
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1126823
This article is part of the Research Topic Cardiometabolic diseases and inflammatory responses View all 16 articles
 Yanan Wang1,2,3†
Yanan Wang1,2,3† Dan Fang1,2,3†
Dan Fang1,2,3† Qinzhi Yang1,2,3†Jingcan You1,2,3
Qinzhi Yang1,2,3†Jingcan You1,2,3 Liqun Wang1,2,3Jianbo Wu1,2,3
Liqun Wang1,2,3Jianbo Wu1,2,3 Min Zeng4*
Min Zeng4* Mao Luo1,2,3,4*
Mao Luo1,2,3,4*Atherosclerosis is an early pathological basis of numerous cardiovascular events that result in death or disability. Recent studies have described PCSK9 as a novel target for the treatment of atherosclerosis; PCSK9 is capable of degrading LDLR on the surface of hepatocytes through the regulation of lipid metabolism, and it can function as a novel inflammatory modulator in atherosclerosis. Inflammasomes are important intracellular multiprotein complexes that promote the inflammatory response in atherosclerosis. Among inflammasomes, the NLRP3 inflammasome is particularly notable because of its important role in the development of atherosclerotic disease. After activation, NLRP3 forms a complex with ASC and pro-caspase-1, converting pro-caspase-1 into activated caspase-1, which may trigger the release of IL-1β and IL-18 and contribute to the inflammatory response. Several recent studies have indicated that there may be interactions between PCSK9 and the NLRP3 inflammasome, which may contribute to the inflammatory response that drives atherosclerosis development and progression. On the one hand, the NLRP3 inflammasome plays an important role via IL-1β in regulating PCSK9 secretion. On the other hand, PCSK9 regulates caspase-1-dependent pyroptosis by initiating mtDNA damage and activating NLRP3 inflammasome signaling. This paper reviews the mechanisms underlying PCSK9 and NLRP3 inflammasome activation in the context of atherosclerosis. Furthermore, we describe the current understanding of the specific molecular mechanism underlying the interactions between PCSK9 and NLRP3 inflammasome signaling as well as the drug repositioning events that influence vascular cells and exert beneficial antiatherosclerotic effects. This review may provide a new therapeutic direction for the effective prevention and treatment of atherosclerosis in the clinic.
Inflammation is an important driver of atherosclerosis, which is an early pathological basis of cardiovascular disease (1). Atherosclerosis is characterized by the excessive accumulation of lipids, extracellular matrix, and cholesterol-laden macrophages under the arterial endothelium, resulting in the formation of atherosclerotic plaques (2, 3). LDL-cholesterol (C) is considered to be a major risk factor for the development of atherosclerotic diseases (4). The mechanism by which proprotein convertase subtilisin/kexin type 9 (PCSK9) regulates LDLR degradation involves both extracellular and intracellular pathways. On the one hand, extracellular PCSK9 can act as a companion protein and bind to LDL-receptor (R) on the cell surface, thus causing the formation of a complex and directing LDLR to lysosomes where the PCSK9/LDLR complex is degraded, thereby promoting further LDL-C accumulation (5). On the other hand, intracellular PCSK9 can also directly bind with LDLR in the Golgi network to induce the lysosomal degradation of LDLR (6). PCSK9 is closely associated with the indirect regulation of lipid metabolism (5) and participates in the direct regulation of atherosclerosis via the accumulation of foam cells and inflammatory mediators as well as apoptosis in vascular walls (7). If PCSK9 inhibition is reduced, there is more PCSK9, more degraded LDLRs and therefore an increase in LDL levels (8). Persistent accumulation of LDL results in not only the formation of foam cells but also the chronic amplification of inflammatory responses, which are major causes of plaque rupture and vascular thrombosis (9). NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasomes are classical receptors of intracellular innate immunity that closely regulate inflammatory responses (10). NLRP3 inflammasome activation is a powerful mediator of the inflammatory response via caspase-1 activation (11). The NLRP3 inflammasome can activate caspase-1 and cleave pro-IL-1β and IL-18 to generate IL-1β and IL-18, which further promote inflammatory responses and play crucial roles in regulating atherosclerotic lesions (12).
Recently, several studies have reported that PCSK9 activates the NLRP3 inflammasome signaling pathway and the associated inflammation (13–15). Conversely, the NLRP3 inflammasome signaling pathway can regulate PCSK9 secretion (16). However, the regulatory mechanisms are still not fully understood, especially in the context of atherosclerosis. In this review, we summarize the current findings related to the interactions between PCSK9 and the NLRP3 inflammasome in atherosclerosis. Furthermore, we describe the specific molecular mechanism underlying the PCSK9 and NLRP3 inflammasome signaling pathway in atherosclerosis and cells related to inflammation, including vascular smooth muscle cells (VSMCs), endothelial cells (ECs) and macrophages (MФ). These findings may provide a novel theoretical basis and important targets for clinical application in the treatment of atherosclerosis.
In 2003, Abifadel M et al. first reported that PCSK9 is highly expressed in the liver and contributes to cholesterol homeostasis (17). Similar to other proprotein convertases, PCSK9 is synthesized as a soluble proenzyme and undergoes autocatalytic cleavage in the endoplasmic reticulum (ER) at residue 152 between its prodomain and catalytic domains (18). Then, a stable heterodimer consisting of a prodomain of approximately 14 kDa and a mature fragment of approximately 57 kDa is formed, as shown in Figure 1. After transportation to the Golgi apparatus, the protein is modified by acetylation and subsequently secreted (18). PCSK9 is mainly secreted by the liver, small intestine, kidney, skin, and cerebrospinal fluid, while PCSK9 in blood is almost exclusively secreted from the liver (19). PCSK9 and LDLR form a tight complex and are targeted to lysosomes for degradation, thereby reducing the level of LDLR on the surface of hepatocytes and decreasing hepatic clearance of LDL-C (20).
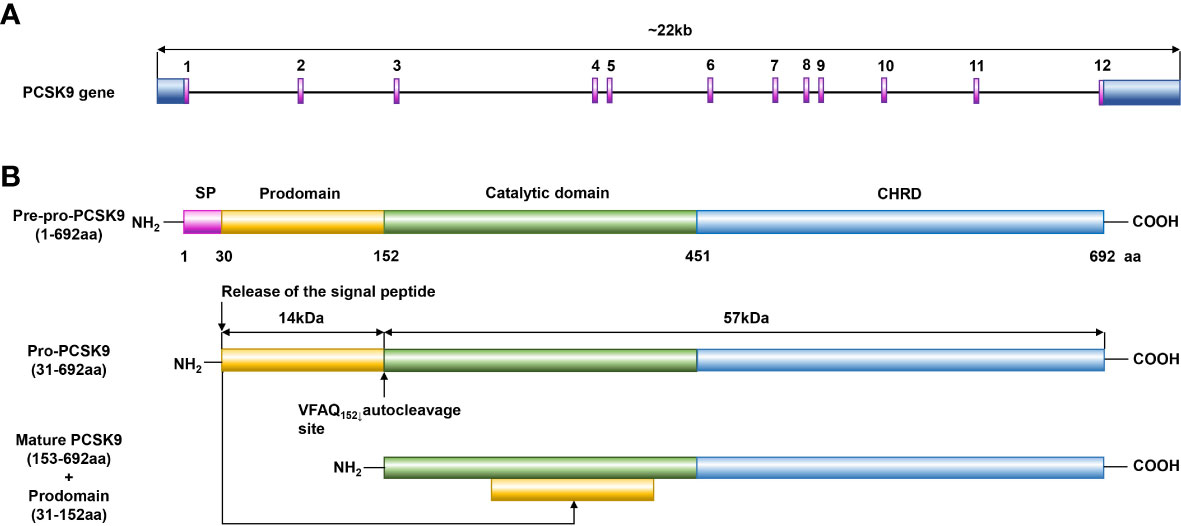
Figure 1 Structure diagram of PCSK9. (A) PCSK9 is the ninth member of the subtilisin serine protease family, and its gene is located on human chromosome 1p32.3, is 22 kb in length, includes 12 exons, and encodes 692 amino acids. (B) PCSK9 consists of a signal peptide (residues 1–30), a prodomain (residues 31–152), a catalytic domain (residues 153–451), and a C-terminal domain (residues 452–692). Similar to other proprotein convertases, PCSK9 is synthesized as a soluble proenzyme and undergoes autocatalytic cleavage in the ER at residue 152 between its prodomain and catalytic domains. Then, a stable heterodimer consisting of a prodomain of approximately 14 kDa and a mature fragment of approximately 57 kDa is formed, and after transportation to the Golgi apparatus, the protein is modified by acetylation and subsequently secreted.
Through its function of degrading the LDLR, PCSK9 is a key player in lipid metabolism by regulating the LDL level in blood responsible for hypercholesterolemia, which is associated with the risk of atherosclerosis (21). Overexpression of PCSK9 was found to inversely downregulate the expression of LDLR and reduce the clearance of cholesterol from plasma, so inhibiting PCSK9 overexpression is significant in the prevention and treatment of atherosclerosis. Moreover, Tavori et al. found that PCSK9 expression is involved in regulating the size and composition of atherosclerotic plaques and significantly enlarges atherosclerotic lesion areas (8). Giunzioni et al. further showed that PCSK9 directly increases the inflammation of atherosclerotic lesion in an LDLR-dependent but cholesterol-independent manner, suggesting that therapeutic PCSK9 inhibition may have vascular benefits that are secondary to reductions in the LDL levels (22). Overall, PCSK9 is expressed at high levels at the sites of atherosclerotic lesions, particularly in intimal plaques (23). PCSK9 inhibition has emerged as a potential novel therapeutic approach to treat hypercholesterolemia and associated diseases, such as atherosclerosis.
