 Mengwen Niu
Mengwen Niu Feng Zhao1
Feng Zhao1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 March 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1127277
This article is part of the Research TopicPain, Immunity, and Neurological and Autoimmune DisordersView all 7 articles
Rheumatoid arthritis (RA) is characterized by the augment of vascular permeability, increased inflammatory cells infiltration, dysregulated immune cells activation, pannus formation and unbearable pain hyperalgesia. Ca2+ affect almost every aspect of cellular functions, involving cell migration, signal transduction, proliferation, and apoptosis. Transient receptor potential channels (TRPs) as a type of non-selective permeable cation channels, can regulate Ca2+ entry and intracellular Ca2+ signal in cells including immune cells and neurons. Researches have demonstrated that TRPs in the mechanisms of inflammatory diseases have achieved rapid progress, while the roles of TRPs in RA pathogenesis and pain hyperalgesia are still not well understood. To solve this problem, this review presents the evidence of TRPs on vascular endothelial cells in joint swelling, neutrophils activation and their trans-endothelial migration, as well as their bridging role in the reactive oxygen species/TRPs/Ca2+/peptidyl arginine deiminases networks in accelerating citrullinated proteins formation. It also points out the distinct functions of TRPs subfamilies expressed in the nervous systems of joints in cold hyperalgesia and neuro-inflammation mutually influenced inflammatory pain in RA. Thus, more attention could be paid on the impact of TRPs in RA and TRPs are useful in researches on the molecular mechanisms of anti-inflammation and analgesic therapeutic strategies.
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease characterized by the infiltration of T cells, B cells, and inflammatory cells such as neutrophils, macrophages, and so on within joints. Inflammatory factors and autoantibodies produced and secreted by immune cells act on cartilage, tendons, ligaments, and bone tissues, causing joint swelling, pain hyperalgesia, pannus formation, and eventually bone destruction (1, 2). It has a high prevalence and disability rate, bringing a huge economic burden and pressure to society and families (3). While the pathogenesis of RA remains unclear, so, it is still a long way to go.
Ca2+ affect almost every aspect of cellular functions, involving cell proliferation, apoptosis, migration as well as signal transduction. Calcium ion as a necessary molecule of physiological signal transduction in eukaryotes, it functioned as a central manipulator of inflammation as well as immune response in RA (4). As non-selective Ca2+ permeable channels, TRPs expressed on neutrophils (5, 6), chondrocytes (7, 8), fibroblast-like cells (FLS) (9) and nervous system (10, 11) have been proven to regulate cation flow and potential changes, helping them participate in RA.
The TRP channel superfamily consist of 28 members (27 in humans) in mammals (3). Based on sequence homology, the superfamily is divided into seven subfamilies in mammals (12). Four subfamilies of TRPs have been well studied which are known as TRPCs, TRPVs, TRPMs, and TRPA1. There are also different categories within each subtype, including the Canonical subfamily (TRPC1-7), the Vanilloid subfamily (TRPV1-6), the Melastatin subfamily (TRPM1-8) (13, 14). As they can be activated by diverse stimuli no matter in extracellular and intracellular circumstances, for example the alteration of the temperature, osmolarity, depletion of calcium stores, as well as cytokines. Therefore, they play a series of important roles in physiological and pathological status (15–17).
Though TRPs have been studied in many other diseases such as cardiovascular, renal, tumor, lung inflammation and other inflammatory diseases, researches on their roles in rheumatoid arthritis are limited. We carefully selected representative articles and explore the different and indispensable roles of these TRPs in RA pathogenesis and in cold and inflammatory stimuli perceptions as well as their influence on neuro-inflammatory interaction outcomes. At the same time, we also show our ideas as we illustrate the relevant issues.
RA patients typically complain of swelling and stiffness of joints. Vascular endothelial cells (ECs) are involved in immune and inflammatory responses because they can produce different cytokines and react with immune-inflammatory factors, causing the augmentation of endothelial permeability (18). The increase of Ca2+ in ECs closely related to endothelial cell permeability (19). However, the exact molecular mechanism involved in the increase of endothelial permeability is not yet completely known.
The inner layer of blood vessels consists of adjacent ECs and their tight connections. The integrity of the endothelial layer was guaranteed by intercellular linkages comprising of adherents and tight junctions (20, 21). The calcium influxes/transients cause adherents junction disassembly, cytoskeletal rearrangements and facilitate ECs retraction. Therefore, the morphology of ECs was transformed to a round shape, and gaps between them were augmented, inducing their permeability increase (21, 22). The concentration of intracellular calcium ion promoted with the increased opening of TRPC1 on ECs, which accelerated the formation of actin stress fibers and enhanced the stretching and deforming ability of ECs, thus leading to increased permeability (19). The TRPC6 expressed in ECs collaborated with platelet/endothelial cell adhesion molecule-1 (PECAM) to surround leukocytes during their trans-endothelial migration (TEM) and could help them in their migration. The down-regulated expression of TRPC6 or shRNA knockdown in ECs arrested neutrophils over the junction, similar to when PECAM was blocked (23).
Among the 28 identified and widespread mammalian TRP channel isoforms, at least 19 are expressed in ECs (24, 25), and the transient receptor potential canonical channels (TRPCs) are the subfamily of TRPs closely related to endothelial cell permeability. ECs express six non-selectively cation permeable TRPCs, which are TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, and TRPC7 (25, 26). Most studies agreed that the influx of Ca2+ into ECs through TRPC1, TRPC4, and TRPC5 was mediated by the store-operated Ca2+ entry (SOCE), starting with the depletion of Ca2+ in endoplasmic reticulum (ER) stores (27, 28). But, some studies had questioned whether the TRPC1/4/5 channels mediated the increase of Ca2+ through SOCE (29). It has been reached an agreement that the influx of Ca2+ through TRPC3, TRPC6 and TRPC7 was induced by the receptor-operated Ca2+ entry (ROCE). An important stimulant of TRPCs is the α-subunits of G-proteins. Thus, inflammatory stimuli cause the activation of G-protein-coupled receptors (GPCR) and the phospholipase C (PLC) promote the opening of TRPC channels (30, 31). Even though TRPCs may also activated by some other stimulates, the mainly same effect is the increase of calcium influx in ECs. The mechanisms of different TRPC subfamilies in mediating calcium ions entering into endothelial cells are shown in Figure 1.
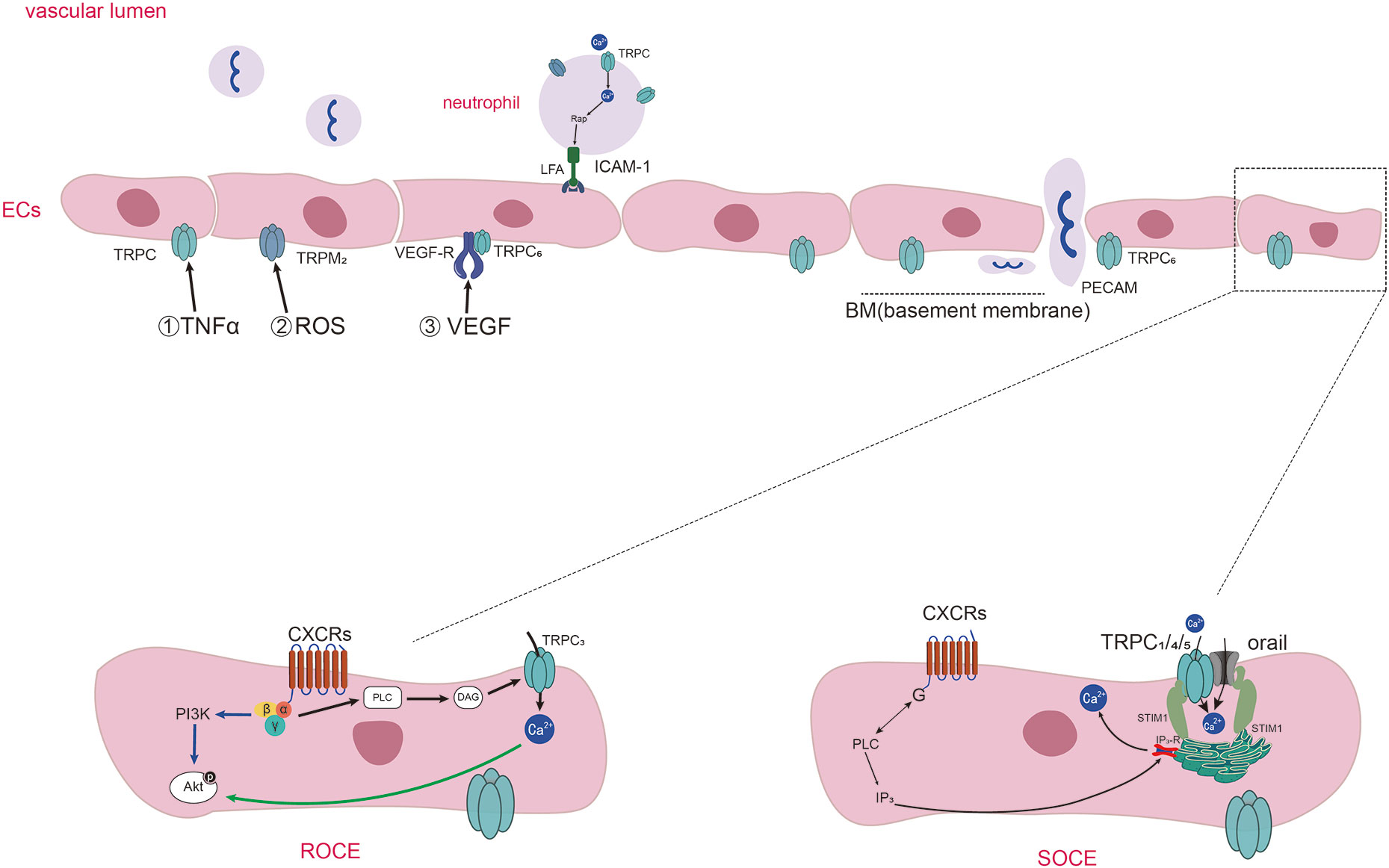
Figure 1 Diagram of TRPs-mediated Ca2+ entering into endothelial cells and neutrophils chemotaxis and adhesion in inflammation. Diagram of TRPs-mediated Ca2+ entering into endothelial cells and neutrophils chemotaxis and adhesion in inflammation. Inflammatory stimuli (such as TNF-α, ROS, VEGF) influence the expression and activation of TRPs expressed on ECs. Activation of specific GPCRs on the cell surface causes the activation of PLC which cleaves the membrane phospholipid PIP2 into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). DAG induce the activation of TRPC3, TRPC6, and TRPC7 channels, and the way of the influx of Ca2+ called receptor operated Ca2+ entry (ROCE). IP3 binds to IP3 receptors (IP3Rs) present on the ER, causing Ca2+ release from intracellular stores. Then the Ca2+ sensor stromal interaction molecule1(STIM1) proteins gather to the membrane of ER and activate the channels of orai1 and TRPC1/4/5, thus promoting extracellular Ca2+ influx which is called as SOCE. TRPCs also mediate the adhesion of neutrophils through regulating the anchor link of neutrophil and epithelial cells. TRPC6 expressed in endothelial cells colocalize with PECAM, surrounding leukocytes and promoting neutrophil trans-endothelial migration.
