 Alexander J. Suptela
Alexander J. Suptela Ian Marriott
Ian Marriott
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 March 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1130172
This article is part of the Research Topic The Molecular Basis of Programmed Cell Death and Neuroinflammation in Neurodegenerative Diseases View all 10 articles
Genomic instability is a key driving force for the development and progression of many neurodegenerative diseases and central nervous system (CNS) cancers. The initiation of DNA damage responses is a critical step in maintaining genomic integrity and preventing such diseases. However, the absence of these responses or their inability to repair genomic or mitochondrial DNA damage resulting from insults, including ionizing radiation or oxidative stress, can lead to an accumulation of self-DNA in the cytoplasm. Resident CNS cells, such as astrocytes and microglia, are known to produce critical immune mediators following CNS infection due to the recognition of pathogen and damage-associated molecular patterns by specialized pattern recognition receptors (PRRs). Recently, multiple intracellular PRRs, including cyclic GMP-AMP synthase, interferon gamma-inducible 16, absent in melanoma 2, and Z-DNA binding protein, have been identified as cytosolic DNA sensors and to play critical roles in glial immune responses to infectious agents. Intriguingly, these nucleic acid sensors have recently been shown to recognize endogenous DNA and trigger immune responses in peripheral cell types. In the present review, we discuss the available evidence that cytosolic DNA sensors are expressed by resident CNS cells and can mediate their responses to the presence of self-DNA. Furthermore, we discuss the potential for glial DNA sensor-mediated responses to provide protection against tumorigenesis versus the initiation of potentially detrimental neuroinflammation that could initiate or foster the development of neurodegenerative disorders. Determining the mechanisms that underlie the detection of cytosolic DNA by glia and the relative role of each pathway in the context of specific CNS disorders and their stages may prove pivotal in our understanding of the pathogenesis of such conditions and might be leveraged to develop new treatment modalities.
In 2022, there were an estimated 1.9 million new cancer cases and over 600,000 deaths due to these diseases in the United States (1). Of these, approximately 25,000 were associated with the brain and the central nervous system (CNS), and these have a poor prognosis with only a 5-year survival rate of 32.5% (2). Additionally, lethal cases of neurodegenerative diseases including Alzheimer’s disease (AD) and Parkinson’s disease (PD) are becoming increasingly prevalent, and this is a serious concern as an estimated 6.5 million Americans were reported to live with AD in 2022 and 1 million having PD in 2019 (3, 4). While multiple mechanisms, including cellular senescence and dysfunctional mitophagy, have been associated with cancer development and age-related diseases (5–8), genomic instability arising from excessive DNA damage and/or DNA repair mechanism dysfunction is recognized as a key driving force for such diseases in general (9), and the development of CNS cancers and neurodegenerative diseases more specifically (10, 11).
High fidelity DNA repair is required to maintain genomic integrity. Breaks in DNA are a relatively common occurrence and can occur naturally up to tens of thousands of times per day, per cell. However, the number of such breaks can be drastically increased by exogenous insults such as ionizing radiation (IR) and oxidative stress. Exposure to high levels of IR can be tremendously detrimental and affects nearly every macromolecule in a cell. These effects can either occur directly or indirectly. For example, IR alone directly disrupts the structure of DNA, creating breaks in its molecular backbone (12). Alternatively, IR can elicit radiolysis of cellular water whereby the chemical bonds of water molecules are broken down resulting in the generation of reactive oxygen species (ROS) including hydroxyl radicals, ionized water, superoxide anions, and hydrogen peroxide (12). While ROS play essential roles in many cellular processes, excessive levels of ROS disrupt redox homeostasis and can induce DNA lesions (13). DNA lesions/breaks initiate DNA damage responses (DDR), a collection of mechanisms that ensure efficient DNA repair and maintain genomic integrity (14, 15). However, DNA repair is not infallible. If cells are permitted to replicate with unrepaired or incompletely repaired DNA, an accumulation of mutations and DNA damage may lead to cancer development, neurodegenerative disorders, and other age-related diseases. Furthermore, DNA damage has recently been implicated in the generation of detrimental inflammation, and this effect is often associated with the presence of self-DNA in the cytoplasm (16–19).
DNA damage due to exposure to IR, oxidative stress, or even chemotherapy, results in cytosolic DNA accumulation, often in the form of micronuclei (20). Micronuclei are small nuclei-like structures containing lagging or damaged chromosome fragments that continue into the interphase following completion of mitosis or meiosis (20) The nuclear envelopes surrounding micronuclei are typically defective and prone to rupture, after which, their DNA cargo is liberated into the cytosol. Additionally, micronuclei have been described as a source of complex genome rearrangements, including one-off catastrophic rearrangement events known as chromothripsis (21). Furthermore, the presence of micronuclei is associated with many autoimmune diseases (22–26), neurodegenerative diseases (27–32), and aggressive cancers (33) in affected tissues. Indeed, the presence of micronuclei has historically been used as a means to assess the genotoxicity of chemicals and mutagens via the cytokinesis-block micronucleus assay, and their contribution to the initiation of innate immune responses is now becoming apparent (34).
While micronuclei are likely to be a major source of cytoplasmic genomic self-DNA, mitochondria may also serve as a source of DNA in the cytosol (35). Mitochondria primarily function to produce the ATP necessary for normal cell activity via oxidative phosphorylation. Outside of oxidative phosphorylation, mitochondria also perform many other metabolic and non-metabolic roles ranging from the regulation of apoptosis to the generation of ROS that are necessary for maintaining redox homeostasis (36, 37). What makes mitochondria truly unique, however, is that they contain their own circular genome due to their endosymbiotic origin (38). The various means by which mitochondrial contents, including mitochondrial DNA (mtDNA), are released have been extensively reviewed elsewhere (39–44), but generally involves passive/accidental release due to mitochondrial stress and dysfunction, and cell death pathways including necrosis and apoptosis (40).
Since DNA is normally sequestered in the nucleus and mitochondria, its presence in the cytosol can function as a damage associated molecular pattern (DAMP) and serve to trigger inflammatory innate immune responses. In the CNS, such responses to both endogenous and exogenous insults must be tightly regulated to avoid damaging, or even lethal, inflammation. While the recruitment of peripheral leukocytes to the CNS and their subsequent activation are important in the development of immune responses in disease states, it is now recognized that resident glial cells, such as microglia and astrocytes, play a critical role in the initiation of detrimental neuroinflammation.
In the healthy brain, microglia and astrocytes are essential for homeostasis. Microglia perform critical housekeeping functions such as synaptic remodeling and pruning (45, 46), and removal of cellular debris and dead or dying cells (47), which is necessary for creating a regenerative environment (48). Astrocytes are the most abundant glial cell in the brain and have a crucial role in synaptogenesis, synaptic transmission, and neurotransmitter recycling, and for the maintenance of the blood brain barrier (BBB) (49–53). Importantly, it is now apparent that microglia and astrocytes both serve as sentinel cells that initiate and exacerbate immune responses associated with CNS pathology (54, 55). Given their wide distribution throughout the CNS, they are ideally positioned to confront and respond to trauma or invading pathogens. In disease states, microglia and astrocytes are activated and produce a wide array of potent proinflammatory mediators, such as IL-6, IL-1β, and TNF, as well as chemokines that promote the recruitment of peripheral leukocytes across the BBB that can further contribute to potentially damaging neuroinflammation (56–59). The initiation of microglial and astrocytic responses is now recognized to be mediated by multiple families of cell surface, endosomal, and cytosolic PRRs that are triggered by DAMPs and pathogen-associated molecular patterns (PAMPs). This subsequently results in the activation of transcription factors that precipitate the production of cell surface and secreted immune mediators.
Of these PRRs, perhaps the best studied are the cell surface and endosomal Toll-like receptors (TLRs) and the cytosolic nucleotide-binding and oligomerization domain-containing (NOD)-like receptors (NLRs), and these sensors have been exhaustively studied for their roles in antimicrobial and antiviral responses (as reviewed in 60–69). The TLR and NLR families consist of at least 10 and 22 members in mammals, respectively, and these receptors are widely expressed throughout the body on/in peripheral leukocytes and non-leukocytic cell types (70–72). Importantly, we, and others, have demonstrated the constitutive and/or inducible expression of TLRs (54, 55, 58, 73–75) and the NLRs, NOD1 and NOD2 (76–78), on/in both microglia and astrocytes.
These TLRs and NLRs can detect a variety of bacterial or viral extra- and intracellular PAMPs and DAMPs to activate downstream signaling cascades and initiate proinflammatory and/or antiviral activity by glial cells. However, NOD1/2 appears to be limited to the detection of bacterial cell wall components (78). Furthermore, while several TLRs are able to detect and respond to nucleic acids in endosomal compartments (72, 79, 80), they are not well suited to detect compromised cytosolic sterility or damage. Rather, it now appears that cells utilize discrete cytosolic PRR families that are capable of responding to the presence of foreign and/or self-nucleic acids in the cytoplasm.
PRR families, in addition to TLRs and NLRs, have recently been identified that serve as cytosolic sensors for foreign or altered self-nucleic acids (81). These include the retinoic acid-inducible gene I (RIG-I)-like family of receptors (RLRs) that can detect bacterial and viral nucleic acids and, more specifically, dsRNA (82–84). There are currently three known RLRs: RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). All three RLRs share a common central helicase domain and carboxy-terminal domain, although only RIG-I and MDA5 possess two caspase activation and recruitment domains (CARDs) that are required for downstream signal transduction (85). In contrast, LGP2 lacks a CARD domain and, instead, negatively regulates RIG-I and MDA5 activity (85, 86). Upon RNA binding, RIG-I and MDA5 interact with the CARD domain found on mitochondrial antiviral-signaling (MAVS), a mitochondria-localized adaptor protein, which subsequently activates TRAF family member-associated nuclear factor-kappa-B activator (TANK)-binding kinase 1 (TBK1) and inhibitor of nuclear factor kappa-B kinase subunit epsilon (IKKϵ). TBK1 and IKKϵ, in turn, activate the transcription factors interferon regulatory factors 3 and 7 (IRF3 and IRF7), which finally induce the transcription of type-I interferons (IFNs) among other antiviral mediators. Importantly, we have shown that RIG-I and MDA5 are constitutively expressed in microglia and astrocytes and that such expression is upregulated following viral infection (87). Furthermore, we have recently demonstrated that RIG-I not only recognizes viral dsRNA in glia, but is also able to respond to bacterial dsRNA (88).
