 Aristeidis E. Boukouris1
Aristeidis E. Boukouris1 Ioannis Michelakis1
Ioannis Michelakis1 Dionysios Metaxas1Gianna Karapati1
Dionysios Metaxas1Gianna Karapati1 George Kanellis2Athina Lioni1
George Kanellis2Athina Lioni1 Vasiliki Tzavara1*
Vasiliki Tzavara1*- 11st Department of Internal Medicine, Korgialenio-Benakio Red Cross General Hospital, Athens, Greece
- 2Hematopathology Department, Evangelismos Hospital, Athens, Greece
Indolent systemic mastocytosis (ISM) represents the most common form of SM, typically following a slow clinical course. While anaphylactic reactions may come up in the life course of an ISM patient, these are often moderate and do not pose a threat to patient’s health. Here, we present an undiagnosed case of ISM with recurrent severe anaphylactic episodes following consumption of food and emotional stress. One of these episodes led to anaphylactic shock, necessitating temporary mechanical ventilation and intensive care unit (ICU) support. Besides hypotension, a diffuse, itchy, red rash was the only notable clinical finding. Upon recovery, we found abnormally high baseline serum tryptase level as well as 10% bone marrow (BM) infiltration by multifocal, dense clusters of CD117+/mast cell tryptase+/CD25+ mast cells (MCs), consolidating the diagnosis of ISM. Prophylactic treatment with a histamine receptor antagonist was initiated, resulting in milder episodes thereafter. Diagnosis of ISM requires a high level of suspicion; its prompt recognition and treatment are important in preventing potentially life-threatening anaphylactic episodes.
1. Introduction
Systemic mastocytosis is a rare (estimated prevalence: 1/10,000) (1) hematologic condition, characterized by increased proliferation and uncontrolled accumulation of mast cells in various organs, including the skin, bone marrow, liver, spleen and GI tract. Common manifestations include anemia, abdominal pain, pruritus/flushing and anaphylactoid reactions as well as neurocognitive symptoms (e.g., depression, mood changes, lack of concentration). Hepatosplenomegaly, lymphadenopathy and signs of anemia are often noted on physical examination (2). Disease severity and course can vary greatly depending on the degree of mast cell infiltration and resulting organ dysfunction. While aggressive SM and mast cell leukemia (MCL) (collectively termed advanced SM) are characterized by rapid progression and poor survival rates, less severe cases of SM (indolent SM; ISM and smoldering SM; SSM) follow a much less impactful course over several years (median OS: 28.4 years according to large ISM patient databases) (3), often making it hard to discern from other diseases. Diagnosis (Table 1) and classification (4) are based on the combination of a number of different findings: a) clinical (B-findings, indicative of high MC burden and involvement of various organs (bone marrow, liver, spleen, lymph nodes) without causing dysfunction vs. C-findings, indicative of SM-induced organ dysfunction (cytopenias, hepatopathy, hypersplenism, malabsorption, osteolysis)), b) serological (measurement of serum tryptase) and c) histological (tissue biopsy). Here, we present a rare case of ISM with fulminant presentation in a 45-year-old male that remained undiagnosed for almost 20 years, eventually leading to a potentially life-threatening event (anaphylactic shock, coma and intubation).

Table 1 SM Diagnostic Criteria (2021 update).
2. Case presentation
A 45-year-old male patient (birthplace and residence: Athens, Greece) with a family and past medical history of arterial hypertension was admitted to the emergency department (ED) in a comatose state (Glascow Coma Scale; GCS: 3) after sudden loss of consciousness at work. The timeline of events and case progress with relevant data are presented in Figure 1.
