 Cristina E. Martina
Cristina E. Martina James E. Crowe Jr.
James E. Crowe Jr. Jens Meiler1,2,6
Jens Meiler1,2,6
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 March 2023
Sec. Vaccines and Molecular Therapeutics
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1126034
This article is part of the Research Topic A year in review: Discussions in Vaccines and Molecular Therapeutics View all 7 articles
Glycan masking is a novel technique in reverse vaccinology in which sugar chains (glycans) are added on the surface of immunogen candidates to hide regions of low interest and thus focus the immune system on highly therapeutic epitopes. This shielding strategy is inspired by viruses such as influenza and HIV, which are able to escape the immune system by incorporating additional glycosylation and preventing the binding of therapeutic antibodies. Interestingly, the glycan masking technique is mainly used in vaccine design to fight the same viruses that naturally use glycans to evade the immune system. In this review we report the major successes obtained with the glycan masking technique in epitope-focused vaccine design. We focus on the choice of the target antigen, the strategy for immunogen design and the relevance of the carrier vector to induce a strong immune response. Moreover, we will elucidate the different applications that can be accomplished with glycan masking, such as shifting the immune response from hyper-variable epitopes to more conserved ones, focusing the response on known therapeutic epitopes, broadening the response to different viral strains/sub-types and altering the antigen immunogenicity to elicit higher or lower immune response, as desired.
Carbohydrates are one of the basic building blocks of the cell, together with nucleotides, amino acids and lipids (1). Complex sugar chains (or glycans) can be covalently attached to proteins (forming glycoproteins), to lipids (glycolipids), and also to RNA (glyco-RNA) (2). The covalent link between a glycan chain and a protein can be mediated by nitrogen atoms (referred as N-glycosylation), by oxygen atoms (O-glycosylation), or, more rarely, by sulfur atoms (3).
In eukaryotes, glycan chains are added to a nascent polypeptide by the glycosyltransferases in the endoplasmic reticulum (ER) (4). These enzymes recognize a pattern of residues on the unfolded protein, called sequon, that is specific for each type of glycosyltransferase and glycosylation (N or O-linked) (5). The sequon for N-glycosylation, used in glycan masking, is formed by the triplet NxS/T, in which the first residue is the asparagine that will be glycosylated, the second can be any amino acid except for proline and the third is a serine or a threonine (6).
Following glycosylation in the ER, the protein is then transported through the Golgi apparatus, in which other glycosyltransferases and glycosidases can add, remove or modify the basal glycosylation (4). After modification, the protein is then exported to the surface of the cell or released in the extracellular matrix.
The mechanism of action of the glycosyltranferases in the ER or in the Golgi is not fully understood. The same asparagine in a protein can be found with different sugar types, a phenomenon referred as glycan heterogeneity (7). For example, the SARS-CoV-2 spike protein presents a total of 183 different glycoforms, 114 of which on the same glycosylation site (8).
Most enveloped viruses such as influenza, HIV and SARS-CoV-2 present one or more glycoproteins exposed on the surface of the viral nanoparticle (9–11). These proteins are involved in receptor binding and cell entry, and they are one of the main target of the immune response during viral infection (12). In order to escape recognition from the immune system and in particular from therapeutic antibodies, viruses undergo rapid mutations which alter their glycoproteins immunogenicity (13). The insertion of new glycosylation sites on the protein surface blocks recognition and antibody binding, leading to lack in protection and increased vulnerability during infection. This natural phenomenon of glycan shielding, or glycan masking, has been adapted to alternative applications, such as immuno-focused vaccine design or protein-protein interface mapping (14, 15).
In this review, we will give an overview of the state-of-the-art in glycan masking, highlighting successes and limitations of this technique. We will describe the pathogens and antigens considered for epitope focused immunogen design and the vector systems used for enhancement of the immune response. Finally we will then explain the different applications that can be accomplished through glycan masking, including immuno-shifting, immuno-focusing, immuno-broadening and immuno-altering.
We selected 25 scientific articles in which glycan masking has been applied to vaccine design. Selection of the papers is based on: 1- the use of N-glycosylation through the insertion of a NxT/S sequon; 2- the addition of non-native glycans to the protein of interest; 3- the creation of immunogen candidates. The 25 selected articles are listed in publication order in Table 1. Articles published by the same research group, and with same pairs of targets and immunogens are grouped within a single row in the table for a total of 19 independent references.
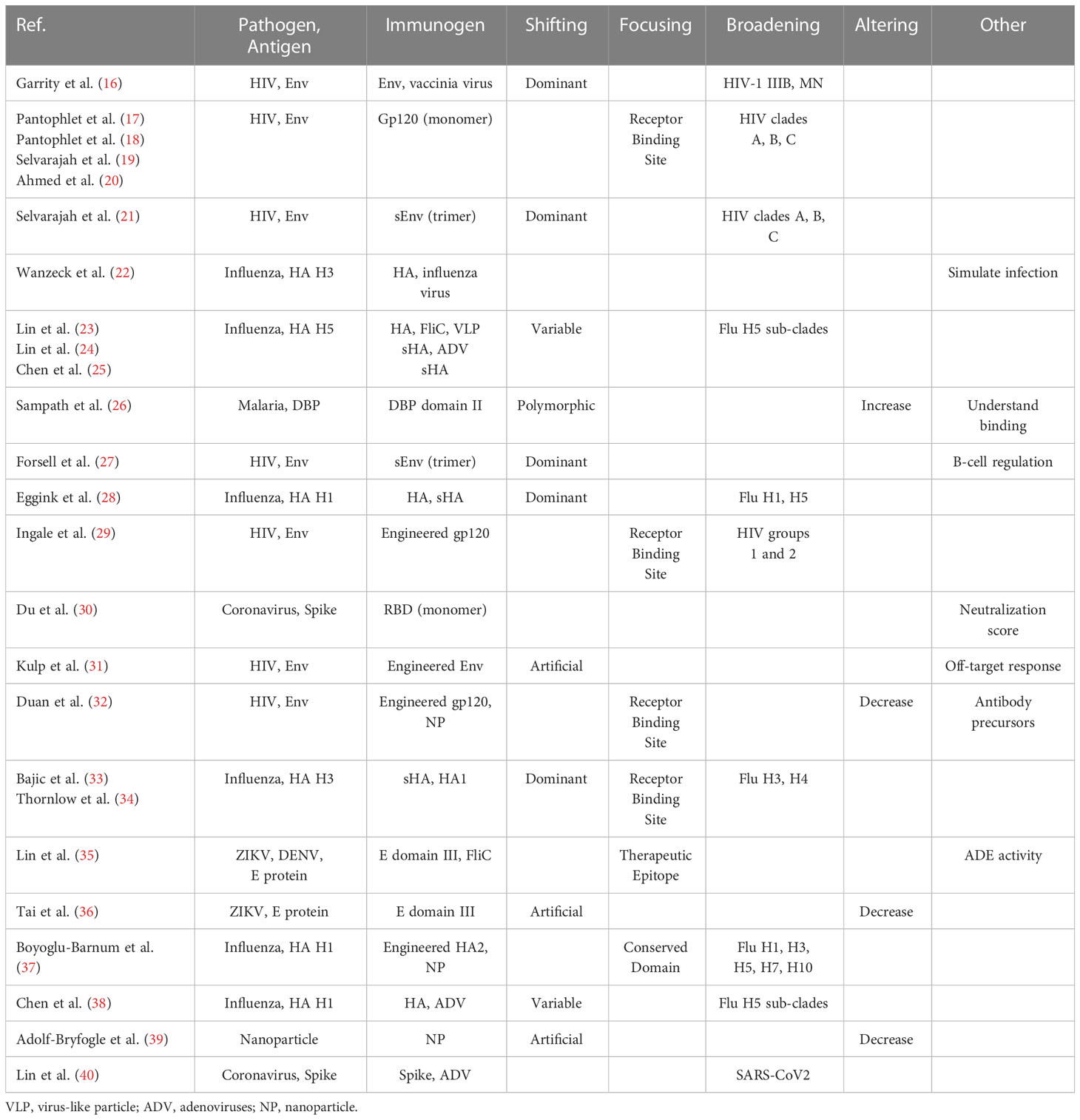
Table 1 Collection of articles on vaccine design through glycan masking.
