LPS, the former resulting in overexpression of IL-8 and the latter leading to the release of IL-12 and IL-1β, which induce differentiation of T helper (Th) 1 and Th17 cells, producers of pro-inflammatory cytokines, IL-2, IL-6, and IL-18, an IL-1β antagonist, thus contributing to immune response evasion (150, 151). IFN-γ is primarily produced by Th1 cells (152). Osaki et al. (153) conducted experiments on mice and human gastric cells and found that this cytokine induced apoptosis, or programmed cell death, in gastric epithelial cells. Using a mouse model of H. pylori infection, the authors found that mice lacking IFN-γ had reduced inflammation and less metaplasia compared to wild-type mice (153). Moreover, its expression was increased in patients with IM (153). Therefore, IFN-γ seems to be required for the progression of IM and its induction of gastric epithelial cell death appears to contribute to this process (153). Atrophy and atrophic gastritis can result from increased expression of IL-1β, which decreases the activity of the gastric proton pump, and release of IL-17A by Th17 cells, inducing parietal cell apoptosis (154–156). Regulatory T cells, also known as Tregs, play a crucial role in maintaining immune homeostasis and preventing excessive immune responses to gastric microbiota, being able to suppress the activation and proliferation of Th7 cells (157–159). However, imbalances in gastric microbiota can directly impact Treg cell number and function, contributing to the perpetuation of inflammation (160). Patients with gastritis and active H. pylori exhibit significantly elevated expression of Tregs, TGF-β, and IL-10 compared to non-infected individuals (161, 162). Therefore, the presence of these cells in the mucosa may inhibit the immune response, resulting in the prolonged presence of infection (163). The exact mechanisms by which Treg cells regulate immune responses and the interplay between these cells, gastric microbiota, and gastric inflammation is still not fully understood and is an active area of research.
Evidence suggests that using TLR4 antagonists, such as the TAK-242 molecule, may contribute to the treatment of H. pylori-associated gastric lesions, since it binds to the receptor and disrupts its interaction with LPS, inhibiting the activation of downstream signaling pathways (164). Concerning TLR9, its expression has been found increased in response to H. pylori DNA, potentiating cellular proliferation, migration and invasion (165). Moreover, in gastric atrophy, intracellular TLR9 is activated due to the release of damage-associated molecular patterns (DAMPs) from cellular and bacterial debris (166).
Numerous studies have focused on the impact and role of TLR polymorphisms in GC and precancerous lesions (167, 168). SNPs in TLR-encoding genes, namely TLR2, TLR4, TLR5, TLR9, and TLR10 have been associated with H. pylori infection risk and/or chronic gastritis (166, 169–173). On the other hand, carriers of the TLR1 rs4833095 T allele presented decreased risk of infection and chronic atrophic gastritis (173, 174). Lower risk of the latter was also observed in individuals carrying the TLR10 rs10004195 T allele, suggesting a potential role of these SNPs in gastric pathogenesis associated with H. pylori (174, 175).
Regarding non-H. pylori bacteria, Neisseria subflava has been shown to interact with TLR4 through LPS, resulting in IL-8 expression, whereas TLR9-KO mice were protected from gastric inflammation and hyperplasia induced by H. felis (176, 177).
3.4 Cyclooxygenase-2 role in gastritis-associated inflammation
COX-2 is an enzyme with dual enzymatic activity, cyclooxygenase and peroxidase, responsible for catalyzing the rate-limiting step in prostaglandins synthesis: the addition of molecular oxygen into arachidonic acid (178). Prostaglandins are biologically active lipids involved in a variety of physiological processes, including inflammation, and PGE2 is the best known and most abundant, appearing to affect virtually all hallmarks of cancer (178). COX-2 is an enzyme with dual enzymatic activity, cyclooxygenase and peroxidase, responsible for catalyzing the rate-limiting step in prostaglandins synthesis: the addition of molecular oxygen into arachidonic acid (178). Prostaglandins are biologically active lipids involved in a variety of physiological processes, including inflammation, and PGE2 is the best known and most abundant, appearing to affect virtually all hallmarks of cancer (178).
The first description of COX-2 expression in GC goes back to 1997 and many studies reported an increase of its levels in H. pylori-mediated gastritis, suggesting that high COX-2 levels is an early event induced by this bacteria in gastric carcinogenesis (67–74). Nevertheless, the publication period of those studies comprised mainly the early 2000’s, whereas most recent reports have been focusing on cancer prevention through COX-2 inhibition and drug repurposing (179). A review on the latter topic is available in the literature (180). In gastric adenocarcinomas, high levels of COX-2 expression are linked to lymph node metastasis and depth of invasion, suggesting that prostanoids generated by this enzyme increase the aggressive behavior of these tumors (16). COX-2 expression is particularly high in GC, and it is also higher in dysplastic lesions compared with normal mucosal tissue, revealing the contribution of COX-2 in gastric carcinogenesis even at the preinvasive state (16). Concerning its association with microbiota, full induction of this enzyme has been demonstrated to be dependent on direct bacterial contact, a functional T4SS system, and epidermal growth factor receptor (EGFR) activation (181). On the other hand, PGE2 signaling, and H. pylori infection are required for the induction of the chemokine C-C motif ligand 2 (CCL2), responsible for attracting macrophages to the site (181).
Dysregulation of the COX-2 pathway has been reported in GC, and genetic variants of these genes have been identified as susceptibility biomarkers for the development of this malignancy, as well as gastritis and IM (182–185). Consequently, higher PGE2 levels have been reported in the gastric juice of patients with chronic AG and IM compared to controls (186). Furthermore, treatment of advanced gastric lesions with a selective COX-2 inhibitor alone, celecoxib, revealed beneficial effects on the regression of the disease, like what was seen with H. pylori eradication (187). There is evidence on the interplay between TLR/MyD88 signaling and COX-2/PGE2 pathway, reviewed in (188), suggesting the former promotes tumorigenesis by activating the latter and generating an inflammatory microenvironment. Additionally, NF-κB has also been shown to mediate COX-2 expression, resulting in higher PGE2 levels (189, 190). Parallelly, the latter pathway has been linked to IL-8 production in gastric epithelial cells (191).
4 Liquid biopsies as a promising source of dysbiosis-derived biomarkers in early gastric cancer development
Liquid biopsies have revolutionized the field of Precision Oncology, easing the collection of samples and biomarker assessment through minimally invasive procedures and allowing continuous monitoring of a tumor over time with increased sensibility (192). Overall, the most frequently used in clinical practice are the serum markers carcinoembryonic antigen (CEA), a glycoprotein, and carbohydrate antigen (CA)19-9, both found overexpressed in GC (193). However, these proteins, as well as other common markers such as alpha-fetoprotein, CA125 and CA72-4, lack sensitivity and sensibility, with the combined detection of CEA, CA19-9 and CA72-4 presenting greater diagnostic value (194–196). The most assessed biomarkers targeting earlier stages of gastric carcinogenesis, namely AG include serum pepsinogen (PG) levels, gastrin, and anti-H. pylori antibody detection (197). The performance of the combination of these three biomarkers as a test for AG diagnosis has been reported in a meta-analysis by Zagari et al. (197), proving to be a reliable tool, but still requiring cost-effectiveness analysis. Other markers include neopterin and C-reactive protein (CRP), early inflammation molecules that increase significantly in IM and atrophy compared with chronic gastritis alone (198).
Choi et al. (199), using a metagenomic approach to characterize gastric juice samples, identified Helicobacter and Streptococcus as the most prevalent bacterial genera in gastric malignancies or ulcers when compared to controls. Furthermore, H. pylori-derived extracellular vesicles (EVs) were abundant and shown to contain CagA and VacA, inducing the production of cytokines, such as TNF-α, IL-6, IL-1β, and IL-8, and promoting inflammation (199). More recently, the gastric mucosa and gastric juice microbial composition were proven to change considerably from superficial gastritis to GC, with a panel including Gemella, Haemophilus, Peptostreptococcus, Streptococcus, and Veillonella, validated in an independent set with robust performance (200). Interestingly, the bacterial community profile in the gastric mucosa converged to the one found in gastric juice throughout disease progression, suggesting an interchange of microbes between the tissue and the liquid biopsy in direct contact (200). Thus, gastric fluid appears as a promising source of biomarkers, since it reflects the functional state of the stomach, potentially complementing and enhancing the accuracy of endoscopy as the gold standard for precancerous stomach and GC detection and surveillance (11). In fact, a novel device that allows H. pylori detection and corpus AG diagnosis during endoscopy from gastric juice, regardless of proton inhibitor therapy or previous eradication, has been presented, positively impacting the use of health resources and patients comfort (201–203). Furthermore, a systematic review was recently published on the role of non-blood liquid biopsies in GC development and detection (11).