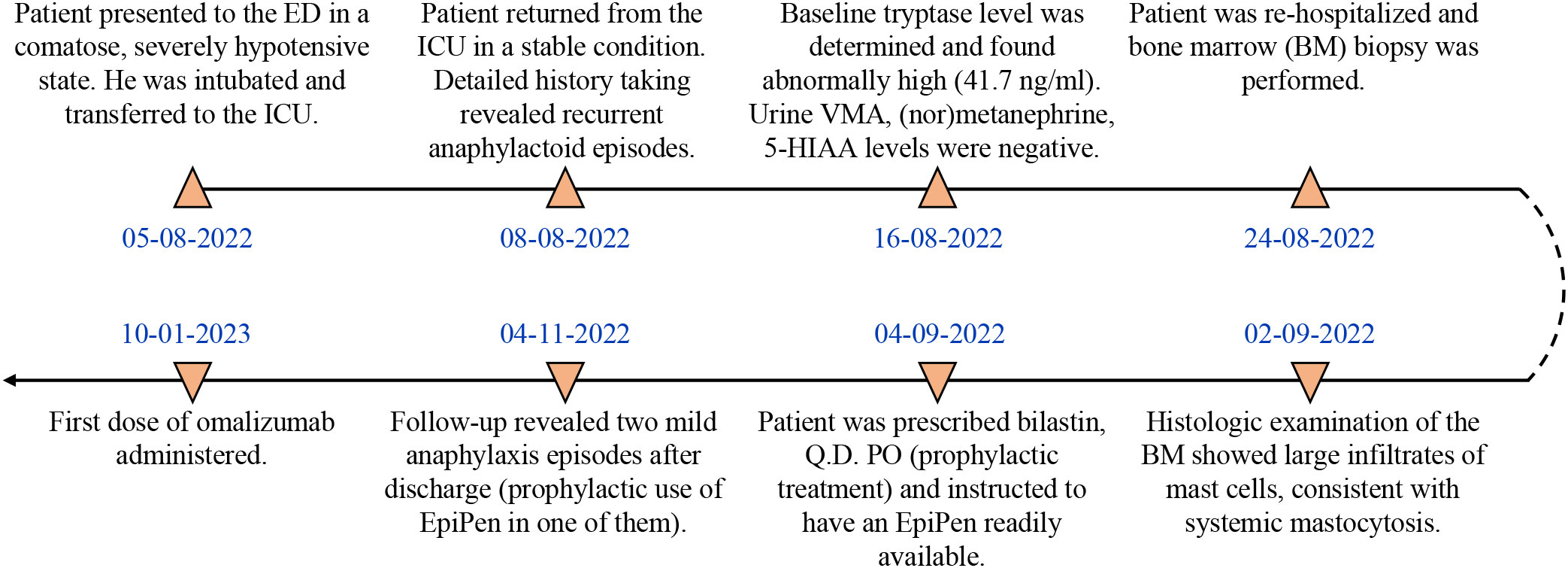
Figure 1 Timeline and case progress with relevant data.
Upon arrival, the patient was hypotensive (BP: 70/40 mmHg) and was intubated shortly thereafter for airway protection. Assessment of cardiac function, including an ECG and echocardiogram, did not reveal any abnormalities, such as arrhythmia or severe hypokinesia that could explain the hypotension. Emergency contrast-enhanced CT scans of the brain (to exclude an intracranial hemorrhage), chest and abdomen (to exclude a dissecting aortic aneurysm) were also unremarkable. Clinical examination revealed mild hepatomegaly and multiple, monomorphic, reddish brown macules of the trunk and extremities (Figure 2A). There was no evidence of tongue biting or urinary incontinence, suggestive of a possible epileptic episode and postictal seizure state. Routine CBC and BMP were grossly unremarkable, except for a mildly elevated troponin level. Urine toxicology screen, consisting of benzodiazepines, barbiturates, opiates, amphetamines and cocaine was also negative. The patient was transferred to the ICU where he fully recovered within 48 hours. Upon recovery, the patient was able to recall several (~20) episodes of flushing (Figure 2B), tachycardia, lightheadedness and hypotension (Figure 2C) in the past, which he could link with emotional stress. Further inquiry about preceding events revealed a fairly consistent pattern of ingestion of certain foods (nuts, legumes, chocolate) or alcoholic beverages, together with emotional stress prior to the onset of the episodes. Intriguingly, these episodes continued even after the patient started establishing a link between them and food, gradually adjusting his diet by himself. Previous extensive work up in tertiary hospitals during the past 20 years, including negative food allergy testing, 24-hour Holter monitoring and electroencephalography, had failed to pinpoint to a specific cause for these episodes, other than psychological factors, thus recommending professional support.
