 Bo Hou
Bo Hou Ting Chen
Ting Chen He Zhang
He Zhang Jiatong Li
Jiatong Li Peter Wang
Peter Wang Guanning Shang
Guanning Shang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 17 January 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1123244
This article is part of the Research Topic Community Series in Manipulating the Immunological Tumor Microenvironment - Volume II View all 20 articles
The tumor microenvironment (TME) is the tumor surrounding environment, which is critical for tumor development and progression. TME is also involved in clinical intervention and treatment outcomes. Modulation of TME is useful for improving therapy strategies. PD-L1 protein on tumor cells interacts with PD-1 protein on T cells, contributing to T cell dysfunction and exhaustion, blockage of the immune response. Evidence has demonstrated that the expression of PD-1/PD-L1 is associated with clinical response to anti-PD-1/PD-L1 therapy in cancer patients. It is important to discuss the regulatory machinery how PD-1/PD-L1 protein is finely regulated in tumor cells. In recent years, studies have demonstrated that PD-1/PD-L1 expression was governed by various E3 ubiquitin ligases in TME, contributing to resistance of anti-PD-1/PD-L1 therapy in human cancers. In this review, we will discuss the role and molecular mechanisms of E3 ligases-mediated regulation of PD-1 and PD-L1 in TME. Moreover, we will describe how E3 ligases-involved PD-1/PD-L1 regulation alters anti-PD-1/PD-L1 efficacy. Altogether, targeting E3 ubiquitin ligases to control the PD-1/PD-L1 protein levels could be a potential strategy to potentiate immunotherapeutic effects in cancer patients.
The tumor microenvironment (TME) is the tumor surrounding environment, including fibroblasts, blood vessels, different immune cells, the extracellular matrix, etc (1, 2). TME connects tumor cells and local normal cells to make them interactions, which is critical for tumor development and progression (3, 4). Moreover, tumor cells and TME influence each other. Because the TME contains some various immune cells, TME is involved in immunotherapy (5–7). TME often exhibits immunosuppressive in cancer patients, and this situation makes tumor cells evade immunologic surveillance (8). In addition, TME is critically involved in clinical intervention and changes treatment outcomes. Modulation of TME is useful for fine-tuning of therapy strategies (9–11).
The post-translational modifications (PTMs) have various types, such as acetylation, ubiquitination, phosphorylation, methylation, SUMOylation, glycosylation, and palmitoylation (12–14). The ubiquitin proteasome system (UPS) is one of PTMs to regulate protein ubiquitination and degradation (15). In general, UPS have several critical elements, such as ubiquitin, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), ubiquitin-protein enzyme (E3), 26S proteasome, and deubiquitinating enzymes (DUBs). E1, E2 and E3 tightly regulate the protein ubiquitination and degradation (16). E3 ligases specifically target substrates and have several classifications based on their structures, such as HECT E3 ligases, RBP E3 ligases and RING E3 ligases (17).
PD-L1 protein on tumor cells interacts with PD-1 protein on T cells, contributing to T cell dysfunction and exhaustion, including the suppression of T lymphocyte proliferation, reduction of cytokine production, blockage of the immune response (18, 19). Indeed, dysfunctional T cells have high expression of PD-1 in TME. Blockade of PD-1/PD-L1 signaling invigorates active T cells and increases immunotherapy efficacy (20, 21). Several antibodies against PD-1, such as cemiplimab (Libtayo), nivolumab (Opdivo), pembrolizumab (Keytruda), and antibodies against PD-L1, including avelumab (Bavencio), durvalumab (Imfinzi), Atezolizumab (Tecentriq), have been used in clinical trials (22–24). However, in clinical set, only a portion of patients with PD-1/PD-L1 positive tumors display a good response to anti-PD-1/PD-L1 immunotherapy (25). The poor response and adaptive immune resistance can hinder the treatment efficacy. One reason is the dynamic expression of PD-1/PD-L1 in cells because PD-1/PD-L1 expression can be induced by cytokines and multiple factors and be regulated by E3 ligases in TME (26–28). In-depth evaluation is necessary to discover how E3 ligases regulate PD-1/PD-L1 expression in TME to affect immunotherapy outcomes.