5 Clinical utility of dysbiosis-derived biomarkers in gastric cancer-targeted screening and personalized surveillance
Although several advanced techniques have been developed aiming to enhance the accuracy of AG diagnosis by improving the visualization of gastric pre-malignant changes, including autofluorescence imaging (AFI), chromoendoscopy, high-definition endoscopy with magnification, and narrow band imaging (NBI), studies exploring the clinical utility of dysbiosis-driven inflammatory biomarkers have been rather scarce (204).
Overall, H. pylori role in inflammation and gastric carcinogenesis has been extensively explored, unlike other gastric bacteria. Nevertheless, a study by Liu et al. (17) reported other relevant bacteria with robust performance to distinguish superficial gastritis from AG (area under the ROC curve [AUC] = 0.86), IM (AUC = 0.71), and GC (AUC = 0.85), despite also including Helicobacter (14). Furthermore, the combination of six bacterial genera from tongue coating, excluding Helicobacter, was able to distinguish GC patients from healthy controls with a median area under the curve (AUC) value of 0.88, proving the potential of non-H. pylori bacteria as powerful diagnostic biomarkers for the detection of gastric pathologies (205).
6 Conclusion
Over a decade ago tumor-promoting inflammation was defined as an enabling characteristic fundamentally triggering the activation of the eight hallmark capabilities necessary for tumor growth and progression. In this review, we highlighted the role of inflammation in bridging the gap between gastric dysbiosis and precancerous stomach with chronic infection with, but not limited to, H. pylori eliciting a strong proinflammatory Th1 cell response in early stages of gastric carcinogenesis with several inflammatory-related biomarkers identified, namely NF-κB, TNF-α, IL-1β and IL-6. Considering most studies offered a mechanistic perspective, the clinical utility of the microbial-triggered inflammatory biomarkers in precancerous stomach tailored detection and management should be warranted in future studies. Furthermore, promising data recently published, albeit sparse, support not only the diagnostic value of microbial biomarkers but also the potential of gastric juice as a liquid biopsy. If validated in future studies, the non-invasive detection of inflammation-related biomarkers can push forward the field towards the concept of Functional Endoscopy and personalization of care in GC early diagnosis and surveillance.
Author contributions
CP, MD-R, and PP-N contributed to the conception and design of this review article. CL and TA prepared the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This article is a result of the project NORTE-01-0145-FEDER-000050, supported by North Portugal Regional Operational Program (NORTE 2020), under the PORTUGAL 2020 partnership agreement, through the European Regional Development Fund (ERDF). TA is a research grant holder in the scope of that project. CL (UI/BD/151488/2021) and CP (SFRH/BPD/114803/2016) are research fellowship holders supported by Fundação para a Ciência e Tecnologia (FCT), co-financed by European Social Funds (ESF) and national funds of MCTES under the Human Strategic Reference Framework (POCH).
Acknowledgments
We would like to acknowledge the Biorender website for their helpful tools and resources in creating the illustrations in this article. Figure 1 was created with BioRender.com. Figure 2 was adapted from “H. Pylori Pathogenesis”, by BioRender.com (2023), retrieved from https://app.biorender.com/biorender-templates.
Conflict of interest
MD-R receives funding from Fujifilm through a scopes loan.
The other authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AG, Atrophic gastritis; AUC, Area under the ROC curve; CA, Carbohydrate antigen; CagA , Cytotoxin-associated gene A; COX-2, Cyclooxygenase-2; GC , Gastric cancer; H. pylori , Helicobacter pylori; IL, Interleukin; IM , Intestinal metaplasia; LPS, Lipopolysaccharide; NF-κB, uclear factor-κB; PG, Prostaglandin; T4SS, Type IV secretion system; TLR, Toll-like receptor; acA, Vacuolating cytotoxin A.
References
1. Ferlay J LM, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, et al. Global cancer observatory: Cancer tomorrow (2020). Available at: https://gco.iarc.fr/tomorrow.
2. Wong MCS, Huang J, Chan PSF, Choi P, Lao XQ, Chan SM, et al. Global incidence and mortality of gastric cancer, 1980-2018. JAMA Netw Open (2021) 4(7):e2118457. doi: 10.1001/jamanetworkopen.2021.18457
3. Correa P. Human gastric carcinogenesis: A multistep and multifactorial process- first american cancer society award lecture on cancer epidemiology and prevention. Cancer Res (1992) 52(24):6735–40.
4. Sipponen P, Maaroos HI. Chronic gastritis. Scand J Gastroenterol (2015) 50(6):657–67. doi: 10.3109/00365521.2015.1019918
5. Holmes K, Egan B, Swan N, O'Morain C. Genetic mechanisms and aberrant gene expression during the development of gastric intestinal metaplasia and adenocarcinoma. Curr Genomics (2007) 8(6):379–97. doi: 10.2174/138920207783406460
6. Tan P, Yeoh KG. Genetics and molecular pathogenesis of gastric adenocarcinoma. Gastroenterology. (2015) 149(5):1153–62.e3. doi: 10.1053/j.gastro.2015.05.059
7. Oh CM, Won YJ, Jung KW, Kong HJ, Cho H, Lee JK, et al. Cancer statistics in Korea: Incidence, mortality, survival, and prevalence in 2013. Cancer Res Treat (2016) 48(2):436–50. doi: 10.4143/crt.2016.089
8. Anderson LA, Tavilla A, Brenner H, Luttmann S, Navarro C, Gavin AT, et al. Survival for oesophageal, stomach and small intestine cancers in Europe 1999-2007: Results from EUROCARE-5. Eur J Cancer. (2015) 51(15):2144–57. doi: 10.1016/j.ejca.2015.07.026
9. Kim JH, Kim SS, Lee JH, Jung DH, Cheung DY, Chung WC, et al. Early detection is important to reduce the economic burden of gastric cancer. J Gastric Cancer. (2018) 18(1):82–9. doi: 10.5230/jgc.2018.18.e7
10. Xia JY, Aadam AA. Advances in screening and detection of gastric cancer. J Surg Oncol (2022) 125(7):1104–9. doi: 10.1002/jso.26844
11. Lopes C, Chaves J, Ortigao R, Dinis-Ribeiro M, Pereira C. Gastric cancer detection by non-blood-based liquid biopsies: A systematic review looking into the last decade of research. United Eur Gastroenterol J (2023) 11(1):114–30. doi: 10.1002/ueg2.12328
12. Areia M, Spaander MC, Kuipers EJ, Dinis-Ribeiro M. Endoscopic screening for gastric cancer: A cost-utility analysis for countries with an intermediate gastric cancer risk. United Eur Gastroenterol J (2018) 6(2):192–202. doi: 10.1177/2050640617722902
13. Rugge M, Fassan M, Pizzi M, Farinati F, Sturniolo GC, Plebani M, et al. Operative link for gastritis assessment vs operative link on intestinal metaplasia assessment. World J Gastroenterol (2011) 17(41):4596–601. doi: 10.3748/wjg.v17.i41.4596
14. Pimentel-Nunes P, Libanio D, Marcos-Pinto R, Areia M, Leja M, Esposito G, et al. Management of epithelial precancerous conditions and lesions in the stomach (MAPS II): European society of gastrointestinal endoscopy (ESGE), European helicobacter and microbiota study group (EHMSG), European society of pathology (ESP), and sociedade portuguesa de endoscopia digestiva (SPED) guideline update 2019. Endoscopy. (2019) 51(4):365–88. doi: 10.1055/a-0859-1883
