
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 31 March 2023
Sec. NK and Innate Lymphoid Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1130933
This article is part of the Research TopicHeterogeneity of ILC2sView all 10 articles
 Patrycja M. Topczewska1
Patrycja M. Topczewska1 Zoe A. Rompe1
Zoe A. Rompe1 Manuel O. Jakob1Anton Stamm1
Manuel O. Jakob1Anton Stamm1 Pierre S. Leclère1
Pierre S. Leclère1 Alexandra Preußer1
Alexandra Preußer1 Claudia U. Duerr1
Claudia U. Duerr1 Linda Marie Laura Thole2
Linda Marie Laura Thole2 Katja Kotsch2David Artis3
Katja Kotsch2David Artis3 Christoph S. N. Klose1*
Christoph S. N. Klose1*The initiation of type 2 immune responses at mucosal barriers is regulated by rapidly secreted cytokines called alarmins. The alarmins IL-33, IL-25 and TSLP are mainly secreted by stromal and epithelial cells in tissues and were linked to chronic inflammatory diseases, such as allergic lung inflammation, or to resistance against worm infections. Receptors for alarmins are expressed by a variety of immune cells, including group 2 innate lymphoid cells (ILC2s), an early source of the type 2 cytokines, such as IL-5 and IL-13, which have been linked to atopic diseases and anti-worm immunity as well. However, the precise contribution of the IL-33 receptor signals for ILC2 activation still needs to be completed due to limitations in targeting genes in ILC2. Using the newly established Nmur1iCre-eGFP mouse model, we obtained specific conditional genetic ablation of the IL-33 receptor subunit ST2 in ILC2s. ST2-deficient ILC2s were unresponsive to IL-33 but not to stimulation with the alarmin IL-25. As a result of defective ST2 signals, ILC2s produced limited amounts of IL-5 and IL-13 and failed to support eosinophil homeostasis. Further, ST2-deficient ILC2s were unable to expand and promote the recruitment of eosinophils during allergic lung inflammation provoked by papain administration. During infection with Nippostrongylus brasiliensis, ILC2-intrinsic ST2 signals were required to mount an effective type 2 immune response against the parasite leading to higher susceptibility against worm infection in conditional knockout mice. Therefore, this study argues for a non-redundant role of cell-intrinsic ST2 signals triggering proper activation of ILC2 for initiation of type 2 immunity.
Barrier surfaces allow for vital exchange with the environment but are also exposed to a broad array of stressors and are exploited by pathogens for invasion. In case the physical, chemical and biological measures to protect the epithelium from invasion do not prevail, the penetration of the pathogen triggers the secretion of danger signals, such as Interleukin (IL)-33, IL-25 and thymic stromal lymphopoietin (TSLP). Such ‘alarming’ signals activate tissue-resident immune cells and initiate the combat against the pathogen (1). Alarmins are produced to a large degree by specialized epithelial, endothelial or stromal cells in tissue. Recent research has exposed stromal cells as the predominant source of IL-33 in tissues, but dendritic cells, mast cells, endothelial and epithelial cells have been described as IL-33 producers as well (2–6). IL-33 has a chromatin binding motif and nuclear localization sequence, which targets the cytokine in the nucleus. Cell death and cellular stress appear to trigger the release of IL-33 from the nucleus, but the entire process is poorly understood (7). Further processing of full-length IL-33 by various proteases, including allergic proteases, significantly increases its biological activity (8).
Expression of the IL-33 receptor was reported from many immune cells, including innate lymphoid cells type 2 (ILC2s), CD4+ and CD8+ T cells, basophils, mast cells, eosinophils and macrophages (3, 7, 9–13). The IL-33 receptor is a member of the IL-1 receptor family, which in addition includes IL-1, IL-18 and IL-36 receptors. The binding of IL-33 to the ST2 chain (Il1rl1) enables the association with the IL-1rap chain, recruitment of the Myd88 adapter for signal transduction and triggering of a signaling cascade which ultimately leads to the activation of NF-κB as the main transcription factor promoting inflammation (7).
Tissue-resident immune cells include innate lymphoid cells, which are enriched at barrier surfaces and which have a similar functional diversity to T cells in terms of lineage-specifying transcription factors and effector functions. The transcription factors GATA-3, GfI-1, RORα and Bcl11b are crucial for ILC2 development and function in general (14, 15). In addition, ILC2 require the lymphoid cytokine IL-7 to develop (9) and ILC2 activation is regulated by a variety of cytokines, inflammatory mediators, neuronal factors, metabolites and hormones. Upon stimulation, ILC2s secrete IL-5, IL-9, IL-13 and amphiregulin as main effector cytokines to regulate the type 2 immune response in tissues (14, 15).
Single nucleotide polymorphisms (SNPs) in the IL-33 gene have been linked to allergic asthma and chronic inflammatory diseases (16, 17). IL-33 plays an essential role in triggering type 2 immune responses in mouse models of lung inflammation (18, 19). However, since ST2 is expressed by many type 2 effector cells, the precise contribution for ILC2 activation and the initiation of type 2 inflammation remains incompletely understood. This study provides evidence for a cell-intrinsic requirement of ST2 in ILC2s in steady state, as well as models of allergic lung inflammation and anti-worm immunity. By genetically ablating ST2 in ILC2s using Nmur1iCre-eGFP mice, we establish a conditional knockout model to investigate the role of ST2 in ILC2 biology. ST2-deficient ILC2s produced limited amounts of type 2 cytokines and, therefore, did not provide sufficient support for proper eosinophil homeostasis. Furthermore, Nmur1iCre-eGFP Il1rl1flox/flox failed to recruit eosinophils during allergic lung inflammation provoked by papain treatment and mount a fully protective type 2 immune response against Nippostrongylus brasiliensis (N. brasiliensis) infection. Taken together, our data demonstrate a cell-intrinsic and non-redundant role for ST2 as a regulator of type 2 immunity via activation of ILC2s.
In order to investigate the importance of the IL-33 receptor for ILC2 regulation in vivo, we aimed to target the ST2 chain of the IL-33 receptor (encoded by the Il1rl1 gene) in ILC2 using the Cre-loxP system. To this end, we crossed the recently developed BAC-transgenic Nmur1iCre-eGFP mouse, which allows gene targeting in ILC2 (20, 21), to Il1rl1flox/flox mice (22). Indeed, Nmur1iCre-eGFP Il1rl1flox/flox lacked ST2 mRNA and protein expression in ILC2s of the small intestine, lung, adipose tissue, mesenteric lymph node, and skin similar to Il1rl1-/- mice but not in CD4+ T cells, CD8+ T cells, eosinophils, basophils or mast cells (Figures 1A–G; Supplementary Figures 1A–E).