Surprisingly, RIG-I may elicit immune responses to cytosolic DNA in addition to RNA, albeit in an indirect manner mediated by the actions of RNA polymerase III (RP3) (88–93), wherein RP3 reverse transcribes cytosolic dsDNA into a 5’ triphosphate-containing dsRNA ligand that then can subsequently be detected by RIG-I. Such a mechanism might explain the earlier, and perhaps erroneous, description of RP3 as a DNA sensor (92, 93).
However, it is now apparent that cells, including glia, possess a number of molecules that specifically serve as cytosolic DNA sensing molecules to initiate responses to foreign or self-DNA. The first of such molecules to be discovered was DNA sensor Z-DNA binding protein (ZBP1; previously known as DNA-dependent activator of IRFs (DAI)) in 2007 (94). Since then, our knowledge of cytosolic DNA sensing PRRs has expanded to include proteins such as cyclic GMP-AMP synthase (cGAS) (95), interferon gamma-inducible 16 (IFI16) (96), and absent in melanoma 2 (AIM2) (97–99). Importantly, we, and others, have described the expression and activity of cGAS (100, 101), IFI16 (102–104), ZBP1 (105–108), and AIM2 (101, 109, 110), in resident CNS cells including human and murine microglia and/or astrocytes.
As shown in Figure 1, cGAS and IFI16 use the endoplasmic reticulum-localized protein stimulator of interferon genes (STING) as an adaptor molecule to exert their responses, while ZBP1 and AIM2 recruit their own adaptor molecules, receptor interacting serine/threonine-protein (RIP) kinases 1 and 3 (RIPK1/3), and apoptosis-associated speck-like protein containing a CARD (ASC), respectively (99, 111). Upon activation, STING facilitates the activation of transcription factors TBK1, IRF3, and nuclear factor kappa-B (NF-κB) that then initiate the transcription of type-I IFNs, proinflammatory cytokines, and chemokines (112). ZBP1 recruits RIPK1 and 3 via interactions with its RIP homotypic interaction motif (RHIM) domains, which subsequently activates NF-κB (113). Both IFI16 and AIM2 can, like other receptors upstream of inflammasome formation, recruit ASC following DNA detection (99, 114). AIM2 and IFI16 can both form an inflammasome complex with ASC to recruit the effector protein, caspase-1, that subsequently cleaves pro-IL-1β and pro-IL18 to their mature forms for release (Figure 1) (99, 114). Additionally, AIM2-ASC inflammasome complex formation can lead to pyroptotic cell death via cleavage of gasdermin-D (GSDMD) (97, 98).

Figure 1 Cytosolic DNA sensors and their signaling pathways. Sensing of dsDNA by ZBP1 induces its association with RIPK3 and subsequent activation of NF-κB to elicit pro-inflammatory cytokine production, and/or its interaction with TBK1 to induce IRF3 activation and type-I IFN expression. Sensing of dsDNA by cGAS catalyzes the production of cGAMP that activates STING and leads to TBK1 activation that induces IRF3 activation and type-1 IFN expression. IFI16 can directly interact with STING following DNA sensing resulting in NF-κB and IRF3 activation, or can associate with ASC to form an inflammasome complex resulting in caspase-1-mediated IL-1β and IL-18 release. AIM2 sensing of dsDNA also leads to inflammasome complex formation with ASC and mature IL-1β and IL-18 release. This figure was created with Biorender.com.
Finally, several other putative DNA sensors such as DNA-dependent protein kinase (DNA-PK) (115), DEAD-box helicase 41 (DDX41) (116), and meiotic recombination 11 homolog A (MRE11) (117) have been described in peripheral cell types, but their functions as PRRs for DNA in the CNS have not been investigated to date.
Peripheral and resident CNS cells exhibit inflammatory phenotypes following DNA damage (118–120), although the mechanisms underlying the initiation of these responses have remained elusive. Interestingly, several cytosolic DNA sensors appear to be capable of detecting mitochondrial and genomic self-DNA resulting from insults such as oxidative stress and IR (41, 121–128). As such, cytosolic DNA sensors may be the link between DNA damage and subsequent innate immune responses.
The presence of DNA in the cytoplasm is indicative of cell compromise as it is normally confined to the nucleus or mitochondria in healthy cells. While cytosolic DNA sensors have been extensively studied with regard to bacterial and viral infections, it has only been recently recognized that they may also play a critical role in the generation of immune responses to cytosolic mtDNA and genomic self-DNA (42, 121, 129–131). In contrast to endosomal nucleic acid sensors such as TLR9 that detect prokaryotic DNA based on their distinct methylation patterns (132), cytosolic DNA sensors seem to be unable to discriminate between foreign and self-DNA, and so can detect and respond to either (133, 134). However, it remains unclear whether such responses to self-DNA are protective or detrimental, especially in the context of the CNS where there is strong potential for damaging neuroinflammation. Regardless, it is now apparent that glial cells express multiple sensors capable of initiating their immune functions in response to the presence of cytosolic DNA as described below.
Of all the DNA sensors, cGAS has risen to the forefront of nucleic acid sensor research since it’s discovery in 2013 (95). Over the past decade, our understanding of its role has expanded from the triggering of antiviral immunity to include the inhibition of homologous recombination-mediated DNA repair (135, 136), control of DNA replication dynamics (137), cellular senescence (35, 138–141), cell death (142–147), and tumorigenesis (136).
As shown in Figure 2A, following DNA binding, cGAS catalyzes the production of the secondary messenger molecule 2’3’cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which subsequently binds to STING that is located on the surface of the endoplasmic reticulum (95, 148, 149). Following interaction with cGAMP, STING undergoes a conformational change that initiates the recruitment of TBK1, which then phosphorylates IRF3 and liberates NF-κB. Activation of this signaling cascade results in the production of potent proinflammatory cytokines and chemokines including IL-6, TNF, and IL-8, and the type-I IFNs such as IFN-β (112, 150–152).
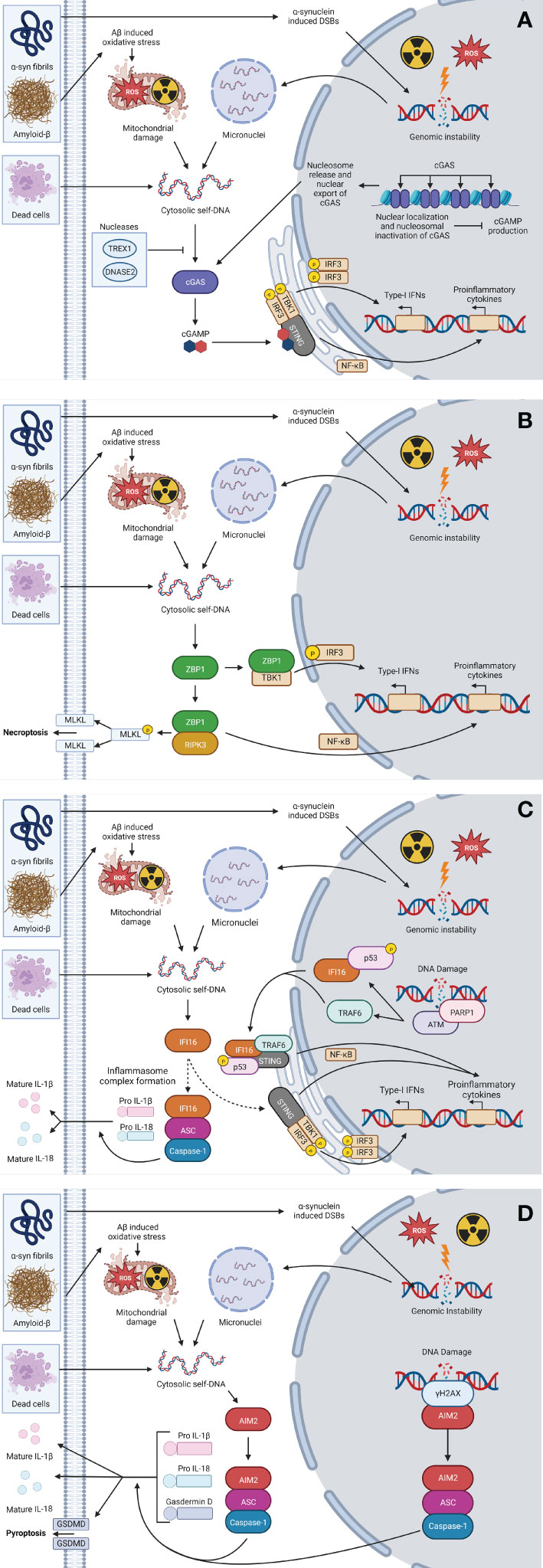
Figure 2 Cytosolic self-DNA sensing pathways. In the CNS, the uptake of α-Syn fibrils, amyloid-β, and/or cellular debris by resident CNS cells, or exposure to exogenous oxidative stress or ionizing radiation (IR), can lead to liberation of self-DNA to the cytosol via mitochondrial damage or genomic instability leading to micronuclei formation. The presence of self-DNA in the cytoplasm can then be perceived via DNA sensing PRRs including cGAS (A), ZBP1 (B), IFI16 (C), and/or AIM2 (D), leading to sensor-specific signaling pathways that precipitate the production of inflammatory cytokines, type-I IFNs and/or immunogenic necroptotic and pyroptotic cell death pathways. This figure was created with Biorender.com.
The discrimination, or lack thereof, between foreign and self-DNA has been recognized as a significant issue regarding cytosolic DNA sensors since indiscriminate DNA binding could lead to the development of autoimmune responses. Indeed, several autoimmune diseases, including Aicardi-Goutières syndrome (AGS), are associated with increased levels of cytoplasmic DNA and inflammatory mediator production (153). While DNA-mediated activation of cGAS occurs in a length dependent manner, with robust activity occurring only with DNA longer than 45 bp or shorter DNA fragments with flayed ends (154, 155), its ability to discriminate between DNA fragments seems to end here.
DNA damage and genomic instability has long been known to induce inflammatory responses (123, 156, 157), but it has only been recently that this phenomenon has been shown to be connected to cytosolic nucleic acid sensors including cGAS. In 2017, Mackenzie et al. (121) discovered that a primary culprit in the induction of inflammatory responses following IR-induced DNA damage was the formation of micronuclei. The breakdown of the micronuclear envelope and subsequent exposure of self-DNA was associated with rapid cGAS translocation to the micronuclei and the onset of proinflammatory immune responses (121), a result that has since been corroborated in other studies (122, 158–163). Additionally, DNA damaging events often precipitate the release of mtDNA to the cytoplasm and this can similarly be recognized by cGAS (19, 164–169).
