
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 April 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1125017
This article is part of the Research Topic The Promise of Immunogenetics for Precision Oncology View all 11 articles
 Franziska Darzentas1†
Franziska Darzentas1† Monika Szczepanowski1†
Monika Szczepanowski1† Michaela Kotrová1
Michaela Kotrová1 Alina Hartmann1,2,3Thomas Beder1,2Nicola Gökbuget4Stefan Schwartz5,6
Alina Hartmann1,2,3Thomas Beder1,2Nicola Gökbuget4Stefan Schwartz5,6 Lorenz Bastian1,2,3Claudia Dorothea Baldus1,2,3
Lorenz Bastian1,2,3Claudia Dorothea Baldus1,2,3 Karol Pál7
Karol Pál7 Nikos Darzentas1‡*
Nikos Darzentas1‡* Monika Brüggemann1,2,3‡
Monika Brüggemann1,2,3‡Introduction: The malignant transformation leading to a maturation arrest in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) occurs early in B-cell development, in a pro-B or pre-B cell, when somatic recombination of variable (V), diversity (D), and joining (J) segment immunoglobulin (IG) genes and the B-cell rescue mechanism of VH replacement might be ongoing or fully active, driving clonal evolution. In this study of newly diagnosed BCP-ALL, we sought to understand the mechanistic details of oligoclonal composition of the leukemia at diagnosis, clonal evolution during follow-up, and clonal distribution in different hematopoietic compartments.
Methods: Utilizing high-throughput sequencing assays and bespoke bioinformatics we identified BCP-ALL-derived clonally-related IGH sequences by their shared ‘DNJ-stem’.
Results: We introduce the concept of ‘marker DNJ-stem’ to cover the entirety of, even lowly abundant, clonally-related family members. In a cohort of 280 adult patients with BCP-ALL, IGH clonal evolution at diagnosis was identified in one-third of patients. The phenomenon was linked to contemporaneous recombinant and editing activity driven by aberrant ongoing DH/VH-DJH recombination and VH replacement, and we share insights and examples for both. Furthermore, in a subset of 167 patients with molecular subtype allocation, high prevalence and high degree of clonal evolution driven by ongoing DH/VH-DJH recombination were associated with the presence of KMT2A gene rearrangements, while VH replacements occurred more frequently in Ph-like and DUX4 BCP-ALL. Analysis of 46 matched diagnostic bone marrow and peripheral blood samples showed a comparable clonal and clonotypic distribution in both hematopoietic compartments, but the clonotypic composition markedly changed in longitudinal follow-up analysis in select cases. Thus, finally, we present cases where the specific dynamics of clonal evolution have implications for both the initial marker identification and the MRD monitoring in follow-up samples.
Discussion: Consequently, we suggest to follow the marker DNJ-stem (capturing all family members) rather than specific clonotypes as the MRD target, as well as to follow both VDJH and DJH family members since their respective kinetics are not always parallel. Our study further highlights the intricacy, importance, and present and future challenges of IGH clonal evolution in BCP-ALL.
The immense diversity of the antibody repertoire in humans is physiologically conveyed by mechanistic editing and assembly of immunoglobulin (IG) genes, particularly in the processes of somatic recombination of variable (V), diversity (D), and joining (J) segment genes. In the early developmental stage of a pro-B cell, one DH and one JH gene segment of the IG heavy (H) chain locus become joint, eventually followed by VH to DJH recombination. Consequently, the somatic VDJH recombination yields a uniquely rearranged IGH DNA sequence of the VDJH joining region in each B lymphocyte. The joining is an imprecise process, in which germline segment ends are cleaved by the RAG1/2 recombinase activity and joined upon random addition of “non-templated” nucleotides (N nucleotides) into the junctions between segments. The resulting DNA sequence variations add to the overall diversity of antigen receptors and constitute a unique “fingerprint” target for the design of leukemia-clone specific MRD markers. They also offer a valuable tool to decipher which mechanism of somatic recombination was active and to gather clonally related clonotypes (1–4).
Malignant transformation is preceded by a premalignant state, in which genetic aberrations accumulate over time until the driver lesion finally complements the transformation process. Expanded malignant cells harbor the successively acquired genetic aberrations allowing for backtracking the mutational trajectories and (sub)clonal architecture of the malignant population. In the case of malignantly transformed immune cells such as B and T lymphocytes, these carry the unique VDJ rearrangement of the preleukemic cell of origin. Importantly, the VDJ recombination is a stepwise and temporarily tightly regulated process, thus, the completeness of the rearrangements basically depends on the developmental state of the malignantly transformed cell of origin.
The malignant transformation in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) occurs at an early stage in the B-cell development, hitting precursor B cells in the stage of a pro-B or a pre-B cell. Therefore, the process of somatic VDJ recombination might still be active in the malignant B cell, driving IGH clonal evolution either through recombination of incomplete DJH rearrangements with multiple VH segments (occasionally with DH segments as DH-DH tandem fusion rearrangements) or by the process of VH replacement (5–11).
The aforementioned processes are mediated by the RAG1/2 recombinase activity, which recognizes the canonical recombination signal sequences (RSS) during VH-DJH recombination as well as cryptic RSS during DH-DJH recombination and VH replacement (10, 12, 13). In the latter process, the cryptic RSS of the original VH segment and the conventional RSS of the incoming VH segment are processed to replace the original VH segment by the incoming with the original DJH assembly kept. VH replacement, which can go on for multiple rounds, may leave a remnant of up to five base pairs of the original VH gene, which can be further diversified by exonuclease activity or N nucleotide addition. VH replacement also occurs in healthy humans in bone marrow immature B cells to rescue B cells with nonfunctional or autoreactive receptors (14–16).
IGH clonal evolution in BCP-ALL might pose a serious diagnostic challenge for IGH-based MRD assessment upon further diversification of leukemic clonotypes during long-term follow-up. MRD monitoring utilizing clone-specific sequences of the uniquely rearranged and edited gene segments is widely used to assess therapy response after induction and consolidation therapy, which has crucial prognostic importance in BCP-ALL patients (17–23). High-throughput sequencing (HTS)-based marker identification and MRD monitoring offers the benefit of providing additional information on background repertoire including minor accompanying clones, which may reflect clonal evolution [reviewed in (24)].
In this study of BCP-ALL diagnostic and follow-up bone marrow (BM) and peripheral blood (PB) materials utilizing HTS techniques to backtrack rearranged leukemic IGH sequences and their trajectories, we sought to understand the mechanistic details of leukemic clonal composition at diagnosis, clonal evolution during follow-up, and clonal distribution in different body or hematopoietic compartments. We link diversified IGH clonal family members and discriminate between the mechanisms driving clonal evolution. We correlate these results with immunophenotypes obtained by multiparameter flow cytometry and with transcriptome-based molecular subtypes. We report on the frequency of the phenomenon of clonal evolution, provide examples of clonal sustainability under immunotherapy and finally discuss the implications of clonal evolution in BCP-ALL with regard to initial diagnostics and MRD monitoring.
A total of 465 diagnostic and selected MRD-positive follow-up samples from 280 patients aged 18-55 years with BCP-ALL were investigated within the frame of a research project associated with the German Multicenter Adult Acute Lymphoblastic Leukemia (GMALL) 08/2013 therapy optimization trial (EudraCT-No.: 2013-003466-13). Initial diagnostic samples were subjected to IGH amplicon-based HTS, multiparameter flow cytometry, and transcriptome sequencing (RNA-Seq) analyses. An overview of patient characteristics is detailed in Supplementary Table 1. From all 280 patients either diagnostic BM (n=217) or PB (n=63) samples were available (Figure 1, cohort #1). Concurrent diagnostic PB and BM samples were available for pairwise comparisons in 46/280 cases (Figure 1, cohort #2). In addition, a total of n=139 MRD-positive early or late follow-up and relapse samples of 63 patients were selected. Clonal IGH evolution was monitored during early therapy phases, in particular during the cyclophosphamide/dexamethasone-containing prephase (Figure 1, cohort #3, 66 paired samples from 33 patients) and after Induction I including one dose of rituximab (Figure 1, cohort #4, 82 paired samples from 41 patients). Ultimately, we studied 149 diagnostic and follow-up samples of 34 patients to track the kinetics of clonal evolution over time in longitudinal case reports (Figure 1, cohort #5). Main results and details on IGH sequences within the cohorts #1-5 are summarized in Supplementary Table 4. All patients provided informed consent. All research described herein was approved by the Frankfurt Research Ethics Board (188/15F) and performed in accordance with the Declaration of Helsinki.

Figure 1 Study overview. A total of 465 samples from 280 patients with BCP-ALL enrolled in the GMALL 08/2013 therapy optimization trial were analyzed in five different cohorts to decipher frequency, mechanisms and kinetics of the leukemic clonal evolution. The bioinformatic analyses on IGH HTS data were carried out on DNJ-stem sequences identified by ARResT/Interrogate (25). Transcriptome sequencing and molecular subtype calling were performed as described by Bastian et al. (26) HTS, high-throughput sequencing; RNA-Seq, RNA next- generation sequencing.
Initial diagnoses were established by standard routine diagnostics. Initial immunophenotype, BCR::ABL1 and KMT2A rearrangement status, initial molecular IG/TR markers, and MRD at follow-up were obtained by GMALL trials central diagnostic reference laboratories (Berlin & Kiel, Germany).
RNA was extracted according to standard procedures recommended by the manufacturer (Trizol, Life Technologies, Carlsbad, CA). Library preps, transcriptome sequencing, and molecular subtype calling were performed as described previously (26). Cases with intermediate or divergent gene expression profiles could not be assigned to any existing molecular profile category and were thus described as “other”.