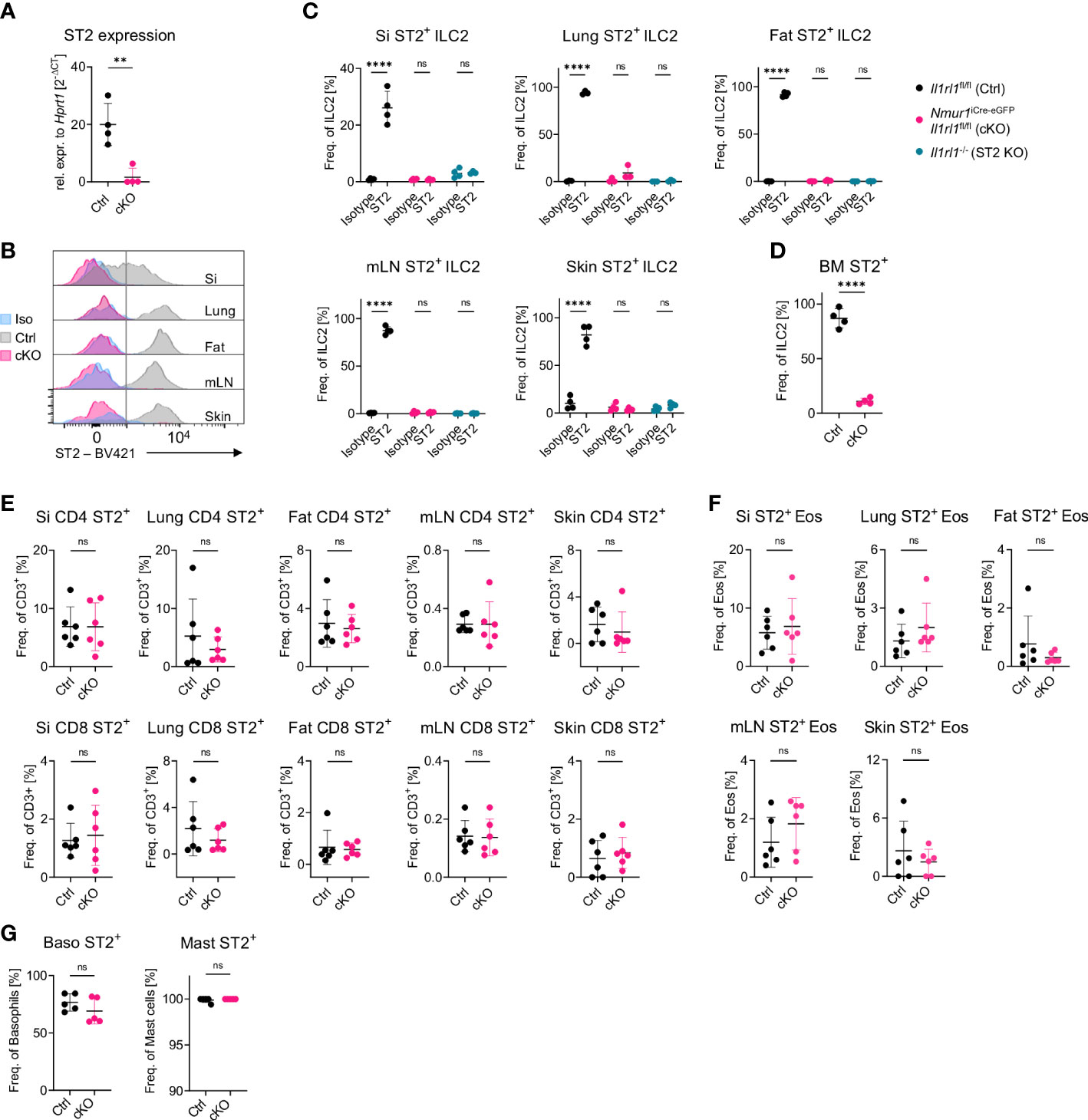
Figure 1 A model for ILC2-specific genetic ablation of ST2. (A), Expression of the Il1rl1 gene in Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) sort-purified ILC2s. (B), Flow cytometric histograms of ST2 expression in ILC2s of Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice across organs, including isotype control on Il1rl1flox/flox (Iso) in the steady state. ILC2s were gated on live CD45+ Lin- (CD3, CD5, CD19, Ly6G, Fcϵr1), CD127+ and KLRG1+. (C–G) Percentage of ST2+ expression in Il1rl1flox/flox (Ctrl), Nmur1iCre-eGFP Il1rl1flox/flox (cKO) (D–G) and Il1rl1-/- (ST2 KO) mice including isotype control (C) across different organs in the indicated immune cells. Basophils from spleen and mast cells from the peritoneal lavage. For (C–G): Each symbol represents data from one mouse, data are representative of two experiments with four to six mice per group. Mean +/- SD, Student’s t-Test. ns, not significant, **p < 0.01, ****p < 0.0001.
To validate whether ST2-deficient ILC2s lost the capacity to sense IL-33, we sort-purified ILC2s from the small intestine, which have low ST2 expression, and from the bone marrow, which have high ST2 expression (Figure 1D), and cultured the cells with IL-7 alone to provide a survival signal, or IL-7 + IL-33 to induce activation. While control ILC2s massively expanded, blasted and secreted type 2 effector cytokines upon IL-33 stimulation, ST2-deficient ILC2s failed to respond to IL-33, indicating efficient deletion of ST2 and unresponsiveness to react to the ligand (Figures 2A–C; Supplementary Figure 2A). Next, we aimed to test if ILC2 still properly responded to stimulation with other alarmins despite the conditional deletion of ST2. To this end, we performed a similar in vitro assay with sort-purified ILC2s from the small intestine but now exposed the cells to IL-7 or IL-7 + IL-25. ST2-deficient ILC2s expanded and blasted to a comparable degree to ST2-sufficient ILC2s upon IL-25 stimulation (Figures 2A, B). We also exposed ILC2s to combinations of TSLP together with IL-7 or IL-25 and IL-33 and assessed blasting and cytokine production of ILC2s (Supplementary Figures 2B–D). However, ST2-deficient ILC2s could still respond to TSLP stimulation. Taken together, these data suggest that ILC2 were selectively unresponsive to IL-33 but could still respond to other alarmins, such as IL-25 and TSLP.
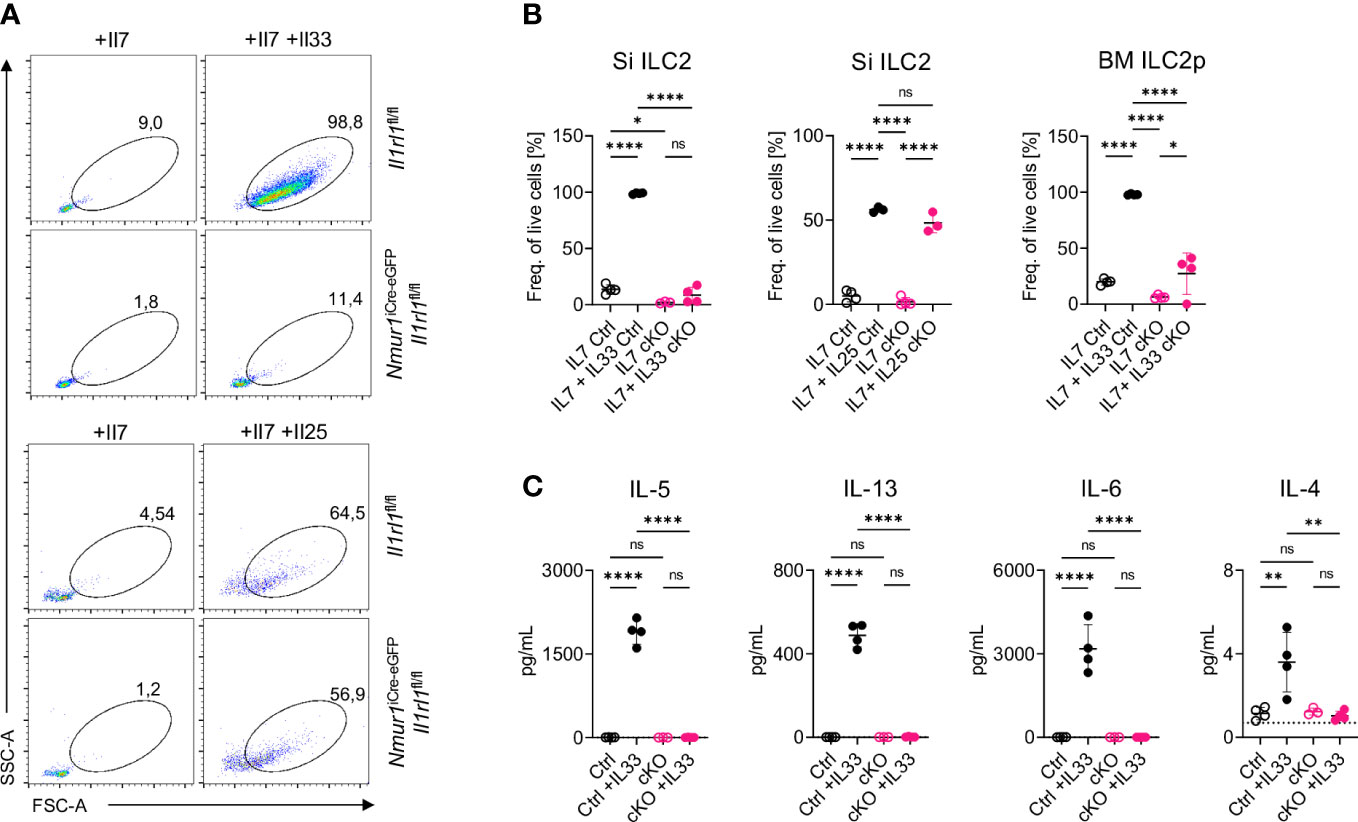
Figure 2 ILC2 of Nmur1iCre-eGFP Il1rl1flox/flox mice are unresponsive to IL-33. (A), Flow cytometric plots of small intestine ILC2s from Il1rl1flox/flox and Nmur1iCre-eGFP Il1rl1flox/flox mice after stimulation with IL-7, IL-7 and IL-33 or IL-7 and IL-25. ILC2s were gated on live CD45+ Lin- (CD3, CD5, CD19, Ly6G, Fcer1), CD127+ and KLRG1+. (B), Quantification of ILC2 activation from (A) and bone marrow ILC2s of Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice after a three day culture with cytokines IL-7 or IL-7, IL-33 or IL-7, IL-25. (C), Concentration of the indicated cytokine as determined in the cell culture supernatant 3 days after stimulation as in (A, B). For (B, C): Each symbol represents data from one mouse, data are representative of two experiments with four mice per group. Mean +/- SD, One-way ANOVA with multiple comparison. ns, not significant, *p < 0.05, **p < 0.01, ****p < 0.0001.