Inflammatory factors interact with TRPs on vascular ECs of the inflammatory sites, leading to increased vascular permeability. TNF-α works as a major inflammatory factor in RA, it is often used as an inflammatory mediator to simulate the inflammatory reaction process of RA (32–35). TNF-α could cause the TRPC1 expression which medicated increased Ca2+ entry through SOCE, representing an important mechanism of endothelial injury through TRPC1 (19, 36). Ca2+ flowing through TRPC1 can also increase TRPC1 expression through positive feedback. Protease-activated receptor-1(PAR-1) activation elicited cellular responses including NF-κB activation and increased Ca2+ influx through TRPC1. Ca2+ then regulated the expression of TRPC1 expression which may be a vicious circle resulting in vascular permeability (36–38). Besides, inflammatory mediators, such as thrombin were reported to be increased in RA (39, 40). Thrombin induced the phosphorylation of TRPC1 by PKCα activation, resulting in Ca2+ entry and the increase in permeability in confluent endothelial monolayers (41). The inflammatory factor thrombin could also promote the association between TRPC6 and phosphatase tensin homologue (PTEN) which worked as a role of the scaffold for TRPC6, enabling the expression of TRPC6 on cell surface, which made the increase of Ca2+ entry (42). Therefore, the increased Ca2+ entry through TRPC1/6 plays an important role in augmenting endothelial permeability in RA.
Reactive oxygen species (ROS) are cellular metabolites that contain at least one oxygen atom and one or more unpaired electrons (43). ROS could damage DNA, proteins, lipids, and many other molecules (44). Phagocytes and recruited immune cells could produce a lot of ROS in the RA synovitis microenvironment (45–47). Increased ROS caused by Oxidative stress can destroy endothelial integrity by leading to Ca2+ influx into ECs, and disrupt the tight junctions between them (48). TRPM2 expressed in ECs can be activated by ROS and play a role as Ca2+-permeable channel. The sub-lytic concentration of H2O2 induced an increase in ECs intracellular Ca2+ by stimulating Ca2+ entry through the TRPM2, and subsequent reduction of transendothelial resistance (49).
VEGF was first isolated and named vascular permeability factor (VPF), a potent vascular permeability-enhancing cytokine and a selective mitogen for endothelial cells (50). VEGF expression was increased in vascular endothelium and synovial tissue in RA joints and was associated with inflammation and angiogenesis (51–54). VEGF could also provoke ROS production with the participation of NOX, followed by the autophosphorylation of VEGFR2 (55). Corroborating with the above paragraph, VEGF and ROS can act as two interactive inflammatory molecules synthesized at the site of inflammation, and weaken the endothelial functions. Decrease expression of TRPC6 in human microvascular endothelial cells (HMVECs) restrained the VEGF-mediated increases in Ca2+ and migration of HMVECs (56). Therefore, TRPC6 is an indispensable cation channels needed in the VEGF-mediated increase of Ca2+ in HMVECs.
VEGFs can identify and combine with their cognate VEGF receptors, including VEGFR1, VEGFR2, and VEGFR3, as well as some co-receptors (57). VEGF medicated endothelial hyperpermeability in a phospholipase C (PLC)-IP3 pathway, which result in extracellular Ca2+ entry via the plasmalemma store-operated TRPC1. VEGF augmented the interaction of IP3R with TRPC1 (58). VEGF could activate a receptor-operated cation current in HMVECs, in which VEGF bound with VEGFR2 which activated heterologously co-expressed TRPC3/6 channels, such as the style of VEGFR2-TRPC3 and VEGFR2-TRPC6 on cells (59). In conclusion, we hypothesized that when VEGF binds to the corresponding VEGFR or co-receptors on ECs, it may cause the activation of TRPC channels in the form of linking the body with these receptors, which leads to the increase of intracellular calcium ions and the increase of endothelial cell permeability.
Neutrophils are the main force of the first line of immune defense and always be recruited to sites of inflammation as soon as possible. The recruitment of neutrophils and their activation against pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) are highly coordinated and tightly regulated processes that involve the activation of various kinds of receptors as well as many ion channels (60, 61). Fluctuations in intracellular Ca2+ levels are hallmarks in the above complex processes of neutrophils. Hence, TRPs, as non-selective permeable Ca2+ channels distributed on neutrophil, have an important role in the functions of neutrophil in RA.
Leukocyte TEM is the first step of their participating in the immune response and is vital to inflammation (62–66). Neutrophils can be activated by plentiful of “neutrophil-active” chemo-attractants, which initiate the TEM process, including different kinds of chemokines and cytokines (such as CXCLs) as well as fragments of complement (65). TRPC6(-/-) neutrophils had decreased Ca2+ transient during initial adhesion, resulting in reduced activation of Rap1 and β2 integrins and reduced binding to ICAM-1. This suggests that the TRPC6 on neutrophils is a key functional channel protein in CXCL1’s recruitment of inflammatory cells from the bloodstream (67). The deletion of TRPC1 led to reduced migration and chemotaxis of neutrophils due to disruption of Ca2+ gradient homeostasis within TRPC1-/- neutrophils, resulting in more disordered migration and reduced directed migration of neutrophils (68).
A large number of activated neutrophils were found in both peripheral blood and synovial tissue from RA patients (69). Different kinds of chemokines are manifested by studies highly expressed in the serum and synovial fluid in RA, for instance, the CXC-chemokines, the CC-chemokines, and the CX3C-chemokine CX3CL1 (70–73). Intracellular Ca2+ flow from the extracellular space induced by CXCR2 as a chemotactic agent is affected in TRPC6(-/-) neutrophils. Deletion of TRPC6 inhibited phosphorylation of AKT and MAPK molecules, which were downstream of CXCR2 receptors, attenuated actin remodeling, and thus impeded the chemotaxis of neutrophils (74). TRPC6 could also be activated by the CXC-type G-protein-coupled chemokine receptors upon stimulation with macrophage inflammatory protein-2 (MIP-2), which was essential for the arrangement of filamentous actin of migrating neutrophils (75).
TRPM7 is a member of the subfamily TRPMs and works as not only an ion channel but also kinase activity. If the TRPM7 channel or kinase activity was blocked, the ability of human neutrophils to migrate along CXCL8 chemokine gradients was significantly reduced. Kinase activity of TRPM7 affected the Akt1/mTOR signaling pathway and regulated neutrophil migration and function (5). Recent researches showed that the expression of CD147 on neutrophils from RA patients was higher than that in healthy group (76). TRPM-7-mediated the increase of Ca2+ and induced chemotaxis, adhesion ability, and invasiveness of RA neutrophils response to CD147. When treated with the transient receptor potential melastatin 7 small interfering RNA(si-TRPM7), the chemotaxis and the relative adhesion rates of neutrophils were reduced significantly (6).
Once neutrophils are activated by inflammatory stimuli, their intracellular Ca2+ concentration will be enhanced. The calcium-dependent peptidyl arginine deiminases (PADs) will be stimulated by the increased concentration of Ca2+ in neutrophil. PADs convert the protein arginine residues to citrulline residues, this process is called protein citrullination (77). The autoantibodies that recognize citrulline protein antigen (ACPAs) may have been present prior to diagnosis, and studies have shown that it is a major pathogenic molecule of RA (78, 79). In the process of citrullination, the PADs are the key enzymes, therefore, it is necessary to explore the influencing factors of PADs activity. So, we consider that TRPs that can cause calcium influx may play a role in promoting the activity of PADs in neutrophil.
It has been manifested that Calcium is the primary factor in the activation of PADs enzymes, because it can motivate structural changes of PADs. PADs have five calcium-binding sites, and the enzyme active sites will be exposed when they bind to calcium ions (80). At the same time, the conformational changes are accompanied by the transition from inactive to active PADs.
It has been shown that three subtypes of TRPs are expressed in human and mouse PMNs, including TRPCs, TRPMs and TRPVs (60). Receptors on neutrophils can be activated by inflammatory stimuli, leading to the activation of PLC which cleaves the membrane phospholipid PIP2 into inositol 1,4,5-trisphosphate (IP3) and DAG. DAG induce the activation of TRPC6 channels, through which the Ca2+ entry into neutrophils (81). IP3 binds to IP3 receptors (IP3Rs) present on the ER, causing Ca2+ release from intracellular stores. Then the Ca2+ sensor stromal interaction molecule 1(STIM1) proteins gather to the membrane of ER and activate the Orai1 and TRPC1, thus promoting extracellular Ca2+ influx neutrophils (82). Increased intracellular Ca2+ concentration in neutrophils promotes PADs activation, which in turn leads to the formation of citrullination proteins or neutrophil extracellular traps (NETs). We have shown evidence that TRPCs offer assistance in Ca2+ influx through SOCE and ROCE during the process of the recruitment and activation of neutrophils, and summarized in Figures 1, 2.
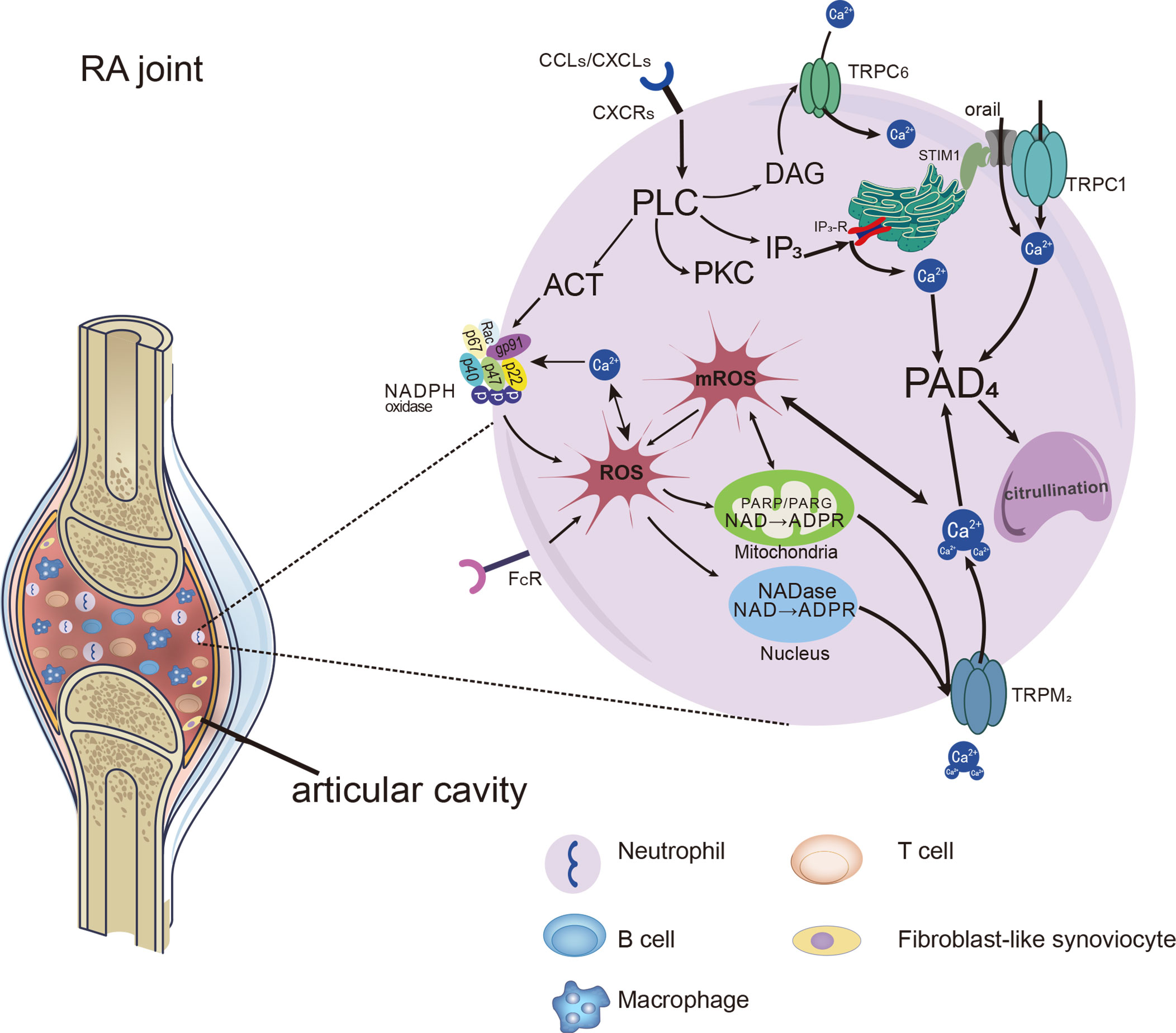
Figure 2 Distinct Ca2+ signals and downstream cellular events associated with TRPs in neutrophils. Distinct Ca2+ signals and downstream cellular events associated with TRPs on neutrophils in RA joints. Chemokines and immune-inflammation factors stimulate the activation of receptors on neutrophils, leading to the activation of PLC and the production of molecules downstream of it. NADPH oxidase can be activated and make an increase in ROS production. ROS leads to the increase of ADPR production, which acts as the activator of the TRPM2 channel and further promotes the entry of Ca2+ into cells. In addition, DAG, as the activator of TRPC6, can induce increased calcium influx through ROCE. With the addition of Ca2+ entering by SOCE, cytosolic Ca2+ level rapidly increased by the SOCE, ROCE and ROS/TRPs mechanisms. Ca2+ catalyzes PADs which facilitate citrullination, and even promote the formation of NETs in neutrophils.
