 Milton Pereira1*
Milton Pereira1* Ricardo T. Gazzinelli1,2,3,4*
Ricardo T. Gazzinelli1,2,3,4*- 1Division of Infectious Diseases and Immunology, Department of Medicine, University of Massachusetts Chan Medical School, Worcester, MA, United States
- 2Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brazil
- 3Instituto René Rachou, Fundação Oswaldo Cruz, Belo Horizonte, MG, Brazil
- 4Plataforma de Medicina Translacional, Fundação Oswaldo Cruz, Ribeirão Preto, SP, Brazil
The Toll-like receptors (TLRs) and interleukin-1 receptors (IL-1R) families are of paramount importance in coordinating the early immune response to pathogens. Signaling via most TLRs and IL-1Rs is mediated by the protein myeloid differentiation primary-response protein 88 (MyD88). This signaling adaptor forms the scaffold of the myddosome, a molecular platform that employs IL-1R-associated kinase (IRAK) proteins as main players for transducing signals. These kinases are essential in controlling gene transcription by regulating myddosome assembly, stability, activity and disassembly. Additionally, IRAKs play key roles in other biologically relevant responses such as inflammasome formation and immunometabolism. Here, we summarize some of the key aspects of IRAK biology in innate immunity.
1. Introduction
Pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and nucleotide oligomerization domain-like receptors (NLRs) are of paramount importance in the innate host resistance to microbial infections. These receptors recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), transducing these signals into biological responses. TLRs accomplishes this by recruiting the signaling adaptors myeloid differentiation primary-response protein 88 (MyD88) and/or TIR domain-containing adaptor protein inducing IFN-β (TRIF) and their respective co-adaptors MyD88-adapter-like (Mal) and TRIF-related adaptor molecule (TRAM) (1–8). Most TLRs employ MyD88 as signaling adaptor, with the exceptions being TLR3 that signals exclusively via TRIF and TLR4 that uses both TRIF and MyD88 (2). In addition to PRRs, many of the early inflammatory responses are modulated by the interleukin (IL)-1 family of cytokines, which include IL-1α, IL-1β, IL-18 and IL-33 (9). Responses to these cytokines are mediated by the IL-1 receptor (IL-1R) and the closely related IL-18R and IL-33R, all of which employs MyD88 as signaling adaptor, similarly to TLRs (9–11).
Engagement of IL-1R or most TLRs results in the hierarchical recruitment of MyD88, IL-1 receptor-associated kinase (IRAK) 4, and IRAK2, or alternatively IRAK1, followed by E3 ubiquitin ligase TNF receptor associated factor 6 (TRAF6) (10–18), forming a supramolecular organizing center (SMOC) termed myddosome (14, 19). This SMOC controls the inflammatory response by mediating transcriptional and non-transcriptional events. These include regulation of transcription factors, control of post-transcriptional mRNA stability and transport, metabolic responses such as glycolysis, oxidative phosphorylation, and inflammasome activation (20–26).
Activation of the ubiquitin ligase TRAF6, in addition to ubiquitin ligases of the Pellino family (17, 27–32), are tightly regulated by upstream IRAK proteins. This event controls the activation of mitogen-activated protein kinases (MAPK), leading to transcription of activation protein 1 (AP-1) target genes, in addition to activation of IκB kinase complex, responsible for activation of transcription factors such as nuclear factor kappa B (NF-κB), interferon regulatory factor (IRF) 5 and IRF7 (8, 33–40) (Figure 1). This review will discuss how IRAK proteins regulate myddosome signaling: from assembly to disassembly, including stability, stimulation and inhibition of transcriptional and non-transcriptional responses.
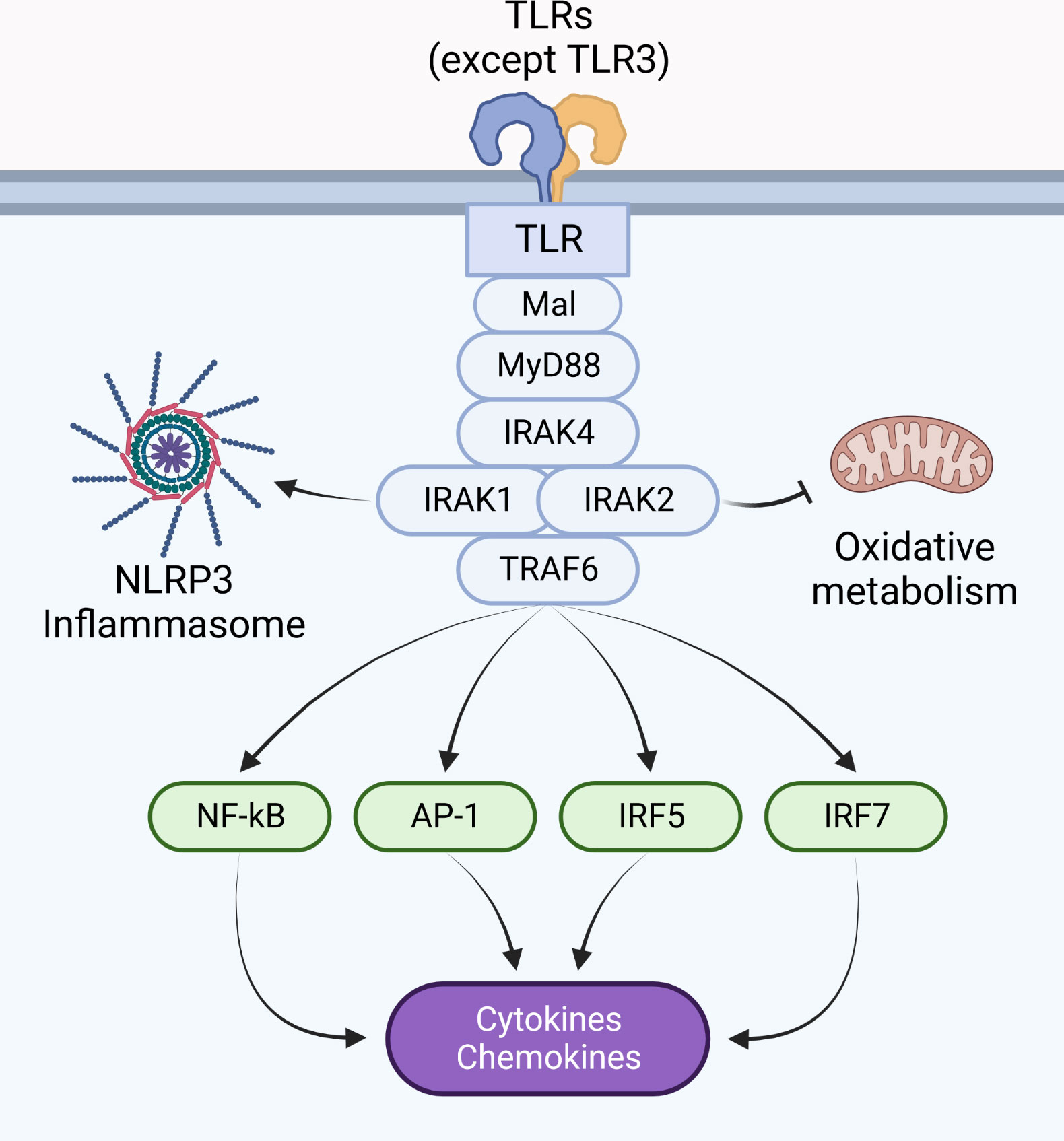
Figure 1 Pro-inflammatory responses are coordinated by the myddosome. The myddosome is a supramolecular signaling complex assembled upon dimerization of IL-1Rs and most TLRs (except TLR3), and is composed of MyD88, IRAK4, IRAK1 and/or IRAK2, in addition to TRAF6. Its activity coordinates the inflammatory response by activating NLRs, modulating metabolic responses, and regulating gene transcription via activation of transcription factors such as NF-κB, AP-1, IRF5, and IRF7.
2. Regulation of cell signaling by the IRAK family
IRAK proteins are serine/threonine kinases originally discovered in the context of IL-1R signaling (10, 12, 18, 41–45). Their recruitment to IL-1Rs is mediated by the signaling adaptor MyD88 (11), which is also shared by most TLRs (except TLR3) (2, 8). These proteins act as the main regulators of myddosome activity and are required for controlling a wide range of infections (12, 46–48). As such, mutations in these proteins are often associated with enhanced susceptibility to infections, sepsis, sterile inflammation, cancer (48–54), etc., and many pathogens have evolved evasion mechanisms that interfere with IRAKs (55–60).
2.1. The structure of IRAK proteins
IRAKs are highly conserved in vertebrates and contain a N-terminal death domain (DD), a ProST domain, a kinase domain (KD), and finally a C-terminal domain containing TRAF6 binding motifs (TBMs) (61, 62). DDs present in IRAKs are involved in homotypic protein-protein interactions, such as the interactions between IRAK4 and MyD88 discussed below. ProST domains (rich in proline, serine and threonine residues) are targeted by autophosphorylation, and are involved in activation of IRAK proteins. Close to its ProST domain, IRAK1 may also contain two PEST domains (63, 64) (rich in proline (P), glutamate (E) or aspartic acid, serine (S), and threonine (T)) which usually target proteins for degradation (65). All IRAKs contain a kinase domain, which is inactive in IRAK-M, due absence of a key aspartate residue (66). IRAK2 also lacks this aspartate residue, but this protein has kinase activity and behaves like an atypical kinase, discussed below (67). Interestingly, IRAK-M contains an active guanylate cyclase (GC) center in its kinase domain (68, 69). Finally, the C-terminal domain contains TBMs: three in IRAK1, two in IRAK2, and one in IRAK-M. IRAK4 does not contain a C-terminal domain and has no TBMs (62, 70) (Figure 2).
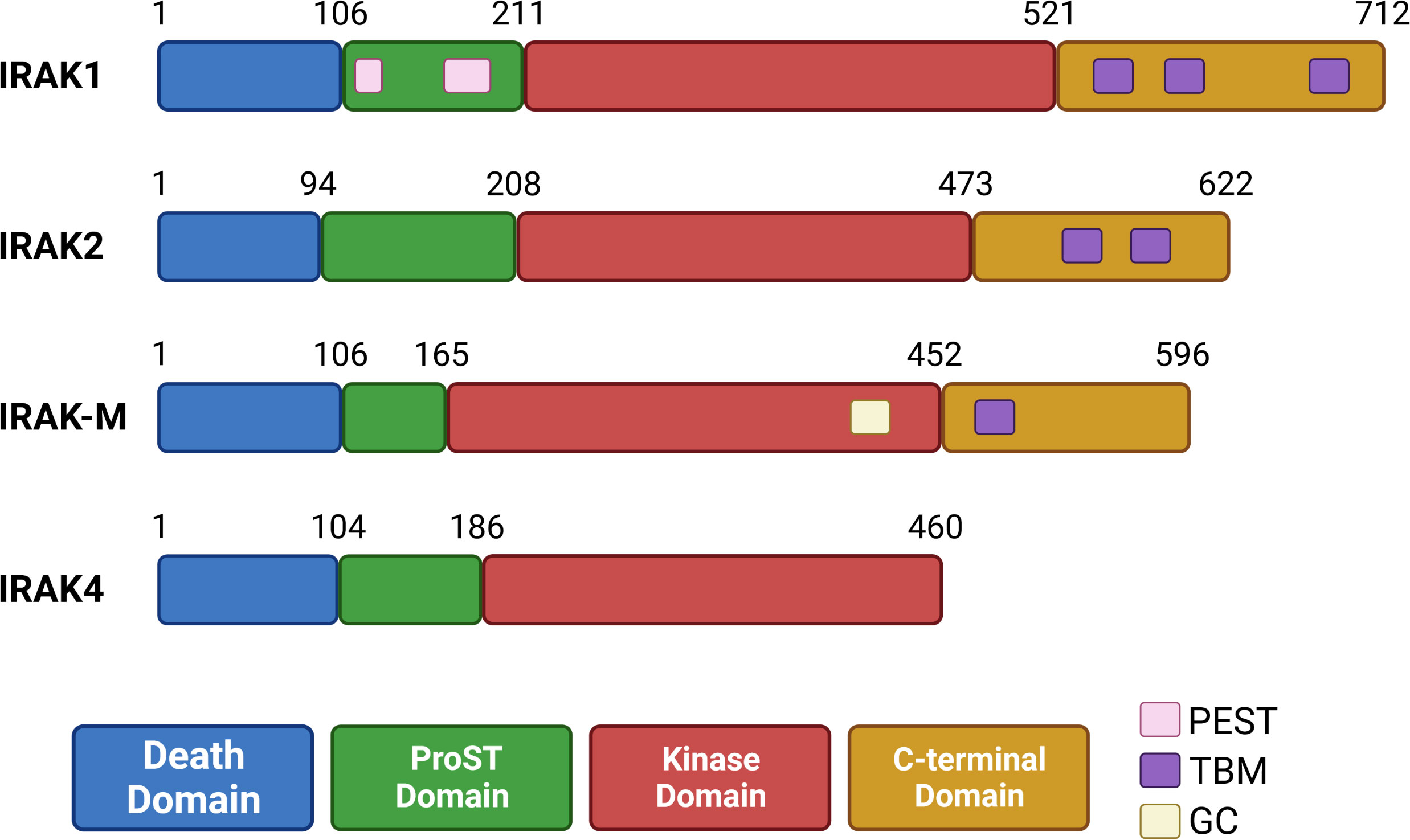
Figure 2 Organization of human IRAK proteins. IRAK proteins contain a death domain (DD), ProST domain, kinase domain (inactive in IRAK-M) and a C-terminal domain (absent in IRAK4). IRAK1 has two putative PEST sequences in its ProST region. IRAK-M contains a guanylate cyclase (GC) center in its kinase domain. The C-terminal domain contains up to three TRAF6 binding motifs (TBMs), and is absent in IRAK4.
