 Alexander Chi
Alexander Chi Nam Phong Nguyen
Nam Phong Nguyen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 June 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1125905
This article is part of the Research TopicThe Role of Radiotherapy in Reshaping Tumor Immune MicroenvironmentView all 5 articles
Immunotherapy consisted mainly of immune checkpoint inhibitors (ICIs) has led to significantly improved antitumor response. However, such response has been observed only in tumors possessing an overall responsive tumor immune micro-environment (TIME), in which the presence of functional tumor-infiltrating lymphocytes (TILs) is critical. Various mechanisms of immune escape from immunosurveillance exist, leading to different TIME phenotypes in correlation with primary or acquired resistance to ICIs. Radiotherapy has been shown to induce antitumor immunity not only in the irradiated primary tumor, but also at unirradiated distant sites of metastases. Such antitumor immunity is mainly elicited by radiation’s stimulatory effects on antigenicity and adjuvanticity. Furthermore, it may be significantly augmented when irradiation is combined with immunotherapy, such as ICIs. Therefore, radiotherapy represents one potential therapeutic strategy to restore anti-tumor immunity in tumors presenting with an unresponsive TIME. In this review, the generation of anti-tumor immunity, its impairment, radiation’s immunogenic properties, and the antitumor effects of combining radiation with immunotherapy will be comprehensively discussed.
Although immunotherapy for cancer has been intensely studied, significant and durable antitumor response with limited severe treatment-related toxicity has only been observed since FDA’s approval of immune checkpoint inhibitors (ICIs) that target specific immune checkpoints (1–3). Over the past decade, ICIs have quickly become the primary systemic treatment option for advanced melanoma and non-small cell lung cancer (NSCLC) with expanding indications for solid tumors arising from other anatomical sites (2–5). ICIs that are currently in clinical use mostly target the PD-1/PD-L1(PD-(L)1) immune checkpoint at the site of peripheral tumor (6). This strategy of targeting the primary mechanism of escape from cancer immunosurveillance at the peripheral tumor sites has led to the restoration of adaptive anti-tumor immunity in an overall immunosuppressive local tumor micro-environment (TME) without causing a significant systemic response in normal tissues (1, 7). Thus, leading to long lasting anti-tumor response and a low incidence of severe toxicities in a number of patients (4, 5). Based on previous studies on the treatment of solid tumors with anti-PD-(L)1 inhibitors, the tumor immune micro-environment (TIME) of responders to ICIs has been characterized in contrast to non-responders (8–13). An overall inflamed TIME in which IFN-γ driven tumor expression of PD-L1, and tumor infiltration by functional CD8+ T cells have been consistently identified in responders (8, 12, 13). On the contrary, a paucity of tumor infiltration by functional CD8+ T cells has been clearly demonstrated in non-responders. Tumor progression after an initial response may also occur after acquiring additional mechanisms of immune evasion, such as loss-of-function mutations or other genomic alterations, which can subsequently induce an overall non-inflamed TIME (14–17). Currently, the response rate to ICI remains low in non-lymphoid solid tumors, such as NSCLC (5, 17). This makes more effective treatment strategies to overcome ICI resistance urgently needed. With increased understanding of the mechanisms of antitumor immunity and its impairment, many strategies to enhance antitumor immunity and overcome immunosuppression within the TIME, subsequently converting a non-inflamed or “cold” TIME into an inflamed one have been proposed (18–20). As one major local treatment strategy for cancer, radiotherapy (RT) has been shown to have immunostimulatory properties, which can be further exploited to serve this purpose (21–25). In this review, the impairments leading to suboptimal antitumor immunity and poor response to ICIs, RT’s immunogenic properties, and the rationales for combining RT with immunomodulatory agents to remodel the TIME in order to restore or augment anti-tumor immunity will be discussed.
Immune surveillance of cancer has long been known to exist, which allows the immune system to eradicate malignant lesions as they arise in the human body (7, 26, 27). To initiate anti-tumor immunity for tumor elimination (Figure 1), a coordinated activation of both innate and adaptive immunity is required (26–29). In the process, tumor antigens and the detection of “danger signals” from the tumor by the innate immune sensors, trigger the recruitment and activation of antigen presentation cells (APCs) (28–33). Subsequently, tumor antigen cross-presentation by APCs that travel to the tumor draining lymph node (tdLN) leads to the priming and activation of T cells (33). Co-stimulation signals, which may be further tuned by co-inhibitory signals, are required for T cells to be fully activated; while sub-optimal activation only leads to T cell anergy (34, 35). Fully activated T cells are able to express a unique set of “homing” chemokine receptors that are accompanied by increased expression of related chemokines in the TME. Thus, allowing for the homing of activated T cells to the tumor and its infiltration (33). One well known example is the CXC receptor 3(CXCR3), and its ligands CXCL9, CXCL10, and CXCL11, which interactions in tumor infiltration may also involve other chemokines, such as CXCL5 (33, 36, 37). At the site of tumor, selectin ligands and other adhesion molecules are also required for the binding of blood vessels and extravasation by T cells (33, 36). After tumor infiltration, cytotoxic CD8+ T cells will recognize tumor cells through recognizing tumor antigens presented by the major histo-compatibility complex class I (MHC-I) molecules and induce T cell mediated cytotoxicity (33).
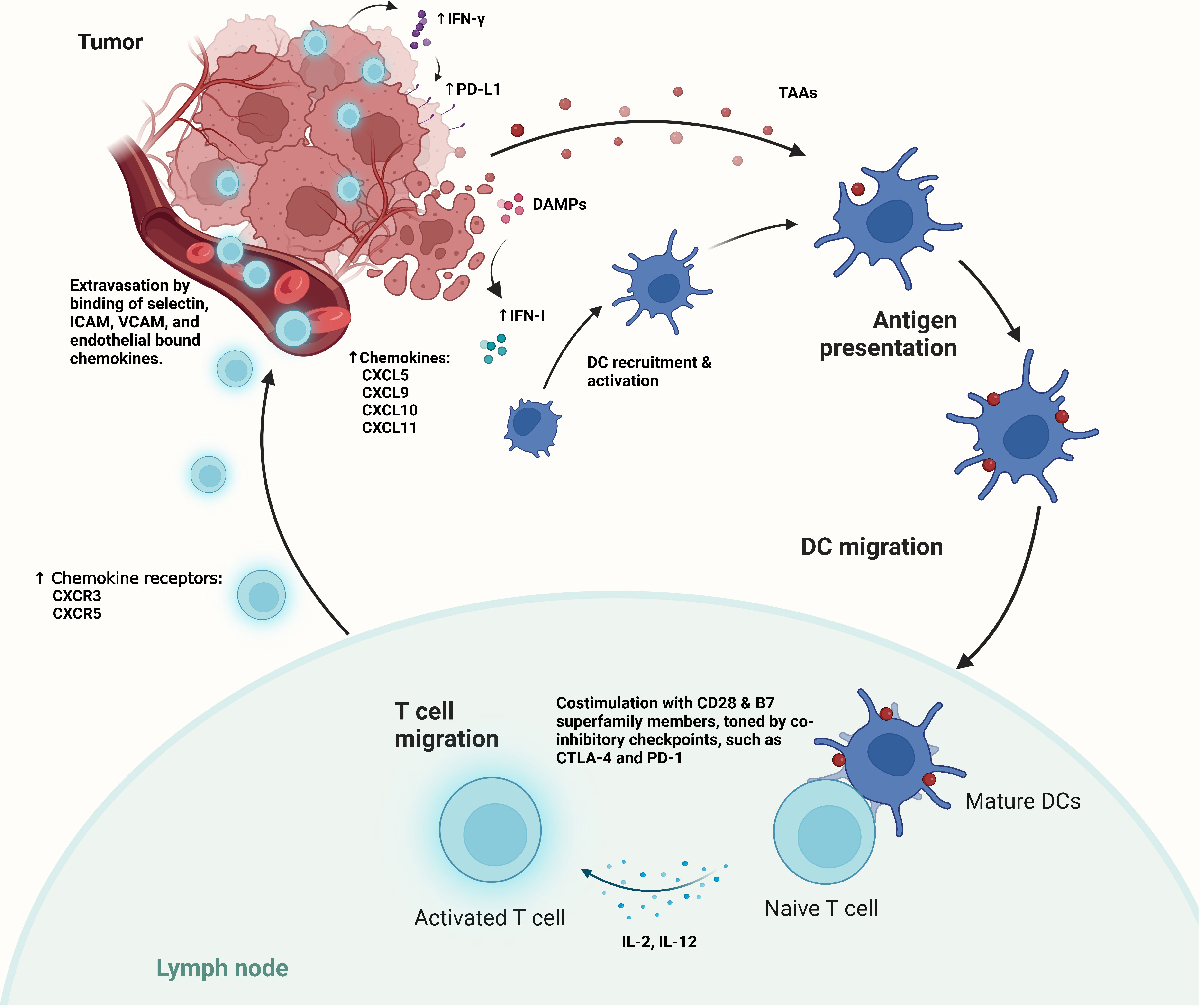
Figure 1 The generation of antitumor immunity relies on the detection of tumor antigens and danger-associated molecular patterns (DAMPs) by antigen presenting cells, such as dendritic cells (DCs). The presence of DAMPs leads to increased IFN-I secretion in the TIME, and the subsequent recruitment and activation of DCs. Activated DCs migrate to the tumor draining lymph nodes (tdLNs), where they cross present tumor antigens, and express additional co-stimulatory checkpoints and cytokines for T cell activation. Such activation may be attenuated by the presence of co-inhibitory checkpoints, such as CTLA-4 and PD-1. Upon activation, tumor-antigen specific T cells with increased expression of chemokine receptors migrate to the peripheral tumor, which have increased expression of T cell specific chemokines. Effector T cells then extravasate from the tumor vasculature through the binding of selectin, as well as the binding and activation of integrins, such as ICAM and VCAM for adhesion and transmigration into the tumor micro-environment.
Immune escape by tumor cells eventually develops. T cell activation within the TIME will stimulate the expression of PD-1 and release of IFN-γ by T cells (2). IFN-γ then induces PD-L1 expression by tumor cells as well as other cells within the TIME, resulting in the inhibition of the local immune response through PD-1/PD-L1 interaction (2, 38, 39). The process of PD-1/PD-L1 mediated effector T cell exhaustion is also called adaptive immune resistance. It serves as a dominant feedback mechanism to maintain peripheral tolerance, and is a common mechanism of immune escape by various cancers (2, 6, 40). Also, it makes re-invigoration of tumor infiltrating cytotoxic T lymphocytes (CTLs) within the TIME through targeting the PD-1 axis a very effective therapeutic strategy for cancer (1–6, 8, 41). An inflamed TIME that is characterized by IFN-γ induced PD-L1 expression by mostly tumor cells, and the presence of functional tumor infiltrating lymphocytes (TILs) is often associated with such re-invigoration, which is only observed in a limited number of patients with cancers such as NSCLC (2, 8, 41). This implies the presence of other impairment(s) within the steps of anti-tumor immunity generation.
Various mechanisms of immune escape by cancer leading to a paucity of T cell infiltration or a lack of activated T cells within the TME have been characterized in recent years (17, 33, 42–44). Major impairments in the generation of anti-tumor immunity, include the paucity of tumor neoantigens, impaired antigen presentation, lack of T cell priming and activation, poor tumor infiltration by activated T cells, poor tumor cell recognition/impaired IFN-γ signaling, and alterations in the composition and properties of immune cells within the peripheral TIME (Figure 2).
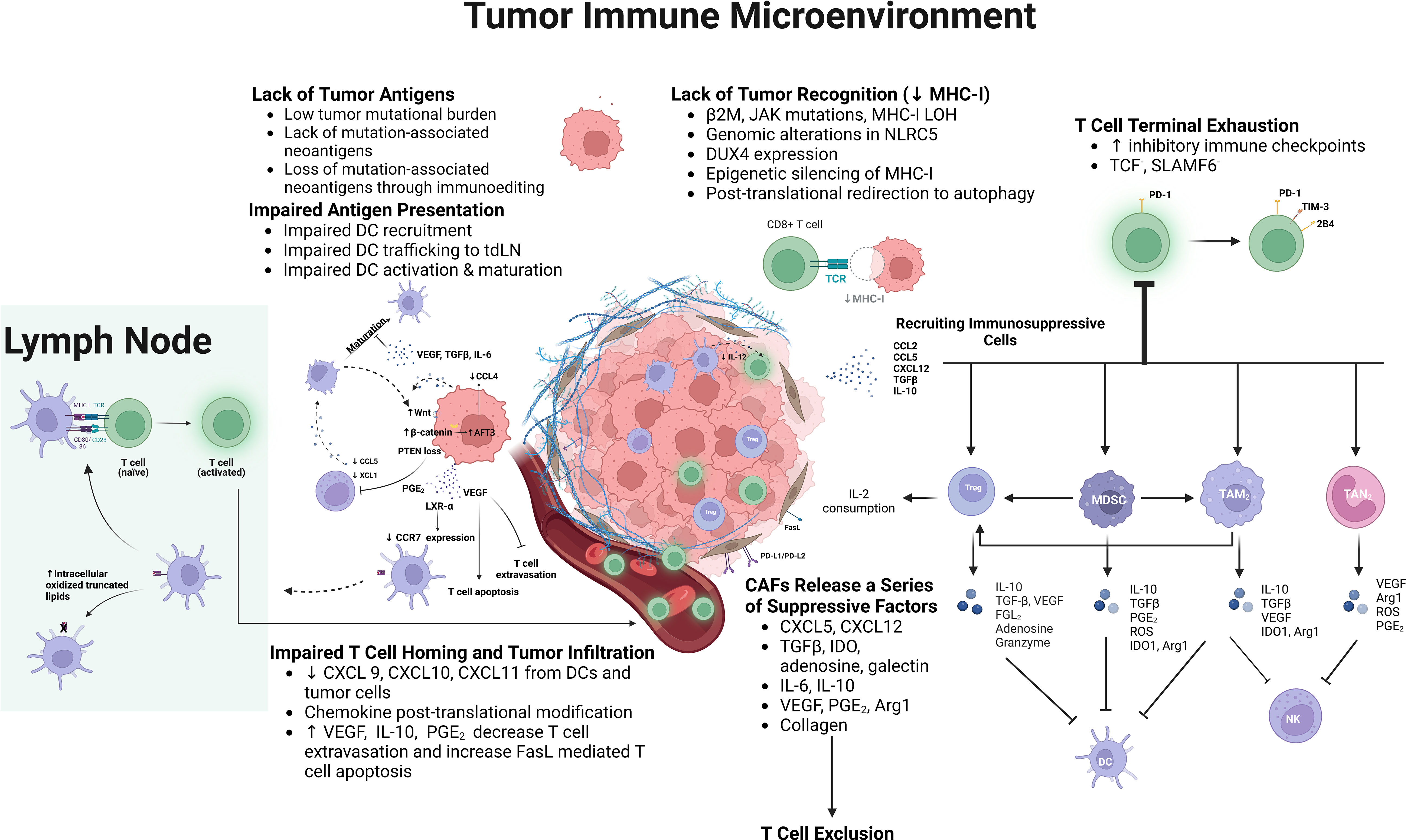
Figure 2 Underlying mechanisms of impaired antitumor immunity generation include: lack of tumor antigens, which develops overtime due to immunoediting; impaired antigen presentation resulting from impaired dendritic cell (DC) trafficking and maturation due to various tumor intrinsic mechanisms, including PTEN loss, Wnt/β-catenin expression, increased expression of LXR-α ligands, as well as increased prostaglandin E2 (PGE2), vascular endothelial growth factor (VEGF), IL-6, and TGFβ expression; suboptimal T cell co-stimulation; impaired cytotoxic T lymphocyte (CTL) homing and tumor infiltration due to decreased chemotactic chemokines in the tumor immune micro-environment (TIME), and down-regulation of adhesion molecules and increased Fas ligand expression in the tumor vasculature; exclusion and inhibition of CTLs by stromal cells, such as cancer associated fibroblasts (CAFs); lack of MHC-I expression by tumor cells resulting from genomic alterations of MHC-I or any component of the antigen presenting machinery, NOD-like receptor family, caspase recruitment domain containing 5 (NLCR5), embryonic transcription factor, DUX4, epigenetic silencing, and post-translational re-direction to autophagy; terminal exhaustion of CD8+ T cells within the TIME characterized by the lack of TCF and SLAMF6 expression, increased expression of PD-1, and additional inhibitory checkpoints, such as TIM-3 and 2B4; and the recruitment of suppressive immune cells, such as Tregs, myeloid derived suppressor cells (MDSCs), as well as macrophages and neutrophils with their polarization toward the M2/ N2 phenotype. These suppressive cells inhibit CTLs, DCs, and NK cells, while the suppressive mediators they release into the TIME further stimulate their recruitment and expansion within the TIME.
