 Qipeng Zhan
Qipeng Zhan Dunfang Zhang
Dunfang Zhang- State Key Laboratory of Biotherapy and Cancer Center, Department of Biotherapy, Collaborative Innovation Center of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China
Sjogren’s syndrome (SS) is a chronic autoimmune disease accompanied by multiple lesions. The main manifestations include dryness of the mouth and eyes, along with systemic complications (e.g., pulmonary disease, kidney injury, and lymphoma). In this review, we highlight that IFNs, Th17 cell-related cytokines (IL-17 and IL-23), and B cell-related cytokines (TNF and BAFF) are crucial for the pathogenesis of SS. We also summarize the advances in experimental treatment strategies, including targeting Treg/Th17, mesenchymal stem cell treatment, targeting BAFF, inhibiting JAK pathway, et al. Similar to that of SLE, RA, and MS, biotherapeutic strategies of SS consist of neutralizing antibodies and inflammation-related receptor blockers targeting proinflammatory signaling pathways. However, clinical research on SS therapy is comparatively rare. Moreover, the differences in the curative effects of immunotherapies among SS and other autoimmune diseases are not fully understood. We emphasize that targeted drugs, low-side-effect drugs, and combination therapies should be the focus of future research.
1 Introduction
Sjogren’s syndrome (SS) is a chronic autoimmune disease associated with functional disorders of the exocrine glands (e.g., parotid and lacrimal glands) and extraglandular manifestations. In 1892, JH Mikulicz reported the first case of SS. In 1933, the Danish ophthalmologist Sjogren reported on 19 female patients with dryness of the mouth and eyes, 13 of whom had rheumatoid arthritis (RA). To distinguish this ailment from xerophthalmia (vitamin A-deficiency-related dryness of the eyes), Sjogren defined the syndrome as keratoconjunctivitis sicca. KJ Bloch presented the clinical features of the currently recognized syndrome and introduced primary Sjogren’s syndrome and secondary Sjogren’s syndrome, which presents without and with an independent connective tissue disease (CTD), respectively (1).
According to a worldwide epidemiological study based on PubMed and Embase data, the incidence rate of SS is 6.92 per 100 000 person-years and the prevalence rate is 60.82 cases per 100 000 inhabitants, or 1 case per 1644 persons. Moreover, the age of patients peaks at 56. In the last 15 years, the disease has affected females more than males (2). Patients with SS experience an enduring and intolerable pain with multiple physical symptoms, such as dental caries, vaginal dryness, and arthralgia (3).
Given the immense social and economic burden caused by SS, we aimed in this review to characterize the current paradigm of the pathogenesis and treatment of SS to motivate and inform the development of efficient treatment strategies, particularly immunological treatments.
2 Brief review of clinical manifestations
Sjogren’s syndrome is a systemic disease with heterogeneous manifestations that involve disorders or damage to the tissues of the exocrine glands (Figure 1). The diagnosis of SS is a multistep process, including the evaluation of oral and ocular dryness, detection of anti-SSA/Ro and anti-SSB/La antibodies, and glandular biopsy. Dryness of the eyes and mouth, which is caused by the dysfunction of salivary and lacrimal glands, is the most salient and common clinical symptom. Severe sicca symptoms of the eyes and mouth profoundly impede quality of life. An aqueous-deficient mouth has a severe effect on oral health and is associated with an increased risk of developing caries (4). A recent study reported that the oral microbiome of patients with SS who have salivary hypofunction was under stress and dysregulated; Veillonella parvula is a potential biomarker of Sjogren’s syndrome (5).
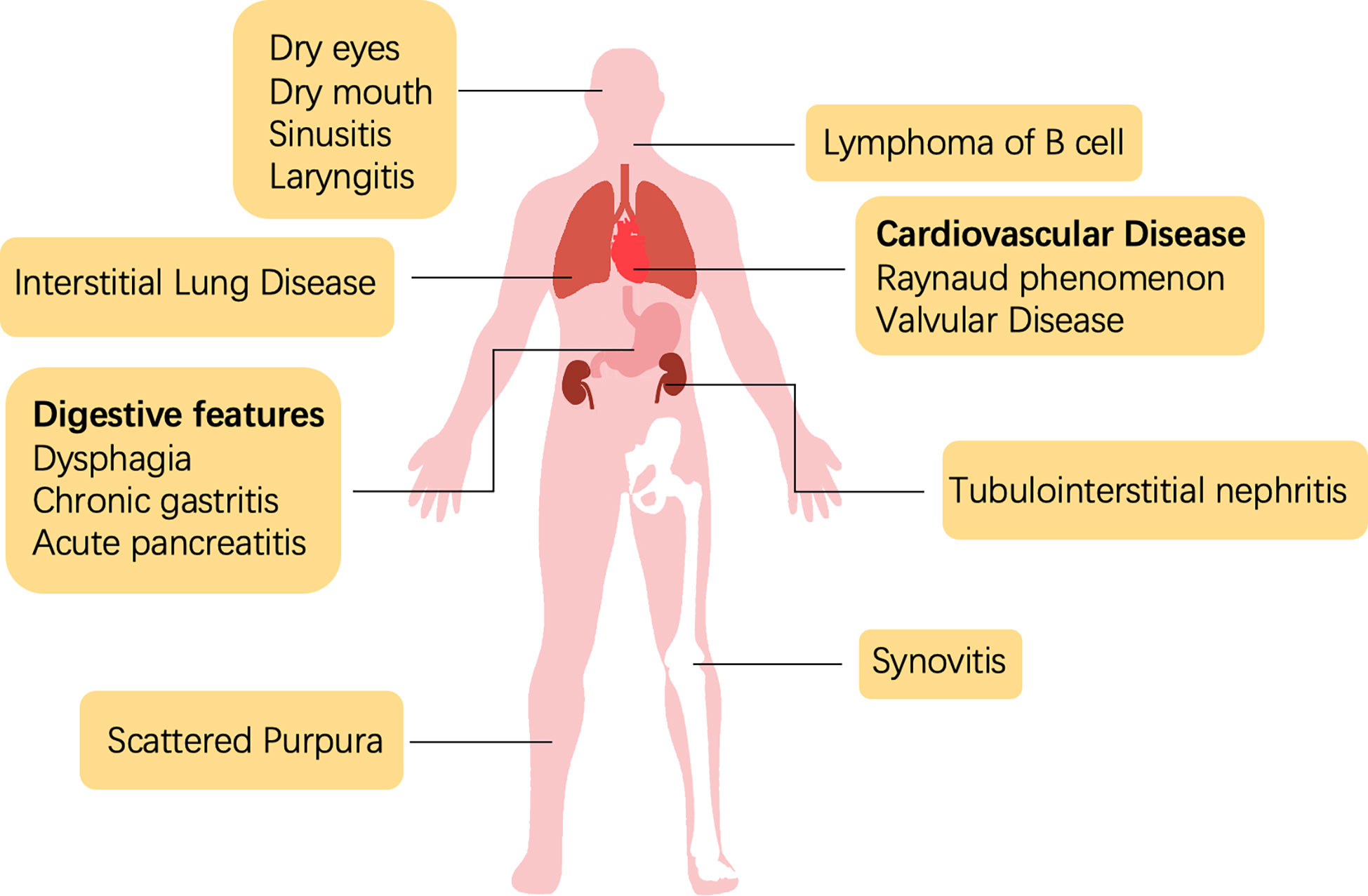
Figure 1 Typical glandular and extraglandular manifestations of SS.
Because SS presents with multiple extraglandular manifestations (Figure 1), the European Alliance of Associations for Rheumatology (EULAR) developed The EULAR SS disease activity index (ESSDAI) to assess disease activity in patients with sicca symptoms and simplify diagnosis. ESSDAI evaluates the severity of disease within 12 clinical domains (i.e., constitutional, lymphadenopathy, glandular, articular, cutaneous, pulmonary, renal, muscular, peripheral nervous system, central nervous system, hematological, biological), and it aims to obtain a standardized evaluation in clinical trials and practice (6).
The involvement of the nervous system was first reported in the 1980s (7). Several neurological diseases have since been associated with SS (8–12), which indicates the importance of precise neurological diagnostic assessments. Interstitial lung disease (ILD) is the most frequent and severe pulmonary complication of SS and contributes substantially to morbidity and mortality. In an Italian cohort, approximately 20% of patients with comorbid SS presented with ILD, and approximately 10% presented with amyloidosis and primary lung lymphoma (13). Efficient clinical examination, including lung biopsy or screening of serological markers, could assist in the early diagnosis and intervention of SS-ILD (13). Unfortunately, no effective treatment strategy exists for SS-ILD (14). As an autoimmune disease, SS can also lead to synovitis and RA, with the latter causing structural damage. A previous study showed that the medication strategy of RA had some success in SS, but the best-performing regimen is unclear (15).
Renal complications have only been observed in less than 10% of patients with SS. Tubulointerstitial nephritis (TIN), caused by lymphocyte infiltration around the renal tubes, occurs in two-thirds of patients with SS and renal dysfunction (16, 17). However, the low prevalence of renal manifestations may be an artefact of the ineffective diagnosis of TIN (18). Non-Hodgkin’s lymphoma (NHL) is the most severe extraglandular complication of SS, with the B cell type being predominant (occurs in approximately 5% of SS cases) (19). Hypergammaglobulinemia or the aberrant expression of other antigens in the blood stimulate the expansion of rheumatoid factor-reactive B cells (20). Meanwhile, B lymphocyte-activating factor (BAFF) and germinal center (GC)-like structures amplify the activation of B cells (21, 22). Some studies have reported that SS-NHL is also associated with abnormal activation of nuclear factor kappa B (NF-κB) (23, 24). Additionally, a multicenter clinical study showed that more than a quarter of patients with SS presented systemic symptoms beyond the current ESSDAI classification, including cardiovascular; digestive; pulmonary; ear, nose, and throat (ENT); cutaneous; and urological features (25).