Recognition of its ligand results in receptor dimerization followed by conformational changes in their cytoplasmic Toll/IL-1 receptor (TIR) domains. These conformational changes allow the receptor TIR domains to interact with 6 to 8 molecules of MyD88 via TIR-TIR homotypic interactions directly or via 2 molecules of Mal (also named TIR domain containing adaptor protein, TIRAP). Next, the MyD88 DDs interact with IRAK4’s DDs, in a stoichiometry of 6 to 8 molecules of MyD88 to 4 molecules of IRAK4. This stable interaction allows IRAK2 molecules (and presumably IRAK1) to interact with IRAK4 in a stoichiometry of 4:4. This forms a three-layered structure, arranged as a single-stranded left-handed helix. The proximity of multiple IRAK4 and downstream IRAKs 1 and 2 allows for sequential activation of their kinase activities to occur (15, 71, 72). IRAK4 molecules bound to MyD88 can auto trans-autophosphorylate, which then recruits IRAK1 to the myddosome and phosphorylates it (44, 73). This initial phosphorylation step activates IRAK1 and stimulates auto-hyperphosphorylation in its ProST region (rich in proline, serine and threonine residues) (Figure 3) (64). In resting cells, Toll-interacting protein (Tollip) interacts with the DD and kinase domain of IRAK1 (and presumably IRAK2), keeping these kinases in an inhibited state (74, 75). Upon receptor activation, PTEN induced protein kinase 1 (PINK1) interacts with Tollip/IRAK1 dimers, facilitating the delivery of IRAK1 to the myddosome (76).
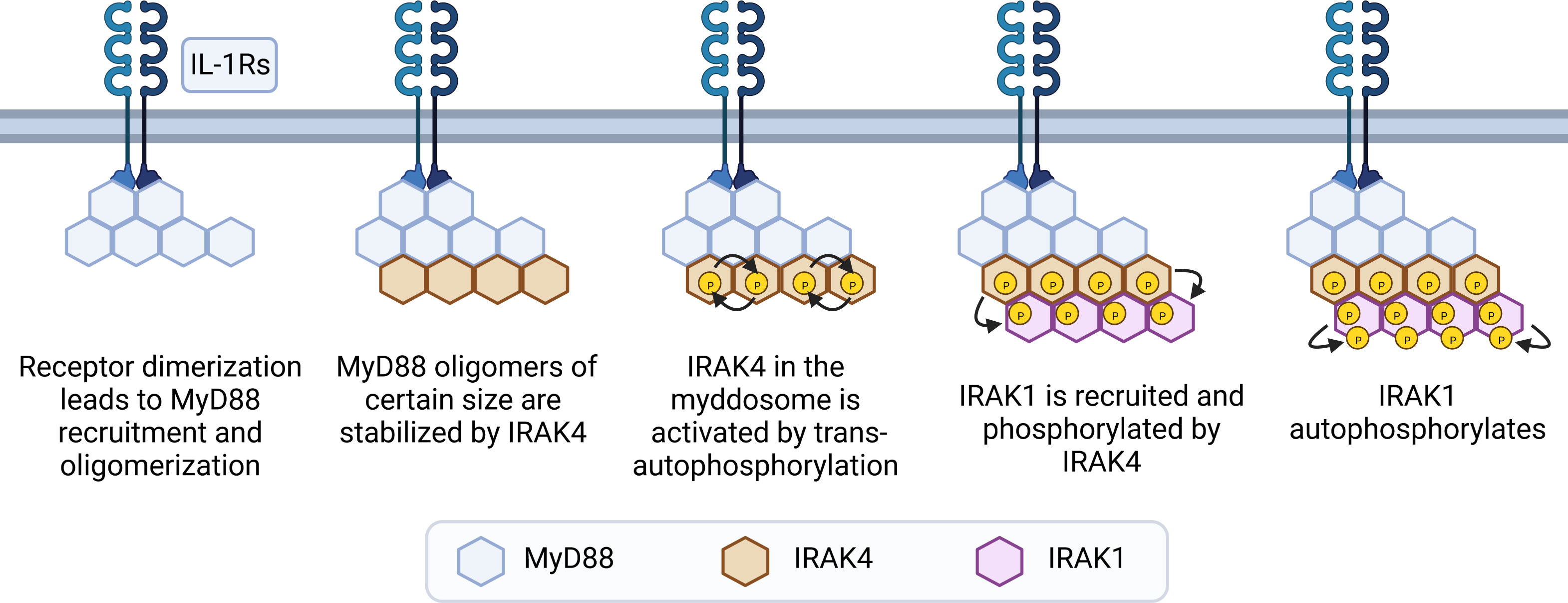
Figure 3 Steps of myddosome assembly. Dimerization of IL-1R or TLRs (except TLR3) leads to conformational changes in their cytoplasmic TIR domains, allowing TIR-TIR interactions with molecules of MyD88 to occur, either directly or via Mal dimers. This leads to formation of small and unstable MyD88 oligomers, which grow if receptor activation is sustained. After reaching a certain size threshold, MyD88 oligomers are stabilized by IRAK4. Interactions with MyD88 allows IRAK4 to trans-autophosphorylate and recruit IRAK1. Next, IRAK1 is activated by phosphorylation events mediated by IRAK4, and then further activated by auto-phosphorylation in its ProST regions, allowing signaling to occur.
The details involved in IRAK2 activation are less clear, but it is generally assumed to be similar to IRAK1. Initially, IRAK2 was thought to be a pseudokinase due its low in vitro kinase activity and substitution of a key aspartate residue to asparagine in its kinase domain (45), but later studies rebutted this claim and demonstrated that IRAK2 does possess kinase activity, which is activated by IRAK4-mediated phosphorylation on its lysine 237. Accordingly, point-mutated IRAK2 K237A fails to induce cytokine production, and no IRAK2 phosphorylation is observed in kinase-deficient IRAK4 macrophages (67, 77). Instead of a pseudokinase, IRAK2 can be classified as an atypical kinase, due substitution of residues 333 (Asp to Asn) and 351 (Asp to His) in its kinase domain (24, 78). Following activation by IRAK4, IRAK2 is autophosphorylated on residues S136 and T140 (24, 79). These autophosphorylation events are involved in IRAK2 translocation to the nuclei and to the mitochondria, where IRAK2 can act as a transporter of mRNAs to the cytoplasm and as an inhibitor of oxidative phosphorylation, respectively (24, 26). It is unclear whether these are the same autophosphorylation events necessary for NF-κB and MAPK activation.
The model of myddosome formation suggested above implies that either IRAK1 or IRAK2 can be recruited by IRAK4. In reality, the details about which IRAK is recruited to the myddosome are largely unknown. Immunoprecipitation experiments from murine macrophages suggests that IRAK2 is the main protein recruited to the myddosome, and IRAK1 is only detectable bound to the myddosome at early time-points and in situations where IRAK4 kinase activity is deficient (77, 80, 81). This could be a consequence of IRAK1 degradation (which requires IRAK4 kinase activity (80–83) and suggests that IRAK1 is recruited to early myddosomes, while later complexes contain IRAK2. This model is consistent with the observation that IRAK1 and IRAK2 are redundant at early time-points, but IRAK2 is required for later responses (Figure 4) (67). However, other cell types require IRAK1 (43). The molecular basis for this preference is unknown but may involve differences in IRAK1 and IRAK2 expression levels, half-life, splice variants, and regulation by additional molecules.
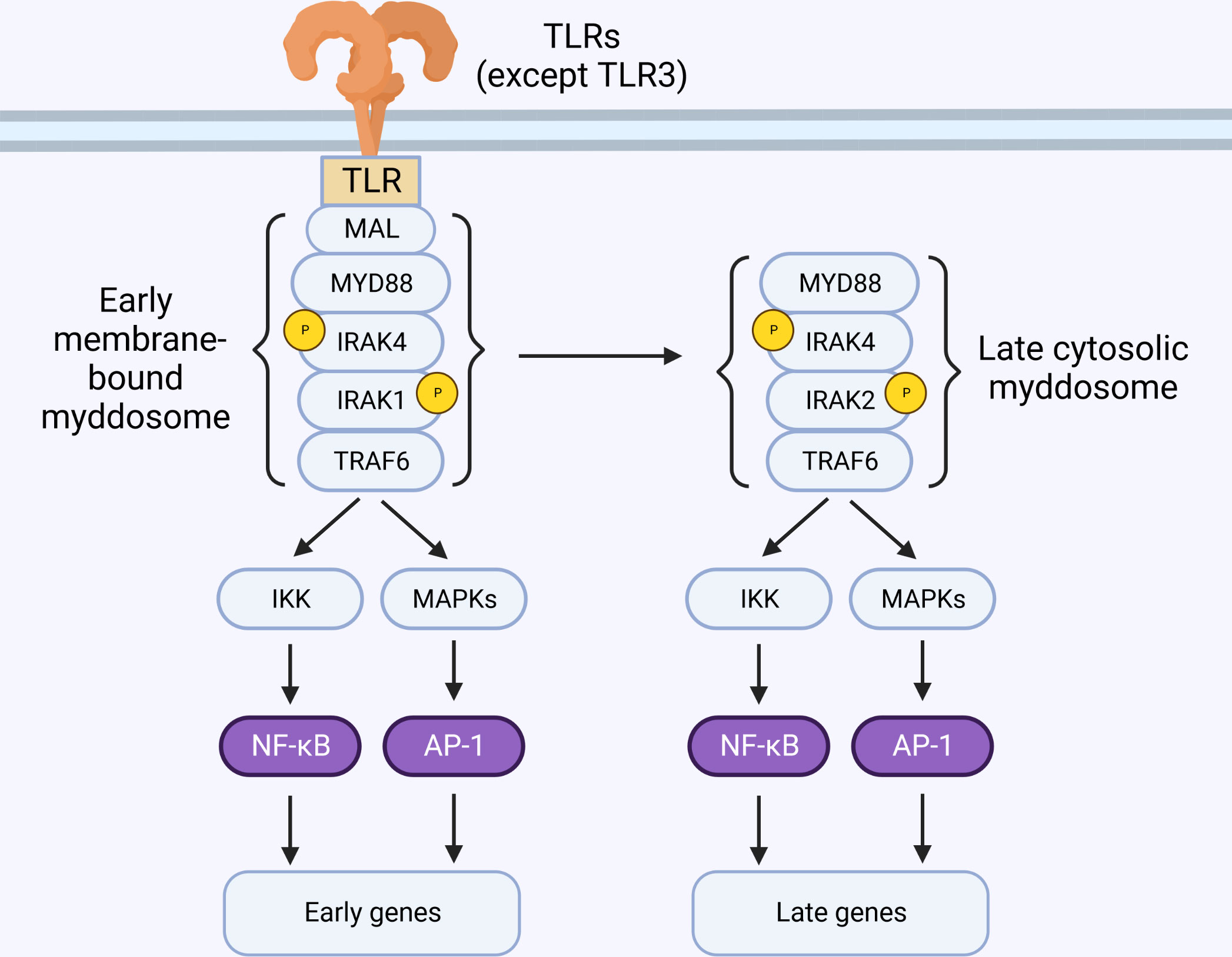
Figure 4 IRAK2 is essential for late-phase TLR signaling. TLR activation at the membrane triggers the sequential recruitment of Mal, MyD88, IRAK4, IRAK1 and TRAF6, forming the myddosome. These early membrane-bound complexes are short-lived and detectable only within minutes of stimulation. Sustained TLR stimulation leads to formation of cytosolic myddosomes containing IRAK2.