Interestingly, most of the stimuli that lead to an increase in the calcium concentration of neutrophils also lead to the production of ROS. ROS generation and Ca2+ concentration in synovial neutrophils from RA patients were increased, and proinflammatory cytokines such as GM-CSF and TNF-α enhanced ROS production in neutrophils by activating NADPH oxidase (83). Moreover, ROS-dependent NETs formation was associated with enhanced PADs activity (84, 85). In line with those described above, RA neutrophils presented increased formation of NETs with elevated ROS and Ca2+ concentrations, accompanied by enhanced PADs mediating citrullination (86). It seems apparent that ROS and Ca2+ play a mutual interaction in promoting PADs enzyme activity.
Notably, TRPM2 cationic channels play a mediating role in regulating Ca2+ mobilization and oxidative stress in neutrophils. They regulate calcium influx in a way that is different from the SOCE and ROCE pathways mentioned above. The activating ligand for TRPM2 is ADP-ribose (ADPR), which is increased when intracellular ROS levels are high and works with intracellular calcium to activate TRPM2 (87–89). H2O2 was shown to prompt ADPR generation, which acted as an activator of TRPM2. The increased calcium through TRPM2 could further induce production of the ROS (90). FcγRIIIb activated by immune complexes evoked protein kinase C (PKC) activation, which promoted NADPH-oxidase in ROS production, leading to activation of TRPM2 channels, allowing significant up-regulation of extracellular Ca2+ into the neutrophil (91). Compared with the control group, the concentration of intracellular free Ca2+ and ROS production in neutrophils were significantly increased in patients with RA. The intracellular Ca2+ concentrations were decreased by anti-TRPM2 therapy (92). Hence, we consider that the TRPM2 work as a bridge of inflammation or oxidative stress and calcium signal in the NADPH oxidase/ROS/TRPs/Ca2+/PADs pathway, as shown in Figure 2.
Angiogenesis/neovascularization is an invasive event in the pathogenesis of RA which is characterized by synovial tissue proliferation. Osteochondral angiogenesis of RA is characterized by fibrovascular tissue expressing VEGF and increased proliferation of ECs and fibroblast-like synovial cells (FLS) (93). VEGF, a key governor of angiogenesis, can regulate angiogenesis via VEGFR2 (94). The overexpression of TRPV4 up-regulated VEGFR2 trafficking and activation, controlling ECs migration and angiogenesis (95). It was also found to induce endothelial progenitor cells (EPC) proliferation and migration, which facilitated angiogenesis and enabled the development of RA (96). TRPC3, TRPC4, and TRPC5 were essential for the formation of tubular structures and revealed an essential contribution of TRPCs to the vitro tubulogenesis of endothelial cell lines (97). TRPC6 was also a cationic channel necessary for the related pathway processes of VEGF-mediated cytoplasmic calcium increase and angiogenesis (56, 98).
Accumulating data has proved that the apoptosis rate of FLS in RA is decreased, and their proliferative and invasive properties lead to progressive destruction of cartilage and bone (99, 100). TRPC6 was found somewhat higher in RA-FLS than that in OA-FLS. TRPC6 deficiency in mice weakened the development of experimental RA and CIA models and inhibited FLS invasiveness and proliferation (101). The expression of TRPM7 was significantly increased in RA-FLS, TRPM7 may have a role in reduced FLS apoptosis because suppression of TRPM7 channels increased RA FLSs apoptosis in vitro, and this was associated with endoplasmic reticulum (ER) stress activation (102). Another study found that TRPA1 activation was associated with decreased proliferation of FLS, an effect that was substantially enhanced by TNF preincubation. The reason may be that activation of TRPA1 increased calcium flux and substantially reduced cell viability by inducing necrosis (103). As we can only retrieve a few studies on the correlation between TRPs and FLS proliferation or apoptosis at present, the assured roles of TRPs in promoting the proliferation or apoptosis of the FLS are not yet clear. We are looking forward to detailed studies in this field.
RA is a serious and persistent painful disease of the distal joints in which neurogenic components are supposed to participate in its pathology. Pain is the most common complaint of patients in the rheumatology department, and the threshold of patients for pain is often lower than that of healthy people, that is, pain hyperalgesia (104). There are a large number of sensory nerve fibers in the synovium of the joint. Located on these sensory nerve fibers, TRPs act as an important superfamily of proteins that mediate heat and mechanical pain. Recent findings regarding the role of TRPs in cold and inflammatory pain hypersensitivity spouted. The mechanisms of TRPs in temperature perception, nociception, sensitization of inflammatory stimuli, and neuro-immune communication will be stated in detail in the following sections.
When the weather gets cold, there are many cases where patients with RA have to visit the clinic because of unbearable joint pain. Although the mechanism of the effect of cold environment on arthritis patients is not clear, it has been shown to be one of the common precursors and aggravating factors of arthritis.
TRPA1 on neurons can perceive the changes of thermal (< 17°C), chemical (menthol, formalin, reactive lipids, etc.), and mechanical (sting, pinch, etc.) (105). Studies established that TRPA1 acted as a cold sensor in experimental models, revealing that TRPA1 was a major sensor for noxious cold (106, 107). TRPA1 protein and mRNA were overexpressed in both peripheral and central nervous system of CFA treated mice, and TRPA1 played a vital role in the development and maintenance of cold pain hyperalgesia. However, pretreatment with TRPA1 antagonist HC-030031 inhibited the above results (108). While, as a opposite, one study argued that WT and TRPA1 KO mice had no difference in the perception of thermal pain in the inflamed claw compared with that of naive mice (109). Additionally, another study hinted that TRPA1 agonists can sensitize the noxious reflex withdrawal to heat, but not cold (110). Why do they have contradictory conclusions? The reasons may be the ways and degrees of cold stimuli and the detection methods they used, and the test time points after stimulation they set. Therefore, different investigational methods may reflect controversial results.
In one study, cold exposed CFA arthritis mice developed bilateral pain sensitivity in their knee joints, which was dependent on the cold sensor, TRPA1. Moreover, blood flow was seen to increase in the joint after cold exposure which also depended on TRPA1, and due to the vasodilatory neuropeptides, including Substance P (SP) and calcitonin gene-related peptide (CGRP) (111). Thus, TRPA1 may act to influence not only its established nociceptive actions within the joint but also blood flow via neuropeptides. Then, why TRPA1 can medicate the pain hyperalgesia and still cause increased blood flow than that of the control group after cold exposure? When the animals were at room temperature, blood flow of CFA-treated joints was lower than that in control joints. However, under the stimulation of a cold environment, the sympathetic nerves of the CFA group were excited and secreted various vasodilatory substances. SP and CGRP participated in the stabilization of blood flow in the synovium after cold stimulation (111). Similarly, blood vessel constriction in the inflammation site was most obvious within 0 - 2 minutes after cold stimulation, and then gradually returned to the baseline level. The initial cold-induced vasoconstriction was mediated via TRPA1-dependent superoxide production, and the subsequent restorative blood flow component was also dependent on TRPA1 activation being mediated by sensory nerve-derived dilator neuropeptides CGRP and SP, and also NOS-derived NO (112, 113). Therefore, blood vessels in the inflamed joint will have a process of the first contraction and then relaxation after being cold, and different experimental detection points will have discrepant results. Increased blood flow is due to the effects of TRPA1 and vasodilatory substances, which also cause hypersensitivity to pain.
TRPM8 is a non-selective cationic channel that can be activated by low temperatures and cooling agents, and plays a role in the perception of cold environments. The response of sensory neurons and intact sensory nerve fibers in TRPM8-deficient mice to cold was significantly weakened in vitro (114, 115). Cold pain associated with CFA-induced inflammation was significantly attenuated in TRPM8(-/-) mice (116). However, one study showed that TRPA1 mediated cold pain in CFA-induced arthritis, but TRPM8 had no effect. The expression of TRPA1, but not TRPM8, increased in CFA-induced inflammation, and knockdown of the TRPA1 gene prevented and reversed inflammation-induced cold hyperalgesia (117). A few experiments found that the threshold of TRPM8 activation was 25°C, consistent with the pleasant/cool feeling induced by menthol products and so, had no effect on cold pain. Different from the above view, TRPM8 and TRPA1 had synergistic effects on cold nociceptive perception (118). A group of researchers found that CFA-induced inflammatory cold pain was selectively mediated by glial cell line derived neurotrophic factor family receptors (GFRα3), which were expressed in a subpopulation of TRPM8 sensory neurons that act as cold nociceptors (119, 120). They then provided new insight that the neurogenic inflammation upstream of the GFRα3 and TRPM8 promoted TRPA1’s perception of cold pain, which was in a TRPM8-dependent manner (121). According to the above, TRPM8 joins hands with TRPA1 and serve an important role in cold pain in CFA-induced arthritis.
Thermal (heat) and mechanical noxious stimuli are detected by specialized nerve endings, which transform the stimuli into electrical signals and convey the stimuli to the central nervous system, causing thermal and mechanical pain. The basis for hyperalgesia is the sensitization of the nociceptive system to stimuli (hypersensitivity), moreover, the threshold for the excitation of nociceptive neurons is lowered(low threshold), and therefore, the responses to noxious stimuli are heightened (122). RA patients suffer from hypersensitivity to heat and mechanical joint pain caused by inflammatory factors and joint movement. In the following sections, we emphasize the introduction of inflammatory thermal (heat) and mechanical pain, mainly on the mechanisms involved in TRPV1 and TRPA1.
The TRPV1 is expressed on sensory neurons and can sense changes of heat (temperature >43°C), proinflammatory substances, lipoxygenase products, endocannabinoids, protons, and so on (123). TRPV1 is involved in two typical inflammatory hyperalgesia: thermal and mechanical pain. Peripheral distribution of TRPV1 played a major role in CFA-induced thermal hyperalgesia, but to a lesser extent in mechanical hyperalgesia. While, spinal cord distribution of TRPV1 act as an important role in the perception of both types of hyperalgesia (124).