We acknowledge that successful immunogen design examples were obtained also through removal of native glycans from the antigen of interest, referred as glycan unmasking (41, 42). Moreover, an interesting alternative in epitope masking is the nanopatterning technique, in which polyethylene glycol (PEG) molecules are used instead of glycans (43, 44). Finally, glycan masking was also successfully applied to the design of protein-based drugs such as interferon alpha 2 (45), human coagulation factor IX (46, 47) human erythropoietin (48) and human hormones (49, 50). Glycan unmasking, nanopatterning and glycosylated drugs however are out of the scope of this review and thus are not included in our collection.
Immunogen design through glycan masking is not trivial: prior knowledge on protein structures and antibody response is essential, and multiple rounds of experimental validation are needed to obtain valid vaccine candidates. For these reasons, glycan masking is applied only to historically challenging pathogens, for which conventional vaccination methods are not available or failed to completely eradicate them. The majority of the pathogens described in our collection are viruses: seven out of the 19 references target human immunodeficiency virus (HIV), six influenza virus, two coronavirus and two Zika and Dengue viruses. The only non-viral pathogen included in our collection is the parasite P. vivax, involved in malaria disease. These pathogens are extremely challenging due to a variety of reasons, including the ability to quickly evolve, the propensity to escape the immune system, the existence of a pathogenic reservoir in different host species and the absence or inefficacy of current vaccines.
The selected antigens for glycan masking are proteins exposed on the surface of the pathogens in 18 out of the 19 references. This is not surprising, since the surface proteins can directly be recognized and targeted by the antibodies produced by the immune system. The only reference not associated to human pathogens is (39), in which the glycosylated protein is a self-assembling nanoparticle developed to better expose immunogen candidates to the immune system (39).
HIV has been discovered in 1983 and it caused more than 36 million deaths worldwide (51). HIV-1 group M is responsible for the majority of HIV infections and it includes nine genetically different sub-types (A, B, C, D, F, G, H, J and K) and multiple hybrids (circulating recombinant forms, CRFs) (52). Sub-types A, B and C account for more than 70% of the global prevalence for HIV world-wide and represent the major focus in the development of therapeutics.
Efforts in producing an effective vaccine against HIV have been taken for decades (53), however no vaccine has been approved by the Food and Drug Administration (FDA) yet. Major difficulties in vaccine development have been associated to: 1- the co-existence of multiple HIV groups and sub-types; 2- the viral ability to rapidly mutate and escape the immune system and 3- the persistency of viral reservoir in the host immune system (54).
The HIV surface glycoprotein is Env, a trimer of heterodimers composed by a N-terminal ectodomain (gp120) and a C-terminal membrane domain (gp41) (55). Env is initially produced as a single polypeptide of 160 kDa, referred as gp160, that is then cleaved during viral maturation. Gp120 includes five conserved regions (C1-C5), which mediates binding to the host receptor CD4, and five variable regions (V1-V5). Gp41 is composed by a cytoplasmic domain, a transmembrane domain and an extracellular domain which drives cell fusion and entry. The Env protein is heavily glycosylated, with 18 to 33 native glycans per monomer (56). Glycosylation and mutations accumulated in the variable regions of gp120 mainly contribute to viral escape (54).
Influenza virus caused countless epidemics and three major worldwide pandemics in the last century: the Spanish flu in 1918, with 50 million deaths; the Asian flu in 1957, with 1 million deaths and the Hong Kong flu in 1968, also with 1 million deaths (57). Influenza virus is classified in four types, A, B, C and D, with only the first three causing illness in humans. Influenza A and B are cause of multiple epidemics, but only type A is cause of the pandemics. Influenza A is divided in sub-types based on the two surface glycoproteins: hemagglutinin (HA) and neuraminidase (NA). There are 18 different HA sub-types (H1-H18) and 11 NA sub-types (N1-N11), but more than 130 combinations of HA and NA have been identified (57). Influenza virus can undergo to two main evolutionary processes: antigenic drift, in which mutations are accumulated overtime leading to viral escape, and antigenic shift, in which co-infection of avian, swine or human influenza variants led to gene reassortment (59). Antigenic drift and shift highly contribute to the long-term inefficiency of current vaccinations against influenza virus. Efforts in vaccine design have mainly focused on HA, since it is responsible for cell attachment and entry (60).
HA, similarly to the Env protein of HIV, is a trimer of heterodimers formed by a head domain (HA1) and a stem domain (HA2), also referred as stalk. The head domain engages and binds the host receptor (sialic acid) through the conserved Receptor Binding Site (RBS), while the stem domain is involved in membrane fusion and cell entry mediated by the fusion loop (61).
The majority of the antibodies elicited by influenza infection or vaccination target the HA head domain (62). For this reason, the head domain is considered immuno-dominant, while the stalk domain is considered immuno-recessive. However, the head domain is also highly variable and mutagenic, and antibodies directed toward the head domain are in general less broadly reactive to other viral strains than the antibodies directed towards the more conserved stalk.
N-glycosylation of HA plays a key-role in receptor binding, membrane fusion, virulence and escape (63). Glycans located in the stem domain are necessary for proper folding and trimerization, and are highly conserved among different viral strains. On the other hand, glycans located on the head domain are mainly involved in viral escape and they widely differ in number and location: for example the HA head domain of influenza H3N2 counts from two to six glycosylation sites, while H7N9 has a single glycosylation site (64).
The coronavirus family includes three main viruses that caused disease in humans: SARS-CoV, which originated the SARS outbreak in 2002-2003 with 8000 infections and at least 774 deaths; MERS-CoV, which caused outbreaks in 2012, 2015 and 2018 with more than 2500 cases and 880 deaths (65) and SARS-CoV-2, which caused the global pandemic in 2019, currently ongoing, with more than 600 million cases and 6,4 million deaths worldwide (66).
In only two years SARS-CoV2 accumulated hundreds of different mutations in its genome, and five variants are considered a major concern: Alpha, first described in UK in December 2020; Beta, South Africa in December 2020; Gamma, Brazil in January 2021; Delta, India in December 2020 and Omicron, South Africa in November 2021 (67).
In 2020-2022, 18 different vaccines have been developed against the original strain of SARS-CoV2 (Wuhan-Hu-1): three are RNA-based, four protein-based, four non-replicating viral vectors, and seven are inactivated virus (68). Most vaccines result effective against newer viral variants including Alpha, Beta, Gamma, Delta and Omicron (69), but it is not clear yet if they will be protective against new emerging strains of SARS-CoV2.
The spike protein, also referred as protein S, is the major target in antibody response upon both natural infection and vaccination protocols (70). As for HIV and influenza virus, the spike protein is a trimer of heterodimers composed by the S1 and S2 domains. The receptor binding domain (RBD), mediates cell attachment by binding to the ACE2 receptor (SARS-CoV and SARS-CoV2) or to the DPP4 receptor (MERS-CoV). The RBD is located in S1, together with the N-terminal domain (NTD) and the C-Terminal domain (CTD). The S2 subunit includes the Fusion Peptide (FP), two heptad repeat regions, the single-pass transmembrane domain and the cytoplasmic tail. FP mediates fusion of the host and virus membranes, allowing viral entry in the cells (11).
The SARS-CoV2 Spike protein contains native 22 glycosylation sites per monomer, mostly located at the NTD domain in the S1 sub-unit (71).
Dengue and Zika viruses belong to the same Flaviviridae family and, together with malaria, they represent the major health concern for mosquito-borne diseases.
Dengue infection rates increased dramatically in the past decades, with 50-100 million cases every year. Mortality is lower than HIV or influenza virus, with a global average of more than 9000 cases per year (72). Dengue virus is divided in four main serotypes (DENV1-4) and 19 genotypes with different transmission rates, cellular targets and immune responses. Antibodies produced after primary infection are able to cross-react, but not neutralize, other Dengue strains, and during secondary infection they might cause more severe symptoms and illness. This phenomenon, called Antibody Dependent Enhancement (ADE), represents one of the main limitation in the production of effective vaccines against dengue virus (73). There is a single licensed vaccine against dengue virus, named Denvaxia, but it is currently administered only to individuals that have already been infected naturally by the virus, while seronegative individuals have higher risk of developing severe illness after secondary infection (74).
Zika virus outbreaks were recorded in 2007 in the Pacific Islands and in 2015-2016 in Latin America, with an estimation of 1.62 million cases (75). Zika infections in adults lead generally to mild symptoms, however the virus can be transmitted from pregnant women to fetuses causing miscarriages, congenital microcephaly, brain malformations or eye abnormalities in the newborns (76). No vaccine is currently available against Zika virus.