Via their C-terminal TBM, activated IRAK1 or IRAK2 can interact and activate TRAF6 (17, 84–86). IRAK2 binding to TRAF6 requires its E528 residue (mouse E525) (84, 87), and full activation requires its last 55 amino acids (88), while IRAK1 binding requires either of its two TBMs (70). Although the molecular mechanism of how IRAKs 1 and 2 activate TRAF6 are not completely understood, evidence suggests that IRAK-TRAF6 interactions results in TRAF6 auto-activation via auto-K63-ubiquitination (89), followed by its translocation from the myddosome to the cytosol (86), where it catalyzes the K63-ubiquitination of various targets such as IκB kinase (IKK) and IRAK1 (90, 91).
Recently, the IRAK4 scaffold was implicated in TRIF-mediated TRAF6 activation (77), and previously it was found that IRAK4 and IRAK1 can physically interact with TRAM (92). IRAK4 lacks the C-terminal TBM, which is present in IRAKs 1 and 2 (85), thus making it possible that the TRIF-mediated TRAF6 activation requires either IRAK1 or IRAK2 in addition to IRAK4, in a MyD88-independent way (77). While experimental evidence is still required to confirm the existence of the triffosome, these data raise the possibility that IRAKs are part of the putative triffosome and can lead to TRAF6 activation in the TLR4-TRIF pathway.
3. Control of myddosome stability and termination of signal transduction
The myddosome is a multifunctional SMOC formed at the cell membrane within minutes of TLR or IL-1R stimulation, and its assembly is essential for regulating biological responses such as production of inflammatory cytokines and metabolic responses (16, 19). As such, tight regulation of myddosome formation, stability, and disassembly is of paramount importance. We find IRAK4 at the center of these tightly regulated processes.
3.1. MyD88 oligomerization and myddosome stability is controlled by IRAK4
Stimulation of IL-1R results in rapid formation of small and unstable MyD88-oligomers. Prolonged exposure to IL-1β, however, favors the formation of larger oligomers that are sensed by IRAK4 (80). This mechanism ensures that only bona fide receptor activation can propagate signal. Live-cell microscopy suggests that MyD88 signaling is required for sustained activation of transcription factors (93), but early membrane-bound complexes containing MyD88, IRAK4 and IRAK1 are short-lived and detectable on a scale of a few minutes after stimulation (80, 94). Interestingly, biochemical isolation of endogenous myddosomes are often accomplished at time-points of 30 minutes or more, and these complexes predominantly contain IRAK2 instead of IRAK1 (77, 81, 95). Evidence suggests that stable MyD88-containing helical complexes occur in the cytosol, and can recruit IRAK4 after stimulation (96). Indeed, transfection of MyD88L265P (gain-of-function mutation) triggers myddosome formation and signaling in the cytosol, bypassing assembly in the membrane (97). Thus, it is feasible that early myddosome assembly in the membrane is a process regulated by IRAK4, and act as a nucleating step that results in the formation of stable signaling complexes in the cytosol, detectable by immunoprecipitation at later time-points (Figure 4) (25, 80, 94, 96, 98, 99).
Formation of longer MyD88 oligomers with enhanced stability is observed in IRAK4 knockout cells, while signal transduction is severely inhibited (12, 80). Similarly, IRAK4 inhibition or presence of kinase-dead IRAK4 increases myddosome stability, with severe deficiency in signal transduction. This is accomplished without increasing the size of MyD88 oligomers, as this activity is kinase-independent (80–82). Interestingly, IRAK4 inhibition does not affect IRAK2 recruitment to the myddosome, while recruitment of IRAK1 is in fact enhanced (25, 77, 80, 81, 98). In this case, neither IRAK1 or IRAK2 appear to be active (77), and the enhanced IRAK1 recruitment could be a consequence of enhanced myddosome stability and/or deficient IRAK1 degradation, normally triggered by IRAK4 kinase activity (64, 77, 83). Recruitment of IRAK4 to the myddosome leads to recruitment and phosphorylation of IRAK 1 and 2, turning on their autophosphorylation activities (67, 73, 100). In humans, however, IRAK1 can auto-phosphorylate when IRAK4 kinase activity is lost, and its activation likely involves an allosteric mechanism triggered by its interaction with IRAK4 in the myddosome (101, 102). No experimental evidence exists for similar compensatory mechanism involving IRAK2. Although in murine macrophages these IRAK4 kinase-deficient myddosomes fail to lead to cytokine production (77), it is possible that recruitment of IRAK1 and/or IRAK2 to this complex could compensate for the lack of IRAK4 kinase activity in other cell type or species (101, 103).
3.2. IRAK4 activation initiates a negative feedback loop
Myddosome formation leads to induction of pro-inflammatory and other adaptations, all of which are required for an efficient response. However, uncontrolled myddosome activation is detrimental, as evidenced by gain-of-function mutations linked to diseases (104, 105). Thus, termination of the response can be as important as its initiation. IRAK4 activity is central to the myddosome-mediated pro-inflammatory responses, but it also initiates a series of adaptations that inhibits myddosome signaling, forming a negative feedback loop. Amongst these adaptations are induction of antagonists such as IRAK-M and A20, post-translational modifications such as S-Nitrosylation, and the degradation of myddosome components such as Mal, IRAK1, TRAF6 and MyD88 itself (64, 66, 83, 106–109). This ensures tight regulation of the pro-inflammatory response initiated by TLRs and IL-1Rs (Figure 5).
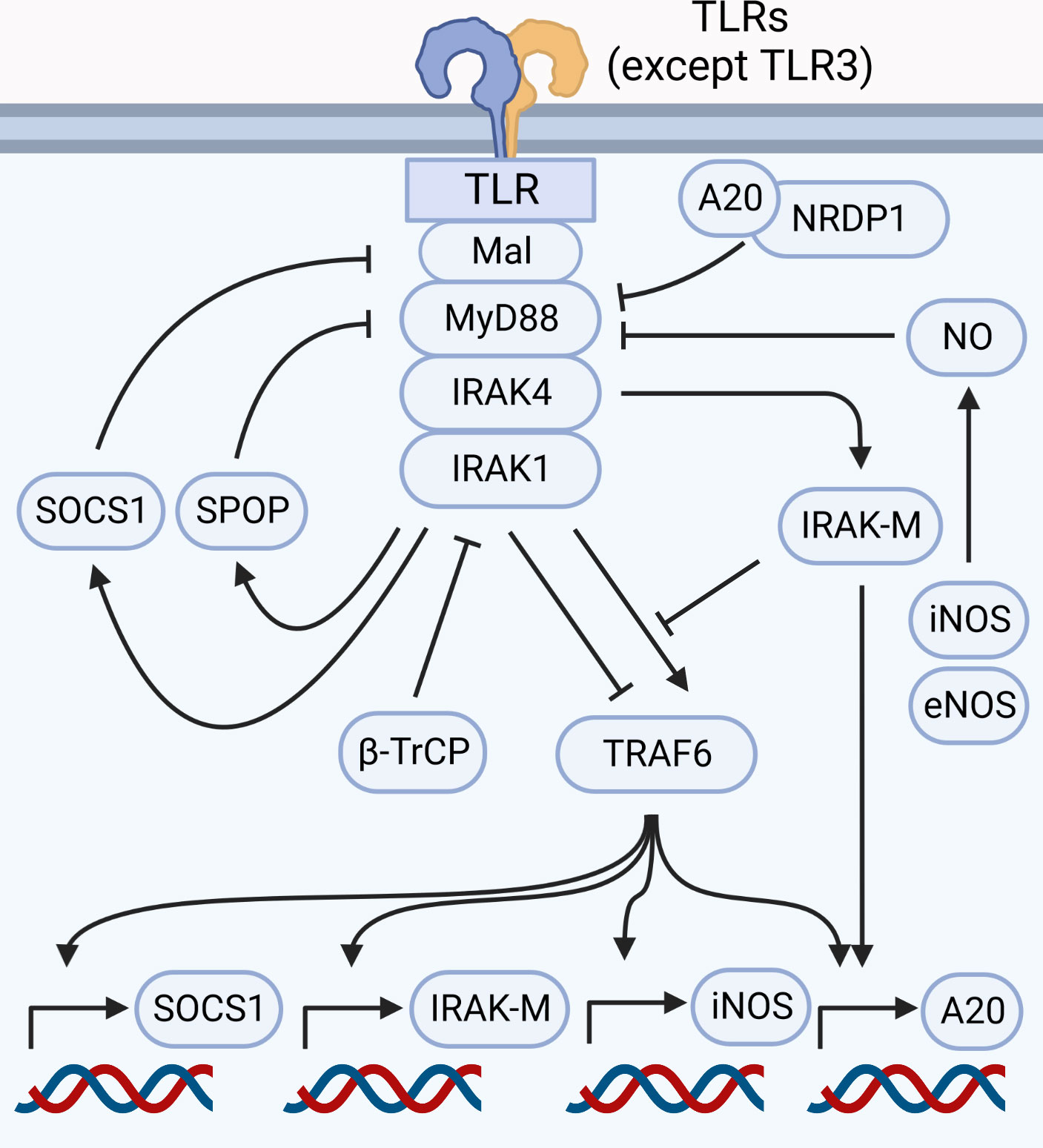
Figure 5 Myddosome signaling is regulated at nearly every step. Signaling through the myddosome leads to transcription of negative regulators such as SOCS1, iNOS, IRAK-M and A20. Proteasomal degradation of components such as Mal (controlled by SOCS1), MyD88 (controlled by A20 and NRDP1, in addition to SPOP in HSC), IRAK1 (controlled by β-TrCP), and TRAF6 (triggered by IRAK1) are likely involved in myddosome disassembly and signal termination. IRAK-M antagonizes the myddosome by inhibiting the binding between IRAK1 and TRAF6, besides stimulating A20 expression. Production of NO by eNOS and iNOS leads to S-Nitrosylation of MyD88, inhibiting signal transduction.
Study of neuregulin receptor degradation protein-1 (NRDP1) revealed that this E3 ubiquitin-ligase modulates cytokine production via K48 ubiquitination of MyD88 and K63 ubiquitination of tank binding kinase 1 (TBK1). This results in production of type I interferons due TBK1 activation, and inhibition of pro-inflammatory cytokines due proteasomal degradation of MyD88 (108). This MyD88 ubiquitination requires physical interactions between myddosome components, NRDP1 and A20 (108, 110). Although no direct links between NRDP1 and IRAKs were established, A20 expression is induced by NF-κB (111), suggesting that MyD88 degradation requires a functional myddosome capable of driving A20 expression. Additionally, other molecules were linked to MyD88 ubiquitination and degradation (112, 113), including Speckle-type BTB–POZ protein (SPOP) (114, 115). SPOP is a ubiquitin ligase highly expressed in hematopoietic stem cells (HSC), and while the details on how SPOP regulates MyD88 degradation are largely unknown, evidence suggests that IRAK4 is required for SPOP-mediated K48 ubiquitination of MyD88 (114).
TLR4 activation leads to ubiquitination and degradation of the co-adaptor Mal, likely playing a role in termination of signal and myddosome disassembly (106). Experiments involving overexpression or pharmacological inhibition of IRAK1 and IRAK4 demonstrates that these IRAKs are required for Mal phosphorylation and subsequent ubiquitination and degradation (106). This is likely mediated by suppressor of cytokine signaling (SOCS) 1 and 3, as evidence suggests that upon TLR activation SOCS1 (and potentially SOCS3) ubiquitinate Mal leading to its proteasomal degradation and suppression of cytokine production (116–118). While Mal degradation is observed within 15 to 30 minutes of TLR2 or TLR4 activation (117), TLR-dependent production of nitric oxide (NO) can upregulate SOCS1 expression, contributing to hypo-responsiveness upon long lipopolysaccharide (LPS) stimulations (119). High concentrations of NO produced by either endothelial nitric oxide synthase (eNOS) or inducible nitric oxide synthases (iNOS) can directly inhibit myddosome signaling, due nitrosylation of cysteine residues in the MyD88 protein (109).
