 Maisha F. Jabeen
Maisha F. Jabeen Timothy S. C. Hinks
Timothy S. C. Hinks- 1Respiratory Medicine Unit, Experimental Medicine Division, Nuffield Department of Medicine, University of Oxford, John Radcliffe Hospital, Oxford, United Kingdom
- 2National Institute for Health Research Oxford Biomedical Research Centre, John Radcliffe Hospital, Oxford, United Kingdom
Mucosal associated invariant T (MAIT) cells are innate-like T lymphocytes, strikingly enriched at mucosal surfaces and characterized by a semi-invariant αβ T cell receptor (TCR) recognizing microbial derived intermediates of riboflavin synthesis presented by the MHC-Ib molecule MR1. At barrier sites MAIT cells occupy a prime position for interaction with commensal microorganisms, comprising the microbiota. The microbiota is a rich source of riboflavin derived antigens required in early life to promote intra-thymic MAIT cell development and sustain a life-long population of tissue resident cells. A symbiotic relationship is thought to be maintained in health whereby microbes promote maturation and homeostasis, and in turn MAIT cells can engage a TCR-dependent “tissue repair” program in the presence of commensal organisms conducive to sustaining barrier function and integrity of the microbial community. MAIT cell activation can be induced in a MR1-TCR dependent manner or through MR1-TCR independent mechanisms via pro-inflammatory cytokines interleukin (IL)-12/-15/-18 and type I interferon. MAIT cells provide immunity against bacterial, fungal and viral pathogens. However, MAIT cells may have deleterious effects through insufficient or exacerbated effector activity and have been implicated in autoimmune, inflammatory and allergic conditions in which microbial dysbiosis is a shared feature. In this review we summarize the current knowledge on the role of the microbiota in the development and maintenance of circulating and tissue resident MAIT cells. We also explore how microbial dysbiosis, alongside changes in intestinal permeability and imbalance between pro- and anti-inflammatory components of the immune response are together involved in the potential pathogenicity of MAIT cells. Whilst there have been significant improvements in our understanding of how the microbiota shapes MAIT cell function, human data are relatively lacking, and it remains unknown if MAIT cells can conversely influence the composition of the microbiota. We speculate whether, in a human population, differences in microbiomes might account for the heterogeneity observed in MAIT cell frequency across mucosal sites or between individuals, and response to therapies targeting T cells. Moreover, we speculate whether manipulation of the microbiota, or harnessing MAIT cell ligands within the gut or disease-specific sites could offer novel therapeutic strategies.
Introduction
The seminal discovery that the mucosal associated invariant T (MAIT) T cell receptor (TCR) recognizes riboflavin metabolites derived from bacteria, mycobacteria and fungi (1), revealed a prime role in sensing and responding to the microbiome at mucosal surfaces. The MAIT TCR is a semi-invariant TCR-α chain (typically TRAV1-2-TRAJ33 or TRAV1-2-TRAJ12 or TRAV1-2-TRAJ20), predominantly associated with the β-chains TRBV20 or TRBV6 in humans and TRBV19 or TRBV13 in mice and specifically recognizes the naturally-occurring activating ligand 5-(2-oxopropylideneamino)-6-dribityllumazine (5-OP-RU) presented on MHC-related protein 1 (MR1) (2). Whilst initially these cells were understood to play a role in antimicrobial host defense, the more recent discoveries of separate antiviral and ‘tissue repair’ responses have revealed a more nuanced complexity in their functional repertoire. Nonetheless the microbiome remains absolutely essential to the development and peripheral expansion of MAIT cells as a source of TCR ligand, such that the nature of the early life microbiome can mediate life-long changes in the MAIT cell repertoire. In this review we therefore make a specific focus on the role of the microbiome in the ontogeny of MAIT cells. We then review how microbial dysbiosis, often marked by compositional shifts in specific phyla, alongside changes in intestinal permeability and inflammatory cytokine milieus, are together involved in the potential pathogenicity of MAIT cells. Though human data on the influence of MAIT cell deficiency on the microbiome are relatively scarce, we explore the insights given by specific clinical instances of acquired MAIT cell deficiency, in particular those of hematopoietic stem cell transplantation and of human immunodeficiency virus (HIV)-induced MAIT cell loss, which provide experimental windows into MAIT cell biology. We review human data from the gut, lung and skin, which comprise the body’s largest barrier surfaces, and conclude with thoughts on the potential for therapeutic manipulation of the microbiome or MAIT cells populations directly.
Ontogeny of MAIT cells
Whilst conventional CD4+ or CD8+ T cells exit the thymus as naïve cells, gaining effector functionality following antigen exposure in secondary lymphoid organs, MAIT cells acquire effector functions intra-thymically (3–5). We review the ontogeny of MAIT cells in mouse and human and consider how commensal derived bacterial metabolites are needed at each stage of their development and acquisition of antimicrobial functionality.
Mouse
MAIT cells arise in the thymic cortex following the development of CD4+CD8+ double positive (DP) thymocytes possessing a T cell receptor (TCR) specific to the MR1:5-OP-RU complex, surveying their vicinity for subsequent positive selection of MR1-expressing cells (6). Using a murine bone marrow chimera model and single cell RNA sequencing, it has been shown that positive selection of 5-OP-RU : MR1 specific thymocytes can occur on different cell types destined for divergent outcomes. These are heat stable antigen high (HSAhi) precursors undergoing positive selection by thymic epithelial cells (TEC) and differentiating into naïve CD4+ T cells or, thymocytes selected by hematopoietic cells differentiating into CD44hi effector cells (7).
Regardless of their mode of selection there is resultant expression of the survival factor Bcl2 with induction of Ccr7 and loss of Ccr9 expression to suggest migration of these cells from the thymic cortex to the medulla. Following positive selection in mouse and human, MAIT cells progress through three stages of intrathymic development summarized in Figure 1. Like iNKT cells (8), effector differentiation of murine MAIT cell precursors selected by hematopoietic cells in the thymus is reliant upon expression of the signaling lymphocyte activation molecule (SLAM) adaptor protein (SAP) (4, 7, 9). This shared characteristic reflects ZBTB16 expression (encoding the master transcription factor PLZF) in both cell types, conferring innate-like functionality. PLZF suppresses the naïve T cell program and is required for expression of effector genes, alongside CD44 expression (3, 4). There is concurrent downregulation of Bach2 (a transcription factor seen in conventional T lymphocytes) and then Klf2, involved in regulating thymic egress (4). The population of naïve MR1-restricted T cells, having undergone positive selection on MR1 expressing TECs, patrol secondary lymphoid organs whereas their counterparts selected on MR1 expressing hematopoietic cells undergo proliferation and differentiation in preparation for migration to peripheral tissue to support mucosal immunity (7).
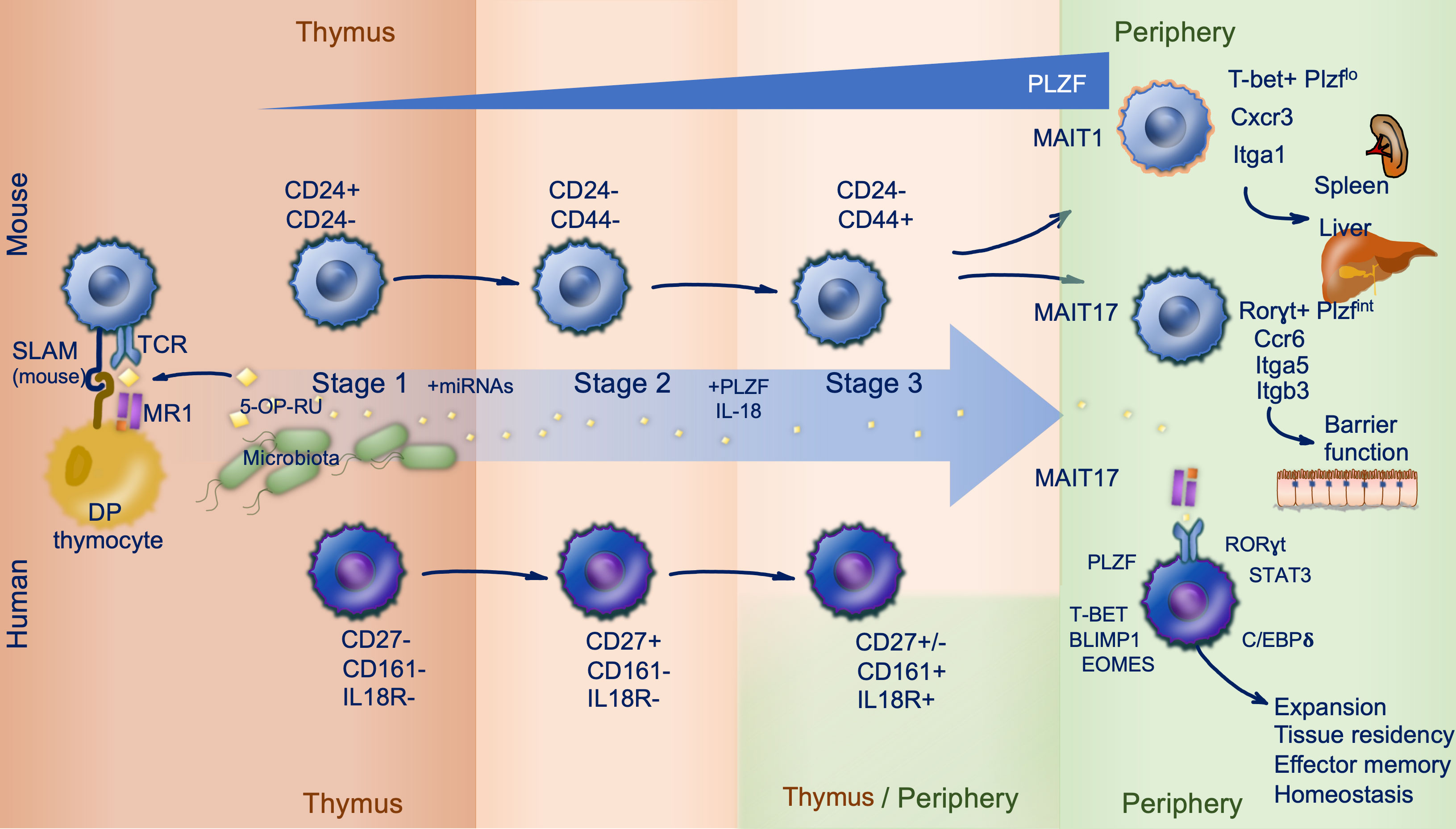
Figure 1 MAIT cell ontogeny. Mouse (top) and human (bottom) mucosal associated invariant T (MAIT) cell development follows three stages, largely within the thymus. Microbially derived 5-OP-RU is trafficked to the thymus and loaded onto MR1 expressed on double positive (DP) thymocytes. MAIT cells are subsequently positively selected on DP thymocytes in a process reliant upon signaling lymphocytic activation molecule (SLAM) interactions in mice (stage 1). During stage 2 and 3 MAIT cells acquire effector functions and their prototypic phenotype driven by PLZF expression. This occurs intrathymically in mice, but in human stage 3 can also occur peripherally. Distinguishing markers and important co-factors regulating differentiation are shown.
In mice two clear MAIT cell subsets with distinct effector properties are identified by their expression factors T-bet (MAIT1) and RORγt (MAIT17) enabling them to readily produce IFN-γ and IL-17 respectively (7). Whist single cell RNAseq has been informative in describing early transcriptional event in MAIT cell development, the signaling pathways and mechanisms responsible for adoption of a MAIT1 versus MAIT17 phenotype are yet to be fully elucidated. It has been speculated that choice of lineage may be directed at random by positively selected MAIT cell precursors, shaped by environmental cues such as cytokines or relate to TCR affinity for the selecting ligand but this remains to be confirmed experimentally (10). MAIT1 and MAIT17 cells express distinct patterns of chemokine receptors and integrins; MAIT1 cells express Cxcr3 and Itga1, while MAIT17 cells express Ccr6, Itga5 and Itgb3 and respond more efficiently to TCR-stimulation (7, 11, 12). PLZF expression in MAIT cells confers a broad tissue homing capacity whereas the transcription factors T-bet and RORγt likely fine tune tissue targeting, promoting residency of MAIT1 cells preferentially in spleen and liver, and MAIT17 cells in barrier tissue including the lung, skin and gut (11, 12).
Human
Mature T cell development begins in utero in humans. MAIT cell precursors appear early during gestation at low frequencies in the thymus and cord blood (5, 13). MR1 is expressed on hematopoietic double positive thymocytes suggesting a similar manner of selection to mouse MAIT cells (14). In human cord blood a minority of Vα7.2+CD161+ have high avidity for the MR1:5-OP-RU complex; only these cells go on to acquire a memory phenotype in the first few weeks of life and expand to provide the adult MAIT cell pool over the following 5 to 6 years (13). Thus, the adult MAIT cell clonal size is antigen driven and results in a restricted TCR repertoire in adults (13). Key similarities and differences between mouse and human MAIT cell development are discussed below.