We employed the EuroClonality-NGS assay and the IGH-VJ-FR1 and IGH-DJ primer sets to sequence diagnostic and follow-up samples. In general, 100 ng of DNA, extracted according to standard procedures recommended by the manufacturer (AllPrep, Qiagen, Hilden, Germany), was used for the analysis of the diagnostic samples. In the longitudinal analysis of diagnostic and follow-up samples, we used 500 ng DNA to ensure adequate sensitivity and direct comparability of results. The EuroClonality-NGS central in-tube quality/quantification control (cIT-QC) was spiked into most diagnostic samples and all follow-up samples as a library-specific quality control and for the normalization of abundance from reads to cells (25, 27, 28). (www.euroclonality.org/protocols). Samples were sequenced on a MiSeq (Illumina, San Diego, CA, USA) with 2x250bp reads (sequencing depth analysis in Supplementary Material S1).
IGH sequences were analyzed with ARResT/Interrogate (25). Usable reads, the denominator for percentage abundance calculations, were defined as sample reads with identified junctions after the exclusion of cIT-QC reads, in other words, reads with patient-only IGH rearrangements. For better readability, especially in figures, we will refer to IGH segment rearrangements using name acronyms omitting ‘IGH’ from gene names, e.g., VH instead of IGHV or DJH instead of IGHD-IGHJ.
For IGH clonal evolution assessment, we isolated the ‘DNJ-stem’ of nucleotide junctions - a subsequence of the junction that consists of the last maximum of 3 DH nucleotides (or if none are identifiable in a VJH rearrangement, of the N region), any N nucleotides between DH and JH, and all JH nucleotides (Figure 2). The DNJ-stem definition is adapted to the understanding that the corresponding junction subsequence is the one remaining generally stable after multiple DH/VH to DJH (DH/VH-DJH) recombination and VH replacement events, and it is therefore used as the link, a shared denominator, between potential family members – whether those clonotypes should indeed be considered related and whether the DNJ-stem should be considered evolving was thoroughly examined by deploying further rules.
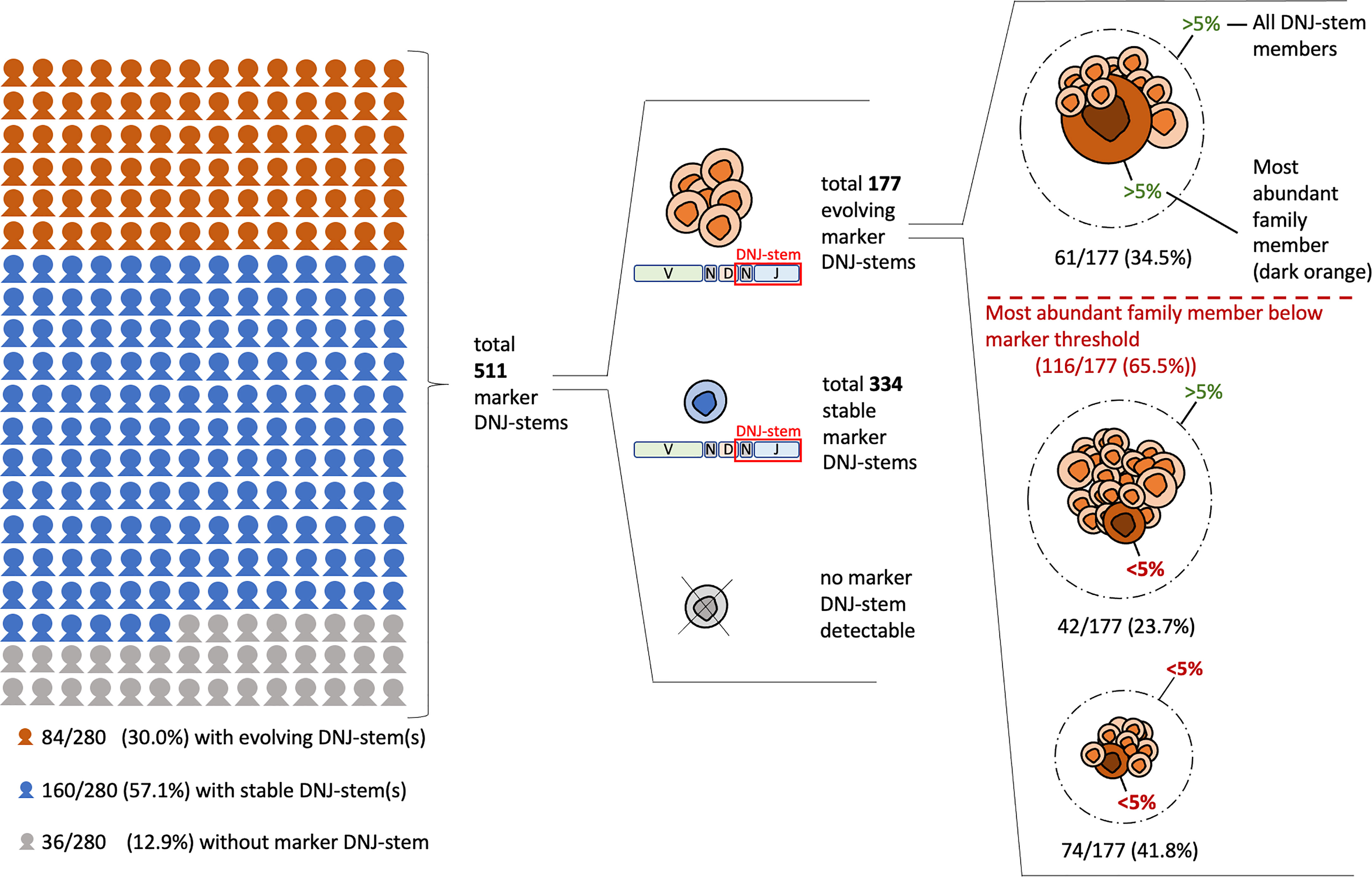
Figure 2 Clonal evolution and clonotypic abundances in diagnostic BCP-ALL samples (cohort #1). The figure is displaying the prevalence of clonal evolution in reference to all 280 patients and in reference to the total of 511 detected marker DNJ-stems. Additionally, the evolving DNJ-stems are itemized by their most abundant family member abundance and total DNJ-stem abundance relating to the 5% marker threshold.
We initially removed - as potential noise - clonotypes highly similar to the most abundant DNJ-stem family member, as well as of abundance below 3 reads or below 0.01% of usable reads. We then considered the number of clonotypes removed above (a high number may indicate an overall noisy DNJ-stem), the absolute and relative (to the sample total) number of remaining DNJ-stem family members, the number of their different 5’ genes and junction lengths as a sign of expected junctional variability, as well as the DNJ-stem’s N region complexity to increase confidence in its specificity. Combined consideration of all these data points led to a consensus call on whether a DNJ-stem was evolving or stable. Please see Supplementary Material S2 for the detailed pseudocode of this algorithm.
VH replacement was identified by considering (i) the general stability of the DH region’s 5’-site and its preceding N1 region’s 3’-site and (ii) the presence of a sequence remnant (fingerprint) of the replaced VH in the new junction. Evolving DNJ-stems with the absence of aforementioned signs were categorized as DH/VH-DJH recombinations.
If a DNJ-stem was detected in both the IGH-VJ and IGH-DJ library, the respective DNJ -stem was termed ‘rooted’, indicating that the underlying root DJH rearrangement was also detectable.
A marker DNJ-stem was defined if any of the following conditions were fulfilled: (i) abundance as a sum of all DNJ-stem family members ≥ 5% of usable reads or ≥ 1% of cells (normalized abundance); (ii) presence of clonal evolution (‘evolving’) at any abundance. We recovered the locus IGHV and IGHD gene order from IMGT (29).
Statistical analyses were performed in R version 4.1 using Fisher’s exact test with default settings, meaning the significance threshold was set at 0.05.
We studied DNJ-stems and the respective VDJH and DJH rearrangements in the diagnostic cohort of 280 BCP-ALL patients (Figure 1, cohort #1). DNJ-stems with only DJH rearrangements were further analyzed only if evolving or otherwise noteworthy.
Clonal evolution, i.e. at least one evolving DNJ-stem, was found in 84/280 (30%) patients. Stable (non-evolving) DNJ-stems were found in 160/280 (57.1%) patients and 36/280 (12.9%) patients had no detectable IGH rearrangements to constitute a marker DNJ-stem – the latter being a rate comparable to previous studies (30, 31). In summary, clonal evolution was prevalent in our cohort affecting one-third of the cases (Figure 2).
In total, 511 marker DNJ-stems (i.e., of ≥ 5% usable reads or evolving at any abundance) were identified in 244/280 (87.1%) patients (range 1-11, average 2.1 per patient). In 116 (65.5%) out of the overall detected 177 evolving DNJ-stems, their most abundant DNJ-stem family member stayed below the generally accepted marker threshold of 5% usable reads and therefore may have been ignored on its own. In other words, applying the standard marker screening approach (by following single clonotypes and not DNJ-stems and using the conventional 5% threshold), 65.5% of markers would not have been reported. By screening for DNJ-stem instead of clonotypes, but still applying the 5% threshold, two-thirds (74) of those 65.5% of markers would still not have been reported. A visual breakdown of the DNJ-stems and family member distribution is depicted in Figure 2.