IRAK1 activity rapidly decreases after TLR stimulation, which correlates with its degradation. Interestingly, IRAK2 is not degraded and can sustain signaling for longer (67, 77). One proposed mechanism for this behavior involves the presence of two putative PEST sequences in IRAK1 (ranging from amino acids 117 to 133 and 153 to 180), which are absent in IRAK2 (63, 64). It was suggested that IRAK4 kinase activity triggers hyperphosphorylation in or adjacent to the IRAK1 PEST sequences, targeting it for degradation (64, 83). It is presently unclear whether the proteasome is responsible for IRAK1 degradation: while some reports suggests that IRAK1 is K48-ubiquitinated by β-transducin repeats-containing proteins (β-TrCP) and degraded by the proteasome following IL-1R or TLR stimulation (120–122), other studies suggest that IRAK1 is mainly targeted by K63-ubiquitination and its degradation is proteasome-independent (83, 123). Whatever the case may be, the PEST-hypothesis cannot be ruled-out, as PEST-containing proteins can also be targeted by other proteases such as calpain (65) and functional validation of PEST regions in IRAK proteins is still required.
The E3-ubiquitin ligase TRAF6 is a key mediator of TLR and IL-1R responses, and downregulation of its expression is one of the reported mechanisms involved in termination of MyD88-dependent responses. TRAF6 expression levels decreases after long periods of TLR activation (around 24 hours in vitro) due proteasomal degradation. The presence of IRAK1, but not its kinase activity, is involved in downregulating TRAF6. One suggested mechanism is that IRAK1 bound to TRAF6 is K48 and K63 ubiquitinated, and this interaction directs both proteins to the proteasome (107). Accordingly, mutations on IRAK1’s TBM can inhibit TRAF6 degradation. IRAK1 contains three C-terminal TBMs, and while deletion of all TBMs fail to transduce signal and induce cytokine production due deficient TRAF6 interaction, TRAF6 degradation, however, requires the DD in addition to one TBM (70). This suggests that depending on context IRAK1 can induce TRAF6 activation or degradation, but the molecular mechanisms for these antagonic activities are presently unknown.
In resting cells, Tollip is bound to IRAK1, supressing its activity (74, 75). Upon stimulation, Tollip releases IRAK1, which is then recruited to the myddosome. Interestingly, Tollip itself is a target of IRAK1-mediated phosphorylation (75), and is involved in the degradation of IL-1R and signal termination (124). It is presently unclear if Tollip phosphorylation triggers IL-1R degradation, but it is tempting to speculate that release and phosphorylation of Tollip is another negative feedback mechanism.
3.3. Myddosome inhibition by IRAK-M
While IRAKs 4, 1 and 2 are, in most conditions, responsible for amplifying the signal from activated TLRs or IL-1Rs, the pseudokinase IRAK-M (also known as IRAK3) is a negative regulator of this response (66). In humans, IRAK-M expression is restricted to cells of the myeloid lineage (45), but murine IRAK-M is detectable in a wider range of cell types (125). In macrophages, IRAK-M represses NF-κB and MAPK by a variety of mechanism, such as inhibition of the interaction between IRAK4 and IRAK1 with TRAF6 (66), stabilization of MAPK phosphatase 1 (MKP-1, a phosphatase responsible for inactivating the MAPK p38), and transcription of inhibitory molecules (66, 126, 127).
Despite negligible kinase activity, IRAK-M can induce a specific subset of NF-κB target-genes, which collectively inhibit the inflammatory response. Indeed, overexpression experiments originally described IRAK-M as a positive regulator of NF-κB signaling, not unlike IRAK1 and IRAK2 (45). Evidence suggests that in physiological conditions, IRAK-M can interact with MyD88 and IRAK4, forming a myddosome that activates transforming growth factor-β-activated kinase 1 (TAK1) and stimulates inhibitory genes such as A20 and NF-κB inhibitor-α (IκB-α) (127). Surprisingly, IRAK-M can also, in specific contexts, induce the expression of pro-inflammatory genes. Upon IL-33 stimulation of murine dendritic cells, the prolyl cis-trans isomerase PIN1 interacts with IRAK-M, promoting its nuclear translocation and transcription of genes involved in type 2 immunity and airway inflammation such as Il6, Csf3, Cxcl2 and Ccl5 (128). It is unclear how the pseudokinase IRAK-M activates gene transcription, but one intriguing possibility involves cGMP as second messenger: bioinformatics studies suggested that IRAK-M possess guanylate-cyclase activity (68), which was later experimentally confirmed (69). Transduction experiments demonstrates that wild type IRAK-M attenuates LPS-induced NF-κB activity, but a IRAK-M mutant with impaired guanylate cyclase activity does not (69). The details regarding this pathway are currently unknown, and further studies are required to confirm its importance in physiological conditions.
Notably, TLR activation leads to a NF-κB-dependent IRAK-M upregulation, forming a negative feedback loop that contributes to signal termination and tolerance (66, 129). This is not exclusive to TLRs and IL-1Rs, and is likely part of a more general mechanism for resolving inflammation, as IRAK-M can also be induced by other transcription factors including glucocorticoid receptors (130, 131). Due its generally anti-inflammatory activity, the induction of IRAK-M can be exploited by different pathogens as an evasion mechanism (59, 132).
4. IRAK4 scaffold and kinase activities
While IRAK4 is the main regulator of myddosome activity, its roles can be divided into “scaffold activity” and “kinase activity”, as suggested by various studies where cells expressing kinase-deficient IRAK4 do not phenocopy IRAK4 knockout cells (77, 133–135). Another surprising observation is that the IRAK4 scaffold, but not its kinase activity, is required for the production of pro-inflammatory cytokines in TLR-stimulated human cells (136). Understanding the subtle differences between IRAK4 scaffold and kinase activities is relevant from a medical standpoint, as IRAK4 inhibitors and degraders are currently under investigation (137).
IRAK4 is a key component of the myddosome, and it is believed that loss of IRAK4 or inhibition of its kinase activity phenocopies the loss of MyD88 (12, 46–49, 77, 133–135, 138). This is, however, context-dependent, as there are important differences between the IRAK4 scaffold and kinase activities depending on stimuli, cell type, species, etc. Kinase-deficient IRAK4 murine macrophages fail to produce pro-inflammatory cytokines upon stimulation of IL-1R and TLRs 2, 7, and 9 (77, 133–135). Interestingly, inhibition of IRAK4 kinase activity in human macrophages or fibroblasts does not impact cytokine production (101, 136). In murine macrophages inhibition of IRAK4 kinase activity impacts NF-κB and MAPK activation (77, 133–135), while in human monocytes this inhibition has no impact on NF-κB and MAPK, and only IRF5 activation is impaired (139).
In TLR4-stimulated mouse macrophages, inhibition of IRAK4 kinase activity does not completely impairs cytokine production, while loss of IRAK4 scaffold does (77, 133–135). This is likely due the fact that TLR4 signals via MyD88 and TRIF, and suggests that the putative triffosome employs the IRAK4 scaffold to activate TRAF6, which is the hub that links MyD88 and TRIF in TLR4 signaling (17, 77, 140). Although physical interactions between TRAM, TRAF6, IRAK4 and IRAK1 were previously described (92, 141), little is known on how the triffosome is assembled and how it activates TRAF6.
In the absence of IRAK4 kinase activity, an enhancement in IRAK1 recruitment is observed as long as the IRAK4 scaffold is present (77, 81). Previous studies suggest IRAK1 can bypass the initial phosphorylation performed by IRAK4, in situations where IRAK4 kinase activity is deficient (64, 102). While this is unlikely the case in murine macrophages, this compensatory mechanism could play a role in human fibroblasts (101, 103, 136, 142). This is further suggested by the D329A mutant in human IRAK4. This mutant lacks kinase activity, but IL-1β-stimulated IRAK4D329A fibroblasts has partial cytokine production likely caused by the decreased interaction between IRAK4 and IRAK1, and not by the lack of IRAK4 kinase activity (82). This compensatory mechanism is not perfect, as IRAK4 kinase activity is required for IRAK1 degradation (83), suggesting that IRAK4 enzymatic activity controls both the initiation and termination of myddosome signaling.
5. IRAK1 and IRAK2 are not redundant in every context
Early studies on IL-1R demonstrated that the cytoplasmic fraction of this receptor physically interacts with a serine-threonine kinase responsible for signal transduction (41, 42). It was postulated at the time that a protein kinase termed IRAK was the responsible for this activity (42). Further study in human cells identified IRAK, a protein that shared high similarity to Pelle, responsible for activation of a NF-κB homolog in Drosophila (18). Generation of IRAK knockout mice, however, revealed that in vivo responses to IL-1β or IL-18 were only partially blocked, suggesting the existence of an alternative pathway (43). Additionally, Irak1-/- mice challenged with LPS showed only a subtle improvement in survival, further suggesting an alternative pathway (143). While these developments occurred, another Pelle-like protein termed IRAK2 was identified, also capable of interacting with IL-1R and the then discovered adaptor MyD88, and required for NF-κB activation (10). Development of IRAK2 knockout mice strongly suggested that this protein was involved in production of cytokines in response to TLR4 and TLR9 stimulation, but NF-κB activation was not completely deficient in IRAK2 knockout cells (67, 144). Finally, study of TLR and IL-1R responses in murine macrophages lacking both IRAK1 and IRAK2 demonstrated that these proteins are somewhat redundant in those cells, albeit with key differences in their behavior (67).
The crystal structure of the myddosome reveals a helical architecture containing multiple copies of MyD88, IRAK4 and IRAK2. While this model does not include IRAK1 due technical limitations (the authors were unable to express IRAK1), the IRAK2 residues required for interaction with IRAK4 in the myddosome are also found in IRAK1 (15). This, in addition to immunoprecipitation data showing that IRAK1 is found in the myddosome when IRAK4 is present, strongly suggests that either IRAK1 or IRAK2 are recruited to this SMOC and can play similar roles (41, 42, 103).
The history of the discoveries of these IRAKs, as well as the myddosome structure, suggests that these proteins are somewhat redundant, and only double knockout cells show a clear phenotype. This is, however, an oversimplification. The roles played by IRAK1 and IRAK2 on cytokine production can be context dependent, varying according to species, cell type, duration of stimuli, etc. Additionally, other biologically relevant activities such as inflammasome activation and metabolism regulation appears to employ one IRAK and not the other.
5.1. IRAK2 is required for sustained signaling in murine macrophages
In murine macrophages, IRAK1 and IRAK2 show redundancy at early time-points. Knockout or knockdown of either IRAKs in those cells fail to decrease NF-κB and MAPK activation, while IRAK1/IRAK2 double knockout macrophages are highly deficient. Despite this early redundancy, IRAK2 knockout cells are critically impaired in inflammatory cytokine production while IRAK1 knockouts behave like wild type controls (67, 77, 87). One possible explanation for this phenomenon is that IRAK1 is degraded after stimulation, making IRAK2 essential for sustained signaling. This hypothesis is further corroborated by the existence of IRAK1b, a splice variant with prolonged stability and capable of sustaining NF-κB activation in human cells (145). This partial redundancy, however, appears to be context dependent. For instance, Irak1-/- mouse embryonic fibroblasts fail to activate NF-κB in response to IL-1β or IL-18 (43), whereas Irak1-/- macrophages activate NF-κB unimpaired in response to a variety of TLR agonists (67, 77). In humans, IRAK1-deficient macrophages are greatly deficient in TNF production, while IRAK2 deficiency has little to no impact, opposite to what is observed in mouse macrophages (136).
5.2. IRAK1 activates IRFs 1, 5 and 7
The activation of transcription factors and control of gene transcription are possibly the most important responses mediated by the myddosome. Although NF-κB activation is often used as surrogate for myddosome activity, this is not the only family of transcription factors controlled by this SMOC. The IRF family, in particular IRFs 1, 5 and 7, are also activated downstream MyD88 in a variety of conditions (2). As discussed above, NF-κB activation often displays redundancy regarding the use of IRAK1 and IRAK2. This redundancy, however, is not observed on the activation of IRF proteins.