Consistent with previous reports investigating mice with germline mutations of ST2, ILC2 were not reduced in frequencies or absolute cell numbers in Nmur1iCre-eGFP Il1rl1flox/flox mice, suggesting that IL-33-ST2 signals are not essential for proper expansion and survival of ILC2s (23, 24) (Figure 3A). Similar results were obtained for bone marrow ILC2 progenitors, indicating that lack of ST2 signals did not lead to overt developmental defects (Figure 3B) (25).
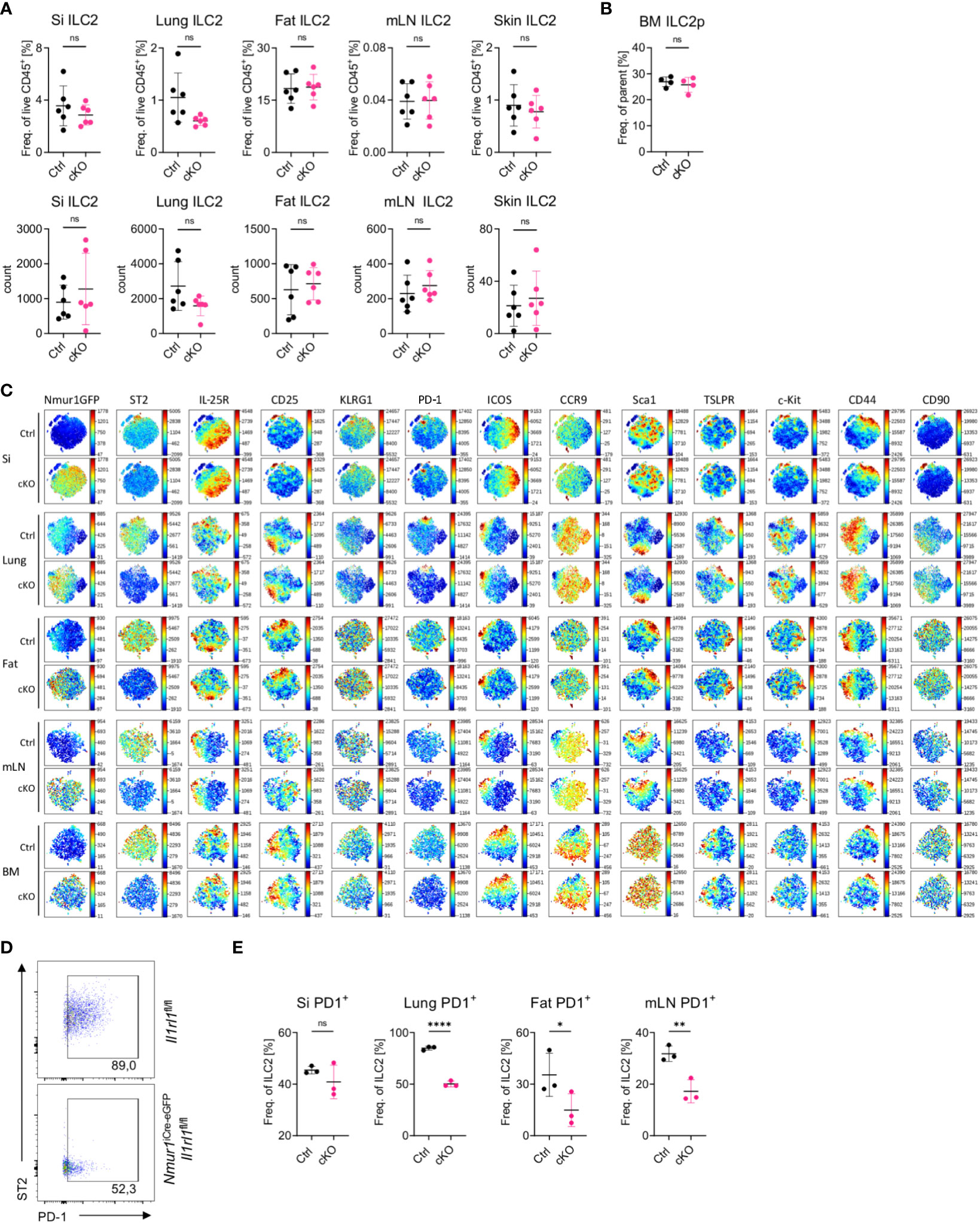
Figure 3 ILC2 develop in normal proportions without ST2 but have reduced PD-1 expression (A), Quantification of relative and absolute ILC2 numbers in Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice across different organs in steady state. (B), Quantification of relative ILC2 progenitor numbers in Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice in the BM. (C), t-distributed stochastic neighbour embedding plots showing the expression level of different ILC2 markers. ILC2s from naïve Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice are shown. Each dot represents a single cell. Data are representative of two experiments with four to five mice per group. (D), Flow cytometric plots of PD-1 expression in lung ILC2s from Nmur1iCre-eGFP Il1rl1flox/flox and Il1rl1flox/flox. (E), Quantification of PD-1 expression in Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice across different organs in steady state. For (A, B, D): Each symbol represents data from one mouse, data are representative of two experiments with three to eight mice per group. Mean +/- SD, student’s t-Test. ns not significant, *p < 0.05, **p < 0.01, ****p < 0.0001.
To test whether deletion of ST2 altered the heterogeneity of ILC2s in tissues, we performed multicolor phenotyping of ILC2s by flow cytometry. Overall, the heterogeneity of ILC2s in tissue was not perturbed and most surface markers used for ILC2 phenotyping, in particular IL-25R or TSLPR were expressed equally in ILC2s from Il1rl1flox/flox and Nmur1iCre-eGFP Il1rl1flox/flox mice (Figure 3C). In contrast, the marker PD-1 was consistently downregulated in ST2-deficient ILC2s (Figures 3C–E; Supplementary Figure 3). Since PD-1 was described as an activation marker on ILC2s (26), these data suggest that ILC2 fail to terminally mature without ST2 signals. In summary, Nmur1iCre-eGFP Il1rl1flox/flox mice are a suitable model to investigate the role of ST2 for ILC2 function in vivo.
Several publications have highlighted the essential role of ILC2 and ILC2-derived IL-5 for eosinophil homeostasis (21, 27). Additional evidence suggests that IL-5 secretion is under the control of alarmin signals (23, 28) and that PD-1+ ILC2 are potent cytokine producers (26). Therefore, we asked if ST2 signals are required for type 2 cytokine production by ILC2. Indeed, IL-5 and IL-13 mRNA levels were significantly reduced in sort-purified ILC2s from Nmur1iCre-eGFP Il1rl1flox/flox compared to control ILC2s (Figure 4A). Next, we assessed the number of eosinophils in Nmur1iCre-eGFP Il1rl1flox/flox mice at steady state due to the IL-5 cytokine being crucial for eosinophil homeostasis (29, 30). Consistent with reduced IL-5 levels in ILC2s, we detected a significant reduction of eosinophils in the lung, adipose tissue, mesenteric lymph nodes and skin, suggesting that ILC2-intrinsic sensing of IL-33 via ST2 is necessary for type 2 cytokine production regulating proper eosinophil homeostasis (Figures 4B, C).