The envelope protein of dengue and Zika virus is the E protein, and it is involved in receptor binding and cell fusion. Differently from the Env, HA and Spike proteins which belong to class-I fusion proteins, E protein belongs to the class-II, and in pre-fusion state it forms a head-to-tail homodimer. Each monomer is formed by five domains: a finger-like dimerization domain containing the fusion loop (DII), a beta-barrel domain (DI), an immunoglobuline like domain (DIII), a stem domain and a transmembrane domain (77). DIII, from both dengue and Zika, has been reported to elicit neutralizing antibodies (nAbs) in animals and represent a major target for immunogen design (78, 79). Dengue E protein contains two glycosylation sites per monomer, while Zika E protein only one. Glycosylation plays a major role in cell attachment viral replication, transmission in both mammalian and insect cells and overall pathogenesis (80).
The only parasite included in our collection is Plasmodium vivax, one of the six Plasmodium variants responsible for malaria disease (81). Malaria circulated globally since millennia, with 300 million deaths in the 20th century and over 600000 in 2020 (82). Challenges in the eradication of malaria disease include the parasite complex multi-stage life cycle, its persistency in mosquito reservoir and the development of resistance against drugs and insecticides (83).
Antibody-mediated immune response against P. vivax was shown in 1961 when purified immunoglobulines were passively transferred from adult survivors to young children, showing protection (84). This discovery raised interest in the development of vaccines, however only one candidate, PfSPZ, reached clinical trial phase IV and is currently in trial until 2023 (85).
The Duffy Binding Protein (DBP) is the surface protein selected for malaria-based immunogen design described in Sampath et al. (26). DBP mediates attachment, entry and invasion of red blood cells, essential steps in the parasite life cycle. DBP binds to the host DARC receptor with region II, formed by 3 subdomains (86). Epitopes reported to be highly immunogenic are the dimeric interface, the conserved subdomain 3 and the subdomain 2 (87).
Epitope-focused immunogen design through glycan masking is based on the idea that specific epitopes, known for eliciting therapeutic antibodies, are fully accessible for recognition by the immune system, while other non-relevant epitopes are hidden with glycan chains. To mask a specific epitope, selected residues have to be substituted with the sequon for N-glycosylation, NxS/T where x can be anything except for a proline (6). The sequon is necessary for insertion of glycan chains, however refined databases for glycoproteins showed that not all sequons are glycosylated once the protein is produced within the cell (88). This possibility has to be kept in mind while designing immunogen candidates.
Moreover, careful preparation of the immunogens is essential to maximize the exposition of therapeutic epitopes and to minimize the availability of non-therapeutic ones. Three main categories of immunogens are used in our collection: native-like, corresponding to the full extracellular antigen exposed by the pathogens; domain-based, in which only a portion of the antigen is selected for glycan masking and engineered antigens. All types of immunogens can be “loaded” onto a supra-molecular vector systems, which better expose the epitopes to the immune system. All four cases will be described in this chapter.
Native-like immunogens created through glycan masking include both the full antigen proteins or their soluble versions, which lack the intracellular and transmembrane domains. The advantage to use native-like immunogens is that the overall structure closely mimic the one exposed on the pathogens surface, including their oligomeric state. Moreover, the risk to present artificial epitopes is minimal, thus avoiding the production of off-target antibodies. Disadvantages however are multiple: some antigens are poorly stable when lacking the transmembrane and intracellular domains, or they fail in forming the correct oligomeric complex with high efficiency. Moreover many viral glycoproteins are heavily glycosylated in their native form (i.e., HIV). The addition of numerous non-native glycans during glycan masking might lead to misfolding and non-expression of the immunogen candidate (89), or to significant decrease in immune response upon immunization.
Immunogens created against HIV presents the trimeric env protein in its native conformation, using the full gp160 gene (16), or gp140 (referred also as sEnv), a soluble version of gp160 lacking the intracellular and transmembrane domains (21, 27). Immunogens against influenza virus present the full trimeric HA antigen, including the intracellular C-terminal region and the transmembrane domain (22), or the soluble ectodomain version sHA (33, 38), or both (23–25, 28). Immunogens against SARS-CoV2 are also composed by the full length Spike protein in its native conformation (40).
Differently from the native-like immunogens, the domain-based ones are composed by a sub-portion of the full antigen: the gp120 domain of Env of HIV (17–20), the subdomain II of DBP of P. vivax (malaria) (26), the RBD of the spike protein of MERS (30), the HA head domain (HA1) of hemagglutinin of influenza virus (33, 34) and the EDIII domain of (35, 36) viruses.
Domain-based immunogens are generally easier to produce due to their smaller size and do not require in vitro oligomerization; however they might present artificial epitopes which can elicit off-target antibodies. Generally, the choice of a domain-based immunogen is driven by prior knowledge on the overall antibody response against the full antigen: if a domain is associated with higher protection, removing the other poorly therapeutic domains contributes to better focus the immune system and to obtain more protective antibodies.
In some cases, the choice of a domain-based immunogen is driven by the lack of structural knowledge for the full antigen, and only individual domains are available to guide the epitope-focused immunogen design through glycan masking.
Engineered versions of the antigens are used as immunogens in four cases out of our 19 references. These variants are mostly used for increased stability and expression yields, for decreased immunogenicity, or to mimic specific conformations. Extensive research is necessary to generate engineered proteins prior to glycan masking.
The glycan masked candidates designed in (29) against HIV-1 are based on Ds12F123 core, and engineered version of gp120 in which the major variable loops V1, V2 and V3 have been deleted and mutations have been introduced to adopt a CD4-bound conformation (90).
Similarly, the immunogen candidates against HIV created by (31) are based on BG505 SOSIP.664, an engineered version of gp140 with superior expression yields and stability, and with mutations in the variable loop V3 to reduce reactivity (91). Immunogens from Duan et al. against HIV (32) are based on eOD-GT8 60-mer, a circularly permutated version of gp120 in fusion with a self assembling nanoparticle subunit (92).
Finally, Boyoglu-Barnum et al. developed an engineered version of HA in which only the stem portion is displayed in a trimeric conformation on a nanoparticle vector (37, 93). The HA stem is more conserved across different influenza strains than the HA head domain, and thus could elicit more broadly reactive antibodies.
One key factor to consider in vaccine design through glycan masking is how immunogens will be presented to the immune system. Although domain-based immunogen candidates are able to elicit an immune response, the use of adjuvants or delivery systems was shown to enhance the immune response (94).
Full native-like immunogens containing the transmembrane domain require a membrane environment for stability. Using the enveloped virus itself is a valid option when the risks of infection lead to less severe outcomes. For example, the glycosylated version of the full HA of influenza virus developed by Wanzeck et al. has been reintroduced in the influenza genetic material, and the full virus has been used for animal immunization (22).
However, this would not be possible with pathogens that cause highly severe symptoms or terminal illness, such as HIV. To overcome this limitation, Garrity et al. used recombinant vaccinia virus, an enveloped virus widely used in human vaccination (95), to expose the full glycosylated Env protein and immunize animal models (16).
Adenoviruses (ADVs) are another class of non-enveloped viruses widely used for vaccination (96). They are used to expose the full glycosylated version of the Spike protein of SARS-CoV2 (40), however, due to the fact they do not have a membrane envelope, they are more often used with soluble versions of the native antigen (lacking the transmembrane domain) or with domain-based immunogens. The soluble glycosylated versions of HA have also been exposed on an adenovirus particle (24, 38).
Virus-like particles (VLPs) are great alternatives to viral vectors for vaccination purposes (97). VLPs are nanoparticles formed from viral proteins, but lacking the genetic material, and thus not able to replicate. They better mimic the dimension of a viral pathogen and are safer than live virus systems. VLPs are used to expose the full glycosylated HA and immunize mice (23).
Similarly to VLPs, self-assembling non-viral nanoparticles (NPs), are used as vector system to better expose immunogen candidates. This is the case for the engineered Env immunogen against HIV (32) and for the engineered HA stem against influenza virus (37).
A major difficulty in using non-viral systems or proteins, is that the immune response upon immunization can be directed not only against the immunogen itself but also against the vector system. In particular, a fraction of the antibodies produced by the immune system would be directed toward an artificial site, which would not be present on the pathogen of interest. To overcome this problem, Adolf-Bryfogle et al. performed glycan masking on a two-component self-assembling nanoparticle specifically designed to expose immunogen candidates (98, 99). Although immunogen candidates are not present on the nanoparticle itself, the glycosylated vector system elicited a lower immune response after immunization in comparison to the unglycosylated version, showing that glycans were able to hide the artificial epitopes (39).
Finally, the only other vector system used in our collection is the soluble form of the flagellin protein (FliC), which has been considered as vaccine adjuvant to enhance the immune response (100). FliC has been used in fusion with the full HA and exposed on a VLP (39), and in fusion to the DIII domain of the E protein of Dengue and Zika viruses (35).