It has been clearly established that PCSK9 plays a crucial role in the development of atherosclerosis. While the liver is the major source of circulating PCSK9, PCSK9 is also expressed in various cellular components of atherosclerotic plaques, including monocytes/MФs, VSMCs, and ECs, that directly participate in the progression of atherosclerotic lesions by exacerbating vascular inflammation (Figure 2 and Table S1) (24–31). Moreover, the secretion of PCSK9 in ECs, SMCs and MФs were strongly induced by LPS treatment with or without ATP compared with the control (23, 32). PCSK9 secreted by VSMCs, which express more PCSK9 than ECs in a paracrine manner, downregulating LDLR expression on the cell surface of MФs and preventing the formation of foam cells, thus reducing atherosclerosis progression. However, native LDL molecules are not the major source of cholesterol accumulation in macrophages and LDLR is not the main receptor for lipoprotein uptake in cells within the atherosclerotic plaque (5). Scavenger receptors expressed by MФ in vessel walls take up LDL-cholesterol, resulting in the transformation of MФs into foam cells that secrete a large amount of proinflammatory cytokines and mature into activated MФ, contributing to the acceleration of arterial inflammation and atherosclerosis (33, 34). Cluster of differentiation 36(CD36) is a scavenger receptor that is highly expressed in macrophages and macrophage-derived foam cells in atherosclerotic plaques. Levy et al. found that gain-of-function PCSK9 mutants could significantly increase CD36 expression (35). In addition, Ding et al. also showed that PCSK9 can enhance the uptake of ox-LDL in MФs by targeting lectin-like ox-LDL receptor 1(LOX-1) (23). Moreover, Giunzioni et al. found that PCSK9 exerts a direct effect on the monocyte/MФ phenotype during the progression of atherosclerosis (22). Given that LDL-R-related protein 1(LRP1) deficiency induces nuclear factor kappa-B (NF-κB) activation and promotes a proinflammatory phenotype in MФs, some studies have suggested that PCSK9 induces macrophage inflammation by targeting LRP1 for degradation. Tang et al. found that PCSK9 overexpression upregulates toll-like receptors 4 (TLR4) expression and promotes inhibitor kappa B alpha (IκBα) degradation, p-IκBα expression, and NF-κB nuclear translocation (36). The findings show that PCSK9 enhances inflammatory cytokine secretion through TLR4/NF-κB pathway activation in MФs. Another mechanism underlying the PCSK9-mediated promotion of inflammation might involve the PCSK9-induced LDLR-independent degradation of apolipoprotein E receptor 2 (ApoER2) (37), which plays a critical role in maintaining the anti-inflammatory phenotype of MФs; these results indicate that PCSK9 can regulate macrophage inflammation through inflammasome activation.
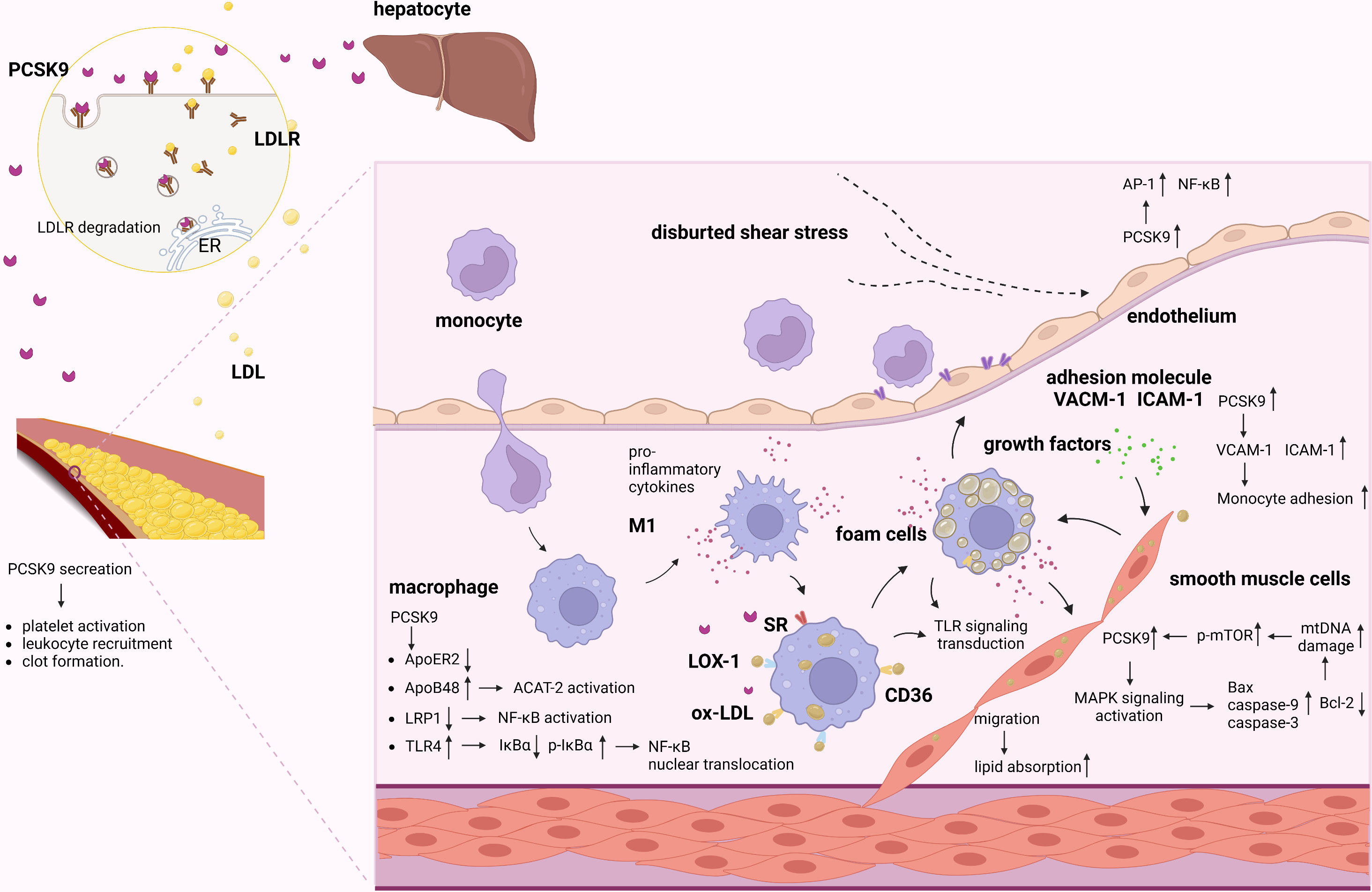
Figure 2 Role of PCSK9 in atherosclerosis. PCSK9 and LDLR form a complex and are targeted to lysosomes for degradation. Secreted PCSK9 by the liver reduces the level of LDLR not only on hepatocytes surface but also on other cells. PCSK9 promotes platelet activation, leukocyte recruitment and clot formation both in the plasma and in vascular cells. Proinflammatory stimuli activate ECs and increase adhesion molecules, promoting the adhesion and migration of monocytes. Migrated monocytes and ox-LDL-C accumulate in the subintimal region. Meanwhile, migrated monocytes transforms into MФs. In inflammatory milieu, PCSK9 increases scavenger receptors to increase ox-LDL uptake. In addition, PCSK9 mediated inflammation via activating LRP1, TLR4/NF-κB pathway. PCSK9 increased cholesterol synthesis by activating ACAT-2/ApoB48. Furthermore, migrated SMCs can absorb lipid and exacerbate the inflammatory responses and myogenic foam cell formation caused by the effects of growth factors and proinflammatory cytokines. In VSMCs, PCSK9 is enhanced by mtDNA damage via the upregulation of p-mTOR. In turn, PCSK9 increases mtDNA damage by regulating the apoptotic proteins via MAPK signaling pathway. Besides, low shear stress increases PCSK9, which induces EC dysfunction and upregulates AP-1 and NF-κB.
VSMCs are important for the maintenance of vascular homeostasis and play a key role in atherosclerosis (38). PCSK9 is reported to be expressed in VSMCs in atherosclerotic plaques (39), and its expression is markedly increased by inflammatory stimuli, indicating that proinflammatory factors that are associated with atherosclerosis stimulate PCSK9 expression in VSMCs and suggesting a critical role for PCSK9 in the development of atherosclerotic lesions (40). For example, migration of SMCs from the tunica media of the vessel wall can lead to increased lipid absorption and exacerbate the inflammatory responses and myogenic foam cell formation caused by the effects of growth factors and proinflammatory cytokines on the fatty streak (41). Previous studies have shown that there is a positive feedback interplay between VSMC-derived PCSK9 and mitochondrial DNA (mtDNA) damage in the proinflammatory milieu that involves mitochondrial reactive oxygen species (mtROS); this feedback loop results in inflammation, oxidative stress, and apoptosis, which directly contribute to atherosclerosis (42). In VSMCs, PCSK9, which increases mtDNA damage by upregulating the expression of the pro-apoptotic proteins Bax, caspase-9, and caspase-3 and downregulating the expression of Bcl-2, is correlated with the activation of the MAPK signaling pathway. Moreover, PCSK9 expression is enhanced by mtDNA damage via the upregulation of p-mTOR expression in a dose-dependent manner, while inhibition of PCSK9 reduces mammalian target of rapamycin (mTOR) phosphorylation and decreases VSMC autophagy.
Vascular endothelial dysfunction and the inflammatory response contribute to the initiation and progression of atherosclerosis (43). Landlinger et al. found that treatment of mice with AT04A obviously suppressed the inflammatory response and activated ECs and stimulated monocyte/macrophage migration, suggesting that PCSK9 inhibition also suppresses endothelial inflammation (44). Although PCSK9 expression and secretion by ECs may be lower than that by VSMCs, the effects of PCSK9 on EC biology in atherosclerosis cannot be ignored. The development of atherosclerosis is caused by a variety of factors, including biochemical factors and physical factors. Hemodynamic shear stress regulates EC functions and influences the pathobiology of atherosclerosis (45). Laminar shear stress is thought to exert atheroprotective effects (46). Conversely, low shear stress significantly increases PCSK9 expression, induces EC dysfunction, and upregulates the expression of transcription factors, including AP-1 and NF-κB, that promote pro-oxidant and proinflammatory states (40, 46); these results indicate that PCSK9 may play an important role in the EC inflammation that is induced by low shear stress.
Hosts regulate the release of intracellular inflammatory mediators and the initiation of inflammatory responses mainly through two types of pattern recognition receptors (PRRs), namely, membrane-bound Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (47, 48); these receptors recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) and thus promote the maturation and release of tightly regulated, highly inflammatory cytokines. NLRP3 is a recognized PRR, and it is a cytoplasmic receptor that responds to danger signals and can be activated to form the NLRP3 inflammasome (49).