In plasmacytoid dendritic cells (pDCs), activation of TLRs leads to a MyD88-dependent production of interferon-α (IFN-α), tumor necrosis factor (TNF), and IL-12 (146), with the IFN-α output primarily regulated by the transcription factor IRF7 (37). Similar to what is observed in murine macrophages, IRAK1 knockout pDCs are not deficient in MAPK and NF-κB activation upon stimulation of TLRs 7 or 9. Accordingly, these Irak1-/- pDCs produced normal levels of TNF and IL-12. Surprisingly, the production of IFN-α was highly deficient in Irak1-/- pDCs (147). TLR stimulation of pDCs leads to physical interactions between MyD88, IRAK4, IRAK1, TRAF6 and IRF7 (36, 147). IRAK1 kinase activity is required for IRF7 activation, possibly by controlling IKK-α, responsible for IRF7 phosphorylation. This leads to IRF7 dimerization and nuclear translocation (34, 147–150). Contrary to NF-κB and MAPK activation, IRAK1, but not IRAK2, is responsible for IRF7 stimulation (84, 151). In fact, TLR9-stimulated IRAK2 knockout pDCs shows higher IFN-α production than WT controls, suggesting that this kinase inhibits IRF7 by an yet unknown mechanism (151).
In TLR9-stimulated macrophages and myeloid dendritic cells (mDCs), production of IFN-β is controlled by the transcription factor IRF1 (152). IRF1 activation requires physical interaction with the myddosome, and most likely involves phosphorylation mediated directly or indirectly by IRAK1 (152, 153). Although it is tempting to speculate that IRF1 activation is, like IRF5 and IRF7, a process that requires IRAK1 and not IRAK2, experimental evidence is still required to rule out the involvement of IRAK2 in this pathway.
IRF5 activation occurs downstream MyD88-dependent TLRs, with MyD88, TRAF6, and IRAK1 required for its activation (34, 35). This suggests that myddosome formation and physical interaction between IRF5 and its components is essential. Indeed, IRAK1:IRF5 interaction precedes IRF5 ubiquitination by TRAF6 and activation (34, 154). In addition to ubiquitination, IRF5 phosphorylation by IKK-β is required to its dimerization and nuclear translocation (39, 40). This activity appears to be mediated by IRAK1 exclusively, as inhibition of IRAK2 signaling by the poxvirus A52 protein failed to impact IRF5 activation (55, 84). The precise timing of all the sequential steps involved in IRF5 activation is unclear, and whether ubiquitination by TRAF6 occurs in physiological conditions remains an open question. Regardless, studies on human monocytes expressing physiological concentrations of IRAK1 and IRF5 also suggests that this transcription factor requires IRAK1, as loss of this IRAK antagonizes IRF5 signaling (155).
While IRFs 1 and 7 regulate the production of type I interferons (37, 38, 156), IRF5 is involved in the control of various pro-inflammatory cytokines (35). Indeed, the set of genes regulated by IRF5 and NF-κB show overlap, and physical interaction between IRF5 and NF-κB is a potential mechanism for gene regulation (157). How IRF5:NF-κB interactions occur and how this process is regulated remains poorly understood.
5.3. IRAK1 controls rapid inflammasome activation
Activation of the NLR family pyrin domain containing 3 (NLRP3) Inflammasome occurs in two phases. In the first phase, a signal primes the cell, enabling it to respond to a DAMP. In the second phase, recognition of a DAMP triggers the assembly of the inflammasome, a macromolecular structure responsible for triggering pyroptosis via caspase-1 activation and maturation of pro-IL-1β and pro-IL-18 (158). The priming phase includes both transcriptional and non-transcriptional responses, such as upregulation of key-components (pro-IL-1β and NLRP3 itself for example), and control of post-translational modifications that enables the inflammasome to respond.
It is well established that transcriptional priming in many situations involves the myddosome (and consequentially IRAKs) (2), but study of non-transcriptional priming of the NLRP3 inflammasome revealed an additional role for IRAK4 and IRAK1. IRAK1-deficient mouse macrophages fails to activate caspase-1, whereas IRAK2-deficient cells behave like wild type controls (23). This response is rapid, precedes transcriptional priming, and allows the NLRP3 inflammasome to quickly release pre-synthesized IL-18 in response to DAMPs (22, 23). While it is unclear how this happens, it is tempting to speculate that IRAK1 phosphorylates components of the inflammasome to fine-tune its assembly. Phosphorylation in the NLRP3 leucine rich repeat controls inflammasome assembly, and mutations on a key serine residue blocks non-transcriptional priming (159). This is further suggested by the observation that kinase-deficient IRAK1 and IRAK4 fail to prime the inflammasome, and physical interactions between IRAK1 and components of the inflammasome can be observed (23).
While IRAK1 appears to stimulate NLRP3 activity in the context of rapid inflammasome activation, it can also act as a negative regulator in other conditions. Upon TLR and IL-1R activation, IRAK1 is ubiquitinated by the ubiquitin ligase Pellino 2 (28, 32). Ubiquitinated IRAK1 cannot interact with NLRP3, which allows Pellino 2 to ubiquitinate and activate NLRP3. In IRAK1-deficient macrophages (or containing a kinase-dead IRAK1), NLRP3 ubiquitination occurs at a higher rate and inflammasome activation is increased. When macrophages are deficient for Pellino 2, however, interactions between IRAK1 and NLRP3 are enhanced, and inflammasome activation is deficient (160).
These seemingly contradictory reports present an incomplete view on how IRAK1, as well as other IRAKs, fine-tunes inflammasome priming and activation. Further investigation is required to understand the different roles that these kinases have on inflammasome activity.
5.4. IRAK2 inhibits oxidative metabolism
While this review has primarily focused on how IRAKs regulates cytokine production, a large amount of recent studies have highlighted another important aspect involved in innate immune responses: modulation of cell metabolism (161). We are only beginning to understand the links between IRAKs and metabolism, with recent studies suggesting that these kinases both regulate and are regulated by cell metabolism.
TLR activation in myeloid cells leads to rapid transition to aerobic glycolysis and increase in succinate concentration. Succinate then acts as a positive regulator of the transcription factor hypoxia inducible factor-1α (HIF-1α) which stimulates the expression of genes such as IL1B and IRAKM (162–164). HIF-1α also controls the transcription of microRNA-146a, which act as a negative regulator of pro-inflammatory gene expression by downregulating the expression of TRAF6 and IRAK1 (165, 166). Another important player in the rapid stimulation of glycolysis via TLRs is TBK1 (167). This protein kinase is known to mediate IRF3 activation downstream TRIF (168), but surprisingly, induction of glycolysis by TBK1 requires myddosome assembly and physical interaction with its components (25). It is presently unknown which IRAKs, if any, mediate myddosome-dependent TBK1 activation.
A more direct link between IRAKs and metabolism comes from the observation that IL-1β inhibits mitochondrial oxidative phosphorylation in adipocytes (26). Upon IL-1R activation, MyD88, IRAK4 and IRAK2 translocate to the mitochondrial outer membrane. From there, IRAK2 further translocates into the mitochondrial inner membrane and inner membrane space via the translocators TOM20 and TIMM50. IRAK2 then inhibits oxidative phosphorylation by disrupting the interaction between prohibitin (PHB) and optic atrophy protein 1 (OPA1), key in the formation of the mitochondrial respiratory chain complex. Importantly, myddosome formation triggers IRAK2 auto-phosphorylation at residues S134 and T140 (24, 79), which are required for IRAK2 translocation and interaction with PHB-OPA1. Interestingly, in adipocytes IRAK1 deficiency did not disrupt oxidative phosphorylation, and IRAK2 deficiency did not affect pro-inflammatory gene expression, further evidence that IRAK1 and IRAK2 play different role in different cell types (26).
6. IRAK mutations in human diseases
IRAK proteins are the main actors coordinating myddosome activity and play essential roles in the innate immune response in mammals. However, important details of their biology differ between humans and other animals. For instance, the kinase activity of IRAK4 does not impair the responses in human macrophages (136), suggestive that in humans this kinase might be involved in other pathways. Similarly, IRAK1 has a more prominent role in humans, while IRAK2 appears to be redundant (136). As such, mutations affecting IRAK1 and IRAK4 can have dramatic clinical impact in humans.
IRAK4 mutations are clinically relevant and life-threatening in childhood, with a mortality rate estimated at 38% (169). Most of the studied IRAK4 disorders are autosomal recessive, often caused by point mutations introducing an early stop codon, leading to virtually absent IRAK4 expression or production of a non-functional protein (49, 50, 169, 170). This results in high susceptibility to pyogenic bacterial infections, specially Staphylococcus aureus, Streptococcus pneumoniae, and Pseudomonas aeruginosa (169). However, cases have been reported linking IRAK4 mutations to susceptibility to other bacterial agents such as Salmonella serogroup C1, Listeria monocytogenes, Shigella sonnei and non-bacterial agents such as herpesvirus (51, 171–173).
Little is known about IRAK1 loss-of-function mutations in humans, but evidence suggests that such mutations potentially lead to increased susceptibility to infections. For instance, investigation of a patient who died as result of pulmonary infection at the age of 7 months revealed complete IRAK1 deletion. This deletion may cause a X-linked recessive disorder with severely impaired TLR responses in fibroblasts, but not in peripheral blood mononuclear cells (54). IRAK-M has several anti-inflammatory activities, and single nucleotide polymorphisms in the IRAK3 gene were associated with higher susceptibility to asthma, likely due deficient anti-inflammatory responses in airway epithelial cells (52).
IRAK1 gain-of-function mutations are more common and may lead to enhanced sepsis-induced injury due increased pro-inflammatory responses (174). Interestingly, various reports suggest that IRAK1 mutations causing enhanced protein expression or spontaneous activation act as an oncogene and are involved in development of various cancers (53, 175–178). Similarly, IRAK4 gain-of-function mutations are associated with cancers and linked to poor prognosis and resistance to chemotherapy (179–181).
7. Conclusion and perspectives
Since the discovery that IL-1R signaling requires a protein kinase (41, 42), many aspects of the IRAK family were studied. After decades of efforts, we understand in broad strokes their biology, but many of the finer details remain elusive. For instance, the myddosome is assembled and signal downstream IL-1Rs and most TLRs, which is both a blessing and a curse: lessons learned from a specific TLR or IL-1R are applied to all other receptors, but important details of specific receptors might be missed.
The myddosome can recruit either IRAK1 or IRAK2 to transduce signals (15), and different cell types appear to employ one IRAK preferentially. What causes the preferential use of one over the other is largely unknown. Study of cell-specific responses may lead to better understanding on how these kinases affect and are affected by different microenvironments. Similarly, human and mouse cells often diverge regarding the use of IRAKs 1 and 2, but the reasons for these species-specific phenotypes are unknown.
The links between pattern recognition receptors and metabolism are only beginning to be understood, but it is clear that they deeply influence each other (161). For example, myddosome assembly upon TLR activation triggers glycolysis via TBK1 (25). It is unclear whether any IRAKs are important in this pathway, but since TBK1 interacts with the myddosome it is tempting to speculate that IRAKs are upstream TBK1. Another report suggests that IRAK2 directly inhibits oxidative phosphorylation in IL-1R-stimulated adipocytes (26). The study of how the myddosome and IRAKs are involved immunometabolism is still in its early days and remain an exciting prospect.
The observation that inhibition of IRAK4 kinase activity has less impact in human than in murine macrophages is particularly interesting (136), as it suggests that in humans this kinase might be involved in other pathways, and only the IRAK4 scaffold is required for TLR signaling. This scaffold, and potentially other IRAKs, might also be involved in TRAF6 activation mediated by the putative triffosome by yet unknown mechanisms (77). A deeper understanding of the differences between IRAK4 kinase and scaffold activities is likely to be clinically relevant, as both IRAK4 inhibitors and degraders are currently in clinical trials (137) and can potentially impact the treatment of a wide range of inflammatory and autoimmune diseases.