15. Pimenta-Melo AR, Monteiro-Soares M, Libanio D, Dinis-Ribeiro M. Missing rate for gastric cancer during upper gastrointestinal endoscopy: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol (2016) 28(9):1041–9. doi: 10.1097/MEG.0000000000000657
16. Ortigão R, Figueirôa G, Frazzoni L, Pimentel-Nunes P, Hassan C, Dinis-Ribeiro M, et al. Risk factors for gastric metachronous lesions after endoscopic or surgical resection: A systematic review and meta-analysis. Endoscopy (2022) 54(09):892–901. doi: 10.1055/s-0041-1724574
17. Liu C, Ng S-K, Ding Y, Lin Y, Liu W, Wong SH, et al. Meta-analysis of mucosal microbiota reveals universal microbial signatures and dysbiosis in gastric carcinogenesis. Oncogene. (2022) 41(28):3599–610. doi: 10.1038/s41388-022-02377-9
18. Cho I, Blaser MJ. The human microbiome: At the interface of health and disease. Nat Rev Genet (2012) 13(4):260–70. doi: 10.1038/nrg3182
19. Li TH, Qin Y, Sham PC, Lau KS, Chu KM, Leung WK. Alterations in gastric microbiota after h. pylori erradication and in different histological stages of gastric carcinogenesis. Sci Rep (2017) 7:44935. doi: 10.1038/srep44935
20. Ramaswamy K, Correa M, Koshy A. Non-healing gastric ulcer associated with candida infection. Indian J Med Microbiol (2007) 25(1):57–8. doi: 10.1016/S0255-0857(21)02236-2
21. Rickes S, Schultze U, Monkemuller K, Malfertheiner P. [Walter krienitz–his life and intuitive description of bacteria in the stomach]. Dtsch Med Wochenschr (2006) 131(23):1341–3. doi: 10.1055/s-2006-946579
22. Dias-Jacome E, Libanio D, Borges-Canha M, Galaghar A, Pimentel-Nunes P. Gastric microbiota and carcinogenesis: the role of non-Helicobacter pylori bacteria - a systematic review. Rev Esp Enferm Dig. (2016) 108(9):530–40. doi: 10.17235/reed.2016.4261/2016
23. Stringer AM, Gibson RJ, Bowen JM, Keefe DM. Chemotherapy-induced modifications to gastrointestinal microflora: evidence and implications of change. Curr Drug Metab (2009) 10(1):79–83. doi: 10.2174/138920009787048419
24. Thomas RM, Jobin C. The microbiome and cancer: Is the 'oncobiome' mirage real? Trends Cancer (2015) 1(1):24–35. doi: 10.1016/j.trecan.2015.07.005
25. Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. (2018) 67(6):1024–32. doi: 10.1136/gutjnl-2017-314281
26. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut. (2018) 67(2):226–36. doi: 10.1136/gutjnl-2017-314205
27. Cover TL, Peek RM Jr. Diet, microbial virulence, and Helicobacter pylori-induced gastric cancer. Gut Microbes (2013) 4(6):482–93. doi: 10.4161/gmic.26262
28. Wang Z, Gao X, Zeng R, Wu Q, Sun H, Wu W, et al. Changes of the gastric mucosal microbiome associated with histological stages of gastric carcinogenesis. Front Microbiol (2020) 11:997. doi: 10.3389/fmicb.2020.00997
29. Kadeerhan G, Gerhard M, Gao JJ, Mejias-Luque R, Zhang L, Vieth M, et al. Microbiota alteration at different stages in gastric lesion progression: a population-based study in linqu, China. Am J Cancer Res (2021) 11(2):561–75.
30. Liu D, Chen S, Gou Y, Yu W, Zhou H, Zhang R, et al. Gastrointestinal microbiota changes in patients with gastric precancerous lesions. Front Cell Infect Microbiol (2021) 11:749207. doi: 10.3389/fcimb.2021.749207
31. Sung JJY, Coker OO, Chu E, Szeto CH, Luk STY, Lau HCH, et al. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut. (2020) 69(9):1572–80. doi: 10.1136/gutjnl-2019-319826
32. Pimentel-Nunes P, Barros A, Pita I, Miranda I, Conceicao G, Borges-Canha M, et al. Gastric microbiome profile throughout gastric carcinogenesis: Beyond helicobacter. Scand J Gastroenterol (2021) 56(6):708–16. doi: 10.1080/00365521.2021.1902560
33. Deng Y, Ding X, Song Q, Zhao G, Han L, Ding B, et al. Alterations in mucosa-associated microbiota in the stomach of patients with gastric cancer. Cell Oncol (Dordr). (2021) 44(3):701–14. doi: 10.1007/s13402-021-00596-y
34. Park JY, Seo H, Kang CS, Shin TS, Kim JW, Park JM, et al. Dysbiotic change in gastric microbiome and its functional implication in gastric carcinogenesis. Sci Rep (2022) 12(1):4285. doi: 10.1038/s41598-022-08288-9
35. Sohn S-H, Kim N, Jo HJ, Kim J, Park JH, Nam RH, et al. Analysis of gastric body microbiota by pyrosequencing: Possible role of bacteria other than Helicobacter pylori in the gastric carcinogenesis. J Cancer Prev (2017) 22(2):115–25. doi: 10.15430/JCP.2017.22.2.115
36. Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter. (2014) 19(6):407–16. doi: 10.1111/hel.12145
37. Gong J, Li L, Zuo X, Li Y. Change of the duodenal mucosa-associated microbiota is related to intestinal metaplasia. BMC Microbiol (2019) 19(1):275. doi: 10.1186/s12866-019-1666-5
38. Sun QH, Zhang J, Shi YY, Zhang J, Fu WW, Ding SG. Microbiome changes in the gastric mucosa and gastric juice in different histological stages of Helicobacter pylori-negative gastric cancers. World J Gastroenterol (2022) 28(3):365–80. doi: 10.3748/wjg.v28.i3.365
39. Wang L, Zhou J, Xin Y, Geng C, Tian Z, Yu X, et al. Bacterial overgrowth and diversification of microbiota in gastric cancer. Eur J Gastroenterol Hepatol (2016) 28(3):261–6. doi: 10.1097/MEG.0000000000000542
40. Araujo GRL, Marques HS, Santos MLC, da Silva FAF, da Brito BB, Correa Santos GL, et al. Helicobacter pylori infection: How does age influence the inflammatory pattern? World J Gastroenterol (2022) 28(4):402–11. doi: 10.3748/wjg.v28.i4.402
41. O'Rourke J BG. Chapter 6: Morphology and ultrastructure. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and genetics. Washington DC (2001).
42. Dunne C, Dolan B, Clyne M. Factors that mediate colonization of the human stomach by Helicobacter pylori. World J Gastroenterol (2014) 20(19):5610–24. doi: 10.3748/wjg.v20.i19.5610
43. O'Toole PW CM. Cell envelope. In: Mobley, Hazell SL, editors. Helicobacter pylori: Physiology and genetics. Washington (DC);: ASM Press (2001).
44. Monteiro MA, Chan KH, Rasko DA, Taylor DE, Zheng PY, Appelmelk BJ, et al. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides Molecular mimicry between H. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. . J Biol Chem (1998) 273(19):11533–43. doi: 10.1074/jbc.273.19.11533
45. Muotiala A, Helander IM, Pyhala L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun (1992) 60(4):1714–6. doi: 10.1128/iai.60.4.1714-1716.1992
46. Hooi JKY, Lai WY, Ng WK, Suen MMY, Underwood FE, Tanyingoh D, et al. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology. (2017) 153(2):420–9. doi: 10.1053/j.gastro.2017.04.022
47. Park JY, Forman D, Waskito LA, Yamaoka Y, Crabtree JE. Epidemiology of Helicobacter pylori and CagA-positive infections and global variations in gastric cancer. Toxins (Basel). (2018) 10(4):163. doi: 10.3390/toxins10040163
48. Smith S, Fowora M, Pellicano R. Infections with Helicobacter pylori and challenges encountered in Africa. World J Gastroenterol (2019) 25(25):3183–95. doi: 10.3748/wjg.v25.i25.3183
49. Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, et al. Traces of human migrations in Helicobacter pylori populations. Science. (2003) 299(5612):1582–5. doi: 10.1126/science.1080857
50. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett (2014) 345(2):196–202. doi: 10.1016/j.canlet.2013.08.016
51. Enroth H, Kraaz W, Engstrand L, Nyren O, Rohan T. Helicobacter pylori strain types and risk of gastric cancer: A case-control study. Cancer Epidemiol Biomarkers Prev (2000) 9(9):981–5.