Importantly, mammalian cells appear to possess mechanisms that serve to limit cGAS activation to prevent excessive or prolonged activation that could lead to devastating autoimmunity. First, recent studies have determined that cGAS may primarily be localized to the nucleus in cells at rest (170, 171), rather than being cytosolic as was initially thought (95). In the nucleus, cGAS is tightly bound to nucleosomes where its DNA binding sites are prevented from interacting with nucleosomal DNA (172–177). Furthermore, while nuclear cGAS activation can occur, cGAMP production resulting from such activation is approximately 500-fold less than that generated when DNA is administered to the cytoplasm (171).
Second, cytoplasmic nucleases, such as three prime repair exonuclease 1 (TREX1) and deoxyribonuclease II (DNASE2) restrict cytoplasmic DNA accumulation thereby limiting cGAS activation (162). The impact of these nucleases is particularly apparent in inflammatory autoimmune disorders that occur due to mutations that result in their loss of function (141, 178, 179). Together, these studies reinforce the importance of the regulation of DNA sensing proteins, especially cGAS.
ZBP1 was the first cytosolic DNA sensor discovered (94) and its importance in host responses to viral infection have been extensively described (105, 180). ZBP1 was initially found to be a critical component in the detection of the dsDNA of HSV-1 (94) and other viruses in both peripheral and CNS cell types (105, 107, 108, 111, 181). More recently, it appears that this sensor may also recognize RNA motifs in addition to DNA (182). Furthermore, this cytosolic sensor has been implicated in the initiation of the necroptotic pathway of immunogenic cell death (105–108, 111, 182–188).
As shown in Figure 2B, upon binding to DNA, ZBP1 recruits RIPK3 that subsequently activates NF-κB to induce proinflammatory cytokine production (113). Additionally, ZBP1 has been shown to associate with TBK1 and IRF3 to regulate the activation of IRF3 and, thus, drive type-I IFN production (94). However, it is important to note that this receptor may act in a ligand and cell type specific manner, as ZBP1 knockdown was found to have no effect on exogenous DNA-induced IFN production in mouse embryonic fibroblasts (189) or a lung epithelial cell line (190), while similar knockdown significantly reduced B-DNA-induced IFN-β production in an immortalized murine fibroblast cell line (190).
Importantly, our current knowledge of the role of ZBP1 in responses to self-DNA remains limited, especially since the precise identity of the ligand(s) for ZBP1 remains controversial (181, 182, 187, 189, 191). However, there is data to support the notion that mtDNA serves as a ligand for ZBP1 (192, 193). A recent study has indicated that mtDNA release induced by low level oxidative stress, in the absence of detectable damage to nuclear DNA, elicits a type-I IFN response by pulmonary epithelial cells (192). Interestingly, in this study, fragments of mtDNA were shown to be released in exosomes that were capable of initiating further inflammatory responses in naïve epithelial cells (192). Furthermore, a second study reported the ability of glucose deprivation to induce mtDNA release via the actions of NOXA in FVB/NJ mice following implantation of the MVT-1 mammary cancer cell line (193), which subsequently initiated necroptosis in a ZBP1-dependent manner (193).
IFI16 is a DNA sensor that is a member of the pyrin and HIN domain (PYHIN) family. While it was first identified in 1992 (194), it wasn’t until 2010 that it was characterized as a cytosolic DNA sensor capable of inducing IFN-β production in response to transfected exogenous DNA (96), and such responses have been reported to occur following direct interaction with STING (195) (Figure 2C). Interestingly, IFI16 exhibits both nuclear and cytosolic localization (196). In the nucleus, IFI16 has been shown to respond to viral DNA, which leads to the formation of an inflammasome that then translocates to the cytoplasm where it participates in the cleavage of pro IL-1β and IL-18 into their mature forms for release (114, 197, 198).
With regard to self-DNA detection, the murine ortholog of IFI16, IFI204, was found to mediate the detection of DNA released into the cytoplasm following DNA damage resulting from ataxia-telangiectasia mutated (ATM) deficiency in a murine model of ataxia telangiectasia (A-T) (123). Since the work of Mackenzie et. al (121), has indicated that cytosolic self-DNA recognition occurs due to the formation of micronuclei, it is possible that a similar mechanism underlies that ability of IFI204/IFI16 to perceive the presence of self-DNA, although further study will be necessary to confirm such a hypothesis.
While IFI16 is capable of binding self-DNA, it displays a preference for long non-self-DNA due to its ability to oligomerize into clusters, forming foci that are unable to bind nucleosomal self-DNA (199, 200). Alternatively, IFI16 may serve to detect DNA damage indirectly via the formation of a complex with DDR proteins that can subsequently initiate STING signaling (Figure 2C). Following etoposide-induced genotoxic stress, IFI16 has been found to combine with the DDR proteins ATM and poly(ADP-ribose) polymerase 1 (PARP1) to induce the formation of a STING signaling complex that results in the activation of NF-κB and proinflammatory cytokine production (200). This ATM-initiated nuclear mechanism results in far more rapid responses to DNA damage than those mediated by cGAS that require the formation and rupture of micronuclei (200). Furthermore, such responses predominantly result in the activation of NF-κB, rather than IRF3, resulting in a pro-inflammatory response (200). However, it is important to note that most of this work has been performed in keratinocytes and it remains to be determined whether such mechanisms exist in resident CNS cell type.
AIM2 is another member of the PYHIN family that appears to have DNA sensing capabilities. First identified in 1997 (201), its role in dsDNA detection was not recognized until a decade later (97–99, 202). As shown in Figure 2D, AIM2 recruits the inflammasome adaptor protein ASC and procaspase-1 following binding to cytosolic DNA, forming an inflammasome complex that permits IL-1β and IL-18 maturation via the actions of caspase-1 (97–99, 202). While AIM2, like ZBP1, can also initiate immunogenic cell death pathways, AIM2 appears to initiate pyroptosis rather than necroptosis via the cleavage of gasdermin-D and subsequent pore formation (97–99, 202).
Importantly, AIM2 has been shown to detect cytosolic self-DNA and mtDNA (131, 203–205). The interaction between AIM2 and cytosolic self-DNA has primarily been studied in the context of autoimmune diseases that characteristically entail cytosolic DNA accumulation, such as arthritis, psoriasis, and systemic lupus erythematosus (131, 203, 206). Furthermore, pharmacological disruption of the nuclear envelope and the liberation of self-DNA to the cytosol has also been shown to induce AIM2 activation (205). As such, it has been inferred from these observations that AIM2 is capable of recognizing DNA and might be able to do so following micronuclei formation.
In addition, a limited number of studies suggest that AIM2 may also be localized to the nucleus in some cell types, such as macrophages (124), and can mediate responses to nuclear DNA damage (124, 207). For example, AIM2 has been reported to co-localize with the DNA damage marker gamma-H2A histone family member X (γH2AX) at sites of double strand breaks (DSBs) following IR exposure in macrophages, where it subsequently forms an AIM2-ASC-caspase-1 inflammasome complex to trigger pyroptotic cell death (124).
While the relative importance of AIM2-mediated DNA detection in the cytosol versus the nucleus remains unclear, it is noteworthy that the localization of the AIM2 inflammasome complex to sites of nuclear DNA DSBs occurs as rapidly as 8 hours following IR exposure (124). Furthermore, it is interesting that these inflammasome complexes have been observed to accumulate in the perinuclear region (124). However, it remains to be determined whether such accumulations involve additional interactions of AIM2 with cytosolic self-DNA.
To date, the mechanisms by which resident CNS cells can detect and respond to self- DNA are understudied, and it remain unclear whether the net result of such responses are beneficial or detrimental. Neuroinflammation, while crucial in protecting the brain against infection, can result in serious neurological damage if it is of inappropriate intensity or duration. Indeed, neuroinflammation that stems from DNA damage and/or deficient or defective DNA repair can underlie or exacerbate neurodegeneration in CNS disease states (11, 208–210). For example, the detection of cytosolic mtDNA and self-DNA by resident glial cells is a hallmark of CNS pathologies including AGS, AD, PD, A-T, and Huntington’s disease (HD) (44, 109, 160, 179, 211–216), as summarized in Table 1.
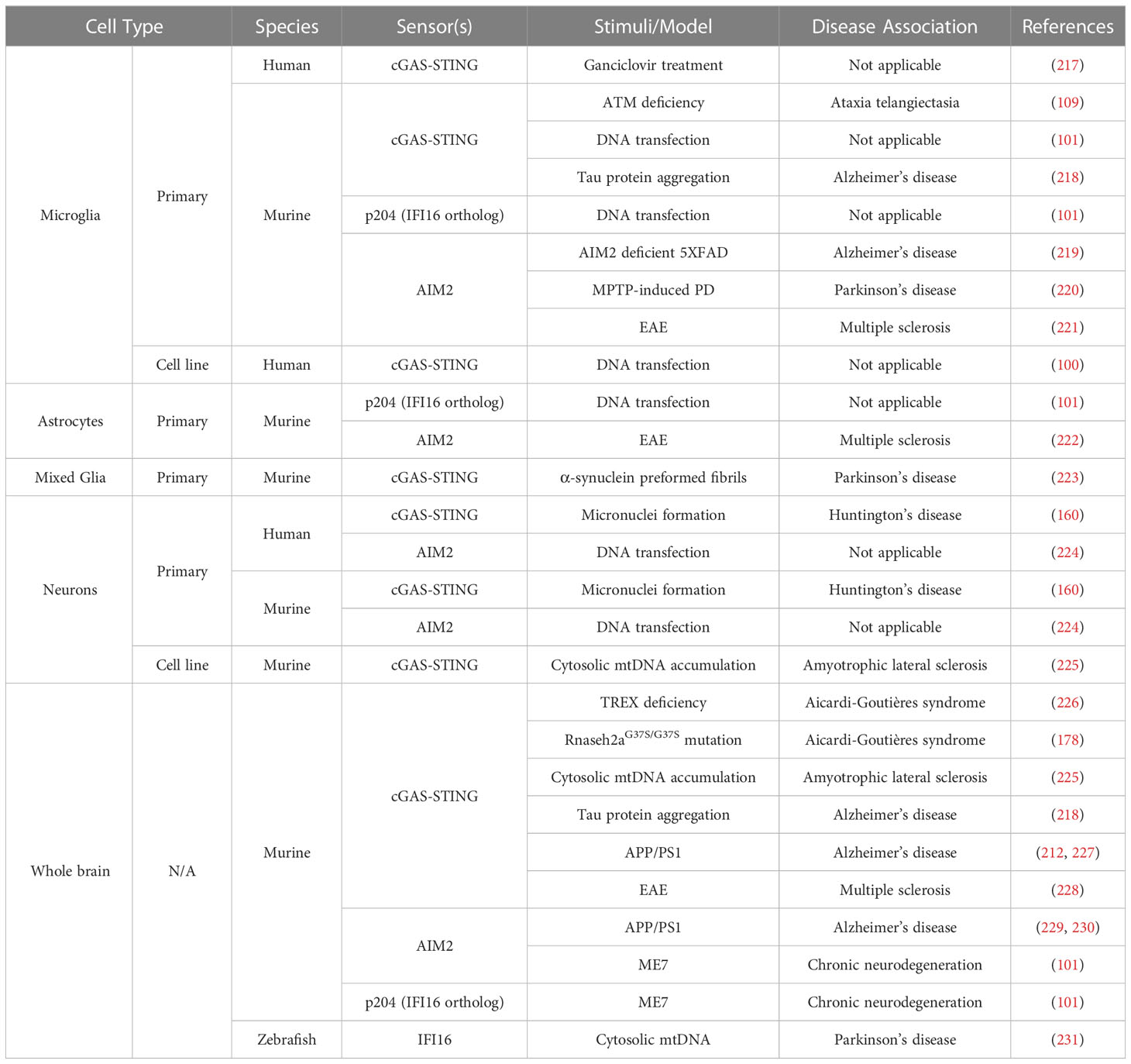
Table 1 Expression of DNA sensors by CNS cells and links to neurogenerative disorders.