There was a significant interaction between the inflammatory state and TRPV1 activation, resulting in inflammatory pain perception (125). Firstly, TRPV1 receptor protein was upregulated during CFA-induced inflammation, and mediated inflammatory hyperalgesia (126–128). Secondly, the activation of TRPV1 is regulated by its phosphorylation status which is dependent on the balanced actions of protein kinases and protein phosphatases (129). The activity of protein kinases and protein phosphatases are often activated or inhibited in inflammatory circumstances. In CFA-induced arthritis models, increased expression of phosphorylated protein kinase A phosphorylated TRPV1 and sensitized it, resulting in inflammatory mechanical and thermal pain (130). Furthermore, evidence showed that TRPV1 phosphorylation by Cdk5(Cyclin-dependent kinase 5, an important serine/threonine kinase) promoted the distribution of TRPV1 in the plasma membrane and contributed to thermal hyperalgesia during inflammation post-CFA (131).
At the beginning of this section, we focused on the role of TRPA1 in cold pain sensitivity in RA patients. In addition, it has been shown to play a role in mediating thermal (heat) and mechanical pain sensations in inflamed joints. One study showed that TRPA1 played a key role in both thermal and mechanical pain in CFA-induced arthritis (132). Similarly, application of the selective TRPA1 antagonist HC-030031 could significantly reduce mechanical hyperalgesia of the CFA-injected joint (108). Furthermore, intraarticular injection of CFA in TRPA1-/- mice inducing ipsilateral mechanical hyperalgesia was maintained for only 24 hours compared to 3 weeks in TRPA1 WT mice (133). These studies indicate that TRPA1 also takes part in inflammatory mechanical hyperalgesia and the transition from acute to chronic pain.
It is well manifested that TRPA1 is co-expressed in 60 – 75% of TRPV1-expressing sensory C-fiber nerves (107, 134). In addition, they interact with each other in pain perception at the central level. The frequency of spontaneous excitatory postsynaptic currents (EPSCs) in lamina I dorsal horn neurons were enhanced by CFA treatment, which could be attenuated with the employment of a specific TRPA1 or TRPV1 antagonist. TRPA1 mediated the increased presynaptic glutamate release to lamina I neurons and TRPV1 contributes to increased glutamatergic input in chronic inflammatory pain perception. Therefore, TRPA1 and TRPV1 cooperate at the spinal cord level, leading to the maintenance of chronic inflammatory pain (135).
Consistent with the above report on the involvement of TRPA1 in inflammatory mechanical hyperalgesia, in the CFA-induced inflammatory pain model, Tmem100 CKO mice showed attenuated mechanical hyperalgesia, indicating that TRPA1 participated in mechanical hyperalgesia. In DRG neurons, Tmem100 could be co-expressed with TRPA1 and TRPV1 and form a complex, which attenuated TRPV1’s inhibition of TRPA1, thereby selectively enhancing TRPA1 activities. Tmem100-3Q, a Tmem100 mutant had the opposite effect which enhanced the association of TRPA1 and TRPV1 and strongly inhibited the activity of TRPA1 in a TRPV1-dependent way. Tmem100-3Q CPP could mimic Tmem100-3Q’s effect and inhibits persistent pain by TRPA1. So, Tmem100-3Q CPP or molecules that have similar functions may be promising pain therapy for alleviating mechanical hyperalgesia (136). However, study also shown that reducing the link between TRPA1 and TRPV1 led to the reduction of TRPA1 function. They found that reduction of the A1-V1 complex significantly reduced TRPA1 sensitization and CFA-induced hypersensitivity (137). These findings suggest that there are certain functional molecules between TRPA1 and TRPV1 during they participate in the development and maintenance of hyperalgesia. Drugs or therapeutic strategies which are able to directly or indirectly target their interactions may shed new lights on potential analgesics and may have minor if any side effects.
Different pro-inflammatory cytokines are not only participants in inflammation, what’s more, each cytokine has its profile of effects on inflammatory pain. And most importantly, the formation of pain perception is determined by a variety of cytokines, and keep in mind that different stimuli and multiple pain receptors exist and work at the same time. Multiple sensory receptors, including TRPs, interact and influence each other to make certain pain perceptions. Due to the important role of TNF-α in RA, we will elaborate on the correlation between TNF-α and TRPs in mediating inflammatory pain. The influences of other inflammatory substances on TRPs in inflammatory pain perception are summarized in Table 1.
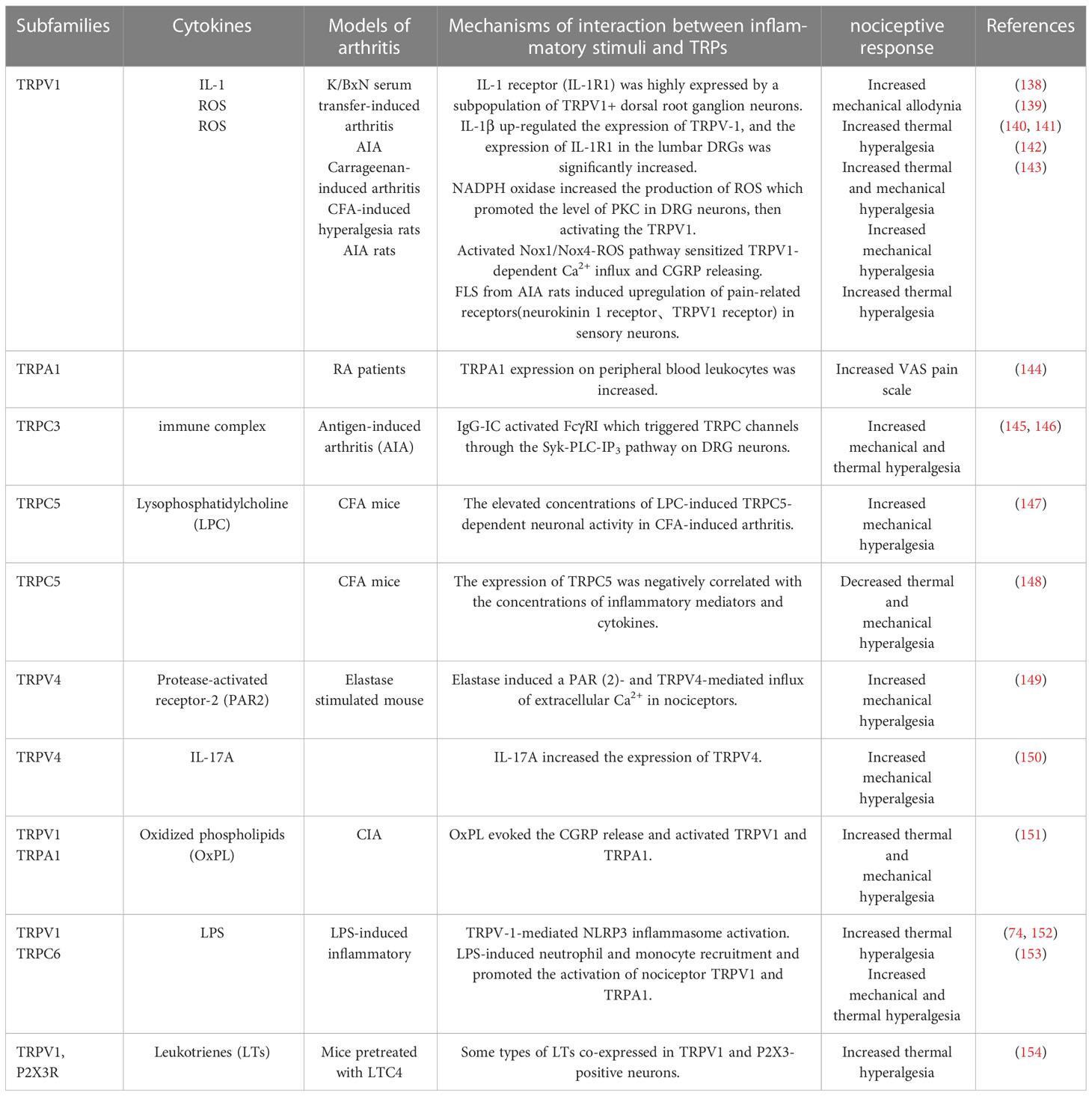
Table 1 Summary of RA related inflammatory stimuli on different subtypes of TRPs in inflammatory pain perception.
TRPV1 are also downstream targets for various pro-inflammatory and pain-producing agents such as IL-1β and TNF-α. These compounds initiate an allosteric modification of the TRPV1 channel protein, resulting in an increase in the probability of channel opening or TRPV1 sensitized.
Paw withdrawal latency (PWL)was always taken as the thermal hyperalgesic threshold (155). TRPV1-/- mice had significantly higher baseline PWL than WT mice post-injection of TNF-a, the WT mice exhibited significantly reduced PWL compared to the TRPV1-/-mice, indicating that TNF-a induced thermal hyperalgesia was TRPV1 dependent. Moreover, contralateral uninjured hind paws also had thermal pain which was TRPV1-dependent, too. In addition to this, the local IL-1β generation in the contralateral paw, PKC and COX-2-derived prostaglandins were essential for the development of bilateral hyperalgesia (10). Researchers also concluded that most of the DRG neurons expressed the TNFR1 also expressed the TRPV1 receptor. TNF-α could up-regulate the expression of TRPV1 receptor, thus causing thermal hyperalgesia (156).
Some researchers completed their study which demonstrated that TNFα-mediated thermal hyperalgesia involves TRPV1 (10). They then wanted to investigate the role of TRPV1 and TRPA1 in the TNF-α induced mechanical hyperalgesia. By studying several models of inflammatory arthritis, they found that central distribution of TRPV1 was involved in TNF-α-induced mechanical hyperalgesia. The peripheral distribution of TRPA1 not only played in the occurrence of TNF-α induced mechanical hyperalgesia, but also act as a key role in the maintenance of CFA-induced mechanical hyperalgesia (133). In line with this result, researchers showed for the first time that TRPA1 has an important peripheral role in TNF-α-mediated mechanical hyperalgesia, which is different from TRPV1 distributed in the central nervous system in mechanical hyperalgesia (157). Thus, these studies present evidence that both TRPV1 and TRPA1 participate in TNF-α medicated inflammatory pain, but they may tend to have their separate strong points in peripheral and central inflammatory perception, which may direct us to explore the corresponding drug. Just as important, anti-TNF-α therapy has also been proven to have analgesic effects.
Neuropeptide-expressing small-diameter sensory neurons are thought to be vital in inducing inflammatory hyperalgesic responses in inflammation. The CGRP peptide expression was increased after CFA injection compared to the control group and contributed to inflammatory hyperalgesia (158, 159). In arthritis mice, Concomitant with the disease progression, there was a significant increase in the density of CGRP+ nerve fibers in the synovium, as well as an enhancement in joint pain-related behaviors compared with the sham-injected mice (104, 160). CGRP-positive neurons partially overlapped with TRPV1 in DRG neurons. As a mutual, activated TRPV1 up-regulated CGRP expression, leading to the induction of inflammation pain (161). TRPV1 expression was also increased in small peptidergic (CGRP positive) neurons after injection of CFA and played a role in chronic thermal hyperalgesia and mechanical pain (126).
In addition to their role in pain perception, CGRP-secreting nerve fibers also contribute to RA inflammation. As highlighted and discussed at the beginning of this paper, CGRP is an extremely potent vasodilator, leading to edema formation and recruitment of inflammatory cells to the local sites. It also significantly elevated the production of inflammatory cytokines (such as IL-1β, IL-6 and TNF-α) in RA patients (162, 163). Neuropeptides may strongly enhance TRPV1 expression in RA synoviocytes. SP or CGRP in combination with TRPV1 could powerfully motivate the expression of pro-inflammatory cytokines IL-6 and IL-8 in RA synoviocytes, having no effects on healthy synoviocytes (164).
