 Chen Chen
Chen Chen Zehua Wang
Zehua Wang Yi Ding
Yi Ding Yanru Qin
Yanru Qin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1133308
This article is part of the Research TopicUnderstanding Convergent Evasion Mechanisms in Cancer and Chronic Infection: Implications for ImmunotherapyView all 14 articles
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy and is the third leading cause of tumor-related mortality worldwide. In recent years, the emergency of immune checkpoint inhibitor (ICI) has revolutionized the management of HCC. Especially, the combination of atezolizumab (anti-PD1) and bevacizumab (anti-VEGF) has been approved by the FDA as the first-line treatment for advanced HCC. Despite great breakthrough in systemic therapy, HCC continues to portend a poor prognosis owing to drug resistance and frequent recurrence. The tumor microenvironment (TME) of HCC is a complex and structured mixture characterized by abnormal angiogenesis, chronic inflammation, and dysregulated extracellular matrix (ECM) remodeling, collectively contributing to the immunosuppressive milieu that in turn prompts HCC proliferation, invasion, and metastasis. The tumor microenvironment coexists and interacts with various immune cells to maintain the development of HCC. It is widely accepted that a dysfunctional tumor-immune ecosystem can lead to the failure of immune surveillance. The immunosuppressive TME is an external cause for immune evasion in HCC consisting of 1) immunosuppressive cells; 2) co-inhibitory signals; 3) soluble cytokines and signaling cascades; 4) metabolically hostile tumor microenvironment; 5) the gut microbiota that affects the immune microenvironment. Importantly, the effectiveness of immunotherapy largely depends on the tumor immune microenvironment (TIME). Also, the gut microbiota and metabolism profoundly affect the immune microenvironment. Understanding how TME affects HCC development and progression will contribute to better preventing HCC-specific immune evasion and overcoming resistance to already developed therapies. In this review, we mainly introduce immune evasion of HCC underlying the role of immune microenvironment, describe the dynamic interaction of immune microenvironment with dysfunctional metabolism and the gut microbiome, and propose therapeutic strategies to manipulate the TME in favor of more effective immunotherapy.
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy and is the third leading cause of cancer-related mortality worldwide in 2020 (1). HCC frequently develops on a background of cirrhosis caused by multiple risk factors, including chronic viral infection of hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol abuse, aflatoxin exposure, non-alcoholic steatohepatitis (NASH), and drug-related liver injury (2). Treatment recommendations differ in various stages of HCC. The choice between locoregional treatments mainly depends on the tumor burden, location, and liver function (3). Based on clinical practice guideline, surgical resection, radiofrequency ablation (RFA), transarterial chemobolization (TACE), and liver transplantation are effective for tumor confined to the liver, whereas systemic therapy targeting the TME is available for unresectable HCC (3, 4). Since the first tyrosine kinase inhibitor (TKI) sorafenib was proven to extend the survival in advanced HCC patients without compromising liver function in 2008 (5), multi-TKIs and vascular endothelial growth factor (VEGF) inhibitors have been integrated into standard systemic therapy for advanced HCC (6–9).
Cancer immunotherapies have greatly revolutionized the clinical management of HCC in recent years, particularly the application of immune checkpoint inhibitor (ICI). It has been proven that the combination of atezolizumab (anti-PD1) and bevacizumab (anti-VEGF) was superior to the first-line treatment sorafenib (10). However, HCC continues one of the worst prognoses due to drug resistance and frequent recurrence. A large percentage of HCC patients still do not benefit from these immunotherapies or undergo immune-related adverse events. A potential explanation is these immune-based approaches primarily aim to reactivate dysfunctional T cell but ignore the immunosuppressive contribution of the tumor microenvironment (TME).
The tumor microenvironment is a complex ecosystem that plays an indispensable role from cancer initiation to distant metastasis (11). It coexists and interacts with various immune cells and their products, referred to the tumor immune environment (TIME). Dysfunctional tumor-immunity cycle can lead to immune evasion by flawed antigen recognition or by immunosuppressive TME (12). Tumor intrinsic mechanism of immune evasion might be attributed to defects of antigen presentation, loss of MHC-I molecules, and epigenetic repression of tumor-associated antigens (TAA) (13). The immunosuppressive TME is an external driver of immune escape due to 1) the presence of immunosuppressive cells; 2) co-inhibitory signals on lymphocytes; 3) the existence of immunosuppressive soluble factors and signaling cascades; 4) metabolically hostile tumor microenvironment, imposing barriers to tumor-infiltrating immune cells; 5) the intra-tumoral microbes that alter the state of the immune microenvironment to prompt HCC progression (14–19). Figure 1 depicts mechanisms of immune evasion mediated by tumor microenvironment in HCC.
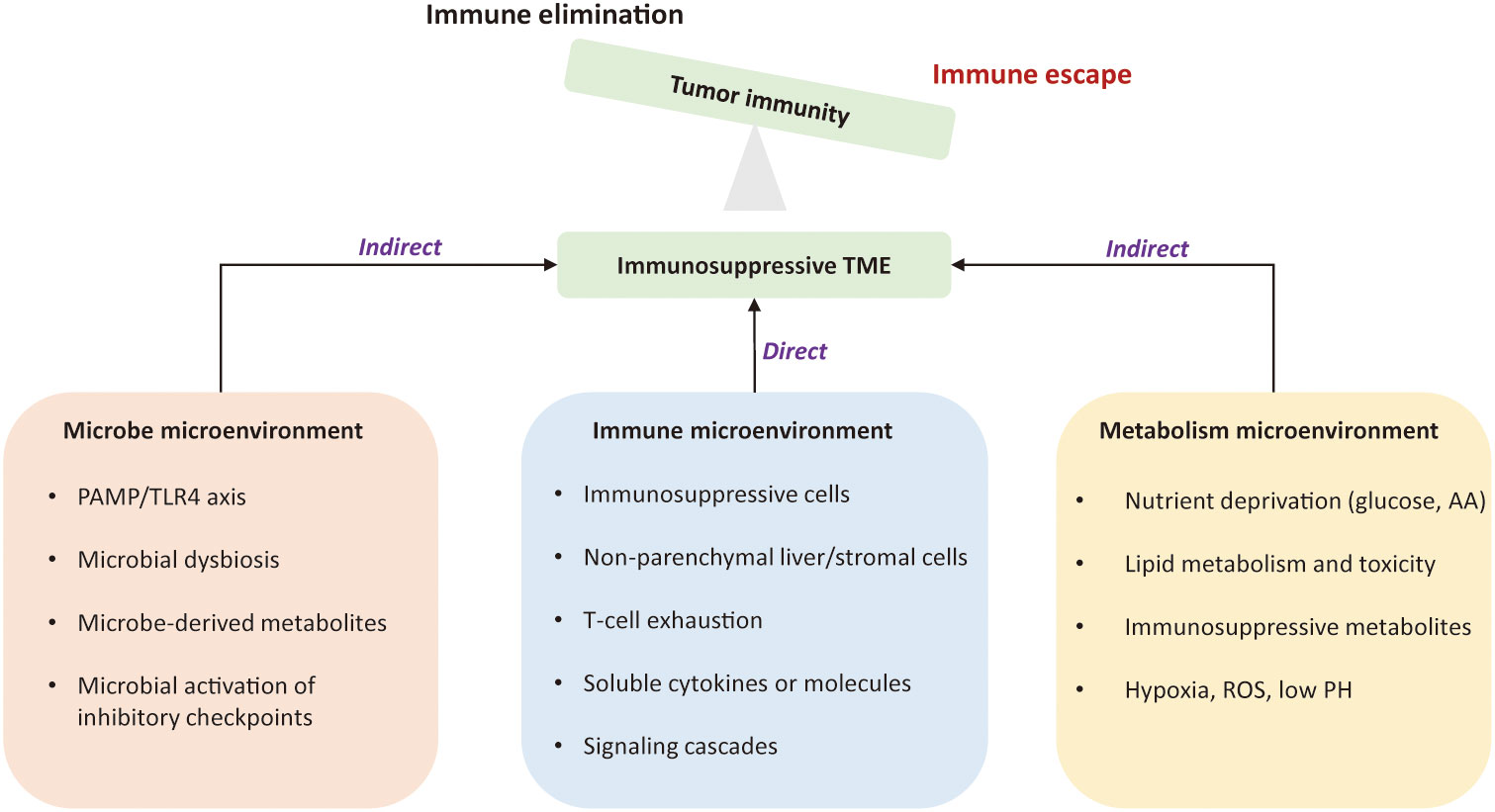
Figure 1 Mechanisms of immune evasion led by the tumor microenvironment in hepatocellular carcinoma. The immunosuppressive tumor microenvironment is an external driver of immune evasion in HCC. The suppressive immune microenvironment is led by intricate interactions among suppressive immune cells, stromal cells, immunoregulatory cytokines, and signaling cascades. Metabolic constraints and gut microbiota also contribute to the immunosuppression. The permissive microenvironment favors tumor cells to proliferate in un uncontrolled manner and is no longer confined by the host immunity. TME, tumor microenvironment; PAMP, pathogen-associated molecular patterns; TLR4, Toll-like receptor 4; AA, amino acid; ROS, reactive oxygen species.
The tumor immune microenvironment can determine whether immunotherapy will be successful. Importantly, gut microbiota and metabolism profoundly affect the immune microenvironment. Understanding their complicated interaction will contribute to better modulating HCC-specific immune response and overcoming resistance to already developed therapies. In this review, we provide an overview of immunosuppressive microenvironment in HCC, mainly introduce mechanisms of immune evasion underlying the role of immune microenvironment, gut microbial microenvironment, and metabolism microenvironment, and propose novel strategies to harness the TME to enhance HCC immunotherapy.
Cancer immunoediting is a dynamic process that includes immune surveillance and tumor progression. It describes the relationship between tumor cells and immune system, proceeding through three phases: elimination, equilibrium, and escape (20). During the elimination phase, immune effector cells are able to recognize and eliminate tumor cells (20). In the equilibrium stage, tumor cells have escaped the elimination stage. But adaptive immunity still prevents the overall growth of the tumor, which keeps tumor cells in a state of functional dormancy (20, 21). In the escape stage, tumor cells continue to grow and proliferate in an uncontrolled manner and is no longer confined by the host immunity (20, 21). Tumor subclones that have acquired alterations could evade detection and destruction (20, 21).
The cancer-immunity cycle is a multistep process (Figure 2). The infinite proliferation and high tumor mutational burden of tumor cells firstly activate innate immune cells, such as natural killer (NK) cells, which target and lyse tumor cells to release tumor-associated antigens into the TME. These molecules are subsequently recognized by antigen-presenting cells (APCs), which travel to secondary lymphoid organs where adaptive immune responses are primed and activated (22). APCs present neoantigens to T cell receptor (TCR) of CD8+ cytotoxic T lymphocytes via the major histocompatibility complex (MHC) class I molecules. These activated T cells migrate and infiltrate into the HCC tissue. The final step is the T lymphocyte-mediated destruction of tumor cells, which in turn allows more tumor-associated antigens released into the TME (23, 24). Of note, the cancer-immunity cycle represents the adaptive aspect of immune surveillance phase (25–27). Tumors can perturb the processes mentioned above to evade immune surveillance by tumor-intrinsic mechanism (acquisition of genetic alterations) or tumor-extrinsic mechanism (generation of an immunosuppressive TME).
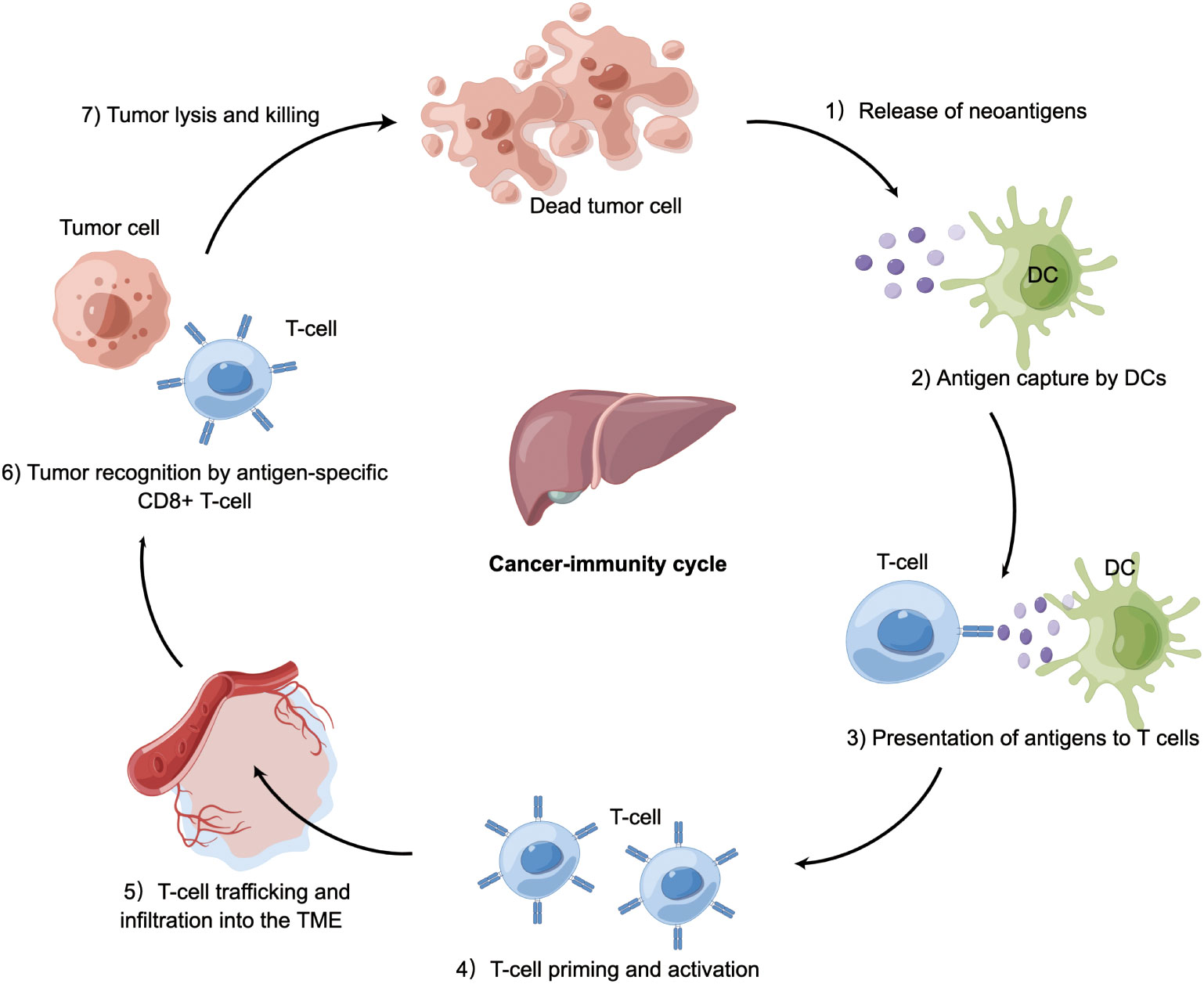
Figure 2 Cancer-immunity in HCC. Tumor cells release antigens into the tumor microenvironment due to necrosis or treatment. Dendritic cells capture cancer antigens and traffic to the lymphoid organs where they present antigens to T cells, followed by T-cell priming and activation. These activated T cells migrate and infiltrate into HCC tissue. CD8+ T cells recognize HCC cells via T cell receptor. The final step is T cell-mediated killing of tumor cells, allowing more cancer-specific antigens to release. Tumor can perturb the processes mentioned above to occur immune evasion. DC, dendritic cell; TME, tumor microenvironment; HCC, hepatocellular carcinoma.
In acute infection, activated T cells can eliminate harmful pathogens. However, during the progression of HCC, these neoantigens are seldom eliminated, leading to the formation of chronic inflammatory stimulation that mediates the silence of the immune response and the loss of cytotoxic capacities of T cells. Previous studies have reviewed the escape of the tumor-intrinsic mechanism (28). The contributions of TME in this issue is usually be ignored. Therefore, the crosstalk among immune microenvironment, gut microbial microenvironment, and metabolic microenvironment is of great importance to HCC immune evasion.
Immune surveillance and evasion are respectively dictated by the opposing activities of effector immune cells and immunosuppressive cells in the TME (Figure 3). The hepatic TME is an intricate ecosystem that is comprised of tumor cells, immune cells, non-parenchymal liver cells, tumor-associated fibroblasts (29). Several lines of evidence suggest that the crosstalk between tumor cells and TIME components is a critical factor for the immune evasion of HCC and for the major cause of resistance to immunotherapies. The immunosuppressive milieu is consisted of immunosuppressive cells, non-parenchymal cells, T-cell exhaustion, soluble cytokines, and signaling cascades (30).