We studied alignments of junction nucleotide sequences of evolving DNJ-stem families from the diagnostic BCP-ALL samples (Figure 1, cohort #1) and observed signs of the two main mechanisms driving IGH clonal evolution, VH to DJH (VH-DJH) recombinations and VH replacements. Additionally, we identified a variant of somatic recombination, in which DH to DJH (DH-DJH) recombination was evident (10, 11). Both mechanisms, DH/VH-DJH recombination and VH replacement, leave distinctive changes in the recombined nucleotide sequences, by which they can be specifically discriminated (Figure 3).
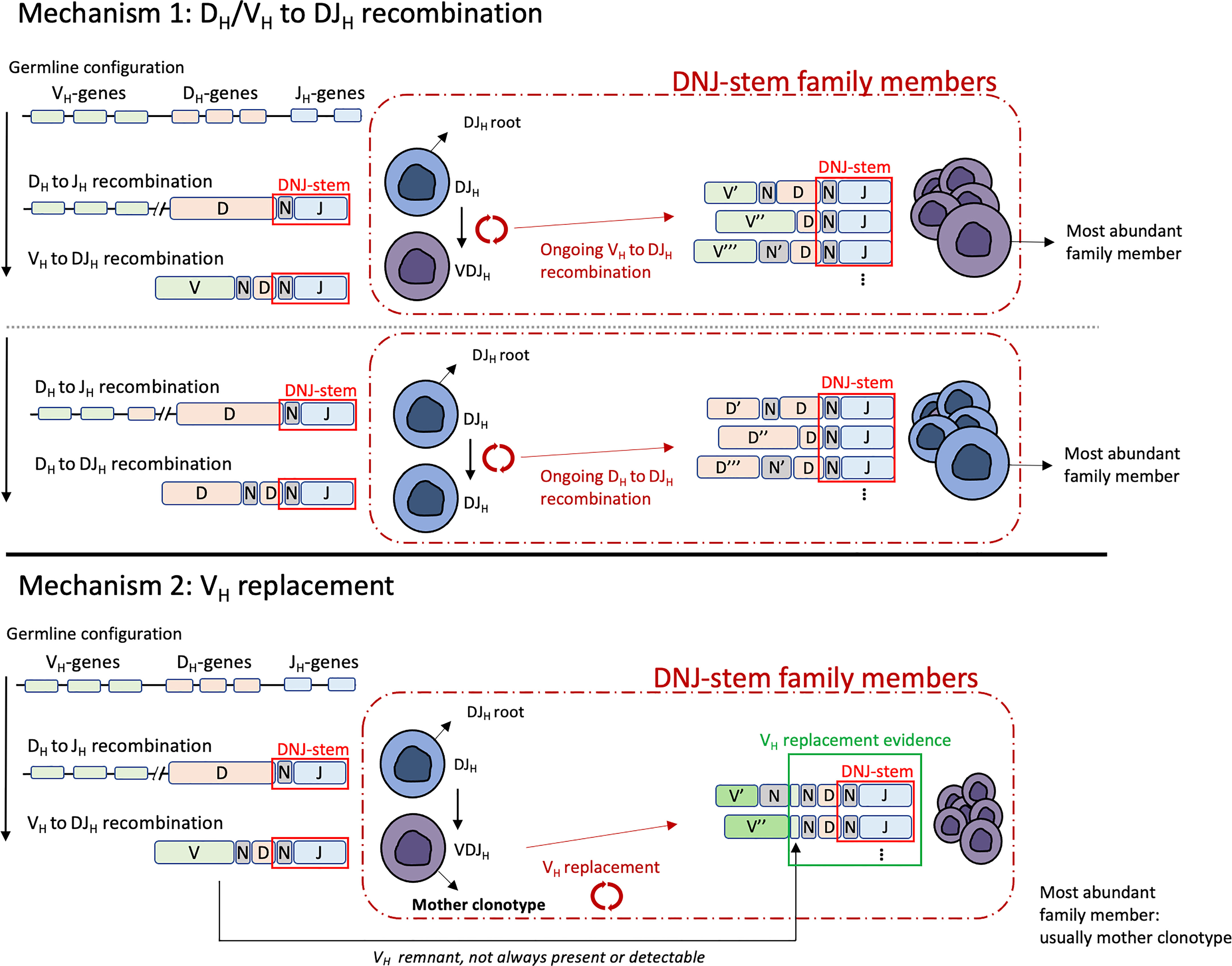
Figure 3 Schematic visualization of the terminology used in this study and the two mechanisms driving clonal evolution: DH/VH to DJH recombination (top) and VH replacement (bottom). All DNJ-stem family members, including the DJH root, the VJH mother clonotype (VH replacement) carry the same DNJ sequence (red box). Evidence of VH replacement (see main text) is not always present or detectable due to further recombination-related nucleotide sequence trimming (green box).
Ongoing DH/VH-DJH recombination was observed in 78% of evolving DNJ-stems. This mechanism results in sequence trimming during the rearrangement of the DH/VH to the existing DJH, which in addition to the random editing of N nucleotides results in sequence variability outside the DNJ-stem. This is exemplified in Figure 4A, case #61 and Figure 4B, lower part, case #126. In the illustration, the top row is the ‘mother’ clonotype for the family of clonotypes to be produced by parallel DH-DJH and VH-DJH recombination (case #61) or solely by VH-DJH recombination (case #126).

Figure 4 Sequence analysis of two cases illustrating the IGH clonal evolution mechanisms. (A) Case #61, ongoing DH-DJH and VH-DJH recombination on the same highly abundant DJH root (55% of reads) (top row, in bold). (B) Case #126, one of two unique cases with DNJ-stems featuring both mechanisms, VH replacement and VH-DJH recombination (VH replacement: top, VH-DJH recombination: bottom). The mother clonotype of the VH replacement and the DJH root of the VH-DJH recombination are each shown on the top row and in bold. The DNJ-stem family members evolving by VH-DJH use ‘late’ D-distal VH genes, which is uncommon for this mechanism (main text, Figure 5). Legend: green = VH, orange = DH, blue = JH, gray/uppercase = N regions, lowercase = germline, dots = deleted nucleotides, black = region affected by the recombination or replacement, yellow = region kept from the mother clonotype (on which the region is boxed), blue/yellow-to-orange (last column) = clonotype abundance in % reads, horizontal bar on top of first sequence = DNJ-stem. Genes are indented in the 1st column to indicate locus order. A number of rows in (B) are ‘squeezed’ and their VH and DH assignment and nucleotide sequences are hidden for space and clarity.
The vast majority of DH/VH-DJH cases featured only VH-DJH recombination-driven clonal evolution, but there were notable exceptions. In particular, of 398 DJH marker DNJ-stems identified in 144/280 (51.4%) patients, 21/398 (5.2%) DNJ-stems were evolving in 15 patients - in 12/15 (80.0%) of these patients this ongoing DH-DJH recombination accompanied the VH-DJH recombination, both driving clonal evolution (Figures 4A, B). The mechanism of VH replacement was not active in those cases. Of note, the only two patients with DH-DJH recombination without accompanying VH-DJH recombination were assigned to the rare molecular subtype CDX2/UBTF, a novel BCP-ALL subgroup with the need of intensified treatment that was identified in 7% of our cohort (26) (see below).
Ongoing VH replacement was observed in 22% of evolving DNJ-stems. T his mechanism is defined by its most distinct signs: (i) the general stability of the 5´-DH region site and its preceding 3´-N1 region site; (ii) the actual nucleotide remnants of the replaced VH in the new junction. There were very few exceptions to the requirement of the incoming VH to be downstream from the mother clonotype segment: 1.6 upstream segments on average (range 1-5) in lowly abundant clonotypes, in 13/39 DNJ-stems evolving by VH replacement, representing 1.9% of all family members. These could be erroneous gene annotations facilitated by sequence errors, false positive DNJ-stem members, or perhaps true members having originated previously. A case with ongoing VH replacement is detailed in Figure 4B (upper part, case #126), where the top row is the ‘mother’ clonotype for the family members to be produced by replacement of its VH segment. The resulting family members underneath show no further deletions in the DH segment, because any sequence loss was buffered by the N1 region and remnants of the replaced VH (highlighted in yellow) and now residing inside the new N1 regions.
Overall, and in contrast to VH replacement that most commonly takes place in the presence of one or more highly abundant mother clonotypes, ongoing VH-DJH recombination results in bursts of numerous lowly abundant family members with the role of the mother clonotype taken by the DJH root. This translates to an average abundance of the most abundant family member of 51% for VH replacement and 7% for VH-DJH recombination in the IGH-VJ library of our diagnostic cohort.
We selected one DNJ-stem from each patient or two DNJ-stems from each of two patients with concomitant VH-DJH recombination and VH replacement - a total of 53 VH-DJH-evolving stems and 33 VH replacement-evolving stems - focusing on evolving DNJ-stems with numerous family members, to highlight specific characteristics related to both mechanisms driving clonal evolution. For each DNJ‐stem, abundances and IGH evolution classifications such as evolving or stable, DJH root detectable or not, DH/VH-DJH recombination or VH replacement were annotated along with its locus-ordered VH gene profile and basic patient metadata. The results are summarized in an overview tabular heatmap (Figure 5).