52. Weel JF, van der Hulst RW, Gerrits Y, Roorda P, Feller M, Dankert J, et al. The interrelationship between cytotoxin-associated gene a, vacuolating cytotoxin, and Helicobacter pylori-related diseases. J Infect Dis (1996) 173(5):1171–5. doi: 10.1093/infdis/173.5.1171
53. Vilaichone RK, Mahachai V, Tumwasorn S, Wu JY, Graham DY, Yamaoka Y. Molecular epidemiology and outcome of Helicobacter pylori infection in Thailand: A cultural cross roads. Helicobacter. (2004) 9(5):453–9. doi: 10.1111/j.1083-4389.2004.00260.x
54. Azuma T, Yamakawa A, Yamazaki S, Ohtani M, Ito Y, Muramatsu A, et al. Distinct diversity of the cag pathogenicity island among Helicobacter pylori strains in Japan. J Clin Microbiol (2004) 42(6):2508–17. doi: 10.1128/JCM.42.6.2508-2517.2004
55. Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, et al. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PloS Genet (2005) 1(4):e43. doi: 10.1371/journal.pgen.0010043
56. Yamaoka Y, Orito E, Mizokami M, Gutierrez O, Saitou N, Kodama T, et al. Helicobacter pylori in north and south America before Columbus. FEBS Lett (2002) 517(1-3):180–4. doi: 10.1016/S0014-5793(02)02617-0
57. Hussein NR. Helicobacter pylori and gastric cancer in the middle East: A new enigma? World J Gastroenterol (2010) 16(26):3226–34. doi: 10.3748/wjg.v16.i26.3226
58. Kalali B, Mejias-Luque R, Javaheri A, Gerhard MH. Pylori virulence factors: Influence on immune system and pathology. Mediators Inflamm (2014) 2014:426309. doi: 10.1155/2014/426309
59. Abadi ATB. Strategies used by Helicobacter pylori to establish persistent infection. World J Gastroenterol (2017) 23(16):2870–82. doi: 10.3748/wjg.v23.i16.2870
60. Baj J, Forma A, Sitarz M, Portincasa P, Garruti G, Krasowska D, et al. Helicobacter pylori virulence factors-mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells (2020) 10(1):27. doi: 10.3390/cells10010027
61. Ishaq S, Nunn L. Helicobacter pylori and gastric cancer: A state of the art review. Gastroenterol Hepatol Bed Bench. (2015) 8(Suppl 1):S6–S14.
62. Chen CC, Liou JM, Lee YC, Hong TC, El-Omar EM, Wu MS. The interplay between Helicobacter pylori and gastrointestinal microbiota. Gut Microbes (2021) 13(1):1–22. doi: 10.1080/19490976.2021.1909459
63. Miftahussurur M, Waskito LA, El-Serag HB, Ajami NJ, Nusi IA, Syam AF, et al. Gastric microbiota and Helicobacter pylori in Indonesian population. Helicobacter. (2020) 25(4):e12695. doi: 10.1111/hel.12695
64. Ndegwa N, Ploner A, Andersson AF, Zagai U, Andreasson A, Vieth M, et al. Gastric microbiota in a low-Helicobacter pylori prevalence general population and their associations with gastric lesions. Clin Transl Gastroenterol (2020) 11(7):e00191. doi: 10.14309/ctg.0000000000000191
65. Rajilic-Stojanovic M, Figueiredo C, Smet A, Hansen R, Kupcinskas J, Rokkas T, et al. Systematic review: Gastric microbiota in health and disease. Aliment Pharmacol Ther (2020) 51(6):582–602. doi: 10.1111/apt.15650
66. Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. (2005) 102(26):9300–5. doi: 10.1073/pnas.0409873102
67. Chan FK, To KF, Ng YP, Lee TL, Cheng AS, Leung WK, et al. Expression and cellular localization of COX-1 and -2 in Helicobacter pylori gastritis. Aliment Pharmacol Ther (2001) 15(2):187–93. doi: 10.1046/j.1365-2036.2001.00918.x
68. Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. (1999) 116(6):1319–29. doi: 10.1016/S0016-5085(99)70496-8
69. Kimura A, Tsuji S, Tsujii M, Sawaoka H, Iijima H, Kawai N, et al. Expression of cyclooxygenase-2 and nitrotyrosine in human gastric mucosa before and after Helicobacter pylori eradication. Prostaglandins Leukot Essent Fatty Acids (2000) 63(5):315–22. doi: 10.1054/plef.2000.0220
70. Sun WH, Yu Q, Shen H, Ou XL, Cao DZ, Yu T, et al. Roles of Helicobacter pylori infection and cyclooxygenase-2 expression in gastric carcinogenesis. World J Gastroenterol (2004) 10(19):2809–13. doi: 10.3748/wjg.v10.i19.2809
71. Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, et al. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol (2000) 157(3):729–35. doi: 10.1016/S0002-9440(10)64586-5
72. Tatsuguchi A, Sakamoto C, Wada K, Akamatsu T, Tsukui T, Miyake K, et al. Localisation of cyclooxygenase 1 and cyclooxygenase 2 in Helicobacter pylori related gastritis and gastric ulcer tissues in humans. Gut. (2000) 46(6):782–9. doi: 10.1136/gut.46.6.782
73. van Rees BP, Saukkonen K, Ristimäki A, Polkowski W, Tytgat GNJ, Drillenburg P, et al. Cyclooxygenase-2 expression during carcinogenesis in the human stomach. J Pathology. (2002) 196(2):171–9. doi: 10.1002/path.1033
74. Yamac D, Ayyildiz T, Coşkun U, Akyürek N, Dursun A, Seckin S, et al. Cyclooxygenase-2 expression and its association with angiogenesis, Helicobacter pylori, and clinicopathologic characteristics of gastric carcinoma. Pathol Res Pract (2008) 204(8):527–36. doi: 10.1016/j.prp.2008.01.002
75. Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol (2012) 2:37. doi: 10.3389/fcimb.2012.00037
76. Matsunari O, Shiota S, Suzuki R, Watada M, Kinjo N, Murakami K, et al. Association between Helicobacter pylori virulence factors and gastroduodenal diseases in Okinawa, Japan. J Clin Microbiol (2012) 50(3):876–83. doi: 10.1128/JCM.05562-11
77. Chauhan N, Tay ACY, Marshall BJ, Jain U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: An overview. Helicobacter. (2019) 24(1):e12544. doi: 10.1111/hel.12544
78. Burkitt MD, Duckworth CA, Williams JM, Pritchard DM. Helicobacter pylori-induced gastric pathology: Insights from in vivo and ex vivo models. Dis Model Mech (2017) 10(2):89–104. doi: 10.1242/dmm.027649
79. Coelho LGV, Marinho JR, Genta R, Ribeiro LT, Passos M, Zaterka S, et al. IVth Brazilian consensus conference on Helicobacter pylori infection. Arq Gastroenterol (2018) 55(2):97–121. doi: 10.1590/s0004-2803.201800000-20
80. Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. The long-term impact of Helicobacter pylori eradication on gastric histology: A systematic review and meta-analysis. Helicobacter. (2007) 12 Suppl 2:32–8. doi: 10.1111/j.1523-5378.2007.00563.x
81. Vinasco K, Mitchell HM, Kaakoush NO, Castano-Rodriguez N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochim Biophys Acta Rev Cancer. (2019) 1872(2):188309. doi: 10.1016/j.bbcan.2019.07.004
82. Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. (2013) 339(6120):708–11. doi: 10.1126/science.1232467
83. Chen Y, Peng Y, Yu J, Chen T, Wu Y, Shi L, et al. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget. (2017) 8(19):31802–14. doi: 10.18632/oncotarget.15992
84. Jo HJ, Kim J, Kim N, Park JH, Nam RH, Seok YJ, et al. Analysis of gastric microbiota by pyrosequencing: Minor role of bacteria other than Helicobacter pylori in the gastric carcinogenesis. Helicobacter. (2016) 21(5):364–74. doi: 10.1111/hel.12293