In some CNS disorders, such as A-T and AGS, the origins of cytosolic DNA accumulation are clear. In A-T, a critical kinase in DDRs, ATM, is defective and results in the accumulation of DSBs leading to the presence of cytosolic DNA (10, 123), while in AGS, mutations in the genes encoding products that process/degrade nucleic acids, such as TREX1 and RNAse H2, lead to cytosolic DNA accumulation and lethal autoimmunity in neonates (178, 226).
However, the origin of cytosolic DNA in other neurodegenerative diseases is either unclear or unknown. For example, in AD, amyloid-β (Aβ) plaques have been shown to induce oxidative stress that can cause mitochondrial dysfunction and subsequent mtDNA release into the cytosol (232). In contrast, the α-synuclein (α-Syn) fibrils that are commonly seen in PD can induce genomic DNA damage (223), while affected striatal neurons in HD patients and murine models of this disease show significantly higher numbers of micronuclei. However, the mechanisms underlying DNA damage in HD remain unknown (160).
We, and others, have shown that exogenous cytosolic DNA elicits reactive astrogliosis and microgliosis, and is associated with the production of proinflammatory and antiviral mediators by these cells (44, 100, 180, 212, 216, 233, 234). Importantly, the recent demonstration that glial cells express multiple cytosolic sensors has provided the means by which they perceive DNA. We showed that ZBP1 is expressed in microglia and astrocytes in an inducible manner following HSV-1 infection (106). In addition, Cox et. al (101), provided evidence that murine microglia and astrocytes express mRNA encoding cGAS, the p204 murine ortholog of IFI16, and AIM2 (101). Subsequently, we, and others, have extended these findings to demonstrate cGAS, IFI16, and AIM2, protein expression in human microglia and astrocytes, and have shown their ability to mediate glial immune responses to exogenous DNA administration (100, 102, 105, 224).
Transfection of exogenous DNA into human and murine primary glia and cell lines elicits the production of type-I IFNs and proinflammatory cytokines including CCL3, CCL5, CXCL2, TNF, IFN-β, and IL-6 (87, 88). The importance of the cytosolic DNA sensors, cGAS, IFI16 (and murine ortholog p204), and AIM2, in the generation of such responses is implied by the demonstration that dsDNA transfection into primary murine microglia and astrocytes significantly upregulates the expression of mRNA encoding these sensors (101). Importantly, we have shown that the responses of a human microglia cell line to intracellular exogenous DNA administration were significantly attenuated following CRISPR/Cas9 knockdown of cGAS expression (102). Furthermore, the secretion of mature IL-1β, downregulated dendritic growth, and enhanced axon extension of primary human and murine neurons elicited by dsDNA transfection was found to be dependent on the presence of AIM2 (224). As such, it is apparent that resident CNS cells are capable of responding to the presence of exogenous cytosolic DNA and do so via various sensor molecules.
With regard to the ability of resident CNS cells to perceive the presence of cytosolic self- DNA, glia have recently been shown to respond to mtDNA and self-DNA accumulation via sensors including cGAS and AIM2 (44, 109, 168, 214, 216, 223, 235, 236), as summarized in Table 1. For example, the addition of α-Syn preformed fibrils (PFF) to primary murine mixed glial cultures to simulate PD pathology resulted in DNA DSBs, an accumulation in cytosolic DNA, and subsequent activation of STING and TBK1 that resulted in type I IFN production (223). Importantly, these responses were attenuated by the pharmacological inhibition of STING activation (223). These findings are consistent with those in an in vivo mouse model where α-Syn-PFF similarly induced DNA damage, TBK1 activation, and IFN production by microglia in situ that preceded PD-like dopaminergic neurodegeneration, and the demonstration that substantia nigra pars compacta tissue from human PD patients show elevated STING protein levels that correlate with α-Syn-PFF accumulation (223).
The cGAS-STING axis has been implicated in the initiation of neurotoxic responses of primary microglial cultures to ATM mutations/deficiency that, again, results in the accumulation of cytosolic self-DNA and models A-T (109). This DNA sensing pathway has also been linked to striatal neuron cell death in HD (160). In primary human and murine HD-affected striatal neurons, Sharma et al. (160) reported a high incidence of micronuclei formation that coincided with cell death due to autophagy, and the increased expression of mRNA encoding CCL5 and CXCL10 that was abolished following cGAS depletion (160). Additionally, studies in mouse models of other neurodegenerative disorders have similarly indicated a role for cGAS in their progression. Most notably, models of AGS development that feature mutations of TREX1 and RNAse H2 have revealed that cGAS is essential for the initiation of the autoimmune responses associated with this disorder (178, 226).
Interestingly, AIM2-mediated responses have also been associated with neurodegenerative diseases but, in contrast to cGAS, such response appear to play a protective rather than a detrimental role, as summarized in Table 1. In primary murine microglia, AIM2 has been shown to alleviate the damaging neuroinflammation seen in the experimental autoimmune encephalitis (EAE) model of multiple sclerosis (MS), and other mouse models of AD and PD (219–221). In EAE, AIM2 was shown to negatively regulate DNA-PK activity in an inflammasome independent manner, and AIM2 deficiency was found to increase levels of microglial activation and peripheral leukocyte recruitment to the CNS (221). Furthermore, the inhibitory activity of AIM2 was found to be comparable to that of pharmacological DNA-PK inhibition (221). However, it should be noted that such findings conflict with other reports suggesting inflammasome components are required for the recruitment of peripheral T-cells to the CNS that exacerbate neuroinflammation in EAE (237, 238). As such, the role of AIM2 in inflammatory CNS diseases appears complex as it relates to both resident glia and peripheral leukocytes and further research is clearly required.
In the 5XFAD model of AD, deletion of AIM2 resulted in a decrease in Aβ deposition, but caused an elevation in the production of the key inflammatory cytokines IL-6 and IL-18, further supporting a negative regulatory role for AIM2 in neuroinflammation (219). Finally, in a neurotoxin (N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine, MPTP)-induced PD mouse model, AIM2 activation served to limit cGAS activity via interference with protein kinase B (AKT)-mediated IRF3 phosphorylation, and conditional knockout of AIM2 in microglia, but not peripheral cells, exacerbated PD-like disease severity (220).
As such, while it is clear that cytosolic DNA sensors play critical roles in the initiation and/or progression of CNS pathologies in animal models (as summarized in Table 1), their specific roles can often appear contradictory and so may be sensor and/or disease condition specific. Additionally, while light has been shed on the beneficial/detrimental effects mediated by these receptors in murine CNS cells and neurodegenerative disorder models, it remains unclear whether cytosolic DNA sensors exert similar functions in human disease. A better understanding of these DNA sensors in the context of human neurons/glia and patients with CNS disorders could identify new targets for therapeutic intervention to limit neuroinflammation and/or to promote beneficial immune responses.
Taken together, it has become apparent that resident CNS cells play a critical role in the protective and detrimental immune responses associated with CNS infection and damage, and the development/progression of neurological disorders. Furthermore, it is increasingly clear that such responses are initiated via the detection of DAMP and PAMP motifs that are associated with cellular damage and infectious agents, respectively. The principal glial cells, microglia and astrocytes, express various cell-surface, endosomal, and cytosolic members of multiple PRR families that trigger immune mediator production to foster neuroinflammation and recruit leukocytes to the CNS. Importantly, glia can constitutively, and/or be induced to, express PRRs that detect the presence of DNA in the cytosolic compartment. While these sensors were initially characterized as components in the detection of viral and bacterial nucleic acids by microglia and astrocytes, it is now recognized that molecules, including cGAS, IFI16, and AIM2, can play important roles in the generation of responses to the presence of self-DNA in the cytosol resulting from DNA damaging insults, such as IR or oxidative stress, or deficient/defective DNA repair. As such, these mechanisms might be targetable to either augment glia-mediated responses that serve to protect against tumorigenesis, or prevent the inflammatory responses of these cells that initiate or exacerbate damaging neuroinflammation.
To date, several small molecule inhibitors and agonists have been developed for cGAS (239) and AIM2 (240), as well as for the downstream adaptor proteins STING (241–245) and RIPK1/3 (246). These agents have shown good efficacy in both peripheral cells and isolated glia where pharmacological inhibition of these components has led to significant reductions in the production of immune mediators including IFN-β, IL-1β, and IL-6, following activation (105, 240, 241, 245, 247–250). Indeed, the STING agonists, TAK-676 (NCT04420884, NCT04879849) and E7766 (NCT04144140), are currently being tested clinically for the treatment of advanced or metastatic solid tumors, lymphomas, and leukemias, as adjunctive agents to conventional chemotherapy. As such, it might be possible in the future to target the DNA sensors or their downstream adaptor proteins associated with CNS cancers and neurodegenerative diseases.