Neural TRPs have the potential role to orchestrate inflammatory signals with neuropeptides released by neurons within the synovial microenvironment of the RA-inflamed joints. Various inflammatory mediators and neuropeptides sensitize and regulate the activation threshold of TRPs, causing inflammatory pain. In turn, TRPs located on neurons not only sense pain, but also boost inflammation by promoting the production of pro-inflammatory neuropeptides. These findings help us understand the neuro-inflammation mutually influenced pain aggravation, as well as inflammation persistence. Moreover, it also proves that TRPs can be used as a new target for clinical treatment of RA.
To sum up, evidence suggests the multiple functions of TRPs in RA pathogenesis, including joint swelling, neutrophil TEM and activation, angiogenesis and pain hyperalgesia. Ca2+ signals or potential induced by TRPs play a central role in transmitting inflammatory signals and amplifying inflammatory pain. TNF-α, VEGF and ROS are inflammatory molecules enriched in the joint microenvironment, acting on TRPs on ECs to increase the vascular permeability, which is the prerequisite for joint edema. TRPs-mediated Ca2+ influx into neutrophils plays an important role in their adhesion and TEM. Ca2+ signals caused by TRPs on neutrophils catalyze PADs activity and NETs formation, which enhancing the amount of auto-antigen in RA and leading to the formation of destructive autoantibodies ACPA. Though the relationship between TRPs and RA-FLS proliferation or apoptosis yet needs further investigation, TRPs can affect ECs migration and angiogenesis. Various inflammatory mediators and neuropeptides can sensitize and modulate the activation threshold of TRPs, resulting in the development of pain behaviors. TRPs can not only perceive cold and inflammatory stimuli but also communicate with pro-inflammation neuropeptides released by neurons within the RA-inflamed joints, resulting in persistent inflammation and pain hyperalgesia.
With scientists’ endeavors to explore TRPs over the years, many drugs targeting TRPs and antagonists interacting with inflammatory factors are in preclinical studies. Preclinical studies on TRPV1 as an analgesic have made fruitful progress. As TRPV1 agonists, capsaicin substances can cause TRPV1 desensitization of nerve endings when applied in a larger dose in the periphery, thus playing an analgesic role. Patches, creams, and sprays containing capsaicin have not only shown good analgesic effects in preclinical studies on neuralgia (165), joint pain (166), etc., but also have relatively few side effects. Antagonists targeting TRPV1 have significant analgesic effects, but blocking TRPV1 impairs the body’s perception of heat, leading to significant hyperthermia side effects (167). Therefore, the current direction of research is toward the development of antagonists that both reduce pain and less affect the body’s perception of temperature changes (168). At present, many synthetic antagonists for TRPA1 are gradually appearing, including Xanthine derivatives (HC-030031) and oxime derivative, which have been described in recent articles (169). From preclinical data, oxidative stress and activation of Schwann cell TRPA1 may serve as new targets for TRPA1 antagonists to relieve chronic pain in models of pain disorders. Studies shown that botulinum toxins (BoNTs) as a kind of neurotoxins had the ability to suppress the increase of neuropeptides (170). BoNTs acted as an analgesic by reducing TRPV1 expression (171). It not only presented analgesic effects in preclinical arthritis pain models, but also had very few side effects (172). Pre-clinical data on drugs derived from natural ingredients targeting TRPs have also made some progress. Tanshinone IIA (TIIA) is an important component of traditional Chinese medicine Danshen, which has achieved good curative effect in the treatment of nervous circulatory system. Studies have proved that it inhibited the release of inflammatory factors and reduced the expression of TRPV1 in the nervous system so as to reduce inflammation and anti-nociceptive pain in inflammatory arthritis (173). In addition, Paeoniflorin inhibited TRPV1 activation, thus exerting anti-inflammatory and analgesic effects without the hyperthermia side effects associated with traditional TRPV1 antagonists (174). S-(+)-dicentrine derived from camphor plants, reduced cold and mechanical pain in inflammatory mice by inhibiting TRPA1 activation (175). Not only has the anti-inflammatory and analgesic effect of traditional Chinese medicine (TCM), but other TCM methods have also shown a pleasing effect in relieving pain. The effect of electroacupuncture on pain relief has been widely recognized, but the specific mechanism is still unclear. Recent studies manifested that electroacupuncture reduced joint pain by interacting with TRPV1 (176). In addition, different acupuncture intensities and the distribution of nerve fibers where TRPV1 was located also vary the analgesic effect and the body parts relieved by electroacupuncture (177). Studies have shown inspiring efficacy in anti-inflammation and analgesic, although there are some side effects. This encourages us to continue to explore the mechanisms of TRPs about the unknown to better relieve the pain and suffering of RA patients.
MN reviewed the literature and wrote the initial draft. MN, PL, and LB designed the article and critically revised the manuscript. FZ and RC collected the data. All authors have approved the manuscript and agreed to the published version of the manuscript.
The authors declare that the review was in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. van de Sande MG, de Hair MJ, van der Leij C, Klarenbeek PL, Bos WH, Smith MD, et al. Different stages of rheumatoid arthritis: Features of the synovium in the preclinical phase. Ann Rheum Dis (2011) 70(5):772–7. doi: 10.1136/ard.2010.139527
2. van Oosterhout M, Bajema I, Levarht EW, Toes RE, Huizinga TW, van Laar JM. Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum (2008) 58(1):53–60. doi: 10.1002/art.23148
3. Myasoedova E, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. Is the incidence of rheumatoid arthritis rising?: Results from olmsted county, minnesota, 1955-2007. Arthritis Rheum (2010) 62(6):1576–82. doi: 10.1002/art.27425
4. Jager E, Murthy S, Schmidt C, Hahn M, Strobel S, Peters A, et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun (2020) 11(1):4243. doi: 10.1038/s41467-020-17749-6
5. Nadolni W, Immler R, Hoelting K, Fraticelli M, Ripphahn M, Rothmiller S, et al. TRPM7 kinase is essential for neutrophil recruitment and function via regulation of Akt/mTOR signaling. Front Immunol (2020) 11:606893. doi: 10.3389/fimmu.2020.606893
6. Wang CH, Rong MY, Wang L, Ren Z, Chen LN, Jia JF, et al. CD147 up-regulates calcium-induced chemotaxis, adhesion ability and invasiveness of human neutrophils via a TRPM-7-mediated mechanism. Rheumatol (Oxford) (2014) 53(12):2288–96. doi: 10.1093/rheumatology/keu260
7. Zhou R, Chen Y, Li S, Wei X, Hu W, Tang S, et al. TRPM7 channel inhibition attenuates rheumatoid arthritis articular chondrocyte ferroptosis by suppression of the PKCalpha-NOX4 axis. Redox Biol (2022) 55:102411. doi: 10.1016/j.redox.2022.102411
8. Ma G, Yang Y, Chen Y, Wei X, Ding J, Zhou RP, et al. Blockade of TRPM7 alleviates chondrocyte apoptosis and articular cartilage damage in the adjuvant arthritis rat model through regulation of the indian hedgehog signaling pathway. Front Pharmacol (2021) 12:655551. doi: 10.3389/fphar.2021.655551
9. Zhu S, Wang Y, Pan L, Yang S, Sun Y, Wang X, et al. Involvement of transient receptor potential melastatin-8 (TRPM8) in menthol-induced calcium entry, reactive oxygen species production and cell death in rheumatoid arthritis rat synovial fibroblasts. Eur J Pharmacol (2014) 725:1–9. doi: 10.1016/j.ejphar.2014.01.001
10. Russell FA, Fernandes ES, Courade JP, Keeble JE, Brain SD. Tumour necrosis factor alpha mediates transient receptor potential vanilloid 1-dependent bilateral thermal hyperalgesia with distinct peripheral roles of interleukin-1beta, protein kinase c and cyclooxygenase-2 signalling. Pain (2009) 142(3):264–74. doi: 10.1016/j.pain.2009.01.021
11. Lowin T, Straub RH. Cannabinoid-based drugs targeting CB1 and TRPV1, the sympathetic nervous system, and arthritis. Arthritis Res Ther (2015) 17(1):226. doi: 10.1186/s13075-015-0743-x
12. Thakore P, Earley S. Transient receptor potential channels and endothelial cell calcium signaling. Compr Physiol (2019) 9(3):1249–77. doi: 10.1002/cphy.c180034
13. Wu LJ, Sweet TB, Clapham DE, International Union of Basic and Clinical Pharmacology, LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev (2010) 62(3):381–404. doi: 10.1124/pr.110.002725
14. Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev (2014) 66(3):676–814. doi: 10.1124/pr.113.008268
15. Pan Z, Yang H, Reinach PS. Transient receptor potential (TRP) gene superfamily encoding cation channels. Hum Genomics (2011) 5(2):108–16. doi: 10.1186/1479-7364-5-2-108
16. Hardie RC. A brief history of trp: Commentary and personal perspective. Pflugers Arch (2011) 461(5):493–8. doi: 10.1007/s00424-011-0922-9
17. Kazandzhieva K, Mammadova-Bach E, Dietrich A, Gudermann T, Braun A. TRP channel function in platelets and megakaryocytes: Basic mechanisms and pathophysiological impact. Pharmacol Ther (2022) 237:108164. doi: 10.1016/j.pharmthera.2022.108164
18. Salvador B, Arranz A, Francisco S, Cordoba L, Punzon C, Llamas MA, et al. Modulation of endothelial function by toll like receptors. Pharmacol Res (2016) 108:46–56. doi: 10.1016/j.phrs.2016.03.038
19. Paria BC, Vogel SM, Ahmmed GU, Alamgir S, Shroff J, Malik AB, et al. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol (2004) 287(6):L1303–13. doi: 10.1152/ajplung.00240.2004
20. Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol (2010) 72:463–93. doi: 10.1146/annurev-physiol-021909-135833
21. Minshall RD, Malik AB. Transport across the endothelium: Regulation of endothelial permeability. Handb Exp Pharmacol (2006) 176(Pt 1):107–44. doi: 10.1007/3-540-32967-6_4
22. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev (2006) 86(1):279–367. doi: 10.1152/physrev.00012.2005
23. Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med (2015) 212(11):1883–99. doi: 10.1084/jem.20150353
24. Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res (2005) 97(9):853–63. doi: 10.1161/01.RES.0000187473.85419.3e
25. Bon RS, Beech DJ. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels – mirage or pot of gold? Br J Pharmacol (2013) 170(3):459–74. doi: 10.1111/bph.12274
26. Yip H, Chan WY, Leung PC, Kwan HY, Liu C, Huang Y, et al. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem Cell Biol (2004) 122(6):553–61. doi: 10.1007/s00418-004-0720-y
27. Ambudkar IS, de Souza LB, Ong HL. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium (2017) 63:33–9. doi: 10.1016/j.ceca.2016.12.009
28. Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, et al. The ca (2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated ca (2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol (2012) 81(4):510–26. doi: 10.1124/mol.111.074658
29. Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev (2015) 95(2):645–90. doi: 10.1152/physrev.00026.2014
30. Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell (2002) 108(5):595–8. doi: 10.1016/s0092-8674(02)00670-0
31. Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient receptor potential canonical (TRPC) channels: Then and now. Cells (2020) 9(9):1983. doi: 10.3390/cells9091983
32. Guo B, Zhao C, Zhang C, Xiao Y, Yan G, Liu L, et al. Elucidation of the anti-inflammatory mechanism of er miao san by integrative approach of network pharmacology and experimental verification. Pharmacol Res (2022) 175:106000. doi: 10.1016/j.phrs.2021.106000
33. Clanchy FIL, Williams RO. Ibudilast inhibits chemokine expression in rheumatoid arthritis synovial fibroblasts and exhibits immunomodulatory activity in experimental arthritis. Arthritis Rheumatol (2019) 71(5):703–11. doi: 10.1002/art.40787
34. Mahnashi MH, Jabbar Z, Alamgeer, Irfan HM, Asim MH, Akram M, et al. Venlafaxine demonstrated anti-arthritic activity possibly through down regulation of TNF-α, IL-6, IL-1β, and COX-2. Inflammopharmacology (2021) 29(5):1413–25. doi: 10.1007/s10787-021-00849-0
35. Yokota K, Sato K, Miyazaki T, Aizaki Y, Tanaka S, Sekikawa M, et al. Characterization and function of tumor necrosis factor and interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73(7):1145–54. doi: 10.1002/art.41666
36. Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, et al. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem (2003) 278(39):37195–203. doi: 10.1074/jbc.M304287200
37. Paria BC, Bair AM, Xue J, Yu Y, Malik AB, Tiruppathi C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J Biol Chem (2006) 281(30):20715–27. doi: 10.1074/jbc.M600722200
38. Luo L, Liu S, Zhang D, Wei F, Gu N, Zeng Y, et al. (CGA)-derived polypeptide (CGA(47-66)) inhibits TNF-α-induced vascular endothelial hyper-permeability through SOC-related ca (2+) signaling. Peptides (2020) 131:170297. doi: 10.1016/j.peptides.2020.170297
39. Solfietti L, Binello GB, Stella S, Bazzan M, Salierno M, Roccatello D. Thrombin generation assay: interactions between chronic inflammation and haemostasis in patients with autoimmune diseases. Clin Exp Rheumatol (2016) 34(5):925–8.