The NLRP3 inflammasome is a multimolecular protein complex that comprises the NOD-like receptor NLRP3, the adaptor ASC and the effector pro-caspase-1, which play fundamental roles in inflammation (50). Among these components, the NOD-like receptor NLRP3 contains a leucine-rich repeat (LRR) domain, nucleotide triphosphatase (NACHT) domain, and pyrin domain (PYD), and it plays fundamental roles in inflammation (51). ASC is composed of the N-terminal PYD domain and the C-terminal CARD domain, which promote oligomeric homotypic interactions (52). Pro-caspase-1 is composed of a CARD and catalytic domain, including p10 and p20 (53). The sensing of various stimuli by TLRs on the cell membrane can induce the activation of the TRIF/NF-κB and TLR4/MyD88 signaling pathways and upregulate the transcription of NLRP3 receptor proteins and proinflammatory cytokines (54, 55). Moreover, the LRR domain of NLRP3 senses stimuli, which leads to NACHT domain oligomerization, most likely by promoting a PYD-ASC interaction, and these processes result in the formation of the NLRP3 inflammasome via the binding of pro-caspase-1, which is another component of the NLRP3 inflammasome, to CARD and ASC (56, 57). Upon activation, the NLRP3 inflammasome results in the proteolytic activation of caspase-1, which facilitates the cleavage of pro-IL-1β and pro-IL-18 and the secretion of the proinflammatory cytokines IL-1β and IL-18 (58) (Figure 3).
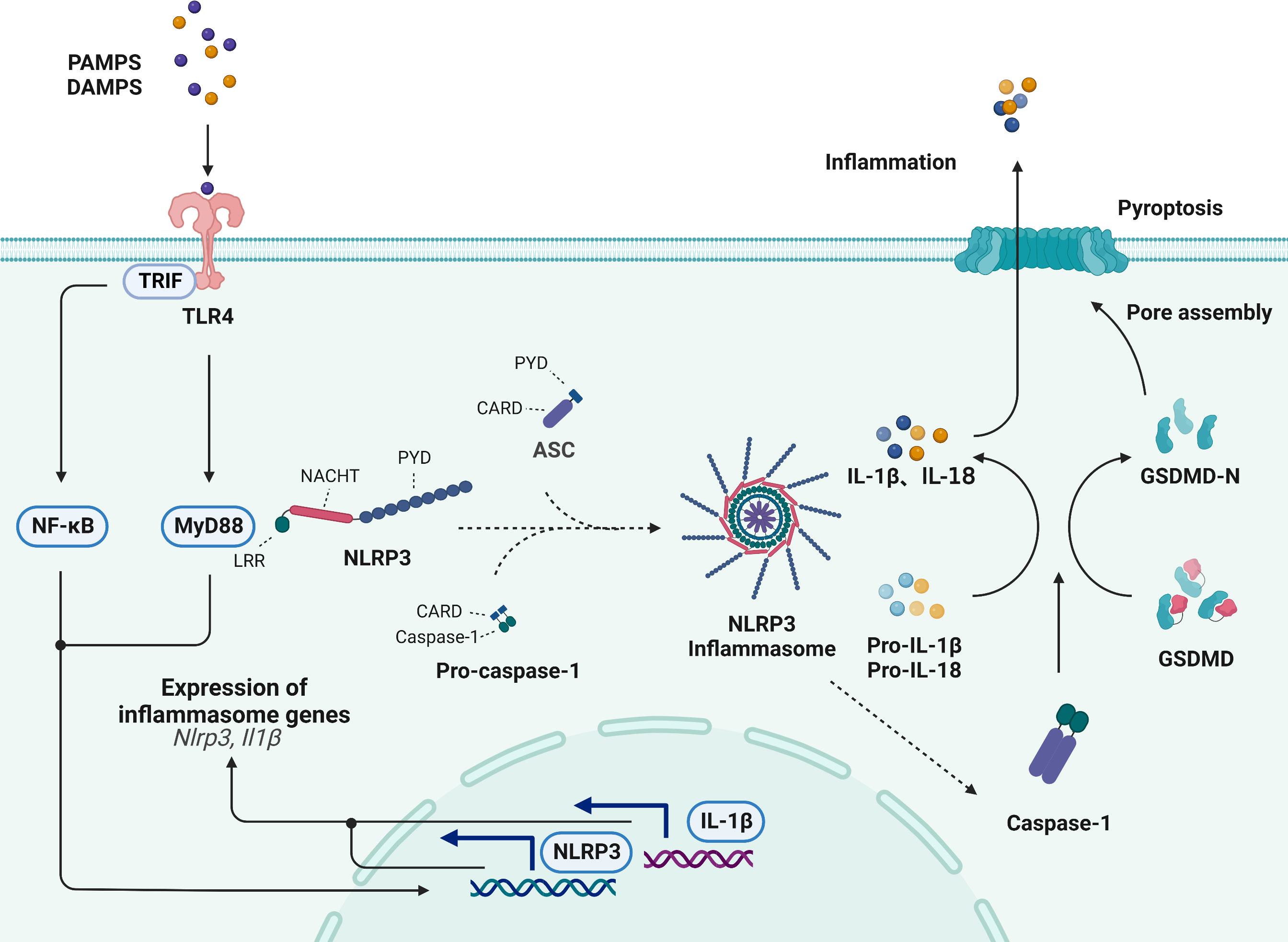
Figure 3 Activation of the NLRP3 inflammasome signaling pathway. The NLRP3 inflammasome is comprised of the NLRP3, ASC and pro-caspase-1. The sensing of various stimuli by TLRs on the cell membrane can induce the activation of the TRIF/NF-κB, the activation of TLR4/MyD88 signaling, the transcription of NLRP3 receptor proteins and lead to formation of NLRP3 inflammasome. Upon activation, NLRP3 induces the proteolytic activation of caspase-1, which facilitates the cleavage of pro-IL-1β and pro-IL-18. Additionally, activated caspase-1 cleaves GSDMD and generates an active GSDMD-NT, which translocates to the plasma membrane and oligomerizes to form a pore that allows the secretion of IL-1β and IL-18, subsequently inducing pyroptotic cell death.
Additionally, the caspase-1-dependent cleavage of gasdermin-D (GSDMD) is thought to be the classic initiator pyroptotic cell death (59). Pyroptosis is a novel form of cell death that is induced in response to cell disruption by the inflammasome-induced GSDMD protein, which promotes the release of proinflammatory cytokines, such as IL-1β and IL-18 (60). Previous studies have revealed that pyroptosis is involved in atherosclerotic plaque formation. Activated caspase-1 cleaves GSDMD and generates an active N-terminal cleaving product (GSDMD-NT), which translocates to the plasma membrane and oligomerizes to form a pore that allows the release of IL-1β and IL-18, subsequently inducing pyroptotic cell death and promoting the occurrence and development of atherosclerosis (61).
Recently, it has been shown that NLRP3 inflammasome/IL-1β signaling-mediated inflammation plays an important role in the development of atherosclerosis (12), as is shown in Table S2 (62–73) and Figure 4. The inflammatory process starts with inflammasome activation, which leads to the release of mature IL-1β. Various PAMPs and DAMPs have been shown to activate macrophages (74). Upon activation, macrophages exacerbate the vascular inflammatory response by releasing cytokines, and activated macrophages are the main source of IL-1β (75). For example, Orecchioni et al. recently found that macrophages express the olfactory receptor Olfr2 and all associated trafficking and signaling molecules, which drive atherosclerosis via NLRP3-dependent IL-1β secretion (76). In addition, a study by Zhang et al. reported that desmosterol suppressed inflammasome activation in macrophages and protected against vascular inflammation and atherosclerosis (77). Decreased desmosterol accumulation in mitochondria promotes mtROS production and NLRP3-dependent inflammasome activation. NLRP3 or ASC deficiency can reverse the increase in inflammasome activity and atherogenesis that is observed in desmosterol-depleted macrophages.