85. Vasapolli R, Schutte K, Schulz C, Vital M, Schomburg D, Pieper DH, et al. Analysis of transcriptionally active bacteria throughout the gastrointestinal tract of healthy individuals. Gastroenterology. (2019) 157(4):1081–92.e3. doi: 10.1053/j.gastro.2019.05.068
86. Zaman C, Osaki T, Hanawa T, Yonezawa H, Kurata S, Kamiya S. Analysis of the microflora in the stomach of Mongolian gerbils infected with Helicobacter pylori. J Gastroenterol Hepatol (2010) 25 Suppl 1:S11–4. doi: 10.1111/j.1440-1746.2009.06215.x
87. Yin YN, Wang CL, Liu XW, Cui Y, Xie N, Yu QF, et al. Gastric and duodenum microflora analysis after long-term Helicobacter pylori infection in Mongolian gerbils. Helicobacter. (2011) 16(5):389–97. doi: 10.1111/j.1523-5378.2011.00862.x
88. Sun YQ, Monstein HJ, Nilsson LE, Petersson F, Borch K. Profiling and identification of eubacteria in the stomach of Mongolian gerbils with and without Helicobacter pylori infection. Helicobacter. (2003) 8(2):149–57. doi: 10.1046/j.1523-5378.2003.00136.x
89. Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett (2018) 414:147–52. doi: 10.1016/j.canlet.2017.11.009
90. Ziebarth D. N-nitrosation of medicinal drugs catalysed by bacteria from human saliva and gastro-intestinal tract, including Helicobacter pylori. Carcinogenesis. (1997) 18(2):383–9. doi: 10.1093/carcin/18.2.383
91. Dong Q, Xin Y, Wang L, Meng X, Yu X, Lu L, et al. Characterization of gastric microbiota in twins. Curr Microbiol (2017) 74(2):224–9. doi: 10.1007/s00284-016-1176-8
92. Mohammadi SO, Yadegar A, Kargar M, Mirjalali H, Kafilzadeh F. The impact of Helicobacter pylori infection on gut microbiota-endocrine system axis; modulation of metabolic hormone levels and energy homeostasis. J Diabetes Metab Disord (2020) 19(2):1855–61. doi: 10.1007/s40200-020-00608-y
93. Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med (2018) 24(10):1526–31. doi: 10.1038/s41591-018-0160-1
94. Brooks AW, Priya S, Blekhman R, Bordenstein SR. Gut microbiota diversity across ethnicities in the united states. PloS Biol (2018) 16(12):e2006842. doi: 10.1371/journal.pbio.2006842
95. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. (2018) 555(7695):210–5. doi: 10.1038/nature25973
96. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci (2006) 103(3):732–7. doi: 10.1073/pnas.0506655103
97. Khosravi Y, Dieye Y, Poh BH, Ng CG, Loke MF, Goh KL, et al. Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients. ScientificWorldJournal. (2014) 2014:610421. doi: 10.1155/2014/610421
98. Kato S, Fujimura S, Kimura K, Nishio T, Hamada S, Minoura T, et al. Non-Helicobacter bacterial flora rarely develops in the gastric mucosal layer of children. Dig Dis Sci (2006) 51(4):641–6. doi: 10.1007/s10620-006-3185-0
99. Hanahan D, Weinberg Robert A. Hallmarks of cancer: The next generation. Cell. (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
100. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin Cancer Res (2009) 15(2):425–30. doi: 10.1158/1078-0432.CCR-08-0149
101. Jaroenlapnopparat A, Bhatia K, Coban S. Inflammation and gastric cancer. Diseases. (2022) 10(3):35. doi: 10.3390/diseases10030035
102. Hoft SG, Pherson MD, DiPaolo RJ. Discovering immune-mediated mechanisms of gastric carcinogenesis through single-cell RNA sequencing. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.902017
103. Zhao X, Wu S, Jing J. Identifying diagnostic and prognostic biomarkers and candidate therapeutic drugs of gastric cancer based on transcriptomics and single-cell sequencing. Pathol Oncol Res (2021) 27:1609955. doi: 10.3389/pore.2021.1609955
104. Kim J, Park C, Kim KH, Kim EH, Kim H, Woo JK, et al. Single-cell analysis of gastric pre-cancerous and cancer lesions reveals cell lineage diversity and intratumoral heterogeneity. NPJ Precis Oncol (2022) 6(1):9. doi: 10.1038/s41698-022-00251-1
105. Rugge M, Sugano K, Sacchi D, Sbaraglia M, Malfertheiner P. Gastritis: An update in 2020. Curr Treat Options Gastroenterology. (2020) 18(3):488–503. doi: 10.1007/s11938-020-00298-8
106. Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, et al. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. (2014) 63(1):54–63. doi: 10.1136/gutjnl-2013-305178
107. Salama NR, Hartung ML, Müller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol (2013) 11(6):385–99. doi: 10.1038/nrmicro3016
108. Backert S, Clyne M, Tegtmeyer N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Communication Signaling (2011) 9(1):28. doi: 10.1186/1478-811X-9-28
109. Bugaytsova JA, Björnham O, Chernov YA, Gideonsson P, Henriksson S, Mendez M, et al. Helicobacter pylori adapts to chronic infection and gastric disease via pH-responsive BabA-mediated adherence. Cell Host Microbe (2017) 21(3):376–89. doi: 10.1016/j.chom.2017.02.013
110. Sánchez-Zauco NA, Torres J, Pérez-Figueroa GE, Álvarez-Arellano L, Camorlinga-Ponce M, Gómez A, et al. Impact of cagPAI and T4SS on the inflammatory response of human neutrophils to Helicobacter pylori infection. PloS One (2013) 8(6):e64623. doi: 10.1371/journal.pone.0064623
111. Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol (2015) 10(6):955–65. doi: 10.2217/fmb.15.32
112. Krakowiak MS, Noto JM, Piazuelo MB, Hardbower DM, Romero-Gallo J, Delgado A, et al. Matrix metalloproteinase 7 restrains Helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization. Oncogene. (2015) 34(14):1865–71. doi: 10.1038/onc.2014.135
113. Yang H, Wei B, Hu B. Chronic inflammation and long-lasting changes in the gastric mucosa after Helicobacter pylori infection involved in gastric cancer. Inflammation Res (2021) 70(10):1015–26. doi: 10.1007/s00011-021-01501-x
114. Sokolova O, Naumann M. Crosstalk between DNA damage and inflammation in the multiple steps of gastric carcinogenesis. Curr Top Microbiol Immunol (2019) 421:107–37. doi: 10.1007/978-3-030-15138-6_5
115. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-κB in development and progression of human cancer. Virchows Archiv. (2005) 446(5):475–82. doi: 10.1007/s00428-005-1264-9
116. Peng C, Ouyang Y, Lu N, Li N. The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: Recent advances. Front Immunol (2020) 11:1387. doi: 10.3389/fimmu.2020.01387
117. Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, et al. H. pylori. activates NF-κB through a signaling pathway involving IκB kinases, NF-κB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. (2000) 119(1):97–108. doi: 10.1053/gast.2000.8540
118. Bontems P, Aksoy E, Burette A, Segers V, Deprez C, Mascart F, et al. NF-κB activation and severity of gastritis in Helicobacter pylori-infected children and adults. Helicobacter. (2014) 19(3):157–67. doi: 10.1111/hel.12118
119. Bauer M, Nascakova Z, Mihai AI, Cheng PF, Levesque MP, Lampart S, et al. The ALPK1/TIFA/NF-κB axis links a bacterial carcinogen to R-loop-induced replication stress. Nat Commun (2020) 11(1):5117. doi: 10.1038/s41467-020-18857-z
120. Hartung ML, Gruber DC, Koch KN, Grüter L, Rehrauer H, Tegtmeyer N, et al. H. pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep (2015) 13(1):70–9. doi: 10.1016/j.celrep.2015.08.074