Author contributions
MP wrote the manuscript. RG revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institutes of Health (R01NS098747 and R01AI079293); Fundação de Amparo a Pesquisa do Estado de São Paulo (Fapesp, 2016/23618-8), Brazilian National Institute of Science and Technology for Vaccines granted by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)/Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig)/Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) (465293/2014-0).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
2. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
3. Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the toll signaling pathway. Nat Immunol (2001) 2:835–41. doi: 10.1038/ni0901-835
4. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for toll-like receptor-4 signal transduction. Nature (2001) 413:78–83. doi: 10.1038/35092578
5. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198:1043–55. doi: 10.1084/jem.20031023
6. Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. TRAM is specifically involved in the toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat Immunol (2003) 4:1144–50. doi: 10.1038/ni986
7. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301:640–3. doi: 10.1126/science.1087262
8. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell (1998) 2:253–8. doi: 10.1016/S1097-2765(00)80136-7
9. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev (2018) 281:8–27. doi: 10.1111/imr.12621
10. Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science (1997) 278:1612–5. doi: 10.1126/science.278.5343.1612
11. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity (1997) 7:837–47. doi: 10.1016/S1074-7613(00)80402-1
12. Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking IRAK-4. Nature (2002) 416:750–4. doi: 10.1038/nature736
13. Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DCG, et al. Ligand-induced conformational changes allosterically activate toll-like receptor 9. Nat Immunol (2007) 8:772–9. doi: 10.1038/ni1479
14. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An oligomeric signaling platform formed by the toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem (2009) 284:25404–11. doi: 10.1074/jbc.M109.022392
15. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature (2010) 465:885–90. doi: 10.1038/nature09121
16. Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, et al. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell (2014) 156:705–16. doi: 10.1016/j.cell.2014.01.019
17. Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature (1996) 383:443–6. doi: 10.1038/383443a0
18. Cao Z, Henzel WJ, Gao X. IRAK: A kinase associated with the interleukin-1 receptor. Science (1996) 271:1128–31. doi: 10.1126/science.271.5252.1128
19. Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol (2014) 14:821–6. doi: 10.1038/nri3757
20. Hartupee J, Li X, Hamilton T. Interleukin 1α-induced NFκB activation and chemokine mRNA stabilization diverge at IRAK1 *. J Biol Chem (2008) 283:15689–93. doi: 10.1074/jbc.M801346200
21. Yin W, Wan Y, Kim TW, Yao P, Xiao H, Zhou H, et al. The kinase activity of interleukin-1 receptor–associated kinase 2 is essential for lipopolysaccharide-mediated cytokine and chemokine mRNA stability and translation. J Interferon Cytokine Res (2011) 31:415–22. doi: 10.1089/jir.2010.0094
22. Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol (2013) 191:3995–9. doi: 10.4049/jimmunol.1301681
23. Lin K-M, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci (2014) 111:775–80. doi: 10.1073/pnas.1320294111
24. Zhou H, Bulek K, Li X, Herjan T, Yu M, Qian W, et al. IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs. eLife (2017) 6:e29630. doi: 10.7554/eLife.29630
25. Tan Y, Kagan JC. Innate immune signaling organelles display natural and programmable signaling flexibility. Cell (2019) 177:384–398.e11. doi: 10.1016/j.cell.2019.01.039
26. Zhou H, Wang H, Yu M, Schugar RC, Qian W, Tang F, et al. IL-1 induces mitochondrial translocation of IRAK2 to suppress oxidative metabolism in adipocytes. Nat Immunol (2020) 21(10):1219–31. doi: 10.1038/s41590-020-0750-1
27. Strickson S, Emmerich CH, Goh ETH, Zhang J, Kelsall IR, MacArtney T, et al. Roles of the TRAF6 and pellino E3 ligases in MyD88 and RANKL signaling. Proc Natl Acad Sci United States America (2017) 114:E3481–9. doi: 10.1073/pnas.1702367114
28. Yu K-Y, Kwon H-J, Norman DAM, Vig E, Goebl MG, Harrington MA. Cutting edge: Mouse pellino-2 modulates IL-1 and lipopolysaccharide Signaling1. J Immunol (2002) 169:4075–8. doi: 10.4049/jimmunol.169.8.4075
29. Xiao H, Qian W, Staschke K, Qian Y, Cui G, Deng L, et al. Pellino 3b negatively regulates interleukin-1-induced TAK1-dependent NFκB activation. J Biol Chem (2008) 283:14654–64. doi: 10.1074/jbc.M706931200
30. Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)-IRAK-Tumor necrosis factor receptor-associated factor 6 (TRAF6) complex *. J Biol Chem (2003) 278:10952–6. doi: 10.1074/jbc.M212112200
31. Jensen LE, Whitehead AS. Pellino2 activates the mitogen activated protein kinase pathway. FEBS Lett (2003) 545:199–202. doi: 10.1016/S0014-5793(03)00533-7
32. Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, et al. The IRAK-catalysed activation of the E3 ligase function of pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J (2008) 409:43–52. doi: 10.1042/BJ20071365
33. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412:346–51. doi: 10.1038/35085597
34. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem (2005) 280:17005–12. doi: 10.1074/jbc.M412584200
35. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature (2005) 434:243–9. doi: 10.1038/nature03308
36. Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in toll-like receptor signaling. Proc Natl Acad Sci (2004) 101:15416–21. doi: 10.1073/pnas.0406933101
37. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi: 10.1038/nature03464
38. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction. Nature (2005) 434:1035–40. doi: 10.1038/nature03547
39. Ren J, Chen X, Chen ZJ. IKKβ is an IRF5 kinase that instigates inflammation. Proc Natl Acad Sci USA (2014) 111:17438–43. doi: 10.1073/pnas.1418516111
40. Lopez-Pelaez M, Lamont DJ, Peggie M, Shpiro N, Gray NS, Cohen P. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc Natl Acad Sci (2014) 111:17432–7. doi: 10.1073/pnas.1418399111
41. Martin M, Böl GF, Eriksson A, Resch K, Brigelius-Flohé R. Interleukin-1-induced activation of a protein kinase co-precipitating with the type I interleukin-1 receptor in T cells. Eur J Immunol (1994) 24:1566–71. doi: 10.1002/eji.1830240717
42. Croston GE, Cao Z, Goeddel DV. NF-κB activation by interleukin-1 (IL-1) requires an IL-1 receptor-associated protein kinase activity *. J Biol Chem (1995) 270:16514–7. doi: 10.1074/jbc.270.28.16514
43. Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, et al. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol (1999) 163:978–84. doi: 10.4049/jimmunol.163.2.978
44. Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. PNAS (2002) 99:5567–72. doi: 10.1073/pnas.082100399
45. Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z. IRAK-m is a novel member of the Pelle/Interleukin-1 receptor-associated kinase (IRAK) family*. J Biol Chem (1999) 274:19403–10. doi: 10.1074/jbc.274.27.19403
46. Béla SR, Dutra MS, Mui E, Montpetit A, Oliveira FS, Oliveira SC, et al. Impaired innate immunity in mice deficient in interleukin-1 receptor-associated kinase 4 leads to defective type 1 t cell responses, b cell expansion, and enhanced susceptibility to infection with toxoplasma gondii. Infection Immun (2012) 80:4298–308. doi: 10.1128/IAI.00328-12
47. Pattabiraman G, Murphy M, Agliano F, Karlinsey K, Medvedev AE. IRAK4 activity controls immune responses to intracellular bacteria listeria monocytogenes and mycobacterium smegmatis. J Leukocyte Biol (2018) 104:811–20. doi: 10.1002/JLB.2A1117-449R
48. von Bernuth H, Picard C, Puel A, Casanova J-L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol (2012) 42:3126–35. doi: 10.1002/eji.201242683
49. Picard C, Puel A, Bonnet M, Ku C-L, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science (2003) 299:2076–9. doi: 10.1126/science.1081902
50. Medvedev AE, Lentschat A, Kuhns DB, Blanco JCG, Salkowski C, Zhang S, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med (2003) 198:521–31. doi: 10.1084/jem.20030701
51. Chapel H, Puel A, von Bernuth H, Picard C, Casanova J-L. Shigella sonnei meningitis due to interleukin-1 receptor–associated kinase–4 deficiency: First association with a primary immune deficiency. Clin Infect Dis (2005) 40:1227–31. doi: 10.1086/428733
52. Balaci L, Spada MC, Olla N, Sole G, Loddo L, Anedda F, et al. IRAK-m is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet (2007) 80:1103–14. doi: 10.1086/518259
53. Yang D, Chen W, Xiong J, Sherrod CJ, Henry DH, Dittmer DP. Interleukin 1 receptor-associated kinase 1 (IRAK1) mutation is a common, essential driver for kaposi sarcoma herpesvirus lymphoma. Proc Natl Acad Sci (2014) 111:E4762–8. doi: 10.1073/pnas.1405423111
54. Della Mina E, Borghesi A, Zhou H, Bougarn S, Boughorbel S, Israel L, et al. Inherited human IRAK-1 deficiency selectively impairs TLR signaling in fibroblasts. Proc Natl Acad Sci (2017) 114:E514–23. doi: 10.1073/pnas.1620139114
55. Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, et al. The poxvirus protein A52R targets toll-like receptor signaling complexes to suppress host defense. J Exp Med (2003) 197:343–51. doi: 10.1084/jem.20021652
56. Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, et al. Hepatitis c virus nonstructural protein 5A modulates the toll-like receptor-MyD88-Dependent signaling pathway in macrophage cell lines. J Virol (2007) 81:8953–66. doi: 10.1128/JVI.00649-07
57. Abu-Dayyeh I, Shio MT, Sato S, Akira S, Cousineau B, Olivier M. Leishmania-induced IRAK-1 inactivation is mediated by SHP-1 interacting with an evolutionarily conserved KTIM motif. PloS Negl Trop Dis (2008) 2:e305. doi: 10.1371/journal.pntd.0000305
58. Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. Kaposi’s sarcoma-associated herpesvirus MicroRNAs target IRAK1 and MYD88, two components of the toll-like Receptor/Interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol (2012) 86:11663–74. doi: 10.1128/JVI.01147-12
59. Jeyanathan M, McCormick S, Lai R, Afkhami S, Shaler CR, Horvath CN, et al. Pulmonary m. tuberculosis infection delays Th1 immunity via immunoadaptor DAP12-regulated IRAK-m and IL-10 expression in antigen-presenting cells. Mucosal Immunol (2014) 7:670–83. doi: 10.1038/mi.2013.86
60. Pantazi I, Al-Qahtani AA, Alhamlan FS, Alothaid H, Matou-Nasri S, Sourvinos G, et al. SARS-CoV-2/ACE2 interaction suppresses IRAK-m expression and promotes pro-inflammatory cytokine production in macrophages. Front Immunol (2021) 12:683800. doi: 10.3389/fimmu.2021.683800
61. Gosu V, Basith S, Durai P, Choi S. Molecular evolution and structural features of IRAK family members. PloS One (2012) 7:e49771. doi: 10.1371/journal.pone.0049771
62. Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem Pharmacol (2010) 80:1981–91. doi: 10.1016/j.bcp.2010.06.020
63. Martin MU, Kollewe C. Interleukin-1 receptor-associated kinase-1 (IRAK-1): A self-regulatory adapter molecule in the signaling cascade of the Toll/IL-1 receptor family. Signal Transduction (2001) 1:37–50. doi: 10.1002/1615-4061(200111)1:1/2<37::AID-SITA37>3.0.CO;2-K
64. Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem (2004) 279:5227–36. doi: 10.1074/jbc.M309251200
65. García-Alai MM, Gallo M, Salame M, Wetzler DE, McBride AA, Paci M, et al. Prat-gay G de. molecular basis for phosphorylation-dependent, PEST-mediated protein turnover. Structure (2006) 14:309–19. doi: 10.1016/j.str.2005.11.012
66. Kobayashi K, Hernandez LD, Galán JE, Janeway CA, Medzhitov R, Flavell RA. IRAK-m is a negative regulator of toll-like receptor signaling. Cell (2002) 110:191–202. doi: 10.1016/S0092-8674(02)00827-9
67. Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, et al. Sequential control of toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol (2008) 9:684–91. doi: 10.1038/ni.1606
68. Freihat L, Muleya V, Manallack DT, Wheeler JI, Irving HR. Comparison of moonlighting guanylate cyclases: roles in signal direction? Biochem Soc Trans (2014) 42:1773–9. doi: 10.1042/BST20140223
69. Freihat LA, Wheeler JI, Wong A, Turek I, Manallack DT, Irving HR. IRAK3 modulates downstream innate immune signalling through its guanylate cyclase activity. Sci Rep (2019) 9:15468. doi: 10.1038/s41598-019-51913-3
70. Watanabe S, Zenke K, Sugiura Y, Muroi M. Minimal structure of IRAK-1 to induce degradation of TRAF6. Immunobiology (2022) 227:152256. doi: 10.1016/j.imbio.2022.152256
71. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of toll-like receptor signalling complexes. Nat Rev Immunol (2014) 14:546–58. doi: 10.1038/nri3713
72. Miguel RN, Wong J, Westoll JF, Brooks HJ, O’Neill LAJ, Gay NJ, et al. A dimer of the toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PloS One (2007) 2:e788. doi: 10.1371/journal.pone.0000788
73. Ferrao R, Zhou H, Shan Y, Liu Q, Li Q, Shaw DE, et al. IRAK4 dimerization and trans-autophosphorylation are induced by myddosome assembly. Mol Cell (2014) 55:891–903. doi: 10.1016/j.molcel.2014.08.006
74. Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, et al. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol (2000) 2:346–51. doi: 10.1038/35014038
75. Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by tollip*. J Biol Chem (2002) 277:7059–65. doi: 10.1074/jbc.M109537200
76. Lee HJ, Chung KC. PINK1 positively regulates IL-1β-mediated signaling through tollip and IRAK1 modulation. J Neuroinflamm (2012) 9:271. doi: 10.1186/1742-2094-9-271
77. Pereira M, Durso DF, Bryant CE, Kurt-Jones EA, Silverman N, Golenbock DT, et al. The IRAK4 scaffold integrates TLR4-driven TRIF and MYD88 signaling pathways. Cell Rep (2022) 40:111225. doi: 10.1016/j.celrep.2022.111225
78. Kanev GK, de Graaf C, de Esch IJP, Leurs R, Würdinger T, Westerman BA, et al. The landscape of atypical and eukaryotic protein kinases. Trends Pharmacol Sci (2019) 40:818–32. doi: 10.1016/j.tips.2019.09.002
79. Weintz G, Olsen JV, Frühauf K, Niedzielska M, Amit I, Jantsch J, et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol Syst Biol (2010) 6:371. doi: 10.1038/msb.2010.29
80. Deliz-Aguirre R, Cao F, Gerpott FHU, Auevechanichkul N, Chupanova M, Mun Y, et al. MyD88 oligomer size functions as a physical threshold to trigger IL1R myddosome signaling. J Cell Biol (2021) 220:e202012071. doi: 10.1083/jcb.202012071
81. De Nardo D, Balka KR, Gloria YC, Rao VR, Latz E, Masters SL. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and toll-like receptor signaling. J Biol Chem (2018) 293:15195–207. doi: 10.1074/jbc.RA118.003314
82. De S, Karim F, Kiessu E, Cushing L, Lin L-L, Ghandil P, et al. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J Biol Chem (2018) 293:15208–20. doi: 10.1074/jbc.RA118.003831
83. Kubo-Murai M, Hazeki K, Nigorikawa K, Omoto T, Inoue N, Hazeki O. IRAK-4-dependent degradation of IRAK-1 is a negative feedback signal for TLR-mediated NF-κB activation. J Biochem (2008) 143:295–302. doi: 10.1093/jb/mvm234
84. Keating SE, Maloney GM, Moran EM, Bowie AG. IRAK-2 participates in multiple toll-like receptor signaling pathways to NFκB via activation of TRAF6 ubiquitination. J Biol Chem (2007) 282:33435–43. doi: 10.1074/jbc.M705266200
85. Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature (2002) 418:443–7. doi: 10.1038/nature00888
86. Qian Y, Commane M, Ninomiya-Tsuji J, Matsumoto K, Li X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFκB *. J Biol Chem (2001) 276:41661–7. doi: 10.1074/jbc.M102262200
87. Pauls E, Nanda SK, Smith H, Toth R, Arthur JSC, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. JI (2013) 191:2717–30. doi: 10.4049/jimmunol.1203268
88. Joshi H, Lunz B, Peters A, Michael Z, Berberich I, Berberich-siebelt F. The extreme c-terminus of IRAK2 assures full TRAF6 ubiquitination and optimal TLR signaling. Mol Immunol (2021) 134:172–82. doi: 10.1016/j.molimm.2021.03.022
89. Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J Biol Chem (2007) 282:4102–12. doi: 10.1074/jbc.M609503200
90. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell (2000) 103:351–61. doi: 10.1016/S0092-8674(00)00126-4
91. Conze DB, Wu C-J, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-κB activation. Mol Cell Biol (2008) 28:3538–47. doi: 10.1128/mcb.02098-07
92. Bin LH, Xu LG, Shu HB. TIRP, a novel Toll/Interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J Biol Chem (2003) 278:24526–32. doi: 10.1074/jbc.M303451200
93. Sakai J, Cammarota E, Wright JA, Cicuta P, Gottschalk RA, Li N, et al. Lipopolysaccharide-induced NF-κB nuclear translocation is primarily dependent on MyD88, but TNFα expression requires TRIF and MyD88. Sci Rep (2017) 7:1428. doi: 10.1038/s41598-017-01600-y
94. Latty S, Sakai J, Hopkins L, Verstak B, Paramo T, Berglund NA, et al. Activation of toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife (2018) 7:1–15. doi: 10.7554/eLife.31377
95. Tan Y, Kagan JC. Biochemical isolation of the myddosome from murine macrophages. Methods Mol Biol (2018) 1714:79–95. doi: 10.1007/978-1-4939-7519-8
96. Moncrieffe MC, Bollschweiler D, Li B, Bryant CE, Klenerman D, Gay NJ, et al. MyD88 death-domain oligomerization determines myddosome Architecture : Implications for toll-like receptor signaling. Structure/Folding Design (2020) 28:1–9. doi: 10.1016/j.str.2020.01.003
97. Manček-Keber M, Lainšček D, Benčina M, Chen JG, Romih R, Hunter ZR, et al. Extracellular vesicle–mediated transfer of constitutively active MyD88L265P engages MyD88wt and activates signaling. Blood (2018) 131:1720–9. doi: 10.1182/blood-2017-09-805499
98. Tan Y, Zanoni I, Cullen TW, Goodman AL, Kagan JC. Mechanisms of toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity (2015) 43:909–22. doi: 10.1016/j.immuni.2015.10.008
99. Balka KR, De Nardo D. Understanding early TLR signaling through the myddosome. J Leukocyte Biol (2019) 105:339–51. doi: 10.1002/JLB.MR0318-096R
100. Cheng H, Addona T, Keshishian H, Dahlstrand E, Lu C, Dorsch M, et al. Regulation of IRAK-4 kinase activity via autophosphorylation within its activation loop. Biochem Biophys Res Commun (2007) 352:609–16. doi: 10.1016/j.bbrc.2006.11.068
101. Qin J, Jiang Z, Qian Y, Casanova J-L, Li X. IRAK4 kinase activity is redundant for interleukin-1 (IL-1) receptor-associated kinase phosphorylation and IL-1 responsiveness*. J Biol Chem (2004) 279:26748–53. doi: 10.1074/jbc.M400785200
102. Vollmer S, Strickson S, Zhang T, Gray N, Lee KL, Rao VR, et al. The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and toll-like receptor agonists. Biochem J (2017) 474:2027–38. doi: 10.1042/BCJ20170097
103. Lye E, Mirtsos C, Suzuki N, Suzuki S, Yeh WC. The role of interleukin 1 receptor-associated kinase-4 (IRAK-4) kinase activity in IRAK-4-mediated signaling. J Biol Chem (2004) 279:40653–8. doi: 10.1074/jbc.M402666200
104. Poulain S, Roumier C, Decambron A, Renneville A, Herbaux C, Bertrand E, et al. MYD88 L265P mutation in waldenstrom macroglobulinemia. Blood (2013) 121:4504–11. doi: 10.1182/blood-2012-06-436329
105. Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, et al. MYD88 L265P mutation in lymphoid malignancies. Cancer Res (2018) 78:2457–62. doi: 10.1158/0008-5472.CAN-18-0215
106. Dunne A, Carpenter S, Brikos C, Gray P, Strelow A, Wesche H, et al. IRAK1 and IRAK4 promote phosphorylation, ubiquitination, and degradation of MyD88 adaptor-like (Mal). J Biol Chem (2010) 285:18276–82. doi: 10.1074/jbc.M109.098137
107. Muroi M, Tanamoto K. IRAK-1-mediated negative regulation of toll-like receptor signaling through proteasome-dependent downregulation of TRAF6. Biochim Biophys Acta - Mol Cell Res (2012) 1823:255–63. doi: 10.1016/j.bbamcr.2011.10.003
108. Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, et al. The E3 ubiquitin ligase Nrdp1 “preferentially” promotes TLR-mediated production of type I interferon. Nat Immunol (2009) 10:744–52. doi: 10.1038/ni.1742
109. Into T, Inomata M, Nakashima M, Shibata K, Häcker H, Matsushita K. Regulation of MyD88-dependent signaling events by s nitrosylation retards toll-like receptor signal transduction and initiation of acute-phase immune responses. Mol Cell Biol (2008) 28:1338–47. doi: 10.1128/MCB.01412-07
110. Meng Z, Xu R, Xie L, Wu Y, He Q, Gao P, et al. A20/Nrdp1 interaction alters the inflammatory signaling profile by mediating K48- and K63-linked polyubiquitination of effectors MyD88 and TBK1. J Biol Chem (2021) 297(1):100811. doi: 10.1016/j.jbc.2021.100811
111. Coornaert B, Carpentier I, Beyaert R. A20: Central gatekeeper in inflammation and immunity. J Biol Chem (2009) 284:8217–21. doi: 10.1074/jbc.R800032200
112. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating syk and promoting degradation of MyD88 and TRIF via cbl-b. Nat Immunol (2010) 11:734–42. doi: 10.1038/ni.1908
113. Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim S-J, et al. Smad6-specific recruitment of smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat Commun (2011) 2:460. doi: 10.1038/ncomms1469
114. Guillamot M, Ouazia D, Dolgalev I, Yeung ST, Kourtis N, Dai Y, et al. The E3 ubiquitin ligase SPOP controls resolution of systemic inflammation by triggering MYD88 degradation. Nat Immunol (2019) 20:1196–207. doi: 10.1038/s41590-019-0454-6
115. Li Q, Wang F, Wang Q, Zhang N, Zheng J, Zheng M, et al. SPOP promotes ubiquitination and degradation of MyD88 to suppress the innate immune response. PloS Pathog (2020) 16:e1008188. doi: 10.1371/journal.ppat.1008188
116. Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity (2002) 17:677–87. doi: 10.1016/S1074-7613(02)00449-1
117. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of cytokine signaling 1 negatively regulates toll-like receptor signaling by mediating mal degradation. Nat Immunol (2006) 7:148–55. doi: 10.1038/ni1299
118. Yong Y-H, Wang P, Jia R-M, Gooneratne R, Robert Wang H-C, Liao M, et al. SOCS3 control the activity of NF-κB induced by HSP70 via degradation of MyD88-adapter-like protein (Mal) in IPEC-J2 cells. Int J Hyperthermia (2019) 36:150–8. doi: 10.1080/02656736.2018.1541484
119. González-León MC, Soares-Schanoski A, del Fresno C, Cimadevila A, Goméz-Piña V, Mendoza-Barberá E, et al. Nitric oxide induces SOCS-1 expression in human monocytes in a TNF-α-dependent manner. J Endotoxin Res (2006) 12:296–306. doi: 10.1177/09680519060120050501
120. Yamin T-T, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation*. J Biol Chem (1997) 272:21540–7. doi: 10.1074/jbc.272.34.21540
121. Hu J, Jacinto R, McCall C, Li L. Regulation of IL-1 receptor-associated kinases by lipopolysaccharide. J Immunol (2002) 168:3910–4. doi: 10.4049/jimmunol.168.8.3910