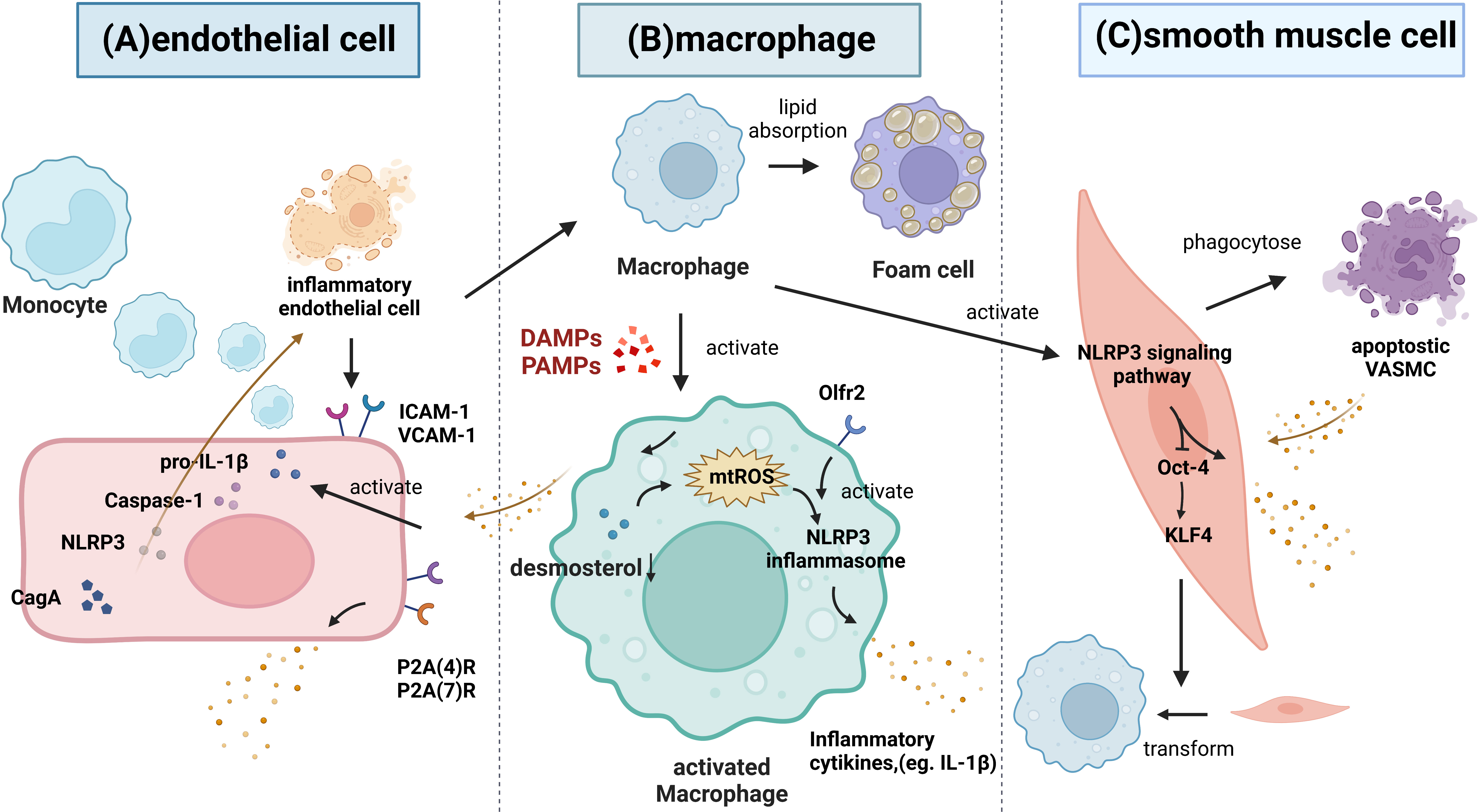
Figure 4 The role of the NLRP3 inflammasome signaling pathway in atherosclerosis. (A) Inflammatory endothelium can improve the recruitment of leucocytes via increasing ICAM-1 and VCAM-1, thus attracting monocytes to migrate and differentiate into macrophages. Then, macrophages phagocytose the modified lipoproteins and increase the foam cells formation. Inflammatory stimuli activate the synthesis of pro-IL-1β and increase the caspase-1. Both of P2X(4)R and P2X(7)R are also significantly upregulated under inflammatory conditions and promote the release of IL-1β and IL-1Ra. Moreover, CagA promotes endothelial inflammation through the NLRP3/caspase-1/IL-1β axis. (B) Various PAMPs and DAMPs can activate macrophages. Macrophages express the Olfr2, which drive atherosclerosis via NLRP3-dependent IL-1β secretion. Desmosterol accumulation in mitochondria suppresses mtROS production and NLRP3 inflammasome activation. (C) VSMC apoptosis can induce the inflammatory response. Normal VSMCs inhibit the inflammation by phagocytosing apoptotic VSMCs in vivo. Monocytes can trigger NLRP3 inflammasome activation and the expression of NLRP3, promoting IL-1β secretion, and these cells can decrease Oct-4 in VSMCs and upregulate klf4 to control the phenotypic transformation of VSMCs into macrophage-like cells.
Endothelium dysfunction is involved in the development of atherosclerotic vascular lesions. It has been shown that ECs synthesize IL-1β in response to inflammatory stimuli; moreover, the atherosclerotic endothelium exhibits increased expression of IL-1β (78). Wilson et al. found that HUVECs express P2X (4) R and P2X(7)R subtypes, and both were significantly upregulated under inflammatory conditions (79). Activation of P2X(7)Rs results in the release of low levels of bioactive IL-1β and the simultaneous release of IL-1Ra (79). Wu et al. demonstrated that cytotoxin-associated gene A (CagA) promotes aortic endothelial inflammation and accelerates atherosclerosis through the NLRP3/caspase-1/IL-1β axis (80). In addition, the activation of the vascular endothelium and the infiltration of circulating monocytes into the vessel wall are considered to be key factors in the occurrence and development of atherosclerosis (81). Hettwer et al. revealed that IL-1β suppression reduces inflammatory leukocyte production and uptake in atherosclerosis (82). When the NLRP3 inflammasome is inhibited in ECs from atherosclerotic aortas, these cells show decreased expression of leukocyte chemoattractants and adhesion molecules, indicating that NLRP3 inflammasome- and IL-1β-targeted therapies may reduce blood leukocyte recruitment to atherosclerotic aortas.
The inflammatory response that is induced by VSMC apoptosis accounts for approximately 15% of the systemic inflammatory response in atherosclerosis. Apoptotic VSMCs are phagocytosed by normal endothelial cells in vivo, which does not require the involvement of macrophages, leading to the inhibition of inflammation and slowing the progression of atherosclerosis (83). In addition, Clarke et al. found that necrotic VSMCs release IL-1a, whereas apoptotic VSMCs undergoing secondary necrosis release both IL-1a and IL-1β. IL-1 from necrotic VSMCs induces the surrounding viable VSMCs to produce proinflammatory cytokines. Thus, the failure to clear apoptotic VSMCs caused by hyperlipidemia in vivo may promote the increases in serum cytokine levels and chronic inflammation associated with atherosclerosis (83). In addition, NLRP3 inflammasome activation and IL-1β signaling play a direct role in VSMC phenotypic switching (84). Fabienne Burger et al. found that monocytes can trigger NLRP3 transcription factors expression, thus promoting NLRP3 inflammasome activation and IL-1β secretion, and these cells can decrease Oct-4 expression in VSMCs and upregulate klf4 expression to control the phenotypic transformation of atherosclerotic VSMCs into macrophage-like cells (85).
Several studies have revealed that both PCSK9 expression and NLRP3 inflammasome-induced pyroptosis occur in atherosclerotic plaques (13). Some studies have reported that there are interactions between PCSK9 and the NLRP3 inflammasome in atherosclerosis (86) (Table S3) (87, 88, Figure 5). PCSK9 is considered to be a key inflammatory signal, and it is also abundantly expressed in cells that are exposed to inflammatory stimuli, such as lipopolysaccharide (LPS) (89). The NLRP3 inflammasome has been considered to be a link between lipid metabolism and inflammation, and it induces pyroptosis and exerts a significant effect on atherogenesis (90). Under hypoxic conditions, PCSK9 promotes the secretion of proinflammatory cytokines by macrophages to exacerbate hypoxia/reoxygenation-induced cardiomyocyte injury by activating the NF-κB signaling pathway (91). Moreover, PCSK9 knockout significantly inhibits hypoxia-induced inflammation and cell death, inhibits macrophage recruitment, and suppresses the migration and proliferation of VSMCs; these effects are related to the inhibition of NLRP3 inflammasome activation (15). Using sepsis models in HUVECs and mice, Huang et al. found that increased PCSK9 expression during sepsis activates the TLR4/MyD88/NF-κB and NLRP3 pathways to induce inflammation, which results in vascular endothelial dysfunction and decreased survival (14). In addition, several studies have also demonstrated that the NLRP3 inflammasome and pyroptosis-related proteins are activated under hypoxia conditions (92). Ding et al. established a LCA ligation model in mice to model the atherosclerotic state and found that PCSK9 expression could be strongly enhanced in the zone bordering the infarcted area (93). Moreover, the study reported that pyroptosis is significantly observed in the zone bordering the myocardial infarcted area (94). These studies further demonstrated a close correlation between PCSK9 and the NLRP3 inflammasome signaling pathway, and both are involved in the process of hypoxia-induced pyroptosis.
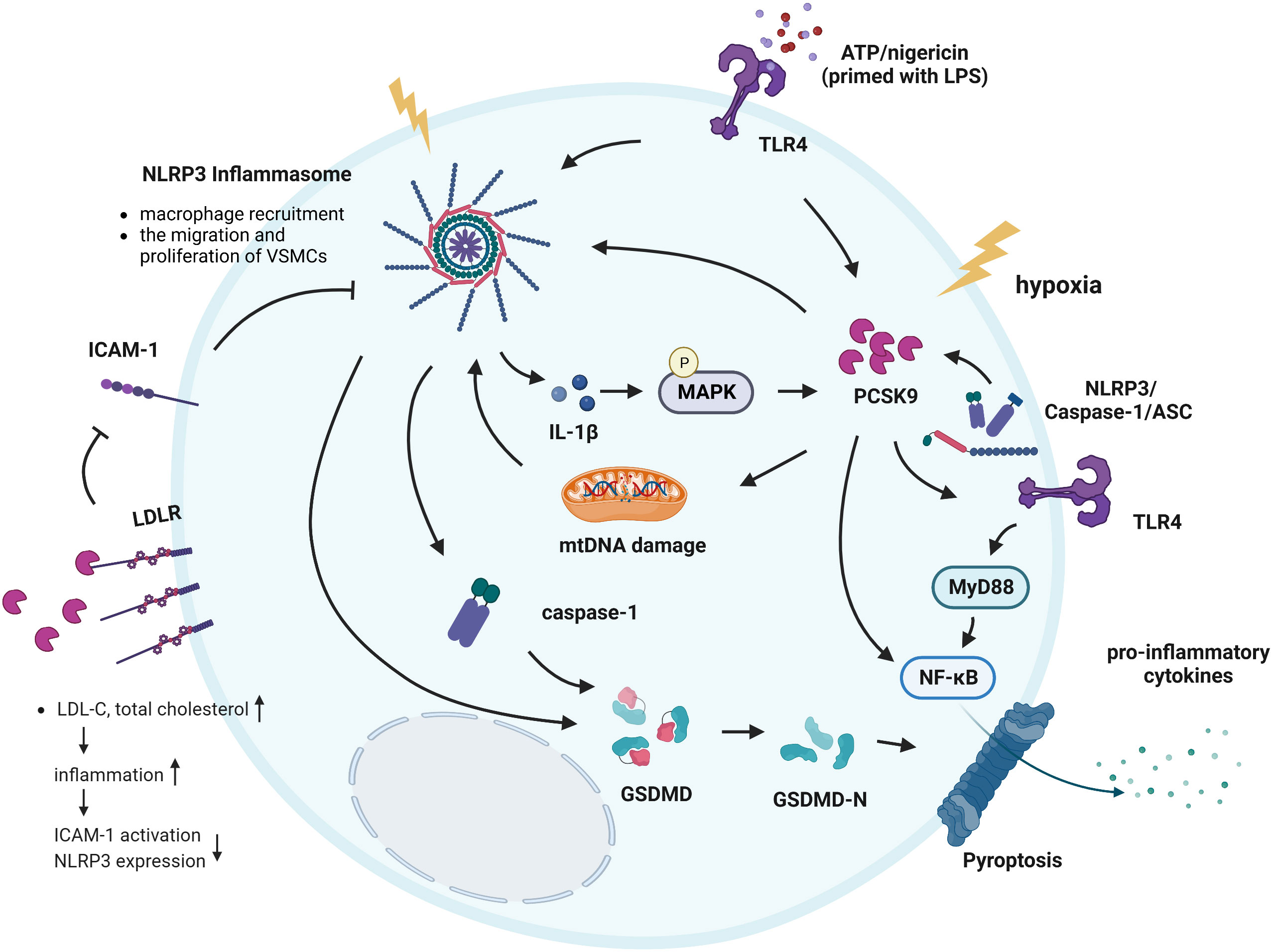
Figure 5 Interactions between PCSK9 and the NLRP3 inflammasome signaling pathway. ATP and nigericin simultaneously induced the NLRP3 inflammasome in cells primed with LPS and induced PCSK9 expression. In addition, hypoxia induces PCSK9 expression and NLRP3 inflammasome activation. The NLRP3 inflammasome and its downstream signals all regulate PCSK9 secretion, while MAPKs play a key role in regulating IL-1β-mediated PCSK9 secretion. PCSK9 can directly activate the NLRP3 inflammasome and promote the secretion of pro-inflammatory cytokines via NF-κB signaling. PCSK9 knockout inhibits macrophage recruitment, suppresses the migration and proliferation of VSMCs; these effects are related to NLRP3 inflammasome. PCSK9 initiates mtDNA damage, activates NLRP3 inflammasome signaling and subsequently induces pyroptosis. Another mechanism underlying the effects of PCSK9 on NLRP3 inflammasome activation involves TLR4/MyD88/NF-κB signaling. PCSK9 binds to LDLR, causing an increase in the total cholesterol and LDL-C levels, inducing inflammation, resulting in ICAM expression and NLRP3 inflammasome activation.
Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. Excess production of mtROS and subsequent mtDNA damage are common triggers of NLRP3 inflammasome activation (95), while GSDMD activation and LDH release are key mediators of the occurrence of pyroptosis. Wang et al. found that PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia (13). PCSK9 initiates mitochondrial DNA (mtDNA) damage, activates NLRP3 inflammasome signaling (NLRP3, ASC, Caspase-1, IL-1β, and IL-18), and subsequently induces Caspase-1-dependent pyroptosis, with intense expression of PCSK9 and the pyroptosis marker GSDMD-NT in the zone bordering the infarct area. Moreover, PCSK9 knockout significantly decreases the NLRP3 inflammasome signaling, GSDMD-NT expression, and LDH release (13, 15). These results suggest that PCSK9 regulates Caspase-1-dependent pyroptosis via mtDNA damage and may reveal proinflammatory processes, including NLRP3 inflammasome signaling and pyroptosis, as potential targets for the treatment of PCSK9-related cardiovascular diseases (13). Additionally, PCSK9 binds to the LDLR and enhances its degradation, which leads to the reduced clearance of LDL-C and a higher risk of atherosclerosis. Landlinger et al. found that AT04A immunization induced high and persistent levels of antibodies against PCSK9, significantly reducing the total cholesterol and LDL-C levels in plasma, decreasing vascular inflammation, and ultimately reducing ICAM expression in activated endothelial cells and caspase-1-activating NLRP3 inflammasome-related protein expression in proinflammatory macrophages (44).
Notably, the expression of NLRP3 inflammasome-related proteins and secretion of PCSK9 were particularly evident when mouse peritoneal macrophages were exposed to LPS, ATP or nigericin, indicating that the NLRP3 inflammasome is connected to PCSK9 secretion (16, 96). Macrophage-derived PCSK9 may also play an important role in atherogenesis via its local effects on atherosclerotic plaques (36). Ding et al. revealed that NLRP3 and its downstream signaling proteins ASC, Caspase-1, IL-18, and IL-1β all regulate PCSK9 secretion by macrophages as well as in a host of tissues, including the liver, small intestine and kidney; on the other hand, MAPKs play a key role in regulating IL-1β-mediated PCSK9 secretion, particularly in the context of high-fat diet consumption (16). The study pointed out that macrophages secrete large amounts of PCSK9 via NLRP3 inflammasome activation. PCSK9 expression is downstream of NLRP3 and IL-1β, which are the major regulators of PCSK9 secretion. Furthermore, HFD-C results in the robust secretion of PCSK9, and this PCSK9 secretion is dependent on IL-1β upregulation, providing a strong link between IL-1β and PCSK9 in the proatherosclerotic and proinflammatory milieu (97, 98).
Inhibiting PCSK9 with inhibitors has been studied as a potential therapeutic approach in the field of atherosclerotic treatment (99). Different forms of PCSK9 inhibitors and their targets, including monoclonal antibodies (mAbs), small interfering RNAs (siRNAs), and vaccines, have recently been explored (100–102). At present, two mAbs against human PCSK9, alirocumab and evolocumab, are approved for listing and are available for use in clinical treatment (103). Ongoing clinical trials have shown that these PCSK9 mAbs are well tolerated, enhance the clearance of LDL-C, and decrease cardiovascular events in patients (104).Therapeutic monoclonal antibodies target PCSK9 in the plasma, while siRNA selective silences the translation of messenger RNA (mRNA) molecules, thus preventing the intracellular translation of PCSK9 mRNA to protein (105). Inclisiran is an siRNA and was discovered several years ago (106). The positive results of the ORION series provide strong supporting evidence for the clinical application of inclisiran in the treatment of atherosclerosis (107, 108). Compared with therapeutic mAbs, siRNA treatment shows a more sustained efficacy and requires fewer injections, thus overcoming some known barriers to treatment compliance compared with mAbs (109). The basic characteristic of a PCSK9 vaccine is the capacity to trigger the generation of anti-PCSK9 antibodies by the host (110). However, research on PCSK9 vaccines, such as PCSK9Qβ-003, AT04A and L-IFPTA+, is still in the animal experimental stage (44, 111, 112).
In terms of NLRP3 inhibitors, currently available clinical agents are targeting IL-1β (113). Although targeting IL-1β is an effective method for treating inflammatory diseases, it seems to have limitations. First, the activated NLRP3 inflammasome produces various inflammatory cytokines, including IL-1β, IL-18 secretion. All of cytokines play crucial roles in the occurrence and development of related diseases (114, 115). Second, IL-1β is produced not only by the NLRP3 inflammasome but also by other inflammasomes or in an inflammasome-independent manner, so inhibition of NLRP3 may have more immunosuppressive effects than inhibition of IL-1β (116, 117). Additionally, MCC950 can specific inhibit classical and nonclassical NLRP3 inflammasome activation and pro-inflammatory cytokines secretion to significantly decrease the maximum degree of aortic stenosis as well as the mean plaque size and volume and increase plaque stability (118–120). Several studies have revealed that MCC950 can directly interact with the Walker B motif in the NACHT domain to block ATP hydrolysis and inhibit NLRP3 inflammasome activation and formation (121, 122). In addition, CY-09, which is an analog of C172, was also found to directly bind to the ATP-binding site of the NLRP3 NACHT domain and inhibit its ATPase activity, inhibiting NLRP3 inflammasome oligomerization and activation (123). Besides, Song et al. revealed that blocking NLRP3 phosphorylation at S194 significantly decreased NLRP3 inflammasome activation, suggesting that the inhibition of NLRP3 phosphorylation may be a potential approach for treating NLRP3-related diseases (124). Melatonin has proven to be effective in treating atherosclerosis by inhibiting NLRP3 inflammasome signaling pathway (125, 126).
Collectively, the research on novel methods for inhibiting PCSK9 and NLRP3 inflammasome is still in an early stage, and the efficacy, safety and feasibility of these approaches require further study.
In summary, PCSK9 can promote the occurrence and development of atherosclerosis by inhibiting the metabolism of LDL-C, enhancing the formation of foam cells and thus promoting the progression of vascular wall inflammation, suggesting that PCSK9 is an important intervention target in the occurrence and development of atherosclerosis. NLRP3 inflammasome activation contributes to the vascular inflammatory response driving atherosclerosis development and progression. Significantly, the effect of PCSK9 and the NLRP3 inflammasome on atherosclerosis seems to be synergistic. Interactions between PCSK9 and the NLRP3 inflammasome may form a positive feedback loop, acting together to drive the inflammatory response and lipid accumulation and thus promoting atherosclerosis. On the one hand, the NLRP3 inflammasome signaling pathway has been shown to promote PCSK9 secretion by regulating IL-1β, and this may be mediated by MAPK signaling. On the other hand, PCSK9 directly induces inflammatory responses in VSMCs as well as the expression and secretion of inflammatory factors and cytokines, including NLRP3 and its downstream molecules ASC, Caspase-1, IL-6, IL-18, and IL-1β, in macrophages. Collectively, these findings suggest a bidirectional positive correlation between the NLRP3 inflammasome signaling pathway and PCSK9. Therapeutic options that target PCSK9 and NLRP3 inflammasome may play an important role in the future treatment of atherosclerosis-related diseases. For example, inclisiran not only decreases LDL-C levels but also reduces Lp(a) concentrations. Given that Lp(a) is an independent risk factor for atherosclerosis, early treatment with inclisiran might result in extensive benefits for patients and decrease the damage caused by atherosclerosis via multiple mechanisms. Additionally, the inhibitors of NLRP3 inflammasome signaling that are currently available are agents that target IL-1β and caspase-1 as well as antagonists of the receptor P2X7.