Evidence has demonstrated that the expression of PD-1/PD-L1 is linked to clinical response to anti-PD-1/PD-L1 therapy in cancer patients. It is important to discover the regulatory machinery how PD-1/PD-L1 protein is finely regulated in tumor cells. In recent years, several studies have demonstrated that PD-1/PD-L1 expression was regulated by various E3 ubiquitin ligases in TME, contributing to resistance of anti-PD-1/PD-L1 therapy in human cancers (29, 30). In the following section, we will discuss the role and molecular mechanisms of E3 ligases-mediated regulation of PD-1 and PD-L1 in TME. Moreover, we will describe how E3 ligases-involved PD-1/PD-L1 regulation alters anti-PD-1/PD-L1 efficacy.
F-box protein is a subunit of SCF E3 ligase complexes and has been characterized to involve in numerous biological functions in human cancer, such as apoptosis, invasion, cell cycle, proliferation, autophagy, drug resistance, EMT, cancer stem cells and metastasis (31–34). SCF E3 ligase consists of adaptor SKP1, scaffold Cullin-1, RBX1 or RBX2 and F-box protein. So far, there are 69 F-box protein in human genome, including 37 FBXO proteins, 10 FBXW proteins, and 22 FBXL proteins (35–37). F-box proteins have been shown to regulate oncogenesis and tumor progression in numerous types of human cancers (38, 39). Liu et al. reported that FBW7-mediated PD-1 protein degradation enhanced anti-PD-1 immunotherapy in non-small cell lung cancer (NSCLC) (40). FBW7 protein is one component of the SCF E3 ubiquitin ligase, and plays a tumor suppressive role in tumorigenesis (41). FBW7 was reported to suppress M2 macrophage polarization and restrict tumor progression via regulation of c-Myc degradation in Lewis lung carcinoma cells (LLCs) (42). FBW7 was also identified as a new E3 ligase for PD-1 protein via enhancement of the K48-linked polyubiquitination of PD-1 at Lys233 site and degradation in NSCLC cells (40). CDK-1-mediated the phosphorylation of Ser261 is necessary for FBW7-involved ubiquitination of PD-1 protein. In vivo study used a natural terpenoids oridonin to activate FBW7 activation (43) and anti-mouse PD-1 monoclonal antibody to treat C57BL/6 mice with LLC cell injection. Combination treatment exhibited more profound tumor suppressive outcomes, which was accompanied with increased apoptosis of tumor cells and increased CD8+ CTLs infiltration. In human NSCLC tissues, FBW7 was negatively associated with PD-1 expression. Overexpression of FBW7 led to PD-1 destruction and in return promoted the blockade of PD-1/PD-L1 evasion pathway (40). High expression of FBW7 in the TME contributed to sensitivity of anti-PD-L1 therapy in NSCLC.
Another study revealed that inactivation of FBW7 effected double-stranded RNA (dsRNA) sensor expression and led to immunotherapy resistance (44). Melanoma patients had heterogeneous reactions to PD-1 blockade therapy. The resistant tumors displayed FBW7 mutations and sensitive tumors did not have the mutations of FBW7. Depletion of FBW7 in murine cancer cells resulted in resistance to PD-1 blockade in mice. Depletion of FBW7 altered TME, downregulated the expression of MDA5 and RIG1, two dsRNA sensors, and reduced the expression of MHC-1 and type I IFN (44). On the contrary, in FBW7-deficient cells, restoring MDA5 and RIG1 sensitized anti-PD-1 therapy. This work indicated that inactivation of FBW7 could be a key driver for anti-PD-1 resistance. Therefore, restoration of FBW7 might improve clinical therapeutic efficacy to anti-PD-1/PD-L1 treatment (44). FBXO38 mediated PD-1 poly-ubiquitination in K48-linked manner and proteasome degradation. In activated T cells, the PD-1 exhibited internalization and degradation (45). T cells with conditional knockout of FBXO38 promoted mouse tumor progression due to upregulation of PD-1 in tumor infiltrating T cells. Moreover, anti-PD-1 treatment abolished the efficacy of FBXO38 depletion on mouse tumor growth. The transcriptional levels of FBXO38 were decreased in tumor infiltrating T cells in mice and human cancer tissues (45). Furthermore, IL-2 treatment restored Fbxo38 transcription and promoted PD-l degradation and reduced PD-1 protein levels in T cells in mice (45). Hence, targeting FBXO38 could be a good choice to reduce PD-1 level and influence immunotherapy.