As seen in mouse, human MAIT cells preferentially locate to non-lymphoid tissue. Thymic MAIT cells express tissue-homing molecules including CCR6, CCR5 and CXCR6 (4). In addition, as they mature, thymic MAIT cells upregulate the transcription factors C/EBPδ (4), important for MAIT cell trafficking (15), and RUNX3 (4), also required by conventional CD8+ tissue resident memory cells to establish niches in diverse tissue environments (16). The shared transcription factors PLZF (ZBTB16), RORγt (RORC) and T-bet (TBX21) are induced differentially in both mouse and human during thymic development (5), but expression of SAP is not necessary for human MAIT cells (5, 7). Mature tetramer positive (MR1:5-OP-RU restricted) thymocytes express PLZF, CD161 and IL-18Rα. However, unlike thymic MAIT cells in mouse, which display a memory phenotype at the point of egress, mature thymic and cord blood MAIT cells in human are negative for the memory marker CD45RO (5, 13). In addition, human MAIT cells are largely T-bet+RORγt+ as they exit the thymus (12) whereas these transcription factors are expressed mutually exclusively in mouse (giving way to MAIT1 and MAIT17 cells) (5). Peripheral MAIT cell expression of transcriptions factors Eomesederin (EOMES) and Blimp-1 (PRDM1) further support type-1 responses, whereas STAT3 supports type-17 responses. Human thymic MAIT cells possess cytotoxic capacity (4) but limited ability to secrete TNF and IFN-γ in response to PMA and ionomycin stimulation when compared to peripheral blood MAIT cells (3). Therefore, in human MAIT cells terminal maturation appears to occur in the periphery following birth suggesting additional peripheral mechanisms support acquisition of full effector functionality.
Microbes and MAIT cells at barrier sites: Development and homeostasis
The microbiome has a fundamental role in the induction, development, and homeostatic function of the host immune system. A symbiotic relationship between the host immune system and microbiota is required to balance regulatory pathways conferring tolerance to innocuous antigens and protective immunity against pathogens (17).
Following their development in the thymus MAIT cells are equipped with a transcriptional program and homing markers to support tissue residency. They localize to sites including the oropharynx, respiratory and GI tracts, skin and female genital mucosa, also hosting uniquely adapted microbial communities (18). MAIT cells thereby occupy a prime position for crosstalk with commensal microorganisms which uniquely synthesize riboflavin at mammalian barrier surfaces (18). The co-evolution of MR1 and TRAV1, and accumulation of MR1 mutations in species following loss of TRAV1, supports the idea that the main function of MR1 is to present antigen to MAIT cells (19). By extension this provides an insight into conserved mechanisms through which barrier surfaces can imprint mucosal immunity. These interactions are further shaped by cellular networks, environmental and metabolic factors within the microenvironment (20). With broad anti-bacterial specificity and a capacity for tissue repair, MAIT cells may be key in restoring homeostasis following infection or tissue injury thereby offering protection from invading pathogens and preserving the microbiome.
Microbes and MAIT cell development
MAIT cell reliance on the microbiome is evidenced by their relative deficiency in the periphery (21) and thymus (3) of germ free (GF) mice, compared with specific pathogen free animals. This pattern of deficiency is similar to other innate and memory T cell populations in GF and antibiotic treated mouse models (22). Metabolites from riboflavin-synthesizing commensals are needed for most stages of MAIT intra-thymic development and subsequent peripheral expansion (3, 23). Riboflavin metabolites secreted by the microbiota travel rapidly to the thymus. In fact 5-OP-RU is trafficked and detected in the thymus within an hour of topical or oral administration (23). MAIT cell development can be promoted following mono-colonization of GF mice with riboflavin synthesizing bacterial species such as Proteus mirabilis or Escherichia coli, but not with species deficient in this biosynthetic pathway such as Lactobacillus johnsonii (21, 23). Using mutant E. coli strains deficient in riboflavin enzymes either upstream (ΔRibD) or downstream (ΔRibE) of 5-A-RU to colonize GF mice has identified the essential role for RibD, and thus 5-OP-RU, in thymic MAIT cell development. The non-stimulatory MR1-ligand Acetyl-6-formyl-pterin (Ac-6-FP) cannot support MAIT cell development (23). Following recolonization of GF mice, there is selective restoration of RORγt+ MAIT17 cells reliant on TCR-triggering for proliferation and function (23). Of note, GF mice retain a small residual population of thymic MAIT cells whereas Mr1-/- are completely lacking in MAIT cells strongly supporting an essential role for MR1 in positive selection of these cells (7).
Microbial recolonization of adult GF mice restores thymic MAIT cell development, but fails to populate peripheral tissue such as the skin (21) or lung (23) with newly differentiated MAIT cells. There is a narrow neonatal window (within first 3 weeks of life) when recolonization of GF mice can restore the MAIT cell population (21). In addition, topical administration of 5-OP-RU is sufficient for MAIT cell development and skin homing in neonates but not adults (21, 23). Thus, adult MAIT cell development is reliant on microbiome-derived co-stimulation. However, preservation of MAIT cells in MyD88- and TLR3- deficient mice rules out TLR or IL-1 receptor family members (IL-1, IL-18 or IL-33) as likely drivers (23). A further explanation for this temporally-restricted reconstitution of MAIT cell populations may be competition for a shared niche imposed by similar innate T cell subsets. MAIT cell frequencies positively correlate with mouse and human iNKT cells (13, 24) and γδT cells (24), all sharing overlapping functions and a reliance on the microbiome. Competitive regulation of individual populations is supported by increased frequency of iNKT and MAIT cells seen in Tcrd-deficient mice (21) and increased splenic and thymic MAIT cells in Cd1d-deficient mice (3). In humans, the R9H mutation in MR1prevents its binding to 5-OP-RU but retains affinity for Ac-6-FP (25); in a rare patient with a homozygous R9H mutation in MR1, MAIT cell deficiency is observed with an expanded γδT cell (Vδ2+) population, again suggesting a compensatory interaction between innate T cell subsets (25). MAIT cells, iNKT and γδT cells do not compete for the same antigen, thus competition for immunological space may be imposed through alternative mechanisms orchestrated by immunoregulatory cytokines (e.g. IL-7, IL-15) (26–28), or host and dietary metabolites regulating shared transcriptional pathways (e.g. via the aryl hydrocarbon receptor) (20, 29). It is yet to be determined if population pressures and temporal restrictions apply to restoring human MAIT cell frequencies. Partial reconstitution of MAIT cells is seen following allogenic hematopoietic stem cell transplantation (HSCT) and correlates with the diversity of gut microbiota (30). It is tempting to speculate that the microbiome offers a key to regulating MAIT cells, however it has not yet been fully elucidated how this interaction could be skewed by neighboring innate T cells or the effects of age and disease.
The microbiome is diverse and heterogenous, varying between mucosal sites with distinct microenvironments. A large in vitro screen of microbiota-associated bacterial species found that the capacity to stimulate MAIT cells correlated with riboflavin secretion as measured by mass spectrometry (31). High stimulator species belonged to Bacteroidetes and Proteobacteria phyla, whereas Actinobacteria and Firmicutes were poor stimulators. Vβ2+ MAIT cells were most activated. Conventional human T cell subsets were able to present MR1-ligand but induced a weaker cytokine response compared to professional APCs. This suggests a capacity for MAIT cells to discriminate between members of the microbiota by TCR signal strength based on antigen load and presenting cell (31). Microbial diversity has been shown to reduce MAIT cell activation in vitro, correlating with net riboflavin secretion in a human intestinal model community. Higher diversity resulted in greater riboflavin consumption and thus less antigen presentation to MAIT cells. Interestingly, introducing microbial stress through environmental acidification reduced activation by impairing availability of riboflavin (32). MAIT cells have also been shown to exhibit microbe-specific responses to bacterial and fungal organisms with differential TCR β-chain bias and MR1-dependent activation, suggesting a further dimension of functional heterogeneity (33).
Microbial diversity varies by tissue, and notably between health and disease, typically marked by dysbiosis with reduced diversity. As discussed below, this may be a mechanism through which MAIT cells contribute to pathology and equally offer a therapeutic opportunity to manipulate MAIT cell function. It is worthwhile considering barrier homeostasis alongside microbial diversity. Pathogen invasion disrupts the mucosa and induces an inflammatory response. Co-stimulation of MAIT cells via TCR and cytokine has been shown in vitro to engage the full antimicrobial repertoire in MAIT cells (34). Therefore, where MAIT cells are implicated in disease pathogenesis it is important to consider any changes to the microbiome in parallel. Further studies, particularly in human, are needed to address this and consider specific mechanisms which shift MAIT cell function from homeostatic to pro-inflammatory.
Microbes and tissue repair
MAIT cells can engage a “tissue repair” program associated with accelerated wound repair in the context of commensal organisms. We and others have described the transcriptome of activated MAIT cells following transcriptomic analysis of MR1:5-OP-RU tetramer positive cells in mouse and human (34–36). Alongside expected pro-inflammatory responses, we identified a TCR-mediated and activation-driven expression of the tissue repair program previously reported in murine skin homing H2-M3 restricted Tc17 cells induced by commensal flora and accelerating repair in an epithelial wound model (35, 37). Key genes expressed in both species included TNF, CSF2, HIF1A, FURIN, VEGFB, PTGES2, PDGFB, TGFB1, MMP25, and HMGB1 (35). Accelerated wound healing could be observed in an intestinal epithelial cell line system following treatment with supernatants from TCR-stimulated MAIT cells and blocked with anti-MR1 antibodies (36). This MAIT tissue repair program is observed following TCR ligation but not cytokine mediated stimulation alone (34, 36). Thus, similarly to H2-M3 restricted Tc17 cells in mouse skin (37) and γδT cells in the lung and gut (38–40), MAIT TCR signaling appears to play a role in tissue homeostasis.
Murine skin-resident MAIT cells also engage a distinct tissue repair transcriptional signature (21) reminiscent of H2-M3 restricted Tc17 cells reactive to S. epidermidis derived N-formylated peptides (37). To unpick the role of MAIT cells from H2-M3 restricted Tc17 or γδT cells, given their overlapping properties, Tcrd-/- mice and S. epidermidis strain incapable of inducing H2-M3 Tc17 cells were utilized (21). This revealed a MAIT cell-dependent tissue repair response to S. epidermidis with accelerated epidermal tongue length growth in a skin punch biopsy model compared with MAIT cell deficient Mr1-/-Tcrd-/- mice. A further observation from this study was that direct topical application of the MAIT cell ligand 5-OP-RU prior to skin injury, in the presence or absence of additional cytokines, was sufficient to induce local expansion of MAIT cells and accelerate tissue repair (21). The importance of this role at human barrier sites and mechanisms through which MAIT cell mediators might exert their homeostatic function on the local environment is yet to be elucidated.
Murine studies have demonstrated reduced intestinal microbial diversity in MR1 deficient animals, which may result from altered IL-17A signaling downregulating tight junction protein expression (41). In non-obese diabetic (NOD) mice deficient in MR1 there is impaired intestinal barrier integrity (42). Further work is needed to understand if human MAIT cells can shape the composition of the healthy microbiome. As discussed below, in disease states this is likely to be closely intertwined with barrier integrity and its effects on the microbiota.
Tissue localization and MAIT cell phenotype
Microbiome-derived signals are likely to contribute to the establishment of tissue resident MAIT cell populations as murine lung and skin MAIT cell frequencies at steady state are cage dependent (21). It is unknown if this is shaped by antigen load or other innate signals. Cytokines appear to have a varied role; in skin IL-23 signaling is necessary for sustaining a MAIT17 cell population (21), yet in the lung normal MAIT cell frequencies are maintained not only in Il23-/- mice but also Ifng-/-, Il6-/-, Il18-/- and Il12-/- deficient animals (43). Cytokine reliance may be imprinted in the thymus as Il23r expression is higher in the MAIT17 thymic subset (4, 44). Tissue MAIT cells undergo terminal differentiation in tissue with unique transcriptomic programs observed between the lung versus the spleen and liver (11), or indeed skin compared with spleen, lung and liver (21).
In humans, peripheral blood MAIT cells respond differently from tissue-derived MAIT cells originating from intestinal mucosa (45), oropharynx (46), nasopharynx (47), lung (48) and female genital tract (49) following TCR ligation, also suggesting tissue-specific imprinting. Colonic MAIT cells acquire a primed phenotype, compared with their peripheral blood counterparts, proportionately to accumulation of antigenic metabolites derived from the microbiome (50). Overall, these adaptive mechanisms are speculated to be favorable for long term residency in tissue. In parabiotic pairs most spleen, liver and lung (except some RORγt- cells) MAIT cells did not recirculate over 5 weeks, implying persistent tissue residency (11). In human, whether MAIT cells are permanently resident or leave tissue and recirculate remains unclear. MAIT cells from matched thoracic duct lymph and blood samples have a shared TCR repertoire but are CCR7-, this could indicate transit through tissue between the two compartments or a CCR7-independent migration mechanism (51). The tissue residency markers CD69 and CD103 are widely expressed by MAIT cells at mucosal surfaces, but rare in blood MAIT cells (20). Therefore, there may be a small pool of recirculating cells in health, although its role in disease is not known.