It is noteworthy that knockout of the PCSK9 gene without inflammatory activation does not affect the levels of inflammatory proteins under physiological conditions; only the presence of factors that induce inflammatory responses, such as a high-fat diet, significantly decrease the levels of inflammatory cytokines. Besides, although the deficiency of the PCSK9 gene partially decreased inflammation in mice with atherosclerosis, it failed to diminish the inflammatory response completely. First, activation of the NLRP3 inflammasome during apoptosis is closely associated with mtROS-mediated mtDNA damage. Second, activated NLRP3-mediated IL-1β secretion in turn serves as a proinflammatory stimulant that can promote inflammatory responses. In addition, Chen et al. indicated that NLRP3, NLRP12 and NLRC4 exerted a significant synergistic effect, suggesting that NLRP3, NLRP12 and NLRC4 could be mutually regulated (127). This synergistic effect can promote caspase-1-dependent GSDMD cleavage-induced pyroptosis during vascular damage and accelerate the secretion of IL-1β. Finally, the synthesis of inflammatory factors and cytokines, such as ASC, Caspase-1, and IL-1β, may not be fully dependent on the NLRP3 inflammasome. NLRP6, NLRP7, NLRP12, NLRC4 and AIM2 have been shown to contribute to proinflammatory cytokine expression. As shown in the study by Liu et al., overexpression of NLRP6 enhanced the activation of caspase-1 and gasdermin-D, induced the pyroptosis of human gingival fibroblasts, and promoted the release of the proinflammatory mediator IL-1β (128).
In conclusion, the NLRP3 inflammasome signaling pathway and PCSK9 play critical roles in atherosclerosis and show promise as essential targets for the prevention and treatment of atherosclerosis. However, the precise mechanisms by which interactions between the NLRP3 inflammasome and PCSK9 affect atherosclerosis have largely remained unclear. Further research is needed to reveal the functions of PCSK9 and NLRP3 inflammasome signaling in atherosclerosis by revealing the underlying molecular mechanisms. Moreover, long-term clinical follow-up with a large sample size will still be necessary for studying PCSK9 and NLRP3 inhibitors since these inhibitors may increase the risk of diabetes and neurological diseases.
YW and DF conceived the idea, analysis of literature, and writing of the manuscript. QY, JY, LW, and JW collected and read the literature and revised the article. MZ and ML read through and corrected the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China [grant numbers 81800434], Grant of Sichuan Province Science and Technology Agency Grant [2019YJ0487].
Figures were created with BioRender software, ©biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1126823/full#supplementary-material
1. Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat Rev Cardiol (2017) 14(3):133–44. doi: 10.1038/nrcardio.2016.185
2. Wu M-Y, Li C-J, Hou M-F, Chu P-Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int J Mol Sci (2017) 18(10):2034. doi: 10.3390/ijms18102034
3. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res (2018) 114(4):565–77. doi: 10.1093/cvr/cvx253
4. Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: Triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev (2019) 40(2):537–57. doi: 10.1210/er.2018-00184
5. Barale C, Melchionda E, Morotti A, Russo I. Pcsk9 biology and its role in atherothrombosis. Int J Mol Sci (2021) 22(11):5880. doi: 10.3390/ijms22115880
6. Poirier S, Mayer G, Poupon V, McPherson PS, Desjardins R, Ly K, et al. Dissection of the endogenous cellular pathways of Pcsk9-induced low density lipoprotein receptor degradation: Evidence for an intracellular route. J Biol Chem (2009) 284(42):28856–64. doi: 10.1074/jbc.M109.037085
7. Stoekenbroek RM, Lambert G, Cariou B, Hovingh GK. Inhibiting Pcsk9 - biology beyond ldl control. Nat Rev Endocrinol (2018) 15(1):52–62. doi: 10.1038/s41574-018-0110-5
8. Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, et al. Human Pcsk9 promotes hepatic lipogenesis and atherosclerosis development Via apoe- and ldlr-mediated mechanisms. Cardiovasc Res (2016) 110(2):268–78. doi: 10.1093/cvr/cvw053
9. Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, Palinski W. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein e-deficient and ldl receptor-deficient mice. J Pathol (2001) 195(2):257–63. doi: 10.1002/path.915
10. Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular mechanisms regulating Nlrp3 inflammasome activation. Cell Mol Immunol (2016) 13(2):148–59. doi: 10.1038/cmi.2015.95
11. Olsen MB, Gregersen I, Sandanger Ø, Yang K, Sokolova M, Halvorsen BE, et al. Targeting the inflammasome in cardiovascular disease. JACC Bas Transl Sci (2022) 7(1):84–98. doi: 10.1016/j.jacbts.2021.08.006
12. Grebe A, Hoss F, Latz E. Nlrp3 inflammasome and the il-1 pathway in atherosclerosis. Circ Res (2018) 122(12):1722–40. doi: 10.1161/CIRCRESAHA.118.311362
13. Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, et al. Pcsk9 regulates pyroptosis Via mtdna damage in chronic myocardial ischemia. Bas Res In Cardiol (2020) 115(6):66. doi: 10.1007/s00395-020-00832-w
14. Huang L, Li Y, Cheng Z, Lv Z, Luo S, Xia Y. Pcsk9 promotes endothelial dysfunction during sepsis via the Tlr4/Myd88/Nf-Kb and Nlrp3 pathways. Inflammation (2022). doi: 10.1007/s10753-022-01715-z
15. Zou Y, Chen Z, Zhang X, Yu J, Xu H, Cui J, et al. Targeting Pcsk9 ameliorates graft vascular disease in mice by inhibiting Nlrp3 inflammasome activation in vascular smooth muscle cells. Front Immunol (2022) 13:894789. doi: 10.3389/fimmu.2022.894789
16. Ding Z, Wang X, Liu S, Zhou S, Kore RA, Mu S, et al. Nlrp3 inflammasome il-1β regulates Pcsk9 secretion. Theranostics (2020) 10(16):7100–10. doi: 10.7150/thno.45939
17. Sundström C, Nilsson K. Cytochemical profile of human haematopoietic biopsy cells and derived cell lines. Br J Haematol (1977) 37(4):489–501. doi: 10.1111/j.1365-2141.1977.tb01022.x
18. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (Narc-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci U.S.A. (2003) 100(3):928–33. doi: 10.1073/pnas.0335507100
19. Moreau F, Thédrez A, Garçon D, Ayer A, Sotin T, Dijk W, et al. Pcsk9 is not secreted from mature differentiated intestinal cells. J Lipid Res (2021) 62:100096. doi: 10.1016/j.jlr.2021.100096
20. Wang Y, Liu Z-P. Pcsk9 inhibitors: Novel therapeutic strategies for lowering ldlcholesterol. Mini Rev Med Chem (2019) 19(2):165–76. doi: 10.2174/1389557518666180423111442
21. Lin X-L, Xiao L-L, Tang Z-H, Jiang Z-S, Liu M-H. Role of Pcsk9 in lipid metabolism and atherosclerosis. Biomed Pharmacother = Biomed Pharmacother (2018) 104:36–44. doi: 10.1016/j.biopha.2018.05.024
22. Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human Pcsk9 on the atherosclerotic lesion. J Pathol (2016) 238(1):52–62. doi: 10.1002/path.4630
23. Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, et al. Cross-talk between lox-1 and Pcsk9 in vascular tissues. Cardiovasc Res (2015) 107(4):556–67. doi: 10.1093/cvr/cvv178
24. Kong N, Xu Q, Cui W, Feng X, Gao H. Pcsk9 inhibitor inclisiran for treating atherosclerosis Via regulation of endothelial cell pyroptosis. Ann Transl Med (2022) 10(22):1205. doi: 10.21037/atm-22-4652
25. Zeng J, Tao J, Xi L, Wang Z, Liu L. Pcsk9 mediates the oxidative Low−Density Lipoprotein−Induced pyroptosis of vascular endothelial cells Via the Uqcrc1/Ros pathway. Int J Mol Med (2021) 47(4):53. doi: 10.3892/ijmm.2021.4886
26. Schuster S, Rubil S, Endres M, Princen HMG, Boeckel J-N, Winter K, et al. Anti-Pcsk9 antibodies inhibit pro-atherogenic mechanisms in Apoe*3leiden.Cetp mice. Sci Rep (2019) 9(1):11079. doi: 10.1038/s41598-019-47242-0
27. Ferri N, Tibolla G, Pirillo A, Cipollone F, Mezzetti A, Pacia S, et al. Proprotein convertase subtilisin kexin type 9 (Pcsk9) secreted by cultured smooth muscle cells reduces macrophages ldlr levels. Atherosclerosis (2012) 220(2):381–6. doi: 10.1016/j.atherosclerosis.2011.11.026
28. Ricci C, Ruscica M, Camera M, Rossetti L, Macchi C, Colciago A, et al. Pcsk9 induces a pro-inflammatory response in macrophages. Sci Rep (2018) 8(1):2267. doi: 10.1038/s41598-018-20425-x
29. Adorni MP, Cipollari E, Favari E, Zanotti I, Zimetti F, Corsini A, et al. Inhibitory effect of Pcsk9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis (2017) 256:1–6. doi: 10.1016/j.atherosclerosis.2016.11.019
30. Jin P, Gao D, Cong G, Yan R, Jia S. Role of Pcsk9 in homocysteine-accelerated lipid accumulation in macrophages and atherosclerosis in apoe mice. Front Cardiovasc Med (2021) 8:746989. doi: 10.3389/fcvm.2021.746989
31. Ragusa R, Basta G, Neglia D, De Caterina R, Del Turco S, Caselli C. Pcsk9 and atherosclerosis: Looking beyond ldl regulation. Eur J Clin Invest (2021) 51(4):e13459. doi: 10.1111/eci.13459
32. Ding Z, Wang X, Liu S, Zhou S, Kore RA, Mu S, et al. Nlrp3 inflammasome Via il-1β regulates Pcsk9 secretion. Theranostics (2020) 10(16):7100–10. doi: 10.7150/thno.45939
33. Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, et al. Pcsk9 regulates expression of scavenger receptors and ox-ldl uptake in macrophages. Cardiovasc Res (2018) 114(8):1145–53. doi: 10.1093/cvr/cvy079
34. Badimon L, Luquero A, Crespo J, Peña E, Borrell-Pages M. Pcsk9 and Lrp5 in macrophage lipid internalization and inflammation. Cardiovasc Res (2021) 117(9):2054–68. doi: 10.1093/cvr/cvaa254
35. Levy E, Ben Djoudi Ouadda A, Spahis S, Sane AT, Garofalo C, Grenier É, et al. Pcsk9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis (2013) 227(2):297–306. doi: 10.1016/j.atherosclerosis.2013.01.023