As implied in the name, one of the main application of glycan masking is to “mask” or “hide” unwanted portions of the protein of interest. However different goals can be reached with the same technique. Among the 19 references, we identified four main categories for which glycan masking is widely used: immuno-shifting, immuno-focusing, immuno-broadening and immuno-altering (Table 1). Goals that are specific for a single reference are grouped in the fifth category “other goals” and are described in the next chapter.
The most common reason to use glycan masking in immunogen design is to shift the immune response from certain epitopes to others. Immuno-shifting can be accomplished also with other techniques, from simple mutagenesis to deletion of larger regions of the protein. For example, glycosylations are added to an engineered version of the env proteins in which the variable regions were previously removed or mutated (29, 31, 90, 91). Immuno-shifting mediated by glycosylation is performed in 11 out of the 19 references presented in this review. Most commonly the goal of glycan masking is to shift away from immuno-dominant epitopes, which elicit high antibody titers but low neutralization activity and thus low protection. Such epitopes are also called “immune decoy epitopes” or “decotopes” by (16).
Immuno-shifting is also used for antigens with highly variable regions, which might elicit potent immune response, but not a broader one. To note, most of the highly variable epitopes considered in our references are also immuno-dominant, however not all the immuno-dominant epitopes are highly variable: the Receptor Binding Site (RBS) of HA is an example of conserved epitope in an immuno-dominant region. Immuno-recessive regions are usually associated to more conserved epitopes, able to elicit broadly neutralizing antibodies.
For the env protein of HIV, immuno-dominant and highly variable epitopes are the V1, V2 and V3 loops. V3 only can be mutated to insert additional glycosylations (16, 27), or all three loops can be glycosylated (21). For influenza virus, the head domain of HA is the immuno-dominant domain and glycosylation was used to shift immune-response to the more conserved stalk (28, 33). The hyper-variable epitopes within the head domain were also mutated for immuno-shifting (23, 38). For malaria, the only non-viral entry in our set, glycan masking was used to shift immune-response away from the polymorphic region (26).
Finally, immuno-shifting is also used to mask artificial sites, generally defined as epitopes which, during natural infection, are not encountered by the immune system. Such epitopes derives in most cases from modification of the natural protein, by truncation or by extraction of individual domains. Glycans were added at the base of the env protein, a region that in the full virus is inaccessible due to the presence of the membrane and of the transmembrane region (31). In the work of Tai et al., the DIII domain of the protein E is used as recombinant immunogen candidate, and glycosylation is used to mask the protein side previously in contact with the other domains in the glycoprotein. Such site is not accessible during natural infection with Zika virus (36). Artificial sites include also proteins that were artificially designed, as for the nanoparticle used in (39). The nanoparticle is intended as a vector to present immunogens to the immune system, not as an immunogen itself, thus representing an artificial site.
Alternative to immuno-shifting is immuno-focusing in which glycans are strategically added to focus the immune system to a particular epitope. More generally, immuno-shifting is best described as eliciting immune response “away from” certain epitopes, while immuno-focusing as eliciting it “to” specific epitopes.
In immuno-focusing, glycosylation sites are inserted outside the epitope of interest, ideally to reduce the production of off-target antibodies. Out of the 19 references we listed, six performs immuno-focusing. The major targets in this category are highly conserved epitopes able to elicit broadly neutralizing antibodies, as for the receptor binding sites CD4bs in HIV Pantophlet et al. (17–20, 29, 32, 33, 37). Glycan masking was used to focus on the DIII domain of Zika virus, previously shown to elicit broad neutralizing antibodies (35).
Immuno-broadening is defined as the ability for a given immunogen to give immune protection among a wider range of viral strains or pathogenic sub-types. Such wider protection is generally obtained when broadly neutralizing antibodies are elicited by highly conserved epitopes. Thus, both immuno-shifting and immuno-focusing can lead to immuno-broadening by respectively shifting or focusing the immune response to conserved epitopes. It is important to note that immuno-shifting or immuno-focusing can also lead to other outcomes rather than immuno-broadening, as seen in the next sections.
From a biomedical point of view, the ability to target multiple viral strains with a single vaccine is of high interest, and in nine out of the 19 references the goal of glycan masking is to broaden the immune response. In five cases immuno-broadening is reached by immuno-shifting: shifting away from the hyper-variable regions of HIV-1 (16, 21) and influenza virus (23–25), or by shifting away from the immuno-dominant epitopes in the HA protein of influenza (28). In three cases immuno-broadening is reached by immuno-focusing: for HIV-1 by focusing on the conserved receptor binding site, CD4bs (17–20, 29), and for influenza virus by focusing on the conserved stalk domain (37). Immuno-broadening for influenza virus is also attempted by both immuno-focusing on the receptor binding site (RBS) with one glycan mutant and by immuno-shifting away from the RBS with another mutant (33, 34).
Immuno-shifting, immuno-focusing and immuno-broadening are intended to change the immune response by altering the antibody recognition pattern, but not the overall amount of elicited antibodies. In other words, antibody titers obtained by immunization with the glycan mutants should remain the same as the ones obtained with the non-glycosylated protein. In immuno-altering the goal is to change the amount of total elicited antibodies. In case of poorly immunogenic proteins, such as the DBP protein of malaria (26), the goal of glycan masking is to increase antibody titers to obtain a more immunogenic protein. In case of immunogens that elicit good antibody titers, but that lack in therapeutic effects, such as the gp120 protein of HIV-1 Kulp et al. (31), the goal is to decrease the overall antibody titers and in particular to decrease the off-target ones. In case of immunogens presenting artificial sites that wouldn’t be encountered by the immune system during natural infection, such as the “hidden” epitope in the Zika virus immunogen (36), the goal is again to decrease the number of off-target antibodies. Finally, for completely artificial constructs such as the nanoparticle designed to better expose viral immunogens (39), the goal is to decrease the overall antibody titers, making it poorly immunogenic and focus the immune response to the loaded immunogens.
Immuno-shifting, focusing, broadening and altering are goals shared by multiple references in our set, and they represent the major applications of immunogen design through glycan masking. However the technique can be applied also to answer very specific questions, as seen in seven references out of the 19.
Wanzeck et al. used glycosylation to study what happens if subjects immunized with hyper-glycosylated viral variant encounter poorly glycosylated ones (22). For example, ancient strains of influenza virus were initially poorly glycosylated and HA accumulated glycosylation during co-evolution with the human host to escape the immune system (64). The risk that ancient (and poorly glycosylated) influenza variants re-emerge among the human population is a major concern. In particular, a novel H1N1 influenza strain caused an outbreak in 2009 in Mexico, Canada and USA, leading to severe symptoms in younger patients and mild ones in elders (101). Wanzeck et al. replicated the H1N1 outbreak in mice, with the hypothesis that older patients encountered poorly glycosylated variants of influenza virus during their youth, and thus were protected, while younger patients were more susceptible because they only encountered hyper-glycosylated viral versions. First they immunized mice with an hyperglycosylated HA variant of influenza virus, and then they infected them with a hypoglycosylated virus. The authors were able to demonstrate that mice were more susceptible to the secondary infection against poorly glycosylated variants despite vaccination (22).
Sampath et al. used glycan masking to both shift and alter the immune response against the DBP protein of malaria, but also to infer the mechanism of binding of DBP to the human DARC receptor (26). The receptor is extremely important for the progress of the infection and DARC-null individuals are less likely to get sick (102). The underlying question is why DBP is poorly immunogenic. Two theories, non mutually exclusive, suggest that 1- antibody recognition is lacking due to a strong affinity between monomeric DBP and the DARC receptor and that 2- interactions with the DARC receptor induce dimerization of DBP thus limiting antibody binding to the conserved epitope (103). With the glycosylated immunogens the authors were able to block DBP binding to DARC receptor and individuate the binding interface (26).
Forsell et al. used glycan masking to study how monoclonal B cell lineages develop, compete and persist in vivo after immunization (27). In particular they want to understand if B cell clones targeting independent epitopes compete with each other and if masking one epitope leads to overall reduction in the total immune response or if it leads to its redistribution to other epitopes on the antigen. Such knowledge is important for the design of epitope-focused vaccines (104). They use as model the soluble trimeric env protein of HIV (105) and insert three glycosylation sites to mask the V3 loop. Before mice immunization, they check binding of the wild-type and glycosylated antigens to plated naive B-cell, showing that the glycosylated immunogens have lower binding compared to the wild-type. This suggest that the available B-cell population for development of potent antibodies is reduced beforehand by the presence of the glycans. After mice immunization they confirm that different epitopes are independently regulated and that there is no competition among their B-cell lineages (27).