In addition, β-TrCP E3 ligase interacted with GSK3β and PD-L1, leading to the phosphorylation-dependent degradation of PD-L1 by β-TrCP. However, PD-L1 glycosylation sites at N192, N200 and N219 blocked the GSK3β binding. Moreover, EGF inactivated GSK3β and stabilized PD-L1 in breast cancer cells (46). Gefitinib suppressed EGF pathway and destabilized PD-L1, resulting in promotion of antitumor T-cell immunity and enhancement of treatment efficacy of anti-PD-1 therapy in mice. Hence, ubiquitination and glycosylation of PD-1 were involved in β-TrCP-mediated degradation of PD-1 and tumor immunotherapy efficacy (46). FBXO22 E3 ligase targeted PD-L1 for ubiquitination and degradation and increased sensitization of tumor cells to DNA damage (47). Inhibition of CDK5 elevated the expression of FBXO22 and subsequently inhibited PD-L1 protein levels, indicating that CDK5 was an upstream regulator of FBXO22 and that CDK5 inhibitors could increase the efficacy of immunotherapy in cancer cells (47).
FGFR3 destabilized PD-L1 via NEDD4 to govern T-cell-involved immune surveillance in bladder cancer (48). FGFR3, a tyrosine kinase, has been known to be overexpressed and activated in human cancers (48–50). FGFR3 alterations play an essential role in the immunotherapy for bladder cancer, including amplifications, fusions, and mutations (51). One study showed that suppression of FGFR3 increased the expression of PD-L1 level via modulating its ubiquitination in bladder cancer cells, contributing to blockade of anticancer activity of CD8+ T cells (48). FGFR3 had an inverse association with PD-L1 in human cancer tissues (48). NEDD4 is a HECT domain family E3 ubiquitin ligase and targets multiple substrates, including ENaC, Notch, Deltex, VEGFR2, HER3, PTEN, AMPA receptor and IGF-1R, for ubiquitination-mediated degradation, leading to regulation of cellular processes (52, 53). NEDD4 can be phosphorylated by FGFR3 and subsequently regulates PD-L1 ubiquitination in K48-linked manner. Mice with NEDD4 knockout bladder cancer displayed impaired CD8+ T cell infiltration and reduced anticancer activity because of upregulation of PD-L1. Therefore, NEDD4 E3 ligase is associated with FGFR3 targeted therapy and PD-L1 immunotherapy. Combination treatment strategy for FGFR3 and NEDD4 could be useful for bladder cancer (48).
It has been known that c-Cbl often acts as a tumor suppressor gene in oncogenesis (54). In immune cells, c-Cbl expression is highly expressed. One study showed that c-Cbl targeted PD-1 for proteasomal degradation and reduced PD-1 level as well as changed TME in colorectal tumors (55). In addition, c-Cbl+/- mice showed increased colorectal tumor growth and more infiltrating immune cells compared to c-Cbl wild-type mice. c-Cbl+/- mice displayed an elevated PD-1 levels in macrophages and CD8+ T lymphocytes (55). Moreover, the tumor phagocytosis in macrophages was reduced in c-Cbl+/- mice; however, this phenotype can be recovered by anti-PD-1 antibody treatment. Mechanistically, the cytoplasmic tail of PD-1 binds to C-terminus of c-Cbl, and causes c-Cbl-mediated degradation of PD-1. Hence, c-Cbl targets PD-1 expression level and alters TME, which could improve immunotherapy (55).