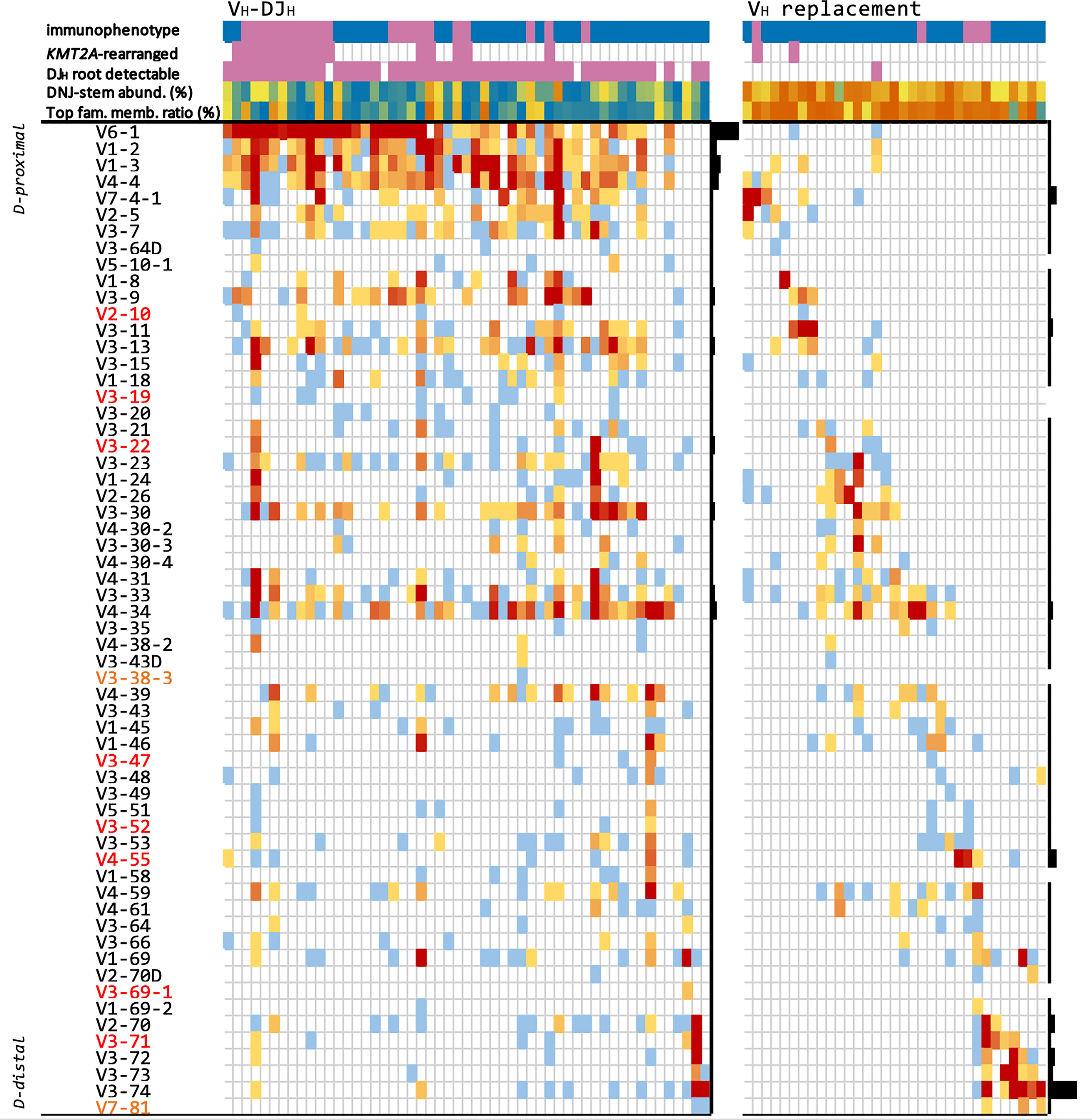
Figure 5 Evolving DNJ-stems in the diagnostic cohort. Each column represents one representative evolving DNJ-stem in one patient, ordered by the locus order of the most frequent gene in that column. VH genes are sorted by their proximity to the DH genes (proximal top to distal bottom), respectively; red gene names: pseudogenes, yellow: ORFs. Top rows: 1) immunophenotype: pro-B ALL purple, common-/pre-B ALL blue; 2) KMT2A molecular subtype status: KMT2Ar purple, non-KMT2Ar no color; 3) DJH root with same DNJ-stem detectable (purple); 4) DNJ-stem abundance (% reads), blue to orange (0.2% to 97%); 5) most abundant family member clonotype to summed DNJ-stem abundance ratio, blue to orange (0.01 to 0.99). Heatmap cells are colored by number of DNJ-stem family members featuring each gene, blue to red (1 to 418); black bar plots on the right of each mechanism group visualize each gene’s relative frequency in that group.
Of the 244 diagnostic samples of cohort #1 with identified marker DNJ-stems, 41 (16.8%) were classified as pro-B ALL and 203 (83.2%) as pre-B/c-ALL by multiparameter flow cytometry.
We detected significantly higher rates of clonal evolution in patients with a pro-B ALL compared to those with a pre-B/c-ALL immunophenotype (61.0% vs. 29.1%, respectively, p=0.0008), suggesting that the maturation arrest at earlier B-cell development stages might increase the frequency of ongoing IGH recombination.
Other noteworthy observations are that (i) VH-DJH overwhelmingly featured ‘early’, J-proximal VH genes, mainly VH6-1 (42/53 (79.2%) DNJ-stems and most family members) - in strong contrast, VH6‐1 featured only twice in VH replacement cases, in which the ‘late’, J-distal VH3-74 segment was the most frequent; (ii) the DJH root was present in the majority of VH-DJH DNJ-stems but in only one VH replacement DNJ-stem; (iii) DH-DJH-driven clonal evolution was observable in VH-DJH cases but not in VH replacement cases; (iv) the two mechanisms, VH-DJH recombination and VH replacement, were almost mutually exclusive (Supplementary Tables 2, 3).
The BCP-ALL molecular subtypes were assigned based on RNA-Seq data available for 167/280 (59.6%) cases and then correlated with the IGH clonal evolution patterns. Overall, 155/167 cases could be assigned to a previously described molecular profile category, while 12 cases showed other gene expression profiles and thus were summarized as “other”. Notably, the aforementioned higher rates of clonal evolution in pro-B ALL were almost exclusively linked to the KMT2A subtype (94.4% evolving), while other molecular subtypes with a pro-B ALL immunophenotype were mostly stable, particularly CDX2/UBTF (cases with UBTF::ATXN7L3 gene fusion, only 22.2% evolving and those only in the IGH-DJ library) and ZNF384 (only 30% evolving) (Figure 6A). Further, the KMT2A subtype showed significantly higher rates of ongoing VH-DJH (16/17 (94.1%) evolving patients, p=3e‐11), prevalently initiating the recombination with ‘early’ J-proximal VH genes, mostly VH6-1 (Figure 5). The preferential usage of ‘early’ J-proximal VH genes was also seen in the molecular subtypes PAX5 P80R and PAX5alt. Although (and in contrast to the KMT2A subtype) most DNJ‐stems in those subtypes were stable (30/40, 75%), the evolving DNJ-stems (10/40, 25%) clearly showed a prevalent (as in 7/10) usage of ‘early’ J-proximal VH segment genes including VH6-1, VH1‐2, VH1-3 and VH4-4. Notably, the three DNJ-stems using more J-distal VH segments belonged to a single patient with concurrent VH replacement and VH-DJH recombination activity (Supplementary Table 2). While the KMT2A subtype was strongly associated with VH-DJH recombination as the prevalent clonal evolution mechanism, the molecular subtypes Ph-like and DUX4 featured no VH-DJH but were instead associated with VH replacement (7/37 and 4/13, p=0.00009 and p=0.067, respectively) (Figure 6A).
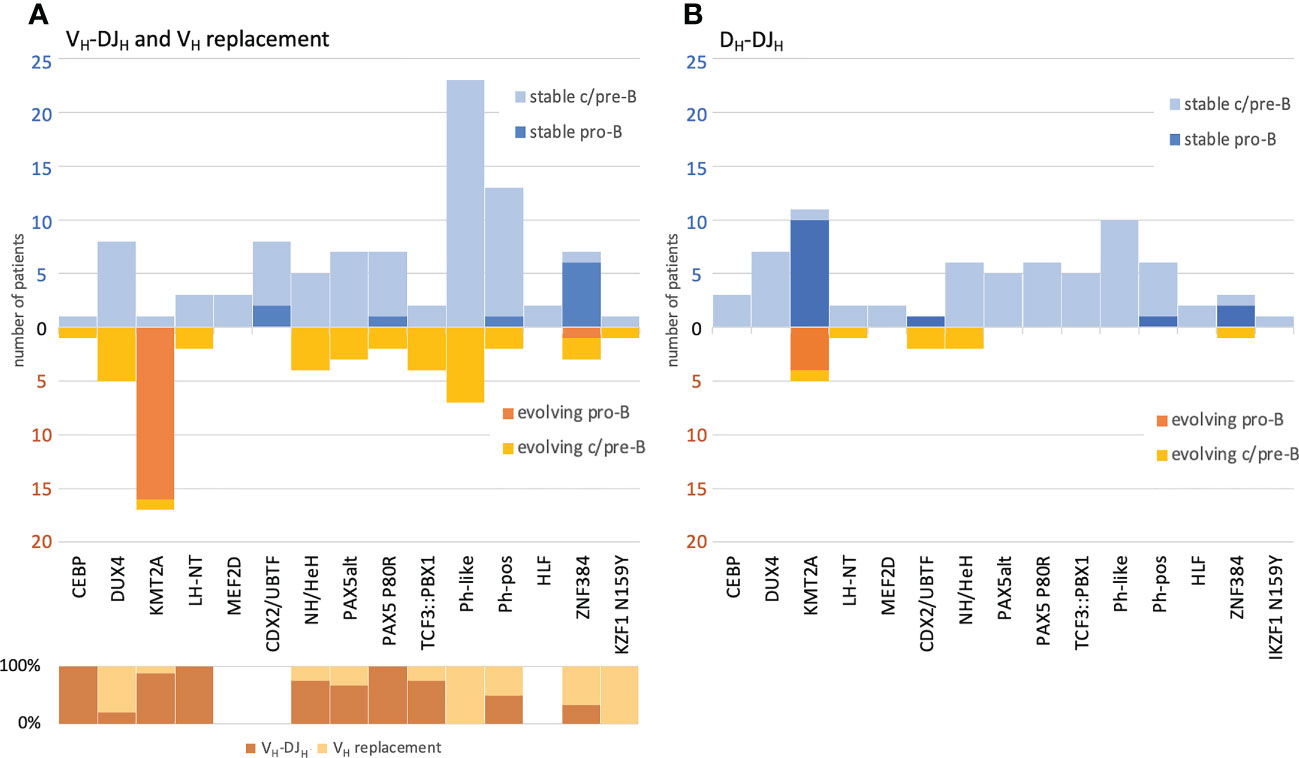
Figure 6 Correlation of BCP-ALL immunophenotype and molecular subtypes versus IGH clonal evolution and its respective mechanisms. VH replacement and VH-DJH recombination (A) and DH-DJH recombination (B). The numbers and classifications are derived from the one-stem-per-case data depicted also in Figure 5. All cases have been labelled by (i) immunophenotype and (ii) their respective molecular subtype as assigned by the transcriptome analysis. Evolving DNJ-stems (warm colors) are shown below the X-axis line and stable DNJ-stems (cold colors) are shown above the X-axis line. For the evolving DNJ-stems, the respective mechanism driving clonal evolution in % of cases has been depicted in the lower left part of the schematic in (A).