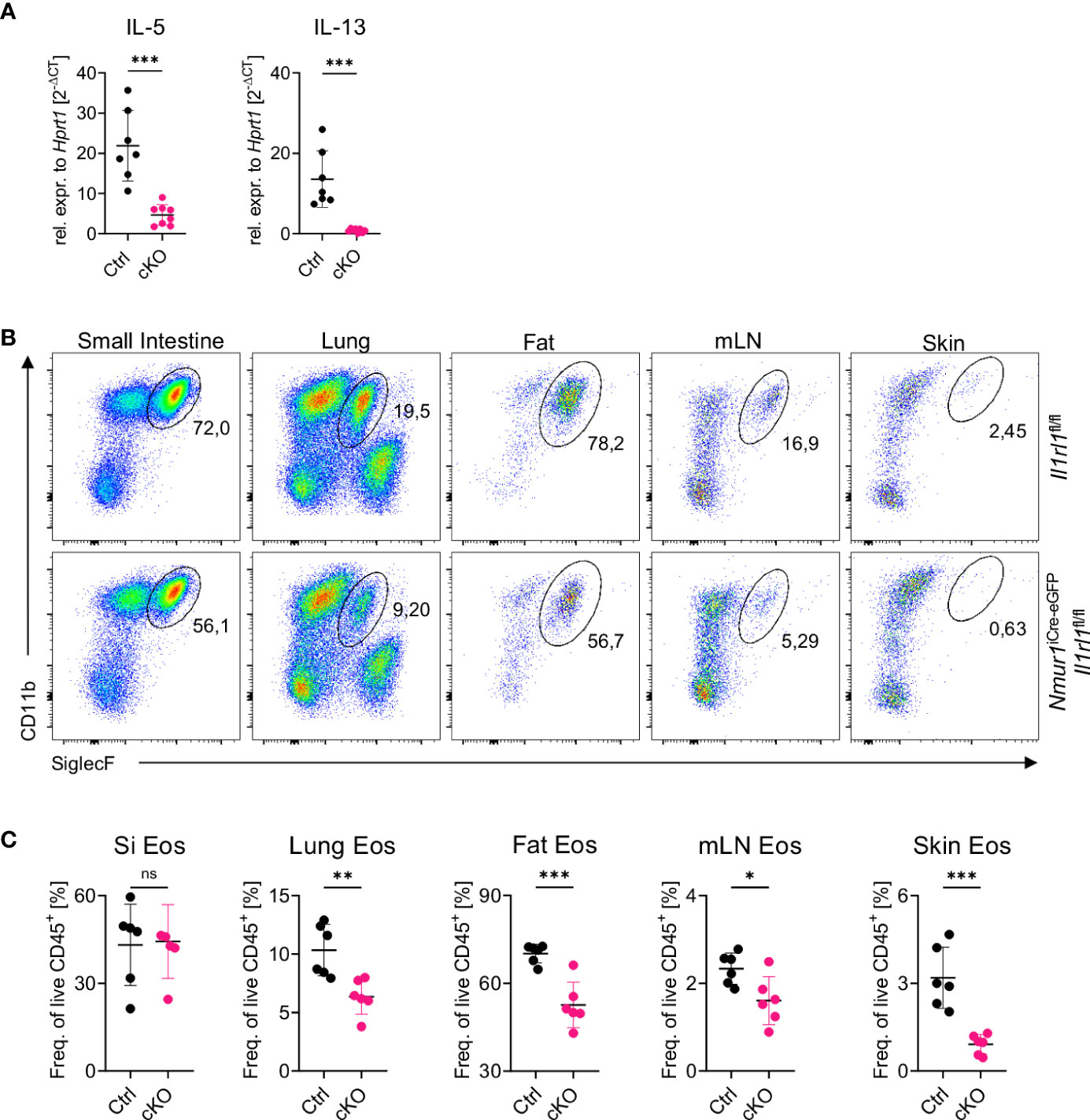
Figure 4 Loss of constant ST2 signaling results in reduced eosinophil counts. (A), Relative expression of the cytokine IL-5 and IL-13 of Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice from the lung at steady state. (B), Flow cytometric plots of eosinophils from Il1rl1flox/flox and Nmur1iCre-eGFP Il1rl1flox/flox mice across different organs in steady state. Eosinophils were gated on live CD45+ Lin- (CD3, CD5, CD19), CD11b+ and SiglecF+. (C), Quantification of (B) in Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice across different organs in steady state. For (A, C): Each symbol represents data from one mouse, data are representative of two experiments with five to seven mice per group. Mean +/- SD, student’s t-Test. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we tested how the lack of ST2 on ILC2s influences the type 2 immune responses in the context of allergic lung inflammation, which was provoked by the administration of the protease allergen papain. Papain administration resulted in expansion of ILC2s, which was significantly reduced in Nmur1iCre-eGFP Il1rl1flox/flox mice (Figures 5A, B). We also found a strong increase in eosinophil counts in lung and BAL fluid upon papain treatment (Figures 5C–E). This increase in eosinophil counts was drastically reduced in papain-treated Nmur1iCre-eGFP Il1rl1flox/flox mice (Figures 5C–E). A more detailed analysis revealed that both CD11b+ SiglecFint tissue-resident and CD11b+ SiglecFhigh inflammatory eosinophils (31) were reduced in Nmur1iCre-eGFP Il1rl1flox/flox mice, suggesting that ST2 signals in ILC2s are required for recruitment and local proliferation of eosinophils (Figures 5C, F).
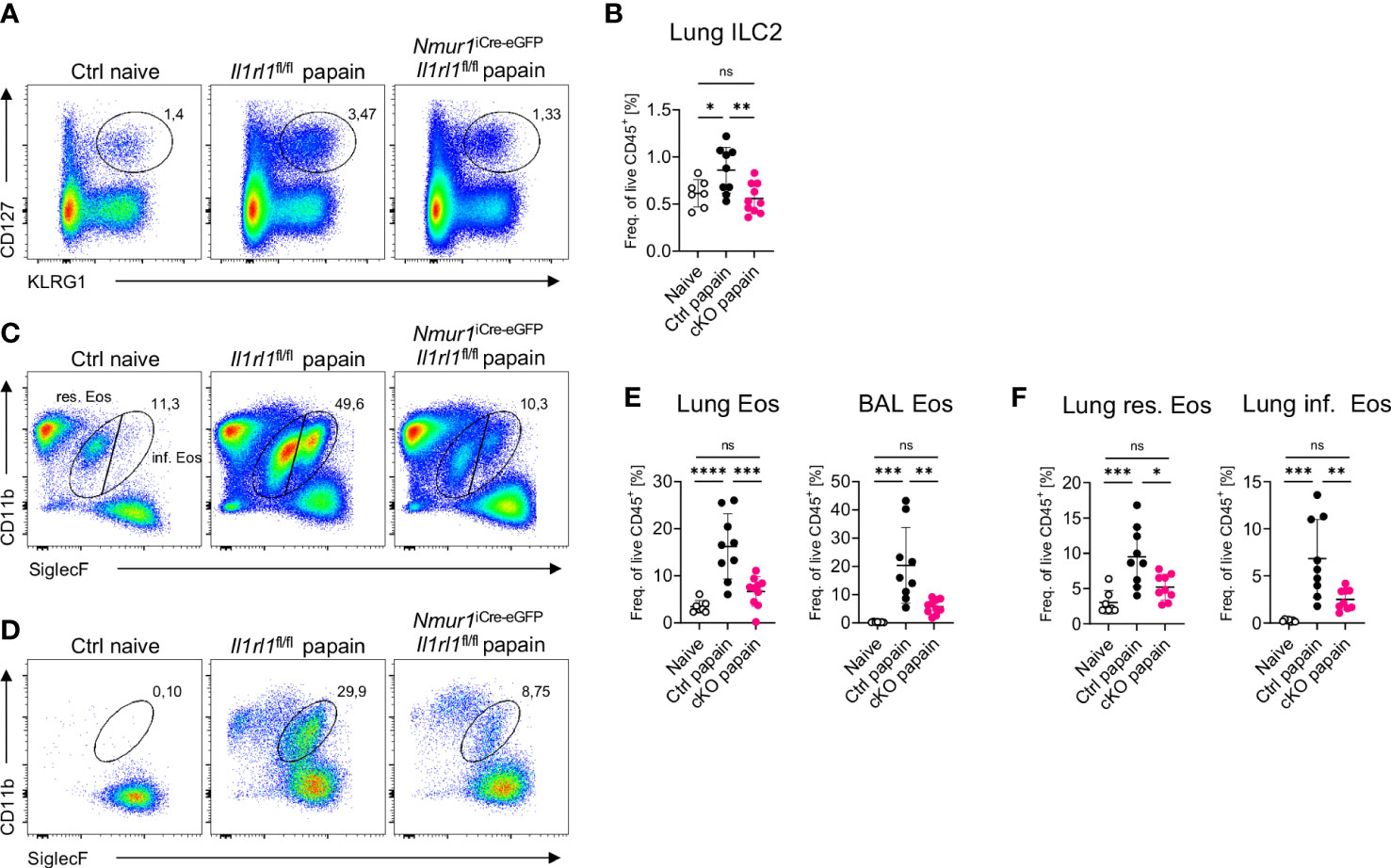
Figure 5 ST2-deficient ILC2s fail to recruit eosinophils in acute lung inflammation. (A, C, D), Flow-cytometric plots showing ILC2s (A) in the Lung, eosinophils (C) in the lung or BAL (D) of untreated Ctrl (naïve) animals and papain treated Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO) and littermate controls (Ctrl). Data are representative of two experiments with four to six mice per group. (B, E), Quantification of relative ILC2 (B) and eosinophil numbers from the lung or BAL (E) in the lung and BAL. (F), Quantification of relative resident and inflammatory eosinophil numbers in the lung. For (B, E, F): One-way ANOVA with multiple comparisons, ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To test whether ST2 is required in a cell-intrinsic manner on ILC2s to mediate resistance against helminth infections, we infected Nmur1iCre-eGFP Il1rl1flox/flox and littermate control mice with the worm N. brasiliensis. Littermate control mice mounted a potent type 2 immune response upon worm infection, which was diminished in Nmur1iCre-eGFP Il1rl1flox/flox mice (Figure 6). The massive expansion of ILC2s upon infection was weakened in Nmur1iCre-eGFP Il1rl1flox/flox mice compared to littermate controls (Figures 6A–C). Furthermore, eosinophil frequencies and tuft cell hyperplasia were reduced in conditional knockout mice, underlining the importance of ST2 signals for a proper type 2 immune response (Figures 6D–I). As a consequence, Nmur1iCre-eGFP Il1rl1flox/flox mice had higher worm counts in the intestine (Figure 6J). Therefore, our data expose cell-intrinsic ST2 signals in ILC2s as a non-redundant stimulatory pathway for triggering protective type 2 immunity.