40. Sokolov AV, Acquasaliente L, Kostevich VA, Frasson R, Zakharova ET, Pontarollo G, et al. Thrombin inhibits the anti-myeloperoxidase and ferroxidase functions of ceruloplasmin: relevance in rheumatoid arthritis. Free Radic Biol Med (2015) 86:279–94. doi: 10.1016/j.freeradbiomed.2015.05.016
41. Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, et al. Protein kinase calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem (2004) 279(20):20941–9. doi: 10.1074/jbc.M313975200
42. Kini V, Chavez A, Mehta D. A new role for PTEN in regulating transient receptor potential canonical channel 6-mediated Ca2+ entry, endothelial permeability, and angiogenesis. J Biol Chem (2010) 285(43):33082–91. doi: 10.1074/jbc.M110.142034
43. Jakubczyk K, Dec K, Kaldunska J, Kawczuga D, Kochman J, Janda K. Reactive oxygen species - sources, functions, oxidative damage. Pol Merkur Lekarski (2020) 48(284):124–7.
44. Khojah HM, Ahmed S, Abdel-Rahman MS, Hamza AB. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic Biol Med (2016) 97:285–91. doi: 10.1016/j.freeradbiomed.2016.06.020
45. Hoffmann MH, Griffiths HR. The dual role of reactive oxygen species in autoimmune and inflammatory diseases: evidence from preclinical models. Free Radic Biol Med (2018) 125:62–71. doi: 10.1016/j.freeradbiomed.2018.03.016
46. Phull AR, Nasir B, Haq IU, Kim SJ. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem Biol Interact (2018) 281:121–36. doi: 10.1016/j.cbi.2017.12.024
47. da Fonseca LJS, Nunes-Souza V, Goulart MOF, Rabelo LA. Oxidative stress in rheumatoid arthritis: What the future might hold regarding novel biomarkers and add-on therapies. Oxid Med Cell Longev (2019) 2019:7536805. doi: 10.1155/2019/7536805
48. He P, Talukder MAH, Gao F. Oxidative stress and microvessel barrier dysfunction. Front Physiol (2020) 11:472. doi: 10.3389/fphys.2020.00472
49. Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res (2008) 102(3):347–55. doi: 10.1161/CIRCRESAHA.107.160176
50. Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med (1994) 180(1):341–6. doi: 10.1084/jem.180.1.341
51. Kim HR, Kim KW, Kim BM, Cho ML, Lee SH. The effect of vascular endothelial growth factor on osteoclastogenesis in rheumatoid arthritis. PloS One (2015) 10(4):e0124909. doi: 10.1371/journal.pone.0124909
52. Biscetti F, Flex A, Pecorini G, Angelini F, Arena V, Stigliano E, et al. The role of high-mobility group box protein 1 in collagen antibody-induced arthritis is dependent on vascular endothelial growth factor. Clin Exp Immunol (2016) 184(1):62–72. doi: 10.1111/cei.12758
53. Ikeda M, Hosoda Y, Hirose S, Okada Y, Ikeda E. Expression of vascular endothelial growth factor isoforms and their receptors flt-1, KDR, and neuropilin-1 in synovial tissues of rheumatoid arthritis. J Pathol (2000) 191(4):426–33. doi: 10.1002/1096-9896 (2000)9999:9999<::Aid-path649>3.0.Co;2-e
54. Chen CY, Su CM, Hsu CJ, Huang CC, Wang SW, Liu SC, et al. CCN1 promotes VEGF production in osteoblasts and induces endothelial progenitor cell angiogenesis by inhibiting miR-126 expression in rheumatoid arthritis. J Bone Miner Res (2017) 32(1):34–45. doi: 10.1002/jbmr.2926
55. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res (2007) 100(6):782–94. doi: 10.1161/01.RES.0000259593.07661.1e
56. Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation (2008) 15(7):605–14. doi: 10.1080/10739680802220323
57. Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res (2010) 87(2):262–71. doi: 10.1093/cvr/cvq105
58. Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, et al. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res (2005) 96(12):1282–90. doi: 10.1161/01.RES.0000171894.03801.03
59. Cheng HW, James AF, Foster RR, Hancox JC, Bates DO. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler Thromb Vasc Biol (2006) 26(8):1768–76. doi: 10.1161/01.ATV.0000231518.86795.0f
60. Immler R, Simon SI, Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur J Clin Invest (2018) 48(Suppl 2):e12964. doi: 10.1111/eci.12964
61. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
62. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med (2011) 17(11):1381–90. doi: 10.1038/nm.2514
63. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
64. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol (2011) 32(10):452–60. doi: 10.1016/j.it.2011.06.008
65. Arancibia SA, Beltran CJ, Aguirre IM, Silva P, Peralta AL, Malinarich F, et al. Toll-like receptors are key participants in innate immune responses. Biol Res (2007) 40(2):97–112. doi: 10.4067/s0716-97602007000200001
66. Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. Induction of cytokines and chemokines by toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol (2010) 30(1):53–67. doi: 10.1615/critrevimmunol.v30.i1.40
67. Lindemann O, Rossaint J, Najder K, Schimmelpfennig S, Hofschröer V, Wälte M, et al. Intravascular adhesion and recruitment of neutrophils in response to CXCL1 depends on their TRPC6 channels. J Mol Med (Berl) (2020) 98(3):349–60. doi: 10.1007/s00109-020-01872-4
68. Lindemann O, Strodthoff C, Horstmann M, Nielsen N, Jung F, Schimmelpfennig S, et al. TRPC1 regulates fMLP-stimulated migration and chemotaxis of neutrophil granulocytes. Biochim Biophys Acta (2015) 1853(9):2122–30. doi: 10.1016/j.bbamcr.2014.12.037
69. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
70. Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum (2005) 52(10):3004–14. doi: 10.1002/art.21301
71. Meeuwisse CM, van der Linden MP, Rullmann TA, Allaart CF, Nelissen R, Huizinga TW, et al. Identification of CXCL13 as a marker for rheumatoid arthritis outcome using an in silico model of the rheumatic joint. Arthritis Rheum (2011) 63(5):1265–73. doi: 10.1002/art.30273
72. Watanabe K, Penfold ME, Matsuda A, Ohyanagi N, Kaneko K, Miyabe Y, et al. Pathogenic role of CXCR7 in rheumatoid arthritis. Arthritis Rheum (2010) 62(11):3211–20. doi: 10.1002/art.27650
73. Yellin M, Paliienko I, Balanescu A, Ter-Vartanian S, Tseluyko V, Xu LA, et al. Randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum (2012) 64(6):1730–9. doi: 10.1002/art.34330
74. Lindemann O, Umlauf D, Frank S, Schimmelpfennig S, Bertrand J, Pap T, et al. TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J Immunol (2013) 190(11):5496–505. doi: 10.4049/jimmunol.1201502
75. Damann N, Owsianik G, Li S, Poll C, Nilius B. The calcium-conducting ion channel transient receptor potential canonical 6 is involved in macrophage inflammatory protein-2-induced migration of mouse neutrophils. Acta Physiol (Oxf) (2009) 195(1):3–11. doi: 10.1111/j.1748-1716.2008.01918.x
76. Wang CH, Dai JY, Wang L, Jia JF, Zheng ZH, Ding J, et al. Expression of CD147 (EMMPRIN) on neutrophils in rheumatoid arthritis enhances chemotaxis, matrix metalloproteinase production and invasiveness of synoviocytes. J Cell Mol Med (2011) 15(4):850–60. doi: 10.1111/j.1582-4934.2010.01084.x
77. Nakashima K, Hagiwara T, Ishigami A, Nagata S, Asaga H, Kuramoto M, et al. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3). J Biol Chem (1999) 274(39):27786–92. doi: 10.1074/jbc.274.39.27786
78. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5(178):178ra40. doi: 10.1126/scitranslmed.3005580
79. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol (2015) 67(12):3135–45. doi: 10.1002/art.39313
80. Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M. Structural basis for ca (2+)-induced activation of human PAD4. Nat Struct Mol Biol (2004) 11(8):777–83. doi: 10.1038/nsmb799
81. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature (1999) 397(6716):259–63. doi: 10.1038/16711
82. Brechard S, Melchior C, Plancon S, Schenten V, Tschirhart EJ. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium (2008) 44(5):492–506. doi: 10.1016/j.ceca.2008.03.002
83. Dang PM, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, et al. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J Clin Invest (2006) 116(7):2033–43. doi: 10.1172/jci27544
84. Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of neutrophil extracellular traps triggered by leishmania parasites. Sci Rep (2015) 5:18302. doi: 10.1038/srep18302
85. Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, et al. Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep (2018) 8(1):14166. doi: 10.1038/s41598-018-32576-y
86. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther (2014) 16(3):R122. doi: 10.1186/ar4579
87. Xia S, Wang L, Fu TM, Wu H. Mechanism of TRPM2 channel gating revealed by cryo-EM. FEBS J (2019) 286(17):3333–9. doi: 10.1111/febs.14939
88. Wang M, Li J, Dong S, Cai X, Simaiti A, Yang X, et al. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part Fibre Toxicol (2020) 17(1):23. doi: 10.1186/s12989-020-00353-3
89. Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell (2005) 18(1):61–9. doi: 10.1016/j.molcel.2005.02.033
90. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol (2015) 6:260–71. doi: 10.1016/j.redox.2015.08.010
91. Aleman OR, Mora N, Rosales C. The antibody receptor fc gamma receptor IIIb induces calcium entry via transient receptor potential melastatin 2 in human neutrophils. Front Immunol (2021) 12:657393. doi: 10.3389/fimmu.2021.657393
92. Dogru A, Naziroglu M, Cig B. Modulator role of infliximab and methotrexate through the transient receptor potential melastatin 2 (TRPM2) channel in neutrophils of patients with rheumatoid arthritis: A pilot study. Arch Med Sci (2019) 15(6):1415–24. doi: 10.5114/aoms.2018.79485
93. Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Fransès RE, Mapp PI, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatol (Oxford) (2010) 49(10):1852–61. doi: 10.1093/rheumatology/keq188
94. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med (2003) 9(6):669–76. doi: 10.1038/nm0603-669
95. Kanugula AK, Adapala RK, Midha P, Cappelli HC, Meszaros JG, Paruchuri S, et al. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J (2019) 33(1):195–203. doi: 10.1096/fj.201800509R