36. Tang Z-H, Peng J, Ren Z, Yang J, Li T-T, Li T-H, et al. New role of Pcsk9 in atherosclerotic inflammation promotion involving the Tlr4/Nf-Kb pathway. Atherosclerosis (2017) 262:113–22. doi: 10.1016/j.atherosclerosis.2017.04.023
37. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, et al. The proprotein convertase Pcsk9 induces the degradation of low density lipoprotein receptor (Ldlr) and its closest family members vldlr and Apoer2. J Biol Chem (2008) 283(4):2363–72. doi: 10.1074/jbc.M708098200
38. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res (2016) 118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361
39. Ferri N, Marchianò S, Tibolla G, Baetta R, Dhyani A, Ruscica M, et al. Pcsk9 knock-out mice are protected from neointimal formation in response to perivascular carotid collar placement. Atherosclerosis (2016) 253:214–24. doi: 10.1016/j.atherosclerosis.2016.07.910
40. Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress Via ros modulates Pcsk9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal (2015) 22(9):760–71. doi: 10.1089/ars.2014.6054
41. Zhang M-J, Zhou Y, Chen L, Wang X, Pi Y, Long C-Y, et al. Impaired Sirt1 promotes the migration of vascular smooth muscle cell-derived foam cells. Histochem Cell Biol (2016) 146(1):33–43. doi: 10.1007/s00418-016-1408-9
42. Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, et al. Cross-talk between Pcsk9 and damaged mtdna in vascular smooth muscle cells: Role in apoptosis. Antioxid Redox Signal (2016) 25(18):997-1008. doi: 10.1089/ars.2016.6631
43. Martínez GJ, Celermajer DS, Patel S. The Nlrp3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis (2018) 269:262–71. doi: 10.1016/j.atherosclerosis.2017.12.027
44. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The At04a vaccine against proprotein convertase Subtilisin/Kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in Apoe*3leiden.Cetp mice. Eur Heart J (2017) 38(32):2499–507. doi: 10.1093/eurheartj/ehx260
45. Chistiakov DA, Orekhov AN, Bobryshev YV. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol (Oxf) (2017) 219(2):382–408. doi: 10.1111/apha.12725
46. Ding Z, Pothineni NVK, Goel A, Lüscher TF, Mehta JL. Pcsk9 and inflammation: Role of shear stress, pro-inflammatory cytokines, and lox-1. Cardiovasc Res (2020) 116(5):908–15. doi: 10.1093/cvr/cvz313
47. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
48. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: A cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
49. Song N, Li T. Regulation of Nlrp3 inflammasome by phosphorylation. Front In Immunol (2018) 9:2305. doi: 10.3389/fimmu.2018.02305
50. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the Nlrp3 inflammasome. Front In Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
51. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res (2020) 126(9):1260–80. doi: 10.1161/CIRCRESAHA.120.315937
52. Shiohara M, Taniguchi Si, Masumoto J, Yasui K, Koike K, Komiyama A, et al. Asc, which is composed of a pyd and a card, is up-regulated by inflammation and apoptosis in human neutrophils. Biochem Biophys Res Commun (2002) 293(5):1314–8. doi: 10.1016/S0006-291X(02)00384-4
53. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med (2018) 215(3):827–40. doi: 10.1084/jem.20172222
54. Zhang H, Du Y, Guo Y, Wang Z, Li H, Lv Z, et al. Tlr4-Nlrp3-Gsdmd-Mediated pyroptosis plays an important role in aggravated liver injury of Cd38 sepsis mice. J Immunol Res (2021) 2021:6687555. doi: 10.1155/2021/6687555
55. Palová-Jelínková L, Dáňová K, Drašarová H, Dvořák M, Funda DP, Fundová P, et al. Pepsin digest of wheat gliadin fraction increases production of il-1β Via Tlr4/Myd88/Trif/Mapk/Nf-Kb signaling pathway and an Nlrp3 inflammasome activation. PloS One (2013) 8(4):e62426. doi: 10.1371/journal.pone.0062426
56. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
57. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: A supramolecular assembly of asc dimers mediating inflammatory cell death Via caspase-1 activation. Cell Death Different (2007) 14(9):1590–604. doi: 10.1038/sj.cdd.4402194
58. Chen G, Shaw MH, Kim Y-G, Nuñez G. Nod-like receptors: Role in innate immunity and inflammatory disease. Annu Rev Pathol (2009) 4:365–98. doi: 10.1146/annurev.pathol.4.110807.092239
59. Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural mechanism for gsdmd targeting by autoprocessed caspases in pyroptosis. Cell (2020) 180(5):941-955.e20. doi: 10.1016/j.cell.2020.02.002
60. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. Nlrp3 inflammasome activation triggers gasdermin d-independent inflammation. Sci Immunol (2021) 6(64):eabj3859. doi: 10.1126/sciimmunol.abj3859
61. Qian Z, Zhao Y, Wan C, Deng Y, Zhuang Y, Xu Y, et al. Pyroptosis in the initiation and progression of atherosclerosis. Front Pharmacol (2021) 12:652963. doi: 10.3389/fphar.2021.652963
62. Liu Y, Tie L. Apolipoprotein m and sphingosine-1-Phosphate complex alleviates tnf-A-Induced endothelial cell injury and inflammation through Pi3k/Akt signaling pathway. BMC Cardiovasc Disord (2019) 19(1):279. doi: 10.1186/s12872-019-1263-4
63. Jin H, Zhu Y, Wang X-D, Luo E-F, Li Y-P, Wang B-L, et al. Bdnf corrects Nlrp3 inflammasome-induced pyroptosis and glucose metabolism reprogramming through Klf2/Hk1 pathway in vascular endothelial cells. Cell Signal (2021) 78:109843. doi: 10.1016/j.cellsig.2020.109843
64. Li Y, Niu X, Xu H, Li Q, Meng L, He M, et al. Vx-765 attenuates atherosclerosis in apoe deficient mice by modulating vsmcs pyroptosis. Exp Cell Res (2020) 389(1):111847. doi: 10.1016/j.yexcr.2020.111847
65. Wen C, Yang X, Yan Z, Zhao M, Yue X, Cheng X, et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol (2013) 168(3):2242–7. doi: 10.1016/j.ijcard.2013.01.211
66. Schönbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P. Ligation of Cd40 activates interleukin 1beta-converting enzyme (Caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1beta. J Biol Chem (1997) 272(31):19569–74. doi: 10.1074/jbc.272.31.19569
67. Lin S-J, Yen H-T, Chen Y-H, Ku H-H, Lin F-Y, Chen Y-L. Expression of interleukin-1 beta and interleukin-1 receptor antagonist in oxldl-treated human aortic smooth muscle cells and in the neointima of cholesterol-fed endothelia-denuded rabbits. J Cell Biochem (2003) 88(4):836–47. doi: 10.1002/jcb.10431
68. Gage J, Hasu M, Thabet M, Whitman SC. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein e-null mice. Can J Cardiol (2012) 28(2):222–9. doi: 10.1016/j.cjca.2011.10.013
69. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of Nlrp3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein e-deficient mice. Mediators Inflammation (2014) 2014:507208. doi: 10.1155/2014/507208
70. Liu W, Yin Y, Zhou Z, He M, Dai Y. Oxldl-induced il-1 beta secretion promoting foam cells formation was mainly Via Cd36 mediated ros production leading to Nlrp3 inflammasome activation. Inflammation Res (2014) 63(1):33–43. doi: 10.1007/s00011-013-0667-3
71. Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Deng X, et al. Lox-1, mtdna damage, and Nlrp3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc Res (2014) 103(4):619–28. doi: 10.1093/cvr/cvu114
72. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, et al. Serum amyloid a activates the Nlrp3 inflammasome Via P2x7 receptor and a cathepsin b-sensitive pathway. J Immunol (2011) 186(11):6119–28. doi: 10.4049/jimmunol.1002843
73. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464(7293):1357–61. doi: 10.1038/nature08938
74. Di Gioia M, Spreafico R, Springstead JR, Mendelson MM, Joehanes R, Levy D, et al. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat Immunol (2020) 21(1):42–53. doi: 10.1038/s41590-019-0539-2
75. Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular metabolism and macrophage functional polarization. Int Rev Immunol (2015) 34(1):82–100. doi: 10.3109/08830185.2014.969421
76. Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by Nlrp3-dependent il-1 production. Sci (New York NY) (2022) 375(6577):214–21. doi: 10.1126/science.abg3067
77. Zhang X, McDonald JG, Aryal B, Canfrán-Duque A, Goldberg EL, Araldi E, et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci U.S.A. (2021) 118(47):e2107682118. doi: 10.1073/pnas.2107682118
78. Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arteriosclerosis Thrombosis Vasc Biol (1996) 16(8):1000–6. doi: 10.1161/01.ATV.16.8.1000
79. Wilson HL, Varcoe RW, Stokes L, Holland KL, Francis SE, Dower SK, et al. P2x receptor characterization and il-1/Il-1ra release from human endothelial cells. Br J Pharmacol (2007) 151(1):115–27. doi: 10.1038/sj.bjp.0707213
80. Li B-W, Liu Y, Zhang L, Guo X-Q, Wen C, Zhang F, et al. Cytotoxin-associated gene a (Caga) promotes aortic endothelial inflammation and accelerates atherosclerosis through the Nlrp3/Caspase-1/Il-1β axis. FASEB J (2021) 35(11):e21942. doi: 10.1096/fj.202100695RR
81. Tedgui A, Mallat Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev (2006) 86(2):515–81. doi: 10.1152/physrev.00024.2005
82. Hettwer J, Hinterdobler J, Miritsch B, Deutsch M-A, Li X, Mauersberger C, et al. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc Res (2022) 118(13):2778–91. doi: 10.1093/cvr/cvab337
83. Clarke MCH, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-Directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res (2010) 106(2):363–72. doi: 10.1161/CIRCRESAHA.109.208389
84. Hallenbeck WH, Markey DR, Dolan DG. Analyses of tissue, blood, and urine samples from a baboon gavaged with chrysotile and crocidolite asbestos. Environ Res (1981) 25(2):349–60. doi: 10.1016/0013-9351(81)90037-2