Mutations arise and accumulate during carcinogenesis and cancer progression, leading to the generation of tumor neoantigens (45, 46). Such neoantigens induce T cell specific reactivity, especially in patients who respond to immune checkpoint blockade (47–49). A high tumor mutation burden (TMB) consistently correlated with a higher incidence of durable clinical response (DCR) and improved survival after treatment with anti-CTLA-4 or anti-PD-L1 ICIs for cancers which are known to harbor a higher level of somatic mutations (50–54). This observation is due to the higher likelihood of a high neoantigen load in the presence of a high TMB (55). As shown in a cohort of melanoma patients, DCR to ICIs is not guaranteed by the presence of a high TMB (50). Instead, specific mutation associated neo-epitopes are required to elicit a cytotoxic anti-tumor T cell response in long-term responders to ICIs, and their occurrence is low even in the presence of a high TMB. This is due to immunoediting or the loss of tumor clones which harbored these mutant neoantigens over time (50, 52, 55). Subsequently, immune ignorance develops with poor TIL infiltration of the tumor. As shown in NSCLC, the repression of clonal neoantigen expression is ongoing in an inflamed TIME through various mechanisms, such as DNA copy number loss, suppression of gene transcription, epigenetic, as well as post-translational modification (56). Loss of immunogenic tumor mutation associated neoantigens has also been identified as one mechanism of acquired resistance to ICIs in NSCLC patients who underwent treatment with anti-PD-1 or combined anti-PD-1 and anti-CTLA-4 ICIs (57). Additional strategies to induce or maintain an adequate immunogenic mutant neoantigen repertoire in cancer patients are needed. However, high TMB and increased PD-L1 expression together represent key characteristics of an immunogenic tumor that has developed PD-1 axis mediated adaptive immune resistance, which may be used to select patients for treatment with anti-PD-1 or anti-CTLA-4 ICIs (50, 52).
As evidenced by a lack of DCR to ICIs in the presence of very high TMB, additional mechanisms of immune escape leading to a paucity of TILs within the TIME exist (50, 52). The antigen cross-presentation process, a key step in the generation of adaptive anti-tumor immunity, can be impaired in multiple ways, leading to a non-inflamed TIME. Among them, the repression of type I conventional dendritic cell (cDC1) trafficking into the TME due to tumor-intrinsic activation of the WNT/β-catenin signaling pathway has been well studied (58–61). A paucity of CD103+ DCs was observed when WNT/β-catenin was expressed in melanoma, which was due to the higher expression of transcriptional repressor AFT3. As a result, AFT3 inhibited the expression of CCL4, which is the key chemokine for guiding cDC1 migration into the tumor (58). Activated b-catenin signaling due to mutations or somatic copy number alteration is frequently observed in non-inflamed tumors (59). As shown in NSCLC, b-catenin expression is associated with shorter overall survival (OS) and significantly less tumor infiltrating CD8+ T cells, especially in the presence of a high TMB (60, 61). The trafficking of DCs into the tdLN can also be suppressed by tumor intrinsic alterations, such as increased expression of liver X receptor-α (LXR-α) ligands that would bind to LXR-α on DCs to inhibit CCR7 expression, which is required for DC migration to the tdLN (62, 63).
Other than the disruption of DC trafficking, activation and maturation of DCs can also be impaired. The maturation of DCs is induced through the detection of danger associated molecular patterns (DAMPs), which are mostly composed of intracellular proteins, and pieces of cytosolic tumor DNA from dying tumor cells by innate immune sensors (29, 64, 65). This leads to increased IFN-β signaling, which induces downstream stimulation of antigen presentation, DC maturation and trafficking to the tdLN. The innate immunity induced by DAMP or cytosolic DNA may be attenuated in tumors with increased cyclooxygenase-driven prostaglandin E2 (PGE2) production, leading to decreased DC recruitment and maturation (66, 67). On the other hand, decreased innate immunity may also result from the neutralization or inhibition of DAMP signaling, which inhibits subsequent DC activation (68, 69).
DC recruitment into the tumor, and differentiation are also mediated through NK cells, which produce cDC1 chemo-attractants CCL5, CXCL1, and survival stimulating cytokine FLT3LG. However, NK cells’ viability and chemokine production may be suppressed by tumor PGE2 overexpression, which demonstrates how the tumor may exert its influence on the TIME (70, 71). Other intrinsic tumor factors may also suppress DC maturation, such as vascular endothelial growth factor (VEGF); or induce a tolerogenic DC phenotype (DCreg), such as interleukin (IL)-6, transforming growth factor (TGF)β (72). At last, cross presentation by DCs can also be impaired due to the accumulation of oxidized lipids as a result of oxidatively truncated lipids’ binding the heat shock protein (hsp) 70, which prevents the translocation of peptide-MHC complex to the cell surface (73, 74). Activation of CD8+ T cells also requires additional stimulatory signals from the APCs, including co-stimulatory signals and APC-released cytokines (64, 75). Peripheral tolerance can develop with suboptimal co-stimulation, which may be alleviated by agonist antibodies to the co-stimulation checkpoints, activating cytokines, or inhibitors to inhibitory checkpoints, such as CTLA-4 (33).
Chemo-taxis of effector T cells to the tumor depends on the expression of CXCR3 on T cells and the presence of its ligands, CXCL9, CXCL10, and CXCL11 within the TIME (76). Within the tumor, Batf3-dependent cDC1s that express CXCL9 and CXCL10 are required for effector T cell chemo-taxis (77). A lack of tumor infiltration by T cells is observed in their absence. On the contrary, their presence is associated with a state of T cell exhaustion, and response to ICIs (78). More recently, macrophage-derived CXCL9 and CXCL10 were also found to be required for CD8+ T cell infiltration into the TIME, and any response to ICIs (79). Chemokines can be modified by post-translational modifications, which have been associated with impaired T cell chemo-taxis to the tumor (33, 80).
In addition, T cell extravasation and tumor infiltration can be suppressed by endothelial cells and stromal cells. CTLs must adhere to the endothelial lining for extravasation, and this may be inhibited by the vascular endothelial growth factor (VEGF) mediated down-regulation of adhesion molecules, or the induction of CTL apoptosis through Fas ligand (FasL) overexpression that are induced by VEGF, IL-10, or PGE2 within the TME (81, 82).
The tumor stroma has largely been known to be immunosuppressive, which can form dense areas of fibrosis in advanced cancer, leading to a state of poor T cell infiltration into the tumor (83). Increased collagen density has been associated with decreased CD8+ T cell infiltration into the tumor (84). Similarly, cancer associated fibroblasts (CAFs) have been shown to induce an immunologically excluded phenotype mediated by increased TGFβ signaling within fibroblasts (85). Other mechanisms of CD8+ T cell inhibition by CAFs include CXCL12 mediated CTL exclusion; PD-L1/L2, FasL upregulation; generation of adenosine; IL-6 mediated DC conversion to DCreg; and blunting of TCR signal transduction (86). In addition, CAFs can secret immunosuppressive factors and stimulate the recruitment of suppressive immune cells into the TIME (87). Tumor stroma along with the ill-formed tumor vasculature represent areas of great therapeutic potential for the generation of a more immunogenic TIME (83, 87, 88).
Reduced tumor neoantigen recognition may result from decreased MHC-I expression caused by the presence of genomic alterations of MHC-I or any component of the antigen processing machinery (APM), such as β2-microglobulin (B2M) (72, 89). Alternatively, it may result from the reduced MHC-I up-regulation due to impaired IFN-γ signaling caused by JAK1/2 mutations (14). Complete loss of the B2M gene causes complete loss of MHC-I expression (14, 15). Genomic alterations of B2M and JAK1/2 have been associated with acquired resistance to ICIs along with MHC-I loss of homozygosity (LOH) (14, 15, 90, 91). Other mechanisms leading to decreased MHC-I expression also exist, which include genomic alterations of the transactivator of MHC-I related genes, NLCR5 (NOD-like receptor family, caspase recruitment domain containing 5); the expression of embryonic transcription factor, DUX4; epigenetic silencing; and post-translational re-direction to autophagy (72, 92, 93).
At last, alterations of the immune cell composition within the TIME can lead to suppression of anti-tumor immunity. One such alteration is the increased presence of terminally exhausted CD8+ T cells. Initially observed after chronic antigen exposure, T cell exhaustion is characterized by increased PD-1 expression its co-expression with additional inhibitory checkpoint receptors, and hierarchical loss of cytolytic function in CD8+ T cells (94–97). TILs appear to be at different stages of exhaustion with effector functions preserved in progenitor exhausted CD8+ T cells, but lost in terminally exhausted CD8+ T cells (95–97). Progenitor exhausted T cells appear to have greater chromatin accessibility; higher expression of stimulatory cytokines, co-stimulatory checkpoints, and survival/memory molecules; while terminally exhausted T cells had more accessibility to and expression of co-inhibitory receptors, effector molecules, and transcription factors associated with exhaustion (98). As shown in NSCLC, increased degree of CD8+ T cell exhaustion is correlated with increased PD-1 expression and the co-expression of additional co-inhibitory receptors, such as TIM-3, TIGIT, and CTLA-4 at later stages of cancer progression (99, 100). PD-1 appears to be the primary mediator of CD8+ T cell exhaustion. However, anti-tumor response to anti-PD-1 therapy alone is poor in tumors infiltrated by terminally exhausted TIL’s, which mostly reside in the TIME (97–100). On the contrary, combined blockade to PD-1 and other co-inhibitory checkpoints was shown to restore effector function in terminally exhausted TILs, and generate significant anti-tumor activity when PD-1 and additional co-inhibitory checkpoints are co-expressed (97, 100).
The immune cell composition may also be sculpted to have an increased presence of suppressive immune cells within the TIME. The inhibitory roles of regulatory T cells (Tregs), and myeloid derived suppressor cells (MDSCs) have been well characterized, while tumor associated macrophages (TAMs) and tumor associated neutrophils (TANs) can develop pro-tumor or anti-tumor properties (101–106). Tregs, Forkhead box P3 (FOXP3) expressing CD4+ T cells, are recruited into the TIME through up-regulation of CCR4, CCR5, CCR8, and CCR10 mediated chemotaxis in inflamed tumors by both tumor and immune cells; and CCR4 mediated chemotaxis due to increased CCL22 expression in EGFR mutant non-inflamed tumors, exert their inhibitory effects on CTLs through IL-2 depletion, binding and capturing of co-stimulatory signals on dendritic cells, and the production of immunosuppressive factors, such as TGFβ, IL-10, FGL2, VEGF, and granzymes (101, 102). MDSCs are derived from polymorphonuclear (PMN) or monocytic myeloid cells in the presence of cancer induced inflammation (103, 104). They secrete a series of immunosuppressive mediators, such as IL-10, PGE2, TGFβ, free radicals; inducing M2 macrophages and Tregs, while suppressing CTL adhesion, TCR expression, activation, and survival (105, 106). TAM and TAN may polarize into immunostimulatory/anti-tumor, or immunosuppressive/pro-tumor types by stimulatory signals, such as IFN-γ, or suppressive signals, such as, TGFβ (105–107). While type 1(M1) macrophages have potent antigen presentation and phagocytotic properties, type 2 (M2) macrophages are immunosuppressive for which they express inhibitory immune checkpoints, such as PD-L1; and secrete a series of suppressive factors, such as TGFβ, IL-10, IDO, and arginase 1 (Arg1), which stimulate Treg function, inhibit DC maturation, and suppress CTL function (105, 106). Similar to TAMs, TANs can secrete a series of immunosuppressive mediators and express a series of inhibitory immune checkpoints, once induced by suppressors, such as TGFβ (107).
Various mechanisms of immune escape by cancer may lead to poor response to ICIs, which shed light on the development of additional therapeutic strategies to induce an inflamed TIME. As a major local treatment modality for cancer, RT has been shown to have immunogenic properties (21). Ablative doses of radiation can cause immunogenic cell death (ICD) through the stimulation of antigen presentation, leading to increased maturation and recruitment of effector CD8+ T cells (Figure 3) (108–112). Its immunostimulatory properties may lead to the restoration of an overall inflamed TIME and enhanced antitumor immunity. Furthermore, such immunogenic antitumor effects may be significantly augmented when RT is combined with immunomodulatory agents, such as ICIs (21, 22).