121. Chen P, Guo H, Wu X, Li J, Duan X, Ba Q, et al. Epigenetic silencing of microRNA-204 by Helicobacter pylori augments the NF-κB signaling pathway in gastric cancer development and progression. Carcinogenesis. (2020) 41(4):430–41. doi: 10.1093/carcin/bgz143
122. Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A, et al. Loss of TFF1 is associated with activation of NF-κB-mediated inflammation and gastric neoplasia in mice and humans. J Clin Invest. (2011) 121(5):1753–67. doi: 10.1172/JCI43922
123. Soutto M, Chen Z, Katsha AM, Romero-Gallo J, Krishna US, Piazuelo MB, et al. Trefoil factor 1 expression suppresses Helicobacter pylori–induced inflammation in gastric carcinogenesis. Cancer. (2015) 121(24):4348–58. doi: 10.1002/cncr.29644
124. Katsha A, Soutto M, Sehdev V, Peng D, Washington MK, Piazuelo MB, et al. Aurora kinase a promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology. (2013) 145(6):1312–22.e8. doi: 10.1053/j.gastro.2013.08.050
125. Maubach G, Lim MCC, Sokolova O, Backert S, Meyer TF, Naumann M. TIFA has dual functions in Helicobacter pylori-induced classical and alternative NF-κB pathways. EMBO Rep (2021) 22(9):e52878. doi: 10.15252/embr.202152878
126. Zhang Z, Fan B, Liu F, Song N, Peng Y, Ma W, et al. HOX transcript antisense RNA is elevated in gastric carcinogenesis and regulated by the NF-κB pathway. J Cell Biochem (2019) 120(6):10548–55. doi: 10.1002/jcb.28340
127. Cao L, Zhu S, Lu H, Soutto M, Bhat N, Chen Z, et al. Helicobacter pylori-induced RASAL2 through activation of nuclear factor-κB promotes gastric tumorigenesis via β-catenin signaling axis. Gastroenterology. (2022) 162(6):1716–31.e17. doi: 10.1053/j.gastro.2022.01.046
128. Byun E, Park B, Lim JW, Kim H. Activation of NF-κB and AP-1 mediates hyperproliferation by inducing β-catenin and c-myc in Helicobacter pylori-infected gastric epithelial cells. Yonsei Med J (2016) 57(3):647–51. doi: 10.3349/ymj.2016.57.3.647
129. Zhu S, Soutto M, Chen Z, Peng D, Romero-Gallo J, Krishna US, et al. Helicobacter pylori-induced cell death is counteracted by NF-κB-mediated transcription of DARPP-32. Gut. (2017) 66(5):761–2. doi: 10.1136/gutjnl-2016-312141
130. Yang F, Xu Y, Liu C, Ma C, Zou S, Xu X, et al. NF-κB/miR-223-3p/ARID1A axis is involved in Helicobacter pylori CagA-induced gastric carcinogenesis and progression. Cell Death Dis (2018) 9(1):12. doi: 10.1038/s41419-017-0020-9
131. Shen L, Zeng J, Ma L, Li S, Chen C, Jia J, et al. Helicobacter pylori induces a novel NF-κB/LIN28A/let-7a/hTERT axis to promote gastric carcinogenesis. Mol Cancer Res (2021) 19(1):74–85. doi: 10.1158/1541-7786.MCR-19-0678
132. Burkitt MD, Williams JM, Duckworth CA, O'Hara A, Hanedi A, Varro A, et al. Signaling mediated by the NF-κB sub-units NF-κB1, NF-κB2 and c-rel differentially regulate Helicobacter felis-induced gastric carcinogenesis in C57BL/6 mice. Oncogene. (2013) 32(50):5563–73. doi: 10.1038/onc.2013.334
133. Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, Rogers AB, et al. Conditional deletion of IκB-kinase-β accelerates Helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology. (2010) 138(3):1022–34.e10. doi: 10.1053/j.gastro.2009.11.054
134. Hwang JW, Baek Y-M, Yang KE, Yoo H-S, Cho C-K, Lee Y-W, et al. Lactobacillus casei extract induces apoptosis in gastric cancer by inhibiting NF-κB and mTOR-mediated signaling. Integr Cancer Therapies. (2013) 12(2):165–73. doi: 10.1177/1534735412442380
135. Blaser N, Backert S, Pachathundikandi SK. Immune cell signaling by Helicobacter pylori: Impact on gastric pathology. Adv Exp Med Biol (2019) 1149:77–106. doi: 10.1007/5584_2019_360
136. Kutikhin AG. Impact of toll-like receptor 4 polymorphisms on risk of cancer. Hum Immunol (2011) 72(2):193–206. doi: 10.1016/j.humimm.2010.11.003
137. Pimentel-Nunes P, Afonso L, Lopes P, Roncon-Albuquerque R Jr., Gonçalves N, Henrique R, et al. Increased expression of toll-like receptors (TLR) 2, 4 and 5 in gastric dysplasia. Pathol Oncol Res (2011) 17(3):677–83. doi: 10.1007/s12253-011-9368-9
138. Schmausser B, Andrulis M, Endrich S, Müller-Hermelink HK, Eck M. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: An implication for interaction with Helicobacter pylori. Int J Med Microbiol (2005) 295(3):179–85. doi: 10.1016/j.ijmm.2005.02.009
139. Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, Afonso L, Lopes P, Roncon-Albuquerque R Jr., et al. Helicobacter pylori induces increased expression of toll-like receptors and decreased toll-interacting protein in gastric mucosa that persists throughout gastric carcinogenesis. Helicobacter. (2013) 18(1):22–32. doi: 10.1111/hel.12008
140. Pimentel-Nunes P, Soares JB, Roncon-Albuquerque R Jr., Dinis-Ribeiro M, Leite-Moreira AF. Toll-like receptors as therapeutic targets in gastrointestinal diseases. Expert Opin Ther Targets. (2010) 14(4):347–68. doi: 10.1517/14728221003642027
141. Uno K, Kato K, Shimosegawa T. Novel role of toll-like receptors in Helicobacter pylori-induced gastric malignancy. World J Gastroenterol (2014) 20(18):5244–51. doi: 10.3748/wjg.v20.i18.5244
142. Pachathundikandi SK, Lind J, Tegtmeyer N, El-Omar EM, Backert S. Interplay of the gastric pathogen Helicobacter pylori with toll-like receptors. BioMed Res Int (2015) 2015:192420. doi: 10.1155/2015/192420
143. Varga MG, Peek RM. DNA transfer and toll-like receptor modulation by Helicobacter pylori. Curr Top Microbiol Immunol (2017) 400:169–93. doi: 10.1007/978-3-319-50520-6_8
144. Meliţ LE, Mărginean CO, Mărginean CD, Mărginean MO. The relationship between toll-like receptors and Helicobacter pylori-related gastropathies: Still a controversial topic. J Immunol Res (2019) 2019:8197048. doi: 10.1155/2019/8197048
145. Hu Y, Liu JP, Zhu Y, Lu NH. The importance of toll-like receptors in NF-κB signaling pathway activation by Helicobacter pylori infection and the regulators of this response. Helicobacter. (2016) 21(5):428–40. doi: 10.1111/hel.12292
146. Schmausser B, Andrulis M, Endrich S, Lee SK, Josenhans C, Müller-Hermelink HK, et al. Expression and subcellular distribution of toll-like receptors TLR4, TLR5 and TLR9 on the gastric epithelium in Helicobacter pylori infection. Clin Exp Immunol (2004) 136(3):521–6. doi: 10.1111/j.1365-2249.2004.02464.x
147. Uno K, Kato K, Atsumi T, Suzuki T, Yoshitake J, Morita H, et al. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol (2007) 293(5):G1004–12. doi: 10.1152/ajpgi.00096.2007
148. Lagunes-Servin H, Torres J, Maldonado-Bernal C, Pérez-Rodríguez M, Huerta-Yépez S, Madrazo de la Garza A, et al. Toll-like receptors and cytokines are upregulated during Helicobacter pylori infection in children. Helicobacter. (2013) 18(6):423–32. doi: 10.1111/hel.12067
149. Pachathundikandi SK, Tegtmeyer N, Arnold IC, Lind J, Neddermann M, Falkeis-Veits C, et al. T4SS-dependent TLR5 activation by Helicobacter pylori infection. Nat Commun (2019) 10(1):5717. doi: 10.1038/s41467-019-13506-6