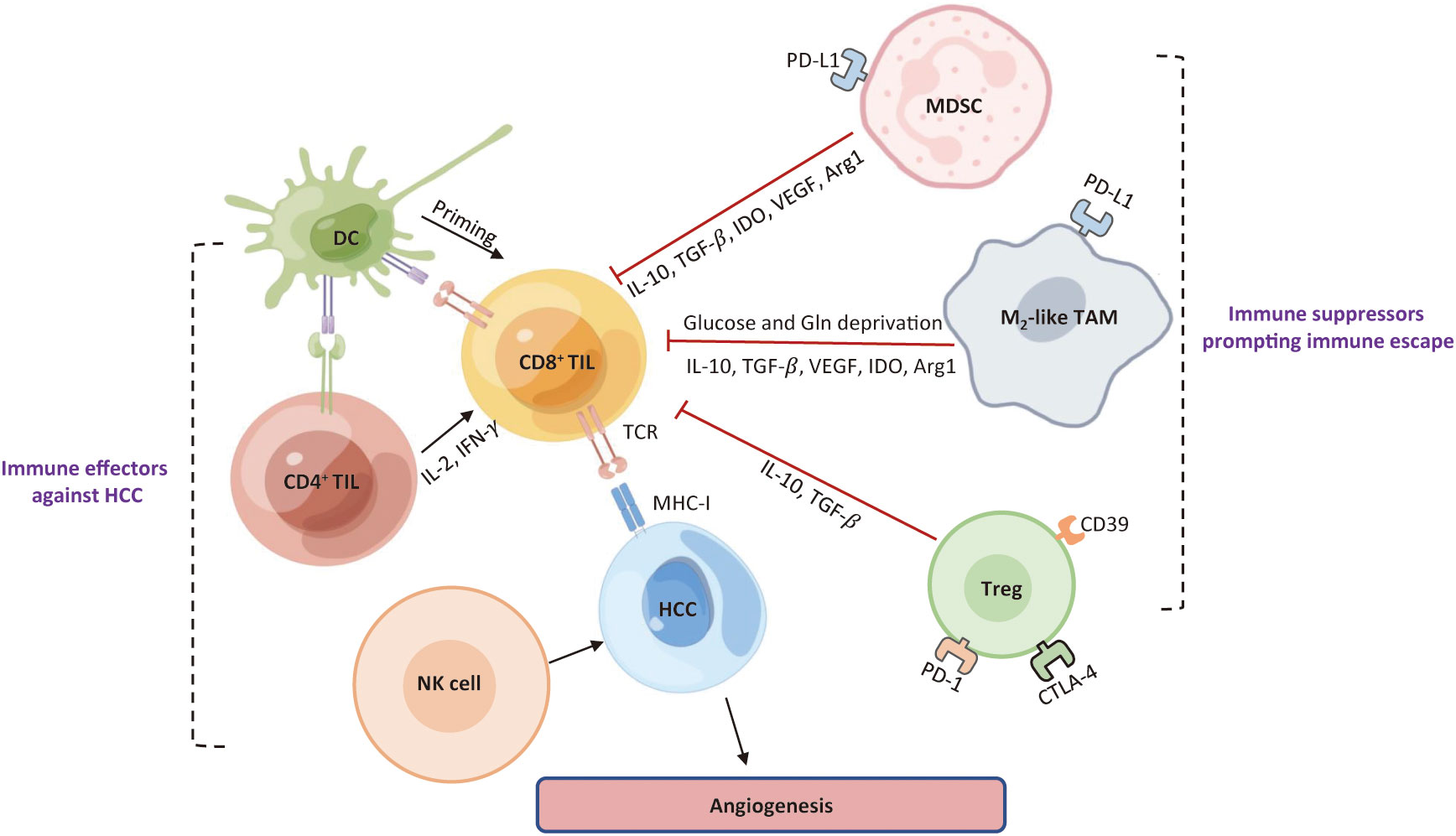
Figure 3 Roles of major immune cells in the HCC immune microenvironment. Immune cells existing in HCC can be roughly classified into one group that prompts an effective anti-tumor response, and the other group that limits immune response against HCC cells and contribute to an immunosuppressive TME. DC, dendritic cell; TIL, tumor-infiltrating lymphocytes; NK, natural killer; MDSC, myeloid-derived suppressor cell; Treg, regulatory T; TAM, tumor-associated macrophage; VEGF, vascular endothelial growth factor; TGF-β, transforming growth factor-Β; IDO, indoleamine 2, 3-dioxygenase; Arg1, arginase 1; Gln, glutamine; TCR, T cell receptor; MHC-I, major histocompatibility complex I; TME, tumor microenvironment; HCC, hepatocellular carcinoma.
Cytotoxic CD8+ T cells, CD4+ T cells, and NK cells work together to maintain immune surveillance, whereas abundant immune cells that resident in HCC contribute to immune evasion to prompt tumor progression, such as myeloid-derived suppressor cells (MDSC), regulatory T (Treg) cells, and tumor-associated macrophages (TAMs). Under physiological conditions, all populations participate in the manipulation of immune response, and thereby preserving homeostasis and self-tolerance (30, 31). However, both adaptive and innate immune response are blunted in HCC, as demonstrated by the TIME with dysfunctional TILs and NK cells (32–34).
MDSCs is a heterogenous group of immature myeloid cells that dampen CTL and NK cell effector functions, displaying a strong immunosuppressive activity in tumor-bearing hosts (35, 36). Several tumor-originated cytokines, such as IL-6, IL-1β, GM-CSF, G-CSF, VEGF, and MCP-1, have been reported to induce MDSC accumulation in preclinical models of HCC (37). An HCC-specific cell cycle-related kinase (CCRK) could upregulate IL-6 production via EZH2/NF-KB signaling, resulting in an extensive infiltration of polymorphonuclear MDSCs (38). Hypoxemia is a key regulatory factor that induces MDSCs accumulation via the chemokine C-C motif Ligand 26 (CCL26)/CX3CR1 pathway (39). Hypoxia-inducible factor 1α (HIF-1α) mediates ENTPD2 overexpression to convert ATP to 5’-AMP, which recruits a great quantity of MDSCs into the TME (40). Tumor-associated fibroblasts (CAFs) also facilitate the production of MDSCs by activating IL-6/STAT3 pathway (41). MDSCs accumulated in HCC could damage effector T cell function, reduce NK cell cytotoxicity, and expand immune checkpoint signaling, which blunt both innate and adaptive immune responses. The liver contains a large number of MDSCs that up-regulate the secretion of VEGF, TGF-β, and arginase, which inhibit T cell activation (42). MDSCs were found to deprive essential amino acids that are critical to T cell proliferation (43), and they release reactive oxygen and nitrogen species (iNOS or NOS2) that disrupt T cell receptor (TCR) signaling (44). Galectin-9 expressed on MDSCs binds to TIM-3 on T cells, which is associated with T cell apoptosis (45). Furthermore, a high infiltration of MDSCs in HCC is able to facilitate the conversion of naïve T cells into Treg cells (30). MDSCs also foster an immune escape status by reducing NK cell cytotoxicity. In senescent hepatocytes, MDSCs are recruited via the CCR2-CCL2 signaling, followed by differentiating into macrophages and blocking HCC initiation. However, once the tumor is initiated and developed, they would lose the ability of differentiation and cause inhibition of NK cell responses (46). Specifically, MDSCs can impair NK cell cytotoxicity by the NKp30 receptor and interact with Kupffer cells to enhance PD-L1 expression (47).
The physiological role of Treg cells is to inhibit excessive immune response to maintain homeostasis and autoimmune tolerance. However, hyperactive work of Treg cells in HCC supports tumor invasiveness, triggering a compromised T-cell immune response through several mechanisms (48–50). More CD4+ CD25+ Treg cells are enriched in the TME relative to that in in healthy individuals (51, 52). Treg cells are recruited by the chemokine receptor 6 (CCR6) and chemokine ligand 20 (CCL20) axis and activated by the binding of TCR with IL-10 and TGF-β signaling (53). Sorafenib, a multi-kinase inhibitor for HCC, has been proven to reduce hepatic Treg infiltration via suppressing TGF-β signaling (54). Long noncoding RNAs (lncRNA) are also involved in Treg cell differentiation (55). Specifically, the lncRNA-EGFR links an immunosuppressive state to HCC by augmenting activation of AP-1/NF-AT1 axis in Treg cells, thus prompting immune evasion (55). Overexpression of IL-35 has been shown to positively correlate with CD39+ FoxP3+ Treg cell infiltration, which may be another independent predictor for treatment efficacy among HCC patients (56). Mechanistically, CD4+ CD25+ FoxP3+ Treg cells could damage CD8+ T cell cytotoxicity by reducing the release of granzyme A, B, and perforin (57). Treg cells selectively inhibit some molecules that are essential in CD8+ T cell activation, such as TNF-α and IFN- γ (57, 58). Treg cell constitutively express CTLA-4 and secrete inhibitory molecules, such as IL-10 and TGF-β (59, 60).
As a significant component in the TME, TAM frequently portends a worse prognosis in HCC (61). TAMs arise from marrow-derived monocytes and obtain versatile immunosuppressive functions at each stage of differentiation. M1 and M2 are two polarizing phenotypes of TAMs with high plasticity in response to different stimuli. Substantial findings support that M1-polarized macrophages create pro-inflammatory cytokines and prevent malignancy development, whereas M2-polarized cells are able to produce tumor growth factor (IL-6), angiogenic molecules (VEGF), and immunosuppressive factors (Arg1, IL-10, TGF-β, and IDO) (62). Several HCC-originated cytokines, including IL-4, IL-13, CSF-1, CCL2, CXCL12, and CTG, promote CCR2+ inflammatory monocytes differentiation into TAMs in the TME (63–65). Moreover, TME-derived TGF-β facilitates TIM-3 expression on TAMs, fostering HCC development and immune tolerance (66). Osteopontin (OPN) correlates with PD-L1 upregulation and prompts TAM chemotaxis through the CSF1-CSF1 pathway (67). Under persistent hypoxia, HIF-1α/IL-1β loop between tumor cells and TAMs fosters epithelial-mesenchymal transition (EMT) and immune evasion (68). TAMs also produce cytokines and chemokines to drive immune suppression in HCC. For example, TAMs-derived CCL17, CCL18, and CCL22 could attract Treg cell infiltration into the TME (69, 70). The interplay between MDSCs and TAMs downregulates the production of IL-6, IL-12, and MHC-II but upregulates IL-10 secretion. TAM-derived IL-10 damages downstream CD8+ T cell and NK cell cytotoxicity but increases CD4+ CD25+ FOXP3+ Treg cell frequency (71, 72). Activated TAMs in the peritumoral stroma of HCC secrete a set of pro-inflammatory cytokines, such as IL-6, IL-23, IL-β, and TNF-α. These cytokines trigger the expansion of T helper 17 (Th17) cells that overexpress PD-1, CTLA-4, and GITR to exert an immunosuppressive function (73). Overall, TAMs might be a promising target for future HCC treatment.
Less common immunosuppressive cell types in human HCC consist of B cell population expressing PD-1, Th17 cells, CD4+ T cells expressing CCR4 and CCR6, CD14+ DCs expressing CTLA-4 and PD-1, tumor-associated neutrophils, tumor-associated fibroblasts, and type-II T helper cells (Th2) (74–77). These cells cooperate in the formation of immunosuppressive milieu and their presence usually manifests a poor prognosis in HCC.
Liver is an immune organ with a number of immunocompetent cells. Non-parenchymal resident cells, such as Kupffer cells, hepatic stellate cells (HSC), and liver sinusoidal endothelial cells (LSEC), cooperate in the maintenance of immune tolerance.
Kupffer cells are liver-resident macrophages that act as antigen-presenting cells (APC) to form the first line of defense against pathogens (78, 79). Kupffer cells can contribute to hepatocarcinogenesis and immune escape underlying several mechanisms: 1) secretion of immunosuppressive cytokines (IL-10) (80); 2) upregulation of inhibitory immune checkpoint ligand PD-1 (81); 3) downregulation of costimulatory molecules (CD80 and CD86) (42, 82); 4) production of Indoleamine 2-3 dioxygenase (IDO) (83); 5) recruitment of Treg cells and T helper 17 (TH17) cells (42, 81, 82). The interaction of PD-L1 expressed by Kupffer cells and PD-1 expressed by T cells leads to T-cell exhaustion in human HCC (84). HSCs can secrete hepatocyte growth factor (HGF) that enables MDSC and Treg cells to accumulate inside the liver (85). Also, HSCs express high levels of PD-L1 to induce T cell apoptosis (86). LSECs not only motivate Treg cell activation via TGF-β but also highly express PD-L1 (87). Tumor-associated fibroblasts (TAF) can trigger NK cell dysfunction by secreting prostaglandin E2 (PGE2) and IDO, and prompt MDSC production by releasing IL-16 and CXCL12 (41).
Immune checkpoints involve co-inhibitory molecules preventing T-cell overactivation. Liver tumor cells and stromal cells express corresponding ligands to evade anti-tumor immunity (88). Co-inhibitory checkpoints include programmed cell death-1 (PD-1), cytotoxic T lymphocyte protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG3), T-cell immunoglobulin and mucin-domain containing 3 (TIM3), and others (88), acting as pivotal regulators of T-cell exhaustion (30, 31, 89).
CTLA-4 is expressed by activated T cells and is constitutively present on Treg cells. It prevents T cell proliferation and induces Treg cell activity inside HCC tissues (75, 90). PD-1 is expressed by activated T cells, NK cells, Treg cells, MDSCs, monocytes, and DCs, while its ligand, PD-L1, is mainly expressed by tumor and stromal cells. The interaction of PD-1/PD-L1 is suppressive for antigen-specific T cell activation (91–93). In HCC, high infiltration of PD-1+ CD8+ T cells predicts a worse prognosis and a higher risk of recurrence (94). In turn, overexpression of PD-L1 in tumor cells prompts CD8+ T cell apoptosis (94). The immune microenvironment of HCC also involves the overexpression of PD-L1 and PD-L2 in Kupffer cells, LSECs, and leukocytes (95).
The immunosuppressive roles of LAG3 and TIM3 have recently been uncovered in HCC. LAG3 that binds MHC-II molecules with high affinity, is upregulated upon T cell activation and is a molecular signature of T cell exhaustion (96). LAG3 expression is significantly higher on CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs) than in other immune constituents among HCC patients (97). Similarly, TIM3 is expressed on CD4+ and CD8+ TILs, TAMs, NK cells in human HCC models (98). TIM3 interacts with its ligand galectin-9 mediating T-cell dysfunction (99), whereas its expression on Treg cells leads to enhanced suppressive activity (100). Notably, TIM3 is highly expressed in less differentiated tumor cells (101), which predicts poor prognosis in HBV-associated HCC (102).
Overall, immune checkpoints are expressed on the surface of T cells in different phases. Tumor cells evade immune-mediated destruction not only by expressing ligands to activate these receptors but also favor a suppressive TME by recruiting non-neoplastic cells to express these ligands. Immune checkpoint inhibitors (ICIs) are monoclonal antibodies designed to specifically disrupt inhibitory ligand-receptor interaction, removing T-cell exhaustion and recovering immune elimination (103–105) (Table 1). LAG3, TIM3, and PD-1 may function synergistically to facilitate HCC immune evasion and develop drug-resistance to PD1 or PD-L1 blockades (66, 106). Preclinical data support LAG3 and TIM3 inhibitors in combination with PD1 or PD-L1 ICIs, though their clinical values still require further elucidation.