Microbial dysbiosis and MAIT cells in immune mediated diseases
MAIT cell dysfunction and dysbiosis often feature together in immune mediated diseases driven by autoimmune, atopic, and metabolic processes, alongside chronic infection. We review how MAIT cell-microbial interactions change from homeostatic to potentially harmful within this context and summarise this in Figure 2. Conditions characterised by shifts in the gut microbiome that provide insight into the potential symbiosis between microbiota and MAIT cell biology are considered first, before exploring other mucosal niches and their potential effects on MAIT cell phenotype. MAIT cell-microbiome interactions in specific autoimmune conditions is reviewed more extensively elsewhere (52),
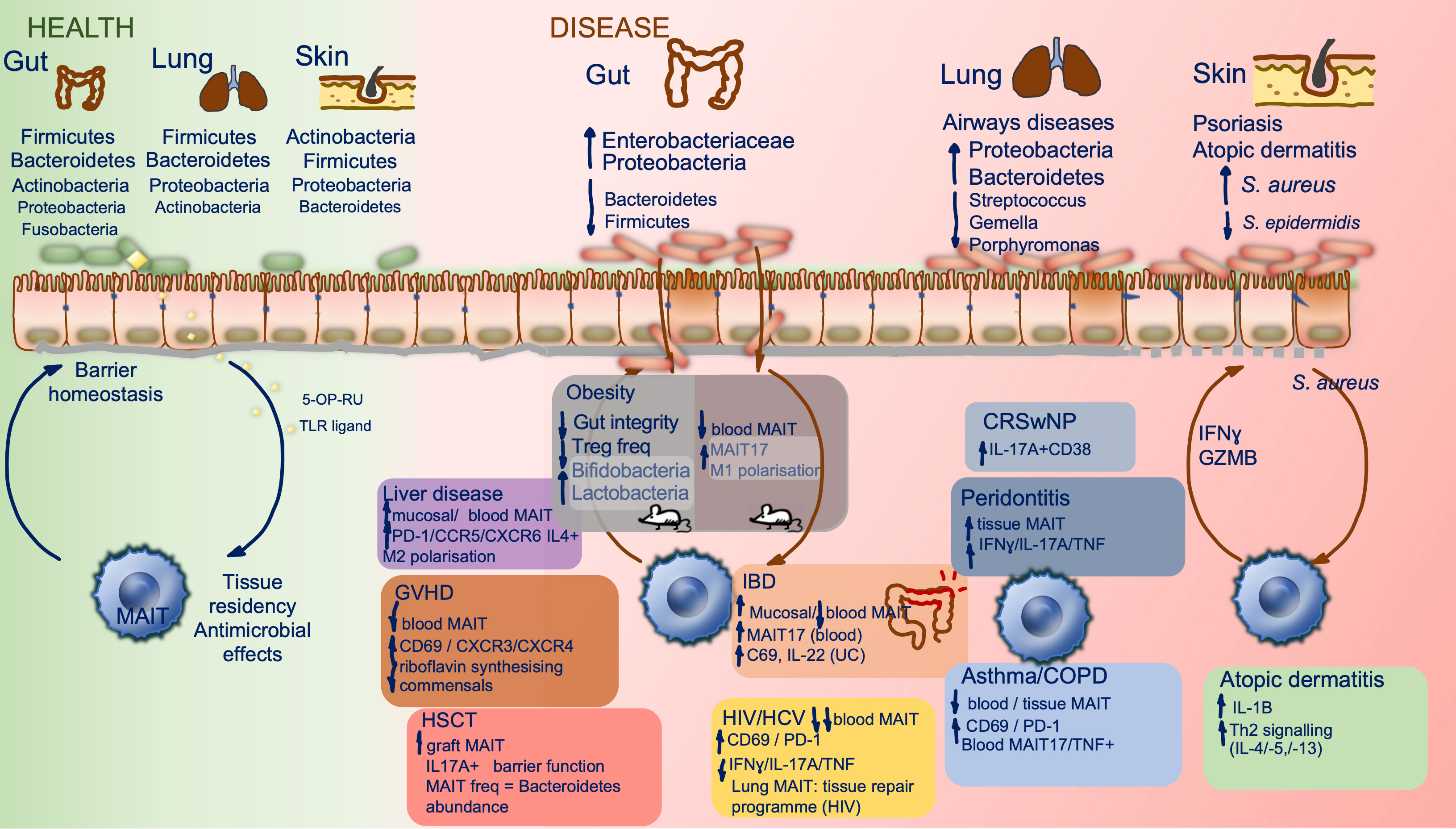
Figure 2 MAIT cells and the microbiome in health and disease. Constituents of the human microbiome in health, at different barrier sites, and interactions with tissue resident MAIT cells are shown (left). Key compositional shifts in human microbiota and their resultant effects in a range of disease states are summarized by barrier site (right), data are derived from human studies except within obesity where mouse studies have been included and highlighted within the figure. There is relative paucity of data on direct MAIT cell effect on the microbiome. CRSwNP, chronic rhinosinusitis with nasal polyposis; COPD, chronic obstructive pulmonary disease; GVHD, graft versus host disease; HSCT, hematopoietic stem cell transplant; HCV, hepatitis C virus; HIV, human immunodeficiency virus; IBD, inflammatory bowel disease.
The gut microbiome and MAIT cells
The gut microbiome has been most widely studied in health and disease. It is estimated that the bacterial density of the colon is 1011-1012/milliliter making it the most densely populated microbial habitat in the human body (53). Human gut microbiota is composed of Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Fusobacteria, and Verrucomicrobia, with Firmicutes and Bacteroidetes accounting for 90% of total species (54). Its principal function is to protect against colonization of exogenous pathogens and potentially pathogenic indigenous organisms, through competition for finite nutrients and modulation of the host immune response (55). Throughout life the gut microbiota is required for the development of innate and adaptive arms of the immune response, providing education in host/pathogen discrimination and sustaining barrier homeostasis (55). As discussed, MAIT cell development is reliant on the gut microbiome. Bacteroidetes, highly abundant in the gut, are the strongest stimulators of MAIT cells and likely further influence their phenotype in the intestinal mucosa (31). In dysbiosis (related to localized pathology or systemic inflammation) MAIT cells, enriched in the lamina propria, are likely to be one of the first cells exposed to translocated gut microbes and inflammatory signals following the resultant barrier compromise, however their direct role in pathological processes caused by dysbiosis is yet to be fully elucidated.
Most mechanistic studies exploring MAIT cell-microbiome interactions have utilized animal models, often comparing to MR1-/- strains devoid of MAIT cells. The fecal microbiota in MR1-/- mice is distinct from wild-type animals with unique organisms belonging to Bacteroidaceae, Desulfovibrionaceae and unclassified Burkholderiales families, conferring greater overall richness. It is also resistant to antibacterial killing and Clostridium difficile colonization (56). Furthermore, intestinal barrier function has been reported to be compromised in the absence of MR1 (41, 42). Gut microbiota vary between mouse strains (57) and to date no studies have compared animals with MAIT cell deficiency or excess across common background laboratory strains to determine if shifts in microbiome are linked to their genetic background. Thus challenges arise when elucidating MAIT cell mechanisms in animal models and it is important to corroborate findings in human studies given fundamental differences not only in microbiome but also MAIT cell tissue distribution.
Inflammatory bowel diseases
Inflammatory bowel diseases (IBD), consisting of the subtypes Crohn’s disease (CD) and ulcerative colitis (UC), are multifactorial chronic inflammatory conditions of the gastrointestinal tract associated with dramatic changes to the microbiota and local metabolic landscape (58). Several studies have investigated the relationship between genetic and immune susceptibility to IBD alongside the role of the gut microbiome in pathogenesis. This has been reviewed elsewhere (59–61). MAIT cells and their derived cytokines, particularly IL-17, have been considered as pathogenic drivers however the evidence for this is conflicting. MAIT cells are depleted in children with early onset inflammatory bowel disease under the age of six (62). Adult peripheral blood MAIT cell are activated (Ki67+) and decline in frequency in CD and UC (63, 64), unrelated to therapeutic intervention (anti-TNF or corticosteroids). There is a concurrent enrichment of ileal mucosa MAIT cells in CD and colonic mucosa MAIT cells in UC, correlating with disease activity (45). Blood MAIT cells in CD demonstrate a shift in cytokine production with greater IL-17 and reduced IFN-γ secretion following ex vivo stimulation (64), while UC MAIT cells upregulate CD69 (45) and secrete more IL-22 (64) and IL-17 (45). In oxazolone colitis, a murine model of UC which histologically resembles UC and is predominantly mediated by type-2 cytokines, it has been proposed that MAIT cell play a directly pathogenic role as disease severity is reduced in MR1-/- animals or with MR-1 antagonist isobutyl 6-formyl pterin (65). However, oxazolone can induce a very severe colitis with a systemic inflammatory response resembling sepsis, thus likely to reflect only the most severe forms of disease, making it challenging to draw parallels with the full spectrum of IBD (66). A recent study has shown that whilst a significant expansion is seen in the Tc17 population during active CD, this is largely due to induction of conventional T cells and not MAIT cells. Disease associated Tc17 cells acquire a distinct phenotype (CD6high, CD39, CD69, PD-1, CD27low). MAIT cells were the major IL-17 producing CD8+ population in blood during health or remission but not active CD, when their frequency declines in blood but is maintained in tissue (67). A subpopulation of predominantly CD8+ Crohn’s-associated invariant T (CAIT) cells have also been described, resembling NKT type II cells and enriched in blood of CD patients concurrently with decline in MAIT cell clonotypes. This could represent a compensatory expansion of an innate-like cell population, but the role of these cells is yet to determined (68). IL-17 is reported to have an additional protective role in the intestinal mucosa (69, 70) and clinical trials with anti-IL-17 therapy (secukinumab) have lacked efficacy in CD (71). IL-12 and IL-18 are upregulated in the intestinal mucosa of CD patients and polymorphisms of IL-23R, NLRP3, IL-18R and IL-12B2 significantly associate with CD implicating these cytokines in its pathophysiology (72). It remains to be seen if phenotypic changes reported in tissue MAIT cells in IBD are an epiphenomenon or directly implicated in pathogenesis. Taken together with barrier disruption and dysbiosis in IBD, MAIT cell may need to balance conflicting roles; their capacity to produce IL-17 and engage a tissue repair program following exposure to microbial antigens provides homeostatic capacity (35), however in a dysbiotic landscape with high antigen burden and pro-inflammatory cytokine co-stimulus a pro-inflammatory program may be engaged (36).
The gut-liver axis and chronic liver diseases
In animal models the liver has been shown to remain sterile in the presence of an intact intestinal mucosa, with immune responses to gut commensals confined to the mesenteric lymphoid system. In the context of infection or inflammation the liver acts as an immunological ‘firewall’, clearing bacteria or their derived products breaching intestinal or vascular barriers (73). MAIT cells are the dominant population of innate like T cells in the liver (up to 50% of CD3+ cells). They are adapted for tissue homing (high expression of CXCR6 and CCR6) and poised for host defense (CD69, HLA-DR, CD38high). Compared with mucosal barrier MAIT cells, their steady state responses are less skewed to type 17 functionality and require IL-7 licensing for sustained IL-17 production following TCR ligation (74). This distinction may be driven by the lack of interaction with commensal organisms in health. Below we consider changes to the microbiome in prevalent chronic liver diseases in which MAIT cells have been implicated.
Fatty liver diseases associated with alcohol, obesity or metabolic syndromes continue to grow in prevalence. High fat diet and alcohol induced dysbiosis can disrupt host-microbe interactions through metabolic dysregulation and mucosal barrier disruption. In this setting MAIT cell activating bacteria and microbe derived metabolites can translocate to the liver (75). Non-alcoholic fatty liver disease (NAFLD) affects approximately 40% of all adults worldwide and can range from benign hepatic steatosis to progressive non-alcoholic steatohepatitis (NASH) (76). Peripheral blood MAIT cells are depleted in NAFLD, with their enrichment in the liver (77). A negative correlation has been reported between circulating MAIT cell frequency and serum glycated hemoglobin (HbA1c), gamma-glutyl transferase or total triglycerides. These cells upregulate PD-1, CCR5 and CXCR6 (77). Upon stimulation, an IL-4 dominant response is seen over IFN-γ and TNF production. In vitro, activated MAIT cells can induce monocyte/macrophage differentiation into a M2 phenotype (77). Furthermore, in a mouse model of NASH, induced with methionine and choline deficient diet, MAIT cells localize to the liver and display a Th2 profile (IL-4+ and IL-10+ > IFN-γ+ cells) in wild-type animals. In the MR1-/- counterparts there was greater steatohepatitis with an accompanying increase in the proportion of CD11c+ pro-inflammatory M1 macrophages relative to CD206+ M2 macrophages suggesting a potentially protective role for MAIT cells (77). However, in the context of fibrosis, a more pro-inflammatory and fibrogenic function has been proposed given the observation of more progressive carbon tetrachloride (CCl4) induced liver fibrosis in animal models using MAIT cell enriched (Vα19TCRTg) over MAIT cell deficient (MR1-/-) mice (78). Both in alcoholic and non-alcoholic fatty liver disease MAIT cells have been shown to accumulate in liver fibrotic septa and demonstrate increased proliferative capacity with upregulation of Ki-67 (78). With progression to cirrhosis these cells are closely positioned to fibrogenic myofibroblasts; in vitro co-culture experiments have shown MR1 and contact-dependent mitogenic effects of MAIT cells on human myofibroblasts (78). Blood MAIT frequency declines with liver fibrosis, and remaining MAIT cells are activated (CD25 and CD69 high). Interestingly long-term prophylactic antibiotic therapy (norfloxacin or rifaximin) is significantly associated with less decline in MAIT cell frequency and lower CD25 expression (78). The human gut microbiome in NAFLD and NASH is devoid of Bacteroidetes, with expansion of Prevotella and Porphyromonas species. In NASH the proportion of ethanol producing bacteria also increases (75). Rifaxamin is the most widely used antimicrobial prophylaxis agent for the prevention of hepatic encephalopathy in end stage liver disease. It reduces bacterial translocation, has anti-inflammatory properties and modulates the microbiota, specifically reducing the abundance of harmful bacteria (e.g. Klebsiella, Streptococcus, Clostridium) relative to probiotic organisms (e.g. Bacteroides) (79).