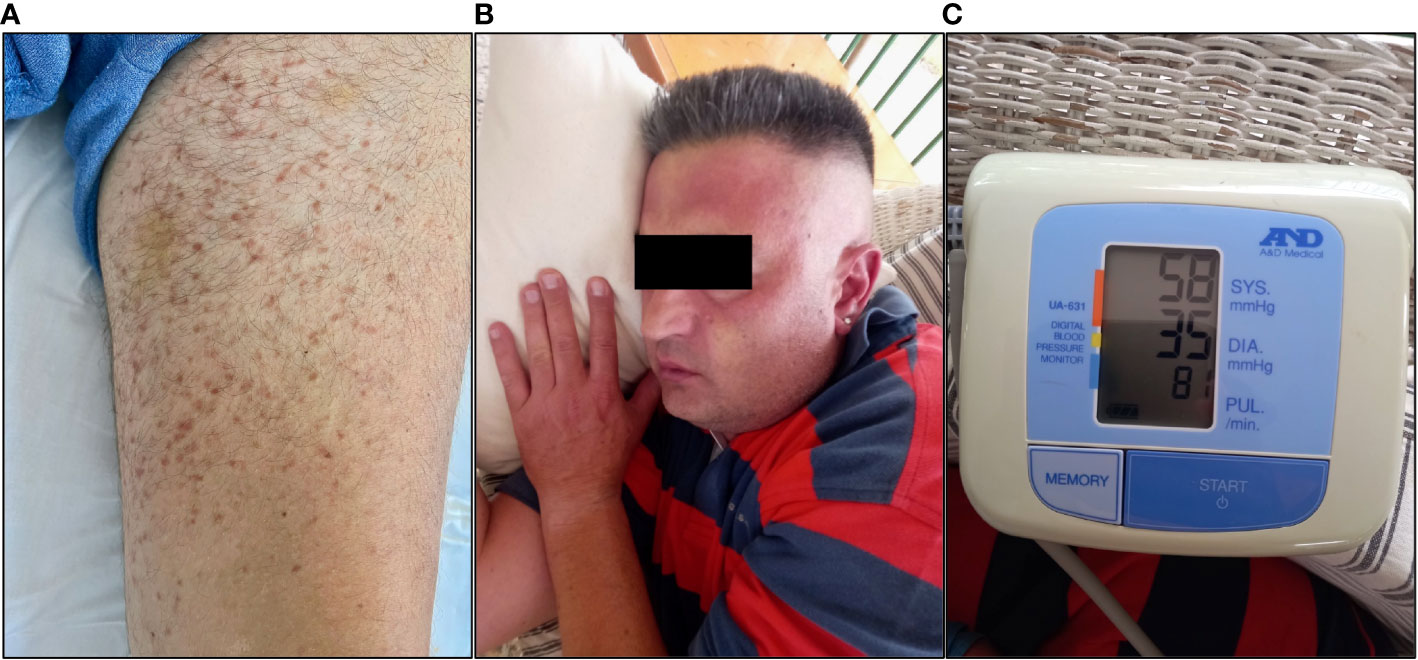
Figure 2 Photographs showing (A) the chronic urticarial rash, (B) facial flushing and (C) profound hypotension during an anaphylaxis episode in our patient (Courtesy of K.T.; written informed consent was obtained from the individual shown).
Based on clinical presentation, imaging findings and previous workup, we assumed a non-cardiogenic, non-neurogenic cause for the recurrent anaphylactoid episodes, and turned our attention to endocrine disorders (pheochromocytoma, carcinoid syndrome) that can elicit such episodes, as well as SM. Testing for these rare diseases required external collaboration, as they were not routinely performed in our hospital. Urine VMA, (nor)metanephrine and 5-Hydroxyindoleacetic acid (5-HIAA) levels were normal, significantly reducing the probability of pheochromocytoma and carcinoid syndrome, respectively. Intriguingly, total serum tryptase level 10 days after the episode (baseline) was abnormally high (41.7 ng/ml; NR: < 11.4 ng/ml), fulfilling a minor SM criterion (Table 1). Subsequent bone marrow (BM) biopsy and histologic examination revealed an approximately 10% BM infiltration (multifocal dense infiltrates) by partially spindle-shaped CD117+/mast cell tryptase+/CD25+ MCs (Figures 3A, B) (major SM criterion; Table 1), establishing the diagnosis of SM. The observed mast cells had oval or elongated nuclei and were mostly located around bone marrow trabeculae (Figure 3C) and, to a lesser extent, blood vessels. The overall BM cellularity was normal (normal representation of the lymphoid lineage, mild elevation of the erythroid lineage and lower than normal myeloid lineage). Low grade paratrabecular fibrosis was evident in the stroma. Next generation sequencing on DNA extracted from neoplastic CD25+/CD2+ MCs following cell sorting did not reveal point mutations of the KIT (e.g. D816V mutation) and other genes (e.g. ASXL1, CBL, JAK2, RAS, RUNX1, SRSF2) that have been associated with SM, especially the advanced forms. The presence of typical chronic skin lesions (urticaria pigmentosa), along with the identification of only one B-finding (hepatomegaly without impairment of liver function) and no C-findings classified our SM case as ISM (favorable prognosis).