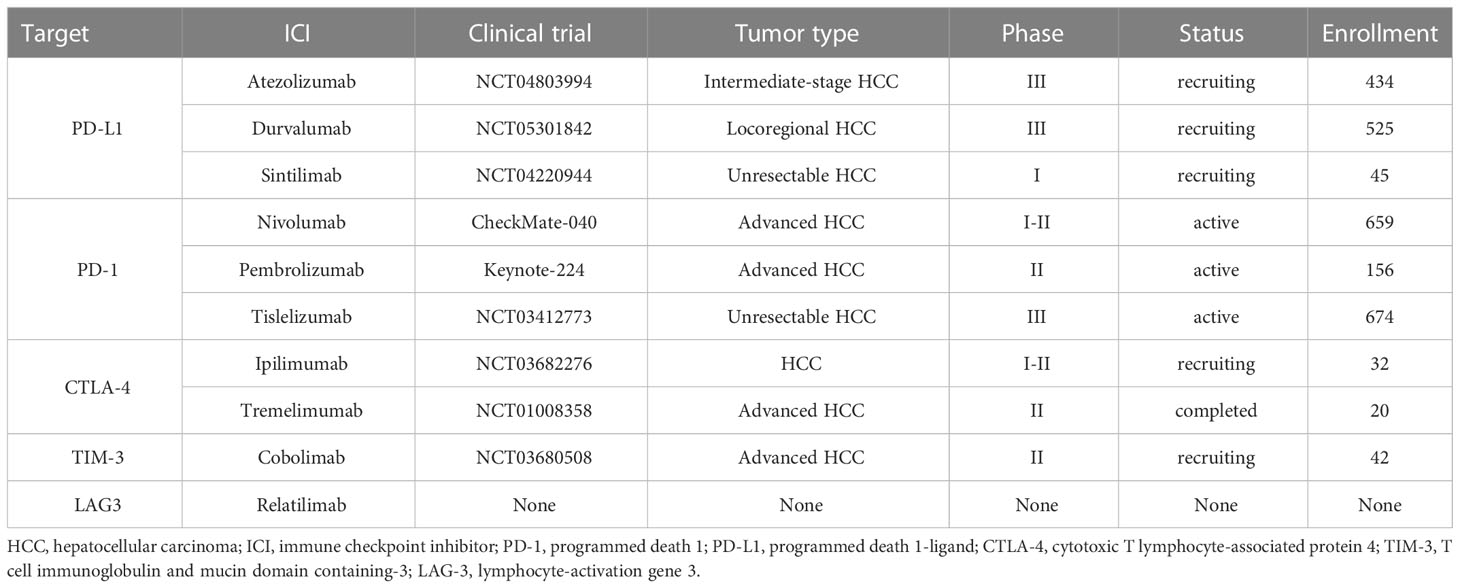
Table 1 Immune checkpoint inhibitors and their targets in HCC.
The local milieu of cytokines and soluble mediators partly dictate the immune microenvironment of HCC. Considering a more complex layer, effects of these pleiotropic molecules greatly differ in their target immune cell population, or in acute or chronic inflammatory milieu (107). Non-parenchymal cells and infiltrating immune cells could secrete several cytokines and concurrently keep sensitive to these cytokines (108, 109). Secretion of TGF-β, IL-10, and VEGF into the TME all contributes to immunosuppression (42).
A well-identified example is TGF-β that is abundant in the TME of HCC. It could be generated by tumor cells, TAMs, and Treg cells and downregulates anti-tumor immunity at varying levels. Explicitly, TGF-β drives the polarization of TAMs into pro-tumorigenic M2-phenotype (110); favors the differentiation of naïve CD4+ T cells into Treg cells (111); impairs effector CD8+ T cell and NK cell cytotoxicity (112, 113); inhibits DC cell activation (114); and exert inhibitory effects on B cells (115). High serum TGF-β might predict poor anti-cancer response to sorafenib and pembrolizumab in HCC patients (116, 117). Evidently, TGF-β plays multitude effects on immune and tumor cells, hindering the inflammatory reaction and supporting immune evasion in HCC.
IL-10, a tolerance-inducing molecule in the HCC TME, is produced by tumor cells, TAMs, Treg cells, and DCs (118). It dampens the recruitment of tumor-infiltrating T cells (119) and upregulates PD-L1 expression in monocytes (120). High circulating levels of IL-10 have been shown to induce decreased TIL activity (121) and increased MDSCs (122). Increased plasma level of IL-10 portends to a poor prognosis in HCC patients (49, 123).
VEGF, a well-known regulator driving tumor angiogenesis, is mainly secreted by both tumor cells and the surrounding stroma (124). In addition to prompt angiogenesis, VEGF attenuates anti-tumor response by negatively affecting antigen-presenting cells (APCs) and effector T cells while maintains immunotolerant TME by positively increasing MDSCs and Tregs recruitment (125). Also, VEGF increases PD-1 expression on T cells and PD-L1 expression on TAMs. Focal gains at chromosome 6p21 leads to overexpression of VEGFA and thereby foster an immunosuppressive TME (126, 127). Overall, these findings build the fundamental to test the efficacy of drugs that counteract the immunosuppressive actions of TGF-β, VEGF, or IL-10 in HCC.
Tumor-intrinsic signaling cascades also affect the composition and function of HCC immune infiltrates. In a mouse model of HCC, CTNNB1 mutation or activation of WNT-β-catenin pathway could downregulate CCL5 expression and dampen DC recruitment, leading to immune escape and resistance to anti-PD-1 therapy (128). The expression of NKG2D ligand on HCC cells is also downregulated by β-catenin signaling, which is detrimental to the MHC-dependent immune response responsible by NK cells (129). Loss of p53 function facilitates the recruitment of immunosuppressive cells, and hepatoma CDK20 activation prompts the recruitment of MDSCs (38). In addition, overexpression of MYC, accounting for around 50-70% HCC cases, has been associated with PD-L1 upregulation (130). Finally, chronic HBV infection also results in overexpression of PD-L1 on Kupffer cells, leukocytes, and LSECs, and thus enhancing inhibitory signals in HCC TME (95, 131).
The microbes reside within the tumor cells and immune cells. Increasing evidence suggests a critical link between the microbiota and the immune system (132–134). Intra-tumoral microbes and their products, defined as the tumor microbe microenvironment, have the potential to affect the tumor immune microenvironment. Gut microbiota is termed as a collection of microorganisms that colonize the intestine (135). Of note, the gut microbiota could repress immunosurveillance and prompt hepatocarcinogenesis. Understanding how gut microbes affect hepatic immune escape creates therapeutic innovations to improve HCC immunotherapy. The negative roles of microbes on TIME are multifaceted: 1) microbial activation of TLR4; 2) microbial dysbiosis; 3) microbe-derived metabolites; 4) microbial stimulation of inhibitory checkpoints.
Microbial adjuvanticity is explained as the immunomodulatory function of the pathogen-associated molecular patterns (PAMP), which could be sensed by pattern recognition receptors (PRR). The most well-elaborated subtype of PRR is Toll-like receptor (TLR) (136). Microbial activation of TLRs contributes to the formation of immunosuppressive TME. TLR4 is considered to be one of the most important receptors to prompt hepatocarcinogenesis, which is expressed by hepatocytes, Kupffer cells, HSCs, LESCs, DCs, NKs, B cells, and T cells (137). Overexpression of TLR4 has been identified in HCC tumor samples (138, 139). TLR4 primarily recognizes lipopolysaccharide (LPS) that is a constituent of the cell wall of Gram-negative bacteria. LPS-induced TLR4 signaling is associated with microvascular invasion, early recurrence, and shortened survival in HCC patients (140).
Microbes mediate immune escape of HCC through direct or indirect TLR4-dependent manners. Firstly, TLR4 affects the recruitment and differentiation of various tolerance-inducing cells. Bacterial LPS recognized by TLR4 could stimulate hepatocytes to express CXCL1 that is a chemokine recruiting CXCR2+ polymorphonuclear MDSCs (141). Similarly, Fusobacterium recognized by TLR4 regulates IL-6/STAT3/C-MYC signaling pathway, facilitating TAM polarization into M2 phenotype (142). The interaction of TLR4 with macrophages indirectly prompts the accumulation of Treg cells in hepatoma cell lines, along with the upregulation of IL-10 and CCL22 (138). Secondly, LPS-induced TLR4 directly activates JNK/MAPK signaling to enhance the invasive ability and EMT of HCC cells (143). EMT enables epithelial cells to obtain mesenchymal characters to favor the formation of an immunosuppressive TME via upregulating co-inhibitory checkpoints and inducing resistance to NK cell-mediated lysis (144–146). The association between EMT and immunosuppression has been widely reported in HCC (147). Thirdly, LPS-mediated TLR4-AKT pathway upregulates the expression of Sox2, a stemness marker gene, thereby increasing the number of cancer stem cells (CSCs) of HCC (148). It is well known that CSCs are involved in immune evasion through certain intrinsic and extrinsic mechanisms (149). There is a tight association between TLR4 expression and CSC characteristics, contributing to the failure of immune surveillance (150). Furthermore, TLR4 is a direct target of microRNA-122 (miR-122), a tumor suppressor that inhibits the expression and activities of cytokines, such as VEGF, IL-6, COX-2, prostaglandin E2, and MMP-9 (151). Downregulation of miR-122 is linked to immune escape of HCC by targeting TLR4, which is associated with PI3K/AKT/NF-KB signaling pathway (151). Additionally, LPS-activated STAT3 signaling upregulates VEGF production for HCC angiogenesis (152). As discussed previously, VEGF is a key negative regulator of anti-tumor immunity.
Overall, these findings suggest that microbial stimulation of TLR4 can change the TIME. Intriguingly, drugs targeting TLR4 might be adjuvants to immune checkpoint inhibitors. Besides interacting with TLR4, a specific gut microbe can exert immunomodulatory effect via many different PRR-mediated signaling pathways, while some of them await further exploration (153).
Maintenance of a balanced microbiota composition is crucial to forming an ecological barrier to insults from the external stimuli. The gut microbiota and mucosal immunity interact with each other to maintain intestinal homeostasis. Once this balance is disrupted, microbial dysbiosis would provide survival advantages for pathogenic bacteria along with decreased number of beneficial ones (154). An imbalance in gut microbiota composition is detected in HCC, with a significant increase of E. coli and Atopobium cluster while a significant regression of Lactobacillus species, Bifidobacterium species, and Enterococcus species (154). A recent study pointed out that a high cholesterol diet could induce gut microbial dysbiosis (depleted Bifidobacterium and Bacteroides) while altered flora metabolites in HCC patients (155).
Dysbiosis-mediated immune escape refers to a variety of mechanisms. Firstly, microbial dysbiosis can affect the content of immunogenic substances participating in intestinal homeostasis maintenance. High levels of lipopolysaccharide (LPS) have been detected in both pre-clinical models and HCC patients (154, 156), which is likely attributed to the leaky gut and bacterial translocation (157). Accumulation of circulating LPS from Bacteroides can prompt immune tolerance and hepatocarcinogenesis (156, 158). Likewise, TLR2 agonist lipoteichoic acid (LTA) can act on HSC to prompt senescence-associated secretory phenotype and enhance hepatocyte proliferation (159, 160). Secondly, microbial dysbiosis may alter the intracellular tight junction, thereby enhancing the interaction of dangerous signals with immune cells and facilitating the chronic inflammation (161–164). Previous studies supported that HCC often occurs in the context of chronic inflammation (165–167). Explicitly, some microbiota can invade colonic epithelial cells and activate intrinsic signaling pathways, aggravating the host inflammatory responses and releasing more cytokines (168, 169). Dysbiosis-driven chronic inflammatory can trigger oxidative stress that can deplete sensitive microbes and leave resistant strains (170). More importantly, it can mediate immune evasion by prompting angiogenesis, disrupting adaptive immunity, and altering the expression of pathogen recognition receptors (such as TLRs) and downstream signaling (171, 172). Overall, changes in microbiome composition are associated with the leaky gut (160, 173), endotoxemia, and systemic inflammation (174–176), predisposing the affected individuals more sensitive to developing HCC.
Microbial metabolites could enter the blood circulation and their receptors spread over both tumor cells and tumor infiltrating lymphocytes. Gut microbe-mediated bile acid metabolism regulates immune escape via decreasing the recruitment of NK T cells. Secondary bile acids (SBA) are derived from primary bile acids, of which process is mediated by gut microbes (177). SBA could downregulate the secretion of chemokine CXCL16 that interacts with CXCR6 to recruit NK T cells. Therefore, a reduced number of NK T cells through SBA via downregulating CXCR6-CXCL16, is beneficial for immune escape and HCC progression. Conversely, antibiotics that eliminate gut microbes could revert the above effects (178).
Deoxycholic acid (DCA) belongs to a gut bacterial metabolite that can induce DNA damage. A research confirmed that dietary or genetic obesity could result in microbial dysbiosis, thereby leading to an increasing level of DCAs (179). DCA has been shown to induce hepatic stellate cell senescence, thereby provoking the secretion of multiple cytokines that prompt hepatocarcinogenesis in mice model exposed to chemical carcinogen (179). Therefore, decreasing DCA level or targeting gut microbiota can specifically prevents immune evasion and inhibits HCC progression. Some other microbial-derived metabolites, such as N-acetylmuramic acid and N-acetylglucosamine, also exert their immunosuppressive effects on the TME (180).
The interactions between microbes and immune checkpoints could protect tumors from immune attack. The well-known inhibitory checkpoints include PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, CEACAM1. Fap2 protein of Fusobacterium mucleatum binds to inhibitory receptor TIGIT or CEACAM1, repressing the activity of NK cells and effector T cells (181–183). The helicobacter pylori HopQ outer membrane protein interacts with CEACAM1 to inhibit immune cell activities (184). In addition, CD47 expressed by tumor cells can recognize its ligand SIRPα expressed by DCs and macrophages. CD47-SIRPα interaction could repress antigen presentation activity and phagocytosis (185). However, Bifidobacterium can upregulate the production of IFN-I in DCs, enhancing antigen presentation and T cell activation. Emerging evidence indicates that intravenous injection of Bifidobacterium could improve the efficacy of CD47 blockade (186). Overall, microbial stimulation of inhibitory checkpoints could manipulate HCC immune escape, but connections between microbes and inhibitory checkpoint deserve more investigation.
In response to external stress, such as nutrient competition, hypoxia, suppressive metabolites, tumor cells occur metabolic adaptions for survival from senescence and immune evasion. Understanding additional immunosuppressive mechanisms led by metabolic constraints would create a promising avenue to shift immune evasion to immune elimination (187). Figure 4 introduces mechanisms of metabolism-mediated immune escape in HCC.
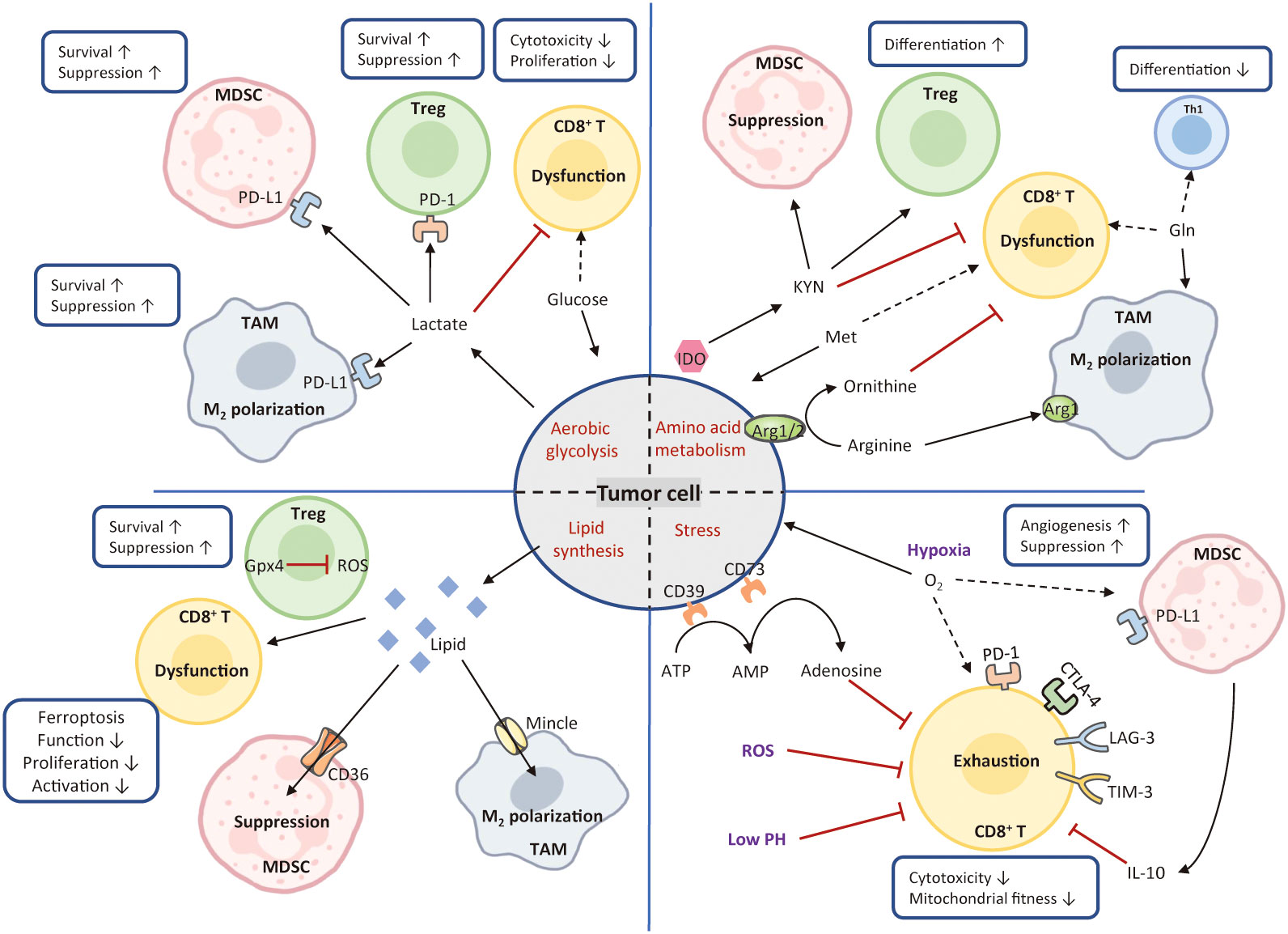
Figure 4 Mechanisms of metabolism-mediated immune escape. In the TME, hypermetabolic tumor cells interfere with immune cell function by depriving nutrients and produce various types of metabolic stress. Tumor cells utilize large amounts of glucose and amino acids to fuel their glycolysis and amino acid metabolism. These activities greatly limit nutrient availability to T cells, leading to the formation of immunosuppressive TME. Tumor cells also release excessive lipids into the TME, resulting in the enhanced lipid metabolism, high oxidative stress, and T-cell dysfunction. Conversely, Treg cells express high levels of glutathione peroxidase 4, avoiding ROS accumulation and the induction of ferroptosis. Cancer metabolism produces various metabolic stimuli, including hypoxia, low PH, and ROS, all of which impede CD8+ cytotoxicity and fitness. The solid black arrows present that the majority of nutrients are consumed by the cells, whereas the dashed black arrows indicate a paucity of molecule available to the cells. The red arrows represent inhibited metabolic pathways. MDSC, myeloid-derived suppressor cell; Treg, regulatory T; TAM, tumor-associated macrophage; Th1, T helper 1; IDO, indoleamine 2, 3-dioxygenase; Arg1, arginase; ROS, reactive oxygen species; KYN, kynurenine; Met, methionine; Gln, glutamine; Gpx4, glutathione peroxidase 4.