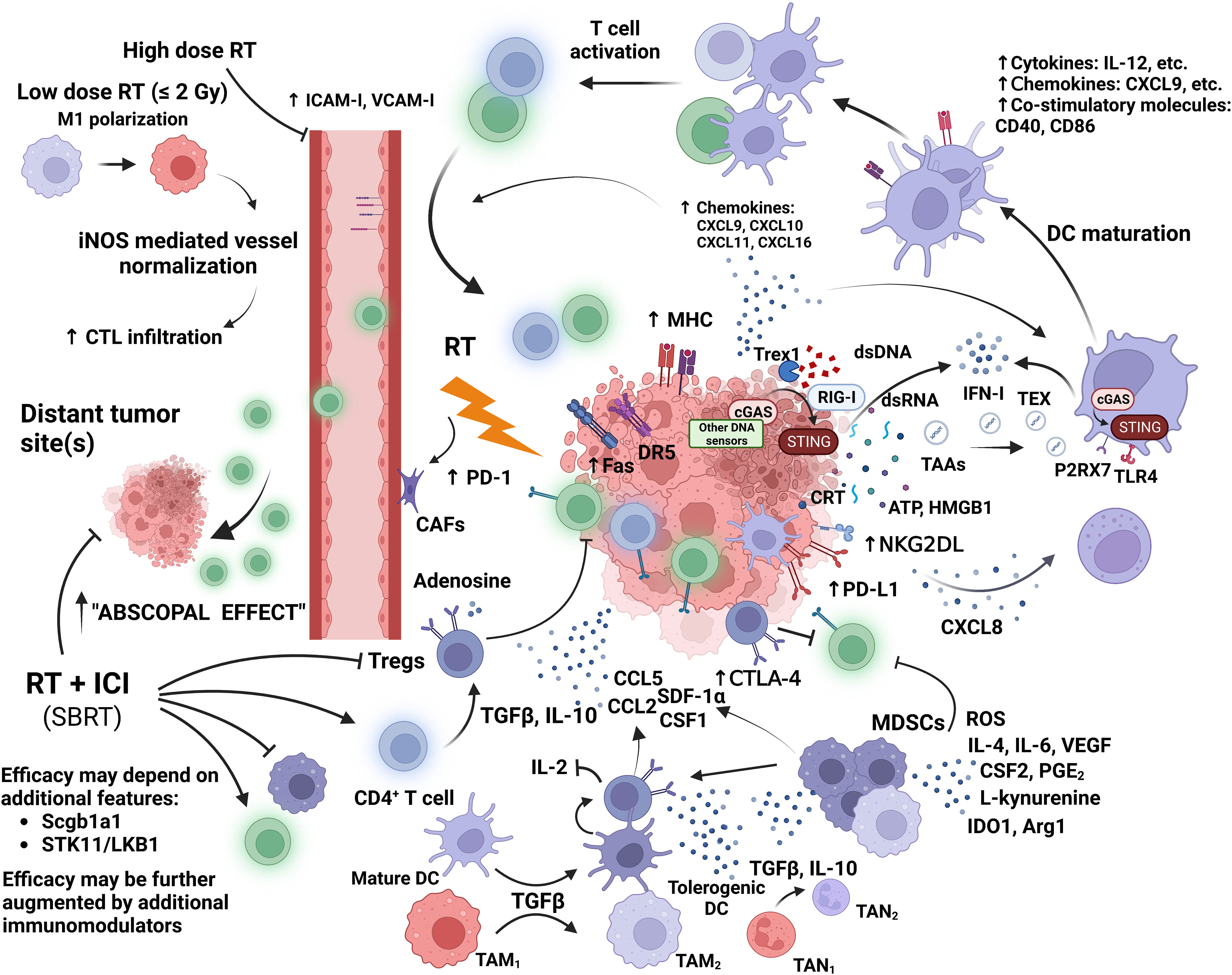
Figure 3 Moderate to high dose irradiation induces the release of tumor associated antigens (TAAs); DNA and RNA fragments; and danger associated molecular patterns (DAMPs) such as calreticulin (CRT), ATP, and HMGB1 by tumor cells. Double-stranded (ds) DNA activates sensors of innate immunity, such as, cyclic GMP-AMP synthase (cGAS); while retinoic acid inducible gene-I (RIG-I) is activated by RNA fragments. These activate the stimulator of IFN genes (STING), which induces IFN-mediated DC recruitment and activation. Cytosolic DNA fragments may be transferred to DCs via tumor derived exomes (TEX). Activated DCs migrate to tumor draining lymph nodes (tdLNs) for cross priming of T cells. Cytosolic dsDNA is degraded by exonuclease Trex1 which is expressed after high dose irradiation of 12-18 Gy. CRT expressed on dying tumor cells leads to DC mediated phagocytosis, while ATP and HMGB1 stimulate DC cross presentation. DCs express a series of cytokines, chemokines, and costimulatory molecules to induce T cell activation. RT upregulates tumor expression of chemokines for T cell homing, and adhesion molecules in the tumor vasculature for T cell extravasation. RT also increases tumor expression of MHC-I and Fas. Fas and DR5 may enhance direct tumor cell killing by CTLs. NK cell cytotoxicity and recruitment are also increased by RT. At low doses, M1 polarization is enhanced along with macrophage mediated cytotoxic T lymphocyte (CTL) infiltration due to increased expression of inducible nitric oxide synthase (iNOS) by irradiated, tumor-infiltrating macrophages. These stimulatory effects are offset by the increased tumor-cell release of suppressive cytokines and chemokines, which result in the recruitment of suppressive immune cells and the induction of suppressive immune phenotypes. Leading to the consumption of stimulatory cytokines (e.g., IL-2), further release of suppressive cytokines, and the inhibition of CTLs and NK cells. RT increases the expression of inhibitory checkpoints on effector T cells (Teff), DCs, tumor cells, and Tregs within the TIME. High dose RT also induces aberrant tumor vasculature formation, increased fibrosis, hypoxia, and the stimulation of cancer-associated fibroblasts (CAFs). Significant augmentation of CTL infiltration into the local and distant TIME has been observed when RT delivering ablative doses was combined with an immune checkpoint inhibitor (ICI). This was found to be associated with the suppression of Tregs and MDSCs, and the stimulation of CD8+ T cell (green) function. The synergy between RT and ICI also depends on specific features of the tumor residing tissue and the tumor itself.
RT’s ability to induce ICD has been well characterized (21–25). This phenomenon largely depends on its ability to increase the release of tumor-associated antigens (TAAs), double stranded (ds) DNA fragments, and various danger associated molecular patterns (DAMPs) by tumor cells (Figure 3). Subsequently, leading to increased antigen presentation, T cell activation and augmented antitumor immunity (22–24). As shown in vitro, irradiation increased intracellular protein breakdown and mammalian target of rapamycin (mTOR) mediated peptide production (113). Some of the peptides produced are TAAs generated from radiation-induced immunogenic mutations, which are expressed in a radiation dose dependent manner (114). These TAAs can be CD8+ T cell specific, or Th1 CD4+ T cell specific. Therefore, radiation-upregulated TAAs activate both CD8+ and Th1 CD4+ T cells in a poorly immunogenic tumor, which result in enhanced antitumor response to ICIs and vaccines (114, 115). CD4+ neoantigens are essential in radiation-induced antitumor immune response, as they not only result in enhanced CD8+ T cell activation and cyto-toxicity, but also engage in direct tumor cell killing through interactions with Fas and the death receptor DR5 on tumor cells (115, 116).
Ionizing radiation (IR) introduces base and sugar damage, crosslinks, and sing or double stranded breaks (SSBs or DSBs) in the DNA (117). The dsDNA breaks represent the most common and lethal radiation induced DNA damage (23). A DNA damage response (DDR) is subsequently elicited by ss or dsDNA breaks, which is mediated by DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia-mutated (ATM), and ataxia telangiectasia and rad-3-related protein (ATR) (24). DNA-PK facilitates non-homologous end-joining (NHEJ), while ATM and ATR facilitate homologous recombination (HR) repair and stabilization of stalled replication forks. Whereas the error-prone NHEJ, and the more precise HR for simple dsDNA breaks are fast, the repairment of more complex DNA damage characterized by multiple oxidative base damage, basic sites, or SSBs around a DSB, is both slower and more error-prone (117).
Defective DNA damage repair leads to the formation of micronuclei from chromosomal fragments through mitosis. The micronuclei are easily ruptured with dsDNA fragments released into the cytosol and detected by the cytosolic dsDNA sensor, cyclic GMP-AMP synthase (cGAS), which localizes to the micronuclei (24). cGAS then activates its adaptor, stimulator of interferon genes (STING), which leads to the secretion of IFN-I and the recruitment of DCs into the TIME (23, 24). Many other sensors of cytosolic nucleic acid exist, such as DDX41, ZBP1, IFI16, MRE11, and HDP-RNP, which can all activate the STING pathway, leading to the initiation of IRF3-dependent or NF-κB-dependent transcriptional programs (118). RNA sensors, such as retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5), can also be activated by radiation, leading to increased IFN-I signaling and DC recruitment (23, 119, 120).
Upon dsDNA sensing within the TIME, intra-tumoral DCs upregulates IFN-β release in a STING dependent manner, which stimulates T cell cross presentation in the tdLNs and T cell activation through the expression of stimulatory cytokines (e.g., IL-6, IL-12, IL-15, TNF), chemokines (e.g., CXCL9), and costimulatory molecules (e.g., CD40, CD86) (121, 122). It is not entirely clear how the cytosolic dsDNA was transported into the cytoplasm of DCs. One mechanism may be through tumor-derived exomes (TEX) (123). The accumulation of cytosolic dsDNA within the tumor cell is regulated by the DNA exonuclease Trex1, which degrades dsDNA. In vitro, Trex1 expression is increased at fractional doses between 12-18 Gy, which correlated with significant reduction of cytosolic dsDNA (124).
Irradiation induced cell death and stress also cause the release of many other DAMPs, which are recognized by pattern recognition receptors (PRRs) on APCs, such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and the receptor for advanced glycation end products (RAGE) (125–127). The most commonly identified DAMPs associated with IR induced cell death are cell surface calreticulin, ATP, and the high mobility group box 1 (HMGB1) (127). Calreticulin is a chaperone protein residing in the ER. It translocate to the surface of dying tumor cells to signal to DCs for phagocytosis as an early event in IR-induced ICD (125, 127). As later events during ICD, ATP and HMGB1 are released into the TIME (125). ATP leads to the activation of the NLRP3/ASC/caspase-1 inflammasome by stimulating the purinergic P2RX7 receptors on DCs (128). This leads to the release of IL-1β, which induces CD4+ and CD8+ T cell cross priming (129, 130). On the contrary, HMGB1 stimulates T cell cross priming through binding to TLR4 on DCs (131).
Other than enhancing antigen presentation, RT also plays a stimulatory role in other key steps of antitumor immunity generation. In vitro, the expression of chemokines essential for CD8+ T cell recruitment, CXCL9, CXCL10, CXCL11, CXCL16, is significantly elevated after high fractional doses between 10-12 Gy (132–134). Diminished T cell extravasation from aberrant tumor vasculature can be improved by RT due to increased expression of adhesion molecules (108, 135, 136). Increased VCAM-1, and ICAM-1 expression after irradiation with fractional doses of 8-15 Gy have been detected, which resulted in persistent T cell infiltration into the tumor (108, 136). In murine colon and breast cancer mouse models, ICAM-1 expression was significantly elevated after 8 Gy x 3 fractions, which was shown to stimulate a strong CD8+ T cell mediated systemic response (136). Multi-fractional(MF) dose regimens with moderate doses may be more suitable for the induction of such effects as they usually lead to more moderate vascular damage that will allow for T cell extravasation. On the contrary, single-fraction (SF) high dose irradiation may lead to significant vessel damage, which prohibits any significant amount of T cell extravasation into the TIME (21, 137).
Normalization of tumor vasculature has been reported at both low or moderate fractional doses (138, 139). Significant T cell recruitment mediated by inducible nitric oxide synthase (iNOS), which is secreted by tumor infiltrating M1 macrophages, is observed at doses as low as 2 Gy, due to iNOS regulated T cell transmigration (138). Low dose (LD) irradiation of 1 Gy was further shown to induce a significant increase in the IFN mediated influx of CTLs into a non-inflamed TIME in vivo, which represents a potential strategy to induce an inflamed TIME that will lead to increased response to ICIs and other immunomodulators (140). Also, RT can alleviate the impaired tumor cell recognition by CTLs through inducing MHC-I expression on tumor cell surface; and stimulate tumor cell lysis by upregulating Fas expression on tumor cells (113, 141).
RT can stimulate innate anti-immunity mediated by NK cells. Irradiation increased NK cell cytotoxicity and homing in a canine sarcoma model (142). NK cell viability and cytotoxicity are increased with increased fractionation (143). Increased NK cell cytotoxicity post-radiation is due to increased expression of ligands to activating receptor NKG2D on tumor cells that is mediated by STING-dependent dsDNA sensing pathways (144, 145). Secondly, the increased post-radiation NK cell homing is due to increased tumor cell expression and secretion of CXCL8 mediated by NF-κB and mTOR, respectively (146).
Among all immune cells, CD8+ T cells are the most radiosensitive, whereas CD4+ T cells and myeloid cells are more radioresistant with macrophages and granulocytes being the most radioresistant (147). This partially accounts for the increased level of suppressive immune cells, such as Tregs, within the TIME post-radiation (148, 149). However, tumor residing CD8+ T cells are more radioresistant than naïve and peripheral CD8+ T cells, which has attributed to IR-induced upregulation of TGFβ within the TIME (150). This is accompanied by increased CD8+ T cell exhaustion, which is evidenced by increased Teff expression of PD-1 and IFN-γ mediated upregulation of tumor PD-L1 expression (151–154).
After irradiation, many cytokines, chemokines and metabolites are upregulated within the TIME, leading to increased recruitment of suppressive immune cells, such as Tregs; which is accompanied by decreased CTL cross priming, recruitment, and function (Figure 3) (21–25). As a major mediator of immunosuppression that is being upregulated by irradiation within the TIME, TGFβ is involved in the suppression of CD8+ T cell mediated adaptive antitumor immunity in multiple ways (153, 155–157). These mainly involve inhibiting CD8+ T cell’s cytolytic function and recruitment; blocking Th1 differentiation and inducing a Treg phenotype; directing DC differentiation towards a tolerogenic phenotype; suppressing NK cell function; recruiting monocytes; polarizing macrophages toward an M2 phenotype; and mediating anti-CD 8+ T cell function by MDSCs (157, 158). Together with IL-10 secreted by T cells and other immune cells within the TIME post-radiation, more CD4+ T cells are being converted to Tregs (159–162). In vivo, irradiation also increased tumor cell expression of CCL2, which increased Treg recruitment into the TIME (163).
IR-induced increase in Tregs are affected by radiation dose fractionation with higher levels of Tregs observed in the TIME after SF high-dose irradiation of > 10 Gy (164–166). Although MF regimens of lower fractional dose can induce an increase in T cell and NK cell activation early, more latent T cell and NK cell activation was observed with SF ablative doses (166). IR-induced Tregs have higher expression of CTLA-4, which leads to increased inhibition of T cell cross presentation (102, 165, 166). Also, Tregs consume IL-2 within the TIME; release additional suppressive cytokines, including TGFβ, IL-10, IL-35, Fgl2; and express ectonucleotidases, such as CD39 and CD73, which increase adenosine production (102).
Increased recruitment of myeloid cells into the TIME post-radiation has been consistently observed. One common mechanism of radiation induced myeloid cell tumor infiltration is increased homing through the upregulation of chemokine expression in tumor cells, such as CCL2, CCL5, and HIF-1 induced stromal-derived factor 1α (SDF-1α), which interacts with CCR2, CCR5, and CXCR4 (163, 167–170). Increased CCL2-CCR2 mediated chemotaxis is found to be at least partially induced by STING mediated IFN-β signaling (171). Irradiation also stimulates DNA damage-induced kinase ABL1 mediated upregulation of macrophage colony-stimulating factor (M-CSF or CSF1) in tumor cells (172). This subsequently increases CSF1-CSF1R mediated trafficking of myeloid cells into the TIME (172, 173).
Suppressive myeloid cells originate from myeloid progenitor cells in the bone marrow, which migrate to peripheral organs to develop into macrophages, DCs, or granulocytes (174). However, soluble factors produced within the TME promote local accumulation and activation of MDSCs. While MDSCs play a suppressive role within the TIME, functional differentiation TAMs and TANs depends on the balance of immunostimulatory and immunosuppressive signals within the TIME (175). Upon irradiation, increased suppressive myeloid cell infiltration generally impairs CTL mediated anti-tumor immunity through nutrient depletion, increasing oxidative stress, impairing CTL trafficking and function, and stimulating Treg recruitment and function (174, 175).
MDSCs within the TIME are primarily categorized as granulocytic (g)/polymorphonuclear (PMN)-MDSCs or monocytic (M)-MDSCs (176). MDSCs secrete a series of suppressive solutes, such as IL4, IL10, TGFβ1, CSF2, VEGFA, PGE2 and L-kynurenine. VEGFA, TGFβ1, IL-10, and IL-6 upregulate Treg expansion and TAM M2 polarization. MDSCs overexpress IDO1 and Arg1, leading to depletion of key amino acids, such as arginine, cystine, and tryptophan within the TIME. MDSCs also produce reactive oxygen species (ROS) and reactive nitrogen species (e.g., nitric oxide, NO), leading to modified MHC-I and receptors for antigens and chemokines on T cells (175, 176). PD-L1 expressed on MDSCs directly suppresses CTLs and NK cells (176).