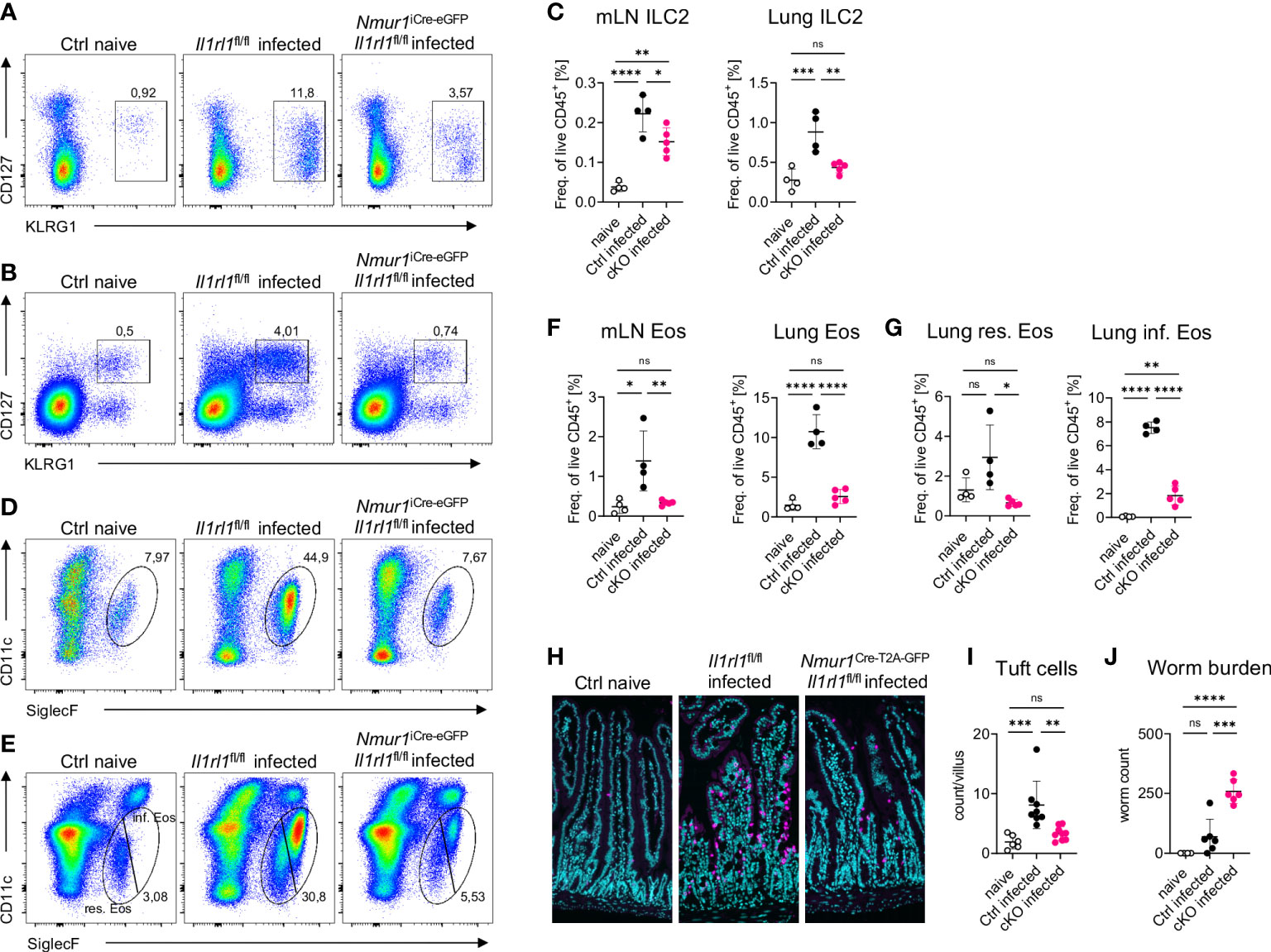
Figure 6 ILC2 intrinsic ST2 is required for immunity against N. brasiliensis infection. (A, B), Flow-cytometric plots showing ILC2s in the mLN (A) and in the lung (B) of untreated Ctrl (naïve) animals and infected Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO) and littermate controls (Ctrl). Mice were infected with N. brasiliensis. (C), Quantification of (A, B). (D, E) Flow-cytometric plots showing eosinophils in the mLN (D) and in the lung (E) of untreated Ctrl (naïve) animals and infected Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO) and littermate controls (Ctrl). Mice were infected with N. brasiliensis. (F), Quantification of (D) and (E, G), Quantification of relative resident and inflammatory eosinophil numbers in the lung of untreated Ctrl (naïve) animals and infected Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO) and littermate controls (Ctrl). (H), Immunofluorescence migrographs of histological sections of the murine small intestine, tuft cells stained with DCLK1 (magenta), DAPI (blue). (I), Quantification of (H). Tuft cell numbers per villus. Data combined from two independent experiments. (J), Worm burden in untreated animals (naïve) and infected Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO) or littermate controls (Ctrl). For (C, F, G, I, J): Data are representative of two experiments with four to six mice per group. One-way ANOVA with multiple comparisons, ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Alarmins are important mediators of inflammation and molecules targeting these pathways have already been or are about to be tested in clinical trials (32–35). However, since alarmins are sensed by a variety of immune cells, the mechanism of how they modulate immune response is incompletely understood. In this study, we characterized the IL-33-ST2 signaling pathway for ILC2 activation in detail, which could not be addressed so far because of limitations in genetic targeting strategies, using a novel Cre deleter strain based on the Nmur1 promoter.