Glycan masking has been used to develop the Neutralizing Immunogenicity Index (NII), a novel concept to quantitatively determine how different epitopes contributes to viral neutralization (30). This concept is very important in the field of immunogen design, since it can differentiate epitopes with a strong therapeutic response from the ones without (106). To calculate the NII value for each epitope, the neutralization activity of post-immunization sera is measured for both a wild-type and single glycan immunogens. Negative values of NII indicates that the epitope contributes negatively to the overall neutralization capability of the immunogen, while positive values indicates that the epitope contribute substantially to the therapeutic effect. Knowing the NII for each epitope in an antigen can easily guide the design of strong immunogens, since the ones contributing negatively in the NII scale can be masked or altered.
Glycan-masked variants have also been used to probe the immune response elicited with standard (non glycosylated) immunogens, and in particular to identify the species-specific off-target response (31). A gp41-dependent, non neutralizing antibody was elicited in rabbits during immunization (107), and the glycan masked variants were able to partially or completely block its binding. A specific class of neutralizing antibodies (VRC01-like) targeting the CD4bs epitope were elicited in mice after immunization, together with off-target antibodies (108). The glycan masked variants were shown to block this off-target response without affecting the therapeutic response obtained by VRC01-like antibodies. Finally, sera obtained after immunization of non-human primates was tested in a competition assay with the glycan variants previously shown to block off-target response (109). Only 77% to 63% of reactivity was retained by the masked variants, suggesting that an off-target response might be elicited also in non-human primates.
Duan et al. designed glycosylated variants to test for binding with both revertants and precursor of the broadly-neutralizing antibody VRC01 (32). The rational is that to elicit VRC01-like antibodies, the immunogen candidate must be able to also bind the VRC01 precursors. Immunogens based on the monomeric version of gp120 and on the native env trimer fail to bind VRC01 precursors, and consequently are not able to elicit this class of potent antibodies (110). Engineered antigen variants such as eOD-GT8 are able to bind both VRC01 precursors and revertants, however they display high off-target immunogenicity (91). To reduce the number of off-target antibodies glycans were strategically added on the surface of the engineered env trimer and tested for binding to VRC01 precursor and revertants. The glycosylated proteins were shown to produce up to 4 times more VRC01-like antibodies compared to the unmasked variant (32). The concept of testing for precursor binding is innovative in the glycan masking field and its integration in the immunogen design protocol could lead to better vaccine candidates.
Glycosylated immunogen candidates designed to mask the highly conserved fusion loop (FL) epitope in the DII domain of protein E of DENV and ZIKV were developed by (35). The FL epitope was shown to elicit cross-reactive non-neutralizing antibodies able to facilitate viral entry in the cell in secondary infections, thus enhancing the disease instead of preventing it Halstead (111). This phenomenon is called antibody-dependent enhancement (ADE), since the worsening of the disease is mediated by antibodies. The ADE activity has been shown for multiple pathogens, including DENV and ZIKV (73), and it is a major problem in the development of vaccine candidates (112). To mask the FL epitope, four immunogens were designed, each with one glycosylation. One out of the four immunogens was able to reduce ADE activity compared to the wild-type variant, while the remaining three were able to completely abolish it. This experiment demonstrates that glycan masking can be successfully used to prevent disease enhancing ADE (35).
Glycan masking in epitope-focused immunogen design is a powerful technique, as shown in the 19 successful examples reported in this review. Upon immunization in animal models, the glycosylated immunogens were able to shift the immune response from immuno-dominant and hyper-variable epitopes to more conserved ones, to focus it on the epitopes of interest, to broaden it to alternative viral strains and to alter it by increasing or decreasing the overall antibody response. Glycan masking has also been used to reduce ADE activity, to elicit antibody precursors, to test off-target response, to evaluate the neutralizing capabilities for different epitopes, to inspect the B-cell regulation mechanism and to simulate potential viral outbreaks.
Immunogens in glycan masking span from native-like antigens, sub-domains or engineered candidates. Vector systems can be used to better display the immunogens, to improve their stability, or to enhance the immune response. Any pathogen could potentially be selected for glycan masking, however the current cases are limited to pathogens for which conventional vaccination failed or is not approved yet.
There are various reasons for which glycan masking cannot represent the first attempt of vaccine design given a new pathogen outbreak. First, knowledge on the immunogenicity of the pathogen extra-cellular proteins is necessary. Proteins associated with a strong immune response, i.e., potent neutralizing antibodies, are the ideal candidates for glycan masking. Second, glycan masking often requires multiple rounds of glycosylation to obtain successful candidates. Extra glycosylations can lead to misfolding, and protein expression is inversely correlated to the number of glycosylation added to the protein of interest. Moreover, the sequon sequence can be present at the correct location on the antigen, but the glycosylation could be missing. In this situation, the only solution is to re-try masking by picking nearby residues.
A third limitation in immunogen design through glycan masking is the availability of structural information. Knowledge on the overall atomic structure of the antigen is recommended to successfully identify potential glycosylation sites on the protein surface, and thus avoid misfolding. The lack of structural information, i.e., when only a domain is experimentally determined in a protein, might lead to introduction of artificial sites that can trigger an off-target immune response. This limitation can however be overcome with the advances in protein structure prediction lead mainly by AlphaFold (113) and RoseTTA Fold (114), in which accurate protein models can be obtained through machine learning methods.
CM conceived and wrote the initial draft of the review. CM, JC and JM contributed to manuscript revision and all authors approved the submitted version.
This work was supported by a grant from the National Institutes of Health (NIH) U01 AI150739-01.
JC has served as a consultant for Luna Labs USA, Merck Sharp & Dohme Corporation, Emergent Biosolutions, GlaxoSmithKline and BTG International Inc, is a member of the Scientific Advisory Board of Meissa Vaccines, a former member of the Scientific Advisory Board of Gigagen (Grifols) and is founder of IDBiologics. The laboratory of JC received unrelated sponsored research agreements from AstraZeneca, Takeda, and IDBiologics.
The remaining authors declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Marth JD. A unified vision of the building blocks of life. Nat Cell Biol (2008) 10(9):1015–5. doi: 10.1038/ncb0908-1015
2. Flynn RA, Pedram K, Malaker SA, Batista PJ, Smith BAH, Johnson AG, et al. Small RNAs are modified with n-glycans and displayed on the surface of living cells. Cell (2021) 184:3109–24.e22. doi: 10.1016/j.cell.2021.04.023
3. Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology (2002) 12:43R–56R. doi: 10.1093/glycob/12.4.43R
4. Rini JM, Esko JD. Glycosyltransferases and glycan-processing enzymes. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al, editors. Essentials of glycobiology, 3rd ed. Cold Spring Harbor (NY: Cold Spring Harbor Laboratory Press (2015). Available at: http://www.ncbi.nlm.nih.gov/books/NBK453021/.