Intestinal microbiota has also been linked to susceptibility in alcoholic liver disease (ALD) (80). The gut microbiome shifts with disease progression from steatohepatitis, through to fibrosis then cirrhosis and with patterns of alcohol consumption (binge drinking vs chronic consumption). The microbiome had been largely characterized in animal models, with limited human studies (81). Overall, there is lower abundance of Bacteroidetes and a higher proportion of Enterobacteriaceae and Proteobacteria (75, 81). Humanized germ-free mice, following transplantation of intestinal microbiota from patients with severe alcoholic hepatitis, develop hepatitis and intestinal barrier impairment (80). In a murine model of acute binge on the background of chronic alcohol exposure, MAIT cells became depleted in the intestine, liver and lung. Liver and lung MAIT cells upregulated CD69, IFN-γ, TNF and the transcription factor T-bet. It is worth noting that female animals were exclusively used in this study, given the variable effects of ethanol consumption and differences in microbiome driven by sex (82). Alcohol exposure reduced intestinal microbiome beta diversity (a measure of community variation between animals), with resultant reduction in riboflavin production capacity. Adoptive transfer of cecal microbiota to antibiotic pre-treated alcohol naïve mice produced a decline in pulmonary and hepatic MAIT cell frequency, with increased IFN-γ+/T-bet+ cells at these sites and TNF+ cells in the lung. This change in MAIT cell frequency could be abolished with antibiotic therapy following alcohol exposure. Serum levels of intestinal fatty acid binding protein (iFABP), a biomarker of intestinal epithelial damage, and bacterial 16S rRNA gene copies were elevated following alcohol exposure. In vitro treatment of human MAIT cells with this serum reduced viability, driving apoptotic death and upregulation of CD38, IFN-γ and granzyme B. Direct ethanol exposure did not produce the same effect (82). Similarly, blood MAIT cells are depleted in patients with severe alcoholic hepatitis and alcohol related cirrhosis with altered transcriptional programming (reduced RORγt and PLZF expression), activated phenotype (CD69high) and altered responses (reduced IL-17 and granzyme B with E. coli challenge). Plasma endotoxin and D-lactate (measure of gut permeability) are increased with ALD. Faecal extracts derived from patient with ALD reproduce the abnormal MAIT cell phenotype in vitro with accelerated cell death, upregulation of CD69 and HLA-DR and diminished antibacterial capacity (reduced IFN-γ, TNF, IL-17, granzyme B and perforin with E. coli infection) (83).
The role of MAIT cells in chronic liver disease pathogenesis is yet to be fully defined; indeed, their deficiency in advanced disease could contribute to increased susceptibility towards systemic infection, particularly with shifts seen in the microbiome. Conversely these changes in the context of wider inflammatory and pro-fibrotic signals could account for MAIT cell phenotypes observed in cirrhosis. It is not known if MAIT cells directly interact with and shape the microbiome in homeostasis, by extension further studies are needed to determine if following barrier disruption MAIT cells are bystanders responding to changes in their microenvironment or lose an innate capacity to support commensal communities.
Obesity and metabolic syndromes
There is evidence that changes in the gut microbiome may contribute to the development of obesity (84), with murine and human studies suggesting gut microbe-derived lipopolysaccharide and translocating gut bacteria may contribute to systemic inflammation in obesity (85–87), which can drive insulin resistance associated with the development of type 2 diabetes (88). Obesity is associated with a decrease in overall circulating MAIT cells (89–91), but an increase in circulating, activated, IL-17 producing MAIT cells, which may be correlated with translocated bacteria such as Bacteroidetes (89, 90). Obesity is also associated with reduction in glycolytic metabolism, mTORC1 signaling, and SLC7A5 aa transport in circulating MAIT cells (92). IL-17-producing and Granzyme B+ MAIT cells are increased in omental adipose tissue in human obesity (89, 91). Moreover, whilst circulating MAIT cells are deficient in obesity, weight loss after bariatric surgery is associated with both a restoration of circulating MAIT cell numbers, and an increased diversity of the gut microbiome, including increased in Bacteroidetes and Fusobacteria (93).
Most human studies are correlative, but in murine models, with leptin deficient or high-fat diet-fed mice, MAIT cells were likewise decreased in peripheral blood and ileal and epididymal adipose tissue, due to a shift towards a pro-apoptotic phenotype, with relative increase in activated, IL-17-producing MAIT cells (88). These changes were correlated with a decreased expression of MR1 ligands and of riboflavin pathway genes within the cecal microbiome. These MAIT cells may have been contributing to insulin resistance, as MR1-/- mice had greater insulin sensitivity to oral insulin tolerance testing. Adipose MAIT cells were also contributing to enhanced pro-inflammatory cytokine and chemokine signaling in adipose tissue, and to a reduction in FoxP3+ Treg and a shift towards M1 rather than M2 polarization of macrophages (88). The presence of MAIT cells was also associated with decreased gut epithelial barrier integrity, measured by FITC-dextran translocation and by expression of tight junction proteins, which would favor bacterial translocation. Thus, these data suggest that MAIT cells promote inflammation in obesity. Might these changes in MAIT cells also affect the gut microbiome in a deleterious manner? In the same mouse model fecal transfer was performed into C57BL/6 mice from MAIT-deficient MR1-/- or MAIT over-expressing Vα19+/- mice, and showed MAIT cells promoted a microbiome lower in Bifidobacteriaceae and Lactobacteriaceae, and these changes were again associated with a decrease in gut epithelial barrier integrity and in frequencies of mucosal Treg, innate lymphoid cell(ILC)2 and ILC3 cells, which implies the MAIT-dependent effects on the microbiome are not simple epiphenomena, but can have significant immunological effects.
Haematopoietic stem cell transplantation
Allogenic HSCT can offer curative therapy for a wide range of hematological disorders but can be complicated by inappropriate immune reconstitution, resulting in inflammatory sequelae such as acute or chronic graft versus host disease (GVHD), and increased risk of infection (94). Post-transplantation intestinal microbiome is associated with survival and complications related to HSCT (95–97). Through their interaction, the intestinal microbiome and mucosal T cells, particularly human MAIT cells, are thought to play a protective role post-transplant (30, 94, 98). In a murine major MHC (class I and II) mismatched allogenic stem cell transplant model, MAIT cell frequency was higher in tissue compared with peripheral blood (41). MAIT cells preferentially localized to the colon, producing high concentration of IL-17A to maintain barrier integrity and limit alloantigen presentation after bone marrow transplantation. By comparison IL-17A-/- and MR1-/- animals displayed relatively accelerated GVHD, associated with altered fecal microbiota and downregulation of tight junction proteins claudin 4 and claudin 8 (41). In human HSCT, a diverse intestinal microbiome early after transplant is associated with a higher MAIT cell frequency (30), reduced incidence of acute GVHD (99, 100) and improved survival (100). Circulating Vδ2 cell frequency correlated with MAIT cells and intestinal alpha diversity, a measure of species diversity and abundance (100). Specifically, a higher abundance of Bacteroidetes was seen with higher MAIT cell numbers, whereas abundance of Firmicutes was associated with fewer MAIT cell (100). Blautia spp. abundance is also predictive of MAIT reconstitution (98). No increased abundance of the riboflavin biosynthesis pathways is however observed between individuals based on MAIT cell frequency (100). Single cell RNA sequencing of peripheral blood MAIT cells revealed a pro-inflammatory phenotype, upregulating genes linked to effector function (GNLY, PRF1, CCL4) and migration (ITGB2) with concurrent downregulation of NfκB signaling inhibitors (NFKBIA, TNFAIP3). Vδ2 cells also display a complementary activated phenotype. As expected, a tissue repair signature was not detected in peripheral blood MAIT cells (100). High MAIT cell frequency in infused grafts is linked to higher abundance of intestinal flora post-transplant and lower incidence of acute GVHD. Following the onset of GVHD, circulating MAIT cell frequency declines as these cells become activated (upregulating CD69, CXCR3, CXCR4 and transcription factors RORγt and T-bet). This coincides with a decline in riboflavin synthesising gut microbiota, perhaps reflecting a component of microbial regulation by MAIT cells in health (99). These studies provide insight into the interdependence of MAIT cells and the microbiome in a unique setting of immune reconstitution in human adults. However, due to the lack of tissue MAIT cell profiling several questions remain unanswered including whether MAIT cells can repopulate tissues at similar baseline frequencies and if their phenotype is altered upon their return thereby affecting homeostatic functions, barrier integrity and microbiome composition.
Chronic infections and immunodeficiency
In a similar pattern to the systemic inflammatory conditions discussed above, chronic infection with HIV (101–104) and HCV (105–107) cause a decline in circulating MAIT cell frequency (108) which cannot be fully restored with treatment (101, 109) and is associated with intestinal dysbiosis (108). Circulating MAIT cells upregulate perforin and granzyme B in HIV and HCV infection, with high CD69 and PD-1 expression in viral co-infection. Following ex vivo stimulation with E. coli there is impaired IFN-γ, TNF and IL-17 generation in presence of mono- or co-infection, compared with health. Virally-infected patients have relatively low endotoxin core antibodies, implying a diminished capacity to control translocating bacteria. There is a compositional change in the fecal microbiome with higher abundance of Bacteroidetes and lower abundance of Firmicutes following viral infection. Bacteroides spp. abundance has been shown to correlate positively with MAIT cell frequency but negatively with TNF production following bacterial challenge, whereas Firmicutes abundance negatively correlated with PD-1+ MAIT cell frequency (108). Gut dysbiosis is not reversed with antiretroviral therapy (ART) in HIV (110) and there is partial recovery following HCV eradication (111) except in cirrhotic patients (112). The lung microbiome is also less diverse in HIV and only partially restored with ART (113). Intriguingly, compared with blood, lung MAIT cells better retain function and transcriptional features in HIV, including a tissue repair capacity (48). These studies further support a role for the microbiome in shaping MAIT cell function; circulating MAIT cells are likely reflective of the state of the gut microbiome given its association with barrier disruption and potential microbial translocation into circulation, whereas at different barrier sites (e.g. lung) the local microbiome has the capacity to uniquely modify tissue resident MAIT cell phenotype. It also raises further questions regarding the requirements for reconstitution of MAIT cells following their depletion – how essential are individual constituents of the microbiome and their capacity for riboflavin synthesis? What other co-stimulatory signals are required and potentially deficient in these viral infections? And, is complete re-population of tissue with MAIT cells restricted by a temporal window as in mice? These questions need to be addressed in human studies to best translate therapeutic strategies for re-establishing tissue homeostasis with MAIT cells.
The respiratory microbiome and MAIT cells
The respiratory tract encompasses the upper (anterior nares, nasal passages, paranasal sinuses, nasopharynx, oropharynx, and laryngeal segment proximal to the vocal cords) and lower (larynx distal to vocal cords, trachea, small airways, and alveoli) tracts. The total surface area of the airways is approximately 70m2 making it the second largest barrier site after the gut mucosa in human (114). Culture independent methods of microbial detection have dispelled the long-held theory of lung sterility and instead demonstrated the presence of diverse communities of microbiota in the lower airway (115). MAIT cells are enriched in the airways and form the largest population of antibacterial T cells in the lungs (116); they express tissue residency markers (CD69 and CD103) and display polycytotoxic potential (117). Thus, MAIT cells occupy a prime position to support pulmonary immunity, and there has been growing interest in studying their role in airways diseases. Airways inflammation and common treatments (corticosteroids and antimicrobials) significantly modify the local microbiome and alter barrier function, therefore careful consideration of the interdependence between MAIT cells and the microbiome is crucial when studying pulmonary pathology as discussed below.
The upper airway
The oropharynx harbors a diverse microbiome characterized by the genera Streptococcus, Neisseria, Rothia, and anaerobes, including Veillonella., Prevotella and Leptotrichia (114). It is thought to be the primary source of lung microbiota, introduced through subclinical microaspiration (118). MAIT cells are present in the buccal mucosa (up to 50% CD8αα T cells), displaying a tissue resident effector memory phenotype (CD69, CD103, HLA-DR and PD-1 high) and IL-17 skewed response to PMA-ionomycin stimulation (46). One study has considered oromucosal MAIT cell function in relation to the microbiome in apical periodontitis, characterized by inflammation of the periodontal tissue leading to translocation and dissemination of opportunistic organisms. In this condition a MAIT cell signature appears in affected tissue with increased MAIT TCR, TNF, IFN-γ and IL-17A transcripts compared to healthy adjacent gingiva. There is an expansion of riboflavin producing taxa in the local microbiome and using a sparse partial least squares discriminant analysis the authors report several bacterial genera negatively correlated with MAIT TCR and IL-17A transcripts (119). Whilst this is an interesting approach, a large variation in abundance and diversity of bacterial taxa was observed in this study within a small population (n=25), thus there is limited power to derive conclusions regarding mechanisms of microbiome-MAIT cell interaction.
The nasopharynx is similarly enriched with MAIT cells (47) but is home to a unique microbiome compared with the oropharynx and lower respiratory tract. As a transition site between keratinized squamous epithelium and stratified squamous epithelium it hosts skin colonizers from the genera Staphylococcus, Propionibacterium and Corynebacterium alongside Moraxella, Corynebacterium, Dolosigranulum, Haemophilus and Streptococcus (114). As seen in the oropharynx, sinonasal MAIT cells possess a tissue resident effector memory phenotype. They have been linked to disease severity in allergic rhinitis with nasal polyposis; in this condition MAIT cell appear more activated with CD38 upregulation and IL-17A skewed response to stimulation. Both markers correlate with disease severity (47). Allergic rhinitis and nasal polyposis are often accompanied by asthma. In these conditions, changes to the nasopharyngeal microbiome have been reported with expansion of MAIT cell activating organisms belonging to Bacteroidetes and Proteobacteria taxa in asthma (120) and predominantly Firmicutes, Proteobacteria and Actinobacteria in chronic rhinosinusitis with nasal polyposis. Interestingly in children, nasal Corynebacterium sp. and S. epidermidis abundance is associated with absence of pet allergen sensitization (121); Corynebacterium have been shown to negatively correlate with inflammatory gene expression in the nose (122) whilst S. epidermidis can engage a tissue repair program in skin resident murine MAIT cells (21). It is tempting to speculate that the microbiome in health supports a homeostatic phenotype in MAIT cells whereas disease driven changes to the microbiome supply the antigenic and co-stimulatory signals to sustain an inflammatory IL-17 dominant response. It is yet to be determined if MAIT cells can in fact interact with the microbiome in this manner and indeed if any potentially pathogenic activity can be reversed by manipulating the microbial community.