3 Pathogenesis
3.1 Brief introduction
In a 2013 review by G Nocturne and X Mariette of the pathogenesis of SS (26), three key steps were identified based on the initial genome-wide association study (GWAS): aberrant activation of the innate immune response, especially through the interferon (IFN) and NF-κB pathways, atypical recruitment to lymphoid follicles mediated by CXCR5, and T cell activation with ascending HLA susceptibility along the IL-12–IFN-γ axis. BAFF was considered to be vital in coordinating the innate and adaptive immune responses to the disease. They also highlighted the pathophysiological role of natural killer (NK) and epithelial cells as well as the dysfunction of the neuroendocrine system.
Mavragani et al. reviewed the treatment strategies and molecular targets of the innate and adaptive immunity pathways (27). Regarding the regulation of innate immunity, previous research focused on inhibiting the production of proinflammatory factors, such as IL-1, IL-6, and tumor necrosis factor-α (TNF-α), which has proven to be effective in other autoimmune diseases. IFN-associated pathway inhibitors were another research topic of interest. For example, downregulating the expression of the primary dendritic cell surface receptor ILT7 to reduce TLR7/9-mediated IFN production was considered a potential treatment route. Regarding adaptive immunity, previous research focused on antigen presentation, co-stimulation, B-cell activation, T-cell proliferation, and germinal center formation. Overall, most strategies were aimed at regulating aberrant inflammation.
3.2 IFN
IFN is an immunoregulatory protein that promotes innate and acquired immunity and antiviral activation. IFN is categorized into three types based on structure and origin, i.e., I, II, and III. In 1981, researchers detected type-I IFN in the blood of patients with autoimmune disease, and linked its expression to the clinical manifestations (28). IFN-I plays an important role in the progression of SS by promoting the activity of immune cells, such as NK cells, CD8+ T cells, and even macrophages. In addition, dendritic cells, the main producers of IFN-I, were observed in the salivary glands of patients with SS, which suggests a role for IFN-I in the formation of salivary gland lesions (29, 30) (31, 32).
IFN activates the overexpression of canonical interferon-stimulated genes (ISGs) through the Janus kinase (JAK)-STAT signaling pathway, which is defined as the “interferon signature.” IFN phosphorylates STAT1, STAT2, STAT3, and STAT5, which activate downstream signals leading to the activation of immune cells (33–35). This signature in gene expression is considered a biomarker of autoimmune diseases (36).
Under physiological conditions, in vitro-derived pathogens or in vivo-derived apoptotic cells can trigger a rapid innate immune response through pattern recognition receptors (PRRs), including TLRs, RLRs, and NLRs (37). PRRs can recognize nucleic acids and induce the production of numerous proinflammatory cytokines and type I IFNs; thus, aberrant activation of the self-antigen recognition Toll-like receptor (TLR) leads to the development of autoimmune disease (38).
Some studies have reported the enhanced expression of the cell adhesion molecules VACM-1, ICAM-1, and programmed death ligand-1 (PD-L1) in patients with SS (39–41). The aberrant expression of these cytokines is mediated by IFN-I and IFN-II through the JAK-STAT pathway (42, 43). A recent study used reactive oxygen species (ROS) and N-acetylcysteine (NAC) to induce or block the expression of ICAM-1 and PD-L1 and revealed that the IFN signature that regulates the expression of ICAM-1 and PD-L1 in SS was related to oxidative stress (43–45).
Several recent studies have suggested that IFN-III contributes to SS. Type III IFNs, which consist of IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4, are mainly produced by plasmacytoid dendritic cells (pDCs) (46, 47). pDCs respond to the secretion of IFN-III and show improved survival under stimulation with IFN-III in vitro. IFN-III enhances the production of IFN-I and TNF-α in pDCs and promotes the expression of CD80 and CD86, which contribute to the maturation of pDCs (35). IFN-III regulates the immune response by upregulating the polarization of Th1 and CD8+ T cells and downregulating Th2 cytokines and Tregs (48).
3.3 Genome loci associated with SS
Etiological research has revealed the pathogenesis of SS at the genomic level. A GWAS of autoimmune diseases identified an association between HLA regions and SS, including HLA-DR, HLA-DQB1, and HLA-DQA1 (Table 1). The allele with the strongest association was HLA-DQB1*0201 (Pmeta = 1.38 × 10–95). All HLA alleles were correlated with the expression of rs115575857. In addition, six non-HLA regions that surpassed the suggestive threshold (Pmeta < 5 × 10–5) were also shown to be involved in SS, including IRF5, STAT4, BLK, IL-12A, TNIP1, and CXCR5, with the expression of IRF5 and STAT4 being the most significant contributors after HLA regions (Table 1) (49).
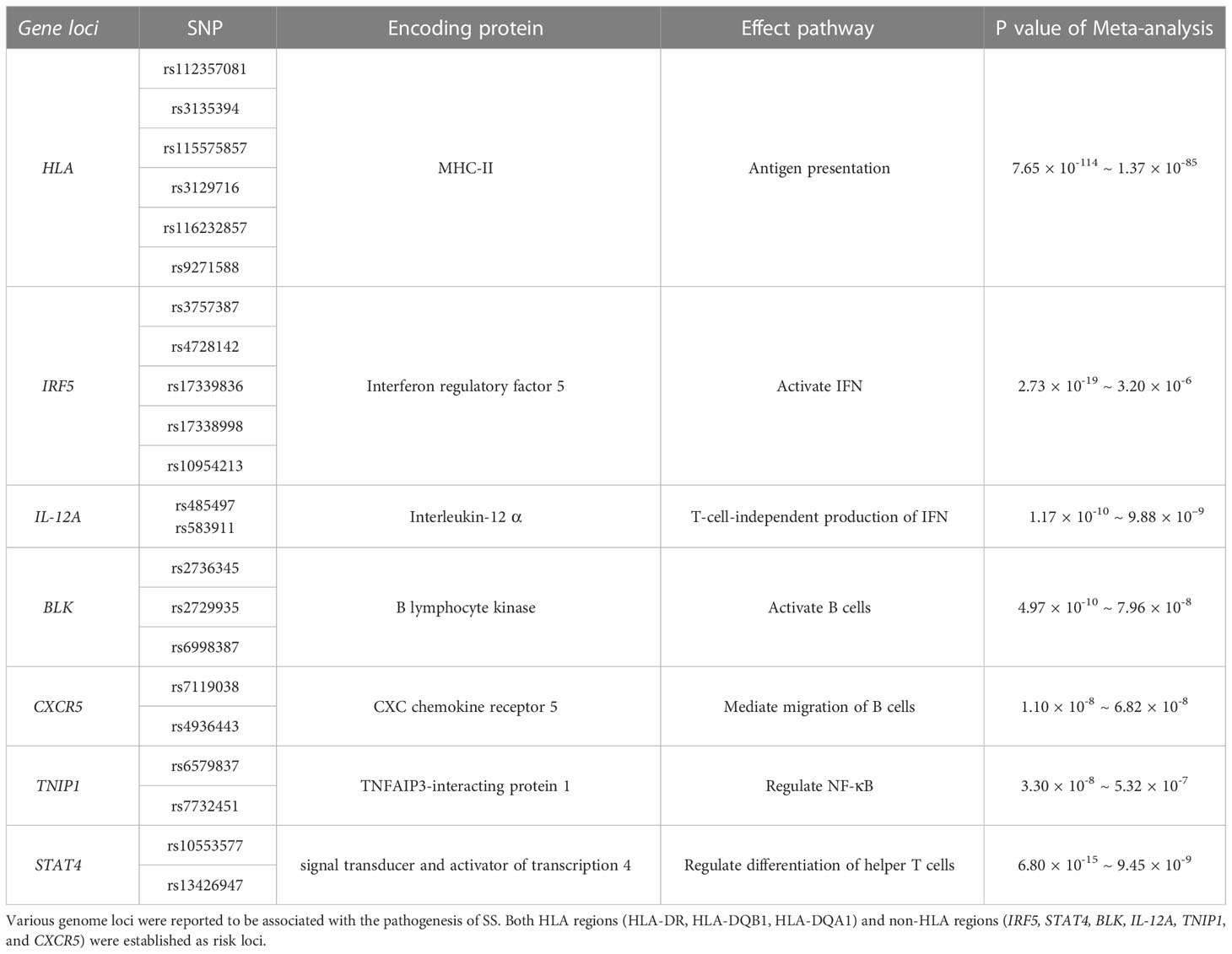
Table 1 Genome loci associated with SS.
3.4 Type 17 helper T (Th17) cells/IL-17
Th17 cells are distinct from Th1/Th2 cells and regulate immune responses independently (50, 51). Th17 cells polarize naïve T cells after stimulation by TGF-β and IL-6 from antigen-presenting cells (APCs). IL-1β secreted from the ductal epithelium and IL-23 secreted from DCs also participate in Th17 cell polarization. IL-17 and IL-22 are produced by and are the main effective cytokines of Th17 cells. Th17 cells mediate inflammation by producing the proinflammatory cytokines TNF-α and IL-6 (52). Previous research revealed that IL-17/IL-23 expression was enhanced in mouse models with SS, indicating that Th17 participated in lymphocytic infiltration of salivary glands and contributed to lesion formation (53, 54). Another study showed that IL-22, IL-23, and IL-17 were increased in the peripheral blood of patients with SS, both at the protein and mRNA levels. Notably, in addition to Th17 cells, NKp44+ NK cells can also produce IL-17 in patients with SS (55). Besides, Th17 cells are potent inducers of matrix metalloproteinase 1 (MMP1) and MMP3 (56), and a study has shown that SS is related to disorders of MMP3/tissue inhibitor of metalloproteinase 1 (TIMP1) and MMP9/TIMP1 ratios (57).