150. Săsăran MO, Meliţ LE, Dobru ED. MicroRNA modulation of host immune response and inflammation triggered by Helicobacter pylori. Int J Mol Sci (2021) 22(3):1406. doi: 10.3390/ijms22031406
151. Ito N, Tsujimoto H, Ueno H, Xie Q, Shinomiya N. Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer. J Clin Med (2020) 9(11):3699. doi: 10.3390/jcm9113699
152. Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol (2010) 184(9):5121–9. doi: 10.4049/jimmunol.0901115
153. Osaki LH, Bockerstett KA, Wong CF, Ford EL, Madison BB, DiPaolo RJ, et al. Interferon-gamma directly induces gastric epithelial cell death and is required for progression to metaplasia. J Pathol (2019) 247(4):513–23. doi: 10.1002/path.5214
154. Yin S, Lan C, Pei H, Zhu Z. Expression of interleukin 1β in gastric cancer tissue and its effects on gastric cancer. Onco Targets Ther (2016) 9:31–5. doi: 10.2147/OTT.S94277
155. Li Z, Wu C, Li L, Wang Z, Xie H, He X, et al. Effect of long-term proton pump inhibitor administration on gastric mucosal atrophy: A meta-analysis. Saudi J Gastroenterol (2017) 23(4):222–8. doi: 10.4103/sjg.SJG_573_16
156. Bockerstett KA, Osaki LH, Petersen CP, Cai CW, Wong CF, Nguyen T-LM, et al. Interleukin-17A promotes parietal cell atrophy by inducing apoptosis. Cell Mol Gastroenterol Hepatology. (2018) 5(4):678–90.e1. doi: 10.1016/j.jcmgh.2017.12.012
157. Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. Regulatory T-cell homeostasis: Steady-state maintenance and modulation during inflammation. Immunol Rev (2014) 259(1):40–59. doi: 10.1111/imr.12170
158. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol (2015) 6:184. doi: 10.3389/fphar.2015.00184
159. Maruyama T, Kono K, Mizukami Y, Kawaguchi Y, Mimura K, Watanabe M, et al. Distribution of Th17 cells and FoxP3(+) regulatory T cells in tumor-infiltrating lymphocytes, tumor-draining lymph nodes and peripheral blood lymphocytes in patients with gastric cancer. Cancer Sci (2010) 101(9):1947–54. doi: 10.1111/j.1349-7006.2010.01624.x
160. Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, Huehn J. Microbiome dependent regulation of t(regs) and Th17 cells in mucosa. Front Immunol (2019) 10:426. doi: 10.3389/fimmu.2019.00426
161. Jang TJ. The number of Foxp3-positive regulatory T cells is increased in Helicobacter pylori gastritis and gastric cancer. Pathol - Res Practice. (2010) 206(1):34–8. doi: 10.1016/j.prp.2009.07.019
162. Gil JH, Seo JW, Cho MS, Ahn JH, Sung HY. Role of treg and TH17 cells of the gastric mucosa in children with Helicobacter pylori gastritis. J Pediatr Gastroenterol Nutr (2014) 58(2):245–51. doi: 10.1097/MPG.0000000000000194
163. Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun (2005) 73(1):523–31. doi: 10.1128/IAI.73.1.523-531.2005
164. Tsai CC, Chen TY, Tsai KJ, Lin MW, Hsu CY, Wu DC, et al. NF-κB/miR-18a-3p and miR-4286/BZRAP1 axis may mediate carcinogenesis in Helicobacter pylori-associated gastric cancer. BioMed Pharmacother. (2020) 132:110869. doi: 10.1016/j.biopha.2020.110869
165. Qin XR, Wu J, Yao XY, Huang J, Wang XY. Helicobacter pylori DNA promotes cellular proliferation, migration, and invasion of gastric cancer by activating toll-like receptor 9. Saudi J Gastroenterol (2019) 25(3):181–7. doi: 10.4103/sjg.SJG_309_18
166. Ding L, Chakrabarti J, Sheriff S, Li Q, Thi Hong HN, Sontz RA, et al. Toll-like receptor 9 pathway mediates schlafen(+)-MDSC polarization during Helicobacter-induced gastric metaplasias. Gastroenterology. (2022) 163(2):411–25.e4. doi: 10.1053/j.gastro.2022.04.031
167. Chen J, Hu S, Liang S, Chen Q, Yang Q, Zheng W, et al. Associations between the four toll-like receptor polymorphisms and the risk of gastric cancer: A meta-analysis. Cancer Biother Radiopharm. (2013) 28(9):674–81. doi: 10.1089/cbr.2012.1395
168. Zou TH, Wang ZH, Fang JY. Positive association between toll-like receptor 4 gene +896A/G polymorphism and susceptibility to gastric carcinogenesis: A meta-analysis. Tumour Biol (2013) 34(4):2441–50. doi: 10.1007/s13277-013-0795-y
169. Eed EM, Hawash YA, Khalifa AS, Alsharif KF, Alghamdi SA, Almalki AA, et al. Association of toll-like receptors 2, 4, 9 and 10 genes polymorphisms and Helicobacter pylori-related gastric diseases in Saudi patients. Indian J Med Microbiol (2020) 38(1):94–100. doi: 10.4103/ijmm.IJMM_20_164
170. Ravishankar Ram M, Goh KL, Leow AH, Poh BH, Loke MF, Harrison R, et al. Polymorphisms at locus 4p14 of toll-like receptors TLR-1 and TLR-10 confer susceptibility to gastric carcinoma in Helicobacter pylori infection. PloS One (2015) 10(11):e0141865. doi: 10.1371/journal.pone.0141865
171. Castaño-Rodríguez N, Kaakoush NO, Pardo AL, Goh KL, Fock KM, Mitchell HM. Genetic polymorphisms in the toll-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer. Hum Immunol (2014) 75(8):808–15. doi: 10.1016/j.humimm.2014.06.001
172. Tongtawee T, Simawaranon T, Wattanawongdon W, Dechsukhum C, Leeanansaksiri W. Toll-like receptor 2 and 4 polymorphisms associated with Helicobacter pylori susceptibility and gastric cancer. Turk J Gastroenterol (2019) 30(1):15–20. doi: 10.5152/tjg.2018.17461
173. Kalkanli Tas S, Kirkik D, Tanoglu A, Kahraman R, Ozturk K, Esen MF, et al. Polymorphisms in toll-like receptors 1, 2, 5, and 10 are associated with predisposition to Helicobacter pylori infection. Eur J Gastroenterol Hepatol (2020) 32(9):1141–6. doi: 10.1097/MEG.0000000000001797
174. Tang FB, Li ZX, Wang YM, Zhang L, Ma JL, Zhou T, et al. Toll-like receptor 1 and 10 polymorphisms, Helicobacter pylori susceptibility and risk of gastric lesions in a high-risk Chinese population. Infect Genet Evol (2015) 31:263–9. doi: 10.1016/j.meegid.2015.02.005
175. Tongtawee T, Bartpho T, Wattanawongdon W, Dechsukhum C, Leeanansaksiri W, Matrakool L, et al. Role of toll-like receptor 10 gene polymorphism and gastric mucosal pattern in patients with chronic gastritis. Turk J Gastroenterol (2017) 28(4):243–7. doi: 10.5152/tjg.2017.16673
176. Miyata N, Hayashi Y, Hayashi S, Sato K, Hirai Y, Yamamoto H, et al. Lipopolysaccharides from non-Helicobacter pylori gastric bacteria potently stimulate interleukin-8 production in gastric epithelial cells. Clin Transl Gastroenterol (2019) 10(3):e00024. doi: 10.14309/ctg.0000000000000024
177. Tang K, McLeod L, Livis T, West AC, Dawson R, Yu L, et al. Toll-like receptor 9 promotes initiation of gastric tumorigenesis by augmenting inflammation and cellular proliferation. Cell Mol Gastroenterol Hepatol (2022) 14(3):567–86. doi: 10.1016/j.jcmgh.2022.06.002
178. Kaur B, Singh P. Inflammation: Biochemistry, cellular targets, anti-inflammatory agents and challenges with special emphasis on cyclooxygenase-2. Bioorganic Chem (2022) 121:105663. doi: 10.1016/j.bioorg.2022.105663
179. Kwon S, Ma W, Drew DA, Klempner SJ, Leonardo BM, Flynn JJ, et al. Association between aspirin use and gastric adenocarcinoma: A prospective cohort study. Cancer Prev Res (2022) 15(4):265–72. doi: 10.1158/1940-6207.CAPR-21-0413