The lower airway
The healthy adult lung microbiome is dominated by the genera Prevotella, Veillonella, and Streptococcus (123). These belong to the phyla Bacteroidetes and Firmicutes, both stimulators of MAIT cells, with a stronger capacity in the former group (31). In lung diseases there are notable changes to the microbiome likely provoked by host inflammatory responses often leading to increased airway wall permeability and mucus production modifying growth conditions (124–126). A sustained bidirectional relationship between the mucosal immune system and disordered respiratory microbiota is likely to be a key driver of disease progression. We propose that the shift in community membership towards species with greater MAIT cell antigenic load, immunogenicity and capacity for epithelial disruption can overwhelm MAIT cell homeostatic barrier defenses, particularly with numeric and functional deficiencies seen in these conditions as exemplified below.
Airways diseases carry a huge global burden; asthma is the commonest chronic respiratory disease affecting 262 million people worldwide (127) and chronic obstructive pulmonary (COPD) is the third leading cause of mortality (128). These conditions have uniquely disordered airway microbiome but share a pulmonary MAIT cell deficiency proportionate to inhaled corticosteroid (ICS) dose (116, 129), a mainstay of therapy with increasing disease severity.
Asthma is a clinically and immunologically heterogenous condition. Treatment approaches in asthma have been revolutionized by identifying and targeting ‘treatable traits’ (130), notably in type-2 cytokine (IL-5, IL-4, IL-13) mediated eosinophilic airways inflammation for which novel biologics have emerged (131, 132). ‘T2-low’ non-eosinophilic disease is refractory to corticosteroids and existing biologics (130). It affects ~30% of severe asthmatics (133), and is associated with airways neutrophilia alongside high IL-17 (133) expression, and may be driven by chronic bacterial airways infection (118, 134–138). Epithelial barrier disruption is central to pathogenesis, regardless of inflammatory phenotype (139). An altered naso-/hypopharyngeal microbiome can predict development of allergic asthma in childhood (134, 140). Circulating MAIT cell frequency at 1 year of age is associated with reduced risk of asthma diagnosis within the first 7 years of life and a Th1 dominant response in CD4+ T cells (141). In pediatric asthma, small studies have reported increased frequency of IL-17+ MAIT cells in bronchoalveolar lavage (142) and blood (143) of patients presenting with severe exacerbations, however no comparisons were made with health when sampling the lower airway (142). In adult disease Haemophilus influenzae has emerged as the commonest potentially pathogenic organism in the airway, associated with sputum neutrophilia and altered microbial diversity, namely reduction in Streptococcus, Gemella and Porphyromonas taxa (135, 136, 144, 145). In a large bronchoscopy study no evidence of increased IL-17A in serum, sputum or BAL was found in asthma nor was there an increase in IL-17+ T cell populations (Th17 or γδT cells). This study identified a striking deficiency in MAIT cell frequency with increasing disease severity and ICS use (116). Corticosteroid exposure has been demonstrated to impair MAIT cell IFN-γ responses in vitro to non-typeable H. influenzae (NTHi), the most prevalent strains linked to exacerbations of airways diseases (129). Sputum MAIT cells are CD69 and PD-1 high, and peripheral blood cells from patients with asthma are skewed towards IL-17/TNF production (over IFN-γ) following activation. IL-7 levels in sputum and serum are elevated in neutrophil dominant airways inflammation and induces greater IL-17 response to PMA-Ionomycin ex vivo stimulation (146). MAIT cell frequencies have been reported to correlate with NK, ILC1, ILC2, ILC3 cells in severe asthma, declining with airflow obstruction (reduction in FEV1%) (147). In a murine model of allergic airway inflammation using Alternaria inhalation, MAIT cells are proposed to repress ILC2 driven inflammation and airway hyperresponsiveness via expression of interleukin-4-induced gene 1 (IL4I1) (148). In asthma, MAIT cells are thus depleted and exposed to a Proteobacteria dominated microbiome in the setting of epithelial barrier disruption. Translocation of bacterial antigen and products may account for the activated MAIT cell phenotype however ICS-disabled anti-bacterial responses could cause susceptibility to airways infection seen in severe asthma. Moreover, their absence could deprive the airway of tissue repair and type-2 immune regulatory mechanisms. The lines of causality in severe treatment refractory asthma, particularly with dominance of pathogenic organisms in the airway and neutrophilic infiltration, are very complex. It likely involves barrier dysfunction and mucosal immune disarmament; in the case of MAIT cells it is yet to be determined if a pathogenic role can be ascribed or if these cells are stripped of their homeostatic antibacterial function due to the wider spanning inflammatory landscape or indeed treatment. Future studies therefore need to examine their function in well characterized patient cohorts and compare tissue resident cells in disease affected and unaffected locations with due consideration to the local microbiome.
COPD is also heterogenous in its pathogenesis and manifests with predominantly neutrophilic airways inflammation, mucus hypersecretion, emphysema and variable vascular dysfunction (149). Unlike asthma, alterations in airway microbiome appear late with more severe disease. The microbiome declines in diversity, it is depleted of Bacteroidetes with a relative expansion in Proteobacteria, particularly Haemophilus and Moraxella (123, 150, 151). MAIT cells are depleted in blood and endobronchial biopsies of corticosteroid treated (but not ICS naïve) COPD patients (129). A further study has reported MAIT cell depletion in blood with enrichment in lung parenchyma and accumulation around alveolar epithelial cells in less clinically severe disease, however a limitation of these data is the lack of reporting on ICS use in lung tissue donors (152). There is a higher frequency of IL-17+ lung MAIT cells in COPD compared to health, and blood MAIT cells generate IL-17 (over IFN-γ) following PMA-ionomycin stimulation in vitro with diminishing magnitude of response seen with worsening airflow obstruction (152). Lower MAIT cell frequencies are associated with elevated serum C-reactive protein (CRP) levels (153) and increased frequency of exacerbations requiring hospitalization (154). At the time of exacerbation peripheral blood MAIT cells are activated with upregulation of CD38 and LAG-3 (154). The above studies hint towards an impaired antibacterial defense with MAIT cell deficiency in COPD sufficient to cause clinical exacerbation events as disease severity increases. The relationship with ICS in COPD and asthma is important and raises the question whether we should supplement such therapies with counteractive strategies to boost barrier MAIT cell frequencies either directly with ligand (5-OP-RU) or indirectly by manipulating the microbiome (e.g. with probiotics). Further work is also needed to understand the significance of MAIT cell derived IL-17 in the setting of neutrophilic airways inflammation – is it a sufficient signal to perpetuate neutrophil recruitment or are MAIT cells attempting to re-establish tissue homeostasis? These questions need to be considered in the context of the microbiome and dysregulated epithelium (155).
The skin microbiome and MAIT cells
The skin microbiome is critical for homeostasis and shaping of the mucosal immune system. As previously discussed, cutaneous commensals have been shown to induce a homeostatic tissue repair program and functionality in murine skin MAIT cells. In human skin, MAIT cells are a tissue resident population upregulating skin homing marker CLA and CD103 and not enriched in common skin lesions (except dermatitis herpetiformis) (156). The colonizing microbial population in human skin is composed of a core group of species and variation is seen with change in topology, introduced by structures such as hair follicles, sebaceous glands and ducts, alongside environmental factors (20, 157). The core phyla comprising the epidermal microbiome are Actinobacteria (up to 50% of organisms), Firmicutes, followed by Proteobacteria and Bacteroidetes. The gut microbiome has also been implicated in shaping skin health given common cutaneous manifestations of GI disorders such as IBD and coeliac disease. The mechanisms underlying gut-skin microbial interactions are not known but it is postulated that gut dysbiosis and resultant systemic inflammation or intestinal microbial translocation may contribute to disrupted skin homeostasis (157).
Skin dysbiosis is recognized in multiple chronic inflammatory conditions including psoriasis, atopic dermatitis, rosacea and acne vulgaris. In psoriasis there are complex patterns of change in microbial composition with variation in reporting between studies (158). Within psoriatic lesions increased S. aureus abundance and decreased S. epidermidis abundance have been observed (158), this pattern is also reported in atopic dermatitis (157). MAIT cells can mount a cytotoxic response to S. aureus infected dendritic cells in an IL-12 reliant and partially MR-1 dependent manner, with IFN-γ and Granzyme B upregulation (159). Thus, they are equipped to provide cutaneous antibacterial defenses against S. aureus. MAIT cells originally garnered interest in psoriasis as a source of IL-17A, which is a key driver of inflammation; but Teunissen et al. have shown that the majority of IL-17A+CD8+ T cells are in fact conventional CD8+ T cells rather than MAIT cells (160). In atopic dermatitis abundance of S. aureus induces a host transcriptomic signature characterized by upregulation of genes encoding antimicrobial factors, tryptophan metabolites, immune activation (IL1B, CCL2, CCL19) and Th2 signaling mediators (IL4R, IL5, IL13, PI3, TNFRSF4, CCR4) (161). Interestingly in a murine model of atopic dermatitis MAIT cells have been implicated in eosinophil activation and recruitment of IL-4/-13 producing type 2 effector cells in a MR1 dependent fashion (162). The skin therefore reflects another major barrier site at which the local microbiome may shape not only antimicrobial type 1 immunity exercised by MAIT cells but also influence the regulation of type 2 allergic inflammation. There is a relative paucity of human data examining the balance between tissue repair and proinflammatory functionality in MAIT cells in the context of primary skin disorders or cutaneous manifestations of systemic inflammatory disorders. Murine and human in vitro data have shown successful harnessing of MAIT cells to accelerate wound healing through TCR ligation (21, 36). It is yet to be seen if this capacity can be utilized as an add-on therapy to address barrier disruption in skin diseases through direct application of MAIT cell ligand or manipulation of the skin/gut microbiome with targeted topical or oral probiotics respectively. This approach may even be relevant within the female genital tract, as an extension of the skin barrier and a mucosal site at which MAIT cells are enriched (49).
Discussion and future directions
A common theme within this expanding body of data is that dysbiosis at any mucosal surface can affect MAIT cell frequency and function, both locally and at distant sites, with dysbiosis typically associated with a decrease in circulating MAIT cell frequencies, but a relative increase in activated, IL-17-producing, pro-inflammatory MAIT cells. Conversely changes in MAIT cell frequency have also been shown to drive changes in the host microbiome, with evidence these can in turn have direct immunological consequences. Given the incredible diversity of bacteria within human microbiomes, full understanding of these complex interactions with the mucosal immune system is hard to achieve, and further research focusing in vivo on changes in defined microbial species and their consequences for the host immune response are warranted.
Nonetheless, already there is scope for investigating the potential for therapeutic manipulation of the microbiome or MAIT cell compartment.
Manipulation of the gut microbiome could be achieved using simple MR1 ligands. Indeed an in vivo murine study showed conceptual proof of principle using exogenous oral administration of the synthetic inhibitory ligand acetyl-6-FP in obese mice. This reduced obesity-associated ileal inflammation and decreased MAIT cell IL-17 production (88). Furthermore, this ligand also led to alteration of the gut microbiome, inducing an increase in Bacteroidetes abundance. From these data a picture emerges of MAIT cells whose functions are very context dependent, and likewise the intent of therapeutic manipulation will be context dependent. In the situation of obesity, changes in the microbiome lead to reduced tissue MAIT cell frequencies, likely through increased apoptosis, but also to increased activation of MAIT cells promoting type 1 biased chronic inflammation, and consequent metabolic dysfunction. In such a situation therapeutic inhibition of MAIT cells has the potential to reduce dysbiosis, improve gut integrity and ameliorate systemic inflammation and metabolic dysfunction.
Conversely in contexts of chronic mucosal infection or epithelial damage, therapeutic stimulation of MAIT cells would be anticipated to promote beneficial antibacterial responses and activate tissue repair programs promoting epithelial wound repair. The use of activating ligands would require selection of synthetic molecules with much greater stability than naturally-occurring 5-OR-RU which degenerates rapidly in aqueous solution.
A key aspect of MAIT cell biology which remains to be elucidated is how it is determined whether the effects of MAIT cell stimulation lead to a dominant pro-inflammatory antimicrobial response, or to a more homeostatic tissue repair activity favoring restoration of epithelial integrity. It is likely that a critical determinant of MAIT cell response is the integrity of the epithelial barrier itself. A damaged epithelium will release a number of factors which may influence MAIT cells directly, including alarmins, such as IL-33, whose receptor IL1R1/ST2 is highly expressed on lung MAIT cells during acute bacterial infection (35). MAIT cells also highly express IL17RE in mice and humans (35), the receptor for IL-17C. IL-17C is abundantly released by epithelia after stimulation by IL-1β, TNF, various pathogens or through cell damage via TLR2 and TLR5, promoting a proinflammatory, Th17 response from T cells (163). A damaged epithelium will also allow translocation of pathogens into proximity to the MAIT cells, directly furnishing danger signals such as lipopolysaccharide, which will activate membrane TLR2, which is particularly highly expressed on stimulated human MAIT cells (35). Conversely, in situations where riboflavin producing commensals are abundant, but the epithelium is intact, the MAIT cell will receive TCR stimulation alone, promoting a dominant homeostatic tissue repair response (35, 36). Therefore, approaches to manipulate MAIT cell biology may need to simultaneously target these danger signal pathways in addition to the MAIT TCR, for instance combining inhibition of TLR2 or IL17RE with inhibitory MAIT cell ligands to reduce inflammation. Theoretically MAIT TCR ligands could be used sequentially or at different stages of an inflammatory response, favoring antagonistic ligands with or without additional immunosuppressants to reduce overt inflammation, followed subsequently by agonistic ligands to support restored barrier integrity. Extensive modelling in experimental systems would be required before clinical trials could be considered.