Both Treg and Th17 cells can be induced by TGF-β from activated T cells, indicating that there might be a balance between these opposing inflammation-related cells. An imbalance in the Th17/Treg ratio has been reported in several other autoimmune diseases, including inflammatory bowel disease (IBD) (58, 59), autoimmune thyroid disease (AITD) (60), psoriasis (61), multiple sclerosis (62), and RA (63, 64). In these diseases, function and stability of Treg cells are impaired, and the aberrant induction and proliferation of Th17 cells result in the activation of other immune cells, which then drive an acute autoimmune response. Metabolic pathways play an important role in the regulation of the Th17-Treg cell network. Th17 cells are glycolysis-dependent; thus, by inhibiting the mammalian target of rapamycin (mTOR) pathway with rapamycin, glycolysis is inhibited and the polarization of Th17 cells is decreased, whereas the expression of Treg cells is increased (65). Tregs tend to increase glycolysis and enhance fatty acid oxidation, while Th17 cells rely on fatty acid synthesis (66). However, current metabolic models of Th17/Treg cell regulation through the glycolysis pathway are inconclusive. Fortunately, Compass (67), a powerful algorithm based on scRNA-sequencing and flux balance analysis, was recently produced to predict the relationship between cellular metabolic states and pathogenicity, and has already been utilized in research on the Th17-Treg network.
Type 17 follicular helper T (Tfh17) and IL-17-producing B (B17) cells also contribute to IL-17 production. A recent study revealed that the number of IL-17-producing cells increased in the peripheral blood and spleen of NOD/ShiLtJ mice with STZ-induced type I diabetes and SS. Surprisingly, the infiltration of IL-17-producing cells in the salivary glands increased in metabolically disordered murine models, and was also associated with greater severity of SS (68). It was subsequently found that the aberrant expression of IL-17 induced by metabolic abnormalities contributed to cell lesions and inhibition of tissue recovery in the salivary glands of patients with SS. Furthermore, retinoic A deficiency can exacerbate the imbalance in the Th17/Treg ratio in patients with SS (69).
3.5 TNF/BAFF
TNF-α is predominantly produced by macrophages and T cells in two forms: soluble TNF-α (sTNF-α) and transmembrane TNF-α (Tm TNF-α). sTNF-α is an effective regulator of inflammation and autoimmune diseases (70). TNF-α can bind with TNFR1 or TNFR2 and mediate inflammation by activating the NF-kB pathway and mitogen-activated protein kinases (MAPKs) (71).
BAFF (or BLyS) is a member of the TNF family that plays a vital role in B cell survival. Usually, BAFF is produced by neutrophils, macrophages, monocytes, DCs, and follicular DCs (72). Increased levels of IFN-I, IFN-γ, IL-10, and G-CSF can induce the expression of BAFF, while TLR3, TLR4, or TLR9 participate in BAFF production (73, 74). B cells express BAFF receptors (BAFF-R or BR3), as well as TACI and BCMA. A previous study reported that BAFF binds to BAFF-R and enhances the conversion of NF-κB2/p100 to p52 (75). Additionally, BAFF binds to BAFF-R, activating the PI3K-AKT1 pathway, which regulates the activation of myeloid cell leukemia sequence 1 (MCL1) and inhibits BCL-2-interacting mediator of cell death (BIM). TNF receptor-associated factor 3 (TRAF3) and TRAF2 are intracellular signaling molecules that bind to BAFF-R or TACI. BAFF-R interacts with BAFF and recruits TRAF3, resulting in the degradation of TRAF3 and inhibition of the NF-κB pathway. Nevertheless, the binding of TACI and BAFF results in the recruitment of TRAF2 or TRAF6 and promotes the activation of the NF-κB pathway (72).
BAFF participates in the pathogenesis of various autoimmune diseases, including RA (76), SLE (77), Graves’ disease (78), and anti-GBM disease (79). Overexpression of BAFF elevates MHC-II expression, enhances lymphocytic infiltration, and increases the number of germinal center (GC)-like structures in SS murine models (80). Increased GC-like structures are associated with enhanced production of rheumatoid factor, anti-RO/SSA, anti-La/SSB, and IgG in patients with SS (81). However, another study claimed that BAFF is unable to mediate the differentiation of B cells from GCs, which suggests the involvement of the inhibitory BAFF-TACI pathway (80). Furthermore, BAFF stimulates monocyte through binding with BAFF-R and fosters the production of IL-6, which induces the aberrant production of IgG from B cells in SS (82).
3.6 Wingless/integrated signaling pathway
The Wnt signaling pathway is involved in several biological processes, including cellular migration, proliferation, differentiation, apoptosis, tissue homeostasis and regeneration, and stem cell self-renewal (83). Dysregulation of the Wnt/β-catenin pathway plays a vital role in the pathogenesis of many cancers and autoimmune diseases (84, 85).
The role of the Wnt signaling pathway in T cell differentiation and immune regulation has been elucidated (86–88). The disorder of Wnt signaling inhibitors in autoimmune diseases was also noticed. Proinflammatory cytokines promote bone damage by fostering the production of Wnt signaling inhibitors, including secreted frizzled-related, Wnt inhibitory factor 1, sclerostin, and Dickkopf (DKK) family proteins (89–91). However, it was found that the role of DKK-1 is different in various autoimmune diseases (92). In a clinical study of 98 SS patients and 165 healthy volunteers, three Wnt/β-catenin signaling pathway-related genes, LRP5, FRZB, and ADIPOQ, were shown to increase the risk of SS, although the biological functions of these genes have not yet been established (91). It implicates Wnt pathway might be involved in the pathogenesis of SS. However, not all studies support this idea. A clinical study reported that serum Dkk-1 and sclerostin levels were decreased in SS and SLE, and the Wnt1 and Wnt3a levels had no significant changes (93).
3.7 IL-33/ST-2
The IL-33-ST2 axis participates in the pathogenesis of SS by promoting transcriptional activation of CD86 and CCL2 in salivary epithelial cells and activation of the NF-κB pathway. IL-33, combined with IL-12 and IL-23, participates in the production of CD4+ T cell-derived and NK/NKT-derived IFN-γ (89, 94). An increase in serum levels of IL-33 and ST2 has been reported in patients with SS (95). IL-33 is a member of the IL-1 family and ST2 is one important member of IL-1 receptor family (90). IL-33 induces phosphorylation of the NF-κB pathway and activates MAP kinases by interacting with ST2 to stimulate downstream Th2-related immune responses. A recent review described IL-33 as an alarmin, that is, a DAMP. Local increases in IL-33 expression can induce an immune response and result in organ lesions (91). The IL-33-ST2 axis is a novel mode in the pathogenesis of SS and a potential therapeutic target in related salivary gland disorders.
4 Experimental therapeutic strategies of SS
4.1 Targeting the Treg/Th17
The Treg/Th17 plays a crucial role in the pathogenesis of autoimmune diseases. Various drugs have been designed to target the molecular mechanisms involved in the polarization and activation of the Treg/Th17, including IL-17-related molecules (IL-17, IL-23), transcription factors (RORγt, STAT3, Foxp3, and FoxO1), and intracellular signaling pathways (ROCK and MAPK) (96). However, compared to RA, SLE, IBD, and psoriasis, clinical or preclinical drugs for SS are rare. Nevertheless, previous pharmacological exploration and clinical trials have provided promising results for SS therapy.
IL-38—a member of the IL-1 family and, which was named as IL-1F10 (97)—inhibits the secretion of Th17 cell-related cytokines, including IL-6, IL-8, IL-17, IL-22, and IL-23, by binding with IL-36 receptors (98). A previous clinical study reported a selective anti-IL-17A mAb, secukinumab, that can alleviate the symptoms of psoriasis by blocking the expression of IL-17A, while that of IL-1 receptor antagonist (IL-1ra) and IL-38 was downregulated (Figure 2) (99). A previous study tested the effect of IL-38 treatment on Th17 cell activity and found that the expression levels of IL-17 and IL-23 were decreased in a murine model of SS; IL-38 inhibited IL-17 expression through the NF-κB and MAPK signaling pathways(Figure 2). They also found that IL-17 can upregulate the expression of IL-38 (54). This hints at a potential approach for the treatment of SS.