SPOP has been reported to participate in tumor development and progression via regulating its multiple substrates, including Cyclin E1, ERG, BRD4, Cdc20, TRIM24, HDAC6, Gli2, and SIRT2 (56, 57). One elegant study revealed that SPOP destructed PD-L1 protein and controlled cancer immune surveillance (58). Zhang et al. reported that PD-L1 abundance was governed by cyclin D-CDK4 and Cullin 3/SPOP. Suppression of CDK4/6 reduced phosphorylation of SPOP, and subsequently promoted APC/C Cdh1-mediated the degradation of SPOP, contributing to high expression of PD-L1 levels (58). Strikingly, loss-of-function mutations in SPOP elevated PD-L1 protein level due to dysregulation of PD-L1 degradation, conferring to reduction of TILs in human prostate cancer tissues and mouse tumor samples (58). Inhibition of CDK4/6 by inhibitors increased the anti-PD-1 immunotherapy efficacy and prolonged overall survival rates and promoted tumor regression in mice (58). Meng et al. demonstrated that Rho-associated protein kinase (ROCK) can lead to moesin phosphorylation and competing SPOP for interacting with PD-L1 (59). Blockade of ROCK by Y-27632 inhibitor or depletion of moesin reduced the expression of PD-L1, contributing to activation of T cells. Y-27632 inhibitor retarded tumor progression and promoted CD8+ and CD4+ T cell infiltration in mice via upregulation of multiple immune response genes (59).
Aldehyde dehydrogenase 2 (ALDH2) blocked SPOP-mediated degradation of PD-L1 via binding with the intracellular segment of PD-L1 (60). Silencing of ALDH2 decreased PD-L1 protein and enhanced TILs infiltration in colorectal cancer cells. Suppression of ALDH2 also caused promotion of anti-PD-1 therapeutic efficacy in colorectal cancer mouse model, indicating that ALDH2 facilitates tumor progression and enhanced immune escape via regulation of SPOP-mediated degradation of PD-L1 (60). Casein kinase 2 (CK2) was reported to phosphorylate PD-L1 at Thr285 and Thr290 and subsequently stabilize the PD-L1 in tumor and dendritic cells. The interaction between PD-L1 and SPOP was blocked by PD-L1 phosphorylation, contributing to protection of PD-L1 degradation. Suppression of CK2 reduced PD-L1 accumulation and increased CD80 release from dendritic cells to reactivate functions of T cells (61).
ATR inhibitor destabilized PD-L1 protein due to activation of CDK1/SPOP axis. ATR inhibitors plus anti-PD-L1 treatment led to increased innate immune activation in mice (62). Sorting nexin 6 (SNX6) can bind with Cullin 3, leading to reduction of interaction between SPOP and Cullin 3, which reduces the PD-L1 degradation. Consistently, depletion of SNX6 reduced PD-L1 protein in cancer cells (63). One research revealed that c-Myb enhanced tumor immune escape via targeting miR-145-5p/SPOP/PD-L1 pathway in esophageal adenocarcinoma cells (64). Specifically, c-Myb increased miR-145-5p expression and in turn reduced SPOP and regulated PD-L1, leading to suppression of T cell functions and induction of immune escape in esophageal adenocarcinoma cells (64). Cancer stem cell-derived exosomal miR-17-5p reduced SPOP expression and increased PD-L1 accumulation, leading to suppression of antitumor immunity in colorectal cancer cells (65). Recently, SPOP was found to increase the movement of PD-1 away from PD-L1 in spatial localization, and enhanced tumor metastasis in cervical cancer (66). In ovarian cancer cells, Cullin 3/SPOP facilitated sensitivity of chemotherapy and blocked immune escape via promoting PD-L1 protein degradation (67). Recently, SPOP mutations were revealed to enhance tumor immune escape through targeting the interferin regulatory factor 1 (IRF1)-PD-L1 axis in endometrial cancer (68). Taken together, SPOP E3 ligase targets PD-L1 degradation and involves in anti-PD-1 immunotherapy in human cancers.