IL-33 and the type 2 effector cytokines IL-4, IL-5 and IL-13 have been linked to atopic diseases in mice and humans (16, 17). Similarly, type 2 effector cells, such as ILC2, Th2, mast cells and basophils were linked to this disease entity and were reported to respond to IL-33 with the release of type 2 effector cytokines and are therefore a potential candidate to mediate the effects of IL-33 in the context of type 2 inflammation. In our study interrogating the role of ST2 on ILC2s, we found that conditional deletion of ST2 rendered ILC2s unresponsive to IL-33 and resulted in decreased production of the effector cytokines IL-5 and IL-13 directly ex vivo. In contrast, ILC2s developed in regular proportions without ST2. These findings are consistent with several studies, which found that the germline deletion of the alarmin receptors IL-25, IL-33 and TSLP did not result in decreased ILC2 numbers but diminished cytokine expression (23, 24). We further detected a reduction in eosinophil numbers at steady state in Nmur1iCre-eGFP Il1rl1flox/flox mice, which is most likely the consequence of diminished IL-5 production by ILC2s, since ILC2s were described being the major source of IL-5 in tissues (27) and the IL-5–IL-5ra signaling was proven indispensable for proper development of eosinophils (29, 30). Finally, ILC2-deficient mice have defects in eosinophil development comparable to the deficiency described in IL-5 or IL5rα knockout mice (21, 29, 30). Of note, similar effects on eosinophil numbers were found in mice with germline deletion of ST2 combined with recombinant IL-33 administration (36). The results of our study allow to conclude that ILC2s are the cell type requiring IL-33 signals via ST2 in order to regulate eosinophil homeostasis at steady state and during allergic lung inflammation.
Despite the well-established role of alarmins in triggering allergic lung inflammation, the target cell types are only partly defined. IL-33 receptor was found to play a pivotal role in activating different effector cells in the context of type 2 inflammation including TH2 cells, eosinophils, basophils and mast cells. The contribution of our study is the demonstration of an indispensable role for the IL-33-ST2 axis to stimulate ILC2 activation during allergic lung inflammation, which cannot be compensated by other signaling pathways or cell types. While we did not examine the role of conditional deletion of ST2 in other type 2 immune cells, our data suggest that IL-33 primarily activates ILC2 to initiate type 2 inflammation.
We used the Nmur1iCre-eGFP Il1rl1flox/flox mouse model to obtain genetic deletion of ST2 in ILC2s but not in other cell types investigated (Figures 1A–G). The Nmur1iCre-eGFP mice have reported highly efficient recombination capabilities (20, 21). To confirm the efficient deletion of ST2, we have compared ST2 levels to isotype control and mice with germline mutation of ST2. While very efficient deletion was obtained in most organs as validated by flow cytometry, a minor fraction of cells remained formally ST2+ in the lung of Nmur1iCre-eGFP Il1rl1flox/flox mice, which might be explained by the limitations in the discriminatory power of the ST2 antibody staining (Figures 1B, C).
Our results with respect to ST2 signaling in ILC2s are comparable to published studies investigating the effect of conditional gene deletion of alarmin receptors in ILC2s. Kabata et al. could show that TSLP receptor is required on ILC2s to trigger allergic lung inflammation provoked by papain using Il7rCre/+ and Il5Cre/+ and conditional alleles of TSLP receptor. The same study described a pivotal role of TSLP receptor on dendritic cells and CD4+ T cells in the ovalbumin-induced airway inflammation model (37). Furthermore, Leyva-Castillo et al. conditionally deleted the IL-25 receptor (IL-25R) by crossing RoraCre/+ and IL17rbflox/flox mice. In a model of skin inflammation provoked by ova treatment, the symptoms were reduced in RoraCre/+ and IL17rbflox/flox mice, suggesting a crucial role for the IL-25R for ILC2 activation (38).
Previous studies reported a pivotal role for IL-25 and IL-33 in regulating worm expulsion of the parasites N. brasiliensis and Strongyloides venezuelensis (9, 28, 39–41). IL-25 together with other inflammatory mediators is secreted by Tuft cells and activates a subsets of IL-25R+ ST2- ILC2s coined inflammatory ILC2s (42–46). IL-33 was also reported to stimulate the generation of inflammatory ILC2s via activation of the enzyme tryptophan hydroxylase in ILC2s (43, 47). A correlation of ILC2 frequencies and worm resistance during N. brasiliensis infection further supports the idea that ILC2s have a crucial function for worm expulsion, most likely activated by alarmin signals. Adoptive transfer experiments of ILC2s in lymphopenic or immunodeficient mice did rescue defects to expel the parasite, and therefore provide additional evidence for a pivotal role of ILC2s (9, 28). Complete depletion of ILC2 using Nmur1iCre-eGFP Ild2flox/flox, Nmur1iCre-eGFP Gata3flox/flox or Klrg1Cre Gata3flox/flox resulted in a severe defect in worm expulsion demonstrating the essential role of ILC2 for resistance to N. brasiliensis infection (21, 48). IL-25R and ST2 fulfill complementary functions to trigger ILC2 activation since genetic ablation of both receptors resulted in a comparable phenotype to ILC2-deficient mice characterized by strongly delayed worm expulsion and ongoing worm infection until day 14 - 20 post infection (28).
The Nmur1iCre-eGFP Il1rl1flox/flox mouse model allows us to more directly address the question of ILC2 redundancy and IL-33 signals without using adoptive cell transfer systems in combination with lymphopenic mice. Overall, our data from the Nmur1iCre-eGFP Il1rl1flox/flox mice establish a cell-intrinsic and non-redundant role of ST2 for ILC2 function in the context of a type 2 immune response in lymphoreplete mice.
C57BL/6 mice were purchased from Janvier. Il1rl1flox/flox mice (22) (provided by the UC Davis/MMRRC repository), Il1rl1-/- mice (28) and Nmur1iCre-eGFP mice (20, 21) bred locally at Charité. Sex and age-matched animals were used for experiments if not otherwise indicated. We did not use randomization to assign animals to experimental groups. All animal experiments were approved and are in accordance with the local animal care committees (Lageso Berlin).
Small intestine was removed, cleaned from remaining fat tissue and washed in ice-cold PBS. Peyer’s patches were eliminated, small intestine was opened longitudinally and washed in ice-cold PBS. Dissociation of epithelial cells was performed by incubation on a shaker at 37°C in HBSS (Sigma-Aldrich) containing 10 mM Hepes (Gibco) and 5 mM EDTA (Roboklon) two times for 15 min. After each step, samples were vortexed and the epithelial fraction discarded. Afterwards, remaining tissue was chopped into small pieces and enzymatic digestion was performed using dispase (0,5 U/ml; Corning), collagenase D (0,5 mg/ml; Roche) and DNaseI (100 μg/ml; Sigma-Aldrich). Leukocytes were further enriched by Percoll gradient centrifugation (GE Healthcare). Lungs were chopped and incubated in the enzyme cocktail described above for 40 min on a shaker at 37°C. The remaining tissues were mashed with a syringe plunger and single cell suspensions were filtered through a 70 μm cell strainer. Leukocytes were then further enriched by Percoll gradient centrifugation. Mesenteric lymph nodes were chopped and incubated in RPMI 1640 medium (Gibco) supplemented with 1% BSA (Sigma-Aldrich), collagenase II (1 mg/ml; Sigma-Aldrich) and DNaseI (100 μg/ml) for 20 min on a shaker at 37°C. Afterwards, cells were dissociated using a pasteur pipette, and filtered through a 70 μm cell strainer. Epididymal white adipose tissue was removed and incubated in the same digestion buffer for 45 min on a shaker at 37°C. After incubation, cells were dissociated using a Pasteur pipette, filtered through a 70 μm cell strainer, spun down and the adipocyte layer was aspirated. For skin preparation, ears were removed, split and put dermal side down in DMEM supplemented with Liberase TL (0,5 mg/ml; Roche) at 37°C for 1,5 h. The tissue was then mashed through a 70 μm cell strainer and the leukocytes further enriched by Percoll gradient centrifugation. For isolation of bone marrow cells, femur and tibia bone were crushed with a pestle, rinsed and cells were filtered through a 70 μm cell strainer. Red cell lysis was performed in ACK lysis buffer for 3 min.
Dead cells were routinely excluded with Fixable Aqua Dead Cell Stain or SYTOX Blue Dead Cell Stain (Thermo Fisher Scientific). Single cell suspensions were incubated on ice with anti-CD16/CD32 antibody and the following conjugated antibodies in PBS (Ca2+ and Mg2+-free, Sigma-Aldrich). If indicated, lineage-positive cells were excluded by staining for CD3e (145-2C11 or 500A2), CD5 (53-7.3), CD19 (1D3 or 6D5), FcϵRIα (Mar-1) and Ly6G (1A8). For surface staining the following antibodies were used: c-Kit (2B8), CCR9 (9B1), CD11b (M1/70), CD11c (N418), CD127 (A7R34), CD25 (PCG1.5), CD4 (GK1.5 and RM4-5), CD44 (IM7), CD45.2 (104), CD49b (DX5), CD64 (X54-5/7.1), CD8a (53-6.7), CD90.2 (53-2.1), F4/80 (BM8), ICOS(C398.4A), IL-25R (Munc33), KLRG1 (2F1 or MAFA), Ly6G (1A8), NK1.1 (PK136), PD-1 (29F.1A12), Sca-1 (D7), SiglecF (E50-2440), ST2 (RMST2-33), TSLPR (22H9), Rat IgG2b kappa Isotype (eB149/10H5). All antibodies used in flow cytometry were purchased from eBioscience, Biolegend or BD Biosciences if not otherwise indicated. All flow cytometry experiments were acquired using a custom configuration Fortessa flow cytometer and the FACS Diva software (BD Biosciences) and were analyzed with FlowJo V9.9.3 or V10.6.2 software (TreeStar) and for the t-SNE plots analyzed with the Cytobank Software using custom configurations (Backman Coulter) or sort-purified by using a custom configuration FACSAria cell sorter (BD Biosciences).