5. Dutta D, Mandal C, Mandal C. Unusual glycosylation of proteins: Beyond the universal sequon and other amino acids. Biochim Et Biophys Acta Gen Subjects (2017) 1861(12):3096–108. doi: 10.1016/j.bbagen.2017.08.025
6. Kohda D. Structural basis of protein asn-glycosylation by oligosaccharyltransferases. Adv Exp Med Biol (2018) 1104:171–99. doi: 10.1007/978-981-13-2158-0_9
7. Strum JS, Nwosu CC, Hua S, Kronewitter SR, Seipert RR, Bachelor RJ, et al. Automated assignments of n- and O-site specific glycosylation with extensive glycan heterogeneity of glycoprotein mixtures. Anal Chem (2013) 85:5666–75. doi: 10.1021/ac4006556
8. Chang D, Klein JA, Nalehua MR, Hackett WE, Zaia J. Data-independent acquisition mass spectrometry for site-specific glycoproteomics characterization of SARS-CoV-2 spike protein. Anal Bioanal Chem (2021) 413(29):1–14. doi: 10.1007/s00216-021-03643-7
9. Swanstrom R, Graham WD, Zhou S. Sequencing the biology of entry: The retroviral env gene. Curr Top Microbiol Immunol (2017) 407:65–82. doi: 10.1007/82_2017_35
10. Russell CJ. Hemagglutinin stability and its impact on influenza a virus infectivity, pathogenicity, and transmissibility in avians, mice, swine, seals, ferrets, and humans. Viruses (2021) 13:746. doi: 10.3390/v13050746
11. Zhang J, Xiao T, Cai Y, Chen B. Structure of SARS-CoV-2 spike protein. Curr Opin Virol (2021) 50:173–82. doi: 10.1016/j.coviro.2021.08.010
12. Wilson IA, Stanfield RL. 50 years of structural immunology. J Biol Chem (2021) 296:100745. doi: 10.1016/j.jbc.2021.100745
13. Vossen MT, Westerhout EM, Soderberg-Nauclér C, Wiertz EJ. Viral immune evasion: a masterpiece of evolution. Immunogenetics (2002) 54:527–42. doi: 10.1007/s00251-002-0493-1
14. Hariharan V, Kane RS. Glycosylation as a tool for rational vaccine design. Biotechnol Bioeng (2020) 117:2556–70. doi: 10.1002/bit.27361
15. Deimel LP, Xue X, Sattentau QJ. Glycans in HIV-1 vaccine design - engaging the shield. Trends Microbiol, (2022) 30(9):S0966–842X(22)00042-7. doi: 10.1016/j.tim.2022.02.004
16. Garrity RR, Rimmelzwaan G, Minassian A, Tsai WP, Lin G, Jong J, et al. Refocusing neutralizing antibody response by targeted dampening of an immunodominant epitope. J Immunol (1997) 159:279–89. doi: 10.4049/jimmunol.159.1.279
17. Pantophlet R, Wilson IA, Burton DR. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J Virol (2003) 77:5889–901. doi: 10.1128/JVI.77.10.5889-5901.2003
18. Pantophlet R, Wilson IA, Burton DR. Improved design of an antigen with enhanced specificity for the broadly HIV-neutralizing antibody b12. Protein Eng Design sSelection: PEDS (2004) 17:749–58. doi: 10.1093/protein/gzh085
19. Selvarajah S, Puffer B, Pantophlet R, Law M, Doms RW, Burton DR. Comparing antigenicity and immunogenicity of engineered gp120. J Virol (2005) 79:12148–63. doi: 10.1128/JVI.79.19.12148-12163.2005
20. Ahmed FK, Clark BE, Burton DR, Pantophlet R. An engineered mutant of HIV-1 gp120 formulated 540 with adjuvant quil a promotes elicitation of antibody responses overlapping the CD4-binding site. Vaccine (2012) 30:922–30. doi: 10.1016/j.vaccine.2011.11.089
21. Selvarajah S, Puffer BA, Lee FH, Zhu P, Li Y, Wyatt R, et al. Focused dampening of antibody response to the immunodominant variable loops by engineered soluble gp140. AIDS Res Hum Retroviruses (2008) 24:301–14. doi: 10.1089/aid.2007.0158
22. Wanzeck K, Boyd KL, McCullers JA. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am J Respir Crit Care Med (2011) 183:767–73. doi: 10.1164/rccm.201007-1184OC
23. Lin SC, Lin YF, Chong P, Wu SC. Broader neutralizing antibodies against H5N1 viruses using prime-boost immunization of hyperglycosylated hemagglutinin DNA and virus-like particles. PloS One (2012) 7:e39075. doi: 10.1371/journal.pone.0039075
24. Lin SC, Liu WC, Jan JT, Wu SC. Glycan masking of hemagglutinin for adenovirus vector and recombinant protein immunizations elicits broadly neutralizing antibodies against H5N1 avian influenza viruses. PloS One (2014) 9:e92822. doi: 10.1371/journal.pone.0092822
25. Chen TH, Liu WC, Lin CY, Liu CC, Jan JT, Spearman M, et al. Glycan-masking hemagglutinin antigens from stable CHO cell clones for H5N1 avian influenza vaccine development. Biotechnol Bioeng (2019) 116:598–609. doi: 10.1002/bit.26810
26. Sampath S, Carrico C, Janes J, Gurumoorthy S, Gibson C, Melcher M, et al. Glycan masking of plasmodium vivax Duffy binding protein for probing protein binding function and vaccine development. PloS Pathogens (2013) 9:e1003420. doi: 10.1371/journal.ppat.1003420
27. Forsell MNE, Soldemo M, Dosenovic P, Wyatt RT, Karlsson MCI, Karlsson Hedestam GB. Independent expansion of epitope-specific plasma cell responses upon HIV-1 envelope glycoprotein immunization. J Immunol (Baltimore Md: 1950) (2013) 191:44–51. doi: 10.4049/jimmunol.1203087
28. Eggink D, Goff PH, Palese P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J Virol (2014) 88:699–704. doi: 10.1128/JVI.02608-13
29. Ingale J, Tran K, Kong L, Dey B, McKee K, Schief W, et al. Hyperglycosylated stable core immunogens designed to present the CD4 binding site are preferentially recognized by broadly neutralizing antibodies. J Virol (2014) 88(24):14002–16. doi: 10.1128/JVI.02614-14
30. Du L, Tai W, Yang Y, Zhao G, Zhu Q, Sun S, et al. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat Commun (2016) 7:13473. doi: 10.1038/ncomms13473
31. Kulp DW, Steichen JM, Pauthner M, Hu X, Schiffner T, Liguori A, et al. Structure-based design of native-like HIV-1 envelope trimers to silence non-neutralizing epitopes and eliminate CD4 binding. Nat Commun (2017) 8:1655. doi: 10.1038/s41467-017-01549-6
32. Duan H, Chen X, Boyington JC, Cheng C, Zhang Y, Jafari AJ, et al. Glycan masking focuses immune responses to the HIV-1 CD4-binding site and enhances elicitation of VRC01-class precursor antibodies. Immunity (2018) 49:301–11.e5. doi: 10.1016/j.immuni.2018.07.005
33. Bajic G, Maron MJ, Adachi Y, Onodera T, McCarthy KR, McGee CE, et al. Influenza antigen engineering focuses immune responses to a subdominant but broadly protective viral epitope. Cell Host Microbe (2019) 25:827–35.e6. doi: 10.1016/j.chom.2019.04.003
34. Thornlow DN, Macintyre AN, Oguin TH, Karlsson AB, Stover EL, Lynch HE, et al. Altering the immunogenicity of hemagglutinin immunogens by hyperglycosylation and disulfide stabilization. Front Immunol (2021) 12:737973. doi: 10.3389/fimmu.2021.737973
35. Lin HH, Yang SP, Tsai MJ, Lin GC, Wu HC, Wu SC. Dengue and zika virus domain III-flagellin fusion and glycan-masking e antigen for prime-boost immunization. Theranostics (2019) 9:4811–26. doi: 10.7150/thno.35919
36. Tai W, Chen J, Zhao G, Geng Q, He L, Chen Y, et al. Rational design of zika virus subunit vaccine with enhanced efficacy. J Virol (2019) 93(17):93. doi: 10.1128/JVI.02187-18
37. Boyoglu-Barnum S, Hutchinson GB, Boyington JC, Moin SM, Gillespie RA, Tsybovsky Y, et al. Glycan repositioning of influenza hemagglutinin stem facilitates the elicitation of protective cross group antibody responses. Nat Commun (2020) 11:791. doi: 10.1038/s41467-020-14579-4
38. Chen TH, Yang YL, Jan JT, Chen CC, Wu SC. Site-specific glycan-Masking/Unmasking hemagglutinin antigen design to elicit broadly neutralizing and stem-binding antibodies against highly pathogenic avian influenza H5N1 virus infections. Front Immunol (2021) 12:692700. doi: 10.3389/fimmu.2021.692700