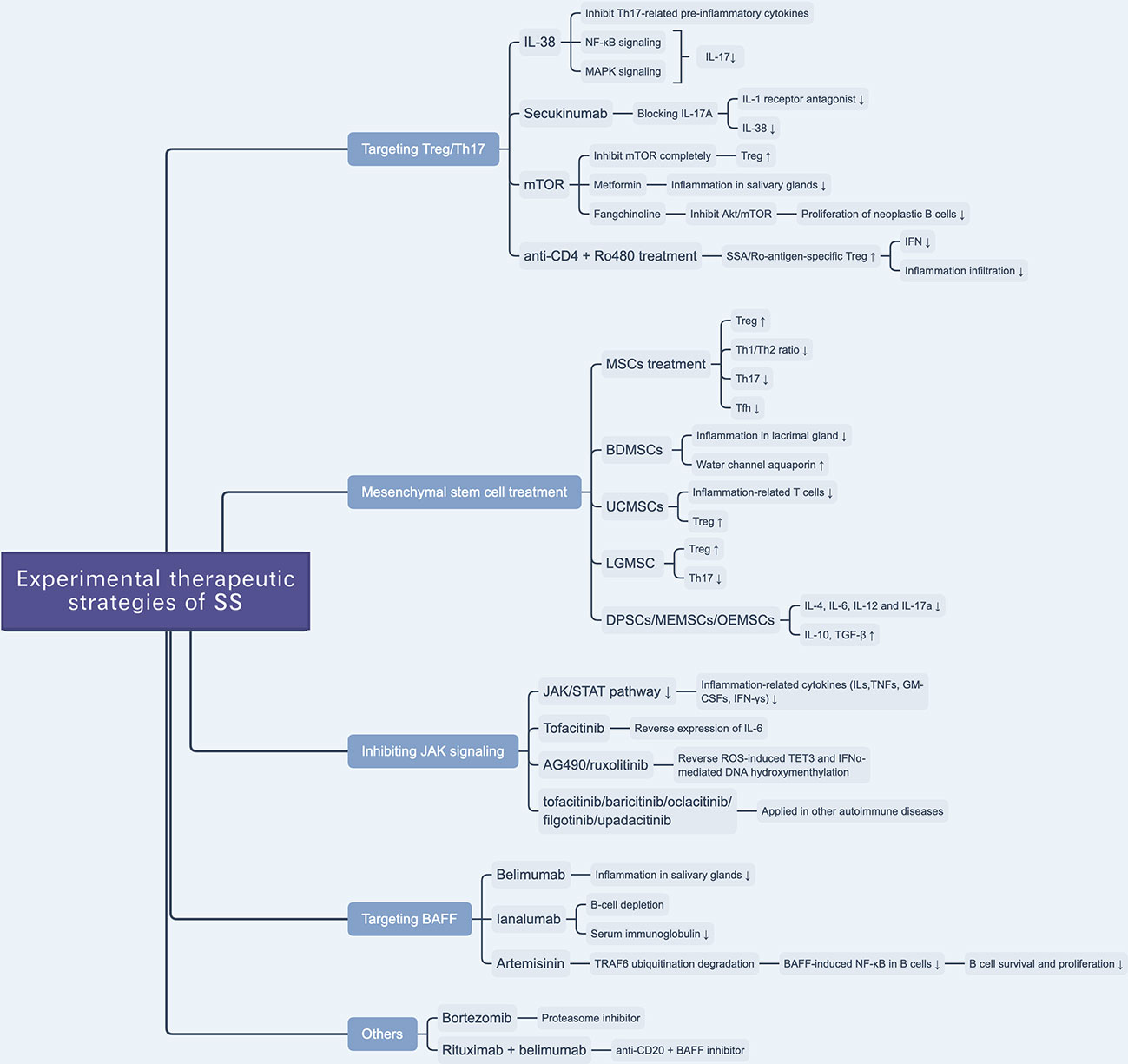
Figure 2 Overview of novel experimental therapeutic strategies of SS. There are various experimental drugs (e.g. JAK inhibitors) or schemes (e.g. targeting Treg/Th17 and BAFF) being investigated for SS treatment. Besides, mesenchymal stem cell treatment is reported to be a promising scheme to treat SS.
mTOR, a member of the phosphoinositol 3-kinase (PI3K) family, is an atypical serine/threonine kinase that plays a vital role in cellular metabolism and activity (100). mTOR likely prevents anergy induction by IL-2 expression in T cells. A previous study found that by blocking mTOR with rapamycin, the cell cycle of clonal T cells was inhibited, while it induced cell anergy even with costimulations (101). In the process of naïve T cell differentiation, mTOR mediates the transformation to Th17 or Treg cells by altering the sensitivity of T cells to TGF-β, which influences the effects of STAT3 signaling (102). The inhibition of different mTOR complexes (including mTORC1 and mTORC2) would activate different pathways of polarization to Th17 cells, whereas a complete inhibition of mTOR can promote polarization to Treg cells (103). mTOR plays a role in Th17/Treg balance, given that mTOR inhibitors interfere with the Th17/Treg ratio, which suggests a potential therapeutic target to ameliorate glandular lesions in patients with SS. Given the anti-inflammatory and immunomodulatory effects of metformin—an AMPK-dependent mTOR-STAT3 inhibitor—researchers examined its therapeutic effect in SS murine models and found that it ameliorated inflammation in the salivary glands and, based on flow cytometry, regulated the Th17/Treg ratio (Figure 2) (104). Yu et al. reported that an alkaloid extracted from the traditional Chinese herbal medicine Stephania tetrandra S. Moore, fangchinoline, can be used to treat SS by inhibiting the Akt/mTOR pathway, which inhibits the proliferation of neoplastic B lymphocytes (Figure 2) (105).
More than that, a recent study reported that SSA/Ro-antigen-specific Treg cells can downregulate the production of CD4+ T cell-derived IFN-γ and suppress inflammatory infiltration of the salivary gland (106). Researchers reported that the combination treatment with anti-CD4 mAb and autoantigen-specific peptide Ro480 induces SSA/Ro-antigen-specific Treg cells in vivo and suppresses CD4+ T cell-related IFN-γ production in salivary glands, providing a potential novel immunotherapeutic strategy for the treatment of SS (Figure 2) (106).
4.2 Mesenchymal stem cell treatment
Mesenchymal stem cells (MSCs) exert immunomodulatory effects on both adaptive and innate pathways. MSC can manipulate the balance between suppressive Treg cells and inflammatory T helper cells (Th1, Th2, Th17, and Tfh) and ameliorate inflammatory infiltration in the salivary glands (107, 108).
Xu et al. revealed that immunomodulatory functions of MSCs are impaired in SS-like murine models, and allogeneic bone marrow mesenchymal stem cells (BMMSCs) infusion can suppress SS-like inflammation, showing therapeutic effects of BMMSCs on SS (109). Furthermore, they also elucidated that the stromal cell-derived factor-1(SDF-1)/C-X-C chemokine receptor 4(CXCR4) axis plays an important role in MSC migration and restoration of salivary glands. More than that, they also treated twenty-four SS patients with umbilical cord-MSCs (UCMSCs), and all patients showed alleviation of SS symptoms and well tolerance of allogeneic UCMSCs (Figure 2) (109).
Zoukhri et al. revealed that a biotherapeutic strategy involving bone-derived MSCs (BDMSCs) alleviated lacrimal glandular manifestation in a SS murine model by inhibiting inflammation and promoting the expression and activation of water channel aquaporin 5 (Figure 2) (110). Li et al. verified the immunomodulatory effects of UCMSCs and found that UCMSCs induced CD4+FoxP3+ Treg cells in vitro and caused anergy of inflammation-related T cells in vivo, accompanied by an increase in Treg cells (111). Hua et al. assessed the effects of labial gland-derived MSCs (LGMSCs) and their exosomes on SS, and found that they ameliorated salivary gland inflammatory infiltration by inhibiting the polarization of Th17 cells and promoting the proliferation of Treg cells (Figure 2) (112). Furthermore, dental pulp stem cells (DPSC) (113), murine embryonic MSCs (MEMSCs) (114), and olfactory ecto-MSCs (OEMSCs) (115) can be used to treat SS by interfering with inflammation-related cytokines (IL-4, IL-6, IL-12, and IL-17a) and suppressive cytokines (IL-10 and TGF-β) (Figure 2) (116).
4.3 Inhibiting JAK pathway
JAK enzymes are involved in the JAK/STAT pathway through the phosphorylation of STAT, which leads to the activation of signals transferred to the nucleus. The JAK family consists of four members, JAK1, JAK2, JAK3, and TYK2 (117). Membrane receptor subunits usually bind to a specific JAK. For example, JAK3 can only selectively binds to the γc chain, which is a common receptor chain of IL-2, IL-4, IL-9, IL-15, and IL-21 (118).
The JAK/STAT pathway regulates the production of ILs, TNFs, GM-CSFs, and IFN-γs, which are associated with inflammation and autoimmunity (119). The JAK inhibitors tofacitinib (120), baricitinib (120), oclacitinib (121), filgotinib (122), and upadacitinib (123) have been applied in the treatment of autoimmune diseases (Figure 2). Renaudineau et al. reported that AG490 and ruxolitinib, two JAK1/2 inhibitors, can reverse ROS-induced production of ten-eleven translocation 3 (TET3) and IFNα-mediated DNA hydroxymethylation and could potentially treat SS (Table 2) (45, 132). Tofacitinib is also a candidate drug for SS, given that it can reverse the expression of IL-6 in ATG5-deficient 3D-acini, which leads to the inhibition of inflammation (Table 2) (133).
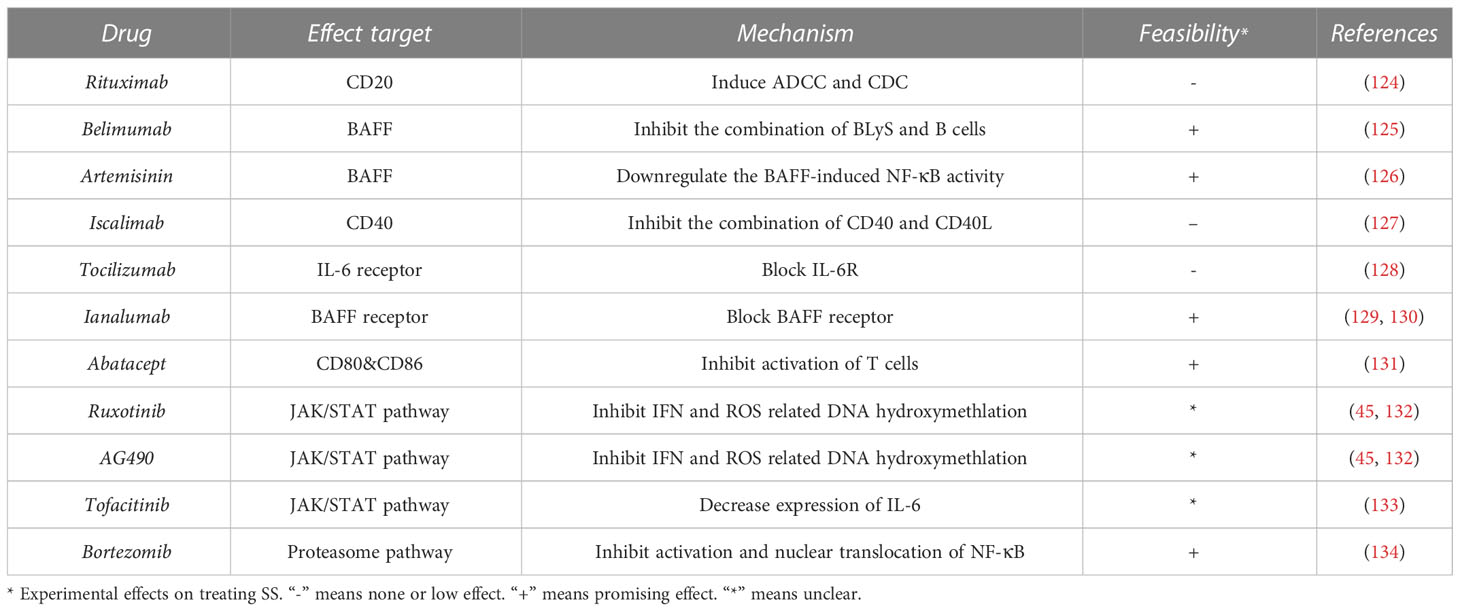
Table 2 Experimental Biotherapeutic Drugs of SS.