An alternative approach to therapeutic manipulation of MAIT cells would be altering the microbiome could be altered directly by the administration of probiotics which favor MAIT ligand producing microbiomes, and might thereby enhance the overall riboflavin-synthetic capacity of the fecal microbial community. The net effects of such an intervention are, however, unpredictable, due to complex symbiotic relationships between microbes, such that increasing riboflavin availability might, paradoxically, favor the growth of otherwise less dominant species which lack this synthetic pathway. The potential impact of a modulated MAIT microbiome to impact the host immune response was demonstrated in work by Toubal et al. (88) which showed that feces from MR1-/- mice triggered more MAIT cell activation than feces from MR1 sufficient mice, suggesting MAIT cells are able to differentially sense microbiome assemblages which differ in riboflavin synthesis. The use of probiotics is controversial as a large proportion of bacteria fail to survive the gastric and upper GI environment, or to become significantly established amongst the complex, diverse microbiome of the lower GI tract which compete for the same niche. Nonetheless it could be possible to engineer bacterial species to over-express riboflavin pathways to enhance MAIT cell activation of the entire microbial assemblage, which might for instance help accelerate reconstitution of mucosal immune cell populations after HSCT or HIV. The expected clinical effect of these will need to be determined using in vivo models because the ultimate effect on MAIT cell activation will be influenced by a number of factors which are hard to predict from in vitro experiments, including differing efficiencies of Rib pathway enzymes, interactions between riboflavin producing and riboflavin scavenging species and the extent to which other microbial factors trigger concomitant innate immune signaling.
Within the airways administration of MAIT cell ligands might be manipulated to enhance immune responses during vaccination, as a component of aerosolized vaccines, or as an adjunctive treatment for persistent microbial infection. Indeed, preliminary work has explored this in the context of chronic infection with Mycobacterium tuberculosis (M.tb) (164). During acute infection administration of 5-OP-RU did not enhance protective responses, but surprisingly rather delayed T cell priming through mechanisms dependent on MAIT cells and TGF-β. However, conversely, during chronic infection intrapulmonary 5-OP-RU administration drove a 30-fold MAIT cell expansion and an IL-17A-dependent 10-fold reduction in pulmonary bacterial loads. Of note this protective effect was local to the mucosa and did not affect bacterial load in the liver, implying the potential to manipulate mucosal MAIT cell populations selectively.
In human airways disease, long term antibiotic therapy with macrolide antibiotics has been shown to reduce inflammation and exacerbations of asthma (165). The mechanism is unknown, but it is postulated that reduction in bacteria such as Haemophilus influenzae colonizing the respiratory mucosa may lead to amelioration of mucosal inflammation. It is known that MAIT cells are deficient in airways diseases (116, 129) so studies using direct airway tissue sampling should explore whether the MAIT cell populations are restored by antibiotics and whether this is associated with restoration of a homeostatic rather than proinflammatory MAIT cell phenotype.
Simpler than both these approaches, and more amenable to very simple clinical trials would be assessment of the utility of topical application of MAIT cell ligands or riboflavin-synthesizing commensal bacteria of low invasive capacity to accelerate wound healing, as has already been demonstrated in mice with H2-M3 restricted commensal bacteria (37) and with topical 5-OP-RU (21).
In the decade since the first MR1 ligands were discovered (1) our knowledge of MAIT cell biology has expanded rapidly. Though there remain many unanswered questions, we should expect to see the field start to progress to the next stage of clinical translation over the next 5 years.
Author contributions
MJ and TH jointly conceived the review, conducted the literature review, and drafted the manuscript. TH created the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from MRC-Oxford Doctoral Training Programme (MJ), the Wellcome Trust (104553/z/14/z, 211050/Z/18/z) (TH) and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) (MJ and TH). The views expressed are those of the authors and not those of the NHS or NIHR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin b metabolites to MAIT cells. Nature (2012) 491:717–23. doi: 10.1038/nature11605
2. Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-Cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature (2014) 509:361–5. doi: 10.1038/nature13160
3. Koay HF, Gherardin NA, Enders A, Loh L, Mackay LK, Almeida CF, et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol (2016) 17:1300–11. doi: 10.1038/ni.3565
4. Koay HF, Su S, Amann-Zalcenstein D, Daley SR, Comerford I, Miosge L, et al. A divergent transcriptional landscape underpins the development and functional branching of MAIT cells. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aay6039
5. Martin E, Treiner E, Duban L, Guerri L, Laude H, Toly C, et al. Stepwise development of MAIT cells in mouse and human. PloS Biol (2009) 7:e54. doi: 10.1371/journal.pbio.1000054
6. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature (2003) 422:164–9. doi: 10.1038/nature01433
7. Legoux F, Gilet J, Procopio E, Echasserieau K, Bernardeau K, Lantz O. Molecular mechanisms of lineage decisions in metabolite-specific T cells. Nat Immunol (2019) 20:1244–55. doi: 10.1038/s41590-019-0465-3
8. Gapin L. Development of invariant natural killer T cells. Curr Opin Immunol (2016) 39:68–74. doi: 10.1016/j.coi.2016.01.001
9. Krovi SH, Loh L, Spengler A, Brunetti T, Gapin L. Current insights in mouse iNKT and MAIT cell development using single cell transcriptomics data. Semin Immunol (2022) 60:101658. doi: 10.1016/j.smim.2022.101658
10. Salou M, Legoux F, Lantz O. MAIT cell development in mice and humans. Mol Immunol (2021) 130:31–6. doi: 10.1016/j.molimm.2020.12.003
11. Salou M, Legoux F, Gilet J, Darbois A, du Halgouet A, Alonso R, et al. A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J Exp Med (2019) 216:133–51. doi: 10.1084/jem.20181483
12. Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med (2015) 212:1095–108. doi: 10.1084/jem.20142110
13. Ben Youssef G, Tourret M, Salou M, Ghazarian L, Houdouin V, Mondot S, et al. Ontogeny of human mucosal-associated invariant T cells and related T cell subsets. J Exp Med (2018) 215:459–79. doi: 10.1084/jem.20171739
14. Gold MC, Eid T, Smyk-Pearson S, Eberling Y, Swarbrick GM, Langley SM, et al. Human thymic MR1-restricted MAIT cells are innate pathogen-reactive effectors that adapt following thymic egress. Mucosal Immunol (2013) 6:35–44. doi: 10.1038/mi.2012.45
15. Lee CH, Zhang HH, Singh SP, Koo L, Kabat J, Tsang H, et al. C/EBPdelta drives interactions between human MAIT cells and endothelial cells that are important for extravasation. Elife (2018) 7. doi: 10.7554/eLife.32532
16. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552:253–7. doi: 10.1038/nature24993
17. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
18. Mondot S, Boudinot P, Lantz O. MAIT. MR1, microbes and riboflavin: a paradigm for the co-evolution of invariant TCRs and restricting MHCI-like molecules? Immunogenetics (2016) 68:537–48. doi: 10.1007/s00251-016-0927-9
19. Boudinot P, Mondot S, Jouneau L, Teyton L, Lefranc MP, Lantz O. Restricting nonclassical MHC genes coevolve with TRAV genes used by innate-like T cells in mammals. Proc Natl Acad Sci U.S.A. (2016) 113:E2983–2992. doi: 10.1073/pnas.1600674113
20. Amini A, Pang D, Hackstein CP, Klenerman P. MAIT cells in barrier tissues: Lessons from immediate neighbors. Front Immunol (2020) 11:584521. doi: 10.3389/fimmu.2020.584521
21. Constantinides MG, Link VM, Tamoutounour S, Wong AC, Perez-Chaparro PJ, Han SJ, et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science (2019) 366. doi: 10.1126/science.aax6624
22. Kennedy EA, King KY, Baldridge MT. Mouse microbiota models: Comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol (2018) 9:1534. doi: 10.3389/fphys.2018.01534
23. Legoux F, Bellet D, Daviaud C, El Morr Y, Darbois A, Niort K, et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science (2019) 366:494–9. doi: 10.1126/science.aaw2719
24. Gherardin NA, Souter MN, Koay HF, Mangas KM, Seemann T, Stinear TP, et al. Human blood MAIT cell subsets defined using MR1 tetramers. Immunol Cell Biol (2018) 96:507–25. doi: 10.1111/imcb.12021
25. Howson LJ, Awad W, von Borstel A, Lim HJ, McWilliam HEG, Sandoval-Romero ML, et al. Absence of mucosal-associated invariant T cells in a person with a homozygous point mutation in MR1. Sci Immunol (2020) 5. doi: 10.1126/sciimmunol.abc9492
26. Park JY, Won HY, DiPalma DT, Kim HK, Kim TH, Li C, et al. In vivo availability of the cytokine IL-7 constrains the survival and homeostasis of peripheral iNKT cells. Cell Rep (2022) 38:110219. doi: 10.1016/j.celrep.2021.110219
27. Baccala R, Witherden D, Gonzalez-Quintial R, Dummer W, Surh CD, Havran WL, et al. Gamma delta T cell homeostasis is controlled by IL-7 and IL-15 together with subset-specific factors. J Immunol (2005) 174:4606–12. doi: 10.4049/jimmunol.174.8.4606
28. Martin CE, Spasova DS, Frimpong-Boateng K, Kim HO, Lee M, Kim KS, et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity (2017) 47:171–182.e174. doi: 10.1016/j.immuni.2017.07.005
29. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun (2019) 10:2123. doi: 10.1038/s41467-019-10015-4
30. Konuma T, Kohara C, Watanabe E, Takahashi S, Ozawa G, Suzuki K, et al. Reconstitution of circulating mucosal-associated invariant T cells after allogeneic hematopoietic cell transplantation: Its association with the riboflavin synthetic pathway of gut microbiota in cord blood transplant recipients. J Immunol (2020) 204:1462–73. doi: 10.4049/jimmunol.1900681
31. Tastan C, Karhan E, Zhou W, Fleming E, Voigt AY, Yao X, et al. Tuning of human MAIT cell activation by commensal bacteria species and MR1-dependent T-cell presentation. Mucosal Immunol (2018) 11:1591–605. doi: 10.1038/s41385-018-0072-x
32. Krause JL, Schape SS, Schattenberg F, Muller S, Ackermann G, Rolle-Kampczyk UE, et al. The activation of mucosal-associated invariant T (MAIT) cells is affected by microbial diversity and riboflavin utilization in vitro. Front Microbiol (2020) 11:755. doi: 10.3389/fmicb.2020.00755
33. Dias J, Leeansyah E, Sandberg JK. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc Natl Acad Sci U.S.A. (2017) 114:E5434–43. doi: 10.1073/pnas.1705759114
34. Lamichhane R, Schneider M, de la Harpe SM, Harrop TWR, Hannaway RF, Dearden PK, et al. TCR- or cytokine-activated CD8(+) mucosal-associated invariant T cells are rapid polyfunctional effectors that can coordinate immune responses. Cell Rep (2019) 28:3061–3076.e3065. doi: 10.1016/j.celrep.2019.08.054
35. Hinks TSC, Marchi E, Jabeen M, Olshansky M, Kurioka A, Pediongco TJ, et al. Activation and In vivo evolution of the MAIT cell transcriptome in mice and humans reveals tissue repair functionality. Cell Rep (2019) 28:3249–3262.e3245. doi: 10.1016/j.celrep.2019.07.039
36. Leng T, Akther HD, Hackstein CP, Powell K, King T, Friedrich M, et al. TCR and inflammatory signals tune human MAIT cells to exert specific tissue repair and effector functions. Cell Rep (2019) 28:3077–3091.e3075. doi: 10.1016/j.celrep.2019.08.050
37. Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell (2018) 172:784–796.e718. doi: 10.1016/j.cell.2017.12.033
38. Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U.S.A. (2002) 99:14338–43. doi: 10.1073/pnas.212290499
39. King DP, Hyde DM, Jackson KA, Novosad DM, Ellis TN, Putney L, et al. Cutting edge: protective response to pulmonary injury requires gamma delta T lymphocytes. J Immunol (1999) 162:5033–6. doi: 10.4049/jimmunol.162.9.5033
40. Guo XJ, Dash P, Crawford JC, Allen EK, Zamora AE, Boyd DF, et al. Lung gammadelta T cells mediate protective responses during neonatal influenza infection that are associated with type 2 immunity. Immunity (2018) 49:531–544.e536. doi: 10.1016/j.immuni.2018.07.011
41. Varelias A, Bunting MD, Ormerod KL, Koyama M, Olver SD, Straube J, et al. Recipient mucosal-associated invariant T cells control GVHD within the colon. J Clin Invest (2018) 128:1919–36. doi: 10.1172/JCI91646
42. Rouxel O, Da Silva J, Beaudoin L, Nel I, Tard C, Cagninacci L, et al. Cytotoxic and regulatory roles of mucosal-associated invariant T cells in type 1 diabetes. Nat Immunol (2017) 18:1321–31. doi: 10.1038/ni.3854