The DH-DJH evolution was a less frequent observation and only detectable in 11 of 155 (7.1%) patients with molecular subtype assignment, with the low number of patients impeding significant associations with molecular subtypes. Despite the fact that the KMT2A subtype not only showed the highest rate of VH-DJH evolution, but also significantly higher rates of DH-DJH evolution (27.8% evolving, p=0.0001), we found that the presence of DH-DJH and VH-DJH was not fully concordant (Figure 6B). Strikingly, two patients of the rare CDX2/UBTF subtype showed only DH-DJ evolution (p=0.04), whereas there were no signs of VH-DJH evolution. Overall, the evolving DH-DJH rearrangements showed a remarkable preference for the usage of DH2-2 as the incoming DH (14/15 (93.3%) of DNJ-stems) (Supplementary Table 3), which was also observed by others but in the context of normal rearrangements in blood B cells from healthy donors (32).
We sought to understand the overlap of DNJ-stems and their family members between diagnostic BM and PB of the 46 patients with both diagnostic materials available (Figure 1, cohort #2). We identified 39 patients with paired BM and PB DNA samples and with marker DNJ-stem(s) in at least one of these two compartments. The total number of DNJ-stems was 93, with 66 DNJ-stems in both compartments as marker DNJ-stems (see Methods for definition), 21 DNJ-stems also in both compartments but appearing in one compartment outside the marker DNJ-stem rules (and almost always at very low abundance), and the remaining 6 appearing only in BM compartment (Figure 7). This translated into 37/39 patients with at least one of the 87 DNJ-stems in both materials, or 36/39 when we demanded marker DNJ-stems in both materials, i.e., considering only the 66 DNJ-stems; all other cases had the DNJ-stem in BM only. Of the 66 DNJ-stems in both the BM and PB compartments of the 36 patients, all but two had concordant clonal evolution status between BM and PB, with the two exceptional cases evolving in BM and stable in PB. In 32/36 patients we saw a complete marker DNJ-stem overlap between BM and PB.
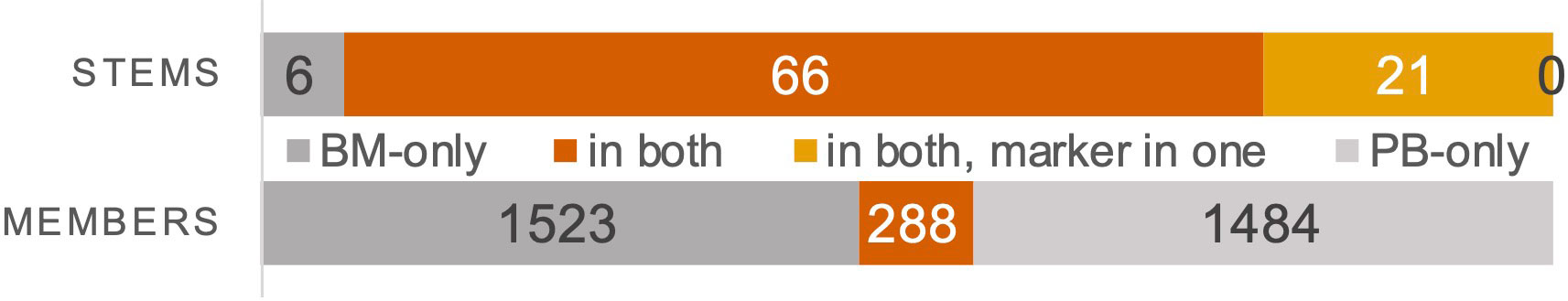
Figure 7 Overlap of marker DNJ-stems and their family members detectable in BM and PB. Total numbers of marker DNJ-stems (top) and DNJ-stem family members (bottom) that were found in only one of the compartments are illustrated in grey. Those that were overlapping between the compartments are illustrated in orange. In case of marker DNJ-stems, we distinguished between concordant marker status (dark orange) and detectable in both, but only marker status in one (light orange).
When we compared the overlap of ~3300 family members of 26 DNJ-stems that were evolving both in BM and PB, we found the overlap to be only 8.7%, with the rest split almost equally between BM and PB (Figure 7). Evolving DNJ-stem abundance in BM was on average 18.7% reads (range 0.6-89.9%), while the average abundance of family members was 0.16% reads (range 0.0008-71.8%). In PB the evolving DNJ-stem abundance was on average 12.9% reads (0.2-76.8%) and the average abundance of family members 0.11% (range 0.0002-73.8%). The fact that the minimum abundance of family members is below the 0.01% reads threshold used for family members when calling clonal evolution, is due to searching for members across materials at any abundance to maximize the sensitivity and information content of the overlap analysis.
In order to understand the longitudinal dynamics of evolving clonotypes during and after treatment, we compared therapy-naive BCP-ALL samples (day 0) and matched samples from different follow-up time points (Figure 1, cohort #3-5): (i) day +6 samples after the 5-day cyclophosphamide/dexamethasone-containing prephase, no rituximab (cohort #3); (ii) day +22 samples after Induction I including one dose of rituximab (cohort #4); and (iii) later time points including additional doses of rituximab and refractory disease/relapse-related secondary immunotherapies, e.g. blinatumomab (cohort #5).
Cohort #3 encompassed 33 BCP-ALL patients with a total of 66 matched peripheral blood (PB) samples taken at diagnosis and at day +6 of the prephase. In 3/33 (9.1%) diagnostic PB samples, no marker DNJ-stem was identified. In one case, the stable marker DNJ-stem was undetectable after prephase therapy. More than two-thirds of the cases were either concordantly stable (19/33, 57.6%) or concordantly evolving (4/33, 12.1%) across day 0 and day +6. Four cases (12.1%) were evolving at day 0 and stable at day +6, with the loss of family members after prephase therapy probably due to the general reduction of tumor burden. Two cases showed clonal evolution after prephase therapy but not at diagnosis, which could be rather attributed to the overall reduction of benign mature B-cell numbers in the post-prephase samples and the resulting higher sensitivity of HTS. In the majority of concordantly evolving DNJ-stems (4/6), the highest abundant family member at day 0 remained the highest abundant family member at day +6. The overlap of evolving clonotypes between the two time points was on average 29.6%.
We further quantified the MRD at day +6 using either the DNJ-stem, or only the most abundant family member as MRD marker and compared the MRD levels. The DNJ-stem-based MRD was on average 3.5-times higher (range 1.1 to 9.7-times) than the most abundant family member-based MRD. In 2/8 evolving DNJ-stems, the discrepancy between DNJ-stem MRD and most abundant family member MRD equaled a log10 difference.
Cohort #4 encompassed 41 BCP-ALL patients with a total of 82 matched bone marrow (BM) samples taken at diagnosis and at day +22 after Induction I with 1 dose of rituximab administered after the prephase. In 6/41 (14.6%) of initial diagnostic samples, no DNJ-stem was identified. The evolving marker DNJ-stem in one case and a stable marker DNJ-stem in another two cases were undetectable after Induction I. In accordance with cohort #1, nearly two-thirds of patients were either concordantly stable (20/41, 48.8%) or concordantly evolving (7/41, 17.1%), with 5 patients evolving at day 0 and stable after Induction I. However, no cases were stable at diagnosis but evolving after Induction I. The average overlap of evolving clonotypes was 28.5%.