96. Tsai CH, Chen CJ, Gong CL, Liu SC, Chen PC, Huang CC, et al. CXCL13/CXCR5 axis facilitates endothelial progenitor cell homing and angiogenesis during rheumatoid arthritis progression. Cell Death Dis (2021) 12(9):846. doi: 10.1038/s41419-021-04136-2
97. Antigny F, Girardin N, Frieden M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J Biol Chem (2012) 287(8):5917–27. doi: 10.1074/jbc.M111.295733
98. Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng T, et al. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett (2009) 283(1):43–51. doi: 10.1016/j.canlet.2009.03.023
99. Korb A, Pavenstadt H, Pap T. Cell death in rheumatoid arthritis. Apoptosis (2009) 14(4):447–54. doi: 10.1007/s10495-009-0317-y
100. Elshabrawy HA, Chen Z, Volin MV, Ravella S, Virupannavar S, Shahrara S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis (2015) 18(4):433–48. doi: 10.1007/s10456-015-9477-2
101. Liu G, Xu D, He Y, Xu J, Huang S, Zhang W, et al. Inhibition of transient receptor potential canonical 6 attenuates fibroblast-like synoviocytes mediated synovial inflammation and joint destruction in rheumatoid arthritis. Clin Exp Rheumatol (2021) 39(1):115–24. doi: 10.55563/clinexprheumatol/rffvyg
102. Li X, Wang X, Wang Y, Li X, Huang C, Li J. Inhibition of transient receptor potential melastatin 7 (TRPM7) channel induces RA FLSs apoptosis through endoplasmic reticulum (ER) stress. Clin Rheumatol (2014) 33(11):1565–74. doi: 10.1007/s10067-014-2599-x
103. Lowin T, Bleck J, Schneider M, Pongratz G. Selective killing of proinflammatory synovial fibroblasts via activation of transient receptor potential ankyrin (TRPA1). Biochem Pharmacol (2018) 154:293–302. doi: 10.1016/j.bcp.2018.05.015
104. Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Coughlin KA, Kaczmarska MJ, Castaneda-Corral G, et al. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum (2012) 64(7):2223–32. doi: 10.1002/art.34385
105. Hwang SW, Oh U. Current concepts of nociception: nociceptive molecular sensors in sensory neurons. Curr Opin Anaesthesiol (2007) 20(5):427–34. doi: 10.1097/ACO.0b013e3282eff91c
106. Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, et al. TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci U.S.A. (2009) 106(4):1273–8. doi: 10.1073/pnas.0808487106
107. Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell (2003) 112(6):819–29. doi: 10.1016/s0092-8674(03)00158-2
108. da Costa DSM, Meotti FC, Andrade EL, Leal PC, Motta EM, Calixto JB. The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain (2010) 148(3):431–7. doi: 10.1016/j.pain.2009.12.002
109. Horváth Á, Tékus V, Boros M, Pozsgai G, Botz B, Borbély É, et al. Transient receptor potential ankyrin 1 (TRPA1) receptor is involved in chronic arthritis: in vivo study using TRPA1-deficient mice. Arthritis Res Ther (2016) 18:6. doi: 10.1186/s13075-015-0904-y
110. Dunham JP, Leith JL, Lumb BM, Donaldson LF. Transient receptor potential channel A1 and noxious cold responses in rat cutaneous nociceptors. Neuroscience (2010) 165(4):1412–9. doi: 10.1016/j.neuroscience.2009.11.065
111. Fernandes ES, Russell FA, Alawi KM, Sand C, Liang L, Salamon R, et al. Environmental cold exposure increases blood flow and affects pain sensitivity in the knee joints of CFA-induced arthritic mice in a TRPA1-dependent manner. Arthritis Res Ther (2016) 18:7. doi: 10.1186/s13075-015-0905-x
112. Aubdool AA, Graepel R, Kodji X, Alawi KM, Bodkin JV, Srivastava S, et al. TRPA1 is essential for the vascular response to environmental cold exposure. Nat Commun (2014) 5:5732. doi: 10.1038/ncomms6732
113. Aubdool AA, Kodji X, Abdul-Kader N, Heads R, Fernandes ES, Bevan S, et al. TRPA1 activation leads to neurogenic vasodilatation: Involvement of reactive oxygen nitrogen species in addition to CGRP and NO. Br J Pharmacol (2016) 173(15):2419–33. doi: 10.1111/bph.13519
114. Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature (2007) 448(7150):204–8. doi: 10.1038/nature05910
115. Colburn RW, Lubin ML, Stone DJ Jr., Wang Y, Lawrence D, D'Andrea MR, et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron (2007) 54(3):379–86. doi: 10.1016/j.neuron.2007.04.017
116. Knowlton WM, Palkar R, Lippoldt EK, McCoy DD, Baluch F, Chen J, et al. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J Neurosci (2013) 33(7):2837–48. doi: 10.1523/JNEUROSCI.1943-12.2013
117. Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest (2005) 115(9):2393–401. doi: 10.1172/JCI25437
118. Pan Y, Thapa D, Baldissera L Jr., Argunhan F, Aubdool AA, Brain SD. Relevance of TRPA1 and TRPM8 channels as vascular sensors of cold in the cutaneous microvasculature. Pflugers Arch (2018) 470(5):779–86. doi: 10.1007/s00424-017-2085-9
119. Lippoldt EK, Ongun S, Kusaka GK, McKemy DD. Inflammatory and neuropathic cold allodynia are selectively mediated by the neurotrophic factor receptor GFRalpha3. Proc Natl Acad Sci U.S.A. (2016) 113(16):4506–11. doi: 10.1073/pnas.1603294113
120. Lippoldt EK, Elmes RR, McCoy DD, Knowlton WM, McKemy DD. Artemin, a glial cell line-derived neurotrophic factor family member, induces TRPM8-dependent cold pain. J Neurosci (2013) 33(30):12543–52. doi: 10.1523/JNEUROSCI.5765-12.2013
121. Yamaki S, Chau A, Gonzales L, McKemy DD. Nociceptive afferent phenotyping reveals that transient receptor potential ankyrin 1 promotes cold pain through neurogenic inflammation upstream of the neurotrophic factor receptor GFRα3 and the menthol receptor transient receptor potential melastatin 8. Pain (2021) 162(2):609–18. doi: 10.1097/j.pain.0000000000002043
122. Schaible HG. Nociceptive neurons detect cytokines in arthritis. Arthritis Res Ther (2014) 16(5):470. doi: 10.1186/s13075-014-0470-8
123. Aghazadeh Tabrizi M, Baraldi PG, Baraldi S, Gessi S, Merighi S, Borea PA. Medicinal chemistry, pharmacology, and clinical implications of TRPV1 receptor antagonists. Med Res Rev (2017) 37(4):936–83. doi: 10.1002/med.21427
124. Kanai Y, Hara T, Imai A, Sakakibara A. Differential involvement of TRPV1 receptors at the central and peripheral nerves in CFA-induced mechanical and thermal hyperalgesia. J Pharm Pharmacol (2007) 59(5):733–8. doi: 10.1211/jpp.59.5.0015
125. Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase c activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol (2001) 534(Pt 3):813–25. doi: 10.1111/j.1469-7793.2001.00813.x
126. Yu L, Yang F, Luo H, Liu FY, Han JS, Xing GG, et al. The role of TRPV1 in different subtypes of dorsal root ganglion neurons in rat chronic inflammatory nociception induced by complete freund's adjuvant. Mol Pain (2008) 4:61. doi: 10.1186/1744-8069-4-61
127. Amaya F, Oh-hashi K, Naruse Y, Iijima N, Ueda M, Shimosato G, et al. Local inflammation increases vanilloid receptor 1 expression within distinct subgroups of DRG neurons. Brain Res (2003) 963(1-2):190–6. doi: 10.1016/s0006-8993(02)03972-0
128. Carlton SM, Coggeshall RE. Peripheral capsaicin receptors increase in the inflamed rat hindpaw: a possible mechanism for peripheral sensitization. Neurosci Lett (2001) 310(1):53–6. doi: 10.1016/s0304-3940(01)02093-6
129. Roskoski R Jr.. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun (2005) 331(1):1–14. doi: 10.1016/j.bbrc.2005.03.012
130. Yen CM, Wu TC, Hsieh CL, Huang YW, Lin YW. Distal electroacupuncture at the LI4 acupoint reduces CFA-induced inflammatory pain via the brain TRPV1 signaling pathway. Int J Mol Sci (2019) 20(18):4471. doi: 10.3390/ijms20184471
131. Liu J, Du J, Yang Y, Wang Y. Phosphorylation of TRPV1 by cyclin-dependent kinase 5 promotes TRPV1 surface localization, leading to inflammatory thermal hyperalgesia. Exp Neurol (2015) 273:253–62. doi: 10.1016/j.expneurol.2015.09.005
132. Shin SM, Itson-Zoske B, Cai Y, Qiu C, Pan B, Stucky CL, et al. Satellite glial cells in sensory ganglia express functional transient receptor potential ankyrin 1 that is sensitized in neuropathic and inflammatory pain. Mol Pain (2020) 16:1744806920925425. doi: 10.1177/1744806920925425
133. Fernandes ES, Russell FA, Spina D, McDougall JJ, Graepel R, Gentry C, et al. A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor α-induced inflammatory hyperalgesia and freund's complete adjuvant-induced monarthritis. Arthritis Rheum (2011) 63(3):819–29. doi: 10.1002/art.30150
134. Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, et al. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol (2005) 493(4):596–606. doi: 10.1002/cne.20794
135. Huang Y, Chen SR, Chen H, Pan HL. Endogenous transient receptor potential ankyrin 1 and vanilloid 1 activity potentiates glutamatergic input to spinal lamina i neurons in inflammatory pain. J Neurochem (2019) 149(3):381–98. doi: 10.1111/jnc.14677
136. Weng HJ, Patel KN, Jeske NA, Bierbower SM, Zou W, Tiwari V, et al. Tmem100 is a regulator of TRPA1-TRPV1 complex and contributes to persistent pain. Neuron (2015) 85(4):833–46. doi: 10.1016/j.neuron.2014.12.065
137. Patil MJ, Salas M, Bialuhin S, Boyd JT, Jeske NA, Akopian AN. Sensitization of small-diameter sensory neurons is controlled by TRPV1 and TRPA1 association. FASEB J (2020) 34(1):287–302. doi: 10.1096/fj.201902026R
138. Mailhot B, Christin M, Tessandier N, Sotoudeh C, Bretheau F, Turmel R, et al. Neuronal interleukin-1 receptors mediate pain in chronic inflammatory diseases. J Exp Med (2020) 217(9):e20191430. doi: 10.1084/jem.20191430
139. Ebbinghaus M, Uhlig B, Richter F, von Banchet GS, Gajda M, Brauer R, et al. The role of interleukin-1beta in arthritic pain: Main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum (2012) 64(12):3897–907. doi: 10.1002/art.34675
140. Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, et al. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci (2008) 28(38):9486–94. doi: 10.1523/jneurosci.1857-08.2008
141. Westlund KN, Kochukov MY, Lu Y, McNearney TA. Impact of central and peripheral TRPV1 and ROS levels on proinflammatory mediators and nociceptive behavior. Mol Pain (2010) 6:46. doi: 10.1186/1744-8069-6-46