85. Burger F, Baptista D, Roth A, da Silva RF, Montecucco F, Mach F, et al. Nlrp3 inflammasome activation controls vascular smooth muscle cells phenotypic switch in atherosclerosis. Int J Mol Sci (2021) 23(1):340. doi: 10.3390/ijms23010340
86. Ruscica M, Ferri N, Macchi C, Corsini A, Sirtori CR. Lipid lowering drugs and inflammatory changes: An impact on cardiovascular outcomes? Ann Med (2018) 50(6):461–84. doi: 10.1080/07853890.2018.1498118
87. Cyr Y, Bissonnette S, Lamantia V, Wassef H, Loizon E, Ngo Sock ET, et al. White adipose tissue surface expression of ldlr and Cd36 is associated with risk factors for type 2 diabetes in adults with obesity. Obes (Silver Spring) (2020) 28(12):2357–67. doi: 10.1002/oby.22985
88. Cyr Y, Lamantia V, Bissonnette S, Burnette M, Besse-Patin A, Demers A, et al. Lower plasma Pcsk9 in normocholesterolemic subjects is associated with upregulated adipose tissue surface-expression of ldlr and Cd36 and Nlrp3 inflammasome. Physiol Rep (2021) 9(3):e14721. doi: 10.14814/phy2.14721
89. Grune J, Meyborg H, Bezhaeva T, Kappert K, Hillmeister P, Kintscher U, et al. Pcsk9 regulates the chemokine receptor Ccr2 on monocytes. Biochem Biophys Res Commun (2017) 485(2):312–8. doi: 10.1016/j.bbrc.2017.02.085
90. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. Nlrp3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol (2018) 233(3):2116–32. doi: 10.1002/jcp.25930
91. Yang C-L, Zeng Y-D, Hu Z-X, Liang H. Pcsk9 promotes the secretion of pro-inflammatory cytokines by macrophages to aggravate H/R-induced cardiomyocyte injury Via activating nf-Kb signalling. Gen Physiol Biophys (2020) 39(2):123–34. doi: 10.4149/gpb-2019057
92. Cheng S-B, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis (2019) 10(12):927. doi: 10.1038/s41419-019-2162-4
93. Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, et al. Pcsk9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res (2018) 114(13):1738–51. doi: 10.1093/cvr/cvy128
94. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif (2019) 52(2):e12563. doi: 10.1111/cpr.12563
95. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ros promote macrophage pyroptosis by inducing gsdmd oxidation. J Mol Cell Biol (2019) 11(12):1069–82. doi: 10.1093/jmcb/mjz020
96. Wu N-Q, Shi H-W, Li J-J. Proprotein convertase Subtilisin/Kexin type 9 and inflammation: An updated review. Front Cardiovasc Med (2022) 9:763516. doi: 10.3389/fcvm.2022.763516
97. Kumar S, Kang D-W, Rezvan A, Jo H. Accelerated atherosclerosis development in C57bl6 mice by overexpressing aav-mediated Pcsk9 and partial carotid ligation. Lab Invest (2017) 97(8):935–45. doi: 10.1038/labinvest.2017.47
98. Frostegård J, Ahmed S, Hafström I, Ajeganova S, Rahman M. Low levels of Pcsk9 are associated with remission in patients with rheumatoid arthritis treated with anti-Tnf-A: Potential underlying mechanisms. Arthritis Res Ther (2021) 23(1):32. doi: 10.1186/s13075-020-02386-7
99. O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), Pcsk9 inhibition, and cardiovascular risk. Circulation (2019) 139(12):1483–92. doi: 10.1161/CIRCULATIONAHA.118.037184
100. Mullard A. Nine paths to Pcsk9 inhibition. Nat Rev Drug Discovery (2017) 16(5):299–301. doi: 10.1038/nrd.2017.83
101. Oyama K, Furtado RHM, Fagundes A, Zelniker TA, Tang M, Kuder J, et al. Effect of evolocumab on complex coronary disease requiring revascularization. J Am Coll Cardiol (2021) 77(3):259–67. doi: 10.1016/j.jacc.2020.11.011
102. Schwartz GG, Steg PG, Szarek M, Bittner VA, Diaz R, Goodman SG, et al. Peripheral artery disease and venous thromboembolic events after acute coronary syndrome: Role of lipoprotein(a) and modification by alirocumab: Prespecified analysis of the odyssey outcomes randomized clinical trial. Circulation (2020) 141(20):1608–17. doi: 10.1161/CIRCULATIONAHA.120.046524
103. Guedeney P, Giustino G, Sorrentino S, Claessen BE, Camaj A, Kalkman DN, et al. Efficacy and safety of alirocumab and evolocumab: A systematic review and meta-analysis of randomized controlled trials. Eur Heart J (2019) 43(2):e17–125. doi: 10.1093/eurheartj/ehz430
104. Xu S, Luo S, Zhu Z, Xu J. Small molecules as inhibitors of Pcsk9: Current status and future challenges. Eur J med Chem (2019) 162:212–33. doi: 10.1016/j.ejmech.2018.11.011
105. Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, et al. Effect of an rna interference drug on the synthesis of proprotein convertase Subtilisin/Kexin type 9 (Pcsk9) and the concentration of serum ldl cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet (2014) 383(9911):60–8. doi: 10.1016/S0140-6736(13)61914-5
106. Warden BA, Duell PB. Inclisiran: A novel agent for lowering apolipoprotein b-containing lipoproteins. J Cardiovasc Pharmacol (2021) 78(2):e157–e74. doi: 10.1097/FJC.0000000000001053
107. Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable rnai therapeutic inhibitor of Pcsk9. New Engl J Med (2017) 376(1):41–51. doi: 10.1056/NEJMoa1609243
108. Ray KK, Wright RS, Kallend D, Koenig W, Leiter LA, Raal FJ, et al. Two phase 3 trials of inclisiran in patients with elevated ldl cholesterol. New Engl J Med (2020) 382(16):1507–19. doi: 10.1056/NEJMoa1912387
109. Hardy J, Niman S, Pereira E, Lewis T, Reid J, Choksi R, et al. A critical review of the efficacy and safety of inclisiran. Am J Cardiovasc Drugs (2021) 21(6):629–42. doi: 10.1007/s40256-021-00477-7
110. Ataei S, Momtazi-Borojeni AA, Ganjali S, Banach M, Sahebkar A. The immunogenic potential of Pcsk9 peptide vaccine in mice. Curr Med Chem (2023) 30:1–8. doi: 10.2174/0929867329666220930114429
111. Wu D, Pan Y, Yang S, Li C, Zhou Y, Wang Y, et al. Pcsk9qβ-003 vaccine attenuates atherosclerosis in apolipoprotein e-deficient mice. Cardiovasc Drugs Ther (2021) 35(1):141–51. doi: 10.1007/s10557-020-07041-6
112. Momtazi-Borojeni AA, Jaafari MR, Badiee A, Banach M, Sahebkar A. Therapeutic effect of nanoliposomal Pcsk9 vaccine in a mouse model of atherosclerosis. BMC Med (2019) 17(1):223. doi: 10.1186/s12916-019-1457-8
113. Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discovery (2012) 11(8):633–52. doi: 10.1038/nrd3800
114. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial il-18 equilibrium controls barrier function in colitis. Cell (2015) 163(6):1444–56. doi: 10.1016/j.cell.2015.10.072
115. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel role of pkr in inflammasome activation and Hmgb1 release. Nature (2012) 488(7413):670–4. doi: 10.1038/nature11290
116. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the Nlrp3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
117. Davis BK, Wen H, Ting JP. The inflammasome nlrs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
118. Wu D, Chen Y, Sun Y, Gao Q, Li H, Yang Z, et al. Target of Mcc950 in inhibition of Nlrp3 inflammasome activation: A literature review. Inflammation (2020) 43(1):17–23. doi: 10.1007/s10753-019-01098-8
119. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. Nlrp3 inflammasome inhibition by Mcc950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arteriosclerosis Thrombosis Vasc Biol (2017) 37(8):1457–61. doi: 10.1161/ATVBAHA.117.309575
120. Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M, et al. The selective Nlrp3 inhibitor Mcc950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep (2021) 11(1):19305. doi: 10.1038/s41598-021-98437-3
121. MacDonald JA, Wijekoon CP, Liao K-C, Muruve DA. Biochemical and structural aspects of the atp-binding domain in inflammasome-forming human nlrp proteins. IUBMB Life (2013) 65(10):851–62. doi: 10.1002/iub.1210
122. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. Mcc950 directly targets the Nlrp3 atp-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15(6):556–9. doi: 10.1038/s41589-019-0277-7
123. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct Nlrp3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214(11):3219–38. doi: 10.1084/jem.20171419
124. Song N, Liu Z-S, Xue W, Bai Z-F, Wang Q-Y, Dai J, et al. Nlrp3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell (2017) 68(1):185–197.e6. doi: 10.1016/j.molcel.2017.08.017
125. Ma S, Chen J, Feng J, Zhang R, Fan M, Han D, et al. Melatonin ameliorates the progression of atherosclerosis Via mitophagy activation and Nlrp3 inflammasome inhibition. Oxid Med Cell Longevity (2018) 2018:9286458. doi: 10.1155/2018/9286458
126. Zhang Y, Liu X, Bai X, Lin Y, Li Z, Fu J, et al. Melatonin prevents endothelial cell pyroptosis Via regulation of long noncoding rna Meg3/Mir-223/Nlrp3 axis. J Pineal Res (2018) 64(2):e12449. doi: 10.1111/jpi.12449
127. Chen H, Deng Y, Gan X, Li Y, Huang W, Lu L, et al. Nlrp12 collaborates with Nlrp3 and Nlrc4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol Neurodegenerat (2020) 15(1):26. doi: 10.1186/s13024-020-00372-w
Keywords: PCSK9, NLRP3 inflammasome signaling, interactions, atherosclerosis, therapy
Citation: Wang Y, Fang D, Yang Q, You J, Wang L, Wu J, Zeng M and Luo M (2023) Interactions between PCSK9 and NLRP3 inflammasome signaling in atherosclerosis. Front. Immunol. 14:1126823. doi: 10.3389/fimmu.2023.1126823
Received: 18 December 2022; Accepted: 08 February 2023;
Published: 22 February 2023.
Edited by:
Wilfried Le Goff, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Charles Thomas, Université de Bourgogne, FranceCopyright © 2023 Wang, Fang, Yang, You, Wang, Wu, Zeng and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mao Luo, bHVvbWFvMjAwNTA5MDhAMTYzLmNvbQ==; Min Zeng, emVuZ21pbmx6QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.