180. El-Malah AA, Gineinah MM, Deb PK, Khayyat AN, Bansal M, Venugopala KN, et al. Selective COX-2 inhibitors: Road from success to controversy and the quest for repurposing. Pharmaceuticals. (2022) 15(7):827. doi: 10.3390/ph15070827
181. Oshima H, Hioki K, Popivanova BK, Oguma K, Van Rooijen N, Ishikawa TO, et al. Prostaglandin E2 signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. (2011) 140(2):596–607.e7. doi: 10.1053/j.gastro.2010.11.007
182. Lopes C, Pereira C, Farinha M, Medeiros R, Dinis-Ribeiro M. Prostaglandin E2 pathway is dysregulated in gastric adenocarcinoma in a Caucasian population. Int J Mol Sci (2020) 21(20):7680. doi: 10.3390/ijms21207680
183. Lopes C, Pereira C, Farinha M, Medeiros R, Dinis-Ribeiro M. Genetic variations in prostaglandin E2 pathway identified as susceptibility biomarkers for gastric cancer in an intermediate risk European country. Int J Mol Sci (2021) 22(2):648. doi: 10.3390/ijms22020648
184. Achyut BR, Ghoshal UC, Moorchung N, Mittal B. Role of cyclooxygenase-2 functional gene polymorphisms in Helicobacter pylori induced gastritis and gastric atrophy. Mol Cell Biochem (2009) 321(1):103–9. doi: 10.1007/s11010-008-9924-0
185. Kodaka Y, Futagami S, Tatsuguchi A, Yamawaki H, Sato H, Hashimoto S, et al. Impact of cyclooxygenase-2 1195 G-carrier genotype associated with intestinal metaplasia and endoscopic findings based on kyoto classification. Digestion. (2017) 96(3):173–83. doi: 10.1159/000479864
186. Dias A, Garcia C, Majewski M, Wallner G, McCallum RW, Poplawski C, et al. Gastric juice prostaglandins and peptide growth factors as potential markers of chronic atrophic gastritis, intestinal metaplasia and gastric cancer: Their potential clinical implications based on this pilot study. Digestive Dis Sci (2011) 56(11):3220–5. doi: 10.1007/s10620-011-1758-z
187. Wong BC, Zhang L, Ma JL, Pan KF, Li JY, Shen L, et al. Effects of selective COX-2 inhibitor and Helicobacter pylori eradication on precancerous gastric lesions. Gut. (2012) 61(6):812–8. doi: 10.1136/gutjnl-2011-300154
188. Echizen K, Hirose O, Maeda Y, Oshima M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and toll-like receptor/MyD88 pathways. Cancer Sci (2016) 107(4):391–7. doi: 10.1111/cas.12901
189. Chang YJ, Wu MS, Lin JT, Sheu BS, Muta T, Inoue H, et al. Induction of cyclooxygenase-2 overexpression in human gastric epithelial cells by Helicobacter pylori involves TLR2/TLR9 and c-src-dependent nuclear factor-kappaB activation. Mol Pharmacol (2004) 66(6):1465–77. doi: 10.1124/mol.104.005199
190. Lim JW, Kim H, Kim KH. Nuclear factor-kappaB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab Invest. (2001) 81(3):349–60. doi: 10.1038/labinvest.3780243
191. Takehara H, Iwamoto J, Mizokami Y, Takahashi K, Ootubo T, Miura S, et al. Involvement of cyclooxygenase-2-prostaglandin E2 pathway in interleukin-8 production in gastric cancer cells. Dig Dis Sci (2006) 51(12):2188–97. doi: 10.1007/s10620-006-9436-2
192. Lone SN, Nisar S, Masoodi T, Singh M, Rizwan A, Hashem S, et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol Cancer. (2022) 21(1):79. doi: 10.1186/s12943-022-01543-7
193. Jelski W, Mroczko B. Molecular and circulating biomarkers of gastric cancer. Int J Mol Sci (2022) 23(14):7588. doi: 10.3390/ijms23147588
194. Liang Y, Wang W, Fang C, Raj SS, Hu W-M, Li Q-W, et al. Clinical significance and diagnostic value of serum CEA, CA19-9 and CA72-4 in patients with gastric cancer. Oncotarget (2016) 7(31):49565–73. doi: 10.18632/oncotarget.10391
195. Wang H, Jin W, Wan C, Zhu C. Diagnostic value of combined detection of CA72-4, CA19-9, and carcinoembryonic antigen comparing to CA72-4 alone in gastric cancer: A systematic review and meta-analysis. Transl Cancer Res (2022) 11(4):848–56. doi: 10.21037/tcr-22-537
196. Matsuoka T, Yashiro M. Biomarkers of gastric cancer: Current topics and future perspective. World J Gastroenterol (2018) 24(26):2818–32. doi: 10.3748/wjg.v24.i26.2818
197. Zagari RM, Rabitti S, Greenwood DC, Eusebi LH, Vestito A, Bazzoli F. Systematic review with meta-analysis: Diagnostic performance of the combination of pepsinogen, gastrin-17 and anti-Helicobacter pylori antibodies serum assays for the diagnosis of atrophic gastritis. Aliment Pharmacol Ther (2017) 46(7):657–67. doi: 10.1111/apt.14248
198. Kutluana U, Kilciler AG, Mizrak S, Dilli U. Can neopterin be a useful immune biomarker for differentiating gastric intestinal metaplasia and gastric atrophy from non-atrophic non-metaplastic chronic gastritis? Gastroenterol Hepatol (2019) 42(5):289–95. doi: 10.1016/j.gastrohep.2019.01.005
199. Choi H-I, Choi J-P, Seo J, Kim BJ, Rho M, Han JK, et al. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp Mol Med (2017) 49(5):e330–e. doi: 10.1038/emm.2017.47
200. He C, Peng C, Shu X, Wang H, Zhu Z, Ouyang Y, et al. Convergent dysbiosis of gastric mucosa and fluid microbiome during stomach carcinogenesis. Gastric Cancer. (2022) 25(5):837–49. doi: 10.1007/s10120-022-01302-z
201. Zullo A, Germanà B, Galliani E, Iori A, De Pretis G, Manfredi G, et al. Real-time EndoFaster improves Helicobacter pylori detection in chronic active gastritis. J. Clin Pathol (2021) 75:572–74. doi: 10.1136/jclinpath-2021-207498
202. Cazzato M, Esposito G, Galli G, Pilozzi E, Lahner E, Corleto VD, et al. Diagnostic accuracy of EndoFaster® and narrow-band imaging endoscopy in patients with impaired gastric acid secretion: A real-time prospective study. Gastroenterol Res Pract (2021) 2021:6616334. doi: 10.1155/2021/6616334
203. Zullo A, Germanà B, Galliani E, Iori A, de Pretis G, Manfredi G, et al. Real-time determination of gastric juice pH with EndoFaster® for atrophic gastritis assessment. Dig Liver Dis (2022) 54(12):1646–48. doi: 10.1016/j.dld.2022.06.014
204. White JR, Banks M. Identifying the pre-malignant stomach: From guidelines to practice. Transl Gastroenterol Hepatol (2022) 7:8. doi: 10.21037/tgh.2020.03.03
Keywords: atrophic gastritis, biomarkers, inflammation, microbiota, functional endoscopy
Citation: Lopes C, Almeida TC, Pimentel-Nunes P, Dinis-Ribeiro M and Pereira C (2023) Linking dysbiosis to precancerous stomach through inflammation: Deeper than and beyond imaging. Front. Immunol. 14:1134785. doi: 10.3389/fimmu.2023.1134785
Received: 30 December 2022; Accepted: 17 March 2023;
Published: 31 March 2023.
Edited by:
Sukanya Raghavan, University of Gothenburg, SwedenReviewed by:
Ricardo Valle-Rios, National Autonomous University of Mexico, MexicoBin Bao, Boston Children's Hospital and Harvard Medical School, United States
Copyright © 2023 Lopes, Almeida, Pimentel-Nunes, Dinis-Ribeiro and Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carina Pereira, YW5hLm1hcnRpbnMucGVyZWlyYUBpcG9wb3J0by5taW4tc2F1ZGUucHQ=
†These authors have contributed equally to this work and share first authorship