However, considerable hurdles remain for the development and use of such agents as the available data regarding the specific role of each cytosolic DNA sensor in CNS disorders can be unclear or even contradictory. This may be due to species-specific differences, such as in the ability of STING to be activated by cGAMP (251), or it may be that the glial responses initiated by the presence of cytosolic DNA are sensor, cell type, or even disease stage, dependent. In addition, the ability of many small molecule inhibitors and agonists that target cytosolic DNA sensors to cross the BBB and their efficacy in the brain have not been determined. Indeed, marked differences in the effectiveness of such agents has been noted between studies in isolated glia and those in murine model or clinical settings, primarily due to poor BBB penetration, as reviewed elsewhere (252). While various methods have been successfully employed to circumvent this issue, including BBB disruption and intracerebral, intrathecal, or intranasal, delivery, each carries its own problems, such as neurotoxicity (BBB disruption) or a high degree of invasiveness (intrathecal delivery) (252). Finally, the potential for adverse side-effects of agents targeting DNA sensor mechanisms remains unclear, as no DNA sensor inhibitors or agonists are currently undergoing clinical trials for their efficacy in the treatment of CNS tumors or neurodegenerative disease.
Clearly, more study of these novel cytosolic DNA sensor pathways is warranted given our current lack of understanding of the role of each in glial functions in the context of specific CNS disorders and their stages. Furthermore, successfully establishing the in situ efficacy of agonists/antagonists of these DNA sensor pathways in the CNS, and solving the issue of delivery across the BBB, could represent an exciting new therapeutic modality that might be used alone or in conjunction with existing approaches to improve the treatment of a wide range of CNS pathologies.
AS and IM co-wrote this literature review article. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. SEER cancer stat facts: Cancer of any site. National Cancer InstituteBethesda, MD (2022). Available at: https://seer.cancer.gov/statfacts/html/all.html.
2. SEER cancer stat facts: Brain and other nervous system cancer. Bethesda MD: National Cancer Institute (2022). Available at: https://seer.cancer.gov/statfacts/html/brain.html.
3. Alzheimer’s disease. In: Centers for disease control and prevention. Available at: https://www.cdc.gov/dotw/alzheimers/index.html.
4. Rong S, Xu G, Liu B, Sun Y, Snetselaar LG, Wallace RB, et al. Trends in mortality from parkinson disease in the united states, 1999–2019. Neurology (2021) 97(20):e1986–93.
5. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest [Internet]. (2018) 128(4):1238–46. https://www.jci.org/articles/view/95148.
6. Childs BG, Durik M, Baker DJ, Van Deursen JM. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat Med (2015) 21(12):1424–35.
7. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Denis Alexander H, Ross OA. Age and age-related diseases: Role of inflammation triggers and cytokines. Front Immunol (2018) 9(APR):1–28.
8. Cai Q, Jeong YY. Mitophagy in alzheimer’s disease and other age-related neurodegenerative diseases. Cells [Internet]. (2020) 89(1):150. https://www.mdpi.com/2073-4409/9/1/150.
9. Zhao Y, Simon M, Seluanov A, Gorbunova V, damage DNA. And repair in age-related inflammation. Nat Rev Immunol [Internet]. (2023) 23(2):75–89. https://www.nature.com/articles/s41577-022-00751-y.
10. Zhu L-S, Wang D-Q, Cui K, Liu D, Zhu L-Q. Emerging perspectives on dna double-strand breaks in neurodegenerative diseases. Curr Neuropharmacol. (2019) 17(12):1146–57.
11. Coppedè F, Migliore L. DNA Damage in neurodegenerative diseases. Mutat Res - Fundam Mol Mech Mutagen [Internet]. (2015) 776:84–97. doi: 10.1016/j.mrfmmm.2014.11.010
12. Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM. Effects of ionizing radiation on biological molecules - mechanisms of damage and emerging methods of detection. Antioxidants Redox Signal (2014) 21(2):260–92.
13. Saikolappan S, Kumar B, Shishodia G, Koul S, Koul HK. Reactive oxygen species and cancer: A complex interaction. Cancer Lett [Internet] (2019) 452:132–43. doi: 10.1016/j.canlet.2019.03.020
14. Giglia-Mari G, Zotter A, Vermeulen W. DNA Damage response. Cold Spring Harb Perspect Biol (2011) 3(1):1–19.
15. Abuetabh Y, Wu HH, Chai C, Al Yousef H, Persad S, Sergi CM, et al. DNA Damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp Mol Med [Internet]. (2022) 54(10):1658–69. doi: 10.1038/s12276-022-00863-4
16. Horn V, Triantafyllopoulou A. DNA Damage signaling and polyploid macrophages in chronic inflammation. Curr Opin Immunol [Internet]. (2018) 50:55–63. doi: 10.1016/j.coi.2017.11.002
17. Taffoni C, Steer A, Marines J, Chamma H, Vila IK, Laguette N. Nucleic acid immunity and dna damage response: new friends and old foes. Front Immunol (2021) 12(April):1–10.
18. Li T, Chen ZJ. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med [Internet]. (2018) 215(5):1287–99. doi: 10.1084/jem.20180139
19. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep [Internet]. (2019) 29(5):1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050
20. Krupina K, Goginashvili A, Cleveland DW. Causes and consequences of micronuclei. Curr Opin Cell Biol [Internet]. (2021) 70:91–9. doi: 10.1016/j.ceb.2021.01.004
21. Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature (2015) 522(7555):179–84.
22. Ballardin M, Barsacchi R, Bodei L, Caraccio N, Cristofani R, Di Martino F, et al. Oxidative and genotoxic damage after radio-iodine therapy of graves’ hyperthyroidism. Int J Radiat Biol (2004) 80(3):209–16.
23. Zúñiga-González GM, Batista-González CM, Gómez-Meda BC, Ramos-Ibarra ML, Zamora-Perez AL, Muñoz-Magallanes T, et al. Micronuclei in diabetes: Folate supplementation diminishes micronuclei in diabetic patients but not in an animal model. Mutat Res - Genet Toxicol Environ Mutagen (2007) 634(1–2):126–34.
24. Karaman A, Binici DN, Melikoĝlu MA. Comet assay and analysis of micronucleus formation in patients with rheumatoid arthritis. Mutat Res - Genet Toxicol Environ Mutagen (2011) 721(1):1–5.
25. Donmez-Altuntas H, Sut Z, Ferahbas A, Hamurcu Z, Demirtas H. Increased micronucleus frequency in phytohaemagglutinin-stimulated blood cells of patients with vitiligo. J Eur Acad Dermatol Venereol [Internet]. (2007) 22(2):162–7. doi: 10.1111/j.1468-3083.2007.02356.x
26. Silva FSG, Oliveira H, Moreiras A, Fernandes JC, Bronze-da-Rocha E, Figueiredo A, et al. Cytotoxic and genotoxic effects of acitretin, alone or in combination with psoralen-ultraviolet a or narrow-band ultraviolet b-therapy in psoriatic patients. Mutat Res - Genet Toxicol Environ Mutagen [Internet]. (2013) 753(1):42–7. doi: 10.1016/j.mrgentox.2012.12.017
27. Sprung CN, Chao M, Leong T, McKay MJ. Chromosomal radiosensitivity in two cell lineages derived from clinically radiosensitive cancer patients. Clin Cancer Res (2005) 11(17):6352–8.
28. Trippi F, Scarpato R, Petrozzi L, Bonuccelli U, Latorraca S, Sorbi S, et al. Spontaneous and induced chromosome damage in somatic cells of sporadic and familial alzheimer’s disease patients. Mutagenesis [Internet]. (2001) 16(4):323–7. http://mutage.oxfordjournals.org/content/16/4/323.short.
29. Sathasivam K, Woodman B, Mahal A, Bertaux F, Wanker EE, Shima DT, et al. Centrosome disorganization in fibroblast cultures derived from R6/2 huntington’s disease (HD) transgenic mice and HD patients. Hum Mol Genet (2001) 10(21):2425–35.
30. Wang Y, Duan H, Dai Y, Bin P, Cheng J, Pan Z, et al. Antaxia-telangiectasia mutated gene polymorphisms and susceptibility to chromosomal damage among polycyclic aromatic hydrocarbons exposed workers. Sci Total Environ [Internet]. (2009) 407(8):2615–20. doi: 10.1016/j.scitotenv.2009.01.002
31. Migliore L, Scarpato R, Coppede F, Petrozzi L, Bonuccelli U, Rodilla V. Chromosome and oxidative damage biomarkers in lymphocytes of parkinson’s disease patients. Int J Hyg Environ Health (2001) 204(1):61–6.
32. Petrozzi L, Lucetti C, Gambaccini G, Bernardini S, Del Dotto P, Migliore L, et al. Cytogenetic analysis oxidative damage in lymphocytes of parkinson’s disease patients. Neurol Sci (2001) 22(1):83–4.
33. Cortés-Ciriano I, Lee JJK, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet (2020) 52(3):331–41.
35. Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell (2021) 184(22):5506–26.
36. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell [Internet]. (2015) 163(3):560–9. doi: 10.1016/j.cell.2015.10.001
37. Andreyev AY, Kushnareva YE. Mitochondrial metabolism of reactive oxygen Species. Biochem [Internet]. (2005) 70(2):200–14. Availablr at: http://www.oxphos.org/staticfile/pubs/Mitochondrialmetabolismofreactiveoxygenspecies.pdf%0A http://protein.bio.msu.r/biokhimiya/contents/v70/pdf/bcm_0200.pdf
38. Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol (2017) 27(21):R1177–92.
39. Carvalho G, Repolês BM, Mendes I, Wanrooij PH. Mitochondrial dna instability in mammalian cells. Antioxidants Redox Signal (2022) 36(13–15):885–905.
40. Wu Z, Sainz AG, Shadel GS. Mitochondrial DNA: cellular genotoxic stress sentinel. Trends Biochem Sci [Internet]. (2021) 46(10):812–21. doi: 10.1016/j.tibs.2021.05.004
41. Moya GE, Rivera PD, Dittenhafer-Reed KE. Evidence for the role of mitochondrial dna release in the inflammatory response in neurological disorders. Int J Mol Sci [Internet]. (2021) 22(13):7030. https://www.mdpi.com/1422-0067/22/13/7030.
42. De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, et al. Molecular mechanisms of mtdna-mediated inflammation. Vol. 10 Cells (2021), 1–21.