Sort-purified cells were homogenized in Trizol (Thermo Fisher Scientific) and stored at -80°C. RNA was extracted with chloroform and RNA concentration was determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription of total RNA was performed using the High Capacity cDNA Reverse Transcription kit according to the protocol provided by the manufacturer (Thermo Fisher Scientific). Reaction was detected on a QuantStudio 5 Real-Time PCR (Thermo Fisher Scientific) using Taqman Gene Expression Assay (Applied Biosystems) with Il5 (Mm00439646_m1), Il13 (Mm00434204_m1) or SYBR Green Master Mix with Il1rl1 (forward: 5’- GGGCACACAGGTCCTACTTG-3’, reverse: 5’- ATGTAGTTGGTTCCATTCTCCG-3’). Gene expression was normalized to the housekeeping gene Hprt1 (Mm00446968_m1) for Taqman and Hprt1 (forward: 5’-GATACAGGCCAGACTTTGTTGG-3’, reverse: 5’-CAACAGGACTCCTCGTATTTGC-3’) for SYBR Green.
Third-stage larvae (L3) of N. brasiliensis were purified with a Baermann apparatus. After washing three times in PBS, larvae were counted and 500 purified larvae were injected subcutaneously in PBS. Mice were killed, organs were analyzed and worm burden was determined in the small intestine 7 days post infection.
For allergic asthma induction, 30 μg of Papain (Roche) in PBS were administered intranasally on three consecutive days. Mice were killed on day seven after initial administration, organs were collected and analyzed.
Sort-purified ILC2s (as live CD45+, Lin-, NK1.1-, CD127+ and KLRG1+ for small intestine or CD25+ for bone marrow) were incubated in DMEM with high glucose supplemented with 10% FCS, 10 mM Hepes, 1 mM sodium pyruvate, non-essential amino acids, 80 μM 2-Mercaptoethanol, 2mM Glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin (all from Gibco) in 96-well U-bottom microtiter plates (Nunc) for three days at 37°C and 5% CO2. As indicated, the culture was supplemented with a cocktail of IL-7 only, IL-7 and IL-33 or IL-7 and IL-25, IL-7 and TSLP or IL-7 and IL-33, IL-25, TSLP (Biolegend, TSLP from R&D, 20 ng/ml each).
Cytokine concentrations in culture supernatants were determined by using the LEGENDplex murine TH2 Panel V03 multiplex beads-based assay (Biolegend) according to the manufacture’s protocol. Samples were recorded on a custom configuration Fortessa flow cytometer and the FACS Diva software (BD Biosciences) and the flow cytometry data files were analyzed using the Legendplex cloud-based analysis software suite (Biolegend).
For immunofluorescence staining, the tissue was fixed in 4% PFA at 4°C until tissue embedding. Paraffin-embedded sections were de-paraffinized and rehydrated. Sections were permeabilized with 0.5% Triton-X in PBS and blocked with PBS 0,5% Triton X-100 and 10% serum and stained with rabbit anti-DCLK1 (Abcam) followed by donkey anti-rabbit antibody coupled to Alexa Fluor 555 (Thermo Fisher Scientific). Nuclei were counterstained with DAPI (Thermo Fisher Scientific). Images were captured on a Zeiss Axio Observer 7 microscope and analysed with Zen software (Zeiss). For tuft cell numbers, five to ten representative villi were counted on four independent images per mouse using ImageJ.
Data is plotted showing the mean +/- standard deviation. P values of data sets were determined by unpaired two-tailed Student’s t-test with 95% confidence interval. If equal variances could not be assumed, Welch test was performed. Ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to analyze several groups. Normal distribution was assumed. Before mentioned statistical tests were performed with Graph Pad Prism V9 software (GraphPad Software, Inc.). (*p <0.05; **p <0.01; ***p <0.001; ****p <0.0001 and ns, not significant).
The original contributions presented in the study are included in the article/Supplementary Material further inquiries can be directed toY2hyaXN0b3BoLmtsb3NlQGNoYXJpdGUuZGU=.
The animal study was reviewed and approved by Lageso Berlin.
PMT and ZAR carried out the experiments and analyzed the data. MOJ, PL, AS, AP and CSNK helped performing experiments. LMLT and KK helped with the t-SNE analysis. CUD and DA provided crucial input and tools for the study. CSNK and PMT conceived the project and wrote the manuscript with input from all co-authors. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the European Research Council Starting Grant (ERCEA; 803087 to CSNK), the German Research Foundation (DFG; Project-ID 259373024 – CRC/TRR 167, FOR2599 project 5 - KL 2963/5-2, SPP1937 - KL 2963/2-1 and KL 2963/3-1 to CSNK, SPP1937 - DU1295/4-1 to CDU); the Swiss National Science foundation (Grant-ID: 184425 to MJ), the US National Institutes of Health (DK126871, AI151599, AI095466, AI095608, AR070116, AI172027, DK132244 to DA), LEO foundation, Cure for IBD, Jill Roberts Institute, the Sanders Family, the Rosanne H. Silbermann Foundation (all to DA).
We thank the UC Davis and MMRRC repository for providing frozen material of Il1rl1flox/flox mice, A. McKenzie for Il1rl1-/- mice, A. Branco and the Benjamin Franklin Flow Cytometry Facility for cell sorting (supported by DFG Instrument Grants INST 335/597-1 FUGG and INST 335/777-1) and L. Paul for technical assistance.
DA has contributed to scientific advisory boards at Pfizer, Takeda, FARE and the KRF.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor HK declared a past co-authorship with the author DA.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1130933/full#supplementary-material
Supplementary Figure 1 | (related to Figure 1): Gating strategy for flow cytometry. (A, B), Gating strategy of ILC2s and eosinophils from the small intestine, lung, mesenteric lymph nodes, fat, skin (A) and bone marrow (B). Lineage: CD3, CD5, CD19, Ly6G, FcϵRIα. (C), Gating strategy of CD4+ and CD8+ T cells from the lung. (D), basophils from the spleen and mast cells from the peritoneal lavage. Lineage: CD3, CD5, CD19, Ly6G. (E), Geometric mean fluorescent intensity measurement of ST2 in ILC2s from Il1rl1flox/flox (Ctrl), Nmur1iCre-eGFP Il1rl1flox/flox (cKO) and Il1rl1-/- (ST2 KO) mice (indicated as ST2) with isotype control (indicated as Isotype) across different organs. For (A-D): Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox mice (cKO). Mean ± SD. Student's t-Test. ns, not significant, **p < 0.01, ****p < 0.0001.
Supplementary Figure 2 | (related to Figure 2): ST2-deficient ILC2s are unresponsive to IL-33 but not to other alarmins. (A), Cytokine measurement from culture supernatants three days after stimulation as in (Figure 2C) (extending). (B), Flow cytometric plots of ILC2s from the small intestine of Il1rl1flox/flox and Nmur1iCre-eGFP Il1rl1flox/flox mice after stimulation with IL-7, IL-7 and IL-33, IL-7 and Il-25, IL-7 and TSLP or IL-7 and IL-33, IL-25 and TSLP (alrmins) as indicated. ILC2s were gated on live CD45+ Lin- (CD3, CD5, CD19, Ly6g, Fcer1), CD127+ and KLRG1+. (C), Quantification of ILC2 activation from (B) of Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice after a three day culture with cytokines IL-7, IL-7 and IL-33, IL-7 and Il-25, IL-7 and TSLP or IL-7 and IL-33, IL-25 and TSLP (alrmins) as indicated. (D), Concentration of the indicated cytokine as determined in the cell culture supernatant three days after stimulation as in (B, C). For (A, C, D): Each symbol represents data from one mouse, data are representative of two experiments with four mice per group. Mean +/- SD. One-way ANOVA with multiple comparisons (A), Student’s t-Test (C, D). ns not significant, * p<0.05, ** p<0.01, **** p<0.0001.