Cohort #5 encompassed 34 BCP-ALL patients with a total of 149 matched PB and/or BM samples taken at diagnosis and at multiple follow-up time points per patient, i.e. at the end of prephase at day +6, after Induction I at day +22, at later time points with refractory disease or/and at relapse during chemotherapy including 4 or 8 doses of rituximab or at relapse after additional immunotherapy with blinatumomab or inotuzumab ozogamicin. In particular, the clonotypic composition of initial diagnostic samples was compared to 97 matched samples obtained during these aforementioned divergent MRD-positive follow-up time points. In order to evaluate in how many cases the conventional MRD detection (which relies on the tracking of the one or two most abundant family members) would have been hampered by the vanishing of the most abundant family member, we analyzed clonal evolution dynamics over time in the 34 patients of cohort #5. At diagnosis 14 of 34 patients (41.2%) had stable DNJ-stems (no clonal evolution), 4 (11.8%) had no IGH marker and 16 (46.4%) were evolving. In those 16 patients with clonal evolution, we identified 45 evolving DNJ-stems in total (Supplementary Table 4). Noteworthy, 17/45 (37.8%) of evolving DNJ-stems including all family members became untraceable in the follow-up samples. In detail, 15/17 evolving DNJ-stems were distributed among two patients, in whom the MRD remained only traceable through the KMT2A fusion-derived MRD marker assay, underscoring the importance of this alternative approach of MRD monitoring in KMT2A-rearranged B-ALL. The remaining 2/17 disappearing evolving DNJ-stems were both related to one patient and presented the most abundant clonotypes at diagnosis but became eradicated during the follow-up. In 7/45 (15.6%) of evolving DNJ-stems, the most abundant family member stayed most abundant across all follow-up time points. In 2/45 (4.4%) evolving DNJ-stems the most abundant family member was traceable at all the time points, but did not stay the most abundant family member at all time points, thus not reflecting the most correct MRD level. In the majority of evolving DNJ-stems (19/45, 42.2%), the most abundant family member disappeared over time, while other family members of the same DNJ-stem remained present and would be available to constitute the MRD signal in the DNJ-stem based MRD approach. The median abundance of the most abundant family member in those 19 evolving DNJ-stems was 0.06% (0.002 -37.7%) cells compared to 20% (0.67 - 87.6%) cells in the 7 evolving DNJ-stems, in which the most abundant family member remained most abundant at later time points. The non-persistence of the most abundant family member was most likely caused by its low abundance in follow-up in combination with a sampling bias. In one case (Supplementary Material S3A, Ph-neg#27), a highly abundant family member (37.7% cells) disappeared in the course of therapy, while only the second most abundant family member persisted. Taken together, substantial changes in the abundance and composition of the initially most abundant family members conventionally chosen as molecular MRD markers were observed in 37/45 (82%) of the evolving DNJ-stems and accordingly in 9/16 (56%) patients with evolving DNJ-stems, in whom the correct assignment of the MRD level could be adversely affected, i.e. underestimated or failing detection. Critically, in 2/34 (5.9%) of patients, the DNJ-stem with the vanishing most abundant family member was the only IGH target for MRD detection.
In the Supplementary Material S3A-D we present four examples of Philadelphia chromosome-negative (Ph-neg) cases with interesting dynamics of family members belonging to the same DNJ-stem, all presenting different kinetics over time, in which the conventional marker identification (5% reads threshold) or the conventional MRD quantification (clonotype based) would have led to impaired MRD detection in follow-up samples.
Of note, in all cases, the mechanism of clonal evolution in all follow-up samples remained the same compared to the respective diagnostic sample. In consideration of the cohorts #3-5, we conclude that applying the following rules could improve MRD monitoring in follow-up samples: (i) consider the DNJ-stem and not specific family members as MRD basis marker, also because new family members of initially stable DNJ-stems could emerge during the course of therapy; (ii) if an evolving DNJ-stem in the IGH-VJ library is also found in the IGH-DJ library sample (and thus rooted), one should also follow the DNJ-stem in the IGH-DJ library for MRD monitoring even if it does not fulfill marker criteria in the diagnostic sample; (iii) use DNJ-stem abundance as the sum of all family members to avoid underestimation of the extent of MRD.
Clonal evolution in the IGH locus and its significance and implications in BCP-ALL patients have all been well documented (5, 33–35). Our research was conducted on a well-studied cohort in order to develop tools for the identification and characterization of this important phenomenon. We provide refined observations with a more detailed insight into clonal evolution processes during BCP-ALL therapy.
We report that IGH clonal evolution is prevalent in BCP-ALL, conservatively identified in one-third of the cases. Clonal evolution has been shown to be a malignancy-related phenomenon of B cells (8). The approach presented in this manuscript to identify IGH clonal evolution and the resulting IGH oligoclonality is not based on the simple detection of more than two dominant clonal IGH rearrangements, which could be present even without oligoclonality in the case of biallelic rearrangements or trisomy of the IGH locus (e.g., in hyperdiploid ALL) (36, 37). Instead, we introduce the concept of ‘marker DNJ-stem’ to cover the entire, clonally-related family of evolving clonotypes. We identified 40% of evolving marker DNJ-stems at levels below conventional clonality thresholds (<5% reads) in 38/280 (14%) patients presenting clonal evolution. In such cases, conventional abundance thresholds produce misleading results and should therefore not be applied. In other words, evolving DNJ-stems at any level should be assumed to be BCP-ALL-related and therefore considered as a marker.
We further investigated whether the degree of clonal evolution and the composition of DNJ-stem family members differs between BM and PB. Comparing the diagnostic sample pairs of 46 patients, we found that the high overlap of marker DNJ-stems and their evolution status indicates that PB is a comparable and reliable source for the determination of the IGH oligoclonality status. The VH usage of DNJ-stem family members did not show a tendency towards more mature clonotypes in PB compared to BM. In line with our previous study, which showed that MRD levels tend to be lower in PB than in BM (38), 22% of marker DNJ-stems were exclusively found in BM, where DNJ-stems and clonotypes are generally more abundant and evolving, indicating that not all malignant family members access the bloodstream in sufficient numbers to be detected. As expected, the combination of the low abundance of evolved family member clonotypes and sampling biases resulted in their low overlap between the two materials - but of note, their total number was not higher in BM compared to PB.
Abandoning the conventional 5% reads clonality threshold for evolving DNJ-stems as suggested above entails the risk of reporting unspecific, malignancy-unrelated markers which can result in impaired MRD detection. Thus, it should be discussed if the DNJ-stem is unique and specific enough to be a reliable MRD target. The insertion of short palindromic P nucleotides during VDJH recombination has been reported in around ten percent of IGH rearrangements (39). These nonrandom sequences might reduce the specificity of the DNJ-stem for MRD detection, especially in cases with short or completely absent N2 nucleotides. Furthermore, it is not clear if the application of DNJ-stem-based MRD detection is of benefit in mature lymphoid malignancies. We, therefore, tested the sensitivity and specificity of this approach using IGH sequences in internal cohorts of two distinct mature B-lymphoid malignancies, i.e. ~100 chronic lymphocytic leukemia (CLL) and ~100 multiple myeloma (MM) cases. While we detected no clonal evolution in the CLL cohort, three DNJ-stems in as many cases in the MM cohort were reported. All three were ‘borderline’ calls with few family members and low variability of genes and junction lengths, calls that in the context of MM could be dismissed. Additionally, we screened our BCP-ALL cohort for marker DNJ-stems that appear in more than one patient at diagnosis. Only one of the 511 marker DNJ-stems detected in the IGH-VJ library was shared between two patients. Overall, we would advise the application of bio/medical context during the interpretation of results, and to examine DNJ-stem sequences for specificity within each cohort and against a large reference cohort.
The study of the junction alignments of the family members of evolving DNJ-stems allowed us to identify the apparent mechanism involved, DH/VH to DJH recombination or VH replacement, and thus provide another perspective into our data and results (Figures 3-5).
DH-DJH segment fusions were first hypothesized in 1982 (40), experimentally verified in 1989 and thereby considered a rare but physiologically legitimate activity adding another combinatorial dimension to the antibody repertoire diversity (11). Since DH-DJH recombination clearly violates the 12-23 rule applying in the process of V(D)J recombination, concerns were raised about a possible aberrant nature of this phenomenon. Further research led to the discovery of hitherto uncharacterized cryptic RSS in the DH genes, confirming the physiological nature of the occasional DH-DJH recombination (10).
Taken together, our data undoubtedly show that all of the described ongoing recombinatory processes, the DH/VH to DJH recombination and VH replacement, drive the clonal evolution in BCP-ALL patients. Moreover, we demonstrate that these mechanisms are almost mutually exclusive and separate patients into clearly delineated groups on different levels:
(i) the DH/VH-DJH group was characterized by an overrepresentation of KMT2A and PAX5 aberrations, the presence of a DJH root, the preferential usage of DH-proximal VH genes and the presence of low-abundant DNJ-stem family members, as well as accompanying DH-DJH evolution in KMT2A rearranged cases;
(ii) the VH replacement group was (less strongly) associated with the DUX4 and Ph-like molecular subtypes, as well as with D-distal VH genes used by DNJ-stem family members, one or more of which playing the role of abundant mother clonotypes.
The two exceptions to the observation that DH-DJH evolution cases associated with VH-DJH evolution, were two cases belonging to the newly described CDX2/UBTF molecular subtype, which otherwise presented no IGH evolution despite the predominance of a pro-B immunophenotype (26).
Our results point towards DH/VH-DJH recombination as an ontogenically earlier-stage and VH replacement as an ontogenically later-stage associated mechanism. In most VH-DJH cases, the DNJ-stem was detectable in the IGH-DJ library, suggesting that VH-DJH is a highly active and ongoing process, resulting in a burst of immature rearrangements. Generally, the biased usage of DH/VH genes, particularly VH6-1 in VH-DJH recombination and DH2-2 in DH-DJH recombination, could be related to the distinctive accessibility of different parts of the VH and DH clusters to the VDJH recombination machinery as postulated by Tsakou and colleagues (41) and previously shown in mice (42).