44. Zhao Y, Liu B, Xu L, Yu S, Fu J, Wang J, et al. ROS-induced mtDNA release: The emerging messenger for communication between neurons and innate immune cells during neurodegenerative disorder progression. Antioxidants [Internet]. (2021) 10(12):1917. https://www.mdpi.com/2076-3921/10/12/1917.
45. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: New roles for the synaptic stripper. Neuron [Internet]. (2013) 77(1):10–8. doi: 10.1016/j.neuron.2012.12.023
46. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Sci (80-). (2011) 333(6048):1456–8.
47. Tremblay M-E, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci [Internet]. (2011) 31(45):16064–9. doi: 10.1523/JNEUROSCI.4158-11.2011
48. Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain [Internet]. (2008) 132(2):288–95. doi: 10.1093/brain/awn109
49. Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Sci (80- ). (2012) 337(6092):358–62.
50. Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature (2013) 504(7480):394–400.
51. Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature (2012) 486(7403):410–4.
52. Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, et al. Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature (2014) 509(7499):189–94.
53. Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Sci (80- ). (2011) 334(6063):1727–31.
54. Carpentier PA, Duncan DS, Miller SD. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun (2008) 22(2):140–7.
55. Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol (2005) 175(7):4320–30.
56. Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J Neuroinflammation. (2012) 9:1–18.
57. Heneka MT, O’Banion MK. Inflammatory processes in alzheimer’s disease. J Neuroimmunol (2007) 184(1–2):69–91.
58. Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol (2002) 61(11):1013–21.
59. Griffiths MR, Gasque P, Neal JW. The multiple roles of the innate immune system in the regulation of apoptosis and inflammation in the brain. J Neuropathol Exp Neurol (2009) 68(3):217–26.
60. Wicherska-Pawłowska K, Wróbel T, Rybka J. Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-Like receptors (RLRs) in innate immunity. TLRs, NLRs, and RLRs ligands as immunotherapeutic agents for hematopoietic diseases. Int J Mol Sci [Internet]. (2021) 22(24):13397. https://www.mdpi.com/1422-0067/22/24/13397.
61. Asami J, Shimizu T. Structural and functional understanding of the toll-like receptors. Protein Sci (2021) 30(4):761–72.
62. Mokhtari Y, Pourbagheri-Sigaroodi A, Zafari P, Bagheri N, Ghaffari SH, Bashash D. Toll-like receptors (TLRs): An old family of immune receptors with a new face in cancer pathogenesis. J Cell Mol Med (2021) 25(2):639–51.
63. Lind NA, Rael VE, Pestal K, Liu B, Barton GM. Regulation of the nucleic acid-sensing toll-like receptors. Nat Rev Immunol (2022) 22(4):224–35.
64. Li L, Acioglu C, Heary RF, Elkabes S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun [Internet] (2021) 91(August 2020):740–55. doi: 10.1016/j.bbi.2020.10.007
65. Johnston EL, Heras B, Kufer TA, Kaparakis-Liaskos M. Detection of bacterial membrane vesicles by NOD-like receptors. Int J Mol Sci (2021) 22(3):1–14.
66. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys [Internet] (2019) 670(February):4–14. doi: 10.1016/j.abb.2019.02.008
67. Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol (2013) 4(October):1–18.
68. Danis J, Mellett M. Nod-like receptors in host defence and disease at the epidermal barrier. Int J Mol Sci [Internet]. (2021) 22(9):4677. https://www.mdpi.com/1422-0067/22/9/4677.
69. Kienes I, Weidl T, Mirza N, Chamaillard M, Kufer TA. Role of nlrs in the regulation of type i interferon signaling, host defense and tolerance to inflammation. Int J Mol Sci (2021) 22(3):1–28.
70. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol (2006) 7(12):1250–7.
71. Takeda K, Takeuchi O, Akira S. Recognition of lipopeptides by toll-like receptors. J Endotoxin Res (2002) 8(6):459–63.
72. Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol (2006) 27(11):525–32.
73. Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia (2003) 43(3):281–91.
74. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol (2004) 173(6):3916–24.
75. Rasley A, Anguita J, Marriott I. Borrelia burgdorferi induces inflammatory mediator production by murine microglia. J Neuroimmunol (2002) 130(1–2):22–31.
76. Sterka D, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia (2006) 53(3):322–30.
77. Chauhan VS, Sterka DG, Furr SR, Marriott I. NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia (2009) 57(4):414–23.
78. Sterka D, Marriott I. Characterization of nucleotide-binding oligomerization domain (NOD) protein expression in primary murine microglia. J Neuroimmunol (2006) 179(1–2):65–75.
79. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nat [Internet]. (2001) 413(6857):732–8. http://www.nature.com/articles/35099560.
80. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol (2004) 5(2):190–8.
81. Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity (2016) 44(4):739–54.
82. Rehwinkel J, Gack MU. RIG-i-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol [Internet]. (2020) 20(9):537–51. doi: 10.1038/s41577-020-0288-3
83. Loo YM, Gale M. Immune signaling by RIG-i-like receptors. Immun [Internet]. (2011) 34(5):680–92. doi: 10.1016/j.immuni.2011.05.003
84. Yoneyama M, Fujita T. Function of RIG-i-like receptors in antiviral innate immunity. J Biol Chem [Internet]. (2007) 282(21):15315–8. doi: 10.1074/jbc.R700007200
85. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol (2005) 175(5):2851–8.
86. Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol (2005) 175(8):5260–8.
87. Furr SR, Chauhan V, Sterka D, Grdzelishvili V, Marriott I. Characterization of retinoic acid-inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus. J Neurovirol. (2008) 14(6):503–13.
88. Johnson MB, Halman JR, Burmeister AR, Currin S, Khisamutdinov EF, Afonin KA, et al. Retinoic acid inducible gene-I mediated detection of bacterial nucleic acids in human microglial cells. J Neuroinflammation. (2020) 17(1):1–14.
89. Zhao Y, Karijolich J. Know thyself: RIG-I-Like receptor sensing of DNA virus infection. J Virol [Internet] (2019) 93(23). doi: 10.1128/JVI.01085-19
90. Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol [Internet]. (2017) 38(3):194–205. doi: 10.1016/j.it.2016.12.004
91. Schlee M, Hartmann G. The chase for the RIG-I ligand–recent advances. Mol Ther [Internet]. (2010) 18(7):1254–62. https://linkinghub.elsevier.com/retrieve/pii/S1525001616310693.
92. Chiu YH, MacMillan JB, Chen ZJ. RNA Polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell [Internet]. (2009) 138(3):576–91. doi: 10.1016/j.cell.2009.06.015
93. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-i-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol (2009) 10(10):1065–72.
94. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448(7152):501–5.
95. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Sci (80-). (2013) 339(6121):786–91.
96. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11(11):997–1004.
97. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8.
98. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13.
99. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Sci (80- ) [Internet]. (2009) 323(5917):1057–60. doi: 10.1126/science.1169841
100. Jeffries AM, Marriott I. Human microglia and astrocytes express cGAS-STING viral sensing components. Neurosci Lett (2017) 658(July):53–6.
101. Cox DJ, Field RH, Williams DG, Baran M, Bowie AG, Cunningham C, et al. DNA Sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia (2015) 63(5):812–25.
102. Jeffries AM, Nitika, Truman AW, Marriott I. The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells. J Neurovirol [Internet]. (2020) 26(4):544–55. doi: 10.1007/s13365-020-00852-1
103. Coulon PG, Dhanushkodi N, Prakash S, Srivastava R, Roy S, Alomari NI, et al. NLRP3, NLRP12, and IFI16 inflammasomes induction and caspase-1 activation triggered by virulent HSV-1 strains are associated with severe corneal inflammatory herpetic disease. Front Immunol (2019) 10(JULY):1–19.
104. Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol (2012) 5(2):173–83.
105. Jeffries AM, Suptela AJ, Marriott I. Z-DNA binding protein 1 mediates necroptotic and apoptotic cell death pathways in murine astrocytes following herpes simplex virus-1 infection. J Neuroinflamm [Internet]. (2022) 19(1):1–13. doi: 10.1186/s12974-022-02469-z
106. Furr SR, Chauhan VS, Moerdyk-Schauwecker MJ, Marriott I. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J Neuroinflammation. (2011) 8:1–12.
107. Daniels BP, Kofman SB, Smith JR, Norris GT, Snyder AG, Kolb JP, et al. The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immun [Internet]. (2019) 50(1):64–76.e4. doi: 10.1016/j.immuni.2018.11.017
108. Rothan HA, Arora K, Natekar JP, Strate PG, Brinton MA, Kumar M. Z-DNA-Binding protein 1 is critical for controlling virus replication and survival in west nile virus encephalitis. Front Microbiol (2019) 10(September):1–10.
109. Song X, Ma F, Herrup K. Accumulation of cytoplasmic DNA due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J Neurosci [Internet]. (2019) 39(32):6378–94. doi: 10.1523/JNEUROSCI.0774-19.2019
110. Yogarajah T, Ong KC, Perera D, Wong KT. AIM2 inflammasome-mediated pyroptosis in enterovirus a71-infected neuronal cells restricts viral replication. Sci Rep [Internet]. (2017) 7(1):1–16. doi: 10.1038/s41598-017-05589-2
111. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe [Internet]. (2012) 11(3):290–7. doi: 10.1016/j.chom.2012.01.016
112. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8.
113. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep (2009) 10(8):916–22.
114. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe [Internet]. (2011) 9(5):363–75. doi: 10.1016/j.chom.2011.04.008
115. Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife (2012) 2012(1):1–17.
116. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol (2011) 12(10):959–65.
117. Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA Damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type i interferon by regulating STING trafficking. Proc Natl Acad Sci U S A. (2013) 110(8):2969–74.
118. Murata M, Kawanishi S. Oxidative DNA damage induced by nitrotyrosine, a biomarker of inflammation. Biochem Biophys Res Commun (2004) 316(1):123–8.
119. Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbeck’s Arch Surg (2006) 391(5):499–510.
120. Kiernan EA, Smith SMC, Mitchell GS, Watters JJ. Mechanisms of microglial activation in models of inflammation and hypoxia: Implications for chronic intermittent hypoxia. J Physiol (2016) 594(6):1563–77.