39. Adolf-Bryfogle J, Labonte JW, Kraft JC, Shapovalov M, Raemisch S, Lutteke T, et al. Growing glycans in Rosetta: Accurate de novo glycan modeling, density fitting, and rational sequon design. J Med Virol Company: Cold Spring Harbor Laboratory Distributor: Cold Spring Harbor Laboratory Label: Cold Spring Harbor Laboratory Section (2021) 94(5):1825–32. doi: 10.1101/2021.09.27.462000v3
40. Lin WS, Chen IC, Chen HC, Lee YC, Wu SC. Glycan masking of epitopes in the NTD and RBD of the spike protein elicits broadly neutralizing antibodies against SARS-CoV-2 variants. Front Immunol (2021) 12:795741. doi: 10.3389/fimmu.2021.795741
41. Liu WC, Jan JT, Huang YJ, Chen TH, Wu SC. Unmasking stem-specific neutralizing epitopes by abolishing n-linked glycosylation sites of influenza virus hemagglutinin proteins for vaccine design. J Virol (2016) 90(19):8496–508. doi: 10.1128/JVI.00880-16
42. Ringe RP, Pugach P, Cottrell CA, LaBranche CC, Seabright GE, Ketas TJ, et al. Closing and opening holes in the glycan shield of HIV-1 envelope glycoprotein SOSIP trimers can redirect the neutralizing antibody response to the newly unmasked epitopes. J Virol (2019) 93:e01656–18. doi: 10.1128/JVI.01656-18
43. Arsiwala A, Varner C, McCaffery JN, Kell A, Pendyala G, Castro A, et al. Nanopatterning protein antigens to refocus the immune response. Nanoscale (2019) 11:15307–11. doi: 10.1039/C9NR05145G
44. Castro A, Carreno JM, Duehr J, Krammer F, Kane RS. Refocusing the immune response to selected ˜ epitopes on a zika virus protein antigen by nanopatterning. Adv Healthcare Mater (2021) n/a:2002140. doi: 10.1002/adhm.202002140
45. Gugliotta A, Leopold MJ, Mufarrege E, Etcheverrigaray M, Kratje R, Ceaglio N, et al. Pharmacokinetics versus In vitro antiproliferative potency to design a novel hyperglycosylated hIFN-α2 biobetter. Pharm Res (2021) 38(1):37–50. doi: 10.1007/s11095-020-02978-7
46. Ghasemi F, Zomorodipour A, Karkhane AA, Khorramizadeh MR. In silico designing of hyper-glycosylated analogs for the human coagulation factor IX. J Mol Graph Model (2016) 68:39–47. doi: 10.1016/j.jmgm.2016.05.011
47. Ghasemi F, Khorramizadeh MR, Karkhane AA, Zomorodipour A. Studying the expression efficiencies of human clotting factor IX analogs, rationally-designed for hyper-glycosylation. Iranian J Pharm Res: IJPR (2021) 20:523–35. doi: 10.22037/ijpr.2020.112027.13503
48. Burgi M, Aparicio GI, Dorella A, Kratje R, Scorticati C, Oggero M. Novel erythropoietin-based therapeutic candidates with extra n-glycan sites that block hematopoiesis but preserve neuroplasticity. Biotechnol J (2021) 16:e2000455. doi: 10.1002/biot.202000455
49. Shafaghi M, Shabani AA, Minuchehr Z. Rational design of hyper-glycosylated human luteinizing hormone analogs (a bioinformatics approach). Comput Biol Chem (2019) 79:16–23. doi: 10.1016/j.compbiolchem.2019.01.002
50. Bahadori Z, Shabani AA, Minuchehr Z. Rational design of hyper-glycosylated human follicle-stimulating hormone analogs (a bioinformatics approach). J Biomol Struct Dyn (2021) 40(19):1–12. doi: 10.1080/07391102.2021.1924268
51. Global HIV & AIDS statistics — fact sheet; Available at: https://www.unaids.org/en/resources/fact-sheet.
52. Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The challenge of HIV-1 subtype diversity. New Engl J Med (2008) 358(15):1590–602. doi: 10.1056/NEJMra0706737
53. Kim J, Vasan S, Kim JH, Ake JA. Current approaches to HIV vaccine development: a narrative review. J Int AIDS Soc (2021) 24:e25793. doi: 10.1002/jia2.25793
54. Hargrave A, Mustafa AS, Hanif A, Tunio JH, Hanif SNM. Current status of HIV-1 vaccines. Vaccines (2021) 9:1026. doi: 10.3390/vaccines9091026
55. Arrildt KT, Joseph SB, Swanstrom R. The HIV-1 env protein: a coat of many colors. Curr HIV/AIDS Rep (2012) 9:52–63. doi: 10.1007/s11904-011-0107-3
56. Lambert GS, Upadhyay C. HIV-1 envelope glycosylation and the signal peptide. Vaccines (2021) 9:176. doi: 10.3390/vaccines9020176
57. Saunders-Hastings PR, Krewski D. Reviewing the history of pandemic influenza: Understanding patterns of emergence and transmission. Pathogens (2016) 5:66. doi: 10.3390/pathogens5040066
58. Freidl GS, Binger T, Muller MA, de Bruin E, van Beek J, Corman VM, et al. Serological evidence of influenza a viruses in frugivorous bats from Africa. PloS One (2015) 10:e0127035. doi: 10.1371/journal.pone.0127035
59. Kim H, Webster RG, Webby RJ. Influenza virus: Dealing with a drifting and shifting pathogen. Viral Immunol (2018) 31:174–83. doi: 10.1089/vim.2017.0141
60. Angeletti D, Yewdell JW. Is it possible to develop a “Universal” influenza virus vaccine? Cold Spring Harbor Perspect Biol (2018) 10:a028852. doi: 10.1101/cshperspect.a028852
61. Wu NC, Wilson IA. Structural biology of influenza hemagglutinin: An amaranthine adventure. Viruses (2020) 12:1053. doi: 10.3390/v12091053
62. Crowe JE. Influenza virus–specific human antibody repertoire studies. J Immunol (2019) 202:368–73. doi: 10.4049/jimmunol.1801459
63. Kim JI, Park MS. N-linked glycosylation in the hemagglutinin of influenza a viruses. Yonsei Med J (2012) 53:886–93. doi: 10.3349/ymj.2012.53.5.886
64. Tate MD, Job ER, Deng YM, Gunalan V, Maurer-Stroh S, Reading PC. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses (2014) 6:1294–316. doi: 10.3390/v6031294
65. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
66. Wu D, Wu T, Liu Q, Yang Z. The SARS-CoV-2 outbreak: What we know. Int J Infect Dis (2020) 94:44–8. doi: 10.1016/j.ijid.2020.03.004
67. Aleem A, Akbar Samad AB, Slenker AK. Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19). In: StatPearls. Treasure Island (FL: StatPearls Publishing (2022). Available at: http://www.ncbi.nlm.nih.gov/books/NBK570580/.
68. Fiolet T, Kherabi Y, MacDonald CJ, Ghosn J, Peiffer-Smadja N. Comparing COVID-19 vaccines for their characteristics, efficacy and effectiveness against SARS-CoV-2 and variants of concern: a narrative review. Clin Microbiol Infect (2022) 28(2):202–21. doi: 10.1016/j.cmi.2021.10.005
69. Araf Y, Akter F, Tang YD, Fatemi R, Parvez MSA, Zheng C, et al. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. (2022). doi: 10.1002/jmv.27588
70. Roltgen K, Boyd SD. Antibody and b cell responses to SARS-CoV-2 infection and vaccination. Cell Host Microbe (2021) 29:1063–75. doi: 10.1016/j.chom.2021.06.009
71. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Sci (New York Ny) (2020) 369:330–3. doi: 10.1126/science.abb9983
72. Harapan H, Michie A, Sasmono RT, Imrie A. Dengue: A minireview. Viruses (2020) 12:829. doi: 10.3390/v12080829
73. Ulrich H, Pillat MM, Tarnok A. Dengue fever, COVID-19 (SARS-CoV-2), and antibody-dependent enhancement (ADE): A perspective. Cytometry (2020) 97(7):662–7. doi: 10.1002/cyto.a.24047
74. Wilder-Smith A. Dengue vaccine development: status and future. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz (2020) 63:40–4. doi: 10.1007/s00103-019-03060-3
75. Pielnaa P, Al-Saadawe M, Saro A, Dama MF, Zhou M, Huang Y, et al. Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development. Virology (2020) 543:34–42. doi: 10.1016/j.virol.2020.01.015
76. Walker CL, Little MT, Roby JA, Armistead B, Gale M, Rajagopal L, et al. Zika virus and the non-microcephalic fetus: Why we should still worry. Am J Obstet Gynecol (2019) 220:45–56. doi: 10.1016/j.ajog.2018.08.035
77. Zhang X, Jia R, Shen H, Wang M, Yin Z, Cheng A. Structures and functions of the envelope glycoprotein in flavivirus infections. Viruses (2017) 9:338. doi: 10.3390/v9110338
78. Chen HW, Liu SJ, Li YS, Liu HH, Tsai JP, Chiang CY, et al. A consensus envelope protein domain III can induce neutralizing antibody responses against serotype 2 of dengue virus in non-human primates. Arch Virol (2013) 158:1523–31. doi: 10.1007/s00705-013-1639-1