Supplementary Figure 3 | (related to Figure 3): ST2 dependent changes in PD-1 expression. Geometric mean fluorescent intensity measurement of PD-1 in ILC2s from Il1rl1flox/flox (Ctrl) and Nmur1iCre-eGFP Il1rl1flox/flox (cKO) mice across different organs in steady state. Each symbol represents data from one mouse, data are representative of two experiments with three to four mice per group. Mean +/- SD, student’s t-Test. ns not significant, * p<0.05, **** p<0.0001.
1. Stanbery AG, Smita S, von Moltke J, Tait Wojno ED, Ziegler SF. TSLP, IL-33, and IL-25: Not just for allergy and helminth infection. J Allergy Clin Immunol (2022) 150:1302–13. doi: 10.1016/j.jaci.2022.07.003
2. Mahlakoiv T, Flamar AL, Johnston LK, Moriyama S, Putzel GG, Bryce PJ, et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci Immunol (2019) 4(35). doi: 10.1126/sciimmunol.aax0416
3. Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science (2012) 335:984–9. doi: 10.1126/science.1215418
4. Rana BMJ, Jou E, Barlow JL, Rodriguez-Rodriguez N, Walker JA, Knox C, et al. A stromal cell niche sustains ILC2-mediated type-2 conditioning in adipose tissue. J Exp Med (2019) 216:1999–2009. doi: 10.1084/jem.20190689
5. Spallanzani RG, Zemmour D, Xiao T, Jayewickreme T, Li C, Bryce PJ, et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol (2019) 4(35). doi: 10.1126/sciimmunol.aaw3658
6. Hung LY, Tanaka Y, Herbine K, Pastore C, Singh B, Ferguson A, et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci Immunol (2020) 5(53). doi: 10.1126/sciimmunol.abc6259
7. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
8. Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev (2018) 281:154–68. doi: 10.1111/imr.12619
9. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
10. Neill DR, McKenzie AN. Nuocytes and beyond: New insights into helminth expulsion. Trends Parasitol (2011) 27:214–21. doi: 10.1016/j.pt.2011.01.001
11. Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An interleukin-33-Mast cell-Interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity (2015) 43:175–86. doi: 10.1016/j.immuni.2015.06.021
12. Magat JM, Thomas JL, Dumouchel JP, Murray F, Li WX, Li J. Endogenous IL-33 and its autoamplification of IL-33/ST2 pathway play an important role in asthma. J Immunol (2020) 204:1592–7. doi: 10.4049/jimmunol.1900690
13. Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol (2010) 185:3472–80. doi: 10.4049/jimmunol.1000730
14. Klose CSN, Artis D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res (2020) 30:475–91. doi: 10.1038/s41422-020-0323-8
15. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
16. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. Consortium-based genomewide association study of asthma. New Engl J Med (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
17. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse north American populations. Nat Genet (2011) 43:887–92. doi: 10.1038/ng.888
18. Hiraishi Y, Yamaguchi S, Yoshizaki T, Nambu A, Shimura E, Takamori A, et al. IL-33, IL-25 and TSLP contribute to development of fungal-associated protease-induced innate-type airway inflammation. Sci Rep (2018) 8:18052. doi: 10.1038/s41598-018-36440-x
19. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
20. Tsou AM, Yano H, Parkhurst CN, Mahlakoiv T, Chu C, Zhang W, et al. Neuropeptide regulation of non-redundant ILC2 responses at barrier surfaces. Nature (2022) 611:787–93. doi: 10.1038/s41586-022-05297-6
21. Jarick KJ, Topczewska PM, Jakob MO, Yano H, Arifuzzaman M, Gao X, et al. Non-redundant functions of group 2 innate lymphoid cells. Nature (2022) 611:794–800. doi: 10.1038/s41586-022-05395-5
22. Hemmers S, Schizas M, Rudensky AY. T Reg cell-intrinsic requirements for ST2 signaling in health and neuroinflammation. J Exp Med (2021) 218(2):e20201234. doi: 10.1084/jem.20201234
23. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol (2018) 19:1093–9. doi: 10.1038/s41590-018-0201-4
24. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med (2018) 215:263–81. doi: 10.1084/jem.20170449
25. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
26. Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature (2016) 539:102–6. doi: 10.1038/nature20105
27. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502:245–8. doi: 10.1038/nature12526
28. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
29. Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, et al. IL-5-deficient mice have a developmental defect in CD5+ b-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity (1996) 4:15–24. doi: 10.1016/S1074-7613(00)80294-0
30. Yoshida T, Ikuta K, Sugaya H, Maki K, Takagi M, Kanazawa H, et al. Defective b-1 cell development and impaired immunity against angiostrongylus cantonensis in IL-5R alpha-deficient mice. Immunity (1996) 4:483–94. doi: 10.1016/S1074-7613(00)80414-8
31. Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest (2016) 126:3279–95. doi: 10.1172/JCI85664
32. Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. New Engl J Med (2021) 384:1800–9. doi: 10.1056/NEJMoa2034975
33. Chen YL, Gutowska-Owsiak D, Hardman CS, Westmoreland M, MacKenzie T, Cifuentes L, et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci Trans Med (2019) 11(515). doi: 10.1126/scitranslmed.aax2945
34. Donovan C, Hansbro PM. IL-33 in chronic respiratory disease: From preclinical to clinical studies. ACS Pharmacol Transl Sci (2020) 3:56–62. doi: 10.1021/acsptsci.9b00099
35. Kelsen SG, Agache IO, Soong W, Israel E, Chupp GL, Cheung DS, et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J Allergy Clin Immunol (2021) 148:790–8. doi: 10.1016/j.jaci.2021.03.044
36. Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol (2016) 197:3445–53. doi: 10.4049/jimmunol.1600611
37. Kabata H, Flamar AL, Mahlakoiv T, Moriyama S, Rodewald HR, Ziegler SF, et al. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol (2020) 13:626–36. doi: 10.1038/s41385-020-0266-x
38. Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, et al. ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol (2020) 145:1606–14.e4. doi: 10.1016/j.jaci.2020.02.026
39. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
40. Hung LY, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical type 2 immunity against hookworms. Proc Natl Acad Sci USA (2013) 110:282–7. doi: 10.1073/pnas.1206587110
41. Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci USA (2012) 109:3451–6. doi: 10.1073/pnas.1201042109
42. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (2018) 359:114–9. doi: 10.1126/science.aam5809
43. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol (2015) 16:161–9. doi: 10.1038/ni.3078
44. Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell (2018) 174:271–84.e14. doi: 10.1016/j.cell.2018.05.014
45. Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature (2016) 529:226–30. doi: 10.1038/nature16527
46. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature (2016) 529:221–5. doi: 10.1038/nature16161
47. Flamar AL, Klose CSN, Moeller JB, Mahlakoiv T, Bessman NJ, Zhang W, et al. Interleukin-33 induces the enzyme tryptophan hydroxylase 1 to promote inflammatory group 2 innate lymphoid cell-mediated immunity. Immunity (2020) 52:606–619 e6. doi: 10.1016/j.immuni.2020.02.009
Keywords: ILC2 - group 2 innate lymphoid cell, IL-33 and ST2, type 2 immune response, mucosal immunity, innate immunity
Citation: Topczewska PM, Rompe ZA, Jakob MO, Stamm A, Leclère PS, Preußer A, Duerr CU, Thole LML, Kotsch K, Artis D and Klose CSN (2023) ILC2 require cell-intrinsic ST2 signals to promote type 2 immune responses. Front. Immunol. 14:1130933. doi: 10.3389/fimmu.2023.1130933
Received: 23 December 2022; Accepted: 08 March 2023;
Published: 31 March 2023.
Edited by:
Hiroki Kabata, Keio University, JapanReviewed by:
Jorg Hermann Fritz, McGill University, CanadaCopyright © 2023 Topczewska, Rompe, Jakob, Stamm, Leclère, Preußer, Duerr, Thole, Kotsch, Artis and Klose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph S. N. Klose, Y2hyaXN0b3BoLmtsb3NlQGNoYXJpdGUuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.