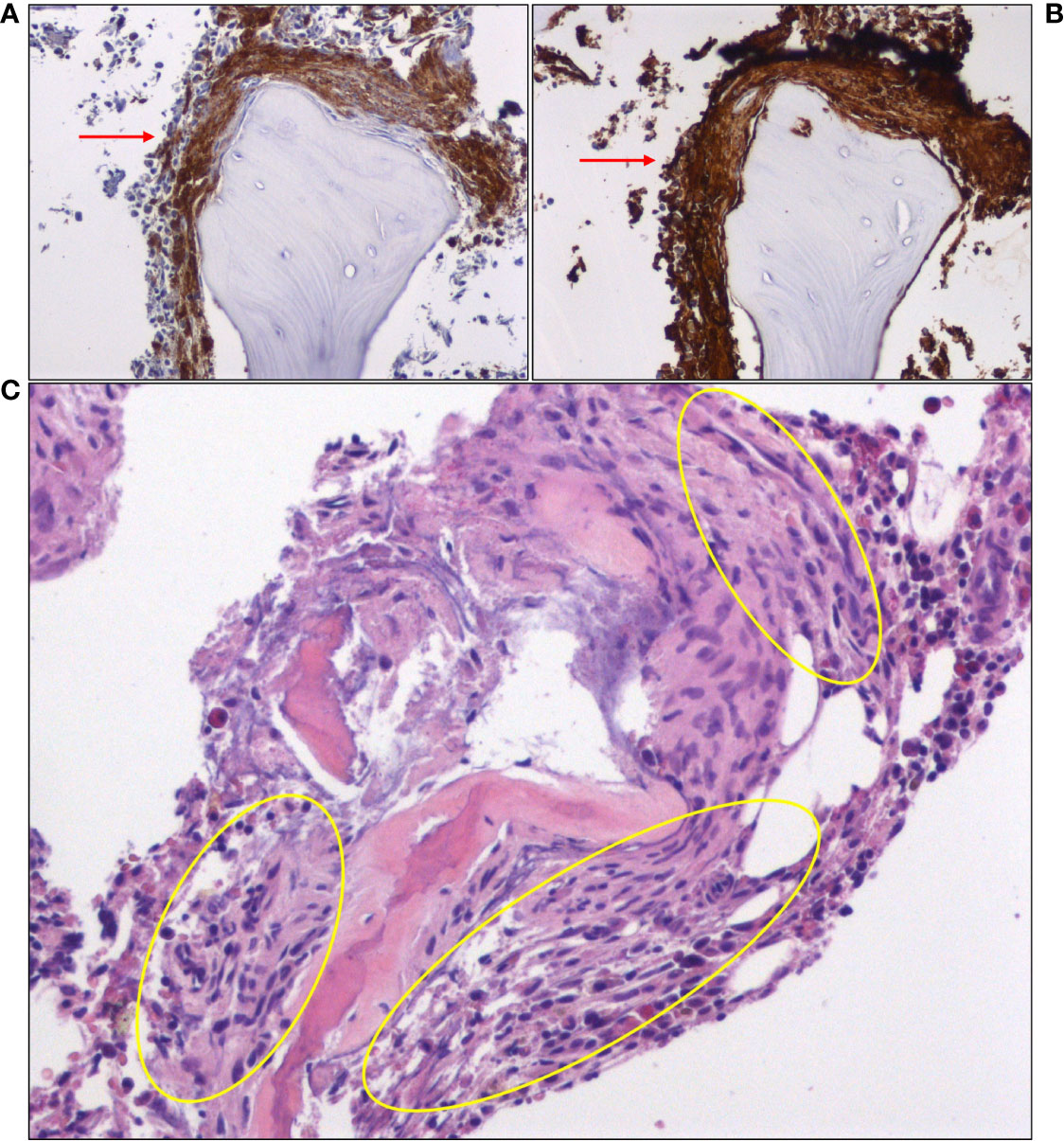
Figure 3 Immunohistochemical staining of bone marrow biopsy showing large clusters of mast cells (red arrows), expressing (A) CD25 and (B) mast cell tryptase. (C) A hematoxylin and eosin (H&E)-stained section of bone marrow core shows areas of paratrabecular fibrosis, in which mast cells can be identified (yellow oval circles).