4.4 Targeting BAFF
Belimumab is an anti-BAFF monoclonal antibody and a potential biotherapeutic drug for SLE and SS (Table 2) (125, 135). A bi-centric clinical trial reported that, after a 28-week regimen of belimumab (10 mg/kg, at weeks 0, 2, 4, and then every 4 weeks), 18 out of 30 patients achieved two of five primary endpoints. The mean and standard deviation of ESSDAI and EULAR Sjogren’s Syndrome Patients Reported Index (ESSPRI) were both reduced (Figure 2) (125).
Ianalumab is a BAFF-blocking monoclonal antibody that leads to B-cell depletion (Figure 2). A previous clinical study found that, in SS, ianalumab reduced the ESSDAI, ESSPRI, and serum immunoglobulin levels (Table 2) (129).
Besides, Zheng et al. reported a Chinese herb-derived drug, Artemisinin (ART), which is used to treat chloroquine-resistant malaria originally, has immunosuppressive effects in the SS-like murine model (Figure 2) (126). The study demonstrated that ART downregulates BAFF-induced NF-κB activity in B cells through targeting TRAF6 ubiquitination, which results in the inhibition of B cell survival and proliferation. Therefore, the levels of B lymphocyte-related immunoglobulin and autoantibody in the SS-like murine model were attenuated and lymphocytic infiltration in the salivary gland was ameliorated (Table 2) (126).
4.5 Others
Bortezomib is a proteasome inhibitor used in the treatment of multiple myeloma (Table 2). A Mexican case report described a female patient that suffered from SS for 16 years and presented with sicca complex, extreme fatigue, Raynaud phenomenon, generalized arthralgia, and heavy headaches. After ineffective conventional glucocorticoid and rituximab therapy, doctors administered an experimental regimen of bortezomib at a dose of 1.3 mg/m2 (2.0 mg/dose) at days 1, 4, 8, 11, 22, 29, 36, 43, 50, and 57. Surprisingly, the patient’s headaches and fatigue were resolved after three months, and serum globulin levels and viscosity decreased significantly (Table 2) (134). However, the efficacy and safety of bortezomib for the treatment of SS are still unconfirmed (Figure 2).
Rituximab—a chimeric monoclonal anti-CD20 antibody—has been reported to induce B-cell depletion and has been used to treat autoimmune diseases (136, 137). It has also been used to treat SS over the last 20 years, but with limited clinical efficacy (Table 2) (124, 138). Researchers have investigated a combination therapy with the anti-BAFF and anti-CD20 [NCT02631538]—belimumab and rituximab. The results of the clinical trial provide evidence that simultaneous targeting of the BAFF axis and B cells is a promising treatment strategy for SS (Figure 2) (139). The drugs in development for SS treatment are summarized in Table 2.
Various biotherapeutic drugs have been used to treat SS experimentally. Some of them have been used to treat other autoimmune diseases (e.g. Rituximab), and the others are novel drugs targeting inflammation-related signaling pathways (e.g. AG490). However, not all of them have prospective effects on treating SS (e.g. Iscalimab, Tocilizumab).
5 Conclusion & discussion
As a systemic autoimmune disease, SS causes multiple organ lesions, especially in salivary and lacrimal glands, which limits endocrine function. Besides focal inflammation in the salivary gland, acinar atrophy, duct dilation, and fibrosis are commonly observed in SS patients. Due to a high disease specificity and limited invasiveness, labial salivary gland biopsy is widely accepted as the best method to diagnose SS currently (140). Lymphocytic infiltration around the striated ducts in salivary glands, or so-called periductal foci, is a critical hallmark for the diagnosis of SS (141). Since adipose tissue replacement in the salivary gland is related to the stages of SS, and adipocytes are detected in IL-6-rich regions, detecting the degree of adipose tissue replacement provides aid to improve diagnosis accuracy (142). In addition, comorbidities, such as secondary pulmonary disease, kidney injury, and lymphoma, further reduce the quality of life of patients. The pathogenesis of SS is characterized by the production of inflammatory cytokines and lymphocyte infiltration. IFN and IL-17/Il-23 play pivotal roles in the formation of inflammatory lesions, and B cells are crucial for infiltrative injury. Th17, B, and dendritic cells play critical roles in the aberrant regulation of the immune system.
Similar to other autoimmune diseases, such as SLE, RA, and psoriasis, the traditional therapeutic strategy for SS is disease-modifying antirheumatic drugs (DMARDs), such as glucocorticoids, while novel biotherapeutic approaches take advantage of neutralizing antibodies and inflammation-related receptor blockers. Compared with the traditional strategy, this new scheme is more targeted, which can promote safety and efficacy. Although considerable progress has recently been made in the treatment of SS, disease-specific drugs are rare. Many SS drugs are currently undergoing clinical trials. As Th17 and B cells play important roles in the pathogenesis of SS, the targeting of Th17 cell- and B cell-related signaling pathways and molecular events has drawn increasing attention.
Research on SS therapies is limited by a lack of systematic clinical trials compared with other autoimmune diseases, such as SLE, RA, and MS. Many potential therapeutic targets have been identified in the pathogenesis of SS, and some targeted drugs have shown reasonable efficacy under experimental conditions in vitro or in vivo. Unfortunately, the translation of these drugs to clinical use is rare. Additionally, it is unclear why immune inhibitors lack pharmacological effect in SS compared to SLE, RA, and MS. Nevertheless, immune inhibitors can be used in the management of complications to improve the prognosis and quality of life of patients with SS. For example, BAFF receptor blockers not only prevent inflammatory lesions but also protect against B-cell lymphoma; however, such therapeutic strategies are rare. Combination therapies have shown some efficacy; however, inappropriate combinations of drugs may cause excessive inhibition of the immune system, resulting in unexpected complications, such as secondary infections. Thus, targeted and low-side-effect drugs should be the focus of future research.
Author contributions
QZ drafted the manuscript. JZ, YL, WC and XF edited the manuscript. DZ supervised the work and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key Project of the Science and Technology Department of Sichuan Province (NO. 2022YFH0100), the National Natural Science Foundation of China (NO. 82171829), the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (NO. ZYYC21012), and the Fundamental Research Funds for the Central Universities (20822041E4084).
Acknowledgments
DZ sincerely wants to commemorate Dr. Sang-A Park, who passed away suddenly on January 22, 2018.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bloch Kj, Buchanan Ww, Wohl Mj, Bunim Jj. SJÖGREN'S SYNDROME: A clinical, pathological, and serological study of sixty-two cases. Medicine (1965) 44(3):187–231. doi: 10.1097/00005792-196505000-00001
2. Qin B, Wang J, Yang Z, Yang M, Ma N, Huang F, et al. Epidemiology of primary sjogren's syndrome: a systematic review and meta-analysis. Ann Rheum Dis (2015) 74(11):1983–9. doi: 10.1136/annrheumdis-2014-205375
3. Cornec D, Devauchelle-Pensec V, Mariette X, Jousse-Joulin S, Berthelot JM, Perdriger A, et al. Severe health-related quality of life impairment in active primary sjogren's syndrome and patient-reported outcomes: Data from a Large therapeutic trial. Arthritis Care Res (Hoboken) (2017) 69(4):528–35. doi: 10.1002/acr.22974
4. Berman N, Vivino F, Baker J, Dunham J, Pinto A. Risk factors for caries development in primary sjogren syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol (2019) 128(2):117–22. doi: 10.1016/j.oooo.2019.04.011
5. Singh M, Teles F, Uzel NG, Papas A. Characterizing microbiota from sjogren's syndrome patients. JDR Clin Trans Res (2021) 6(3):324–32. doi: 10.1177/2380084420940623
6. Seror R, Bowman SJ, Brito-Zeron P, Theander E, Bootsma H, Tzioufas A, et al. EULAR sjogren's syndrome disease activity index (ESSDAI): a user guide. RMD Open (2015) 1(1):e000022. doi: 10.1136/rmdopen-2014-000022
7. Alexander EL, Provost TT, Stevens MB, Alexander GE. Neurologic complications of primary sjogren's syndrome. Med (Baltimore) (1982) 61(4):247–57. doi: 10.1097/00005792-198207000-00004
8. Alexander EL, Malinow K, Lejewski JE, Jerdan MS, Provost TT, Alexander GE. Primary sjogren's syndrome with central nervous system disease mimicking multiple sclerosis. Ann Intern Med (1986) 104(3):323–30. doi: 10.7326/0003-4819-104-3-323
9. Fauchais AL, Magy L, Vidal E. Central and peripheral neurological complications of primary sjogren's syndrome. Presse Med (2012) 41(9 Pt 2):e485–93. doi: 10.1016/j.lpm.2012.06.002
10. Fox RJ. The prevalence of sjogren syndrome in patients with primary progressive multiple sclerosis. Neurology (2002) 58(10):1576. doi: 10.1212/wnl.58.10.1576
11. Griffin JW, Cornblath DR, Alexander E, Campbell J, Low PA, Bird S, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with sjogren's syndrome. Ann Neurol (1990) 27(3):304–15. doi: 10.1002/ana.410270313
12. Misterska-Skora M, Sebastian A, Dziegiel P, Sebastian M, Wiland P. Inclusion body myositis associated with sjogren's syndrome. Rheumatol Int (2013) 33(12):3083–6. doi: 10.1007/s00296-012-2556-4
13. Sambataro G, Ferro F, Orlandi M, Sambataro D, Torrisi SE, Quartuccio L, et al. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary sjgren's syndrome: A systematic review from the Italian society of rheumatology. Autoimmun Rev (2020) 19(2):102447. doi: 10.1016/j.autrev.2019.102447
14. Luppi F, Sebastiani M, Silva M, Sverzellati N, Cavazza A, Salvarani C, et al. Interstitial lung disease in sjogren's syndrome: a clinical review. Clin Exp Rheumatol (2020) 38 Suppl 126(4):291–300.