Glucose is not only the most dependent nutrient for tumor cells, but also an essential energy source for immune cell activation, differentiation, and function (188, 189). Owing to the enhanced aerobic glycolysis, tumor cells consume a large amount glucose. This activity limits the glucose availability and results in lactate accumulation that acidifies the TME, severely impeding CD8+ T cell activation and function (190). Glucose restriction in TILs is found to reduce mTOR activity, glycolytic capacity, and IFN-γ production, and thereby immune cells gradually lose their effector functions (191, 192). By contrast, Treg cells can use lactate to fuel the tricarboxylic acid (TCA) cycle and support their survival under a low glucose environment (193). Moreover, M2-like TAMs and MDSCs can be highly glycolytic and use glucose to reinforce their survival and suppressive activity (194–196). In addition, lactate prompts TAM M2 polarization, MDSC differentiation, as well as PD-L1 expression in TAMs and MDSCs, contributing to immunosuppression (196–200).
Competition uptake for amino acids also contributes to immune escape (201, 202). For example, glutamine (Gln) deficiency in the TME inhibits effector T cell activation and reduces cytokine production (203). Also, Gln deprivation impairs Th1 cell differentiation while favoring Treg cell maintenance (204, 205). Intriguingly, TAM can enhance Gln synthetase to provide Gln and support TAMs in skewing towards the M2 phenotype even within a Gln-deficiency environment (206). Likewise, arginine (Arg) has been reported to be deprived in the TME. Arginase 1 (Arg1) or 2 convert arginine to ornithine that hampers CD8+ T cell activation and cytotoxicity (207). Conversely, Arg1 maintains the immunosuppressive property of MDSCs and facilitate repolarization of M2-like macrophages, consequently maintaining an immunosuppressive TME (208). In addition, tumor cells also outcompete T cells for methionine (Met). Met recycling pathway has been reported to drive T cell exhaustion in HCC (209).
Tumor cells display enhanced lipogenesis and produce a large amount of lipids in the TME. Immune cells uptake excessive lipids by CD36 or Mincle, leading to increased lipid metabolism and high oxidative stress. The direct consequences are T cell dysfunction and ferroptosis. However, Treg cells with high-level of glutathione peroxidase 4, prevents ROS accumulation and ferroptosis. Further, lipid-mediated endoplasmic reticulum stress prompt M2 differentiation and favors their suppressive function. Cholesterol homeostasis is disrupted due to the overexpression of acyl coenzyme A-cholesterol acyltransferase 1 (ACAT1), consequently accelerating the migration of HBV-related tumor cells while inhibiting the function of HCC-specific TILs (210, 211).
Metabolites existing in the HCC TME also hold immunomodulatory properties. Indoleamine-pyrrole 2,3-dioxygenase (IDO) is a heme-containing enzyme catalyzing the conversion of tryptophan to kynurenine. Its activation supports malignant cells to escape from immune clearance (30). Hyperactive IDO leads to the depletion of tryptophan from the TME contributing to T-cell anergy (212). Moreover, kynurenine accumulation upregulates PD-1 expression in effector T cells (213) and induce Treg cell production (214). IDO upregulation plays a role in drug-resistance to ICIs in patients with HCC. It has been confirmed that inhibiting IDO adds therapeutic benefits of ICI (215).
Adenosine is another immunosuppressive metabolite, concurrently impairing T cell functionality and prompting Treg cell proliferation (216, 217). Both tumor cells and MDSCs express ectonucleotidase CD39 and CD73 hydrolyzing ATP/ADP to adenosine (216). HCC patients with high levels of CD39 tend to have increased risk of recurrence and shortened overall survival (218). Overexpression of CD73 has been reported in human HCC cell lines, where it promotes HCC growth and metastasis (219).
It is a common phenomenon that tumor cells consume excessive oxygen leading to an anoxia TME. HIF-1α is a major transcriptional factor that is upregulated in T-cell in response to hypoxia. First, hypoxia prompts the expression of inhibitory checkpoints, such as PD-1, LAG-3, TIM-3, and CTLA-4 (220). It also drives PD-L1 and IL-10 expression on MDSCs, which enhances their suppressive activity (221). Second, HIF-1α-induced EMT could create advantages for hepatoma cells to recruit IDO-overexpressing TAMs to repress T-cell response, and thereby facilitating immune escape via CCL20-dependent manner (147). Third, hypoxia-induced HIF-1α is detrimental to Treg cell differentiation and stability (222). Furthermore, HIF-1α binds to the promoter region of VEGF, followed by enhanced tumor angiogenesis (223). Hypoxia also aggravates the accumulation of lactate, which acidifies the TME and curtails effector immune cell function (224). Lactate contributes to the M2-like TAM polarization and maintains Treg cell function in a glucose-deficiency TME (197, 225). Under hypoxic condition, the COX-2/PGE2 axis stabilizes HIF-2α expression and activity to prompt HCC progression and develop drug-resistance to sorafenib (226). Overall, hypoxia can drive immunosuppression and exacerbate HCC immune escape.
Auspiciously, systemic therapies with molecular and immune therapies have remarkably revolutionized the management of HCC. Five single-agent molecular agents have been adopted by the US Food and Drug Administration (FDA) (3, 4, 227). In 2017 and 2018, two anti-PD-1 blockades, nivolumab and pembrolizumab, are approved as the second-line treatments for HCC (228). Notably, the superior results of atezolizumab plus bevacizumab versus sorafenib for advanced HCC heralded a new orientation of combination therapies (10). Currently, numerous clinical trials are in progress with ICIs, along with combined with anti-VEGF agents or tyrosine kinase inhibitor (TKIs). All approved drugs for HCC have been displayed in Table 2. A more refined understanding of the tumor microenvironment has led to great interests on ICIs. It is well evidenced that the immunosuppressive microenvironment in HCC triggers immune tolerance and escape by different mechanisms. Therefore, harnessing the TME by direct or indirect manners would provide new breakthroughs in HCC clinical treatment.
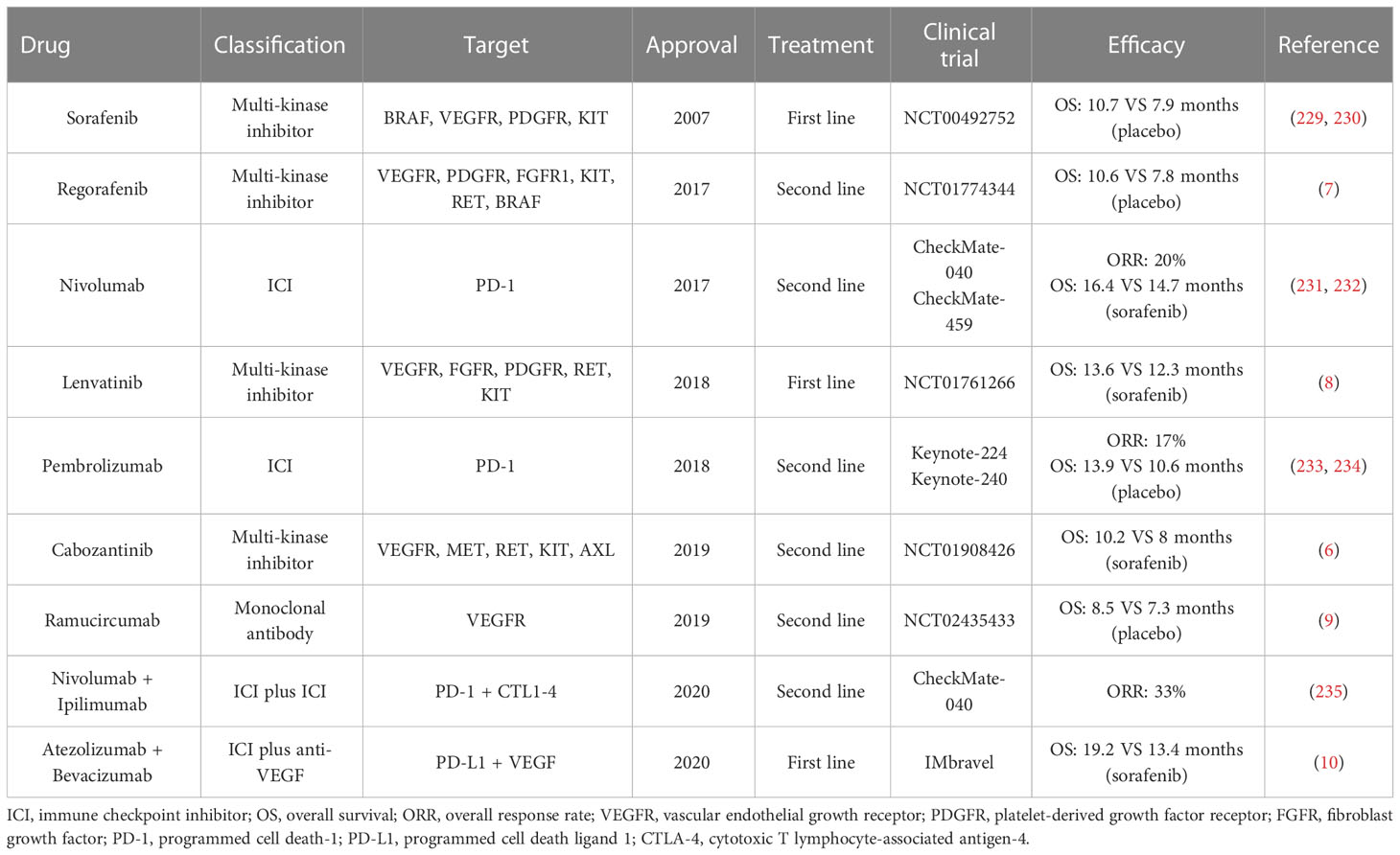
Table 2 FDA-approved drugs for hepatocellular carcinoma.
A promising approach is to deprive or neutralize cells with immunosuppressive functions. MDSCs have been considered as a potential target for resetting the immune tolerance status of HCC. Trabectedin not only targets malignant cells but also induces apoptosis or senescence of bone marrow cells (236). It has been reported to exert a strong cytotoxic effect on HCC cells (237). Another agent is estrogen that reportedly reduces IL-6 stimulation and inhibits STAT6 activation, leading to the disruption of bone marrow cells in HCC models (238, 239). The combination therapy of anti-PD1/PD-L1 and anti-MDSCs (CCRK inhibition, p38 MAPK inhibitor, and C5AR blockade) may exert a synergistical effect on eradicating HCC (38, 240, 241). Also, combination use of radiation and IL-12 could boost anti-tumor immunity by reducing MDSC accumulation and ROS production (242). Many potential targets of MDSCs have been designed to interfere with immature myeloid cells (Table 3), but their combination with anti-PD-1/PD-L1 blockades still require additional validation in preclinical and clinical models. Alternatively, inhibiting Tregs or TAMs is another strategy to restore immune response (258, 259). Treg can be depleted by numerous agents, such as cyclophosphamide, gemcitabine, mitoxantrone, fludarabine, and CCR4-targeted antibodies (253). Sorafenib, a multi-kinase inhibitor for HCC, is able to reduce Treg infiltration into the liver by downregulating the TGF- β signaling (54). It has been shown that WNT-β-catenin signaling induces M2-like polarization of TAM and thereby reinforces malignant behaviors, whereas blocking WNT-β -catenin pathway in TAMs may rescue immune evasion of HCC (260). Overall, the modulation of suppressive immune cells is a possible adjuvant therapy to attenuate HCC progression. As shown in Table 3, treatment of MDSCs, TAMs, and Tregs targets in HCC has been documented and could be a new strategy for treating HCC (254–257, 261).
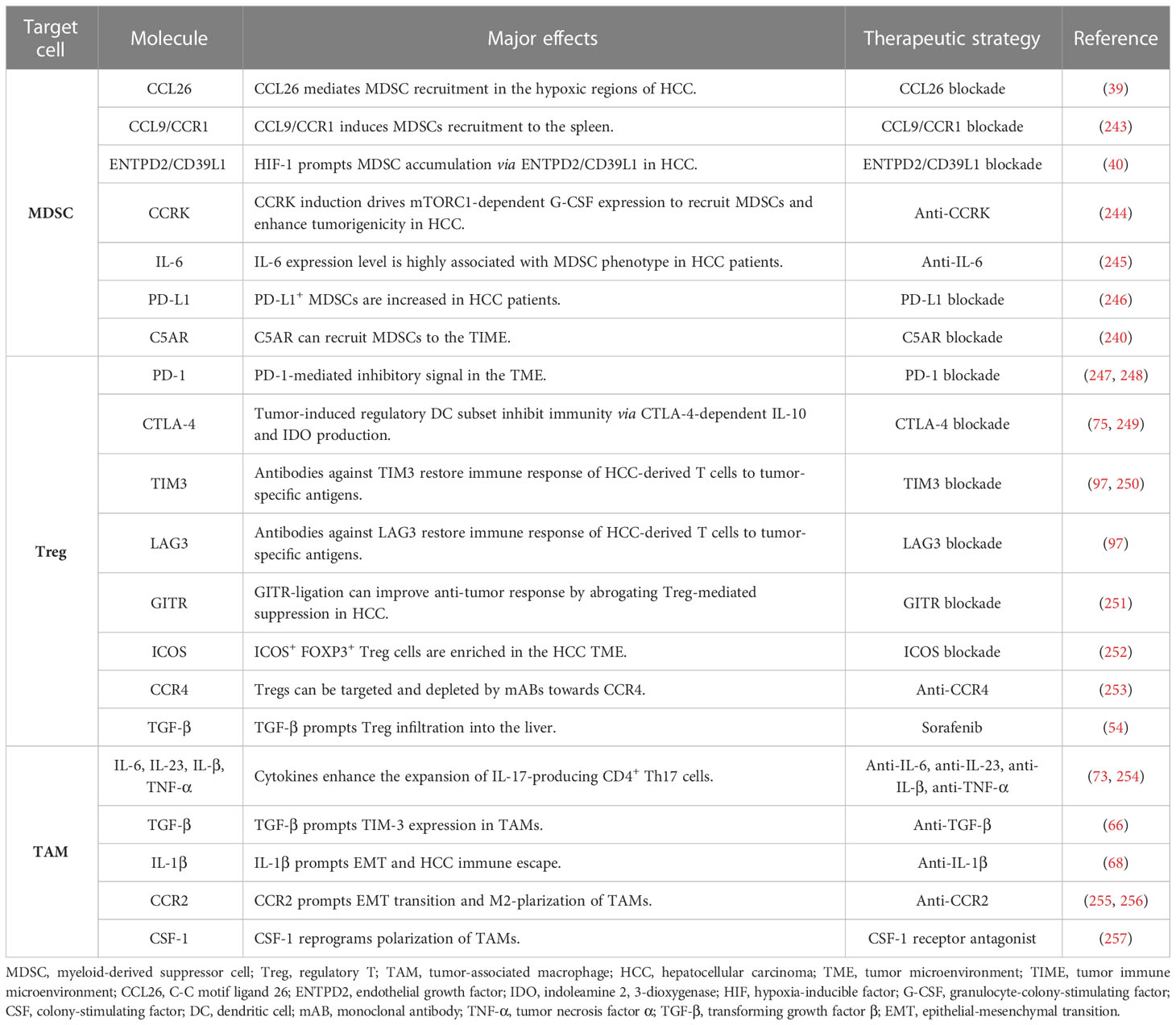
Table 3 A summary of molecular targets in the tumor immune microenvironment of HCC.