He was placed on a low histamine diet and started prophylactic treatment with a generally well-tolerated, non-sedating second-generation histamine H1-receptor antagonist (once daily). Since discharge, he has reported a couple of mild anaphylactic episodes, both in response to emotional stress. He pre-emptively used an EpiPen injection in one of them (as instructed to prevent recurrence of a shock condition). He also very recently started therapy with the anti-IgE monoclonal antibody omalizumab, which has shown promise in patients with ISM (5, 6).
3. Discussion
We describe a case of ISM with an unusually fulminant presentation leading to a comatose state and intubation. Our patient had remained undiagnosed for almost 20 years despite extensive previous neurological and cardiovascular workup, which was associated with a considerable financial burden.
ISM represents the most common form of SM, and is associated with anaphylactic reactions that can cause syncope in up to a third of all cases (7). However, very severe (grade 5) or fatal anaphylaxis (anaphylactic shock, GCS: <13) in ISM patients is rare. Review of the literature identified very few such cases, which occurred in response to either drug injection (iodinated contrast agents (8), anesthetics (9–11)) or insect stings (12–14). Unlike these reports, in our patient, severe anaphylactic reactions were precipitated by a combination of environmental (food) and emotional stimuli, also known risk factors for anaphylaxis in patients with mastocytosis (15). To the best of our knowledge, this is the first report of food- and emotional stress-induced anaphylactic shock in a patient with ISM. Diagnosis of ISM was based on a number of clinical and laboratory criteria, in accordance with the latest consensus guidelines. A limitation of our study is that we were unable to identify the highly prevalent KIT D816V mutation (or mutations in other SM-relevant genes), despite using sensitive molecular techniques, which suggests our patient may fall into the much less common KIT-negative category. Tryptase level could have been measured in the first few hours after the episode and compared with baseline tryptase in order to further strengthen our theory of persistent mast cell activation.
In conclusion, our case emphasizes that caution should be exercised by physicians when approaching conditions with systemic manifestations in order to exclude underlying organic causes, before attributing them solely to psychological factors. It further highlights the importance of detailed history taking, a largely neglected skill in modern medicine, in uncovering the recurrent nature of the episodes and identifying preceding events in diseases, such as SM. Careful physical examination of our patient also provided important clues (observation of the “typical” skin rash, absence of profound hepatosplenomegaly and lymphadenopathy). Lastly, in the case of SM, a high level of suspicion is required for its diagnosis, especially the indolent variant, due to its often-atypical presentation and slowly progressing clinical course.
Overall, our patient has met the developments in his health condition with enthusiasm, realizing he can finally consider an evidence-based approach to his long-standing symptoms. He has already experienced an improvement in his quality of life with the aforementioned interventions, while eagerly anticipating further benefit from omalizumab treatment.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Article design and writing: AB, VT. Collection of clinical data/samples: IM, AB, DM, GiK, AL, VT. Immunohistochemistry procedures and pathology report: GeK. Supervision: AL, VT. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Hellenic Institute for the Study of Sepsis.