The preferential use of VH6-1 could at least be related to hampered PAX5 activity. Wild-type PAX5 was reported to cooperate with interleukin receptor 7 to confer locus contraction and subsequent VH-DJH recombination of distal VH segments, whereas it was not required for the preceding DH to JH recombination (43, 44). Consequently, at least the biased usage of VH6-1 could be related to the inaccessibility of distal VH segments due to absent or hampered activity of PAX5 and/or related factors.
Our results, which clearly demonstrate a correlation of the molecular subtype with different frequencies and even different mechanisms of clonal evolution, entail clinical consequences. Our selected longitudinal case reports show that MRD quantification, in the form in which it is currently routinely performed, can be affected by the degree of clonal evolution and at worst might lead to false negative MRD results. Thus, the reliability of IGH-based MRD quantification varies depending on the molecular subtype. In particular, patients with the KMT2A subtype, of whom more than 94% are evolving, should be monitored with caution.
Besides the high rates of clonal evolution in KMT2A-rearranged BCP-ALL, increasing the susceptibility to errors, studies have shown that IG/TR rearrangements in infant KMT2A-rearranged ALL are more likely oligoclonal or even completely absent, which makes any IG/TR based MRD-monitoring less convenient. It has therefore been recommended to preferably employ the KMT2A gene fusion for more stable and reliable MRD detection (45, 46).
On the basis of our findings, we suggest the following in order to improve IG/TR-based MRD detection in BCP-ALL: i) follow the DNJ-stem (covering all DNJ-stem family members) and not specific family members; ii) follow the DNJ-stem using both IGH-VJ and IGH-DJ assays; iii) express MRD as the DNJ-stem abundance (the sum of all DNJ-stem family members) in order to avoid underestimation of the MRD level in samples with high degree of clonal evolution; iv) be aware that the classical and still widely adopted real-time quantitative PCR (RQ-PCR) often uses primers and probes designed to track only specific clonal rearrangements (unlike HTS), thus potentially missing family members that are either already present at diagnosis or arise during the course of the disease. Crucially, further testing of the above-suggested approach on large well-defined cohorts is necessary to prove its prognostic strength before it can be routinely applied.
Wrapping up, we will outline a number of future perspectives. First, we herein only studied clonal evolution in the IGH locus, but a study on clonal evolution of T-cell receptors might allow deeper insights - particularly as the TRD locus has shown similar recombinase activity as the IGH locus in patients with VH replacement (47). Second, a recent study has shown that BCP-ALL harboring TP53 alterations are associated with immature DJH rearrangements (48). Assuming that TP53 alterations occur already in the pre-leukemic cell compartment, one should study if these cases show clonal evolution rates similar to KMT2A-rearranged cases. Finally, it was recently reported that in childhood BCP-ALL, clonal evolution is not only relevant for reliable MRD detection but also directly correlated to the clinical outcome (33). A similarinvestigation into this critical association of outcome and clonal evolution in adult BCP-ALL would require expanded prospective studies.
The data presented in the study are deposited in the European Nucleotide Archive (ENA) at EMBL-EBI, accession number PRJEB59052 (https://www.ebi.ac.uk/ena/browser/view/PRJEB59052).
The studies involving human participants were reviewed and approved by Frankfurt Research Ethics Board (188/15F). The patients/participants provided their written informed consent to participate in this study.
MB, ND, MK and MS designed the research. NG supervised the clinical trial. SS and MS performed immunophenotypic analyses. MK, ND and FD processed, analyzed and interpreted high-throughput sequencing data. ND and KP worked on ARResT/Interrogate. LB, AH and TB analyzed and interpreted RNA-Seq data. FD, ND and MS performed statistical analyses. MB, ND, MS and CB supervised the project. FD, ND, MS, MK and MB drafted the first version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche José Carreras Leukämie-Stiftung (grants DJCLS R 15/11 and DJCLS 06R/2019) to MS and MB as well as by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - project number 444949889 (KFO 5010/1 Clinical Research Unit ‘CATCH ALL’) to LB, AH, MB, and CDB.
The authors thank the Hematology Laboratory Kiel (HLK) staff for sample, IGH HTS and RNA-Seq processing. The authors are indebted to the GMALL Trial Center (R. Reutzel, C. Fuchs) and participating hospitals for patients´ recruitment, care and logistics.
MB received grants and personal fees from Amgen advisory board, speakers bureau, travel support, and personal fees from Becton Dickinson, Janssen, Pfizer speakers bureau and Jazz travel support, all outside the submitted work. MS and MK received a personal fee from Amgen speakers bureau, outside the submitted work. SS has received personal fees advisory board from AMGEN, Gilead Sciences, Pfizer, SERB SAS, honoraria speaker bureau from Akademie für Infektionsmedizin e.V., AMGEN, AVIR Pharma, CSi Hamburg GmbH, Gilead, Labor28, Novartis, Persberg Group GmbH/DGIM e.V., Pfizer, Vivantes GmbH, research grants from Protherics Medicines Development Ltd, and travel support from Gilead Sciences, Novartis, outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1125017/full#supplementary-material
1. Alt FW, Yancopoulos GD, Blackwell TK, Wood C, Thomas E, Boss M, et al. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO J (1984) 3:1209–19. doi: 10.1002/j.1460-2075.1984.tb01955.x
2. Alt FW, Oltz EM, Young F, Gorman J, Taccioli G, Chen J. VDJ recombination. Immunol Today (1992) 13:306–14. doi: 10.1016/0167-5699(92)90043-7
3. Alt FW, Baltimore D. Joining of immunoglobulin heavy chain gene segments: Implications from a chromosome with evidence of three d-J(H) fusions. Proc Natl Acad Sci USA (1982) 79:4118–22. doi: 10.1073/pnas.79.13.4118
4. Tonegawa S. Somatic generation of antibody diversity. Nature (1983) 302:575–81. doi: 10.1038/302575a0
5. Beishuizen A, Verhoeven M, van Wering E, Hahlen K, Hooijkaas H, van Dongen J. Analysis of ig and T-cell receptor genes in 40 childhood acute lymphoblastic leukemias at diagnosis and subsequent relapse: Implications for the detection of minimal residual disease by polymerase chain reaction analysis. Blood (1994) 83:2238–47. doi: 10.1182/blood.V83.8.2238.2238
6. Beishuizen A, Hählen K, Hagemeijer A, Verhoeven MA, Hooijkaas H, Adriaansen HJ, et al. Multiple rearranged immunoglobulin genes in childhood acute lymphoblastic leukemia of precursor b-cell. Leukemia (1991) 5(8):657–67.
7. Choi Y, Greenberg SJ, Du TL, Ward PM, Overturf PM, Brecher ML, et al. Clonal evolution in b-lineage acute lymphoblastic leukemia by contemporaneous VH-VH gene replacements and VH-DJH gene rearrangements. Blood (1996) 87:2506–12. doi: 10.1182/blood.V87.6.2506.bloodjournal8762506
8. Gawad C, Pepin F, Carlton VEH, Klinger M, Logan AC, Miklos DB, et al. Massive evolution of the immunoglobulin heavy chain locus in children with b precursor acute lymphoblastic leukemia. Blood (2012) 120:4407–17. doi: 10.1182/blood-2012-05-429811
9. Theunissen PMJ, van Zessen D, Stubbs AP, Faham M, Zwaan CM, van Dongen JJM, et al. Antigen receptor sequencing of paired bone marrow samples shows homogeneous distribution of acute lymphoblastic leukemia subclones. Haematologica (2017) 102:1869–77. doi: 10.3324/haematol.2017.171454
10. Safonova Y, Pevzner PA. V(DD)J recombination is an important and evolutionarily conserved mechanism for generating antibodies with unusually long CDR3s. Genome Res (2020) 30:1547–58. doi: 10.1101/gr.259598.119
11. Meek KD, Hasemann CA, Capra JD. Novel rearrangements at the immunoglobulin d locus. inversions and fusions add to IgH somatic diversity. J Exp Med (1989) 170:39–57. doi: 10.1084/jem.170.1.39
12. Reth M, Gehrmann P, Petrac E, Wiese P. A novel VH to VHDJH joining mechanism in heavy-chain-negative (null) pre-b cells results in heavy-chain production. Nature (1986) 322:840–2. doi: 10.1038/322840a0
13. Kleinfield R, Hardy RR, Tarlinton D, Dangl J, Herzenberg LA, Weigert M. Recombination between an expressed immunoglobulin heavy-chain gene and a germline variable gene segment in a ly 1+ b-cell lymphoma. Nature (1986) 322:843–6. doi: 10.1038/322843a0
14. Steenbergen EJ, Verhagen OJHM, van Leeuwen EF, von dem Borne AEGK, van der Schoot CE. Distinct ongoing ig heavy chain rearrangement processes in childhood b- precursor acute lymphoblastic leukemia. Blood (1993) 82:581–9. doi: 10.1182/blood.V82.2.581.581