43. Wang H, Kjer-Nielsen L, Shi M, D'Souza C, Pediongco TJ, Cao H, et al. IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aaw0402
44. Lee DJ, Schnitzlein CW, Wolf JP, Vythilingam M, Rasmusson AM, Hoge CW. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: Systemic review and meta-analyses to determine first-line treatments. Depress Anxiety (2016) 33:792–806. doi: 10.1002/da.22511
45. Haga K, Chiba A, Shibuya T, Osada T, Ishikawa D, Kodani T, et al. MAIT cells are activated and accumulated in the inflamed mucosa of ulcerative colitis. J Gastroenterol Hepatol (2016) 31:965–72. doi: 10.1111/jgh.13242
46. Sobkowiak MJ, Davanian H, Heymann R, Gibbs A, Emgard J, Dias J, et al. Tissue-resident MAIT cell populations in human oral mucosa exhibit an activated profile and produce IL-17. Eur J Immunol (2019) 49:133–43. doi: 10.1002/eji.201847759
47. Rha MS, Yoon YH, Koh JY, Jung JH, Lee HS, Park SK, et al. IL-17A-producing sinonasal MAIT cells in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol (2022) 149:599–609.e597. doi: 10.1016/j.jaci.2021.07.037
48. Khuzwayo S, Mthembu M, Meermeier EW, Prakadan SM, Kazer SW, Bassett T, et al. MR1-restricted MAIT cells from the human lung mucosal surface have distinct phenotypic, functional, and transcriptomic features that are preserved in HIV infection. Front Immunol (2021) 12:631410. doi: 10.3389/fimmu.2021.631410
49. Gibbs A, Leeansyah E, Introini A, Paquin-Proulx D, Hasselrot K, Andersson E, et al. MAIT cells reside in the female genital mucosa and are biased towards IL-17 and IL-22 production in response to bacterial stimulation. Mucosal Immunol (2017) 10:35–45. doi: 10.1038/mi.2016.30
50. Schmaler M, Colone A, Spagnuolo J, Zimmermann M, Lepore M, Kalinichenko A, et al. Modulation of bacterial metabolism by the microenvironment controls MAIT cell stimulation. Mucosal Immunol (2018) 11:1060–70. doi: 10.1038/s41385-018-0020-9
51. Voillet V, Buggert M, Slichter CK, Berkson JD, Mair F, Addison MM, et al. Human MAIT cells exit peripheral tissues and recirculate via lymph in steady state conditions. JCI Insight (2018) 3. doi: 10.1172/jci.insight.98487
52. Mechelli R, Romano S, Romano C, Morena E, Buscarinu MC, Bigi R, et al. And microbiota in multiple sclerosis and other autoimmune diseases. Microorganisms (2021) 9. doi: 10.3390/microorganisms9061132
53. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi: 10.1038/4441022a
54. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature (2011) 473:174–80. doi: 10.1038/nature09944
55. Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol (2013) 14:668–75. doi: 10.1038/ni.2635
56. Smith AD, Foss ED, Zhang I, Hastie JL, Giordano NP, Gasparyan L, et al. Microbiota of MR1 deficient mice confer resistance against clostridium difficile infection. PloS One (2019) 14:e0223025. doi: 10.1371/journal.pone.0223025
57. Guo J, Song C, Liu Y, Wu X, Dong W, Zhu H, et al. Characteristics of gut microbiota in representative mice strains: Implications for biological research. Anim Model Exp Med (2022) 5:337–49. doi: 10.1002/ame2.12257
58. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol (2019) 4:293–305. doi: 10.1038/s41564-018-0306-4
59. Lee M, Chang EB. Inflammatory bowel diseases (IBD) and the microbiome-searching the crime scene for clues. Gastroenterology (2021) 160:524–37. doi: 10.1053/j.gastro.2020.09.056
60. Qiu P, Ishimoto T, Fu L, Zhang J, Zhang Z, Liu Y. The gut microbiota in inflammatory bowel disease. Front Cell Infect Microbiol (2022) 12:733992. doi: 10.3389/fcimb.2022.733992
61. Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol (2020) 145:16–27. doi: 10.1016/j.jaci.2019.11.003
62. Dou Y, Maurer K, Conrad M, Patel T, Shraim R, Sullivan KE, et al. Mucosal invariant T cells are diminished in very early-onset inflammatory bowel disease. J Pediatr Gastroenterol Nutr (2021) 73:529–36. doi: 10.1097/MPG.0000000000003189
63. Rosati E, Pogorelyy MV, Dowds CM, Moller FT, Sorensen SB, Lebedev YB, et al. Identification of disease-associated traits and clonotypes in the T cell receptor repertoire of monozygotic twins affected by inflammatory bowel diseases. J Crohns Colitis (2020) 14:778–90. doi: 10.1093/ecco-jcc/jjz179
64. Serriari NE, Eoche M, Lamotte L, Lion J, Fumery M, Marcelo P, et al. Innate mucosal-associated invariant T (MAIT) cells are activated in inflammatory bowel diseases. Clin Exp Immunol (2014) 176:266–74. doi: 10.1111/cei.12277
65. Yasutomi Y, Chiba A, Haga K, Murayama G, Makiyama A, Kuga T, et al. Activated mucosal-associated invariant T cells have a pathogenic role in a murine model of inflammatory bowel disease. Cell Mol Gastroenterol Hepatol (2022) 13:81–93. doi: 10.1016/j.jcmgh.2021.08.018
66. Meroni E, Stakenborg N, Gomez-Pinilla PJ, De Hertogh G, Goverse G, Matteoli G, et al. Functional characterization of oxazolone-induced colitis and survival improvement by vagus nerve stimulation. PloS One (2018) 13:e0197487. doi: 10.1371/journal.pone.0197487
67. Globig AM, Hipp AV, Otto-Mora P, Heeg M, Mayer LS, Ehl S, et al. High-dimensional profiling reveals Tc17 cell enrichment in active crohn's disease and identifies a potentially targetable signature. Nat Commun (2022) 13:3688. doi: 10.1038/s41467-022-31229-z
68. Rosati E, Rios Martini G, Pogorelyy MV, Minervina AA, Degenhardt F, Wendorff M, et al. A novel unconventional T cell population enriched in crohn's disease. Gut (2022) 71:2194–204. doi: 10.1136/gutjnl-2021-325373
69. O'Connor W Jr., Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol (2009) 10:603–9. doi: 10.1038/ni.1736
70. O'Connor W Jr., Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol (2010) 11:471–6. doi: 10.1038/ni.1882
71. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab in crohn's disease study g. secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
72. Treiner E. Mucosal-associated invariant T cells in inflammatory bowel diseases: bystanders, defenders, or offenders? Front Immunol (2015) 6:27. doi: 10.3389/fimmu.2015.00027
73. Balmer ML, Slack E, de Gottardi A, Lawson MA, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med (2014) 6:237ra266. doi: 10.1126/scitranslmed.3008618
74. Kurioka A, Walker LJ, Klenerman P, Willberg CB. MAIT cells: new guardians of the liver. Clin Transl Immunol (2016) 5:e98. doi: 10.1038/cti.2016.51
75. Betrapally NS, Gillevet PM, Bajaj JS. Changes in the intestinal microbiome and alcoholic and nonalcoholic liver diseases: Causes or effects? Gastroenterology (2016) 150:1745–1755.e1743. doi: 10.1053/j.gastro.2016.02.073
76. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2022) 7:851–61. doi: 10.1016/S2468-1253(22)00165-0
77. Li Y, Huang B, Jiang X, Chen W, Zhang J, Wei Y, et al. Mucosal-associated invariant T cells improve nonalcoholic fatty liver disease through regulating macrophage polarization. Front Immunol (2018) 9:1994. doi: 10.3389/fimmu.2018.01994
78. Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun (2018) 9:2146. doi: 10.1038/s41467-018-04450-y
79. Yu X, Jin Y, Zhou W, Xiao T, Wu Z, Su J, et al. Rifaximin modulates the gut microbiota to prevent hepatic encephalopathy in liver cirrhosis without impacting the resistome. Front Cell Infect Microbiol (2021) 11:761192. doi: 10.3389/fcimb.2021.761192
80. Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut (2016) 65:830–9. doi: 10.1136/gutjnl-2015-310585
81. Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol (2012) 302:G966–978. doi: 10.1152/ajpgi.00380.2011
82. Gu M, Samuelson DR, Taylor CM, Molina PE, Luo M, Siggins RW, et al. Alcohol-associated intestinal dysbiosis alters mucosal-associated invariant T-cell phenotype and function. Alcohol Clin Exp Res (2021) 45:934–47. doi: 10.1111/acer.14589
83. Riva A, Patel V, Kurioka A, Jeffery HC, Wright G, Tarff S, et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut (2018) 67:918–30. doi: 10.1136/gutjnl-2017-314458
84. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444:1027–31. doi: 10.1038/nature05414
85. Massier L, Chakaroun R, Tabei S, Crane A, Didt KD, Fallmann J, et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut (2020) 69:1796–806. doi: 10.1136/gutjnl-2019-320118
86. Anhe FF, Jensen BAH, Varin TV, Servant F, Van Blerk S, Richard D, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab (2020) 2:233–42. doi: 10.1038/s42255-020-0178-9
87. Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, et al. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem (2014) 388:203–10. doi: 10.1007/s11010-013-1911-4
88. Toubal A, Kiaf B, Beaudoin L, Cagninacci L, Rhimi M, Fruchet B, et al. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun (2020) 11:3755. doi: 10.1038/s41467-020-17307-0
89. Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Invest (2015) 125:1752–62. doi: 10.1172/JCI78941
90. Li Y, Yang Y, Wang J, Cai P, Li M, Tang X, et al. Bacteroides ovatus-mediated CD27(-) MAIT cell activation is associated with obesity-related T2D progression. Cell Mol Immunol (2022) 19:791–804. doi: 10.1038/s41423-022-00871-4
91. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol (2015) 194:5775–80. doi: 10.4049/jimmunol.1402945
92. O'Brien A, Loftus RM, Pisarska MM, Tobin LM, Bergin R, Wood NAW, et al. Obesity reduces mTORC1 activity in mucosal-associated invariant T cells, driving defective metabolic and functional responses. J Immunol (2019) 202:3404–11. doi: 10.4049/jimmunol.1801600
93. Fukuda N, Ojima T, Hayata K, Katsuda M, Kitadani J, Takeuchi A, et al. Laparoscopic sleeve gastrectomy for morbid obesity improves gut microbiota balance, increases colonic mucosal-associated invariant T cells and decreases circulating regulatory T cells. Surg Endosc (2022) 36:7312–24. doi: 10.1007/s00464-022-09122-z
94. Andrlova H, van den Brink MRM, Markey KA. An unconventional view of T cell reconstitution after allogeneic hematopoietic cell transplantation. Front Oncol (2020) 10:608923. doi: 10.3389/fonc.2020.608923
95. Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N Engl J Med (2020) 382:822–34. doi: 10.1056/NEJMoa1900623
96. Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal blautia is associated with reduced death from graft-versus-Host disease. Biol Blood Marrow Transplant (2015) 21:1373–83. doi: 10.1016/j.bbmt.2015.04.016
97. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood (2014) 124:1174–82. doi: 10.1182/blood-2014-02-554725
98. Bhattacharyya A, Hanafi LA, Sheih A, Golob JL, Srinivasan S, Boeckh MJ, et al. Graft-derived reconstitution of mucosal-associated invariant T cells after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2018) 24:242–51. doi: 10.1016/j.bbmt.2017.10.003
99. Gao MG, Hong Y, Zhao XY, Pan XA, Sun YQ, Kong J, et al. The potential roles of mucosa-associated invariant T cells in the pathogenesis of gut graft-Versus-Host disease after hematopoietic stem cell transplantation. Front Immunol (2021) 12:720354. doi: 10.3389/fimmu.2021.720354
100. Andrlova H, Miltiadous O, Kousa AI, Dai A, DeWolf S, Violante S, et al. MAIT and Vdelta2 unconventional T cells are supported by a diverse intestinal microbiome and correlate with favorable patient outcome after allogeneic HCT. Sci Transl Med (2022) 14:eabj2829. doi: 10.1126/scitranslmed.abj2829
101. Cosgrove C, Ussher JE, Rauch A, Gartner K, Kurioka A, Huhn MH, et al. Early and nonreversible decrease of CD161++ /MAIT cells in HIV infection. Blood (2013) 121:951–61. doi: 10.1182/blood-2012-06-436436
102. Leeansyah E, Ganesh A, Quigley MF, Sonnerborg A, Andersson J, Hunt PW, et al. Activation, exhaustion, and persistent decline of the antimicrobial MR1-restricted MAIT-cell population in chronic HIV-1 infection. Blood (2013) 121:1124–35. doi: 10.1182/blood-2012-07-445429
103. Eberhard JM, Hartjen P, Kummer S, Schmidt RE, Bockhorn M, Lehmann C, et al. CD161+ MAIT cells are severely reduced in peripheral blood and lymph nodes of HIV-infected individuals independently of disease progression. PloS One (2014) 9:e111323. doi: 10.1371/journal.pone.0111323
104. Fernandez CS, Amarasena T, Kelleher AD, Rossjohn J, McCluskey J, Godfrey DI, et al. MAIT cells are depleted early but retain functional cytokine expression in HIV infection. Immunol Cell Biol (2015) 93:177–88. doi: 10.1038/icb.2014.91
105. Hengst J, Strunz B, Deterding K, Ljunggren HG, Leeansyah E, Manns MP, et al. Nonreversible MAIT cell-dysfunction in chronic hepatitis c virus infection despite successful interferon-free therapy. Eur J Immunol (2016) 46:2204–10. doi: 10.1002/eji.201646447
106. Bolte FJ, O'Keefe AC, Webb LM, Serti E, Rivera E, Liang TJ, et al. Intra-hepatic depletion of mucosal-associated invariant T cells in hepatitis c virus-induced liver inflammation. Gastroenterology (2017) 153:1392–1403.e1392. doi: 10.1053/j.gastro.2017.07.043