TGF-β pathway is a promising target for HCC therapy, as its inhibition tends to reduce the EMT and reactivate NK cells. Galunisertib is a small molecular inhibitor that reduces the phosphorylation of SMAD2, downregulating TGF-β pathway and inhibiting HCC progression (262). Galunisertib monotherapy has been shown to extend overall survival of advanced HCC patients in a phase-II trial (263). Combination of galunisertib and sorafenib demonstrated an improvement of efficacy compared to historical records of sorafenib monotherapy (NCT01246986). The combination strategy of galunisertib and PD-1 blockade is ongoing in clinical trials (NCT02423343 and NCT02947165). The monoclonal anti-TGF-β antibody ascrinvacumab also showed hopeful results among HCC patients in a phase I-II trial (264) and its combinational application with nivolumab is currently under investigation (NCT05178043).
Targeting VEGF enables ICIs more effective through multiple pathways (265, 266). VEGF inhibition not only transiently normalizes abnormal vasculature, but also increases CTL infiltration and modulates checkpoint expression on T lymphocytes (267, 268). Therefore, VEGF inhibition appears to be an ideal combinatorial partner for ICI as a locoregional therapy for HCC. IMbrave150 trial demonstrated that the addition of anti-VEGF inhibitor (Bevacizumab) significantly improved efficacy from ICI (atezolizumab) (10). Other combinations of ramucirumab (anti-VEGFR2) or Lenvatinib (anti-VEGFR and anti-FGFR) with ICIs also have been investigated (269).
Targeting the gut microbiota for HCC is increasingly attractive, including probiotics, prebiotics, fecal microbiota transplantation (FMT), and antibiotics. Since the gut microbiota dynamically regulates the host immunity, manipulating the gut microbiota may be a new orientation to improve anti-HCC immunotherapy.
Probiotics can keep gut microbial balance when given in certain amounts. Probiotic supplement as a dietary approach to repress HCC growth has been demonstrated. Feeding probiotics mixture Prohep (comprising Lactobacillus rhamnosus and Escherichia coli) could reduce liver tumor size, alter gut microbial composition to beneficial bacteria (Oscillibacter and Prevotella), and decrease the secretion of VEGF (270). Supplementing probiotics to Chinese subjects who are exposed to AFB1, such as Lactobacillus rhamnosus LC705 and Propionibacterium, could reduce the urinary excretion of aflatoxin-DNA adduct (AFB1-N7-guanine) (271). This finding kept in line with the protective capacity of probiotics against AFB1-induced HCC (272, 273). In another rat study, probiotics treatment containing Lactobacilli, Bifidobacteria, and Streptococcus thermophilus subsp Salivarius, can alleviate diethylnitrosamine (DEN)-induced hepatocarcinogenesis by preserving intestinal homeostasis and ameliorating chronic inflammation (154). Also, mice models treated with probiotics had a lower level of Th17 cells in gut compared to untreated mice. Therefore, probiotic can improve microecological balance, enhance intestinal barrier function, and prevent immune evasion of HCC.
Prebiotics are foods that selectively accelerate beneficial microorganism growth and suppress harmful bacterial growth, thereby adjusting gut microbial homeostasis (274). Besides, they can result in the production of short-chain fatty acid (SCFA) and ultimately inhibit HCC development. Prebiotics were found to maintain microbial stability and decrease pro-inflammatory pathways that trigger HCC initiation and progression (275). In mice given transplantation of BCR-ABL-transfected BaF3 cells, insulin-type fructans hold the promise to decrease hepatic BaF3 cell infiltration, relieve inflammation, and increase portal propionate content (276). Propionate inhibits BaF3 cell proliferation via cAMP-dependent pathway or by binding with GPR43 (276). Overall, prebiotics supplementation is a novel strategy to treat HCC.
Using antibiotics is another effective strategy to interrupt the tumor-prompting gut-liver axis. Antibiotics can reduce bacteria translocation, decrease pro-inflammatory signals from the leaky gut, and repress the synthesis of bacterial metabolites. For example, intestinal sterilization with antibiotic cocktail (containing neomycin, ampicillin, vancomycin, and metronidazole) has been proven to efficiently reduce the number and size of liver tumors induced by DEN-CCL4 or DMBA-HFD (179, 277). Consistently, the antibiotic cocktail (ABX, including vancomycin, primaxin, neomycin) or vancomycin treatment selectively elicited anti-tumor responses with increased CXCR6+ NK T cells and heightened IFN-γ production in HCC mouse models (178). As mentioned previously, CXCR6 expression level is controlled by gut microbiome-mediated primary-to-secondary bile acid conversion. A recent study suggests that vancomycin can inhibit HCC progression in insulin-fed TLR5-deficient mice (278). Concurrently, vancomycin can lead to selective depletion of gut microbiota, comprising Bifidobacteria, G+ Lachnospiraceae, and Ruminococcaceae.
FMT refers to the infusion of fecal solution from a healthy donor to the recipient intestinal tract to treat a disease associated with altered gut microbiota (279). FMT has successfully been used to treat Clostridium difficile infection via mechanisms including activation of mucosal immune system, maintenance of bile acid metabolism, and repair of the intestinal barrier (280). For example, alcohol-sensitive mice exhibited a decrease in Bacteroidetes and an increase in Actinobacteria following alcohol intake. After FMT, liver injury was relieved and dysregulated flora was partially recovered (281). Bajaj et al. reported that FMT enriched with Lachnospiraceae and Ruminococcaceae is able to restore the disruption of microbial diversity and function led by antibiotics in advanced cirrhosis patients (282). However, there are limited data on FMT in the treatment of HCC, and it is unclear whether microbial dysbiosis can be reverted by FMT (275). More studies are needed to validate the safety and efficacy of FMT in the future.
The significance of gut microbiota in modulating anti-tumor response to ICIs has been widely highlighted (283, 284). On the one hand, the dynamic change of gut microbiota can predict early outcome of immunotherapy. In a study, fecal samples from patients who respond to ICI showed higher taxa richness and more gene counts compared to non-responding patients (285). Stool fecal microbiota transplantation from cancer patients, who respond to ICIs, into germ-free or antibiotic-treated mice, ameliorated the efficacy of PD-1 ICIs, whereas fecal transplantation from non-responders failed to do so (286). This provoking finding is also supported by two other studies, describing different gut microbiota associated with improved response to ICIs (287, 288). Given those HCC patients with microbial dysbiosis, it is reasonable to speculate that the underlying dysbiosis potentially leads to immunotherapy failure. Microbial intervention may produce more profound effects in HCC than in other tumors. A feasible strategy is to combine ICI and selective microbiota manipulation. Recently, a clinical trial (NCT03785210) combining vancomycin treatment with ICI has been initiated, which will answer whether such a combination strategy would benefit patients with HCC. On the other hand, there is an association between the gut microbiota and immune-related toxicity (289). Targeting the specific microbiota may strengthen the effects of CTLA-4 blockade by reducing collateral toxicity (148).
The tumor-immune crosstalk inevitably leads to metabolic modifications in tumor cells and immune cells, serving as one of the most important mechanisms of immune evasion of HCC. Nutritional interventions aim to target immunometabolism in the TME (290). Dietary has been shown to have direct effects on both immune cells and tumor cells.
A ketogenic diet targets the Warburg effect in tumor cells by reducing glucose consumption while reprogramming effector T cells to rely on the OXPHOS (290, 291). In response to an increase of ketone bodies, CD4+ and CD8+ T cell secrete more cytokines, such as IFN-γ, TNF-α, perforin, and granzyme B (290). Nutritional interventions of essential amino acids also affect anti-tumor response. For example, arginine supplementation could switch T-cell metabolism from glycolysis to OXPHOS to enhance their survival (292, 293). Met supplementation might restore anti-tumor immunity by prompting the secretion of IL-2, TNF-α, and IFN-γ from TILs (294). IDO inhibition renders the TME less immunosuppressive by avoiding tryptophan depletion. It has been reported that IDO is involved in drug-resistance to ICI (295). Combinatorial treatments of IDO inhibitor and anti-PD1 or anti-CTLA4 blockades were shown to prolong survival in mouse models (295, 296). A phase I-II clinical trial (NCT03695250) is underway to evaluate IDO1 inhibitor (BMS-986205) in combination with nivolumab in patients with liver cancer. Caloric restriction is an alternative strategy to treat HCC. A study supported that caloric restriction in combination with radiation can decrease the abundance of Treg cells and expand the proliferation of CD8+ TILs in the TME (297). Moreover, it supports an immune signature linked to superior anti-tumor immunity and confers stem cell-like properties to effector T cells (298, 299). Altogether, targeting tumor-associated metabolic pathways is crucial to enhancing response to immune surveillance.
The tumor microenvironment of HCC is a dynamic and complicated network. Intricate interactions among suppressive immune cells, immunoregulatory cytokines or signaling, hostile metabolites, and the unbalanced gut microbiome collectively create a permissive TME that mediates immune evasion to favor HCC growth. In recent years, the combination therapy of atezolizumab and bevacizumab opened a new era for HCC treatment. However, HCC is still one of the worst prognoses and novel strategy targeting the TME is an urgent need. Given the complexity of the TME in HCC, combinatorial therapies can include ICIs, agents targeting immunosuppressive immune cells, anti-VEGF inhibitors, anti-TGF-β antibodies, microbiota manipulation, and metabolism intervention. A more holistic approach should be considered as a standard treatment for patients with advanced HCC. However, the molecular underpinnings governing immune evasion still need further clarification. Profound appreciation of the tumor-stromal interactions will enhance our understanding of the negative drivers of immunosurveillance. Multidimensional analysis, such as single cell analysis and next-generation sequencing technology, contribute to exploring detailed mechanisms behind HCC occurrence and identifying other targets in the TME.
YQ designed the study and reviewed the manuscript. CC participated in study design and wrote the original draft of the manuscript. CC and ZW were mainly responsible for the design of tables and figures. YD contributed to the conception of the paper. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (grant no. 81872264).
We thank Figdraw (www.figdraw.com) for the assistance in creating figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2016) 2:16018. doi: 10.1038/nrdp.2016.18
2. Singal AG, El-Serag HB. Hepatocellular carcinoma from epidemiology to prevention: Translating knowledge into practice. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterological Assoc (2015) 13(12):2140–51. doi: 10.1016/j.cgh.2015.08.014
3. Easl clinical practice guidelines: Management of hepatocellular carcinoma. J Hepatol (2018) 69(1):182–236. doi: 10.1016/j.jhep.2018.03.019
4. Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American association for the study of liver diseases. Hepatol (Baltimore Md) (2018) 68(2):723–50. doi: 10.1002/hep.29913
5. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. New Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
6. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. New Engl J Med (2018) 379(1):54–63. doi: 10.1056/NEJMoa1717002
7. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (Resorce): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London England) (2017) 389(10064):56–66. doi: 10.1016/s0140-6736(16)32453-9
8. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet (London England) (2018) 391(10126):1163–73. doi: 10.1016/s0140-6736(18)30207-1
9. Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased Α-fetoprotein concentrations (Reach-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2019) 20(2):282–96. doi: 10.1016/s1470-2045(18)30937-9
10. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. New Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
11. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Sci (New York NY) (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
12. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
13. Cai L, Michelakos T, Yamada T, Fan S, Wang X, Schwab JH, et al. Defective hla class I antigen processing machinery in cancer. Cancer immunology immunotherapy CII (2018) 67(6):999–1009. doi: 10.1007/s00262-018-2131-2
14. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (Time) for effective therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
15. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. Pd-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol (2021) 18(6):345–62. doi: 10.1038/s41571-021-00473-5
16. Finlay BB, Goldszmid R, Honda K, Trinchieri G, Wargo J, Zitvogel L. Can we harness the microbiota to enhance the efficacy of cancer immunotherapy? Nat Rev Immunol (2020) 20(9):522–8. doi: 10.1038/s41577-020-0374-6
17. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Sci (New York NY) (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
18. Ren D, Hua Y, Yu B, Ye X, He Z, Li C, et al. Predictive biomarkers and mechanisms underlying resistance to Pd1/Pd-L1 blockade cancer immunotherapy. Mol Cancer (2020) 19(1):19. doi: 10.1186/s12943-020-1144-6
19. Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal transduction targeted Ther (2021) 6(1):72. doi: 10.1038/s41392-020-00449-4
20. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi: 10.1038/ni1102-991
21. O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
22. Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
23. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat Rev Cancer (2014) 14(2):135–46. doi: 10.1038/nrc3670
24. Zhang Q, Jia Q, Zhang J, Zhu B. Neoantigens in precision cancer immunotherapy: From identification to clinical applications. Chin Med J (2022) 135(11):1285–98. doi: 10.1097/cm9.0000000000002181
25. Alexander AA, Maniar A, Cummings JS, Hebbeler AM, Schulze DH, Gastman BR, et al. Isopentenyl pyrophosphate-activated Cd56+ {Gamma}{Delta} T lymphocytes display potent antitumor activity toward human squamous cell carcinoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2008) 14(13):4232–40. doi: 10.1158/1078-0432.Ccr-07-4912
26. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. Nk cells stimulate recruitment of Cdc1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
27. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. Γδ T cells support pancreatic oncogenesis by restraining ΑΒ T cell activation. Cell (2016) 166(6):1485–99.e15. doi: 10.1016/j.cell.2016.07.046
28. Lindblad KE, Ruiz de Galarreta M, Lujambio A. Tumor-intrinsic mechanisms regulating immune exclusion in liver cancers. Front Immunol (2021) 12:642958. doi: 10.3389/fimmu.2021.642958
29. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer (2020) 6(7):605–18. doi: 10.1016/j.trecan.2020.02.022
30. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Sci (New York NY) (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
31. Wang F, Wang S, Zhou Q. The resistance mechanisms of lung cancer immunotherapy. Front Oncol (2020) 10:568059. doi: 10.3389/fonc.2020.568059
32. Cariani E, Pilli M, Zerbini A, Rota C, Olivani A, Zanelli P, et al. Hla and killer immunoglobulin-like receptor genes as outcome predictors of hepatitis c virus-related hepatocellular carcinoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2013) 19(19):5465–73. doi: 10.1158/1078-0432.Ccr-13-0986
33. Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, et al. Immunodominance and functional alterations of tumor-associated antigen-specific Cd8+ T-cell responses in hepatocellular carcinoma. Hepatol (Baltimore Md) (2014) 59(4):1415–26. doi: 10.1002/hep.26731
34. Cariani E, Pilli M, Barili V, Porro E, Biasini E, Olivani A, et al. Natural killer cells phenotypic characterization as an outcome predictor of hcv-linked hcc after curative treatments. Oncoimmunology (2016) 5(8):e1154249. doi: 10.1080/2162402x.2016.1154249
35. Li F, Zhao Y, Wei L, Li S, Liu J. Tumor-infiltrating treg, mdsc, and ido expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol Ther (2018) 19(8):695–705. doi: 10.1080/15384047.2018.1450116
36. Bruger AM, Dorhoi A, Esendagli G, Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, et al. How to measure the immunosuppressive activity of mdsc: Assays, problems and potential solutions. Cancer immunology immunotherapy CII (2019) 68(4):631–44. doi: 10.1007/s00262-018-2170-8
37. Kapanadze T, Gamrekelashvili J, Ma C, Chan C, Zhao F, Hewitt S, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol (2013) 59(5):1007–13. doi: 10.1016/j.jhep.2013.06.010
38. Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic ccrk inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut (2018) 67(5):931–44. doi: 10.1136/gutjnl-2017-314032
39. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-c motif) ligand 26. Hepatol (Baltimore Md) (2016) 64(3):797–813. doi: 10.1002/hep.28655
40. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor hif-1 promotes myeloid-derived suppressor cells accumulation through Entpd2/Cd39l1 in hepatocellular carcinoma. Nat Commun (2017) 8(1):517. doi: 10.1038/s41467-017-00530-7
41. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36(8):1090–101. doi: 10.1038/onc.2016.273
42. Ringelhan M, Pfister D, O'Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol (2018) 19(3):222–32. doi: 10.1038/s41590-018-0044-z
43. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4(1):e954829. doi: 10.4161/21624011.2014.954829
44. Lu T, Gabrilovich DI. Molecular pathways: Tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(18):4877–82. doi: 10.1158/1078-0432.Ccr-11-2939
45. Limagne E, Richard C, Thibaudin M, Fumet JD, Truntzer C, Lagrange A, et al. Tim-3/Galectin-9 pathway and mmdsc control primary and secondary resistances to pd-1 blockade in lung cancer patients. Oncoimmunology (2019) 8(4):e1564505. doi: 10.1080/2162402x.2018.1564505
46. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell (2016) 30(4):533–47. doi: 10.1016/j.ccell.2016.09.003
47. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma Via the Nkp30 receptor. Hepatol (Baltimore Md) (2009) 50(3):799–807. doi: 10.1002/hep.23054
48. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25(18):2586–93. doi: 10.1200/jco.2006.09.4565
49. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of Garp(+)Ctla-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res (2013) 73(8):2435–44. doi: 10.1158/0008-5472.Can-12-3381
50. Yuan CH, Sun XM, Zhu CL, Liu SP, Wu L, Chen H, et al. Amphiregulin activates regulatory T lymphocytes and suppresses Cd8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget (2015) 6(31):32138–53. doi: 10.18632/oncotarget.5171
51. Yang XH, Yamagiwa S, Ichida T, Matsuda Y, Sugahara S, Watanabe H, et al. Increase of Cd4+ Cd25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol (2006) 45(2):254–62. doi: 10.1016/j.jhep.2006.01.036
52. Cao M, Cabrera R, Xu Y, Firpi R, Zhu H, Liu C, et al. Hepatocellular carcinoma cell supernatants increase expansion and function of Cd4(+)Cd25(+) regulatory T cells. Lab investigation; J Tech Methods Pathol (2007) 87(6):582–90. doi: 10.1038/labinvest.3700540
53. Chen KJ, Lin SZ, Zhou L, Xie HY, Zhou WH, Taki-Eldin A, et al. Selective recruitment of regulatory T cell through Ccr6-Ccl20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLos One (2011) 6(9):e24671. doi: 10.1371/journal.pone.0024671
54. Wang Q, Yu T, Yuan Y, Zhuang H, Wang Z, Liu X, et al. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing tgf-beta signal. J Surg Oncol (2013) 107(4):422–7. doi: 10.1002/jso.23227
55. Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, et al. The long noncoding rna lnc-egfr stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun (2017) 8:15129. doi: 10.1038/ncomms15129
56. Fu YP, Yi Y, Cai XY, Sun J, Ni XC, He HW, et al. Overexpression of interleukin-35 associates with hepatocellular carcinoma aggressiveness and recurrence after curative resection. Br J Cancer (2016) 114(7):767–76. doi: 10.1038/bjc.2016.47
57. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with Cd8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology (2007) 132(7):2328–39. doi: 10.1053/j.gastro.2007.03.102
58. Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, et al. Tumor-infiltrating Foxp3+ tregs and Cd8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion (2012) 86(4):329–37. doi: 10.1159/000342801
59. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: Mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
60. Sprinzl MF, Galle PR. Immune control in hepatocellular carcinoma development and progression: Role of stromal cells. Semin liver Dis (2014) 34(4):376–88. doi: 10.1055/s-0034-1394138
61. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol (2015) 62(3):607–16. doi: 10.1016/j.jhep.2014.10.029
62. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
63. Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D, et al. Interleukin-8 induces expression of Foxc1 to promote transactivation of Cxcr1 and Ccl2 in hepatocellular carcinoma cell lines and formation of metastases in mice. Gastroenterology (2015) 149(4):1053–67.e14. doi: 10.1053/j.gastro.2015.05.058
64. Cai H, Zhu XD, Ao JY, Ye BG, Zhang YY, Chai ZT, et al. Colony-stimulating factor-1-Induced Aif1 expression in tumor-associated macrophages enhances the progression of hepatocellular carcinoma. Oncoimmunology (2017) 6(9):e1333213. doi: 10.1080/2162402x.2017.1333213
65. Wang TT, Yuan JH, Ma JZ, Yang WJ, Liu XN, Yin YP, et al. Ctgf secreted by mesenchymal-like hepatocellular carcinoma cells plays a role in the polarization of macrophages in hepatocellular carcinoma progression. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie (2017) 95:111–9. doi: 10.1016/j.biopha.2017.08.004
66. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters hcc development by enhancing tgf-Β-Mediated alternative activation of macrophages. Gut (2015) 64(10):1593–604. doi: 10.1136/gutjnl-2014-307671
67. Zhu Y, Yang J, Xu D, Gao XM, Zhang Z, Hsu JL, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-Pd-L1 blockade. Gut (2019) 68(9):1653–66. doi: 10.1136/gutjnl-2019-318419
68. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-inducible factor-1Α/Interleukin-1Β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatol (Baltimore Md) (2018) 67(5):1872–89. doi: 10.1002/hep.29681
69. Guo CL, Yang XH, Cheng W, Xu Y, Li JB, Sun YX, et al. Expression of Fas/Fasl in Cd8+ T and Cd3+ Foxp3+ treg cells–relationship with apoptosis of circulating Cd8+ T cells in hepatocellular carcinoma patients. Asian Pacific J Cancer Prev APJCP (2014) 15(6):2613–8. doi: 10.7314/apjcp.2014.15.6.2613
70. Li L, Yan J, Xu J, Liu CQ, Zhen ZJ, Chen HW, et al. Cxcl17 expression predicts poor prognosis and correlates with adverse immune infiltration in hepatocellular carcinoma. PLos One (2014) 9(10):e110064. doi: 10.1371/journal.pone.0110064
71. Zhou J, Ding T, Pan W, Zhu LY, Li L, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer (2009) 125(7):1640–8. doi: 10.1002/ijc.24556
72. Sharma S, Khosla R, David P, Rastogi A, Vyas A, Singh D, et al. Cd4+Cd25+Cd127(Low) regulatory T cells play predominant anti-tumor suppressive role in hepatitis b virus-associated hepatocellular carcinoma. Front Immunol (2015) 6:49. doi: 10.3389/fimmu.2015.00049
73. Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatol (Baltimore Md) (2010) 51(1):154–64. doi: 10.1002/hep.23291
74. Zhao F, Hoechst B, Gamrekelashvili J, Ormandy LA, Voigtländer T, Wedemeyer H, et al. Human Ccr4+ Ccr6+ Th17 cells suppress autologous Cd8+ T cell responses. J Immunol (Baltimore Md 1950) (2012) 188(12):6055–62. doi: 10.4049/jimmunol.1102918
75. Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, et al. Human Cd14+ ctla-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-Dependent il-10 and indoleamine-2,3-Dioxygenase production in hepatocellular carcinoma. Hepatol (Baltimore Md) (2014) 59(2):567–79. doi: 10.1002/hep.26694
76. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. Pd-1hi identifies a novel regulatory b-cell population in human hepatoma that promotes disease progression. Cancer Discovery (2016) 6(5):546–59. doi: 10.1158/2159-8290.Cd-15-1408
77. Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology (2016) 150(7):1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
78. Ebrahimkhani MR, Mohar I, Crispe IN. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatol (Baltimore Md) (2011) 54(4):1379–87. doi: 10.1002/hep.24508
79. Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J immunotherapy Cancer (2019) 7(1):267. doi: 10.1186/s40425-019-0749-z
80. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human kupffer cells secrete il-10 in response to lipopolysaccharide (Lps) challenge. J Hepatol (1995) 22(2):226–9. doi: 10.1016/0168-8278(95)80433-1
81. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatol (Baltimore Md) (2015) 62(1):279–91. doi: 10.1002/hep.27793
82. Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J Hepatol (2020) 72(1):167–82. doi: 10.1016/j.jhep.2019.08.014
83. Yan ML, Wang YD, Tian YF, Lai ZD, Yan LN. Inhibition of allogeneic T-cell response by kupffer cells expressing indoleamine 2,3-dioxygenase. World J Gastroenterol (2010) 16(5):636–40. doi: 10.3748/wjg.v16.i5.636
84. Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of Cd8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/Programmed death-1 interactions. Cancer Res (2009) 69(20):8067–75. doi: 10.1158/0008-5472.Can-09-0901
85. Höchst B, Schildberg FA, Sauerborn P, Gäbel YA, Gevensleben H, Goltz D, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a Cd44-dependent fashion. J Hepatol (2013) 59(3):528–35. doi: 10.1016/j.jhep.2013.04.033
86. Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol (Baltimore Md 1950) (2013) 190(5):2009–16. doi: 10.4049/jimmunol.1201937
87. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
88. Chen L, Flies DB. Molecular mechanisms of T cell Co-stimulation and Co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
89. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
90. Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: Basics and ongoing clinical trials. Oncology (2017) 92 Suppl 1:50–62. doi: 10.1159/000451016
91. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted Cd8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi: 10.1038/nature04444
92. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. Pd-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
93. Nguyen LT, Ohashi PS. Clinical blockade of Pd1 and Lag3–potential mechanisms of action. Nat Rev Immunol (2015) 15(1):45–56. doi: 10.1038/nri3790
94. Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, et al. Pd-1 and pd-L1 upregulation promotes Cd8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer (2011) 128(4):887–96. doi: 10.1002/ijc.25397
95. Kassel R, Cruise MW, Iezzoni JC, Taylor NA, Pruett TL, Hahn YS. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatol (Baltimore Md) (2009) 50(5):1625–37. doi: 10.1002/hep.23173
96. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of Cd8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27(4):670–84. doi: 10.1016/j.immuni.2007.09.006
97. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology (2017) 153(4):1107–19.e10. doi: 10.1053/j.gastro.2017.06.017
98. Rong YH, Wan ZH, Song H, Li YL, Zhu B, Zang H, et al. Tim-3 expression on peripheral monocytes and Cd3+Cd16/Cd56+Natural killer-like T cells in patients with chronic hepatitis b. Tissue Antigens (2014) 83(2):76–81. doi: 10.1111/tan.12278
99. Mengshol JA, Golden-Mason L, Arikawa T, Smith M, Niki T, McWilliams R, et al. A crucial role for kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis c infection. PLos One (2010) 5(3):e9504. doi: 10.1371/journal.pone.0009504
100. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of Tim-3+ Foxp3+ regulatory T cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
101. Li Z, Li N, Li F, Zhou Z, Sang J, Chen Y, et al. Immune checkpoint proteins pd-1 and Tim-3 are both highly expressed in liver tissues and correlate with their gene polymorphisms in patients with hbv-related hepatocellular carcinoma. Medicine (2016) 95(52):e5749. doi: 10.1097/md.0000000000005749
102. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/Galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis b virus-associated hepatocellular carcinoma. Hepatol (Baltimore Md) (2012) 56(4):1342–51. doi: 10.1002/hep.25777
103. Ribas A. Tumor immunotherapy directed at pd-1. New Engl J Med (2012) 366(26):2517–9. doi: 10.1056/NEJMe1205943
104. Zou W, Wolchok JD, Chen L. Pd-L1 (B7-H1) and pd-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Trans Med (2016) 8(328):328rv4. doi: 10.1126/scitranslmed.aad7118
105. Abdel-Wahab N, Shah M, Lopez-Olivo MA, Suarez-Almazor ME. Use of immune checkpoint inhibitors in the treatment of patients with cancer and preexisting autoimmune disease: A systematic review. Ann Internal Med (2018) 168(2):121–30. doi: 10.7326/m17-2073
106. Yarchoan M, Xing D, Luan L, Xu H, Sharma RB, Popovic A, et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(23):7333–9. doi: 10.1158/1078-0432.Ccr-17-0950
107. Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Ségui B. The tnf paradox in cancer progression and immunotherapy. Front Immunol (2019) 10:1818. doi: 10.3389/fimmu.2019.01818
108. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-Deficient mice. Sci (New York NY) (1996) 274(5291):1379–83. doi: 10.1126/science.274.5291.1379
109. Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, et al. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis b. Int J Cancer (2009) 124(12):2766–70. doi: 10.1002/ijc.24281
110. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. Tgf-Β induces M2-like macrophage polarization Via snail-mediated suppression of a pro-inflammatory phenotype. Oncotarget (2016) 7(32):52294–306. doi: 10.18632/oncotarget.10561
111. Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA. A role for tgf-beta in the generation and expansion of Cd4+Cd25+ regulatory T cells from human peripheral blood. J Immunol (Baltimore Md 1950) (2001) 166(12):7282–9. doi: 10.4049/jimmunol.166.12.7282
112. Okumoto K, Hattori E, Tamura K, Kiso S, Watanabe H, Saito K, et al. Possible contribution of circulating transforming growth factor-Beta1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver Int Off J Int Assoc Study Liver (2004) 24(1):21–8. doi: 10.1111/j.1478-3231.2004.00882.x
113. Ahmadzadeh M, Rosenberg SA. Tgf-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory Cd8 T cells. J Immunol (Baltimore Md 1950) (2005) 174(9):5215–23. doi: 10.4049/jimmunol.174.9.5215
114. Zhong M, Zhong C, Cui W, Wang G, Zheng G, Li L, et al. Induction of tolerogenic dendritic cells by activated tgf-Β/Akt/Smad2 signaling in rig-I-Deficient stemness-high human liver cancer cells. BMC Cancer (2019) 19(1):439. doi: 10.1186/s12885-019-5670-9
115. Eisenstein EM, Williams CB. The T(Reg)/Th17 cell balance: A new paradigm for autoimmunity. Pediatr Res (2009) 65(5 Pt 2):26r–31r. doi: 10.1203/PDR.0b013e31819e76c7
116. Lin TH, Shao YY, Chan SY, Huang CY, Hsu CH, Cheng AL. High serum transforming growth factor-Β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res an Off J Am Assoc Cancer Res (2015) 21(16):3678–84. doi: 10.1158/1078-0432.Ccr-14-1954
117. Feun LG, Li YY, Wu C, Wangpaichitr M, Jones PD, Richman SP, et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer (2019) 125(20):3603–14. doi: 10.1002/cncr.32339
118. Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, et al. Interferon-dependent il-10 production by tregs limits tumor Th17 inflammation. J Clin Invest (2013) 123(11):4859–74. doi: 10.1172/jci65180