15. Mirouse A, Seror R, Vicaut E, Mariette X, Dougados M, Fauchais AL, et al. Arthritis in primary sjogren's syndrome: Characteristics, outcome and treatment from French multicenter retrospective study. Autoimmun Rev (2019) 18(1):9–14. doi: 10.1016/j.autrev.2018.06.015
16. Bossini N, Savoldi S, Franceschini F, Mombelloni S, Baronio M, Cavazzana I, et al. Clinical and morphological features of kidney involvement in primary sjogren's syndrome. Nephrol Dial Transplant (2001) 16(12):2328–36. doi: 10.1093/ndt/16.12.2328
17. Maripuri S, Grande JP, Osborn TG, Fervenza FC, Matteson EL, Donadio JV, et al. Renal involvement in primary sjogren's syndrome: a clinicopathologic study. Clin J Am Soc Nephrol (2009) 4(9):1423–31. doi: 10.2215/CJN.00980209
18. Baldini C, Pepe P, Quartuccio L, Priori R, Bartoloni E, Alunno A, et al. Primary sjogren's syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatol (Oxford) (2014) 53(5):839–44. doi: 10.1093/rheumatology/ket427
19. Alunno A, Leone MC, Giacomelli R, Gerli R, Carubbi F. Lymphoma and lymphomagenesis in primary sjogren's syndrome. Front Med (Lausanne) (2018) 5:102. doi: 10.3389/fmed.2018.00102
20. Koh JH, Park Y, Lee J, Park SH, Kwok SK. Hypergammaglobulinaemia predicts glandular and extra-glandular damage in primary sjogren's syndrome: results from the KISS cohort study. Clin Exp Rheumatol (2021) 39 Suppl 133(6):114–22. doi: 10.55563/clinexprheumatol/volsh1
21. Quartuccio L, Salvin S, Fabris M, Maset M, Pontarini E, Isola M, et al. BLyS upregulation in sjogren's syndrome associated with lymphoproliferative disorders, higher ESSDAI score and b-cell clonal expansion in the salivary glands. Rheumatol (Oxford) (2013) 52(2):276–81. doi: 10.1093/rheumatology/kes180
22. Song H, Tong D, Cha Z, Bai J. C-X-C chemokine receptor type 5 gene polymorphisms are associated with non-Hodgkin lymphoma. Mol Biol Rep (2012) 39(9):8629–35. doi: 10.1007/s11033-012-1717-6
23. Nordmark G, Wang C, Vasaitis L, Eriksson P, Theander E, Kvarnstrom M, et al. Association of genes in the NF-kappaB pathway with antibody-positive primary sjogren's syndrome. Scand J Immunol (2013) 78(5):447–54. doi: 10.1111/sji.12101
24. Johnsen SJ, Gudlaugsson E, Skaland I, Janssen EA, Jonsson MV, Helgeland L, et al. Low protein A20 in minor salivary glands is associated with lymphoma in primary sjogren's syndrome. Scand J Immunol (2016) 83(3):181–7. doi: 10.1111/sji.12405
25. Retamozo S, Acar-Denizli N, Rasmussen A, Horvath IF, Baldini C, Priori R, et al. Systemic manifestations of primary sjogren's syndrome out of the ESSDAI classification: prevalence and clinical relevance in a large international, multi-ethnic cohort of patients. Clin Exp Rheumatol (2019) 37 Suppl 118(3):97–106.
26. Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary sjogren's syndrome. Nat Rev Rheumatol (2013) 9(9):544–56. doi: 10.1038/nrrheum.2013.110
27. Mavragani CP, Moutsopoulos HM. Sjogren's syndrome: Old and new therapeutic targets. J Autoimmun (2020) 110:102364. doi: 10.1016/j.jaut.2019.102364
28. Hooks JJ, Moutsopoulos HM, Notkins AL. Circulating interferon in human autoimmune diseases. Tex Rep Biol Med (1981) 41:164–8.
29. Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary sjogren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum (2005) 52(4):1185–95. doi: 10.1002/art.20998
30. Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary sjogren's syndrome patients from healthy control subjects. Arthritis Rheum (2005) 52(5):1534–44. doi: 10.1002/art.21006
31. Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I interferons in the pathogenesis and treatment of autoimmune diseases. Clin Rev Allergy Immunol (2020) 59(2):248–72. doi: 10.1007/s12016-020-08798-2
32. Yao Y, Liu Z, Jallal B, Shen N, Ronnblom L. Type I interferons in sjogren's syndrome. Autoimmun Rev (2013) 12(5):558–66. doi: 10.1016/j.autrev.2012.10.006
33. Yin Z, Dai J, Deng J, Sheikh F, Natalia M, Shih T, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol (2012) 189(6):2735–45. doi: 10.4049/jimmunol.1102038
34. Finotti G, Tamassia N, Calzetti F, Fattovich G, Cassatella MA. Endogenously produced TNF-alpha contributes to the expression of CXCL10/IP-10 in IFN-lambda3-activated plasmacytoid dendritic cells. J Leukoc Biol (2016) 99(1):107–19. doi: 10.1189/jlb.3VMA0415-144R
35. Finotti G, Tamassia N, Cassatella MA. Interferon-lambdas and plasmacytoid dendritic cells: A close relationship. Front Immunol (2017) 8:1015. doi: 10.3389/fimmu.2017.01015
36. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol (2019) 20(12):1574–83. doi: 10.1038/s41590-019-0466-2
37. Apostolou E, Tzioufas AG. Type-III interferons in sjogren's syndrome. Clin Exp Rheumatol (2020) 38 Suppl 126(4):245–52.
38. Barrat FJ, Elkon KB, Fitzgerald KA. Importance of nucleic acid recognition in inflammation and autoimmunity. Annu Rev Med (2016) 67:323–36. doi: 10.1146/annurev-med-052814-023338
39. Saito I, Terauchi K, Shimuta M, Nishiimura S, Yoshino K, Takeuchi T, et al. Expression of cell adhesion molecules in the salivary and lacrimal glands of sjogren's syndrome. J Clin Lab Anal (1993) 7(3):180–7. doi: 10.1002/jcla.1860070309
40. Zhou J, Jin JO, Kawai T, Yu Q. Endogenous programmed death ligand-1 restrains the development and onset of sjgren's syndrome in non-obese diabetic mice. Sci Rep (2016) 6:39105. doi: 10.1038/srep39105
41. Kobayashi M, Kawano S, Hatachi S, Kurimoto C, Okazaki T, Iwai Y, et al. Enhanced expression of programmed death-1 (PD-1)/PD-L1 in salivary glands of patients with sjogren's syndrome. J Rheumatol (2005) 32(11):2156–63.