Inflammation-related molecule A20 (also known as TNFAIP3) plays an essential role in antitumor immunity and inflammation via negative regulation of NF-κB pathway (69). Melanoma patients with upregulation of A20 displayed poor treatment effect to anti-PD-1 therapy and reduced CD8+ T cell activity. Modulation of A20 regulated PD-L1 expression and invigorated CD8+ T cell infiltration, leading to enhancement of immunotherapy (70). A20, acting as an E3 ubiquitin, ligase, activated STAT3 ad PD-L1 expression due to promotion of prohibitin ubiquitination and degradation (70). CDK5 inhibited the PPARγ E3 ligase activity and protected ESRP1 from degradation. CDK5 triggered Ser 273 phosphorylation of PPARγ and switched CD44 isoform from CD44s to CD44v, leading to promoting TNBC CSCs development (71). Inactivation of CDK5 and phosphorylation of PPARγ suppressed the numbers of CD44v+ breast CSCs in tissues, which inhibited tumor metastasis in TNBC mice. Blockade of stemness transformation facilitated anti-PD-1 treatment outcomes via reversing tumor immunosuppressive microenvironment in TNBC (71). KLHL22 is an adaptor of Cullin 3 E3 ligase and mediated the ubiquitination and degradation of PD-1. Therefore, deficiency of KLHL22 resulted in PD-1 upregulation, conferring to suppression of T cells-mediated antitumor response and facilitated tumor development (72). In clinical colorectal cancer patients, there was a downregulation of KLHL22 in tumor infiltrating T cells. 5-fluorouracil (5-FU) could suppress the KLHL22 transcription and elevate the expression of PD-1 (72). Hence, KLHL22 governed excessive T cell suppression via regulation of PD-1 expression in colorectal cancer. STUB1 was reported to act as an E3 ligase and lead to destabilization of PD-L1. A type-3 transmembrane protein CMTM6 could maintain PD-L1 via blocking ubiquitination in tumor cells (73). CMTM4 displayed the similar function in regulation of PD-L1 protein levels. CMTM6 accelerated the ability of tumor cells with PD-L1 expression to repress T cells (73). TMUB1 was identified as a modulator of PD-L1 PTMs, which competed with HUWE1 to bind with PD-L1 and suppressed its ubiquitination at K281 in the ER. TMUB1 recruited STT3A and accelerated PD-L1 N-glycosylation and stability, resulting in enhancement of PD-L1 maturation and contribution of immune evasion. A peptide that competed with TMUB1 elevated anticancer immunity and retarded tumor growth in mice (74).
DUBs can cleave and remove ubiquitins from molecules, which is classified into two groups: metalloproteases and cysteine proteases. DUBs stabilize the protein levels of PD-1 and PD-L1 in cancer cells. For instance, COP9 signalosome 5 (CSN5) maintained PD-L1 protein accumulation via inhibition of ubiquitination and degradation of PD-L1 (75). TNF-α induced PD-L1 stabilization due to p65-mediated induction of CSN5 in cancer cells. Suppression of CSN5 by natural agent curcumin sensitized tumor cells to anti-CTLA4 blockade because of diminishing PD-L1 expression (75). The deubiquitinase USP22 can bind with PD-L1 and enhance its stability, leading to reduced T cell cytotoxicity in tumor cells (76). Similarly, another group also identified that USP22 can target PD-L1 and resulted in suppression of antitumor immunity (77). USP7 depletion led to downregulation of PD-L1 and caused sensitization of T cells killing in gastric tumor cells (78).