In addition to myeloid cell trafficking, CSF1R signaling also mediates TAM polarization toward an M2 phenotype (177). High dose (HD) irradiation induces the expression of M2 associated genes, such as Arg1, and COX2, whereas LD irradiation of 0.5-2 Gy was shown to induce M1 polarization in TAMs (138, 140, 178). M2 TAMs induce immunosuppression in ways similar to MDSCs. On the contrary, M1 TAMs produce proinflammatory cytokines, such as TNF and IL-12; recruit Th1 CD4+ T cells through CXCL9 and CXCL10 secretion; and induce direct tumor cell killing (175). Similar to TAMs, TANs polarize into the antitumoral N1 or the protumoral N2 phenotypes with N2 TANs having functional overlap with PMH-MDSCs (179, 180). TANs within the TIME have generally been associated with an immunosuppressive role and poor response to immunotherapy or RT with GLUT1 identified to be essential to their protumoral role (180–183).
Increased release of suppressive cytokines post-radiation, such as VEGF and TGFβ, may further stimulate the development of an immunosuppressive TIME by inducing aberrant tumor vasculature formation, increasing fibrosis, and activating CAFs, resulting in poor perfusion, increased hypoxia, increased recruitment of suppressive immune cells; and the inhibition of CTL function, homing, and endothelial adhesion (87, 184–187). However, how RT influences CAFs’ immunomodulatory function remains controversial and needs to be better defined (188).
Immunostimulatory properties of radiation allows it to act synergistically with immunomodulatory agents to induce a CD8+ T cell mediated immunogenic antitumor response that is stronger than from either alone (Figure 3). Strong local and systemic antitumor response (response outside of the irradiated field, which is also known as the “abscopal effect”) were elicited when RT is combined with immunomodulatory agents stimulating the proliferation and/or activation of DCs, Th1 CD4+ T cells, and CD8+ T cells (189–192). This was due to IFN-I mediated DC infiltration, CD8+ cross priming, and subsequent effector CD8+ T cell infiltration into the TIME (191, 192). Such antitumor response was consistently observed when RT was combined with an anti-CTLA-4 ICI in vivo (193–196). The intensity of an abscopal response generated from such combinations are dose fractionation and treatment sequence dependent. As shown by Dewan et al, an abscopal effect was most prominent after combining an anti-CTLA-4 antibody with 8 Gy x 3 fractions vs. 6 Gy x 5 fractions, or 20 Gy x 1 fraction with SF irradiation failing to induce any such effect (194). The three-fraction regimen was further shown to have stronger distant effect than the five-fraction regimen. When compared with concurrent delivery of RT and an anti-CTLA-4 ICI, ICI delivery after RT was shown to have reduced distant therapeutic effect. Increased antitumor immunity with the combined treatment is associated with increased CTLs, CD8/CD4 ratio, reduced Tregs within the TIME, and increased CD8+ T cell clonality (195, 196).
Unlike anti-CTLA-4 ICIs, which suppress the inhibition of CD8+ T cell activation within the more proximal site of tdLNs, anti-PD-(L)1 ICIs are more recognized for their ability to overcome PD-(L)1 mediated inhibition of CTL function within the peripheral TIME (6). PD-L1 expression is upregulated in DCs and tumor cells after irradiation, while acute decrease in PD-1 expression in CD8+ T cells within the TIME is observed after RT with an ablative dose (197, 198). Significant improvement in local and abscopal antitumor responses was observed after combined treatment with RT and an anti-PD-L1 ICI (197–199). Increases in response were more prominent with hypofractionated schedule delivering ablative doses, which results in increased CD8+ T cell infiltration & function; and reduced intratumoral MDSCs resulting from decreased trafficking and CD8+ T cell mediated direct killing requiring TNFα (197, 199). After ablative doses, latent increase in PD-1 expression by tumor-infiltrating T cells and elevated PD-L1 expression by tumor cells at both primary and secondary tumor sites were observed (200, 201). This was accompanied by decreased intratumoral Tregs.
Anti-PD-1 ICIs combined with RT induced significant increases in tumor-antigen specific and memory CD8+ T cells, as well as CD8+/Treg ratio within the peripheral TIME, subsequently leading to significantly amplified local and abscopal antitumor responses (201–204). The increased CD8+/Treg ratio mainly resulted from a reduction of Tregs within the peripheral TIME after the combined treatment, which is accompanied by increased CD8+ T cell clonality at both primary and secondary tumor sites (202, 203). Contrary to the reduction of intratumoral MDSCs observed after ablative doses, increased intratumoral accumulation of MDSCs has been observed after combined treatment with conventionally fractionated radiation and an anti-PD-1 antibody (203). Overall, pre-clinical data supports the use of hypofractionated RT schedules in combined RT and anti-PD-(L)1 treatment strategies.
The benefit of RT and anti-PD-(L)1 combinations also depends on features of the tumor residing tissue and the tumor itself. For example, LD irradiation with 4 Gy x 3 fractions may induce significant CD8+ T cell tumor infiltration and increased CD8+ T cell function when combined with an anti-PD-1 antibody (205). This combination led to significant antitumor response in a KRASG12D mutant orthotopic mouse model of lung adenocarcinoma, which depended on the presence of lung tissue residing club cells that express synaptosome-associated protein 23 (Scgb1a1). This is due to club cell secretory proteins’ immunostimulatory roles, such as suppression of myeloid cells (205). Alternatively, STK11/LKB1 mutations, which frequently co-occurs with KRAS-mutations in lung adenocarcinomas, were associated with poor synergy between RT and anti-PD-1 in vivo (200). This may result from increased CTL exhaustion in the presence of increased CD8+ T cell infiltration and CD8+/Treg ratio after the combined treatment. These examples demonstrate the influences of various tumor-related features on the TIME, which may dictate tumor response to combined RT and ICI treatment. Additional therapeutic strategies are needed to further augment the efficacy of RT and ICI combinations in these situations.
The PD-1/PD-L1 axis is a main mediator of CD8+ T cell exhaustion with in the TIME of resistant melanomas after combined treatment with RT and an anti-CTLA-4 ICI (206). In resistant tumors, increased tumor expression of PD-L1 and the proportion of PD-1+ EOMES+ CD8+ T cells that do not express markers for activation, Ki67 and GzmB were found. Activated CD8+ T cells and CD8+/Treg ratio markedly increased within the TIME after an anti-PD-L1 ICI was added to the RT and anti-CTLA-4 ICI combination. Dual CTLA-4 and PD-(L)1 blockade combined with radiation led to a CR rate of 80% and significant prolongation of survival in vivo. Therefore, significant antitumor immunity may be generated by combining RT and the targeting of multiple stimulatory and/or inhibitory checkpoints. This strategy may be further explored in the context of combining RT with dual immune checkpoint blockade (ICB) (207–212).
Further improvement of antitumor response was observed when an OX40 agonist, an anti-TIM-3, or an anti-TIGIT antibody was added to the RT and anti-PD-(L)1 antibody combination (210–212). Significant increase in CD8+ T cell and CD103+ DC infiltration, as well as decreased CD8+ T cell exhaustion were observed with the addition of an OX40 agonist (210). This led to further improvement in local and abscopal responses, leading to an survival advantage over radiation combined with an anti-PD-1 antibody alone. Similar survival advantage was observed with the addition of an anti-TIM-3, or an anti-TIGIT antibody (211, 212). The added benefit from an anti-TIGIT antibody was dependent on radiation dose fractionation. TIGIT expression by CD8+ T cells was increased by 8 Gy x 3 fractions, but decreased by 2 Gy x 18 fractions (212). Other strategies to further enhance immunostimulatory effects of RT combined with ICIs may include addition of other immunostimulatory agents, such as stimulatory cytokines (e.g., IL-2, IL-12) and activators of innate immunity sensor (213–218). Alternatively, additional therapeutic advantage may be gained by adding agents reducing the level of immunosuppression within the TIME to RT and ICI combinations, such as antibodies against TGFβ, VEGF, or PI3K; M2 TAM or MDSC reducing agents; inhibitors of suppressive metabolite production, such as CD73 antibodies; and inhibitors to other checkpoints, such as CD47 antibodies (199, 209, 219–224).
In case report format, dramatic abscopal response (AR) was initially reported in patients with metastatic melanoma or NSCLC who progressed after multiple courses of systemic therapy (225–227). These patients received single-site hypofractionated RT, or stereotactic body radiotherapy (SBRT) either after progression while on an ICI, or concurrently with an ICI after progression on chemotherapy. Abscopal response rates (ARRs) of 51-53% were further identified in small cohorts of patients with advanced melanoma who progressed an anti-CTLA-4 ICI (Ipilimumab) or an anti-PD-1 ICI (Nivolumab or Pembrolizumab) after they received intra- or extra-cranial hypofractionated RT (228, 229). Furthermore, an AR was associated with improved median survival in patients who progressed on Ipilimumab (22.4 vs. 8.3 months, p = 0.002) (228). This association was also corroborated in a prospective trial testing the efficacy of combining hypofractionated RT with a DC stimulating agent in patients with metastases that progressed on conventional systemic therapy (230). In this study, an ARR of 26.8% was observed in 41patients (4 NSCLC, 5 breast cancer, 2 thymic cancer).
Similar ARR’s were observed in early phase trials evaluating the efficacy and safety of administering SBRT sequentially or concurrently with ICIs in patients with metastatic melanoma, or mixed histology solid tumors refractory to conventional systemic therapy (Table 1) (231–233). Such systemic response was correlated with the expression of IFN-γ associated genes (232). Among responders to such combinations, CD8+ T cells and their expression of 4-1BB & PD-1 were increased in the peripheral blood, reflecting an increase in systemic antitumor immunity after SBRT (233). Also, the intensity of antitumor immunity generated by SBRT combined with an ICI was affected by the choice of site to be irradiated, with liver correlating to higher levels of T cell activation than the lungs.
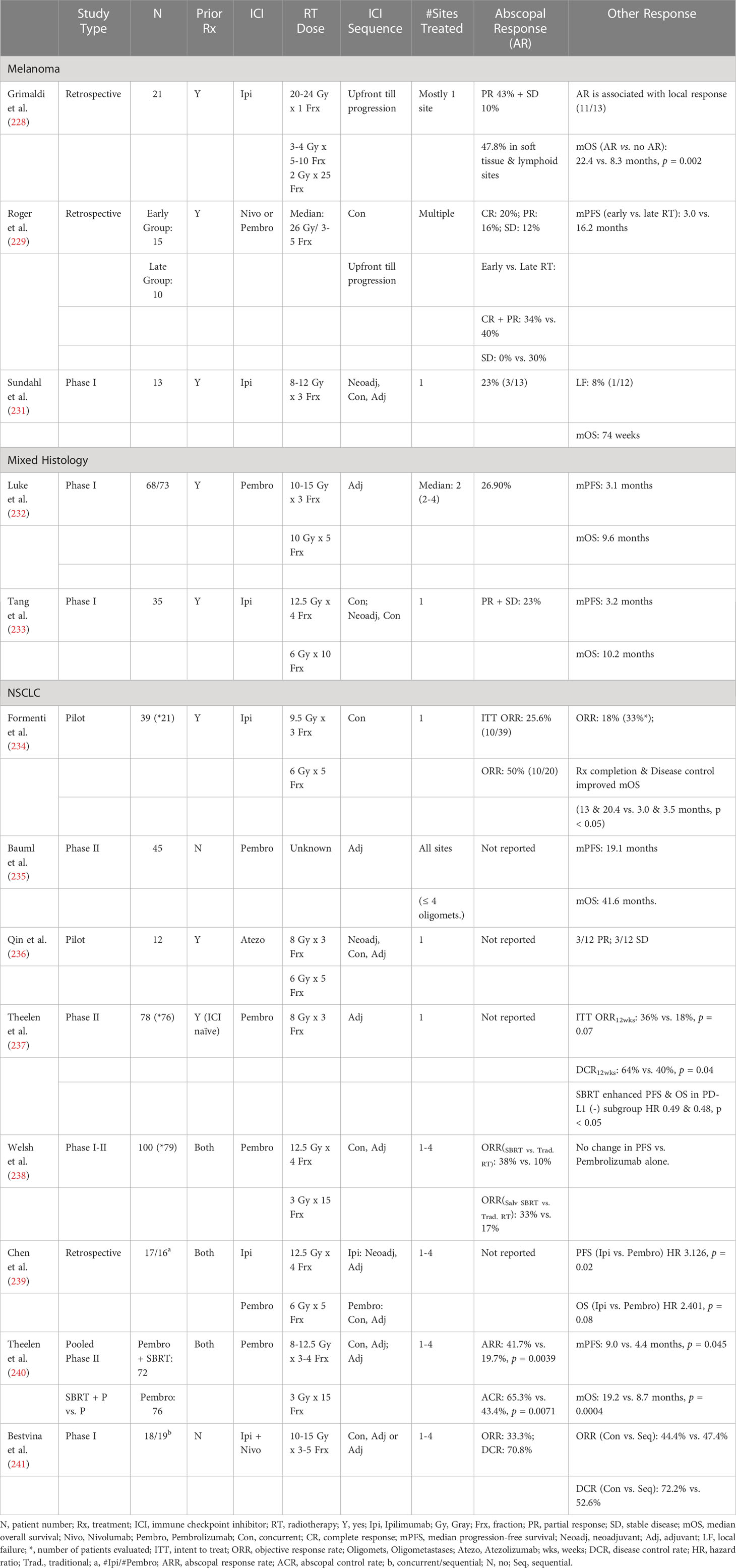
Table 1 Abscopal response following RT + ICIs in metastatic solid tumors.
SBRT and ICI combination’s ability to induce systemic antitumor immunity in mostly treatment refractory metastatic NSCLC has been intensely investigated in early phase clinical trials (234–241). Despite limited sample size, responses at both irradiated and non-irradiated tumor sites were consistently observed in these studies (Table 1). After combing an ICI with RT to mostly 1-2 tumor sites, an ARR between 33-50% were observed (234, 238, 240). Combining dual CTLA-4 and PD-1 blockade with SBRT does not seem to further increase the ARR (241). Clinically, the generation of a robust systemic antitumor response is also associated with the expansion of tumor neoantigen specific CD8+ T cells (234).
Administration of Pembrolizumab after local ablative therapy to all oligometastatic sites has led to impressive median progression-free survival (PFS) and overall survival (OS) of 19.1 and 41.6 months, respectively (235). Although a survival advantage was not demonstrated when SBRT delivering 8 Gy x 3 fractions to 1 tumor site was combined with adjuvant Pembrolizumab compared with Pembrolizumab alone in a phase II trial (PEMBRO-RT), noticeable improvement of the objective response rate (ORR) and disease control rate (DCR) at 12 weeks was observed (237). In the subgroup analysis, significant improvements in the PFS and OS were observed in the PD-L1 negative patients, which will be further confirmed in a separate phase II/III study (242). As shown in a phase I/II study, SBRT induced higher ARR (concurrent: 38% vs. 10%; sequential: 33% vs. 17%) than more protracted course of RT when combined with PD-1 blockade (238). In a small cohort, PD-1 blockade was also found to be more suitable than CTLA-4 blockade for combining with SBRT, as it led to significant improvements in PFS and OS (239). The failure to demonstrate a survival advantage in the PEMBRO-RT trial may stem from the suboptimal sample size of the study (N = 78) (237).
After pooling the PEMBRO-RT trial with another similarly designed phase II trial, significant improvements in the ARR and abscopal control rate (ACR) at 12 weeks were observed when SBRT was combined with Pembrolizumab vs. Pembrolizumab alone, which translated into significant improvements in PFS and OS (Table 1) (240). While early clinical experience does not demonstrate any significant difference in clinical response rates between concurrent or adjuvant ICI administration with SBRT, concurrent treatment was associated with significantly improved global antitumor response, and survival in patients with non-inflamed NSCLC in the presence of high aneuploidy and low TMB (241, 243). Therefore, valid biomarkers for a noninflamed TIME may be used to effectively select patients for concurrent SBRT and ICI.