42. Traore K, Sharma RB, Burek CL, Trush MA. Role of ROS and MAPK in TPA-induced ICAM-1 expression in the myeloid ML-1 cell line. J Cell Biochem (2007) 100(4):1010–21. doi: 10.1002/jcb.21101
43. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights (2016) 7(1):11. doi: 10.1007/s13317-016-0083-0
44. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc Natl Acad Sci U.S.A. (2019) 116(10):4326–35. doi: 10.1073/pnas.1819473116
45. Charras A, Arvaniti P, Le Dantec C, Dalekos GN, Zachou K, Bordron A, et al. JAK inhibitors and oxidative stress control. Front Immunol (2019) 10:2814. doi: 10.3389/fimmu.2019.02814
46. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol (2004) 34(3):796–805. doi: 10.1002/eji.200324610
47. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis c virus. Nat Genet (2013) 45(2):164–71. doi: 10.1038/ng.2521
48. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50(4):907–23. doi: 10.1016/j.immuni.2019.03.025
49. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with sjogren's syndrome. Nat Genet (2013) 45(11):1284–92. doi: 10.1038/ng.2792
50. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
51. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi: 10.1038/ni1254
52. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34(2):149–62. doi: 10.1016/j.immuni.2011.02.012
53. Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary gland tissue expression of interleukin-23 and interleukin-17 in sjogren's syndrome: findings in humans and mice. Arthritis Rheum (2008) 58(3):734–43. doi: 10.1002/art.23214
54. Luo D, Chen Y, Zhou N, Li T, Wang H. Blockade of Th17 response by IL-38 in primary sjogren's syndrome. Mol Immunol (2020) 127:107–11. doi: 10.1016/j.molimm.2020.09.006
55. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with sjogren's syndrome. Ann Rheum Dis (2012) 71(2):295–301. doi: 10.1136/ard.2011.154013
56. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum (2011) 63(1):73–83. doi: 10.1002/art.30093
57. Asatsuma M, Ito S, Watanabe M, Takeishi H, Nomura S, Wada Y, et al. Increase in the ratio of matrix metalloproteinase-9 to tissue inhibitor of metalloproteinase-1 in saliva from patients with primary sjogren's syndrome. Clin Chim Acta (2004) 345(1-2):99–104. doi: 10.1016/j.cccn.2004.03.006
58. Iacomino G, Rotondi Aufiero V, Iannaccone N, Melina R, Giardullo N, De Chiara G, et al. IBD: Role of intestinal compartments in the mucosal immune response. Immunobiology (2020) 225(1):151849. doi: 10.1016/j.imbio.2019.09.008
59. Silva FA, Rodrigues BL, Ayrizono ML, Leal RF. The immunological basis of inflammatory bowel disease. Gastroenterol Res Pract (2016) 2016:2097274. doi: 10.1155/2016/2097274
60. Shao S, Yu X, Shen L. Autoimmune thyroid diseases and Th17/Treg lymphocytes. Life Sci (2018) 192:160–5. doi: 10.1016/j.lfs.2017.11.026
61. Shi Y, Chen Z, Zhao Z, Yu Y, Fan H, Xu X, et al. IL-21 induces an imbalance of Th17/Treg cells in moderate-to-Severe plaque psoriasis patients. Front Immunol (2019) 10:1865. doi: 10.3389/fimmu.2019.01865
62. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T Cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol (2010) 162(1):1–11. doi: 10.1111/j.1365-2249.2010.04143.x
63. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. TNFalpha in the regulation of treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine (2018) 101:4–13. doi: 10.1016/j.cyto.2016.09.001
64. Paradowska-Gorycka A, Wajda A, Romanowska-Prochnicka K, Walczuk E, Kuca-Warnawin E, Kmiolek T, et al. Th17/Treg-related transcriptional factor expression and cytokine profile in patients with rheumatoid arthritis. Front Immunol (2020) 11:572858. doi: 10.3389/fimmu.2020.572858
65. Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol (2007) 7(13):1819–24. doi: 10.1016/j.intimp.2007.08.027
66. Cluxton D, Petrasca A, Moran B, Fletcher JM. Differential regulation of human treg and Th17 cells by fatty acid synthesis and glycolysis. Front Immunol (2019) 10:115. doi: 10.3389/fimmu.2019.00115
67. Wagner A, Wang C, Fessler J, DeTomaso D, Avila-Pacheco J, Kaminski J, et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell (2021) 184(16):4168–85.e21. doi: 10.1016/j.cell.2021.05.045
68. Hwang SH, Park JS, Yang S, Jung KA, Choi J, Kwok SK, et al. Metabolic abnormalities exacerbate sjogren's syndrome by and is associated with increased the population of interleukin-17-producing cells in NOD/ShiLtJ mice. J Transl Med (2020) 18(1):186. doi: 10.1186/s12967-020-02343-7
69. Hwang SH, Woo JS, Moon J, Yang S, Park JS, Lee J, et al. IL-17 and CCR9(+)alpha4beta7(-) Th17 cells promote salivary gland inflammation, dysfunction, and cell death in sjogren's syndrome. Front Immunol (2021) 12:721453. doi: 10.3389/fimmu.2021.721453
70. Bradley JR. TNF-mediated inflammatory disease. J Pathol (2008) 214(2):149–60. doi: 10.1002/path.2287
71. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. The role of tumor necrosis factor alpha (TNF-alpha) in autoimmune disease and current TNF-alpha inhibitors in therapeutics. Int J Mol Sci (2021) 22(5):2719. doi: 10.3390/ijms22052719
72. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9(7):491–502. doi: 10.1038/nri2572
73. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on b cell survival. Annu Rev Immunol (2003) 21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152
74. Scapini P, Bazzoni F, Cassatella MA. Regulation of b-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett (2008) 116(1):1–6. doi: 10.1016/j.imlet.2007.11.009
75. Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, et al. BAFF/BLyS receptor 3 binds the b cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity (2002) 17(4):515–24. doi: 10.1016/s1074-7613(02)00425-9
76. Wei F, Chang Y, Wei W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine (2015) 76(2):537–44. doi: 10.1016/j.cyto.2015.07.014
77. Vincent FB, Morand EF, Mackay F. BAFF and innate immunity: new therapeutic targets for systemic lupus erythematosus. Immunol Cell Biol (2012) 90(3):293–303. doi: 10.1038/icb.2011.111
78. Vannucchi G, Covelli D, Curro N, Dazzi D, Maffini A, Campi I, et al. Serum BAFF concentrations in patients with graves' disease and orbitopathy before and after immunosuppressive therapy. J Clin Endocrinol Metab (2012) 97(5):E755–9. doi: 10.1210/jc.2011-2614
79. Xin G, Cui Z, Su Y, Xu LX, Zhao MH, Li KS. Serum BAFF and APRIL might be associated with disease activity and kidney damage in patients with anti-glomerular basement membrane disease. Nephrol (Carlton) (2013) 18(3):209–14. doi: 10.1111/nep.12032
80. Ding J, Zhang W, Haskett S, Pellerin A, Xu S, Petersen B, et al. BAFF overexpression increases lymphocytic infiltration in sjogren's target tissue, but only inefficiently promotes ectopic b-cell differentiation. Clin Immunol (2016) 169:69–79. doi: 10.1016/j.clim.2016.06.007
81. Carrillo-Ballesteros FJ, Palafox-Sanchez CA, Franco-Topete RA, Munoz-Valle JF, Orozco-Barocio G, Martinez-Bonilla GE, et al. Expression of BAFF and BAFF receptors in primary sjogren's syndrome patients with ectopic germinal center-like structures. Clin Exp Med (2020) 20(4):615–26. doi: 10.1007/s10238-020-00637-0
82. Yoshimoto K, Suzuki K, Takei E, Ikeda Y, Takeuchi T. Elevated expression of BAFF receptor, BR3, on monocytes correlates with b cell activation and clinical features of patients with primary sjogren's syndrome. Arthritis Res Ther (2020) 22(1):157. doi: 10.1186/s13075-020-02249-1
83. Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
84. Duchartre Y, Kim YM, Kahn M. The wnt signaling pathway in cancer. Crit Rev Oncol Hematol (2016) 99:141–9. doi: 10.1016/j.critrevonc.2015.12.005
85. Cici D, Corrado A, Rotondo C, Cantatore FP. Wnt signaling and biological therapy in rheumatoid arthritis and spondyloarthritis. Int J Mol Sci (2019) 20(22):5552. doi: 10.3390/ijms20225552
86. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15(7):808–13. doi: 10.1038/nm.1982
87. Haseeb M, Pirzada RH, Ain QU, Choi S. Wnt signaling in the regulation of immune cell and cancer therapeutics. Cells (2019) 8(11):1380. doi: 10.3390/cells8111380
88. Chae WJ, Bothwell ALM. Canonical and non-canonical wnt signaling in immune cells. Trends Immunol (2018) 39(10):830–47. doi: 10.1016/j.it.2018.08.006
89. Awada A, Nicaise C, Ena S, Schandene L, Rasschaert J, Popescu I, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary sjogren's syndrome. Ann Rheum Dis (2014) 73(6):1259–63. doi: 10.1136/annrheumdis-2012-203187
90. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
91. Soyfoo MS, Nicaise C. Pathophysiologic role of interleukin-33/ST2 in sjogren's syndrome. Autoimmun Rev (2021) 20(3):102756. doi: 10.1016/j.autrev.2021.102756
92. Tao SS, Cao F, Sam NB, Li HM, Feng YT, Ni J, et al. Dickkopf-1 as a promising therapeutic target for autoimmune diseases. Clin Immunol (2022) 245:109156. doi: 10.1016/j.clim.2022.109156
93. Karatas A, Omercikoglu Z, Oz B, Dagli AF, Catak O, Erman F, et al. Wnt signaling pathway activities may be altered in primary sjogren's syndrome. Turk J Med Sci (2021) 51(4):2015–22. doi: 10.3906/sag-2102-367
94. Dong Y, Ming B, Gao R, Mo Q, Wu X, Zheng F, et al. The IL-33/ST2 axis promotes primary sjogren's syndrome by enhancing salivary epithelial cell activation and type 1 immune response. J Immunol (2022) 208(12):2652–62. doi: 10.4049/jimmunol.2101070
95. Zhao L, Yao L, Yuan L, Xia L, Shen H, Lu J. Potential contribution of interleukin-33 to the development of interstitial lung disease in patients with primary sjogren's syndrome. Cytokine (2013) 64(1):22–4. doi: 10.1016/j.cyto.2013.07.006
96. Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic potential of targeting the Th17/Treg axis in autoimmune disorders. Molecules (2017) 22(1):134. doi: 10.3390/molecules22010134
97. Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O'Neill L, et al. IL-1 family nomenclature. Nat Immunol (2010) 11(11):973. doi: 10.1038/ni1110-973
98. Rudloff I, Godsell J, Nold-Petry CA, Harris J, Hoi A, Morand EF, et al. Brief report: Interleukin-38 exerts antiinflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol (2015) 67(12):3219–25. doi: 10.1002/art.39328
99. Kolbinger F, Loesche C, Valentin MA, Jiang X, Cheng Y, Jarvis P, et al. Beta-defensin 2 is a responsive biomarker of IL-17A-driven skin pathology in patients with psoriasis. J Allergy Clin Immunol (2017) 139(3):923–32.e8. doi: 10.1016/j.jaci.2016.06.038
100. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
101. Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol (1999) 162(5):2775–84. doi: 10.4049/jimmunol.162.5.2775
102. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
103. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi: 10.1038/ni.2005
104. Kim JW, Kim SM, Park JS, Hwang SH, Choi J, Jung KA, et al. Metformin improves salivary gland inflammation and hypofunction in murine sjogren's syndrome. Arthritis Res Ther (2019) 21(1):136. doi: 10.1186/s13075-019-1904-0
105. Shao Y, Fu J, Zhan T, Ye L, Yu C. Fangchinoline inhibited proliferation of neoplastic b-lymphoid cells and alleviated sjogren's syndrome-like responses in NOD/Ltj mice via the Akt/mTOR pathway. Curr Mol Pharmacol (2022) 15(7):969–79. doi: 10.2174/1874467215666220217103233
106. Xu J, Liu O, Wang D, Wang F, Zhang D, Jin W, et al. In vivo generating SSA/Ro-antigen specific regulatory T cells improves experimental sjogren's syndrome in mice. Arthritis Rheumatol (2022) 74(10):1699–1705. doi: 10.1002/art.42244
107. Chen W, Yu Y, Ma J, Olsen N, Lin J. Mesenchymal stem cells in primary sjogren's syndrome: Prospective and challenges. Stem Cells Int (2018) 2018:4357865. doi: 10.1155/2018/4357865
108. Chihaby N, Orliaguet M, Le Pottier L, Pers JO, Boisrame S. Treatment of sjogren's syndrome with mesenchymal stem cells: A systematic review. Int J Mol Sci (2021) 22(19):10474. doi: 10.3390/ijms221910474
109. Xu J, Wang D, Liu D, Fan Z, Zhang H, Liu O, et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical sjogren syndrome. Blood (2012) 120(15):3142–51. doi: 10.1182/blood-2011-11-391144
110. Aluri HS, Samizadeh M, Edman MC, Hawley DR, Armaos HL, Janga SR, et al. Delivery of bone marrow-derived mesenchymal stem cells improves tear production in a mouse model of sjogren's syndrome. Stem Cells Int (2017) 2017:3134543. doi: 10.1155/2017/3134543
111. Liu Y, Li C, Wang S, Guo J, Guo J, Fu J, et al. Human umbilical cord mesenchymal stem cells confer potent immunosuppressive effects in sjogren's syndrome by inducing regulatory T cells. Mod Rheumatol (2021) 31(1):186–96. doi: 10.1080/14397595.2019.1707996
112. Li B, Xing Y, Gan Y, He J, Hua H. Labial gland-derived mesenchymal stem cells and their exosomes ameliorate murine sjogren's syndrome by modulating the balance of treg and Th17 cells. Stem Cell Res Ther (2021) 12(1):478. doi: 10.1186/s13287-021-02541-0
113. Ogata K, Matsumura-Kawashima M, Moriyama M, Kawado T, Nakamura S. Dental pulp-derived stem cell-conditioned media attenuates secondary sjogren's syndrome via suppression of inflammatory cytokines in the submandibular glands. Regener Ther (2021) 16:73–80. doi: 10.1016/j.reth.2021.01.006
114. Gong B, Zheng L, Huang W, Pu J, Pan S, Liang Y, et al. Murine embryonic mesenchymal stem cells attenuated xerostomia in sjogren-like mice via improving salivary gland epithelial cell structure and secretory function. Int J Clin Exp Pathol (2020) 13(5):954–63.