107. Barathan M, Mohamed R, Vadivelu J, Chang LY, Saeidi A, Yong YK, et al. Peripheral loss of CD8(+) CD161(++) TCRValpha7.2(+) mucosal-associated invariant T cells in chronic hepatitis c virus-infected patients. Eur J Clin Invest (2016) 46:170–80. doi: 10.1111/eci.12581
108. Merlini E, Cerrone M, van Wilgenburg B, Swadling L, Cannizzo ES, d'Arminio Monforte A, et al. Association between impaired Valpha7.2+CD161++CD8+ (MAIT) and Valpha7.2+CD161-CD8+ T-cell populations and gut dysbiosis in chronically HIV- and/or HCV-infected patients. Front Microbiol (2019) 10:1972. doi: 10.3389/fmicb.2019.01972
109. Khlaiphuengsin A, Chuaypen N, Sodsai P, Reantragoon R, Han WM, Avihingsanon A, et al. Successful direct-acting antiviral therapy improves circulating mucosal-associated invariant T cells in patients with chronic HCV infection. PloS One (2020) 15:e0244112. doi: 10.1371/journal.pone.0244112
110. Ishizaka A, Koga M, Mizutani T, Parbie PK, Prawisuda D, Yusa N, et al. Unique gut microbiome in HIV patients on antiretroviral therapy (ART) suggests association with chronic inflammation. Microbiol Spectr (2021) 9:e0070821. doi: 10.1128/Spectrum.00708-21
111. Honda T, Ishigami M, Yamamoto K, Takeyama T, Ito T, Ishizu Y, et al. Changes in the gut microbiota after hepatitis c virus eradication. Sci Rep (2021) 11:23568. doi: 10.1038/s41598-021-03009-0
112. Bajaj JS, Sterling RK, Betrapally NS, Nixon DE, Fuchs M, Daita K, et al. HCV eradication does not impact gut dysbiosis or systemic inflammation in cirrhotic patients. Aliment Pharmacol Ther (2016) 44:638–43. doi: 10.1111/apt.13732
113. Twigg HL, Knox KS, Zhou J, Crothers KA, Nelson DE, Toh E, et al. Effect of advanced HIV infection on the respiratory microbiome. Am J Respir Crit Care Med (2016) 194:226–35. doi: 10.1164/rccm.201509-1875OC
114. Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat Rev Microbiol (2017) 15:259–70. doi: 10.1038/nrmicro.2017.14
115. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PloS One (2010) 5:e8578. doi: 10.1371/journal.pone.0008578
116. Hinks TS, Zhou X, Staples KJ, Dimitrov BD, Manta A, Petrossian T, et al. Innate and adaptive T cells in asthmatic patients: Relationship to severity and disease mechanisms. J Allergy Clin Immunol (2015) 136:323–33. doi: 10.1016/j.jaci.2015.01.014
117. Meermeier EW, Zheng CL, Tran JG, Soma S, Worley AH, Weiss DI, et al. Human lung-resident mucosal-associated invariant T cells are abundant, express antimicrobial proteins, and are cytokine responsive. Commun Biol (2022) 5:942. doi: 10.1038/s42003-022-03823-w
118. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc (2015) 12:821–30. doi: 10.1513/AnnalsATS.201501-029OC
119. Davanian H, Gaiser RA, Silfverberg M, Hugerth LW, Sobkowiak MJ, Lu L, et al. Mucosal-associated invariant T cells and oral microbiome in persistent apical periodontitis. Int J Oral Sci (2019) 11:16. doi: 10.1038/s41368-019-0049-y
120. Fazlollahi M, Lee TD, Andrade J, Oguntuyo K, Chun Y, Grishina G, et al. The nasal microbiome in asthma. J Allergy Clin Immunol (2018) 142:834–843.e832. doi: 10.1016/j.jaci.2018.02.020
121. Chun Y, Do A, Grishina G, Arditi Z, Ribeiro V, Grishin A, et al. The nasal microbiome, nasal transcriptome, and pet sensitization. J Allergy Clin Immunol (2021) 148:244–249 e244. doi: 10.1016/j.jaci.2021.01.031
122. Chun Y, Do A, Grishina G, Grishin A, Fang G, Rose S, et al. Integrative study of the upper and lower airway microbiome and transcriptome in asthma. JCI Insight (2020) 5. doi: 10.1172/jci.insight.133707
123. Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol (2016) 78:481–504. doi: 10.1146/annurev-physiol-021115-105238
124. Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet (2014) 384:691–702. doi: 10.1016/S0140-6736(14)61136-3
125. Schmidt A, Belaaouaj A, Bissinger R, Koller G, Malleret L, D'Orazio C, et al. Neutrophil elastase-mediated increase in airway temperature during inflammation. J Cyst Fibros (2014) 13:623–31. doi: 10.1016/j.jcf.2014.03.004
126. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway pseudomonas infections of cystic fibrosis patients. J Clin Invest (2002) 109:317–25. doi: 10.1172/JCI0213870
127. Diseases GBD, Injuries C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet (2020) 396:1204–22. doi: 10.1016/S0140-6736(20)30925-9
128. Agusti A, Vogelmeier CF, Halpin DMG. Tackling the global burden of lung disease through prevention and early diagnosis. Lancet Respir Med (2022) 10:1013–5. doi: 10.1016/S2213-2600(22)00302-2
129. Hinks TS, Wallington JC, Williams AP, Djukanovic R, Staples KJ, Wilkinson TM. Steroid-induced deficiency of mucosal-associated invariant T cells in the chronic obstructive pulmonary disease lung. implications for nontypeable haemophilus influenzae infection. Am J Respir Crit Care Med (2016) 194:1208–18. doi: 10.1164/rccm.201601-0002OC
130. Pavord ID, Beasley R, Agusti A, Anderson GP, Bel E, Brusselle G, et al. After asthma: redefining airways diseases. Lancet (2018) 391(10118):350–400. doi: 10.1016/S0140-6736(17)30879-6
131. Singhania A, Rupani H, Jayasekera N, Lumb S, Hales P, Gozzard N, et al. Altered epithelial gene expression in peripheral airways of severe asthma. PloS One (2017) 12:e0168680. doi: 10.1371/journal.pone.0168680
132. Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U.S.A. (2007) 104:15858–63. doi: 10.1073/pnas.0707413104
133. Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Di Stefano A, et al. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur Respir J (2019) 54. doi: 10.1183/13993003.00068-2019
134. Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med (2007) 357:1487–95. doi: 10.1056/NEJMoa052632
135. Simpson JL, Daly J, Baines KJ, Yang IA, Upham JW, Reynolds PN, et al. Airway dysbiosis: Haemophilus influenzae and tropheryma in poorly controlled asthma. Eur Respir J (2016) 47:792–800. doi: 10.1183/13993003.00405-2015
136. Green BJ, Wiriyachaiporn S, Grainge C, Rogers GB, Kehagia V, Lau L, et al. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PloS One (2014) 9:e100645. doi: 10.1371/journal.pone.0100645
137. Goleva E, Jackson LP, Harris JK, Robertson CE, Sutherland ER, Hall CF, et al. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am J Respir Crit Care Med (2013) 188:1193–201. doi: 10.1164/rccm.201304-0775OC
138. Singhania A, Wallington JC, Smith CG, Horowitz D, Staples KJ, Howarth PH, et al. Multi-tissue transcriptomics delineates the diversity of airway T cell functions in asthma. Am J Respir Cell Mol Biol (2017) 58(2):261–70. doi: 10.1165/rcmb.2017-0162OC
139. Carlier FM, de Fays C, Pilette C. Epithelial barrier dysfunction in chronic respiratory diseases. Front Physiol (2021) 12:691227. doi: 10.3389/fphys.2021.691227
140. Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe (2015) 17:704–15. doi: 10.1016/j.chom.2015.03.008
141. Chandra S, Wingender G, Greenbaum JA, Khurana A, Gholami AM, Ganesan AP, et al. Development of asthma in inner-city children: Possible roles of MAIT cells and variation in the home environment. J Immunol (2018) 200:1995–2003. doi: 10.4049/jimmunol.1701525
142. Lezmi G, Abou-Taam R, Garcelon N, Dietrich C, Machavoine F, Delacourt C, et al. Evidence for a MAIT-17-high phenotype in children with severe asthma. J Allergy Clin Immunol (2019) 144:1714–1716 e1716. doi: 10.1016/j.jaci.2019.08.003
143. Lezmi G, Abou Taam R, Dietrich C, Chatenoud L, de Blic J, Leite-de-Moraes M. Circulating IL-17-producing mucosal-associated invariant T cells (MAIT) are associated with symptoms in children with asthma. Clin Immunol (2018) 188:7–11. doi: 10.1016/j.clim.2017.11.009
144. Taylor SL, Leong LEX, Choo JM, Wesselingh S, Yang IA, Upham JW, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol (2018) 141:94–103.e115. doi: 10.1016/j.jaci.2017.03.044
145. Jabeen MF, Sanderson ND, Foster D, Crook DW, Cane JL, Borg C, et al. Identifying bacterial airways infection in stable severe asthma using Oxford nanopore sequencing technologies. Microbiol Spectr (2022) 10:e0227921. doi: 10.1128/spectrum.02279-21
146. Wen X, Nian S, Wei G, Kang P, Yang Y, Li L, et al. Changes in the phenotype and function of mucosal-associated invariant T cells in neutrophilic asthma. Int Immunopharmacol (2022) 106:108606. doi: 10.1016/j.intimp.2022.108606
147. Ishimori A, Harada N, Chiba A, Harada S, Matsuno K, Makino F, et al. Circulating activated innate lymphoid cells and mucosal-associated invariant T cells are associated with airflow limitation in patients with asthma. Allergol Int (2017) 66:302–9. doi: 10.1016/j.alit.2016.07.005
148. Ye L, Pan J, Pasha MA, Shen X, D'Souza SS, Fung ITH, et al. Mucosal-associated invariant T cells restrict allergic airway inflammation. J Allergy Clin Immunol (2020) 145:1469–1473.e1464. doi: 10.1016/j.jaci.2019.12.891
149. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2013) 48:531–9. doi: 10.1165/rcmb.2012-0492TR
150. Diver S, Richardson M, Haldar K, Ghebre MA, Ramsheh MY, Bafadhel M, et al. Sputum microbiomic clustering in asthma and chronic obstructive pulmonary disease reveals a haemophilus-predominant subgroup. Allergy (2020) 75:808–17. doi: 10.1111/all.14058
151. Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax (2018) 73:422–30. doi: 10.1136/thoraxjnl-2017-210408
152. Qiu W, Kang N, Wu Y, Cai Y, Xiao L, Ge H, et al. Mucosal associated invariant T cells were activated and polarized toward Th17 in chronic obstructive pulmonary disease. Front Immunol (2021) 12:640455. doi: 10.3389/fimmu.2021.640455
153. Kwon YS, Jin HM, Cho YN, Kim MJ, Kang JH, Jung HJ, et al. Mucosal-associated invariant T cell deficiency in chronic obstructive pulmonary disease. COPD (2016) 13:196–202. doi: 10.3109/15412555.2015.1069806
154. Pincikova T, Parrot T, Hjelte L, Hogman M, Lisspers K, Stallberg B, et al. MAIT cell counts are associated with the risk of hospitalization in COPD. Respir Res (2022) 23:127. doi: 10.1186/s12931-022-02045-2
155. Huber ME, Larson E, Lust TN, Heisler CM, Harriff MJ. Chronic obstructive pulmonary disease and cigarette smoke lead to dysregulated MAIT cell activation. Am J Respir Cell Mol Biol (2022) 68(1):90–102. doi: 10.1101/2022.02.28.482383
156. Li J, Reantragoon R, Kostenko L, Corbett AJ, Varigos G, Carbone FR. The frequency of mucosal-associated invariant T cells is selectively increased in dermatitis herpetiformis. Australas J Dermatol (2017) 58:200–4. doi: 10.1111/ajd.12456
157. Sinha S, Lin G, Ferenczi K. The skin microbiome and the gut-skin axis. Clin Dermatol (2021) 39:829–39. doi: 10.1016/j.clindermatol.2021.08.021
158. Chen L, Li J, Zhu W, Kuang Y, Liu T, Zhang W, et al. Skin and gut microbiome in psoriasis: Gaining insight into the pathophysiology of it and finding novel therapeutic strategies. Front Microbiol (2020) 11:589726. doi: 10.3389/fmicb.2020.589726
159. Cooper AJR, Clegg J, Cassidy FC, Hogan AE, McLoughlin RM. Human MAIT cells respond to staphylococcus aureus with enhanced anti-bacterial activity. Microorganisms (2022) 10. doi: 10.3390/microorganisms10010148
160. Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA, et al. The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol (2014) 134:2898–907. doi: 10.1038/jid.2014.261
161. Fyhrquist N, Muirhead G, Prast-Nielsen S, Jeanmougin M, Olah P, Skoog T, et al. Microbe-host interplay in atopic dermatitis and psoriasis. Nat Commun (2019) 10:4703. doi: 10.1038/s41467-019-12253-y
162. Naidoo K, Woods K, Pellefigues C, Cait A, O'Sullivan D, Gell K, et al. MR1-dependent immune surveillance of the skin contributes to pathogenesis and is a photobiological target of UV light therapy in a mouse model of atopic dermatitis. Allergy (2021) 76:3155–70. doi: 10.1111/all.14994
163. Hynes GM, Hinks TSC. The role of interleukin-17 in asthma: A protective response? ERJ Open Res (2020) 6. doi: 10.1183/23120541.00364-2019
164. Sakai S, Kauffman KD, Oh S, Nelson CE, Barry CE, Barber DL. MAIT cell-directed therapy of mycobacterium tuberculosis infection. Mucosal Immunol (2021) 14:199–208. doi: 10.1038/s41385-020-0332-4
165. Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): A randomised, double-blind, placebo-controlled trial. Lancet (2017) 390(10095):659–68. doi: 10.1016/S0140-6736(17)31281-3
Keywords: MAIT cell, microbiome and dysbiosis, tissue homeostasis, airways diseases, inflammatory bowel conditions, metabolic syndromes, stem cell transplant (SCT)
Citation: Jabeen MF and Hinks TSC (2023) MAIT cells and the microbiome. Front. Immunol. 14:1127588. doi: 10.3389/fimmu.2023.1127588
Received: 19 December 2022; Accepted: 14 February 2023;
Published: 23 February 2023.
Edited by:
Laurent Gapin, University of Colorado Denver, United StatesReviewed by:
Thierry Mallevaey, University of Toronto, CanadaOlivier Gasser, Victoria University of Wellington, New Zealand
Liyen Loh, University of Colorado, United States
Copyright © 2023 Jabeen and Hinks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy S. C. Hinks, dGltb3RoeS5oaW5rc0BuZG0ub3guYWMudWs=