122. Kong F, Liu Z, Jain VG, Shima K, Suzuki T, Muglia LJ, et al. Inhibition of IRAK1 ubiquitination determines glucocorticoid sensitivity for TLR9-induced inflammation in macrophages. J Immunol (2017) 199:3654–67. doi: 10.4049/jimmunol.1700443
123. Windheim M, Stafford M, Peggie M, Cohen P. Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IκBα kinase. Mol Cell Biol (2008) 28:1783–91. doi: 10.1128/MCB.02380-06
124. Brissoni B, Agostini L, Kropf M, Martinon F, Swoboda V, Lippens S, et al. Intracellular trafficking of interleukin-1 receptor I requires tollip. Curr Biol (2006) 16:2265–70. doi: 10.1016/j.cub.2006.09.062
125. Hubbard LLN, Moore BB. IRAK-m regulation and function in host defense and immune homeostasis. Infect Dis Rep (2010) 2:e9. doi: 10.4081/idr.2010.e9
126. Su J, Xie Q, Wilson I, Li L. Differential regulation and role of interleukin-1 receptor associated kinase-m in innate immunity signaling. Cell Signalling (2007) 19:1596–601. doi: 10.1016/j.cellsig.2007.02.009
127. Zhou H, Yu M, Fukuda K, Im J, Yao P, Cui W, et al. IRAK-m mediates toll-like receptor/IL-1R-induced NFκB activation and cytokine production. EMBO J (2013) 32:583–96. doi: 10.1038/emboj.2013.2
128. Nechama M, Kwon J, Wei S, Kyi AT, Welner RS, Ben-Dov IZ, et al. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat Commun (2018) 9:1603. doi: 10.1038/s41467-018-03886-6
129. Hassan F, Islam S, Tumurkhuu G, Dagvadorj J, Naiki Y, Komatsu T, et al. Involvement of interleukin-1 receptor-associated kinase (IRAK)-m in toll-like receptor (TLR) 7-mediated tolerance in RAW 264.7 macrophage-like cells. Cell Immunol (2009) 256:99–103. doi: 10.1016/j.cellimm.2009.01.013
130. Lyroni K, Patsalos A, Daskalaki MG, Doxaki C, Soennichsen B, Helms M, et al. Epigenetic and transcriptional regulation of IRAK-m expression in macrophages. J Immunol (2017) 198:1297–307. doi: 10.4049/jimmunol.1600009
131. Miyata M, Lee J-Y, Susuki-Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-m. Nat Commun (2015) 6:6062. doi: 10.1038/ncomms7062
132. Sakharwade SC, Mukhopadhaya A. Vibrio cholerae porin OmpU induces LPS tolerance by attenuating TLR-mediated signaling. Mol Immunol (2015) 68:312–24. doi: 10.1016/j.molimm.2015.09.021
133. Tae WK, Staschke K, Bulek K, Yao J, Peters K, Oh KH, et al. A critical role for IRAK4 kinase activity in toll-like receptor-mediated innate immunity. J Exp Med (2007) 204:1025–36. doi: 10.1084/jem.20061825
134. Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H, et al. Essential role of IRAK-4 protein and its kinase activity in toll-like receptor-mediated immune responses but not in TCR signaling. J Exp Med (2007) 204:1013–24. doi: 10.1084/jem.20061523
135. Pennini ME, Perkins DJ, Salazar AM, Lipsky M, Vogel SN. Complete dependence on IRAK4 kinase activity in TLR2, but not TLR4, signaling pathways underlies decreased cytokine production and increased susceptibility to streptococcus pneumoniae infection in IRAK4 kinase–inactive mice. J Immunol (2013) 190:307–16. doi: 10.4049/jimmunol.1201644
136. Sun J, Li N, Oh K-S, Dutta B, Vayttaden SJ, Lin B, et al. Comprehensive RNAi-based screening of human and mouse TLR pathways identifies species-specific preferences in signaling protein use. Sci Signaling (2016) 9:ra3–3. doi: 10.1126/scisignal.aab2191
137. Mullard A. IRAK4 degrader to take on innate immunity. Nat Biotechnol (2020) 38:1221–3. doi: 10.1038/s41587-020-0724-8
138. Bernuth Hv, Picard C, Jin Z, Pankla R, Xiao H, Ku C-L, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321:691–6. doi: 10.1126/science.1158298
139. Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R, et al. IRAK4 kinase activity controls toll-like receptor–induced inflammation through the transcription factor IRF5 in primary human monocytes. J Biol Chem (2017) 292:18689–98. doi: 10.1074/jbc.M117.796912
140. Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the toll-like receptors. J Immunol (2003) 171:4304–10. doi: 10.4049/jimmunol.171.8.4304
141. Verstak B, Stack J, Ve T, Mangan M, Hjerrild K, Jeon J, et al. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J Leukocyte Biol (2014) 96:427–36. doi: 10.1189/jlb.2a0913-487r
142. Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J Biol Chem (2014) 289:10865–75. doi: 10.1074/jbc.M113.544809
143. Swantek JL, Tsen MF, Cobb MH, Thomas JA. IL-1 receptor-associated kinase modulates host responsiveness to Endotoxin1. J Immunol (2000) 164:4301–6. doi: 10.4049/jimmunol.164.8.4301
144. Wan Y, Xiao H, Affolter J, Kim TW, Bulek K, Chaudhuri S, et al. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control*. J Biol Chem (2009) 284:10367–75. doi: 10.1074/jbc.M807822200
145. Jensen LE, Whitehead AS. IRAK1b, a novel alternative splice variant of interleukin-1 receptor-associated kinase (IRAK), mediates interleukin-1 signaling and has prolonged stability*. J Biol Chem (2001) 276:29037–44. doi: 10.1074/jbc.M103815200
146. Hemmi H, Kaisho T, Takeda K, Akira S. The roles of toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell Subsets1. J Immunol (2003) 170:3059–64. doi: 10.4049/jimmunol.170.6.3059
147. Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J Exp Med (2005) 201:915–23. doi: 10.1084/jem.20042372
148. Marié I, Smith E, Prakash A, Levy DE. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol (2000) 20:8803–14. doi: 10.1128/MCB.20.23.8803-8814.2000
149. Sharma S, tenOever BR, Grandvaux N, Zhou G-P, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (2003) 300:1148–51. doi: 10.1126/science.1081315
150. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-α induction through toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol (2004) 5:1061–8. doi: 10.1038/ni1118
151. Wan Y, Kim TW, Yu M, Zhou H, Yamashita M, Kang Z, et al. The dual functions of IL-1 receptor-associated kinase 2 in TLR9-mediated IFN and proinflammatory cytokine production. J Immunol (2011) 186:3006–14. doi: 10.4049/jimmunol.1003217
152. Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M, et al. Interferon-regulatory-factor 1 controls toll-like receptor 9-mediated IFN-β production in myeloid dendritic cells. Eur J Immunol (2007) 37:315–27. doi: 10.1002/eji.200636767
153. Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-γ-induced IFN regulatory factor 1 transcription factor by MyD88 in toll-like receptor-dependent gene induction program. Proc Natl Acad Sci (2006) 103:15136–41. doi: 10.1073/pnas.0607181103
154. Balkhi MY, Fitzgerald KA, Pitha PM. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol Cell Biol (2008) 28:7296–308. doi: 10.1128/MCB.00662-08
155. Moen SH, Ehrnström B, Kojen JF, Yurchenko M, Beckwith KS, Afset JE, et al. Human toll-like receptor 8 (TLR8) is an important sensor of pyogenic bacteria, and is attenuated by cell surface TLR signaling. Front Immunol (2019) 10:1209. doi: 10.3389/fimmu.2019.01209
156. Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-β gene regulatory elements. Cell (1988) 54:903–13. doi: 10.1016/S0092-8674(88)91307-4
157. Saliba DG, Heger A, Eames HL, Oikonomopoulos S, Teixeira A, Blazek K, et al. IRF5:RelA interaction targets inflammatory genes in macrophages. Cell Rep (2014) 8:1308–17. doi: 10.1016/j.celrep.2014.07.034
158. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
159. Niu T, De Rosny C, Chautard S, Rey A, Patoli D, Groslambert M, et al. NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun (2021) 12:5862. doi: 10.1038/s41467-021-26142-w
160. Humphries F, Bergin R, Jackson R, Delagic N, Wang B, Yang S, et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat Commun (2018) 9:1560. doi: 10.1038/s41467-018-03669-z
161. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
162. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
163. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
164. Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity (2015) 42:484–98. doi: 10.1016/j.immuni.2015.02.001
165. Spinello I, Quaranta MT, Paolillo R, Pelosi E, Cerio AM, Saulle E, et al. Differential hypoxic regulation of the microRNA-146a/CXCR4 pathway in normal and leukemic monocytic cells: impact on response to chemotherapy. Haematologica (2015) 100:1160–71. doi: 10.3324/haematol.2014.120295
166. Park H, Huang X, Lu C, Cairo MS, Zhou X. MicroRNA-146a and MicroRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins *. J Biol Chem (2015) 290:2831–41. doi: 10.1074/jbc.M114.591420
167. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15:323–32. doi: 10.1038/ni.2833
168. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol (2003) 4:491–6. doi: 10.1038/ni921
169. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (2010) 89:403. doi: 10.1097/MD.0b013e3181fd8ec3
170. Ku CL, Von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med (2007) 204:2407–22. doi: 10.1084/jem.20070628
171. Liu D, Chen R, Ran L, Zhang R, Chen H. Listeria monocytogenes meningoencephalitis due to IRAK4 deficiency. J Clin Immunol (2021) 41:1677–80. doi: 10.1007/s10875-021-01074-8
172. Gokturk B, Casanova J, Picard C, Cagdas Ayvaz D, Erman B, Tezcan I, et al. A novel homozygous mutation with different clinical presentations in 2 IRAK-4–deficient siblings: First case with recurrent salmonellosis and non-Hodgkin lymphoma. J Investig Allergol Clin Immunol (2018) 28:271–3. doi: 10.18176/jiaci.0261
173. Tepe ZG, Yazıcı YY, Tank U, Köse LI, Özer M, Aytekin C, et al. Inherited IRAK-4 deficiency in acute human herpesvirus-6 encephalitis. J Clin Immunol (2023) 43:192–205. doi: 10.1007/s10875-022-01369-4
174. Liu G, Tsuruta Y, Gao Z, Park Y-J, Abraham E. Variant IL-1 receptor-associated kinase-1 mediates increased NF-κB Activity1. J Immunol (2007) 179:4125–34. doi: 10.4049/jimmunol.179.6.4125
175. Cheng BY, Lau EY, Leung H-W, Leung CO-N, Ho NP, Gurung S, et al. IRAK1 augments cancer stemness and drug resistance via the AP-1/AKR1B10 signaling cascade in hepatocellular carcinoma. Cancer Res (2018) 78:2332–42. doi: 10.1158/0008-5472.CAN-17-2445
176. Zhang X, Dang Y, Li P, Rong M, Chen G. Expression of IRAK1 in lung cancer tissues and its clinicopathological significance: A microarray study. Int J Clin Exp Pathol (2014) 7:8096–104.
177. Wang Y, Wang Y, Duan X, Wang Y, Zhang Z. Interleukin-1 receptor-associated kinase 1 correlates with metastasis and invasion in endometrial carcinoma. J Cell Biochem (2018) 119:2545–55. doi: 10.1002/jcb.26416
178. Li J, Sun Y, Ma Y, Zhao X, Sun X, Wang Y, et al. Comprehensive pan-cancer analysis of IRAK family genes identifies IRAK1 as a novel oncogene in low-grade glioma. J Oncol (2022) 2022:e6497241. doi: 10.1155/2022/6497241
179. Zhang D, Li L, Jiang H, Knolhoff BL, Lockhart AC, Wang-Gillam A, et al. Constitutive IRAK4 activation underlies poor prognosis and chemoresistance in pancreatic ductal adenocarcinoma. Clin Cancer Res (2017) 23:1748–59. doi: 10.1158/1078-0432.CCR-16-1121
180. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, et al. Tumor–stroma IL1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res (2018) 78:1700–12. doi: 10.1158/0008-5472.CAN-17-1366
Keywords: TLR, IRAK, innate immunity, cell signaling, IL-1R, inflammation
Citation: Pereira M and Gazzinelli RT (2023) Regulation of innate immune signaling by IRAK proteins. Front. Immunol. 14:1133354. doi: 10.3389/fimmu.2023.1133354
Received: 28 December 2022; Accepted: 30 January 2023;
Published: 14 February 2023.
Edited by:
Jerrold Weiss, The University of Iowa, United StatesCopyright © 2023 Pereira and Gazzinelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milton Pereira, bWlsdG9uLnBlcmVpcmFAdW1hc3NtZWQuZWR1; Ricardo T. Gazzinelli, cmljYXJkby5nYXp6aW5lbGxpQHVtYXNzbWVkLmVkdQ==