115. Rui K, Hong Y, Zhu Q, Shi X, Xiao F, Fu H, et al. Olfactory ecto-mesenchymal stem cell-derived exosomes ameliorate murine sjogren's syndrome by modulating the function of myeloid-derived suppressor cells. Cell Mol Immunol (2021) 18(2):440–51. doi: 10.1038/s41423-020-00587-3
116. Shi B, Qi J, Yao G, Feng R, Zhang Z, Wang D, et al. Mesenchymal stem cell transplantation ameliorates sjogren's syndrome via suppressing IL-12 production by dendritic cells. Stem Cell Res Ther (2018) 9(1):308. doi: 10.1186/s13287-018-1023-x
117. Cai B, Cai JP, Luo YL, Chen C, Zhang S. The specific roles of JAK/STAT signaling pathway in sepsis. Inflammation (2015) 38(4):1599–608. doi: 10.1007/s10753-015-0135-z
118. Hofmann SR, Ettinger R, Zhou YJ, Gadina M, Lipsky P, Siegel R, et al. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol (2002) 2(6):495–506. doi: 10.1097/00130832-200212000-00004
119. Kolodziejska-Sawerska A, Rychlik A, Depta A, Wdowiak M, Nowicki M, Kander M. Cytokines in canine inflammatory bowel disease. Pol J Vet Sci (2013) 16(1):165–71. doi: 10.2478/pjvs-2013-0025
120. Nakayamada S, Kubo S, Iwata S, Tanaka Y. Chemical JAK inhibitors for the treatment of rheumatoid arthritis. Expert Opin Pharmacother (2016) 17(16):2215–25. doi: 10.1080/14656566.2016.1241237
121. Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia (2013) 27(6):1322–7. doi: 10.1038/leu.2013.71
122. D'Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs (2018) 27(7):595–9. doi: 10.1080/13543784.2018.1492547
123. Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT-494, a selective JAK-1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti-tumor necrosis factor therapy. Arthritis Rheumatol (2016) 68(12):2867–77. doi: 10.1002/art.39801
124. Bowman SJ, Everett CC, O'Dwyer JL, Emery P, Pitzalis C, Ng WF, et al. Randomized controlled trial of rituximab and cost-effectiveness analysis in treating fatigue and oral dryness in primary sjogren's syndrome. Arthritis Rheumatol (2017) 69(7):1440–50. doi: 10.1002/art.40093
125. Mariette X, Seror R, Quartuccio L, Baron G, Salvin S, Fabris M, et al. Efficacy and safety of belimumab in primary sjogren's syndrome: results of the BELISS open-label phase II study. Ann Rheum Dis (2015) 74(3):526–31. doi: 10.1136/annrheumdis-2013-203991
126. Zhan T, Wang B, Fu J, Shao Y, Ye L, Shi H, et al. Artesunate inhibits sjogren's syndrome-like autoimmune responses and BAFF-induced b cell hyperactivation via TRAF6-mediated NF-kappaB signaling. Phytomedicine (2021) 80:153381. doi: 10.1016/j.phymed.2020.153381
127. Fisher BA, Szanto A, Ng W-F, Bombardieri M, Posch MG, Papas AS, et al. Assessment of the anti-CD40 antibody iscalimab in patients with primary sjögren's syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol (2020) 2(3):e142–e52. doi: 10.1016/S2665-9913(19)30135-3
128. Felten R MN, Duffaut P, Saadoun D, Hachulla E, Hatron P, Salliot C, et al. IL-6 receptor inhibition in primary sjögren syndrome : Results from a randomized multicenter academic double blind placebo-controlled trial of tocilizumab in 110 patients. Arthritis Rheumatol (2019) 71(suppl 10).
129. Dorner T, Posch MG, Li Y, Petricoul O, Cabanski M, Milojevic JM, et al. Treatment of primary sjogren's syndrome with ianalumab (VAY736) targeting b cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann Rheum Dis (2019) 78(5):641–7. doi: 10.1136/annrheumdis-2018-214720
130. Smolen J NP, Tahir H, Schulze-Koops H, Li L, Hojnik M, Gellett A, et al. Ianalumab (VAY736), a dual mode of action biologic combining BAFF receptor inhibition with b cell depletion, for treatment of primary sjögren’s syndrome: Results of an international randomized, placebo controlled dose range finding study in 190 patients. Arthritis Rheumatol (2019) 71(suppl 10).
131. Meiners PM, Vissink A, Kroese FG, Spijkervet FK, Smitt-Kamminga NS, Abdulahad WH, et al. Abatacept treatment reduces disease activity in early primary sjogren's syndrome (open-label proof of concept ASAP study). Ann Rheum Dis (2014) 73(7):1393–6. doi: 10.1136/annrheumdis-2013-204653
132. Charras A, Arvaniti P, Le Dantec C, Arleevskaya MI, Zachou K, Dalekos GN, et al. JAK inhibitors suppress innate epigenetic reprogramming: a promise for patients with sjogren's syndrome. Clin Rev Allergy Immunol (2020) 58(2):182–93. doi: 10.1007/s12016-019-08743-y
133. Barrera MJ, Aguilera S, Castro I, Matus S, Carvajal P, Molina C, et al. Tofacitinib counteracts IL-6 overexpression induced by deficient autophagy: implications in sjogren's syndrome. Rheumatol (Oxford) (2021) 60(4):1951–62. doi: 10.1093/rheumatology/keaa670
134. Jakez-Ocampo J, Atisha-Fregoso Y, Llorente L. Refractory primary sjogren syndrome successfully treated with bortezomib. J Clin Rheumatol (2015) 21(1):31–2. doi: 10.1097/RHU.0000000000000210
135. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet (2011) 377(9767):721–31. doi: 10.1016/S0140-6736(10)61354-2
136. Edwards JC, Cambridge G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete b lymphocytes. Rheumatol (Oxford) (2001) 40(2):205–11. doi: 10.1093/rheumatology/40.2.205
137. Godeau B, Porcher R, Fain O, Lefrere F, Fenaux P, Cheze S, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood (2008) 112(4):999–1004. doi: 10.1182/blood-2008-01-131029
138. Sugai S. Report on "the 8th international symposium on sjögren syndrome". Ryumachi (2002) 42(5):765–8.
139. Gandolfo S, De Vita S. Double anti-b cell and anti-BAFF targeting for the treatment of primary sjogren's syndrome. Clin Exp Rheumatol (2019) 37 Suppl 118(3):199–208.
140. Jonsson R, Brokstad KA, Jonsson MV, Delaleu N, Skarstein K. Current concepts on sjogren's syndrome - classification criteria and biomarkers. Eur J Oral Sci (2018) 126 Suppl 1(Suppl Suppl 1):37–48. doi: 10.1111/eos.12536
141. Kroese FGM, Haacke EA, Bombardieri M. The role of salivary gland histopathology in primary sjogren's syndrome: promises and pitfalls. Clin Exp Rheumatol (2018) 36 Suppl 112(3):222–33.
Keywords: sjogren's syndrome, foxp3 + treg, th17 cells, JAK pathway, tumor necrosis factor, interferon
Citation: Zhan Q, Zhang J, Lin Y, Chen W, Fan X and Zhang D (2023) Pathogenesis and treatment of Sjogren’s syndrome: Review and update. Front. Immunol. 14:1127417. doi: 10.3389/fimmu.2023.1127417
Received: 19 December 2022; Accepted: 23 January 2023;
Published: 02 February 2023.
Edited by:
Xiuzhen Sheng, Ocean University of China, ChinaReviewed by:
Dayong Liu, Tianjin Medical University, ChinaJian Li, National Institutes of Health (NIH), United States
Copyright © 2023 Zhan, Zhang, Lin, Chen, Fan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dunfang Zhang, aXpkZkAxNjMuY29t