119. Yi Y, He HW, Wang JX, Cai XY, Li YW, Zhou J, et al. The functional impairment of hcc-infiltrating Γδ T cells, partially mediated by regulatory T cells in a tgfΒ- and il-10-Dependent manner. J Hepatol (2013) 58(5):977–83. doi: 10.1016/j.jhep.2012.12.015
120. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through pd-L1. J Exp Med (2009) 206(6):1327–37. doi: 10.1084/jem.20082173
121. Shi Y, Song Q, Hu D, Zhuang X, Yu S. Tumor-infiltrating lymphocyte activity is enhanced in tumors with low il-10 production in hbv-induced hepatocellular carcinoma. Biochem Biophys Res Commun (2015) 461(1):109–14. doi: 10.1016/j.bbrc.2015.03.177
122. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in Cd14+Hla-Dr -/Low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer immunology immunotherapy CII (2013) 62(8):1421–30. doi: 10.1007/s00262-013-1447-1
123. Chau GY, Wu CW, Lui WY, Chang TJ, Kao HL, Wu LH, et al. Serum interleukin-10 but not interleukin-6 is related to clinical outcome in patients with resectable hepatocellular carcinoma. Ann Surg (2000) 231(4):552–8. doi: 10.1097/00000658-200004000-00015
124. Apte RS, Chen DS, Ferrara N. Vegf in signaling and disease: Beyond discovery and development. Cell (2019) 176(6):1248–64. doi: 10.1016/j.cell.2019.01.021
125. Courau T, Nehar-Belaid D, Florez L, Levacher B, Vazquez T, Brimaud F, et al. Tgf-Β and vegf cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight (2016) 1(9):e85974. doi: 10.1172/jci.insight.85974
126. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of vegfa and molecular classification of hepatocellular carcinoma. Cancer Res (2008) 68(16):6779–88. doi: 10.1158/0008-5472.Can-08-0742
127. Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol (2021) 18(8):527–40. doi: 10.1038/s41571-021-00496-y
128. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. Β-catenin activation promotes immune escape and resistance to anti-Pd-1 therapy in hepatocellular carcinoma. Cancer Discovery (2019) 9(8):1124–41. doi: 10.1158/2159-8290.Cd-19-0074
129. Cadoux M, Caruso S, Pham S, Gougelet A, Pophillat C, Riou R, et al. Expression of Nkg2d ligands is downregulated by Β-catenin signalling and associates with hcc aggressiveness. J Hepatol (2021) 74(6):1386–97. doi: 10.1016/j.jhep.2021.01.017
130. Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med (2019) 25(2):301–11. doi: 10.1038/s41591-018-0321-2
131. Maini MK, Pallett LJ. Defective T-cell immunity in hepatitis b virus infection: Why therapeutic vaccination needs a helping hand. Lancet Gastroenterol Hepatol (2018) 3(3):192–202. doi: 10.1016/s2468-1253(18)30007-4
132. Gorjifard S, Goldszmid RS. Microbiota-myeloid cell crosstalk beyond the gut. J leukocyte Biol (2016) 100(5):865–79. doi: 10.1189/jlb.3RI0516-222R
133. Pabst O. Correlation, consequence, and functionality in microbiome-immune interplay. Immunol Rev (2017) 279(1):4–7. doi: 10.1111/imr.12584
134. Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J immunotherapy Cancer (2019) 7(1):108. doi: 10.1186/s40425-019-0574-4
135. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J (2017) 474(11):1823–36. doi: 10.1042/bcj20160510
136. Di Lorenzo A, Bolli E, Tarone L, Cavallo F, Conti L. Toll-like receptor 2 at the crossroad between cancer cells, the immune system, and the microbiota. Int J Mol Sci (2020) 21(24):9418. doi: 10.3390/ijms21249418
137. Sun L, Dai JJ, Hu WF, Wang J. Expression of toll-like receptors in hepatic cirrhosis and hepatocellular carcinoma. Genet Mol Res GMR (2016) 15(2). doi: 10.4238/gmr.15027419
138. Yang J, Zhang JX, Wang H, Wang GL, Hu QG, Zheng QC. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression Via treg requires Tlr4 signaling. World J Gastroenterol (2012) 18(23):2938–47. doi: 10.3748/wjg.v18.i23.2938
139. Eiró N, Altadill A, Juárez LM, Rodríguez M, González LO, Atienza S, et al. Toll-like receptors 3, 4 and 9 in hepatocellular carcinoma: Relationship with clinicopathological characteristics and prognosis. Hepatol Res Off J Japan Soc Hepatol (2014) 44(7):769–78. doi: 10.1111/hepr.12180
140. Wang L, Zhu R, Huang Z, Li H, Zhu H. Lipopolysaccharide-induced toll-like receptor 4 signaling in cancer cells promotes cell survival and proliferation in hepatocellular carcinoma. Digestive Dis Sci (2013) 58(8):2223–36. doi: 10.1007/s10620-013-2745-3
141. Zhang Q, Ma C, Duan Y, Heinrich B, Rosato U, Diggs LP, et al. Gut microbiome directs hepatocytes to recruit mdscs and promote cholangiocarcinoma. Cancer Discovery (2021) 11(5):1248–67. doi: 10.1158/2159-8290.Cd-20-0304
142. Chen T, Li Q, Wu J, Wu Y, Peng W, Li H, et al. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours Via a Tlr4-dependent mechanism. Cancer immunology immunotherapy CII (2018) 67(10):1635–46. doi: 10.1007/s00262-018-2233-x
143. Li H, Li Y, Liu D, Liu J. Lps promotes epithelial-mesenchymal transition and activation of Tlr4/Jnk signaling. Tumour Biol J Int Soc Oncodevelopmental Biol Med (2014) 35(10):10429–35. doi: 10.1007/s13277-014-2347-5
144. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/jci39104
145. Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery JP, et al. New insights into the role of emt in tumor immune escape. Mol Oncol (2017) 11(7):824–46. doi: 10.1002/1878-0261.12093
146. Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, et al. Targeting the interplay between epithelial-to-Mesenchymal-Transition and the immune system for effective immunotherapy. Cancers (2019) 11(5):714. doi: 10.3390/cancers11050714
147. Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, et al. Hypoxia-induced epithelial-to-Mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res (2016) 76(4):818–30. doi: 10.1158/0008-5472.Can-15-0977
148. Zhou S, Du R, Wang Z, Shen W, Gao R, Jiang S, et al. Tlr4 increases the stemness and is highly expressed in relapsed human hepatocellular carcinoma. Cancer Med (2019) 8(5):2325–37. doi: 10.1002/cam4.2070
149. Dai X, Guo Y, Hu Y, Bao X, Zhu X, Fu Q, et al. Immunotherapy for targeting cancer stem cells in hepatocellular carcinoma. Theranostics (2021) 11(7):3489–501. doi: 10.7150/thno.54648
150. Liu WT, Jing YY, Yu GF, Han ZP, Yu DD, Fan QM, et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett (2015) 358(2):136–43. doi: 10.1016/j.canlet.2014.12.019
151. Wei X, Liu H, Li X, Liu X. Over-expression of mir-122 promotes apoptosis of hepatocellular carcinoma Via targeting Tlr4. Ann Hepatol (2019) 18(6):869–78. doi: 10.1016/j.aohep.2019.07.005
152. Zhang C, Wang N, Tan HY, Guo W, Chen F, Zhong Z, et al. Direct inhibition of the Tlr4/Myd88 pathway by geniposide suppresses hif-1Α-Independent vegf expression and angiogenesis in hepatocellular carcinoma. Br J Pharmacol (2020) 177(14):3240–57. doi: 10.1111/bph.15046
153. Liwinski T, Zheng D, Elinav E. The microbiome and cytosolic innate immune receptors. Immunol Rev (2020) 297(1):207–24. doi: 10.1111/imr.12901
154. Zhang HL, Yu LX, Yang W, Tang L, Lin Y, Wu H, et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J Hepatol (2012) 57(4):803–12. doi: 10.1016/j.jhep.2012.06.011
155. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut (2021) 70(4):761–74. doi: 10.1136/gutjnl-2019-319664
156. Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatol (Baltimore Md) (2010) 52(4):1322–33. doi: 10.1002/hep.23845
157. Almeida J, Galhenage S, Yu J, Kurtovic J, Riordan SM. Gut flora and bacterial translocation in chronic liver disease. World J Gastroenterol (2006) 12(10):1493–502. doi: 10.3748/wjg.v12.i10.1493
158. Telesford KM, Yan W, Ochoa-Reparaz J, Pant A, Kircher C, Christy MA, et al. A commensal symbiotic factor derived from bacteroides fragilis promotes human Cd39(+)Foxp3(+) T cells and treg function. Gut Microbes (2015) 6(4):234–42. doi: 10.1080/19490976.2015.1056973
159. Yu LX, Schwabe RF. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol (2017) 14(9):527–39. doi: 10.1038/nrgastro.2017.72
160. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol (2018) 15(7):397–411. doi: 10.1038/s41575-018-0011-z
161. Thanabalasuriar A, Koutsouris A, Hecht G, Gruenheid S. The bacterial virulence factor nlea's involvement in intestinal tight junction disruption during enteropathogenic e. coli infection is independent of its putative pdz binding domain. Gut Microbes (2010) 1(2):114–8. doi: 10.4161/gmic.1.2.11685
162. Conlon MA, Bird AR. The impact of diet and lifestyle on gut microbiota and human health. Nutrients (2014) 7(1):17–44. doi: 10.3390/nu7010017
163. Capaldo CT, Powell DN, Kalman D. Layered defense: How mucus and tight junctions seal the intestinal barrier. J Mol Med (Berlin Germany) (2017) 95(9):927–34. doi: 10.1007/s00109-017-1557-x
164. Arrazuria R, Pérez V, Molina E, Juste RA, Khafipour E, Elguezabal N. Diet induced changes in the microbiota and cell composition of rabbit gut associated lymphoid tissue (Galt). Sci Rep (2018) 8(1):14103. doi: 10.1038/s41598-018-32484-1
165. Balkwill F, Mantovani A. Inflammation and cancer: Back to virchow? Lancet (London England) (2001) 357(9255):539–45. doi: 10.1016/s0140-6736(00)04046-0
166. Grote VA, Kaaks R, Nieters A, Tjønneland A, Halkjær J, Overvad K, et al. Inflammation marker and risk of pancreatic cancer: A nested case-control study within the epic cohort. Br J Cancer (2012) 106(11):1866–74. doi: 10.1038/bjc.2012.172
167. Wang L, Jiang Y, Zhang Y, Wang Y, Huang S, Wang Z, et al. Association analysis of il-17a and il-17f polymorphisms in Chinese han women with breast cancer. PLos One (2012) 7(3):e34400. doi: 10.1371/journal.pone.0034400
168. Ohkusa T, Yoshida T, Sato N, Watanabe S, Tajiri H, Okayasu I. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: A possible pathogenic mechanism of ulcerative colitis. J Med Microbiol (2009) 58(Pt 5):535–45. doi: 10.1099/jmm.0.005801-0
169. Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell signalling (2013) 25(2):403–16. doi: 10.1016/j.cellsig.2012.10.014
170. Toor D, Wsson MK, Kumar P, Karthikeyan G, Kaushik NK, Goel C, et al. Dysbiosis disrupts gut immune homeostasis and promotes gastric diseases. Int J Mol Sci (2019) 20(10):2432. doi: 10.3390/ijms20102432
171. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
172. Karki R, Kanneganti TD. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer (2019) 19(4):197–214. doi: 10.1038/s41568-019-0123-y
173. Shah A, Shanahan E, Macdonald GA, Fletcher L, Ghasemi P, Morrison M, et al. Systematic review and meta-analysis: Prevalence of small intestinal bacterial overgrowth in chronic liver disease. Semin liver Dis (2017) 37(4):388–400. doi: 10.1055/s-0037-1608832
174. Tarao K, So K, Moroi T, Ikeuchi T, Suyama T. Detection of endotoxin in plasma and ascitic fluid of patients with cirrhosis: Its clinical significance. Gastroenterology (1977) 73(3):539–42.
175. Triger DR, Boyer TD, Levin J. Portal and systemic bacteraemia and endotoxaemia in liver disease. Gut (1978) 19(10):935–9. doi: 10.1136/gut.19.10.935
176. Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol (1987) 4(1):8–14. doi: 10.1016/s0168-8278(87)80003-x
177. Sipe LM, Chaib M, Pingili AK, Pierre JF, Makowski L. Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol Rev (2020) 295(1):220–39. doi: 10.1111/imr.12856
178. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via nkt cells. Sci (New York NY) (2018) 360(6391):eaan5931. doi: 10.1126/science.aan5931
179. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature (2013) 499(7456):97–101. doi: 10.1038/nature12347
180. Rojo D, Hevia A, Bargiela R, López P, Cuervo A, González S, et al. Ranking the impact of human health disorders on gut metabolism: Systemic lupus erythematosus and obesity as study cases. Sci Rep (2015) 5:8310. doi: 10.1038/srep08310
181. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor tigit protects tumors from immune cell attack. Immunity (2015) 42(2):344–55. doi: 10.1016/j.immuni.2015.01.010
182. Gur C, Maalouf N, Shhadeh A, Berhani O, Singer BB, Bachrach G, et al. Fusobacterium nucleatum supresses anti-tumor immunity by activating Ceacam1. Oncoimmunology (2019) 8(6):e1581531. doi: 10.1080/2162402x.2019.1581531
183. Chauvin JM, Zarour HM. Tigit in cancer immunotherapy. J immunotherapy Cancer (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
184. Gur C, Maalouf N, Gerhard M, Singer BB, Emgård J, Temper V, et al. The helicobacter pylori hopq outermembrane protein inhibits immune cell activities. Oncoimmunology (2019) 8(4):e1553487. doi: 10.1080/2162402x.2018.1553487
185. Feng R, Zhao H, Xu J, Shen C. Cd47: The next checkpoint target for cancer immunotherapy. Crit Rev oncology/hematology (2020) 152:103014. doi: 10.1016/j.critrevonc.2020.103014
186. Shi Y, Zheng W, Yang K, Harris KG, Ni K, Xue L, et al. Intratumoral accumulation of gut microbiota facilitates Cd47-based immunotherapy via sting signaling. J Exp Med (2020) 217(5):e20192282. doi: 10.1084/jem.20192282
187. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol (2022) 19(3):151–72. doi: 10.1038/s41571-021-00573-2
188. Krämer B, Körner C, Kebschull M, Glässner A, Eisenhardt M, Nischalke HD, et al. Natural killer P46high expression defines a natural killer cell subset that is potentially involved in control of hepatitis c virus replication and modulation of liver fibrosis. Hepatol (Baltimore Md) (2012) 56(4):1201–13. doi: 10.1002/hep.25804
189. Tian Z, Chen Y, Gao B. Natural killer cells in liver disease. Hepatol (Baltimore Md) (2013) 57(4):1654–62. doi: 10.1002/hep.26115
190. Zhang J, Shi Z, Xu X, Yu Z, Mi J. The influence of microenvironment on tumor immunotherapy. FEBS J (2019) 286(21):4160–75. doi: 10.1111/febs.15028
191. Cham CM, Gajewski TF. Glucose availability regulates ifn-gamma production and P70s6 kinase activation in Cd8+ effector T cells. J Immunol (Baltimore Md 1950) (2005) 174(8):4670–7. doi: 10.4049/jimmunol.174.8.4670
192. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in Cd8+ T cells. Eur J Immunol (2008) 38(9):2438–50. doi: 10.1002/eji.200838289
193. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021) 591(7851):645–51. doi: 10.1038/s41586-020-03045-2
194. Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ros-mediated apoptosis. Cell Death Dis (2017) 8(5):e2779. doi: 10.1038/cddis.2017.192
195. Md-B N, Duncan-Moretti J H, Saldanha-Gama R, Paula-Neto HA, GD G, et al. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim Biophys Acta Mol Cell Res (2020) 1867(2):118604. doi: 10.1016/j.bbamcr.2019.118604
196. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab (2021) 33(10):2040–58.e10. doi: 10.1016/j.cmet.2021.09.002
197. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
198. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. Cox2/Mpges1/Pge2 pathway regulates pd-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci United States America (2017) 114(5):1117–22. doi: 10.1073/pnas.1612920114
199. Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv (2021) 7(46):eabi8602. doi: 10.1126/sciadv.abi8602
200. Zhao JL, Ye YC, Gao CC, Wang L, Ren KX, Jiang R, et al. Notch-mediated lactate metabolism regulates mdsc development through the Hes1/Mct2/C-jun axis. Cell Rep (2022) 38(10):110451. doi: 10.1016/j.celrep.2022.110451
201. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
202. Amobi-McCloud A, Muthuswamy R, Battaglia S, Yu H, Liu T, Wang J, et al. Ido1 expression in ovarian cancer induces pd-1 in T cells Via aryl hydrocarbon receptor activation. Front Immunol (2021) 12:678999. doi: 10.3389/fimmu.2021.678999
203. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of Cd8(+) T cells Via glutamine restriction. Cancer Sci (2018) 109(12):3737–50. doi: 10.1111/cas.13827
204. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter Asct2 facilitation of glutamine uptake and Mtorc1 kinase activation. Immunity (2014) 40(5):692–705. doi: 10.1016/j.immuni.2014.04.007
205. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent Α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signaling (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
206. Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep (2017) 20(7):1654–66. doi: 10.1016/j.celrep.2017.07.054
207. Dröge W, Männel D, Falk W, Lehmann V, Schmidt H, Nick S, et al. Suppression of cytotoxic T lymphocyte activation by l-ornithine. J Immunol (Baltimore Md 1950) (1985) 134(5):3379–83.