79. Yang M, Lai H, Sun H, Chen Q. Virus-like particles that display zika virus envelope protein domain III induce potent neutralizing immune responses in mice. Sci Rep (2017) 7:7679. doi: 10.1038/s41598-017-08247-9
80. Carbaugh DL, Lazear HM. Flavivirus envelope protein glycosylation: Impacts on viral infection and pathogenesis. J Virol (2020) 94:e00104–20. doi: 10.1128/JVI.00104-20
81. Milner DA. Malaria pathogenesis. Cold Spring Harbor Perspect Med (2018) 8:a025569. doi: 10.1101/cshperspect.a025569
82. Boualam MA, Pradines B, Drancourt M, Barbieri R. Malaria in Europe: A historical perspective. Front Med (Lausanne) (2021) 8:691095. doi: 10.3389/fmed.2021.691095
83. Dayanand KK, Achur RN, Gowda DC. Epidemiology, drug resistance, and pathophysiology of plasmodium vivax malaria. J Vector Borne Dis (2018) 55:1–8. doi: 10.4103/0972-9062.234620
84. Cohen S, McGREGOR IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature (1961) 192(4804):733–7. doi: 10.1038/192733a0
85. Richie TL, Billingsley PF, Sim BKL, James ER, Chakravarty S, Epstein JE, et al. Progress with plasmodium falciparum sporozoite (PfSPZ)-based malaria vaccines. Vaccine (2015) 33:7452–61. doi: 10.1016/j.vaccine.2015.09.096
86. Chen E, Salinas ND, Ntumngia FB, Adams JH, Tolia NH. Structural analysis of the synthetic Duffy binding protein (DBP) antigen DEKnull relevant for plasmodium vivax malaria vaccine design. PloS Negl Trop Dis (2015) 9:e0003644. doi: 10.1371/journal.pntd.0003644
87. de Sousa TN, Kano FS, de Brito CFA, Carvalho LH. The Duffy binding protein as a key target for a plasmodium vivax vaccine: lessons from the Brazilian Amazon. Memorias do Inst Oswaldo Cruz (2014) 109(5):608–17. doi: 10.1590/0074-0276130592
88. Kaji H, Shikanai T, Sasaki-Sawa A, Wen H, Fujita M, Suzuki Y, et al. Large-Scale identification of n-glycosylated proteins of mouse tissues and construction of a glycoprotein database, GlycoProtDB. J Proteome Res (2012) 11:4553–66. doi: 10.1021/pr300346c
89. Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of n-linked glycans on glycoproteostasis. Nat Chem Biol (2014) 10:902–10. doi: 10.1038/nchembio.1651
90. Dey B, Svehla K, Xu L, Wycuff D, Zhou T, Voss G, et al. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PloS Pathogens (2009) 5:e1000445. doi: 10.1371/journal.ppat.1000445
91. Steichen J, Kulp D, Tokatlian T, Escolano A, Dosenovic P, Stanfield R, et al. HIV Vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity (2016) 45(3):483–96. doi: 10.1016/j.immuni.2016.08.016
92. Jardine JG, Kulp DW, Havenar-Daughton C, Sarkar A, Briney B, Sok D, et al. HIV-1 broadly neutralizing antibody precursor b cells revealed by germline-targeting immunogen. Sci (New York NY) (2016) 351:1458–63. doi: 10.1126/science.aad9195
93. Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med (2015) 21:1065–70. doi: 10.1038/nm.3927
94. Peek LJ, Middaugh CR, Berkland C. Nanotechnology in vaccine delivery. Adv Drug Deliv Rev (2008) 60:915–28. doi: 10.1016/j.addr.2007.05.017
95. Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, et al. Vaccinia virus vaccines: Past, present and future. Antiviral Res (2009) 84:1–13. doi: 10.1016/j.antiviral.2009.06.006
96. Sakurai F, Tachibana M, Mizuguchi H. Adenovirus vector-based vaccine for infectious diseases. Drug Metab Pharmacokinet (2022) 42:100432. doi: 10.1016/j.dmpk.2021.100432
97. Roldao A, Mellado MCM, Castilho LR, Carrondo MJT, Alves PM. Virus-like particles in vaccine development. Expert Rev Vaccines (2010) 9:1149–76. doi: 10.1586/erv.10.115
98. Marcandalli J, Fiala B, Ols S, Perotti M, de van der Schueren W, Snijder J, et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell (2019) 176(6):1420–31.e17. doi: 10.1016/j.cell.2019.01.046
99. Boyoglu-Barnum S, Ellis D, Gillespie RA, Hutchinson GB, Park YJ, Moin SM, et al. Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature (2021) 592:623–8. doi: 10.1038/s41586-021-03365-x
100. Ajamian L, Melnychuk L, Jean-Pierre P, Zaharatos GJ. DNA Vaccine-encoded flagellin can be used as an adjuvant scaffold to augment HIV-1 gp41 membrane proximal external region immunogenicity. Viruses (2018) 10:E100. doi: 10.3390/v10030100
101. van Ierssel SH, Leven M, Jorens PG. Severe influenza A(H1N1)2009 infection: a single centre experience and review of the literature. Acta Clin Belgica (2012) 67:1–6. doi: 10.2143/ACB.67.1.2062618
102. Menard D, Barnadas C, Bouchier C, Henry-Halldin C, Gray LR, Ratsimbasoa A, et al. Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc Natl Acad Sci (2010) 107:5967–71. doi: 10.1073/pnas.0912496107
103. Batchelor JD, Zahm JA, Tolia NH. Dimerization of plasmodium vivax DBP is induced upon receptor binding and drives recognition of DARC. Nat Struct Mol Biol (2011) 18:908–14. doi: 10.1038/nsmb.2088
104. Tobin GJ, Trujillo JD, Bushnell RV, Lin G, Chaudhuri AR, Long J, et al. Deceptive imprinting and immune refocusing in vaccine design. Vaccine (2008) 26:6189–99. doi: 10.1016/j.vaccine.2008.09.080
105. Yang X, Lee J, Mahony EM, Kwong PD, Wyatt R, Sodroski J. Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J Virol (2002) 76:4634–42. doi: 10.1128/JVI.76.9.4634-4642.2002
106. Ma C, Wang L, Tao X, Zhang N, Yang Y, Tseng CTK, et al. Searching for an ideal vaccine candidate among different MERS coronavirus receptor-binding fragments–the importance of immunofocusing in subunit vaccine design. Vaccine (2014) 32:6170–6. doi: 10.1016/j.vaccine.2014.08.086
107. McCoy LE, van Gils MJ, Ozorowski G, Messmer T, Briney B, Voss JE, et al. Holes in the glycan shield of the native HIV envelope are a target of trimer-elicited neutralizing antibodies. Cell Rep (2016) 16:2327–38. doi: 10.1016/j.celrep.2016.07.074
108. Briney B, Sok D, Jardine JG, Kulp DW, Skog P, Menis S, et al. Tailored immunogens direct affinity maturation toward HIV neutralizing antibodies. Cell (2016) 166:1459–70.e11. doi: 10.1016/j.cell.2016.08.005
109. Pauthner M, Havenar-Daughton C, Sok D, Nkolola JP, Bastidas R, Boopathy AV, et al. Elicitation of robust tier 2 neutralizing antibody responses in nonhuman primates by HIV envelope trimer immunization using optimized approaches. Immunity (2017) 46:1073–88.e6. doi: 10.1016/j.immuni.2017.05.007
110. Hoot S, McGuire AT, Cohen KW, Strong RK, Hangartner L, Klein F, et al. Recombinant HIV envelope proteins fail to engage germline versions of anti-CD4bs bNAbs. PloS Pathogens (2013) 9:e1003106. doi: 10.1371/journal.ppat.1003106
111. Halstead SB. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J Infect Dis (1979) 140:527–33. doi: 10.1093/infdis/140.4.527
112. Martínez-Vega RA, Carrasquila G, Luna E, Ramos-Castaneda J. ADE and dengue vaccination. Vaccine (2017) 35:3910–2. doi: 10.1016/j.vaccine.2017.06.004
113. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596:1–11. doi: 10.1038/s41586-021-03819-2
Keywords: glycan masking, vaccine design, reverse vaccinology, immuno-focusing, immuno-shifting
Citation: Martina CE, Crowe JE Jr. and Meiler J (2023) Glycan masking in vaccine design: Targets, immunogens and applications. Front. Immunol. 14:1126034. doi: 10.3389/fimmu.2023.1126034
Received: 17 December 2022; Accepted: 28 February 2023;
Published: 23 March 2023.
Edited by:
Roberto Adamo, GlaxoSmithKline, ItalyReviewed by:
Maria Agallou, Pasteur Hellenic Institute, GreeceCopyright © 2023 Martina, Crowe and Meiler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina E. Martina, Y3Jpc3RpbmEuZS5tYXJ0aW5hQHZhbmRlcmJpbHQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.