142. Ding R, Jiang H, Sun B, Wu X, Li W, Zhu S, et al. Advanced oxidation protein products sensitized the transient receptor potential vanilloid 1 via NADPH oxidase 1 and 4 to cause mechanical hyperalgesia. Redox Biol (2016) 10:1–11. doi: 10.1016/j.redox.2016.09.004
143. von Banchet GS, Richter J, Huckel M, Rose C, Brauer R, Schaible HG. Fibroblast-like synovial cells from normal and inflamed knee joints differently affect the expression of pain-related receptors in sensory neurones: A co-culture study. Arthritis Res Ther (2007) 9(1):R6. doi: 10.1186/ar2112
144. Pereira I, Mendes SJ, Pereira DM, Muniz TF, Colares VL, Monteiro CR, et al. Transient receptor potential ankyrin 1 channel expression on peripheral blood leukocytes from rheumatoid arthritic patients and correlation with pain and disability. Front Pharmacol (2017) 8:53. doi: 10.3389/fphar.2017.00053
145. Jiang H, Shen X, Chen Z, Liu F, Wang T, Xie Y, et al. Nociceptive neuronal fc-gamma receptor i is involved in IgG immune complex induced pain in the rat. Brain Behav Immun (2017) 62:351–61. doi: 10.1016/j.bbi.2017.03.001
146. Qu L, Li Y, Pan X, Zhang P, LaMotte RH, Ma C. Transient receptor potential canonical 3 (TRPC3) is required for IgG immune complex-induced excitation of the rat dorsal root ganglion neurons. J Neurosci (2012) 32(28):9554–62. doi: 10.1523/JNEUROSCI.6355-11.2012
147. Sadler KE, Moehring F, Shiers SI, Laskowski LJ, Mikesell AR, Plautz ZR, et al. Transient receptor potential canonical 5 mediates inflammatory mechanical and spontaneous pain in mice. Sci Transl Med (2021) 13(595):eabd7702. doi: 10.1126/scitranslmed.abd7702
148. Alawi KM, Russell FA, Aubdool AA, Srivastava S, Riffo-Vasquez Y, Baldissera L C.OMMAJ.R.X.X.X, et al. Transient receptor potential canonical 5 (TRPC5) protects against pain and vascular inflammation in arthritis and joint inflammation. Ann Rheum Dis (2017) 76(1):252–60. doi: 10.1136/annrheumdis-2015-208886
149. Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, et al. Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem (2015) 290(22):13875–87. doi: 10.1074/jbc.M115.642736
150. Segond von Banchet G, Boettger MK, König C, Iwakura Y, Bräuer R, Schaible HG. Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1 receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia. Mol Cell Neurosci (2013) 52:152–60. doi: 10.1016/j.mcn.2012.11.006
151. Oehler B, Kistner K, Martin C, Schiller J, Mayer R, Mohammadi M, et al. Inflammatory pain control by blocking oxidized phospholipid-mediated TRP channel activation. Sci Rep (2017) 7(1):5447. doi: 10.1038/s41598-017-05348-3
152. Yin N, Gao Q, Tao W, Chen J, Bi J, Ding F, et al. Paeoniflorin relieves LPS-induced inflammatory pain in mice by inhibiting NLRP3 inflammasome activation via transient receptor potential vanilloid 1. J Leukoc Biol (2020) 108(1):229–41. doi: 10.1002/JLB.3MA0220-355R
153. Fattori V, Zaninelli TH, Ferraz CR, Brasil-Silva L, Borghi SM, Cunha JM, et al. Maresin 2 is an analgesic specialized pro-resolution lipid mediator in mice by inhibiting neutrophil and monocyte recruitment, nociceptor neuron TRPV1 and TRPA1 activation, and CGRP release. Neuropharmacology (2022) 216:109189. doi: 10.1016/j.neuropharm.2022.109189
154. Okubo M, Yamanaka H, Kobayashi K, Fukuoka T, Dai Y, Noguchi K. Expression of leukotriene receptors in the rat dorsal root ganglion and the effects on pain behaviors. Mol Pain (2010) 6:57. doi: 10.1186/1744-8069-6-57
155. Keeble J, Russell F, Curtis B, Starr A, Pinter E, Brain SD. Involvement of transient receptor potential vanilloid 1 in the vascular and hyperalgesic components of joint inflammation. Arthritis Rheum (2005) 52(10):3248–56. doi: 10.1002/art.21297
156. Schaible HG, von Banchet GS, Boettger MK, Brauer R, Gajda M, Richter F, et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Ann N Y Acad Sci (2010) 1193:60–9. doi: 10.1111/j.1749-6632.2009.05301.x
157. Fernandes ES, Russell FA, Spina D, McDougall JJ, Graepel R, Gentry C, et al. A distinct role for transient receptor potential ankyrin 1, in addition to transient receptor potential vanilloid 1, in tumor necrosis factor alpha-induced inflammatory hyperalgesia and freund's complete adjuvant-induced monarthritis. Arthritis Rheum (2011) 63(3):819–29. doi: 10.1002/art.30150
158. Ishida K, Kawamata T, Tanaka S, Shindo T, Kawamata M. Calcitonin gene-related peptide is involved in inflammatory pain but not in postoperative pain. Anesthesiology (2014) 121(5):1068–79. doi: 10.1097/aln.0000000000000364
159. Fattori V, Pinho-Ribeiro FA, Staurengo-Ferrari L, Borghi SM, Rossaneis AC, Casagrande R, et al. The specialised pro-resolving lipid mediator maresin 1 reduces inflammatory pain with a long-lasting analgesic effect. Br J Pharmacol (2019) 176(11):1728–44. doi: 10.1111/bph.14647
160. Nieto FR, Clark AK, Grist J, Chapman V, Malcangio M. Calcitonin gene-related peptide-expressing sensory neurons and spinal microglial reactivity contribute to pain states in collagen-induced arthritis. Arthritis Rheumatol (2015) 67(6):1668–77. doi: 10.1002/art.39082
161. Nakanishi M, Hata K, Nagayama T, Sakurai T, Nishisho T, Wakabayashi H, et al. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CaMK-CREB cascade: A potential mechanism of inflammatory pain. Mol Biol Cell (2010) 21(15):2568–77. doi: 10.1091/mbc.E10-01-0049
162. Hernanz A, Medina S, de Miguel E, Martín-Mola E. Effect of calcitonin gene-related peptide, neuropeptide y, substance p, and vasoactive intestinal peptide on interleukin-1beta, interleukin-6 and tumor necrosis factor-alpha production by peripheral whole blood cells from rheumatoid arthritis and osteoarthritis patients. Regul Pept (2003) 115(1):19–24. doi: 10.1016/s0167-0115(03)00127-7
163. Raap T, Justen HP, Miller LE, Cutolo M, Scholmerich J, Straub RH. Neurotransmitter modulation of interleukin 6 (IL-6) and IL-8 secretion of synovial fibroblasts in patients with rheumatoid arthritis compared to osteoarthritis. J Rheumatol (2000) 27(11):2558–65.
164. Terenzi R, Romano E, Manetti M, Peruzzi F, Nacci F, Matucci-Cerinic M, et al. Neuropeptides activate TRPV1 in rheumatoid arthritis fibroblast-like synoviocytes and foster IL-6 and IL-8 production. Ann Rheum Dis (2013) 72(6):1107–9. doi: 10.1136/annrheumdis-2012-202846
165. Clifford DB, Simpson DM, Brown S, Moyle G, Brew BJ, Conway B, et al. A randomized, double-blind, controlled study of NGX-4010, a capsaicin 8% dermal patch, for the treatment of painful HIV-associated distal sensory polyneuropathy. J Acquir Immune Defic Syndr (2012) 59(2):126–33. doi: 10.1097/QAI.0b013e31823e31f7
166. Schnitzer TJ, Pelletier JP, Haselwood DM, Ellison WT, Ervin JE, Gordon RD, et al. Civamide cream 0.075% in patients with osteoarthritis of the knee: a 12-week randomized controlled clinical trial with a longterm extension. J Rheumatol (2012) 39(3):610–20. doi: 10.3899/jrheum.110192
167. Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain (2008) 136(1-2):202–10. doi: 10.1016/j.pain.2008.01.024
168. Reilly RM, McDonald HA, Puttfarcken PS, Joshi SK, Lewis L, Pai M, et al. Pharmacology of modality-specific transient receptor potential vanilloid-1 antagonists that do not alter body temperature. J Pharmacol Exp Ther (2012) 342(2):416–28. doi: 10.1124/jpet.111.190314
169. Souza Monteiro de Araujo D, Nassini R, Geppetti P, De Logu F. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin Ther Targets (2020) 24(10):997–1008. doi: 10.1080/14728222.2020.1815191
170. Zhou K, Luo W, Liu T, Ni Y, Qin Z. Neurotoxins acting at synaptic sites: A brief review on mechanisms and clinical applications. Toxins (Basel) (2022) 15(1):18. doi: 10.3390/toxins15010018
171. Fan C, Chu X, Wang L, Shi H, Li T. Botulinum toxin type a reduces TRPV1 expression in the dorsal root ganglion in rats with adjuvant-arthritis pain. Toxicon (2017) 133:116–22. doi: 10.1016/j.toxicon.2017.05.001
172. Blanshan N, Krug H. The use of botulinum toxin for the treatment of chronic joint pain: Clinical and experimental evidence. Toxins (Basel) (2020) 12(5):314. doi: 10.3390/toxins12050314
173. Sun S, Yin Y, Yin X, Cao F, Luo D, Zhang T, et al. Anti-nociceptive effects of tanshinone IIA (TIIA) in a rat model of complete freund's adjuvant (CFA)-induced inflammatory pain. Brain Res Bull (2012) 88(6):581–8. doi: 10.1016/j.brainresbull.2012.06.002
174. Ruan Y, Ling J, Ye F, Cheng N, Wu F, Tang Z, et al. Paeoniflorin alleviates CFA-induced inflammatory pain by inhibiting TRPV1 and succinate/SUCNR1-HIF-1alpha/NLPR3 pathway. Int Immunopharmacol (2021) 101(Pt B):108364. doi: 10.1016/j.intimp.2021.108364
175. Montrucchio DP, Cordova MM, Santos AR. Plant derived aporphinic alkaloid s-(+)-dicentrine induces antinociceptive effect in both acute and chronic inflammatory pain models: evidence for a role of TRPA1 channels. PloS One (2013) 8(7):e67730. doi: 10.1371/journal.pone.0067730
176. Inprasit C, Lin YW. TRPV1 responses in the cerebellum lobules v, VIa and VII using electroacupuncture treatment for inflammatory hyperalgesia in murine model. Int J Mol Sci (2020) 21(9):3312. doi: 10.3390/ijms21093312
Keywords: rheumatoid arthritis, transient receptor potential channels, inflammation, calcium signal, neuro-inflammation, inflammatory pain
Citation: Niu M, Zhao F, Chen R, Li P and Bi L (2023) The transient receptor potential channels in rheumatoid arthritis: Need to pay more attention. Front. Immunol. 14:1127277. doi: 10.3389/fimmu.2023.1127277
Received: 19 December 2022; Accepted: 06 February 2023;
Published: 01 March 2023.
Edited by:
Ping-Heng Tan, Chi Mei Medical Center, TaiwanReviewed by:
Chenhui Wang, Huazhong University of Science and Technology, ChinaCopyright © 2023 Niu, Zhao, Chen, Li and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Li, bGlfcGluZ0BqbHUuZWR1LmNu; Liqi Bi, YmlscUBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.