Combining an ICI with chemoradiation for stage II-III NSCLC has led to significant reduction in distant metastasis or death, leading to prolonged PFS and OS (Table 2) (244–250). Adjuvant Durvalumab after concurrent chemoradiation delivering mostly 60-66 Gy in 30-33 fractions in stage III NSCLC led to remarkable improvement in median PFS from 5.6 to 16.9 months, and median OS from 29.1 to 47.5 months, respectively (244, 251, 252). This was associated with increased ORR (29.8% vs. 18.3%) and a 41% reduction in the risk of death and distant metastasis at 5 years (244). Similar findings were reported when other anti-PD-(L)1 agents were administered after chemoradiation (245–247). As shown in a subgroup analysis of the PACIFIC trial, such OS advantage may be limited to patients with PD-L1 expression ≥ 1% (252). A survival benefit of lesser scale was also observed if patients received sequential chemoradiation prior to adjuvant anti-PD-L1 ICI. ORR in patients receiving sequential chemoradiation prior to ICI administration appears less than that observed in patients who received concurrent chemoradiation (247). This may be one reason for the relatively less survival benefit with adjuvant anti-PD-L1 ICI observed in GEMSTONE-301, in which 33-34% of patients received sequential chemoradiation prior to Sugemalimab (245).
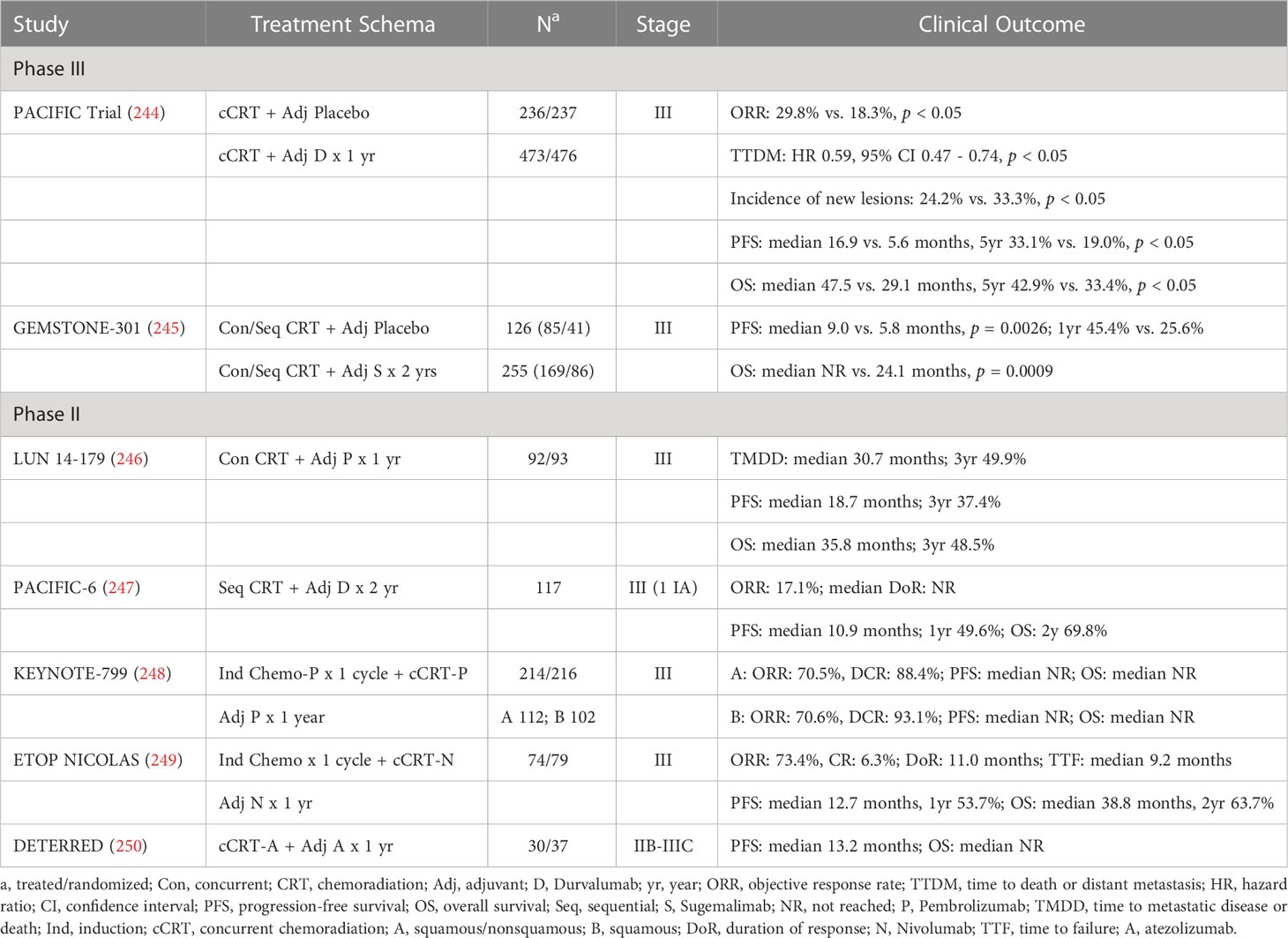
Table 2 Combining chemoradiation with ICI in early-stage NSCLC.
Concurrent administration of an anti-PD-(L)1 agent with chemoradiation has been shown to be tolerable (253). High ORR of approximately 70% was observed with concurrent administration of chemoradiation and an anti-PD-1 agent (248, 249). However, concurrent administration appears to be associated with higher incidences of severe toxicity and/or fatal events when compared with those observed in the PACIFIC trial, which was associated with ≥ grade 3 adverse effects of < 10%, and no fatal toxicity (248–250, 254). This may narrow the therapeutic window associated with the concurrent strategy. How it compares to sequential administration of concurrent chemoradiation and adjuvant ICI is being further tested in the phase III randomized trial, ECOG-ACRIN EA5181 (255). Neoadjuvant administration of moderate dosed SBRT to the primary tumor combined with Durvalumab has also been shown to significantly increase the major pathological response (MPR) rate comparing to Durvalumab alone (53.3% vs. 6.7%, crude odds ratio, OR: 16, p < 0.0001) (256). MPR in the combination group led to an impressive CR rate of 50%. The role of combining SBRT and immunotherapy in very early staged NSCLC is currently being investigated in multiple ongoing trials (257–260).
Combining RT with immunotherapy and chemotherapy has also been tested in small cell lung cancer (SCLC). This strategy was found to be feasible in metastatic or localized stages (261, 262). In limited stage SCLC, concurrent chemoradiation and Pembrolizumab was found to be tolerable, and led to a median OS of 39.5 months, which compares favorably with the median survival of 28.5 months after the same chemoradiation regimen alone in RTOG 0538 (262, 263). In a trial designed similarly to the PACIFIC trial, adjuvant Nivolumab in stage II-III esophageal or gastro-esophageal junction cancer patients with pathological residual disease after neoadjuvant chemoradiation led to improved median disease-free survival (DFS) from 11.0 to 22.4 months (264). On the contrary, combining either SBRT or chemoradiation with an anti-PD-(L)1 antibody has not been shown to be advantageous in head and neck cancer (265, 266). The underlying mechanism for this lack of synergy remains unclear.
LD-RT may induce tumor vasculature normalization and TAM M1 polarization, leading to increased tumor infiltration by CTLs (138, 140). Significant increase in Th1 CD4+ mediated antitumor immunity has been observed when LD-RT is combined with immunotherapy ± chemotherapy (140, 267). Response to such combinations in humans was associated with increased TCR clonality in the peripheral blood, which implies the upregulation of systemic antitumor immunity (140). However, combining LD-RT or moderate doses of 8 Gy x 3 fractions with CTLA-4 and PD-L1 dual blockade did not lead to added clinical benefit in poorly immunogenic colorectal cancer (CRC) and NSCLC in phase II clinical trials (268, 269). This may be due to suboptimal intensity of the antitumor immunity generated by LD-RT to a single site alone, and the induction of suppressive immune cells (270). Multi-site irradiation may provide a solution to this problem.
In vivo, LD-RT with targeted radionuclide therapy (TRT) that targets all malignant lesions was able to induce strong local and abscopal effects when combined with an anti-CTLA-4 ICI, leading to improved survival than either treatment alone (271). In addition to stimulatory effects on NK cells and myeloid cells, CD8+/Treg ratio increased more 1 day after TRT than after either focal LD-RT or moderate dose RT. Combining TRT and ICI led to significant increase in Teff infiltration, CTL activation, and reduction of CD8+ T cell exhaustion & IL-10 within the TIME of poorly immunogenic tumors. This phenomenon was shown to be STING-dependent (271). When combined with focal high-dose (HD) RT and ICI, TRT led to the best response at the primary and secondary sites, which further improved survival compared with either HD-RT or TRT alone combined with ICI. Thus, LD-RT and HD-RT’s different immunostimulatory effects can supplement each other. Comparing to HD-RT alone, further decreases in Tregs, IL-10 secreting macrophages, and TGFβ within the TIME of secondary sites were observed when LD-RT to metastatic sites was added (272, 273).
Combining LD-RT to secondary tumor sites with focal moderate-to-high dose RT to the primary tumor further augmented local and abscopal responses when they are combined with CTLA-4 and PD-1 dual blockade in vivo (273). Such effects were NK and CD4+ T cell dependent, and significantly improved survival. Similarly, enhanced systemic antitumor immunity was observed when LD-RT to secondary sites was combined with moderate-dosed RT and an anti-PD-1 ICI (274). The triple combination increased CD8+ T cell recruitment and reduced intratumoral MDSCs. The benefit of such triple combination was confirmed in small patient cohorts, in which LD-RT to distant lesions led to significant response in irradiated distant lesions (273–275). In a phase II clinical trial, improved response in metastases from NSCLC and melanoma was observed after such treatment in patients who progressed on an anti-PD-(L)1 or anti-CTLA-4 ICI, or their combination (276). Thus, combining LD-RT, HD-RT, and ICI represents a potential strategy to induce an inflamed TIME at both primary and distant tumor sites that are “cold”.
The immunostimulatory effects of hypofractionated RT may be augmented by altering the immune cell composition of the TIME. This may be achieved by directly targeting suppressive immune cells, such as Tregs; or administering stimulatory immune cells, such as Th1 CD4+ T cells (277–279). Adoptive T cell therapy (ACT), which includes chimeric antigen receptor (CAR)-T cell therapy or the administration of engineered-TCR cells, may be further enhanced by RT. This is due to RT’s ability to induce TAA release, IFN-I mediated antigen presentation, and CD8+ recruitment at high doses, as well as its ability to normalize tumor vasculature, alter local stroma, and polarize TAMs toward the M1 phenotype without excessive cytotoxic effect on intratumoral CTLs at low doses (138, 280, 281).
Hypofractionated RT stimulates the proliferation and activation of both tumor antigen specific and adoptively transferred T cells, homing of CD8+ T cells, and tumor cell susceptibility to direct T cell cytotoxicity (134, 282). These stimulatory effects led to improved survival in vivo (134). A major limitation to ACT’s efficacy, T cell recruitment into the TIME, may be improved by RT due to its ability to stimulate CCL5, CXCL9, CXCL10 expression that is at least partially mediated by RT induced recruitment of eosinophils (282). As shown in 3 cohorts of NSCLC and nasopharyngeal cancer patients, RT significantly increased peripheral eosinophil concentration, which correlated with significantly improved PFS.
CAR-T cell therapy’s efficacy relies on tumor expression of specific antigens, which may be lost through immune editing (90). This challenge may be overcome by LD-RT priming, which was shown to induce TRAIL-mediated tumor cell killing in the absence of the CAR specific antigen with excellent local response (283). This makes LD-RT priming a potential therapeutic strategy to overcome CAR-T cell therapy resistance. When used as a debulking regimen in relapsed non-Hodgkin’s lymphoma (NHL), regional RT was shown to be correlated with dramatic response to CAR-T cell therapy and much less toxicity than chemotherapy in a small cohort of 10 patients (284). Synergistic effects between NKG2D-based CAR-T cell therapy and RT were also observed in a glioblastoma mouse model (285). RT may augment ACT’s therapeutic efficacy at either low and high doses, and how to take advantage of their unique immunostimulatory properties remains to be further investigated (286).
The TIME is sculpted by the tumor’s intrinsic features and those of the various immune cells, stromal cells, endothelial cells within the TME. Various mechanisms of failure to generate anti-tumor immunity exist, resulting in a noninflamed or “cold” TIME which is associated with primary or acquired resistance to ICIs and other immunomodulatory agents. Radiotherapy’s immunostimulatory effects are offset by its immunosuppressive effects. Its immunostimulatory effects may be augmented when combined with ICIs and other immunomodulators in a manner that is both dose/fractionation and sequence dependent. This strategy may stimulate the generation of antitumor immunity, and/or reduce immunosuppression caused by suppressive mediators and cells within the TIME. Therefore, combining RT with immunotherapy may help overcome resistance to ICIs and other forms immunotherapy, and enhance established treatments for cancer with promising early clinical evidence emerging. In addition, strategies to enhance the efficacy of such combinations warrant further exploration.