15. Zhang Z. VH replacement in mice and humans. Trends Immunol (2007) 28:132–7. doi: 10.1016/j.it.2007.01.003
16. Zhang Z, Zemlin M, Wang YH, Munfus D, Huye LE, Findley HW, et al. Contribution of VH gene replacement to the primary b cell repertoire. Immunity (2003) 19:21–31. doi: 10.1016/S1074-7613(03)00170-5
17. Bassan R, Brüggemann M, Radcliffe HS, Hartfield E, Kreuzbauer G, Wetten S. A systematic literature review and meta-analysis of minimal residual disease as a prognostic indicator in adult b-cell acute lymphoblastic leukemia. Haematologica (2019) 104:2028–39. doi: 10.3324/haematol.2018.201053
18. Bassan R, Spinelli O, Oldani E, Intermesoli T, Tosi M, Peruta B, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood (2009) 113:4153–62. doi: 10.1182/blood-2008-11-185132
19. Beldjord K, Chevret S, Asnafi V, Huguet F, Boulland ML, Leguay T, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood (2014) 123:3739–49. doi: 10.1182/blood-2014-01-547695
20. Berry DA, Zhou S, Higley H, Mukundan L, Fu S, Reaman GH, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: A meta-analysis. JAMA Oncol (2017) 3(7):e170580. doi: 10.1001/jamaoncol.2017.0580
21. Gökbuget N, Kneba M, Raff T, Trautmann H, Bartram CR, Arnold R, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood (2012) 120:1868–76. doi: 10.1182/blood-2011-09-377713
22. Reiter A, Schrappe M, Ludwig WD, Hiddemann W, Sauter S, Henze G, et al. Chemotherapy in 998 unselected childhood acute lymphoblastic leukemia patients. results and conclusions of the multicenter trial ALL-BFM 86. Blood (1994) 84:3122–33. doi: 10.1182/blood.V84.9.3122.3122
23. Ribera JM, Oriol A, Morgades M, Montesinos P, Sarrà J, González-Campos J, et al. Treatment of high-risk Philadelphia chromosome-negative acute lymphoblastic leukemia in adolescents and adults according to early cytologic response and minimal residual disease after consolidation assessed by flow cytometry: Final results of the PETHEMA ALL-AR-03 trial. J Clin Oncol (2014) 32:1595–604. doi: 10.1200/JCO.2013.52.2425
24. Kotrova M, Darzentas N, Pott C, Baldus CD, Brüggemann M. Immune gene rearrangements: Unique signatures for tracing physiological lymphocytes and leukemic cells. Genes (Basel) (2021) 12:979. doi: 10.3390/genes12070979
25. Bystry V, Reigl T, Krejci A, Demko M, Hanakova B, Grioni A, et al. ARResT/Interrogate: An interactive immunoprofiler for IG/TR NGS data. Bioinformatics (2017) 33:435–7. doi: 10.1093/bioinformatics/btw634
26. Bastian L, Hartmann AM, Beder T, Hänzelmann S, Kässens J, Bultmann M, et al. UBTF::ATXN7L3 gene fusion defines novel b cell precursor ALL subtype with CDX2 expression and need for intensified treatment. Leukemia (2022) 36(6):1676–80. doi: 10.1038/s41375-022-01557-6
27. Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia (2019) 33:2241–53. doi: 10.1038/s41375-019-0496-7
28. Knecht H, Reigl T, Kotrová M, Appelt F, Stewart P, Bystry V, et al. Quality control and quantification in IG/TR next-generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia (2019) 33:2254–65. doi: 10.1038/s41375-019-0499-4
29. Lefranc MP. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res (2001) 29:207–9. doi: 10.1093/nar/29.1.207
30. Kotrova M, Knecht H, Herrmann D, Schwarz M, Olsen K, Trautmann H, et al. The IG/TR next generation marker screening developed within euroclonality-NGS consortium is successful in 94% of acute lymphoblastic leukemia samples. Blood (2018) 132:2830. doi: 10.1182/blood-2018-99-112828
31. Szczepański T, Willemse MJ, Brinkhof B, Van Wering ER, van der Burg M, Van Dongen JJM. Comparative analysis of ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B–ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood (2002) 99:2315–23. doi: 10.1182/blood.V99.7.2315
32. Hansen TØ, Lange AB, Barington T. Sterile DJ h rearrangements reveal that distance between gene segments on the human ig h chain locus influences their ability to rearrange. J Immunol (2015) 194:973–82. doi: 10.4049/jimmunol.1401443
33. Levy G, Kicinski M, van der Straeten J, Uyttebroeck A, Ferster A, de Moerloose B, et al. Immunoglobulin heavy chain high-throughput sequencing in pediatric b-precursor acute lymphoblastic leukemia: Is the clonality of the disease at diagnosis related to its prognosis? Front Pediatr (2022) 10:874771. doi: 10.3389/fped.2022.874771
34. Rosenquist R, Thunberg U, Li AH, Forestier E, Lönnerholm G, Lindh J, et al. Clonal evolution as judged by immunoglobulin heavy chain gene rearrangements in relapsing precursor-b acute lymphoblastic leukemia. Eur J Haematol (1999) 63:171–9. doi: 10.1111/j.1600-0609.1999.tb01765.x
35. Theunissen PMJ, de Bie M, van Zessen D, de Haas V, Stubbs AP, van der Velden VHJ. Next-generation antigen receptor sequencing of paired diagnosis and relapse samples of b-cell acute lymphoblastic leukemia: Clonal evolution and implications for minimal residual disease target selection. Leuk Res (2019) 76:98–104. doi: 10.1016/j.leukres.2018.10.009
36. Fries C, Lee LW, Devidas M, Dai Y, Rabin KR, Gupta S, et al. Prognostic impact of pretreatment immunoglobulin clonal composition in pediatric b-lymphoblastic leukemia. Haematologica (2023) 108:900–4. doi: 10.3324/haematol.2022.281146
37. Szczepanski T, Willemse M, Wering EV, Weerden JFV, Kamps W, Dongen JV. Precursor-B-ALL with d-H-J(H) gene rearrangements have an immature immunogenotype with a high frequency of oligoclonality and hyperdiploidy of chromosome 14. Leukemia (2001) 15:1415–23. doi: 10.1038/sj.leu.2402206
38. Kotrova M, Volland A, Kehden B, Trautmann H, Ritgen M, Wäsch R, et al. Comparison of minimal residual disease levels in bone marrow and peripheral blood in adult acute lymphoblastic leukemia. Leukemia (2019) 34:1154–7. doi: 10.1038/s41375-019-0599-1
39. Jackson KJL, Gaeta B, Sewell W, Collins AM. Exonuclease activity and p nucleotide addition in the generation of the expressed immunoglobulin repertoire. BMC Immunol (2004) 5:19. doi: 10.1186/1471-2172-5-19
40. Kurosawa Y, Tonegawa S. Organization, structure, and assembly of immunoglobulin heavy chain diversity DNA segments. J Exp Med (1982) 155:201–18. doi: 10.1084/jem.155.1.201
41. Tsakou E, Agathagelidis A, Boudjoghra M, Raff T, Dagklis A, Chatzouli M, et al. Partial versus productive immunoglobulin heavylocus rearrangements in chronic lymphocytic leukemia: Implications for b-cell receptor stereotypy. Mol Med (2012) 18:138–45. doi: 10.2119/molmed.2011.00216
42. Sen R, Oltz E. Genetic and epigenetic regulation of IgH gene assembly. Curr Opin Immunol (2006) 18:237–42. doi: 10.1016/j.coi.2006.03.008
43. Hesslein DGT, Pflugh DL, Chowdhury D, Bothwell ALM, Sen R, Schatz DG. Pax5 is required for recombination of transcribed, acetylated, 5′ IgH V gene segments. Genes Dev (2003) 17:37–42. doi: 10.1101/gad.1031403
44. Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldan E, Busslinger M. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev (2004) 18:411–22. doi: 10.1101/gad.291504
45. Darzentas F, Szczepanowski M, Kotrová M, Kelm M, Hartmann A, Beder T, et al. IGH rearrangement evolution in adult KMT2A-rearranged b-cell precursor ALL: implications for cell-of-origin and MRD monitoring. Hemasphere (2022) 7(1):e820. doi: 10.1097/HS9.0000000000000820
46. Stutterheim J, van der Sluis IM, de Lorenzo P, Alten J, Ancliffe P, Attarbaschi A, et al. Clinical implications of minimal residual disease detection in infants with KMT2A-rearranged acute lymphoblastic leukemia treated on the interfant-06 protocol. J Clin Oncol (2021) 39:652–62. doi: 10.1200/JCO.20.02333
47. Steenbergen EJ, Verhagen OJHM, van den Berg H, van Leeuwen EF, Behrendt H, Slater RR, et al. Rearrangement status of the malignant cell determines type of secondary IgH rearrangement (V-replacement or V to DJ joining) in childhood b precursor acute lymphoblastic leukemia. Leukemia (1997) 11:1258–65. doi: 10.1038/sj.leu.2400720
Keywords: acute lymphoblastic leukemia, clonal evolution, minimal residual disease, DNJ-stem, VH replacement, IGH rearrangements, high-throughput sequencing
Citation: Darzentas F, Szczepanowski M, Kotrová M, Hartmann A, Beder T, Gökbuget N, Schwartz S, Bastian L, Baldus CD, Pál K, Darzentas N and Brüggemann M (2023) Insights into IGH clonal evolution in BCP-ALL: frequency, mechanisms, associations, and diagnostic implications. Front. Immunol. 14:1125017. doi: 10.3389/fimmu.2023.1125017
Received: 28 December 2022; Accepted: 27 March 2023;
Published: 18 April 2023.
Edited by:
Christian Kosan, Friedrich Schiller University Jena, GermanyReviewed by:
Marvyn T. Koning, Leiden University Medical Center (LUMC), NetherlandsCopyright © 2023 Darzentas, Szczepanowski, Kotrová, Hartmann, Beder, Gökbuget, Schwartz, Bastian, Baldus, Pál, Darzentas and Brüggemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikos Darzentas, bmlrb3MuZGFyemVudGFzQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.