121. Mackenzie KJ, Carroll P, Martin C-AA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nat [Internet]. (2017) 548(7668):461–5. doi: 10.1038/nature23449
122. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nat [Internet]. (2017) 548(7668):466–70. doi: 10.1038/nature23470
123. Härtlova A, Erttmann SF, Raffi FAM, Schmalz AM, Resch U, Anugula S, et al. DNA Damage primes the type i interferon system via the cytosolic dna sensor sting to promote anti-microbial innate immunity. Immunity (2015) 42(2):332–43.
124. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Sci (80- ). (2016) 354(6313):765–8.
125. Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, et al. Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immun [Internet]. (2018) 49(3):490–503.e4. doi: 10.1016/j.immuni.2018.07.008
126. Chatterjee J, Nairy RK, Langhnoja J, Tripathi A, Patil RK, Pillai PP, et al. ER stress and genomic instability induced by gamma radiation in mice primary cultured glial cells. Metab Brain Dis (2018) 33(3):1–14.
127. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res [Internet]. (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
128. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122(6):877–902.
129. Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci [Internet]. (2015) 112(42):E5699–705. doi: 10.1073/pnas.1516465112
130. Kumar V. The trinity of cGAS, TLR9, and ALRs guardians of the cellular galaxy against host-derived self-DNA. Front Immunol (2021) 11(February):1–31.
131. Jakobs C, Perner S, Hornung V. AIM2 drives joint inflammation in a self-DNA triggered model of chronic polyarthritis. PloS One (2015) 10(6).
132. Hiroaki H, Osamu, Taro K, Tsuneyasu K, Shintaro S, Hideki S, et al. A toll-like receptor recognizes bacterial DNA. Nat [Internet] (2000) 408(DECEMBER):740–5. www.nature.com.
133. Zhang F, Yuan Y, Ma F. Function and regulation of nuclear dna sensors during viral infection and tumorigenesis. Front Immunol (2021) 11(January):1–11.
134. Briard B, Place DE, Kanneganti T-D. DNA Sensing in the innate immune response. Physiol [Internet]. (2020) 35(2):112–24. doi: 10.1152/physiol.00022.2019
135. Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J [Internet]. (2019) 38(21):1–17. doi: 10.15252/embj.2019102718
136. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nat [Internet]. (2018) 563(7729):131–6. doi: 10.1038/s41586-018-0629-6
137. Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv [Internet] (2020) 6(42). doi: 10.1126/sciadv.abb8941
138. Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci [Internet]. (2017) 114(23):E4612–20. doi: 10.1073/pnas.1705499114
139. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol (2017) 19(9):1061–70.
140. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nat [Internet]. (2017) 550(7676):402–6. doi: 10.1038/nature24050
141. Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun [Internet]. (2018) 9(1):1–12. doi: 10.1038/s41467-018-03555-8
142. Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, et al. Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ [Internet]. (2019) 26(2):332–47. doi: 10.1038/s41418-018-0122-7
143. Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe [Internet]. (2014) 15(2):228–38. doi: 10.1016/j.chom.2014.01.009
144. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ [Internet]. (2019) 26(9):1735–49. doi: 10.1038/s41418-018-0251-z
145. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nat [Internet]. (2019) 567(7747):262–6. doi: 10.1038/s41586-019-1006-9
146. Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J Immunol (2018) 200(8):2748–56.
147. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci U S A. (2018) 115(15):3930–5.
148. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. CGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498(7454):380–4.
149. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell (2013) 153(5):1094–107.
150. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. (2009) 106(21):8653–8.
151. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Sci (80- ). (2013) 339(6121):826–30.
152. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immun [Internet]. (2008) 29(4):538–50. doi: 10.1016/j.immuni.2008.09.003
153. Crow YJ, Rehwinkel J. Aicardi-goutie’res syndrome and related phenotypes: Linking nucleic acid metabolism with autoimmunity. Hum Mol Genet (2009) 18(R2):130–6.
154. Herzner AM, Hagmann CA, Goldeck M, Wolter S, Kübler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol (2015) 16(10):1025–33.
155. Andreeva L, Hiller B, Kostrewa D, Lässig C, De Oliveira Mann CC, Jan Drexler D, et al. CGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature (2017) 549(7672):394–8.
156. Paludan SR. Activation and regulation of DNA-driven immune responses. Microbiol Mol Biol Rev (2015) 79(2):225–41.
157. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun [Internet]. (2014) 5(1):5166.
158. Motwani M, Fitzgerald KA. cGAS micro-manages genotoxic stress. Immun [Internet]. (2017) 47(4):616–7. http://linkinghub.elsevier.com/retrieve/pii/S1074761317304314.
159. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature (2018) 553(7689):467–72.
160. Sharma M, Rajendrarao S, Shahani N, Ramírez-Jarquín UN, Subramaniam S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in huntington disease. Proc Natl Acad Sci U S A. (2020) 117(27):15989–99.
161. de Oliveira Mann CC, Kranzusch PJ. cGAS conducts micronuclei DNA surveillance. Trends Cell Biol [Internet]. (2017) 27(10):697–8. doi: 10.1016/j.tcb.2017.08.007
162. Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, Maciejowski J. ER-directed TREX1 limits cGAS activation at micronuclei. Mol Cell (2021) 81(4):724–738.e9.
163. Zhao M, Wang F, Wu J, Cheng Y, Cao Y, Wu X, et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy [Internet]. (2021) 17(12):3976–91. doi: 10.1080/15548627.2021.1899440
164. Guo Y, Gu R, Gan D, Hu F, Li G, Xu G. Mitochondrial DNA drives noncanonical inflammation activation via cGAS–STING signaling pathway in retinal microvascular endothelial cells. Cell Commun Signal (2020) 18(1):1–12.
165. Nadalutti CA, Ayala-Peña S, Santos JH. Mitochondrial DNA damage as driver of cellular outcomes. Am J Physiol - Cell Physiol (2022) 322(2):C136–50.
166. Liu S, Feng M, Guan W. Mitochondrial DNA sensing by STING signaling participates in inflammation, cancer and beyond. Int J Cancer. (2016) 139(4):736–41.
167. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520(7548):553–7.
168. Zhang W, Li G, Luo R, Lei J, Song Y, Wang B, et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp Mol Med (2022) 54(2):129–42.
169. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immun [Internet]. (2020) 52(3):475–486.e5. doi: 10.1016/j.immuni.2020.02.002
170. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife (2019) 8:1–21.
171. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The n-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep [Internet]. (2019) 26(9):2377–2393.e13. doi: 10.1016/j.celrep.2019.01.105
172. Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, et al. Structural basis of nucleosome-dependent cGAS inhibition. Sci (80- ). (2020) 370(6515):450–4.
173. Cao D, Han X, Fan X, Xu RM, Zhang X. Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res [Internet]. (2020) 30(12):1088–97. doi: 10.1038/s41422-020-00422-4
174. Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, et al. Structural basis for the inhibition of cGAS by nucleosomes. Sci (80- ). (2020) 370(6515):455–8.
175. Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nat [Internet]. (2020) 587(7835):678–82. doi: 10.1038/s41586-020-2748-0
176. Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nat [Internet]. (2020) 587(7835):668–72. doi: 10.1038/s41586-020-2750-6
177. Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nat [Internet]. (2020) 587(7835):673–7. doi: 10.1038/s41586-020-2749-z
178. Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core aicardi-goutières syndrome-related mutant invokes cGAS-STING innate immunesensing pathway in mice. J Exp Med (2016) 213(3):329–36.
179. Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet (2017) 26(20):3960–72.
180. Jeffries AM, Marriott I. Cytosolic dna sensors and cns responses to viral pathogens. Front Cell Infect Microbiol (2020) 10(September):1–17.
181. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe [Internet]. (2016) 20(5):674–81. doi: 10.1016/j.chom.2016.09.014
182. Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP 1/ DAI induces necroptosis. EMBO J (2017) 36(17):2529–43.
183. Pham TH, Kwon KM, Kim Y-E, Kim KK, Ahn J-H. DNA Sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol (2013) 87(6):3076–86.
184. Hao Y, Yang B, Yang J, Shi X, Yang X, Zhang D, et al. ZBP1: A powerful innate immune sensor and double-edged sword in host immunity. Int J Mol Sci (2022) 23(18):1–23.
185. Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, et al. ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J Immunol (2019) 203(5):1348–55.
186. Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nat [Internet]. (2016) 540(7631):124–8. doi: 10.1038/nature20558
187. Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol [Internet]. (2016) 1(2):665–76. doi: 10.1126/sciimmunol.aag2045
188. Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis [Internet]. (2018) 9(8):816. doi: 10.1038/s41419-018-0868-3
189. Wang ZC, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci U S A. (2008) 105(14):5477–82.
190. Lippmann J, Rothenburg S, Deigendesch N, Eitel J, Meixenberger K, Van Laak V, et al. IFNβ responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell Microbiol (2008) 10(12):2579–88.
191. Semenova N, Bosnjak M, Markelc B, Znidar K, Cemazar M, Heller L. Multiple cytosolic DNA sensors bind plasmid DNA after transfection. Nucleic Acids Res (2019) 47(19):10235–46.
192. Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyánszki A, Bibli SI, et al. Mitochondrial DNA damage and subsequent activation of z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci Rep (2018) 8(1):1–11.
193. Baik JY, Liu Z, Jiao D, Kwon HJ, Yan J, Kadigamuwa C, et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun [Internet]. (2021) 12(1):1–14. doi: 10.1038/s41467-021-23004-3
194. Trapani JA, Browne KA, Dawson MJ, Ramsay RG, Eddy RL, Shows TB, et al. A novel gene constitutively expressed in human lymphoid cells is inducible with interferon-γ in myeloid cells. Immunogenetics (1992) 36(6):369–76.
195. Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, et al. The mammalian PYHIN gene family: Phylogeny, evolution and expression. BMC Evol Biol [Internet]. (2012) 12(1):140. doi: 10.1186/1471-2148-12-140
196. Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, et al. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet b-exposed keratinocytes as a mechanism of autoantigen processing. Br J Dermatol (2011) 164(2):282–90.
197. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Sci (80- ). (2014) 343(6169):428–32.
198. Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nat [Internet]. (2014) 505(7484):509–14. https://www.nature.com/articles/nature12940.
199. Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci [Internet] (2014) 111(1). doi: 10.1073/pnas.1313577111
200. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the dna sensing adaptor sting by atm and ifi16 mediates nf-κb signaling after nuclear dna damage. Mol Cell [Internet]. (2018) 71(5):745–60. doi: 10.1016/j.molcel.2018.07.034
201. DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, et al. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene [Internet]. (1997) 15(4):453–7. doi: 10.1038/sj.onc.1201206
202. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol [Internet]. (2009) 10(3):266–72. doi: 10.1038/ni.1702
203. Baum R, Sharma S, Carpenter S, Li Q-Z, Busto P, Fitzgerald KA, et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II–deficient mice. J Immunol [Internet]. (2015) 194(3):873–7. doi: 10.4049/jimmunol.1402573
204. Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell [Internet]. (2017) 171(5):1057–1071.e11. doi: 10.1016/j.cell.2017.09.029
205. Micco A, Frera G, Lugrin J, Jamilloux Y, Hsu ET, Tardivel A, et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc Natl Acad Sci U S A. (2016) 113(32):E4671–80.
206. Kimkong I, Avihingsanon Y, Hirankarn N. Expression profile of HIN200 in leukocytes and renal biopsy of SLE patients by real-time RT-PCR. Lupus (2009) 18(12):1066–72.
207. Diner BA, Li T, Greco TM, Crow MS, Fuesler JA, Wang J, et al. The functional interactome of PYHIN immune regulators reveals IFIX is a sensor of viral DNA. Mol Syst Biol [Internet]. (2015) 11(2):787. doi: 10.15252/msb.20145808
208. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol (2019) 94(May):112–20.
209. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Sci (80- ). (2016) 353(6301):777–83.
210. Shiwaku H, Okazawa H. Impaired DNA damage repair as a common feature of neurodegenerative diseases and psychiatric disorders. Curr Mol Med (2015) 15(2):119–28.
211. Xu Q, Xu W, Cheng H, Yuan H, Tan X. Efficacy and mechanism of cGAMP to suppress alzheimer’s disease by elevating TREM2. Brain Behav Immun [Internet] (2019) 81(July):495–508. doi: 10.1016/j.bbi.2019.07.004
212. Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, et al. NAD + supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of alzheimer’s disease via cGAS–STING. Proc Natl Acad Sci [Internet] (2021) 118(37). doi: 10.1073/pnas.2011226118
213. Paul BD, Snyder SH, Bohr VA. Signaling by cGAS–STING in neurodegeneration, neuroinflammation, and aging. Trends Neurosci (2021) 44(2):83–96.
214. Healy LM, Stratton JA, Kuhlmann T, Antel J. The role of glial cells in multiple sclerosis disease progression. Nat Rev Neurol (2022) 18(4):237–48.
215. Zia S, Rawji KS, Michaels NJ, Burr M, Kerr BJ, Healy LM, et al. Microglia diversity in health and multiple sclerosis. Front Immunol 202011(November):1–14.
216. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett (2018) 592(5):728–42.
217. Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, et al. Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron [Internet]. (2017) 96(6):1290–1302.e6. doi: 10.1016/j.neuron.2017.11.032
218. Jin M, Shiwaku H, Tanaka H, Obita T, Ohuchi S, Yoshioka Y, et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat Commun [Internet]. (2021) 12(1):6565. doi: 10.1038/s41467-021-26851-2
219. Wu PJ, Hung YF, Liu HY, Hsueh YP. Deletion of the inflammasome sensor Aim2 mitigates aβ deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer disease mouse model. Neuroimmunomodulation (2017) 24(1):29–39.
220. Rui W, Li S, Yang L, Liu Y, Fan Y, Hu Y, et al. Microglial AIM2 alleviates antiviral-related neuro-inflammation in mouse models of parkinson’s disease. Glia [Internet]. (2022) 70(12):2409–25. doi: 10.1002/glia.24260
221. Ma C, Li S, Hu Y, Ma Y, Wu Y, Wu C, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med [Internet] (2021) 218(5). doi: 10.1084/jem.20201796/211880/AIM2-controls-microglial-inflammation-to-prevent
222. Barclay WE, Aggarwal N, Deerhake ME, Inoue M, Nonaka T, Nozaki K, et al. The AIM2 inflammasome is activated in astrocytes during the late phase of EAE. JCI Insight [Internet]. (2022) 7(8):27710. doi: 10.1172/jci.insight.155563
223. Hinkle JT, Patel J, Panicker N, Karuppagounder SS, Biswas D, Belingon B, et al. STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proc Natl Acad Sci U S A. (2022) 119(15):1–8.
224. Wu PJ, Liu HY, Huang TN, Hsueh YP. AIM 2 inflammasomes regulate neuronal morphology and influence anxiety and memory in mice. Sci Rep2016 6(August):1–12.
225. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell [Internet]. (2020) 183(3):636–649.e18. doi: 10.1016/j.cell.2020.09.020
226. Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of aicardi–goutières syndrome. J Immunol (2015) 195(5):1939–43.
227. Larrick JW, Mendelsohn AR. Modulation of cGAS-STING pathway by nicotinamide riboside in alzheimer’s disease. Rejuvenation Res [Internet]. (2021) 24(5):397–402. doi: 10.1089/rej.2021.0062
228. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol [Internet]. (2014) 192(12):5571–8. doi: 10.4049/jimmunol.1303258
229. Cao L-L, Guan P-P, Zhang S-Q, Yang Y, Huang X-S, Wang P. Downregulating expression of OPTN elevates neuroinflammation via AIM2 inflammasome- and RIPK1-activating mechanisms in APP/PS1 transgenic mice. J Neuroinflamm [Internet]. (2021) 18(1):281. doi: 10.1186/s12974-021-02327-4
230. Chen J, Shu S, Chen Y, Liu Z, Yu L, Yang L, et al. AIM2 deletion promotes neuroplasticity and spatial memory of mice. Brain Res Bull [Internet]. (2019) 152(8):85–94. doi: 10.1016/j.brainresbull.2019.07.011
231. Matsui H, Ito J, Matsui N, Uechi T, Onodera O, Kakita A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of parkinson’s disease. Nat Commun [Internet]. (2021) 12(1):3101. doi: 10.1038/s41467-021-23452-x
232. Yoo SM, Park J, Kim SH, Jung YK. Emerging perspectives on mitochondrial dysfunction and inflammation in alzheimer’s disease. BMB Rep (2020) 53(1):35–46.
233. Gleichman AJ, Carmichael ST. Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol Dis [Internet] (2020) 142(May):104957. doi: 10.1016/j.nbd.2020.104957
234. Booth HDE, Hirst WD, Wade-Martins R. The role of astrocyte dysfunction in parkinson’s disease pathogenesis. Trends Neurosci [Internet]. (2017) 40(6):358–70. doi: 10.1016/j.tins.2017.04.001
235. Soni C, Reizis B. Self-DNA at the epicenter of SLE: Immunogenic forms, regulation, and effects. Front Immunol (2019) 10(JULY):1–18.
236. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature (2018) 561(7722):258–62.
237. Martin BN, Wang C, Zhang C, Kang Z, Gulen MF, Zepp JA, et al. T Cell–intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol [Internet]. (2016) 17(5):583–92. https://www.nature.com/articles/ni.3389.
238. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. (2012) 109(26):10480–5.
239. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol [Internet]. (2021) 21(9):548–69. doi: 10.1038/s41577-021-00524-z
240. Jiao Y, Nan J, Mu B, Zhang Y, Zhou N, Yang S, et al. Discovery of a novel and potent inhibitor with differential species-specific effects against NLRP3 and AIM2 inflammasome-dependent pyroptosis. Eur J Med Chem [Internet]. (2022) 232:114194. https://linkinghub.elsevier.com/retrieve/pii/S0223523422000964.
241. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nat [Internet]. (2018) 559(7713):269–73. doi: 10.1038/s41586-018-0287-8
242. Tian X, Xu F, Zhu Q, Feng Z, Dai W, Zhou Y, et al. Medicinal chemistry perspective on cGAS-STING signaling pathway with small molecule inhibitors. Eur J Med Chem [Internet] (2022) 244(September):114791. doi: 10.1016/j.ejmech.2022.114791
243. Hong Z, Mei J, Li C, Bai G, Maimaiti M, Hu H, et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc Natl Acad Sci [Internet] (2021) 118(24). doi: 10.1073/pnas.2105465118
244. Wu JJ, Zhao L, Hu HG, Li WH, Li YM. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med Res Rev (2020) 40(3):1117–41.
245. Liu K, Lan Y, Li X, Li M, Cui L, Luo H, et al. Development of small molecule inhibitors/agonists targeting STING for disease. BioMed Pharmacother [Internet]. (2020) 132:110945. doi: 10.1016/j.biopha.2020.110945
246. Speir M, Djajawi TM, Conos SA, Tye H, Lawlor KE. Targeting rip kinases in chronic inflammatory disease. Biomolecules (2021) 11(5):1–22.
247. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun [Internet]. (2017) 8(1):1–12. doi: 10.1038/s41467-017-00833-9
248. Wiser C, Kim B, Vincent J, Ascano M. Small molecule inhibition of human cGAS reduces total cGAMP output and cytokine expression in cells. Sci Rep [Internet]. (2020) 10(1):1–11. doi: 10.1038/s41598-020-64348-y
249. Zhang H, You Q-D, Xu X-L. Targeting stimulator of interferon genes (STING): A medicinal chemistry perspective. J Med Chem [Internet]. (2020) 63(8):3785–816. doi: 10.1021/acs.jmedchem.9b01039.
250. Zorman J, Sušjan P, Hafner-Bratkovič I. Shikonin suppresses NLRP3 and AIM2 inflammasomes by direct inhibition of caspase-1. PloS One [Internet]. (2016) 11(7):e0159826. doi: 10.1371/journal.pone.0163887.
251. Chin K-H, Tu Z-L, Su Y-C, Yu Y-J, Chen H-C, Lo Y-C, et al. Novel c-di-GMP recognition modes of the mouse innate immune adaptor protein STING. Biol Crystallogr [Internet]. (2013) 69(3):352–66. https://scripts.iucr.org/cgi-bin/paper?S0907444912047269.
Keywords: genomic integrity, DNA sensors, neuroinflammation, neurodegeneration, glia
Citation: Suptela AJ and Marriott I (2023) Cytosolic DNA sensors and glial responses to endogenous DNA. Front. Immunol. 14:1130172. doi: 10.3389/fimmu.2023.1130172
Received: 22 December 2022; Accepted: 13 February 2023;
Published: 14 March 2023.
Edited by:
Antariksh Tyagi, Yale University, United StatesReviewed by:
Anetta Härtlova, University of Gothenburg, SwedenCopyright © 2023 Suptela and Marriott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian Marriott, SU1hcnJpb3RAdW5jYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.