AC and NN contributed to conception and design of the study. AC wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
The figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell (2018) 175:313–26. doi: 10.1016/j.cell.2018.09.035
2. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
3. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2021) 184:5309–37. doi: 10.1016/j.cell.2021.09.020
4. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (2021) 398:1002–14. doi: 10.1016/S0140-6736(21)01206-X
5. Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res (2019) 25:4592–602. doi: 10.1158/1078-0432.CCR-18-1538
6. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
7. Dunn GP, Old LJ. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
8. Herbst RS, Soria J, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
9. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75:2139–45. doi: 10.1158/0008-5472.CAN-15-0255
10. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T, et al. The immune landscape of cancer. Immunity (2018) 48:812–30. doi: 10.1016/j.immuni.2018.03.023
11. Chi A, He X, Hou L, Nguyen NP, Zhu G, Cameron RB, et al. Classification of non-small cell lung cancer’s tumor immune micro-environment and strategies to augment its response to immune checkpoint blockade. Cancers (2021) 13:2924. doi: 10.3390/cancers13122924
12. Shirasawa M, Yoshida T, Shimoda Y, Takayanagi D, Shiraishi K, Kubo T, et al. Differential immune-related microenvironment determines PD-1/PD-L1 blockade efficacy in advanced non-small cell lung cancer patients. J Thorac Oncol (2021) 16:2078–90. doi: 10.1016/j.jtho.2021.07.027
13. Kim TK, Vandsemb EN, Herbst RS, Chen L. Adaptive immune resistance at the tumour site: mechanisms and therapeutic implications. Nat Rev Drug Disc (2022) 21:529–40. doi: 10.1038/s41573-022-00493-5
14. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
15. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
16. Trujillo JA, Luke J, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer (2019) 7:295. doi: 10.1186/s40425-019-0780-0
17. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37:443–55. doi: 10.1016/j.ccell.2020.03.017
18. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
19. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer (2020) 6:605–18. doi: 10.1016/j.trecan.2020.02.022
20. de Olza MO, Rodrigo BN, Zimmermann S, Coukos G. Turning up the heat on non-immunoreactive tumours: opportunities for clinical development. Lancet Oncol (2020) 21:e419–430. doi: 10.1016/S1470-2045(20)30234-5
21. Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol (2015) 1:1325–32. doi: 10.1001/jamaoncol.2015.2756
22. Weichselbaum RR, Liang H, Deng L, Fu Y. Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol (2017) 16:365–79. doi: 10.1038/nrclinonc.2016.211
23. Rodríguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol (2018) 39:644–55. doi: 10.1016/j.it.2018.06.001
24. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodeling by tumour cells after radiotherapy. Nat Rev Cancer (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1
25. Charpentier M, Spada S, van Nest SJ, Demaria S. Radiation therapy-induced remodeling of the tumor immune microenvironment. Sem Cancer Biol (2022) 86:737–47. doi: 10.1016/j.semcancer.2022.04.003
26. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117:1137–46. doi: 10.1172/JCI31405
27. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331:1565–70. doi: 10.1126/science.1203486
28. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16:343–53. doi: 10.1038/ni.3123
29. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
30. Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res (2017) 27:96–108. doi: 10.1038/cr.2016.149
31. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest (2015) 125:3413–21. doi: 10.1172/JCI80008
32. Lee CH, Yelensky R, Jooss K, Chan TA. Update on tumor neoantigens and their utility: why it is good to be different. Trends Immunol (2018) 39:536–48. doi: 10.1016/j.it.2018.04.005
33. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol (2016) 27:1492–504. doi: 10.1093/annonc/mdw217
34. Hodge JW, Greiner JW, Tsang KY, Sabzevari H, Kudo-Saito C, Grosenbach DW, et al. Costimulatory molecules as adjuvants for immunotherapy. Front Biosci (2006) 11:788–803. doi: 10.2741/1837
35. Chen L, Flies D. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi: 10.1038/nri3405
36. van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM. Migrating into the tumor: a roadmap for T cells. Trends Cancer (2017) 3:797–808. doi: 10.1016/j.trecan.2017.09.006
37. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell (2019) 35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004
38. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
39. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol (2022) 19:287–305. doi: 10.1038/s41571-022-00601-9
40. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med (2016) 375:1767–78. doi: 10.1056/NEJMra1514296
41. Tumeh PC, Harview CL, Yearley JH, IP S, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi: 10.1038/nature13954
42. Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Sig Transduct Target Ther (2021) 6:72. doi: 10.1038/s41392-020-00449-4
43. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
44. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
45. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
46. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
47. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482:400–4. doi: 10.1038/nature10755
48. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31:e439–442. doi: 10.1200/JCO.2012.47.7521
49. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi: 10.1038/nature13988
50. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
51. Le DT, Uram JN, Wang H, Barlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
52. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
53. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1(PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol (2018) 36:633–41. doi: 10.1200/JCO.2017.75.3384
54. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med (2018) 24:1441–8. doi: 10.1038/s41591-018-0134-3
55. van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350:207–11. doi: 10.1126/science.aad0095
56. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature (2019) 567:479–85. doi: 10.1038/s41586-019-1032-7
57. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery (2017) 7:264–76. doi: 10.1158/2159-8290.CD-16-0828
58. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
59. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res (2019) 25:3074–83. doi: 10.1158/1078-0432.CCR-18-1942
60. Muto S, Ozaki Y, Yamaguchi H, Mine H, Takagi H, Watanabe M, et al. Tumor β-catenin expression in non-small cell lung cancer with high tumor mutation burden. Oncol Lett (2021) 21:203. doi: 10.3892/ol.2021.12464
61. Takeuchi Y, Tanegashima T, Sato E, Irie T, Sai A, Itahashi K, et al. Highly immunogenic cancer cells require activation of the wnt pathway for immunological escape. Sci Immunol (2021) 6:eabc6424. doi: 10.1126/sciimmunol.abc6424
62. Villablanca E, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-mediated liver X receptor-α activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med (2010) 16:98–105. doi: 10.1038/nm.2074
63. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
64. Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer (2019) 7:109. doi: 10.1186/s40425-019-0580-6
65. Jang GY, Lee JW, Kim YS, Lee SE, Han HD, Hong KJ, et al. Interactions between tumor-derived proteins and toll-like receptors. Exp Mol Med (2020) 52:1926–35. doi: 10.1038/s12276-020-00540-4
66. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162:1257–70. doi: 10.1016/j.cell.2015.08.015
67. Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci USA (2016) 113:3844–9. doi: 10.1073/pnas.1602023113
68. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity (2017) 47:363–73. doi: 10.1016/j.immuni.2017.07.016
69. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune response through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi: 10.1038/ni.2376
70. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172:1022–37. doi: 10.1016/j.cell.2018.01.004
71. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
72. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer (2021) 21:298–312. doi: 10.1038/s41568-021-00339-z
73. Cao W, Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol (2014) 192:2920–31. doi: 10.4049/jimmunol.1302801
74. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun (2017) 8:2122. doi: 10.1038/s41467-017-02186-9
75. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death Diff (2015) 22:237–46. doi: 10.1038/cdd.2014.134
76. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-redundant requirement for CXCR3 signaling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Comm (2015) 6:7458. doi: 10.1038/ncomms8458
77. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell (2017) 31:711–23. doi: 10.1016/j.ccell.2017.04.003
78. Hoch T, Schulz D, Eling N, Gómez JM, Levesque MP, Bodenmiller B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci Immunol (2022) 7:eabk1692. doi: 10.1126/sciimmunol.abk1692
79. House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res (2020) 26:487–504. doi: 10.1158/1078-0432.CCR-19-1868
80. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208:1949–62. doi: 10.1084/jem.20101956
81. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res (1996) 56:1111–7.
82. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagermann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
83. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol (2018) 15:366–81. doi: 10.1038/s41571-018-0007-1
84. Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbæk MS, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer (2019) 7:68. doi: 10.1186/s40425-019-0556-6
85. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGF-β attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
86. Freeman P, Mielgo A. Cancer-associated fibroblasts mediated inhibition of CD8+ cytotoxic T cell accumulation in tumours: mechanisms and therapeutic opportunities. Cancers (2020) 12:2687. doi: 10.3390/cancers12092687
87. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer (2021) 20:131. doi: 10.1186/s12943-021-01428-1
88. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis (2018) 9:115. doi: 10.1038/s41419-017-0061-0
89. Maggs L, Sadagopan A, Moghaddam AS, Ferrone S. HLA class I antigen processing machinery defects in antitumor immunity and immunotherapy. Trends Cancer (2021) 7:1089–101. doi: 10.1016/j.trecan.2021.07.006
90. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell (2017) 171:1259–71. doi: 10.1016/j.cell.2017.10.001
91. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (2018) 359:582–7. doi: 10.1126/science.aao4572
92. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci USA (2016) 113:5999–6004. doi: 10.1073/pnas.1602069113
93. Chew GL, Campbell AE, De Neef E, Sutliff NA, Shadle SC, Tapscott SJ, et al. DUX4 suppresses MHC class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev Cell (2019) 50:658–71. doi: 10.1016/j.devcel.2019.06.011
94. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
95. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15:486–99. doi: 10.1038/nri3862
96. Budimir N, Thomas GD, Dolina JS, Salek-Ardakani S. Reversing T-cell exhaustion in cancer: lessons learned from PD-1/PD-L1 immune checkpoint blockade. Cancer Immunol Res (2022) 10:146–53. doi: 10.1158/2326-6066.CIR-21-0515
97. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi: 10.1084/jem.20100643
98. Miller B, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
99. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res (2015) 3:1344–55. doi: 10.1158/2326-6066.CIR-15-0097
100. Kim CG, Kim G, Kim KH, Park S, Shin S, Yeo D, et al. Distinct exhaustion features of T lymphocytes shape the tumor-immune microenvironment with therapeutic implication in patients with non-small-cell lung cancer. J Immunother Cancer (2021) 9:e002780. doi: 10.1136/jitc-2021-002780
101. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/nri2343
102. Koyama S, Nishikawa H. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer (2021) 9:e002591. doi: 10.1136/jitc-2021-002591
103. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
104. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumor progression. Br J Cancer (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
105. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
106. Li C, Xu X, Wei S, Jiang P, Xue L, Wang J. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer (2021) 9:e001341. doi: 10.1136/jitc-2020-001341
107. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
108. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol (2005) 174:7516–23. doi: 10.4049/jimmunol.174.12.7516
109. Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. J Immunol (2008) 180:3132–9. doi: 10.4049/jimmunol.180.5.3132
110. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol (2012) 189:558–66. doi: 10.4049/jimmunol.1200563
111. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114:589–95. doi: 10.1182/blood-2009-02-206870
112. Filatenkov A, Baker J, Mueller AMS, Kenkel J, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824
113. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203:1259–71. doi: 10.1084/jem.20052494
114. Lussier DM, Alspach E, Ward JP, Miceli AP, Runci D, White JM, et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc Natl Acad Sci USA (2021) 118:e2102611118. doi: 10.1073/pnas.2102611118
115. Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J Clin Invest (2021) 131:e138740. doi: 10.1172/JCI138740
116. Salomon N, Vascotto F, Selmi A, Vormehr M, Quinkhardt J, Bukur T, et al. A liposomal RNA vaccine inducing neoantigen-specific CD4+ T cells augments the antitumor activity of local radiotherapy in mice. Oncoimmunology (2020) 9:1771925. doi: 10.1080/2162402X.2020.1771925
117. Huang RX, Zhou PK. DNA Damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther (2020) 5:60. doi: 10.1038/s41392-020-0150-x
118. Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell (2018) 34:361–78. doi: 10.1016/j.ccell.2018.05.013
119. Widau RC, Parekh AD, Ranck MC, Golden DW, Kumar KA, Sood RF, et al. RIG-i-like receptor LGP2 protects tumor cells from ionizing radiation. Proc Natl Acad Sci USA (2014) 111:E484–491. doi: 10.1073/pnas.1323253111
120. Min X, Zheng M, Yu Y, Wu J, Kuang Q, Hu Z, et al. Ultraviolet light induces HERV expression to activate RIG-I signalling pathway in keratinocytes. Exp Dermatol (2022) 31:1165–76. doi: 10.1111/exd.14568
121. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
122. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
123. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, et al. Exosomes shuttle Trex1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res (2018) 6:910–20. doi: 10.1158/2326-6066.CIR-17-0581
124. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Comm (2017) 8:15618. doi: 10.1038/ncomms15618
125. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene (2016) 35:5931–41. doi: 10.1038/onc.2016.104
126. Shekarian T, Valsesia-Wittmann S, Brody J, Michallet MC, Depil S, Caux C, et al. Pattern recognition receptors: immune targets to enhance cancer immunotherapy. Ann Oncol (2017) 28:1756–66. doi: 10.1093/annonc/mdx179
127. Golden EB, Frances D, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic death. Oncoimmunology (2014) 3:e28518. doi: 10.4161/onci.28518
128. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
129. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA (2009) 106:7119–24. doi: 10.1073/pnas.0902745106
130. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med (2013) 210:491–502. doi: 10.1084/jem.20122006
131. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
132. Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol (2008) 181:3099–107. doi: 10.4049/jimmunol.181.5.3099
133. Matsumura S, Demaria S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat Res (2010) 173:418–25. doi: 10.1667/RR1860.1
134. Lai JZ, Zhu YY, Ruan M, Chen L, Zhang QY. Local irradiation sensitized tumors to adoptive T cell therapy via enhancing the cross-priming, homing, and cytotoxicity of antigen-specific CD8 T cells. Front Immunol (2019) 10:2857. doi: 10.3389/fimmu.2019.02857
135. Hallahan DE, Virudachalam S. Accumulation of p-selectin in the lumen of irradiated blood vessels. Radiat Res (1999) 152:6–13. doi: 10.2307/3580044
136. Zhao Y, Zhang T, Wang Y, Lu D, Du J, Feng X, et al. ICAM-1 orchestrates the abscopal effect of tumor radiotherapy. Proc Natl Acad Sci USA (2021) 118:e2010333118. doi: 10.1073/pnas.2010333118
137. Kozin SV. Vascular damage in tumors: a key player in stereotactic radiation therapy? Trends Cancer (2022) 8:806–19. doi: 10.1016/j.trecan.2022.06.002
138. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
139. Tong F, Xiong CJ, Wei CH, Wang Y, Liang ZW, Lu H, et al. Hypo-fractionation radiotherapy normalizes tumor vasculature in non-small cell lung cancer xenografts through the p-STAT3/HIF-1 alpha signaling pathway. Ther Adv Med Oncol (2020) 12:1758835920965853. doi: 10.1177/1758835920965853
140. Herrera FG, Ronet C, de Olza MO, Barras D, Crespo I, Andreatta M, et al. Low-dose radiotherapy reverses tumor immune desertification and resistance to immunotherapy. Cancer Disc (2022) 12:108–33. doi: 10.1158/2159-8290.CD-21-0003
141. Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman N, et al. Irradiation of tumor cells up-regulates fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol (2003) 170:6338–47. doi: 10.4049/jimmunol.170.12.6338
142. Canter RJ, Grossenbacher SK, Foltz JA, Sturgill IR, Park JS, Luna JI, et al. Radiotherapy enhances natural killer cell cytotoxicity and localization in pre-clinical canine sarcomas and first-in-dog clinical trial. J Immunother Cancer (2017) 5:98. doi: 10.1186/s40425-017-0305-7
143. Hietanen T, Pitkänen M, Kapanen M, Kellokumpu-Lehtinen PL. Effects of single and fractionated irradiation n natural killer cell populations: radiobiological characteristics of viability and cytotoxicity in vitro. Anticancer Res (2015) 35:5193–200.