208. Maisonneuve C, Tsang DKL, Foerster EG, Robert LM, Mukherjee T, Prescott D, et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep (2021) 34(4):108677. doi: 10.1016/j.celrep.2020.108677
209. Hung MH, Lee JS, Ma C, Diggs LP, Heinrich S, Chang CW, et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun (2021) 12(1):1455. doi: 10.1038/s41467-021-21804-1
210. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature (2019) 567(7747):257–61. doi: 10.1038/s41586-019-0987-8
211. Schmidt NM, Wing PAC, Diniz MO, Pallett LJ, Swadling L, Harris JM, et al. Targeting human acyl-Coa:Cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat Commun (2021) 12(1):2814. doi: 10.1038/s41467-021-22967-7
212. Audrito V, Managò A, Gaudino F, Sorci L, Messana VG, Raffaelli N, et al. Nad-biosynthetic and consuming enzymes as central players of metabolic regulation of innate and adaptive immune responses in cancer. Front Immunol (2019) 10:1720. doi: 10.3389/fimmu.2019.01720
213. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
214. Munn DH, Mellor AL. Ido in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37(3):193–207. doi: 10.1016/j.it.2016.01.002
215. Brown ZJ, Yu SJ, Heinrich B, Ma C, Fu Q, Sandhu M, et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer immunology immunotherapy CII (2018) 67(8):1305–15. doi: 10.1007/s00262-018-2190-4
216. Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: The ultimate identity crisis. Front Immunol (2014) 5:276. doi: 10.3389/fimmu.2014.00276
217. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity (2017) 46(4):675–89. doi: 10.1016/j.immuni.2017.03.019
218. Cai XY, Ni XC, Yi Y, He HW, Wang JX, Fu YP, et al. Overexpression of Cd39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine (2016) 95(40):e4989. doi: 10.1097/md.0000000000004989
219. Shali S, Yu J, Zhang X, Wang X, Jin Y, Su M, et al. Ecto-5'-Nucleotidase (Cd73) is a potential target of hepatocellular carcinoma. J Cell Physiol (2019) 234(7):10248–59. doi: 10.1002/jcp.27694
220. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of Cd8(+) T cells to persistent antigen. Nat Immunol (2013) 14(11):1173–82. doi: 10.1038/ni.2714
221. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. Pd-L1 is a novel direct target of hif-1Α, and its blockade under hypoxia enhanced mdsc-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
222. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting edge: Hypoxia-inducible factor 1 negatively regulates Th1 function. J Immunol (Baltimore Md 1950) (2015) 195(4):1372–6. doi: 10.4049/jimmunol.1402552
223. Ping YF, Bian XW. Consice review: Contribution of cancer stem cells to neovascularization. Stem Cells (Dayton Ohio) (2011) 29(6):888–94. doi: 10.1002/stem.650
224. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLos Biol (2015) 13(7):e1002202. doi: 10.1371/journal.pbio.1002202
225. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
226. Dong XF, Liu TQ, Zhi XT, Zou J, Zhong JT, Li T, et al. Cox-2/Pge2 axis regulates Hif2Α activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(13):3204–16. doi: 10.1158/1078-0432.Ccr-17-2725
227. Vogel A, Cervantes A, Chau I, Daniele B, Llovet JM, Meyer T, et al. Hepatocellular carcinoma: Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol Off J Eur Soc Med Oncol (2018) 29(Suppl 4):iv238–iv55. doi: 10.1093/annonc/mdy308
228. Nivolumab approved for liver cancer. Cancer Discovery (2017) 7(11):Of3. doi: 10.1158/2159-8290.Cd-nb2017-138
229. Lang L. New organ allocation criteria and decreased mortality on liver transplant waiting list. Gastroenterology (2008) 134(2):379–80. doi: 10.1053/j.gastro.2007.12.038
230. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-pacific region with advanced hepatocellular carcinoma: A phase iii randomised, double-blind, placebo-controlled trial. Lancet Oncol (2009) 10(1):25–34. doi: 10.1016/s1470-2045(08)70285-7
231. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (Checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (London England) (2017) 389(10088):2492–502. doi: 10.1016/s0140-6736(17)31046-2
232. Edeline J, Yau T, Park J-W, Kudo M, Han K-H, Mathurin P, et al. Checkmate 459: Health-related quality of life (Hrqol) in a randomized, multicenter phase iii study of nivolumab (Nivo) versus sorafenib (Sor) as first-line (1l) treatment in patients (Pts) with advanced hepatocellular carcinoma (Ahcc). J Clin Oncol (2020) 38(4_suppl):483. doi: 10.1200/JCO.2020.38.4_suppl.483
233. Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (Keynote-224): A non-randomised, open-label phase 2 trial. Lancet Oncol (2018) 19(7):940–52. doi: 10.1016/s1470-2045(18)30351-6
234. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in keynote-240: A randomized, double-blind, phase iii trial. J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(3):193–202. doi: 10.1200/jco.19.01307
235. Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Nivolumab (Nivo) + ipilimumab (Ipi) combination therapy in patients (Pts) with advanced hepatocellular carcinoma (Ahcc): Results from checkmate 040. J Clin Oncol (2019) 37(15_suppl):4012. doi: 10.1200/JCO.2019.37.15_suppl.4012
236. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23(2):249–62. doi: 10.1016/j.ccr.2013.01.008
237. Beumer JH, Schellens JH, Beijnen JH. Hepatotoxicity and metabolism of trabectedin: A literature review. Pharmacol Res (2005) 51(5):391–8. doi: 10.1016/j.phrs.2004.12.001
238. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in Myd88-dependent il-6 production. Sci (New York NY) (2007) 317(5834):121–4. doi: 10.1126/science.1140485
239. Yang W, Lu Y, Xu Y, Xu L, Zheng W, Wu Y, et al. Estrogen represses hepatocellular carcinoma (Hcc) growth Via inhibiting alternative activation of tumor-associated macrophages (Tams). J Biol Chem (2012) 287(48):40140–9. doi: 10.1074/jbc.M112.348763
240. Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, et al. Blocking C5ar signaling promotes the anti-tumor efficacy of pd-1/Pd-L1 blockade. Oncoimmunology (2017) 6(10):e1349587. doi: 10.1080/2162402x.2017.1349587
241. Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut (2020) 69(2):365–79. doi: 10.1136/gutjnl-2018-317257
242. Wu CJ, Tsai YT, Lee IJ, Wu PY, Lu LS, Tsao WS, et al. Combination of radiation and interleukin 12 eradicates Large orthotopic hepatocellular carcinoma through immunomodulation of tumor microenvironment. Oncoimmunology (2018) 7(9):e1477459. doi: 10.1080/2162402x.2018.1477459
243. Li B, Zhang S, Huang N, Chen H, Wang P, Yang J, et al. Ccl9/Ccr1 induces Myeloid−Derived suppressor cell recruitment to the spleen in a murine H22 orthotopic hepatoma model. Oncol Rep (2019) 41(1):608–18. doi: 10.3892/or.2018.6809
244. Xu M, Zhao Z, Song J, Lan X, Lu S, Chen M, et al. Interactions between interleukin-6 and myeloid-derived suppressor cells drive the chemoresistant phenotype of hepatocellular cancer. Exp Cell Res (2017) 351(2):142–9. doi: 10.1016/j.yexcr.2017.01.008
245. Sun H, Yang W, Tian Y, Zeng X, Zhou J, Mok MTS, et al. An inflammatory-ccrk circuitry drives Mtorc1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun (2018) 9(1):5214. doi: 10.1038/s41467-018-07402-8
246. Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, et al. Pd-L1(+)Mdscs are increased in hcc patients and induced by soluble factor in the tumor microenvironment. Sci Rep (2016) 6:39296. doi: 10.1038/srep39296
247. Chen ML, Yan BS, Lu WC, Chen MH, Yu SL, Yang PC, et al. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int J Cancer (2014) 134(2):319–31. doi: 10.1002/ijc.28362
248. Li G, Liu D, Cooper TK, Kimchi ET, Qi X, Avella DM, et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol (2017) 66(1):75–85. doi: 10.1016/j.jhep.2016.07.044
249. Wei R, Hu Y, Dong F, Xu X, Hu A, Gao G. Hepatoma cell-derived leptin downregulates the immunosuppressive function of regulatory T-cells to enhance the anti-tumor activity of Cd8+ T-cells. Immunol Cell Biol (2016) 94(4):388–99. doi: 10.1038/icb.2015.110
250. Li Z, Li N, Li F, Zhou Z, Sang J, Jin Z, et al. Genetic polymorphisms of immune checkpoint proteins pd-1 and Tim-3 are associated with survival of patients with hepatitis b virus-related hepatocellular carcinoma. Oncotarget (2016) 7(18):26168–80. doi: 10.18632/oncotarget.8435
251. Pedroza-Gonzalez A, Zhou G, Singh SP, Boor PP, Pan Q, Grunhagen D, et al. Gitr engagement in combination with ctla-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells ex vivo. Oncoimmunology (2015) 4(12):e1051297. doi: 10.1080/2162402x.2015.1051297
252. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T cells, especially icos(+) Foxp3(+) regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep (2016) 6:35056. doi: 10.1038/srep35056
253. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-Ccr4 mab selectively depletes effector-type Foxp3+Cd4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci United States America (2013) 110(44):17945–50. doi: 10.1073/pnas.1316796110
254. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal Via Stat3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
255. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages Via Ccl2/Ccr2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut (2017) 66(1):157–67. doi: 10.1136/gutjnl-2015-310514
256. Li H, Li H, Li XP, Zou H, Liu L, Liu W, et al. C−C chemokine receptor type 2 promotes Epithelial−to−Mesenchymal transition by upregulating matrix Metalloproteinase−2 in human liver cancer. Oncol Rep (2018) 40(5):2734–41. doi: 10.3892/or.2018.6660
257. Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, et al. Colony-stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor-associated macrophages in hepatocellular carcinoma. Mol Cancer Ther (2017) 16(8):1544–54. doi: 10.1158/1535-7163.Mct-16-0866
258. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res (2005) 65(6):2457–64. doi: 10.1158/0008-5472.Can-04-3232
259. Dong P, Ma L, Liu L, Zhao G, Zhang S, Dong L, et al. Cd86⁺/Cd206⁺, diametrically polarized tumor-associated macrophages, predict hepatocellular carcinoma patient prognosis. Int J Mol Sci (2016) 17(3):320. doi: 10.3390/ijms17030320
260. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages Via Wnt/Β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis (2018) 9(8):793. doi: 10.1038/s41419-018-0818-0
261. Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA, et al. Blocking the Ccl2-Ccr2 axis using Ccl2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther (2017) 16(2):312–22. doi: 10.1158/1535-7163.Mct-16-0124
262. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (Ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug design Dev Ther (2015) 9:4479–99. doi: 10.2147/dddt.S86621
263. Faivre SJ, Santoro A, Kelley RK, Merle P, Gane E, Douillard J-Y, et al. A phase 2 study of a novel transforming growth factor-beta (Tgf-Β1) receptor I kinase inhibitor, Ly2157299 monohydrate (Ly), in patients with advanced hepatocellular carcinoma (Hcc). J Clin Oncol (2014) 32(3_suppl):LBA173–LBA. doi: 10.1200/jco.2014.32.3_suppl.lba173
264. Hsu C, Chang Y-F, Yen C-J, Lu L-C, Zhu X, Xu Y, et al. Safety and efficacy of combination of Gt90001, an anti-activin receptor-like kinase-1 (Alk-1) antibody, and nivolumab in patients with metastatic hepatocellular carcinoma (Hcc). J Clin Oncol (2021) 39(3_suppl):326. doi: 10.1200/JCO.2021.39.3_suppl.326
265. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol (2018) 15(5):310–24. doi: 10.1038/nrclinonc.2018.9
266. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.Ccr-18-1543
267. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. Vegf-a modulates expression of inhibitory checkpoints on Cd8+ T cells in tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
268. Missiaen R, Mazzone M, Bergers G. The reciprocal function and regulation of tumor vessels and immune cells offers new therapeutic opportunities in cancer. Semin Cancer Biol (2018) 52(Pt 2):107–16. doi: 10.1016/j.semcancer.2018.06.002
269. Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, et al. Phase ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(26):2960–70. doi: 10.1200/jco.20.00808
270. Li J, Sung CY, Lee N, Ni Y, Pihlajamäki J, Panagiotou G, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc Natl Acad Sci United States America (2016) 113(9):E1306–15. doi: 10.1073/pnas.1518189113
271. de Moreno de LeBlanc A, Matar C, Perdigón G. The application of probiotics in cancer. Br J Nutr (2007) 98 Suppl 1:S105–10. doi: 10.1017/s0007114507839602
272. El-Nezami HS, Polychronaki NN, Ma J, Zhu H, Ling W, Salminen EK, et al. Probiotic supplementation reduces a biomarker for increased risk of liver cancer in young men from southern China. Am J Clin Nutr (2006) 83(5):1199–203. doi: 10.1093/ajcn/83.5.1199
273. Kumar M, Verma V, Nagpal R, Kumar A, Gautam SK, Behare PV, et al. Effect of probiotic fermented milk and chlorophyllin on gene expressions and genotoxicity during Afb₁-induced hepatocellular carcinoma. Gene (2011) 490(1-2):54–9. doi: 10.1016/j.gene.2011.09.003
274. Fotiadis CI, Stoidis CN, Spyropoulos BG, Zografos ED. Role of probiotics, prebiotics and synbiotics in chemoprevention for colorectal cancer. World J Gastroenterol (2008) 14(42):6453–7. doi: 10.3748/wjg.14.6453
275. Zhou A, Tang L, Zeng S, Lei Y, Yang S, Tang B. Gut microbiota: A new piece in understanding hepatocarcinogenesis. Cancer Lett (2020) 474:15–22. doi: 10.1016/j.canlet.2020.01.002
276. Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC, et al. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer (2012) 107(8):1337–44. doi: 10.1038/bjc.2012.409
277. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and Tlr4. Cancer Cell (2012) 21(4):504–16. doi: 10.1016/j.ccr.2012.02.007
278. Singh V, Yeoh BS, Abokor AA, Golonka RM, Tian Y, Patterson AD, et al. Vancomycin prevents fermentable fiber-induced liver cancer in mice with dysbiotic gut microbiota. Gut Microbes (2020) 11(4):1077–91. doi: 10.1080/19490976.2020.1743492
279. Cammarota G, Ianiro G, Tilg H, Rajilić-Stojanović M, Kump P, Satokari R, et al. European Consensus conference on faecal microbiota transplantation in clinical practice. Gut (2017) 66(4):569–80. doi: 10.1136/gutjnl-2016-313017
280. Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol (2016) 13(9):508–16. doi: 10.1038/nrgastro.2016.98
281. Ferrere G, Wrzosek L, Cailleux F, Turpin W, Puchois V, Spatz M, et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J Hepatol (2017) 66(4):806–15. doi: 10.1016/j.jhep.2016.11.008
282. Bajaj JS, Kakiyama G, Savidge T, Takei H, Kassam ZA, Fagan A, et al. Antibiotic-associated disruption of microbiota composition and function in cirrhosis is restored by fecal transplant. Hepatol (Baltimore Md) (2018) 68(4):1549–58. doi: 10.1002/hep.30037
283. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Sci (New York NY) (2013) 342(6161):971–6. doi: 10.1126/science.1240537
284. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-Pd-L1 efficacy. Sci (New York NY) (2015) 350(6264):1084–9. doi: 10.1126/science.aac4255
285. Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, et al. Gut microbiome affects the response to anti-Pd-1 immunotherapy in patients with hepatocellular carcinoma. J immunotherapy Cancer (2019) 7(1):193. doi: 10.1186/s40425-019-0650-9
286. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of pd-1-Based immunotherapy against epithelial tumors. Sci (New York NY) (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
287. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-Pd-1 immunotherapy in melanoma patients. Sci (New York NY) (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
288. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-Pd-1 efficacy in metastatic melanoma patients. Sci (New York NY) (2018) 359(6371):104–8. doi: 10.1126/science.aao3290
289. McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol (2019) 20(2):e77–91. doi: 10.1016/s1470-2045(18)30952-5
290. Hirschberger S, Strauß G, Effinger D, Marstaller X, Ferstl A, Müller MB, et al. Very-Low-Carbohydrate diet enhances human T-cell immunity through immunometabolic reprogramming. EMBO Mol Med (2021) 13(8):e14323. doi: 10.15252/emmm.202114323
291. Weber DD, Aminazdeh-Gohari S, Kofler B. Ketogenic diet in cancer therapy. Aging (2018) 10(2):164–5. doi: 10.18632/aging.101382
292. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
293. Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature (2021) 598(7882):662–6. doi: 10.1038/s41586-021-04003-2
294. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer Slc43a2 alters T cell methionine metabolism and histone methylation. Nature (2020) 585(7824):277–82. doi: 10.1038/s41586-020-2682-1
295. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting ctla-4. J Exp Med (2013) 210(7):1389–402. doi: 10.1084/jem.20130066
296. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of ctla-4, pd-1/Pd-L1, or ido blockade involves restored il-2 production and proliferation of Cd8(+) T cells directly within the tumor microenvironment. J immunotherapy Cancer (2014) 2:3. doi: 10.1186/2051-1426-2-3
297. Manukian G, Kivolowitz C, DeAngelis T, Shastri AA, Savage JE, Camphausen K, et al. Caloric restriction impairs regulatory T cells within the tumor microenvironment after radiation and primes effector T cells. Int J Radiat oncology biology Phys (2021) 110(5):1341–9. doi: 10.1016/j.ijrobp.2021.02.029
298. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
Keywords: hepatocellular carcinoma, immune evasion, tumor immune microenvironment, metabolism, gut microbiota, immunotherapy
Citation: Chen C, Wang Z, Ding Y and Qin Y (2023) Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma. Front. Immunol. 14:1133308. doi: 10.3389/fimmu.2023.1133308
Received: 28 December 2022; Accepted: 02 February 2023;
Published: 10 February 2023.
Edited by:
Hakim Echchannaoui, Johannes Gutenberg University Mainz, GermanyReviewed by:
Longyun Ye, Fudan University, ChinaCopyright © 2023 Chen, Wang, Ding and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanru Qin, eWFucnVxaW5AMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.