Acknowledgments
We would like to thank Drs. Maria Garofalaki (Molecular Hematology Laboratory, Evangelismos General Hospital, Athens, Greece) and Katherina Psarra (Department of Immunology-Histocompatibility, Evangelismos General Hospital, Athens, Greece) for their valuable help with molecular analysis of the bone marrow aspirate.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brockow K. Epidemiology, prognosis, and risk factors in mastocytosis. Immunol Allergy Clin North Am (2014) 34(2):283–95. doi: 10.1016/j.iac.2014.01.003
2. Pardanani A. Systemic mastocytosis in adults: 2021 update on diagnosis, risk stratification and management. Am J Hematol (2021) 96(4):508–25. doi: 10.1002/ajh.26118
3. Trizuljak J, Sperr WR, Nekvindova L, Elberink HO, Gleixner KV, Gorska A, et al. Clinical features and survival of patients with indolent systemic mastocytosis defined by the updated WHO classification. Allergy (2020) 75(8):1927–38. doi: 10.1111/all.14248
4. Valent P, Akin C, Hartmann K, Alvarez-Twose I, Brockow K, Hermine O, et al. Updated diagnostic criteria and classification of mast cell disorders: A consensus proposal. Hemasphere (2021) 5(11):e646. doi: 10.1097/HS9.0000000000000646
5. Constantine GM, Bressler PB, Petroni D, Metcalfe DD, Carter MC. Twelve-year follow-up of omalizumab therapy for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract (2019) 7(4):1314–6. doi: 10.1016/j.jaip.2018.07.041
6. Slapnicar C, Trinkaus M, Hicks L, Vadas P. Efficacy of omalizumab in indolent systemic mastocytosis. Case Rep Hematol (2019) 2019:3787586. doi: 10.1155/2019/3787586
7. Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood (2013) 121(16):3085–94. doi: 10.1182/blood-2013-01-453183
8. Weingarten TN, Volcheck GW, Sprung J. Anaphylactoid reaction to intravenous contrast in patient with systemic mastocytosis. Anaesth Intensive Care (2009) 37(4):646–9. doi: 10.1177/0310057X0903700415
9. Chatterjee M, Sengupta S, Chakravarty C, Ramasubban S, Bhartia S, Khan S, et al. Indolent systemic mastocytosis manifesting as protracted anaphylactic shock. Indian J Crit Care Med (2018) 22(4):311–3. doi: 10.4103/ijccm.IJCCM_497_17
10. Desborough JP, Taylor I, Hattersley A, Garden A, Wolff A, Bloom SR, et al. Massive histamine release in a patient with systemic mastocytosis. Br J Anaesth. (1990) 65(6):833–6. doi: 10.1093/bja/65.6.833
11. Vaughan ST, Jones GN. Systemic mastocytosis presenting as profound cardiovascular collapse during anaesthesia. Anaesthesia (1998) 53(8):804–7. doi: 10.1046/j.1365-2044.1998.00536.x
12. Florian S, Krauth MT, Simonitsch-Klupp I, Sperr WR, Fritsche-Polanz R, Sonneck K, et al. Indolent systemic mastocytosis with elevated serum tryptase, absence of skin lesions, and recurrent severe anaphylactoid episodes. Int Arch Allergy Immunol (2005) 136(3):273–80. doi: 10.1159/000083954
13. Oude Elberink JN, de Monchy JG, Kors JW, van Doormaal JJ, Dubois AE. Fatal anaphylaxis after a yellow jacket sting, despite venom immunotherapy, in two patients with mastocytosis. J Allergy Clin Immunol (1997) 99(1 Pt 1):153–4. doi: 10.1016/s0091-6749(97)70314-2
14. Wagner N, Fritze D, Przybilla B, Hagedorn M, Rueff F. Fatal anaphylactic sting reaction in a patient with mastocytosis. Int Arch Allergy Immunol (2008) 146(2):162–3. doi: 10.1159/000113520
Keywords: case report, indolent systemic mastocytosis, fulminant presentation, coma, symptom mitigation
Citation: Boukouris AE, Michelakis I, Metaxas D, Karapati G, Kanellis G, Lioni A and Tzavara V (2023) Case report: The devil was hidden in the mastocytes – an unusually fulminant case of indolent systemic mastocytosis in a 45-year-old patient, missed for almost 20 years. Front. Immunol. 14:1134587. doi: 10.3389/fimmu.2023.1134587
Received: 30 December 2022; Accepted: 30 January 2023;
Published: 10 February 2023.
Edited by:
Tracy George, The University of Utah, United StatesReviewed by:
Anton Rets, The University of Utah, United StatesMili Shum, The University of Utah, United States
Copyright © 2023 Boukouris, Michelakis, Metaxas, Karapati, Kanellis, Lioni and Tzavara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasiliki Tzavara, dnR6YXZhcmEyMDE1QGdtYWlsLmNvbQ==