144. Le Bert N, Lam AR, Ho SS, Shen YJ, Liu MM, Gasser S. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology (2014) 3:e29259. doi: 10.4161/onci.29259
145. Kim JY, Son YO, Park SW, Bae JH, Chung JS, Kim HH, et al. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp Mol Med (2006) 38:474–84. doi: 10.1038/emm.2006.56
146. Walle T, Kraske JA, Liao B, Lenoir B, Timke C, von Bohlen Und Halbach E, et al. Radiotherapy orchestrates natural killer cell dependent antitumor immune responses through CXCL8. Sci Adv (2022) 8:eabh4050. doi: 10.1126/sciadv.abh4050
147. Cytlak UM, Dyer DP, Honeychurch J, Williams KJ, Travis MA, Illidge TM. Immunomodulation by radiotherapy in tumor control and normal tissue toxicity. Nat Rev Immunol (2022) 22:124–38. doi: 10.1038/s41577-021-00568-1
148. Reijmen E, De Mey S, De Mey W, Gevaert T, De Ridder K, Locy H, et al. Fractionated radiation severely reduces the number of CD8+ T cells and mature antigen presenting cells within lung tumors. Int J Radiat Oncol Biol Phys (2021) 111:272–83. doi: 10.1016/j.ijrobp.2021.04.009
149. Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys (2011) 81:1128–35. doi: 10.1016/j.ijrobp.2010.09.034
150. Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Comm (2019) 10:3959. doi: 10.1038/s41467-019-11906-2
151. Yoon YN, Choe MH, Kong M, Chung WK, Kim JS, Lim YJ. Dynamic alterations in PD-1/PD-L1 expression level and immune cell profiles based on radiation response status in mouse tumor model. Front Oncol (2022) 12:989190. doi: 10.3389/fonc.2022.989190
152. Kim KJ, Kim JH, Lee SJ, Lee EJ, Shin EC, Seong J. Radiation improves antitumor effect of immune checkpoint inhibitor in murine hepatocellular carcinoma model. Oncotarget (2017) 8:41242–55. doi: 10.18632/oncotarget.17168
153. Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res (2015) 75:2232–42. doi: 10.1158/0008-5472.CAN-14-3511
154. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124:2246–59. doi: 10.1172/JCI73639
155. Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA. Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Invest (1994) 93:892–9. doi: 10.1172/JCI117045
156. Dancea HC, Shareef MM, Ahmed MM. Role of radiation-induced TGF-beta signaling in cancer therapy. Mol Cell Pharmacol (2009) 1:44–56. doi: 10.4255/mcpharmacol.09.06
157. Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A, et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat Comm (2020) 11:1749. doi: 10.1038/s41467-020-15404-8
158. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
159. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, et al. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am J Transplant (2004) 4:1614–27. doi: 10.1111/j.1600-6143.2004.00566.x
160. Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol (2013) 25:637–45. doi: 10.1097/CCO.0000000000000006
161. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol (2015) 195:3665–74. doi: 10.4049/jimmunol.1402898
162. Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, et al. STAT3 modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J Natl Cancer Inst (2019) 111:djz036. doi: 10.1093/jnci/djz036
163. Mondini M, Loyher PL, Hamon P, Gerbé de Thoré M, Laviron M, Berthelot K, et al. CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol Res (2019) 7:376–87. doi: 10.1158/2326-6066.CIR-18-0633
164. Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys (2012) 83:1306–10. doi: 10.1016/j.ijrobp.2011.09.049
165. Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, et al. Stereotactic radiotherapy increases functionally suppressive regulatory T cells in the tumor microenvironment. Cancer Immunol Res (2017) 5:992–1004. doi: 10.1158/2326-6066.CIR-17-0040
166. Sia J, Hagekyriakou J, Chindris I, Albarakati H, Leong T, Schlenker R, et al. Regulatory T cells shape the differential impact of radiation dose-fractionation schedules on host innate and adaptive antitumor immune defenses. Int J Radiat Oncol Biol Phys (2021) 111:502–14. doi: 10.1016/j.ijrobp.2021.05.014
167. Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res (2010) 70:5679–85. doi: 10.1158/0008-5472.CAN-09-4446
168. Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest (2010) 120:694–705. doi: 10.1172/JCI40283
169. Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, et al. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget (2016) 7:86522–35. doi: 10.18632/oncotarget.13287
170. Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res (2016) 23:137–48. doi: 10.1158/1078-0432.CCR-16-0870
171. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Comm (2017) 8:1736. doi: 10.1038/s41467-017-01566-5
172. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res (2013) 73:2782–94. doi: 10.1158/0008-5472.CAN-12-3981
173. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol (2016) 18:797–806. doi: 10.1093/neuonc/nov272
174. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
175. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol (2015) 25:18–27. doi: 10.1016/j.semradonc.2014.07.008
176. Jiménez-Cortegana C, Galassi C, Klapp V, Gabrilovich DI, Galluzzi L. Myeloid-derived suppressor cells and radiotherapy. Cancer Immunol Res (2022) 10:545–57. doi: 10.1158/2326-6066.CIR-21-1105
177. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19:1264–72. doi: 10.1038/nm.3337
178. Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys (2007) 68:499–507. doi: 10.1016/j.ijrobp.2007.01.041
179. Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many faces of tumor-associated neutrophils and macrophages: considerations for targeting innate immune cells in cancer. Trends Cancer (2019) 5:789–98. doi: 10.1016/j.trecan.2019.10.013
180. Faget J, Peters S, Quantin X, Meylan E, Bonnefoy N. Neutrophils in the era of immune checkpoint blockade. J Immunother Cancer (2021) 0:e002242. doi: 10.1136/jitc-2020-002242
181. Wisdom AJ, Hong CS, Lin AJ, Xiang Y, Cooper DE, Zhang J, et al. Neutrophils promote tumor resistance to radiation therapy. Proc Natl Acad Sci USA (2019) 116:18584–9. doi: 10.1073/pnas.1901562116
182. Matsumoto Y, Mabuchi S, Kozasa K, Kuroda H, Sasano T, Yokoi E, et al. The significance of tumor-associated neutrophil density in uterine cervical cancer treated with definitive radiotherapy. Gyne Oncol (2017) 145:469–75. doi: 10.1016/j.ygyno.2017.02.009
183. Ancey PB, Contat C, Boivin G, Sabatino S, Pascual J, Zangger N, et al. GLUT1 expression in tumor-associated neutrophils promotes lung cancer growth and resistance to radiotherapy. Cancer Res (2021) 81:2345–57. doi: 10.1158/0008-5472.CAN-20-2870
184. Hovinga KE, Stalpers LJA, van Bree C, Donker M, Verhoeff JJC, Rodermond HM, et al. Radiation-enhanced vascular endothelial growth factor (VEGF) secretion in glioblastoma multiforme cell lines – a clue to radioresistance? J Neuro Oncol (2005) 74:99–103. doi: 10.1007/s11060-004-4204-7
185. Chen YH, Pan SL, Wang JC, Kuo SH, Cheng JCH, Teng CM. Radiation-induced VEGF-c expression and endothelial cell proliferation in lung cancer. Strahlenther Onkol (2014) 190:1154–62. doi: 10.1007/s00066-014-0708-z
186. Munn LL, Jain RK. Vascular regulation of antitumor immunity. Science (2019) 365:544–5. doi: 10.1126/science.aaw7875
187. Ware MB, El-Rayes BF, Lesinski GB. Mirage or long-awaited oasis: reinvigorating T-cell responses in pancreatic cancer. J Immunother Cancer (2020) 8:e001100. doi: 10.1136/jitc-2020-001100
188. Hellevik T, Berzaghi R, Lode K, Islam A, Martinez-Zubiaurre I. Immunobiology of cancer-associated fibroblasts in the context of radiotherapy. J Trans Med (2021) 19:437. doi: 10.1186/s12967-021-03112-w
189. Chakravarty PK, Alfieri A, Thomas EK, Beri V, Tanaka KE, Vikram B, et al. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res (1999) 59:6028–32.
190. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys (2004) 58:862–70. doi: 10.1016/j.ijrobp.2003.09.012
191. Pilones KA, Charpentier M, Garcia-Martinez E, Daviaud C, Kraynak J, Aryankalayil J, et al. Radiotherapy cooperates with IL15 to induce antitumor immune responses. Cancer Immunol Res (2020) 8:1054–63. doi: 10.1158/2326-6066.CIR-19-0338
192. Walker JM, Rolig AS, Charych DH, Hoch U, Kasiewicz MJ, Rose DC, et al. NKTR-214 immunotherapy synergizes with radiotherapy to stimulate systemic CD8+ T cell responses capable of curing multi-focal cancer. J Immunother Cancer (2020) 8:e000464. doi: 10.1136/jitc-2019-000464
193. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res (2005) 11:728–34. doi: 10.1158/1078-0432.728.11.2
194. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
195. Rudqvist NP, Pilones KA, Lhuillier C, Wennergberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res (2018) 6:139–50. doi: 10.1158/2326-6066.CIR-17-0134
196. Yamamoto J, Takahashi Y, Minami K, Tamari K, Katsuki S, Takenaka W, et al. High dose local photon irradiation is crucial in anti-CTLA-4 antibody therapy to enhance the abscopal response in a murine pancreatic carcinoma model. Cancers (2022) 14:2087. doi: 10.3390/cancers14092087
197. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124:687–95. doi: 10.1172/JCI67313
198. Gong X, Li X, Jiang T, Xie H, Zhu Z, Zhou F, et al. Combined radiotherapy and anti-PD-L1 antibody synergistically enhances antitumor effect in non-small cell lung cancer. J Thorac Oncol (2017) 12:1085–97. doi: 10.1016/j.jtho.2017.04.014
199. Lan J, Li R, Yin LM, Deng L, Gui J, Chen BQ, et al. Targeting myeloid-derived suppressor cells and programmed death ligand 1 confers therapeutic advantage of ablative hypofractionated radiation therapy compared with conventional fractionated radiation therapy. Int J Radiat Oncol Biol Phys (2018) 101:74–87. doi: 10.1016/j.ijrobp.2018.01.071
200. Herter-Sprie GS, Koyama S, Korideck H, Hai J, Deng J, Li YY, et al. Synergy of radiotherapy and PD-1 blockade in kras-mutant lung cancer. JCI Insight (2016) 1:e87415. doi: 10.1172/jci.insight.87415
201. Park SS, Dong H, Liu X, Harrington SM, Krco CJ, Grams MP, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res (2015) 3:610–9. doi: 10.1158/2326-6066.CIR-14-0138
202. Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francia BJ, Velarde E, et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res (2014) 3:345–55. doi: 10.1158/2326-6066.CIR-14-0196
203. Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res (2017) 23:5514–26. doi: 10.1158/1078-0432.CCR-16-1673
204. Zhang X, Niedermann G. Abscopal effects with hypofractinated schedules extending into the effctor phase of the tumor-specific T-cell response. Int J Radiat Oncol Biol Phys (2018) 101:63–73. doi: 10.1016/j.ijrobp.2018.01.094
205. Ban Y, Markowitz GJ, Zou Y, Ramchandani D, Kraynak J, Sheng J, et al. Radiation-activated secretory proteins of Scgb1a1+ club cells increase the efficacy of immune checkpoint blockade in lung cancer. Nat Cancer (2021) 2:919–31. doi: 10.1038/s43018-021-00245-1
206. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi: 10.1038/nature14292
207. Martin AL, Powell C, Nagy MZ, Innamarato P, Powers J, Nichols D, et al. Anti-4-1BB immunotherapy enhances systemic immune effects of radiotherapy to induce b and T cell-dependent anti-tumor immune activation and improve tumor control at unirradiated sites. Cancer Immunol Immunother (2022) 72:1445–60. doi: 10.1007/s00262-022-03325-y
208. Aguilera TA, Elghonaimy EA, Shehade H, Rafat M, Castellini L, Jiang D, et al. Induced tumor heterogeneity reveals factors informing radiation and immunotherapy combinations. Clin Cancer Res (2020) 26:2972–85. doi: 10.1158/1078-0432.CCR-19-4220
209. Pilones KA, Hensler M, Daviaud C, Kraynak J, Fucikova J, Galluzzi L, et al. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: contribution of VISTA blockade. Oncoimmunology (2020) 9:e1830524. doi: 10.1080/2162402X.2020.1830524
210. Han MG, Wee CW, Kang MH, Kim MJ, Jeon SH, Kim IA. Combination of OX40 co-stimulation, radiotherapy, and PD-1 inhibition in a syngeneic murine triple-negative breast cancer model. Cancers (2022) 14:2692. doi: 10.3390/cancers14112692
211. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res (2016) 23:124–36. doi: 10.1158/1078-0432.CCR-15-1535
212. Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J Immunother Cancer (2019) 7:160. doi: 10.1186/s40425-019-0634-9
213. Pieper AA, Rakhmilevich AL, Spiegelman DV, Patel RB, Birstler J, Jin WJ, et al. Combination of radiation therapy, bempegaldesleukin, and checkpoint blockade eradicates advanced solid tumors and metastasis in mice. J Immunother Cancer (2021) 9:e002715. doi: 10.1136/jitc-2021-002715
214. Pimentel VO, Marcus D, van der Wiel AM, Lieuwes NG, Biemans R, Lieverse RI, et al. Releasing the brakes of tumor immunity with anti-PD-L1 and pushing its accelerator with L19-IL2 cures poorly immunogenic tumors when combined with radiotherapy. J Immunother Cancer (2021) 9:e001764. doi: 10.1136/jitc-2020-001764
215. Wu CJ, Tsai YT, Lee IJ, Wu PY, Lu LS, Tsao WS, et al. Combination of radiation and interleukin 12 eradicates large orthotopic hepatocellular carcinoma through immunomodulation of tumor microenvironment. Oncoimmunology (2018) 7:e1477459. doi: 10.1080/2162402X.2018.1477459
216. Jagodinsky JC, Bates AM, Clark PA, Sriramaneni RN, Havighurst TC, Chakravarty I, et al. Local TLR4 stimulation augments in situ vaccination induced via local radiation and anti-CTLA-4 checkpoint blockade through induction of CD8 T-cell independent Th1 polarization. J Immunother Cancer (2022) 10:e005103. doi: 10.1136/jitc-2022-005103
217. Sheng H, Huang Y, Xiao Y, Zhu Z, Shen M, Zhou P, et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer (2020) 8:e000340. doi: 10.1136/jitc-2019-000340
218. Patin EC, Dillon MT, Nenclares P, Grove L, Soliman H, Leslie I, et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage-response inhibition and immune checkpoint blockade. J Immunother Cancer (2022) 10:e004306. doi: 10.1136/jitc-2021-004306
219. Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, et al. Simultaneous targeting of TGFβ/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell (2021) 39:1–16. doi: 10.1016/j.ccell.2021.08.008
220. Chen JL, Pan CK, Huang YS, Tsai CY, Wang CW, Lin YL, et al. Evaluation of antitumor immunity by a combination treatment of high-dose irradiation, anti-PD-L1, and anti-angiogenic therapy in murine lung tumors. Cancer Immunol Immunother (2021) 70:391–404. doi: 10.1007/s00262-020-02690-w
221. Han MG, Jang BS, Kang MH, Na D, Kim IA. PI3Kγδ inhibitor plus radiation enhances the antitumour immune effect of PD-1 blockade in syngenic murine breast cancer and humanised patient-derived xenograft model. Eur J Cancer (2021) 157:450–63. doi: 10.1016/j.ejca.2021.08.029
222. He K, Barsoumian HB, Puebla-Osorio N, Hu Y, Sezen D, Wasley MD, et al. Inhibition of STAT6 with antisense oligonucleotides enhances the systemic antitumor effects of radiotherapy and anti-PD1 in metastatic non-small cell lung cancer. Cancer Immunol Res (2023) 11:486–500. doi: 10.1158/2326-6066.c.6534807.v1
223. Wennerberg E, Spada S, Rudqvist NP, Lhuillier C, Gruber S, Chen Q, et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol Res (2020) 8:465–78. doi: 10.1158/2326-6066.CIR-19-0449
224. Nishiga Y, Drainas AP, Baron M, Bhattacharya D, Barkal AA, Ahrari Y, et al. Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat Cancer (2022) 3:1351–66. doi: 10.1038/s43018-022-00456-0
225. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med (2012) 366:925–31. doi: 10.1056/NEJMoa1112824
226. Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res (2013) 1:365–72. doi: 10.1158/2326-6066.CIR-13-0115
227. Yuan Z, Fromm A, Ahmed KA, Grass GD, Yang GQ, Oliver DE, et al. Radiotherapy rescue of a nivolumab-refractory immune response in a patient with PD-L1-negative metastatic response in a patient with PD-L1-negative metastatic squamous cell carcinoma of the lung. J Thorac Oncol (2017) 12:e135–136. doi: 10.1016/j.jtho.2017.04.029
228. Grimaldi AM, Simeone E, Giannarelli D, Muto P, Falivene S, Borzillo V, et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology (2014) 3:e28780. doi: 10.4161/onci.28780
229. Roger A, Finet A, Boru B, Beauchet A, Mazeron JJ, Otzmeguine Y, et al. Efficacy of combined hypo-fractionated radiotherapy and anti-PD-1 monotherapy in difficult-to-treat advanced melanoma patients. Oncoimmunology (2018) 7:. doi: 10.1080/2162402X.2018.1442166
230. Golden EB, Chhabra A, Chachoua A, Adams S, Donach M, Fenton-Kerimian M, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol (2015) 16:795–803. doi: 10.1016/S1470-2045(15)00054-6
231. Sundahl N, De Wolf K, Kruse V, Meireson A, Reynders D, Goetghebeur E, et al. Phase 1 dose escalation trial of ipilimumab and stereotactic body radiation therapy in metastatic melanoma. Int J Radiat Oncol Biol Phys (2018) 100:906–15. doi: 10.1016/j.ijrobp.2017.11.029
232. Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol (2018) 36:1611–8. doi: 10.1200/JCO.2017.76.2229
233. Tang C, Welsh JW, de Groot P, Massarelli E, Chang JY, Hess KR, et al. Ipilimumab with stereotactic ablative radiation therapy: phase I results and immunologic correlates from peripheral T cells. Clin Cancer Res (2017) 23:1388–96. doi: 10.1158/1078-0432.CCR-16-1432
234. Formenti SC, Rudqvist N, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med (2018) 24:1845–51. doi: 10.1038/s41591-018-0232-2
235. Bauml JM, Mick R, Ciunci C, Aggarwal C, Davis C, Evans T, et al. Pembrolizumab after completion of locally ablative therapy for oligometastatic non-small cell lung cancer: a phase 2 trial. JAMA Oncol (2019) 5:1283–90. doi: 10.1001/jamaoncol.2019.1449
236. Qin A, Rengan R, Lee S, Santana-Davila R, Goulart BHL, Martins R, et al. A pilot study of atezolizumab plus hypofractionated image-guided radiotherapy for the treatment of advanced non-small cell lung cancer. Int J Radiat Oncol Biol Phys (2020) 108:170–7. doi: 10.1016/j.ijrobp.2019.10.047
237. Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs. pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol (2019) 5:1276–82. doi: 10.1001/jamaoncol.2019.1478
238. Welsh J, Menon H, Chen D, Verma V, Tang C, Altan M, et al. Pembrolizumab with or without radiation therapy for metastatic non-small cell lung cancer: a randomized phase I/II trial. J Immunother Cancer (2020) 8:e001001. doi: 10.1136/jitc-2020-001001
239. Chen D, Menon H, Verma V, Guo C, Ramapriyan R, Barsoumian H, et al. Response and outcomes after anti-CTLA4 versus anti-PD1 combined with stereotactic body radiation therapy for metastatic non-small cell lung cancer: retrospective analysis of two single-institution prospective trials. J Immunother Cancer (2020) 8:e000492. doi: 10.1136/jitc-2019-000492
240. Theelen WSME, Chen D, Verma V, Hobbs BP, Peulen HMU, Aerts JGJV, et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: a pooled analysis of two randomised trials. Lancet Respir Med (2021) 9:467–75. doi: 10.1016/S2213-2600(20)30391-X
241. Bestvina CM, Pointer KB, Karrison T, Al-Hallaq H, Hoffman PC, Jelinek MJ, et al. A phase I trial of concurrent or sequential ipilimumab, nivolumab, and stereotactic body radiotherapy in patients with stage IV non-small cell lung cancer (COSINR study). J Thorac Oncol (2022) 17:130–40. doi: 10.1016/j.jtho.2021.08.019
242. Schild SE, Wang X, Bestvina CM, Williams T, Masters G, Singh AK, et al. Alliance A082002-a randomized phase II/III trial of modern immunotherapy-based systemic therapy with or without SBRT for PD-L1-negative, advanced non-small-cell lung cancer. Clin Lung Cancer (2022) 23:e317–320. doi: 10.1016/j.cllc.2022.04.004
243. Spurr LF, Martinez CA, Kang W, Chen M, Zha Y, Hseu R, et al. Highly aneuploid non-small cell lung cancer shows enhanced responsiveness to concurrent radiation and immune checkpoint blockade. Nat Cancer (2022) 3:1498–512. doi: 10.1038/s43018-022-00467-x
244. Spigel DR, Faivre-Finn C, Gray JE, Vicente D, Planchard D, Paz-Ares LG, et al. Five-year survival outcomes from the PACIFIC trial: durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. J Clin Oncol (2022) 40:1301–11. doi: 10.1200/JCO.21.01308
245. Zhou Q, Chen M, Jiang O, Pan Y, Hu D, Lin Q, et al. Sugemalimab versus placebo after concurrent or sequential chemoradiotherapy in patients with locally advanced, unresectable, stage III non-small-cell lung cancer in China (GEMSTONE-301): interim results of a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol (2022) 23:209–19. doi: 10.1016/S1470-2045(21)00630-6
246. Durm GA, Jabbour SK, Althouse SK, Liu Z, Sadiq AA, Zon RT, et al. A phase 2 trial of consolidation pembrolizumab following concurrent chemoradiation for patients with unresectable stage III non-small cell lung cancer: Hoosier cancer research network LUN 14-179. Cancer (2020) 126:4353–61. doi: 10.1002/cncr.33083
247. Garassino MC, Mazieres J, Reck M, Chouaid C, Bischoff H, Reinmuth N, et al. Durvalumab after sequential chemoradiotherapy in stage III, unresectable NSCLC: the phase PACIFIC-6 trial. J Thorac Oncol (2022) 17:1415–27. doi: 10.1016/j.jtho.2022.07.1148
248. Jabbour SK, Lee KH, Frost N, Breder V, Kowalski DM, Pollock T, et al. Pembrolizumab plus concurrent chemoradiation therapy in patients with unresectable, locally advanced, stage III non-small cell lung cancer: the phase 2 KEYNOTE-799 nonrandomized trial. JAMA Oncol (2021) 7:1–9. doi: 10.1001/jamaoncol.2021.2301
249. Peters S, Felip E, Dafni U, Tufman A, Guckenberger M, Álvarez R, et al. Progression-free and overall survival for concurrent nivolumab with standard concurrent chemo-radiotherapy in locally advanced stage IIIA/B NSCLC: results from the European thoracic oncology platform NICHOLAS phase II trial (ETOP 6-14). J Thorac Oncol (2020) 16:278–88. doi: 10.1016/j.jtho.2020.10.129
250. Lin SH, Lin Y, Yao L, Kalhor N, Carter BW, Altan M, et al. Phase II trial of concurrent atezolizumab with chemoradiation for unresectable NSCLC. J Thorac Oncol (2020) 15:248–57. doi: 10.1016/j.jtho.2019.10.024
251. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
252. Paz-Ares L, Spira A, Raben D, Planchard D, Cho BC, Özgüroğlu M, et al. Outcomes with durvalumab by tumour PD-L1 expression in unresectable, stage III non-small-cell lung cancer in the PACIFIC trial. Ann Oncol (2020) 31:798–806. doi: 10.1016/j.annonc.2020.03.287
253. Jabbour SK, Berman AT, Decker RH, Lin Y, Feigenberg SJ, Gettinger SN, et al. Phase 1 trial of pembrolizumab administered concurrently with chemoradiotherapy for locally advanced non-small cell lung cancer: a nonrandomized controlled trial. JAMA Oncol (2020) 6:848–55. doi: 10.1001/jamaoncol.2019.6731
254. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med (2017) 377:1919–29. doi: 10.1056/NEJMoa1709937
255. Varlotto JM, Sun Z, Ky B, Upshaw J, Fitzgerald TJ, Diehn M, et al. A review of concurrent chemo/radiation, immunotherapy, radiation planning, and biomarkers for locally advanced non-small cell lung cancer and their role in the development of ECOG-ACRIN EA5181. Clin Lung Cancer (2022) 23:547–60. doi: 10.1016/j.cllc.2022.06.005
256. Altorki NK, McGraw TE, Borczuk AC, Saxena A, Port JL, Stiles BM, et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: a single-centre, randomised phase 2 trial. Lancet Oncol (2021) 22:824–35. doi: 10.1016/S1470-2045(21)00149-2
257. Hallqvist A, Koyi H, de Petris L, Lindberg K, Farooqi S, Helland Å, et al. Safety analysis of durvalumab following stereotactic body radiotherapy (SBRT) in early-stage non-small cell lung cancer (NSCLC) patients: a first report of a randomized phase II trial (ASTEROID). J Thorac Oncol (2021) 16:s729–736. doi: 10.1016/S1556-0864(21)01905-5
258. Robinson C, Xing L, Tanaka H, Tasaka S, Badiyan SN, Nasrallah H, et al. 122TiP phase III study of durvalumab with SBRT for unresected stage I/II, lymph-node negative NSCLC (PACIFIC-4/RTOG 3515). Ann Oncol (2022) 33:S88. doi: 10.1016/j.annonc.2022.02.149
259. Jabbour SK, Houghton B, Robinson AG, Quantin X, Wehler T, Kowalski D, et al. Phase 3, randomized, placebo-controlled study of stereotactic body radiotherapy (SBRT) with or without pembrolizumb in patients with unresected stage I or II non-small cell lung cancer (NSCLC): KEYNOTE-867. J Clin Oncol (2022) 40:TPS8597. doi: 10.1200/JCO.2022.40.16_suppl.TPS8597
260. Daly ME. Inoperable early-stage non-small cell lung cancer: stereotactic ablative radiotherapy and rationale for systemic therapy. J Clin Oncol (2022) 40:539–45. doi: 10.1200/JCO.21.01611
261. Perez BA, Kim S, Wang M, Karimi AM, Powell C, Li J, et al. A prospective single arm phase 1 and 2 study: ipilimumab and nivolumab with thoracic radiotherapy after platinum chemotherapy in extensive-stage small cell lung cancer. Int J Radiat Oncol Biol Phys (2021) 109:425–35. doi: 10.1016/j.ijrobp.2020.09.031
262. Welsh JW, Heymach JV, Guo C, Menon H, Klein K, Cushman TR, et al. Phase 1/2 trial of pembrolizumab and concurrent chemoradiation therapy for limited stage small cell lung cancer. J Thorac Oncol (2020) 15:1919–27. doi: 10.1016/j.jtho.2020.08.022
263. Bogart J, Wang X, Masters G, Gao J, Komaki R, Gaspar LE, et al. High-dose once-daily thoracic radiotherapy in limited-stage small-cell lung cancer: CALGB 30610 (Alliance)/RTOG 0538. J Clin Oncol (2023) 41:2394–402. doi: 10.1200/JCO.22.01359
264. Kelly RJ, Ajani JA, Kuzdzal J, Zander T, Van Cutsem E, Piessen G, et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med (2021) 384:1191–203. doi: 10.1056/NEJMoa2032125
265. McBride S, Sherman E, Tsai CJ, Baxi S, Aghalar J, Eng J, et al. Randomized phase II trial of nivolumab with stereotactic body radiotherapy versus nivolumab alone in metastatic head and neck squamous cell carcinoma. J Clin Oncol (2020) 39:30–7. doi: 10.1200/JCO.20.00290
266. Lee NY, Ferris RL, Psyrri A, Haddad RI, Tahara M, Bourhis J, et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol (2021) 22:450–62. doi: 10.1016/S1470-2045(20)30737-3
267. Frank MJ, Reagan PM, Bartlett NL, Gordon LI, Friedberg JW, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discovery (2018) 8:1258–69. doi: 10.1158/2159-8290.CD-18-0743
268. Monjazeb AM, Giobbie-Hurder A, Lako A, Thrash EM, Brennick RC, Kao KZ, et al. A randomized trial of combined PD-L1 and CTLA-4 inhibition with targeted low-dose or hypofractionated radiation for patients with metastatic colorectal cancer. Clin Cancer Res (2021) 27:2470–80. doi: 10.1158/1078-0432.CCR-20-4632
269. Schoenfeld JD, Giobbie-Hurder A, Ranasinghe S, Kao K, Lako A, Tsuji J, et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-small-cell lung cancer refractory to previous PD(L)-1 therapy: an open-label, multicentre, randomised, phase 2 trial. Lancet Oncol (2022) 23:279–91. doi: 10.1016/S1470-2045(21)00658-6
270. Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol (2010) 28:4324–32. doi: 10.1200/JCO.2010.28.9793
271. Patel RB, Hernandez R, Carlson P, Grudzinski J, Bates AM, Jagodinsky JC, et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci Transl Med (2021) 13:eabb3631. doi: 10.1126/scitranslmed.abb3631
272. Savage T, Pandey S, Guha C. Postablation modulation after single high-dose radiation therapy improves tumor control via enhanced immunomodulation. Clin Cancer Res (2020) 26:910–21. doi: 10.1158/1078-0432.CCR-18-3518
273. Barsoumian HB, Ramapriyan R, Younes AI, Caetano MS, Menon H, Comeaux NI, et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J Immunother Cancer (2020) 8:e000537. doi: 10.1136/jitc-2020-000537
274. Yin L, Xue J, Li R, Zhou L, Deng L, Chen L, et al. Effect of low-dose radiotherapy on abscopal responses to hypofractionated radiotherapy and anti-PD1 in mice and NSCLC patients. Int J Radiat Oncol Biol Phys (2020) 108:212–24. doi: 10.1016/j.ijrobp.2020.05.002
275. Menon H, Chen D, Ramapriyan R, Verma V, Barsoumian HB, Cushman TR, et al. Influence of low-dose radiation on abscopal responses in patients receiving high-dose radiation and immunotherapy. J Immunother Cancer (2019) 7:237. doi: 10.1186/s40425-019-0718-6
276. Patel RR, He K, Barsoumian HB, Chang JY, Tang C, Verma V, et al. High-dose irradiation in combination with non-ablative low-dose radiation to treat metastatic disease after progression on immunotherapy: results of a phase II trial. Radiother Oncol (2021) 162:60–7. doi: 10.1016/j.radonc.2021.06.037
277. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer (2020) 8:e000826. doi: 10.1136/jitc-2020-000826
278. Knitz MW, Bickett TE, Darragh LB, Oweida AJ, Bhatia S, Van Court B, et al. Targeting resistance to radiation-immunotherapy in cold HNSCCs by modulating the treg-dendritic cell axis. J Immunother Cancer (2021) 0:e001955. doi: 10.1136/jitc-2020-001955
279. Takeshima T, Chamoto K, Wakita D, Ohkuri T, Togashi Y, Shirato H, et al. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res (2010) 70:2697–706. doi: 10.1158/0008-5472.CAN-09-2982
280. Hauth F, Ho AY, Ferrone S, Duda DG. Radiotherapy to enhance chimeric antigen receptor T-cell therapeutic efficacy in solid tumors: a narrative review. JAMA Oncol (2021) 7:1051–9. doi: 10.1001/jamaoncol.2021.0168
281. Laurent PA, Morel D, Meziani L, Depil S, Deutsch E. Radiotherapy as a means to increase the efficacy of T-cell therapy in solid tumors. Oncoimmunology (2022) 12:2158013. doi: 10.1080/2162402X.2022.2158013
282. Cheng JN, Luo W, Sun C, Jin Z, Zeng X, Alexander PB, et al. Radiation-induced eosinophils improve cytotoxic T lymphocyte recruitment and response to immunotherapy. Sci Adv (2021) 7:eabc7609. doi: 10.1126/sciadv.abc7609
283. DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther (2018) 26:2542–52. doi: 10.1016/j.ymthe.2018.09.008
284. Qu C, Ping N, Kang L, Liu H, Qin S, Wu Q, et al. Radiation priming chimeric antigen receptor T-cell therapy in relapsed/ refractory diffuse large b-cell lymphoma with high tumor burden. J Immunother (2020) 43:32–7. doi: 10.1097/CJI.0000000000000284
285. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res (2017) 78:1031–43. doi: 10.1158/0008-5472.CAN-17-1788
Keywords: radiotherapy, immunotherapy, immune checkpoint inhibitors, tumor immune micro-environment, cancer
Citation: Chi A and Nguyen NP (2023) Mechanistic rationales for combining immunotherapy with radiotherapy. Front. Immunol. 14:1125905. doi: 10.3389/fimmu.2023.1125905
Received: 16 December 2022; Accepted: 24 May 2023;
Published: 12 June 2023.
Edited by:
Yun Chen, Nanjing Medical University, ChinaReviewed by:
Nahum Puebla-Osorio, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Chi and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Chi, YWNoaWF6MjAxMEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.