USP8 depletion enhanced immunotherapy by regulation of TME via targeting PD-L1 ubiquitination and activating the infiltrated CD8+ T cells (79). In line with this report, USP8 deubiquitinated PD-L1 and upregulated its expression in pancreatic cancer. Anti-PD-L1 in combination with a USP8 inhibitor attenuated tumor growth via activation of cytotoxic T cells (80). OTUB1 blocked ER-associated degradation of PD-L1 and triggered immunosuppression in tumor cells (81). Interestingly, depletion of USP12 established a tumor-promoting TME due to insufficient deubiquitination of PPM1B and activation of NF-κB in cancer cells, which contributed to desensitization of anti-PD-1 immunotherapy in lung cancer cells (82). In addition, USP14 stabilized indoleamine 2,3 dioxygenase 1 (IDO1) and enhanced immune suppression in colorectal cancer (83). Depletion of USP14 promoted anti-PD-1 responsiveness in mice and reversed inhibition of cytotoxic T cells due to inhibition of IDOI (83).
Evidence has revealed that several compounds regulate the expression of E3 ubiquitin ligases and modulate the expression of PD-1/PD-L1 and change the immunotherapy efficacy in human cancers. For example, 2,5-dimethylcelecoxib (DMC) induced hepatitis B virus X (HBx)-mediated PD-L1 ubiquitination and improved TIME in HCC (84). DMC, an inhibitor of mPGES-1, has been reported to repress HBV-involved HCC progression. DMC elevated the CD8+ T cell infiltrations in HCC mouse model, and mouse tumor tissues displayed the downregulation of PD-L1 and CD163. The combination of DMC and atezolizumab exhibited more significant anticancer efficacy. DMC enhanced RBX1 E3 ligase-mediated PD-1 degradation via activation of AMPK pathway in HCC cells (84). Avadomide is cereblon E3 ligase modulator and upregulates the expression of PD-L1 in CLL cells (85). Avadomide activated interferon (IFN) signaling in T cells and reactivated T cell responses and promoted chemokine expression. PDX mice displayed CD8+ T cell-inflamed TME after avadomide treatment and increased the sensitivity of anti-PD-1/PD-L1 therapy (85). Hence, avadomide could enhance sensitivity of immunotherapy in CLL cells via targeting IFN signaling pathway.
APG-115, an inhibitor of MDM2 E3 ligase, synergized with anti-PD-1 therapy via promoting anticancer immunity in the TME (86). It has been shown that p53 activation inhibited M2 macrophage polarization in myeloid. APG-115 treatment increased activation of p53 and p1 in bone marrow-derived macrophages and decreased c-Myc and c-Maf and caused a reduction number of immunosuppressive M2 macrophage (86). Mice with APG-115 treatment elevated M1 macrophage polarization in the spleen. Moreover, APG-115 in combination with anti-PD-1 therapy contributed to enhanced tumor suppressive activity in mice (86). MET inhibitors, capmatinib and tivantinib, enhanced tumor evasion of the immune response by stabilization of PD-L1 in HCC (87). MET inhibitors elevated PD-L1 expression and blocked the antitumor ability of T cells. Mechanistically, suppression of MET blocked GSK3β phosphorylation and promoted the interaction between TRAF6 and GSK3β, leading to inactivation of GSK3β because of TRAF6-induced GSK3β K63 ubiquitination, which facilitated the PD-L1 expression in HCC cells (87). Metformin attenuated the stability and membrane localization of PD-L1 and elevated the activity of CTL. Metformin activated AMPK and phosphorylated PD-L1 at S195 site, resulting in abnormal PD-L1 glycosylation and its ER accumulation and ERAD (88).
PROTACs is a new technology for regulating a protein of interest (POI) by degradation by specific E3 ligases (89, 90). Numerous of E3 ligases, including cereblon, MDM2, β-TrCP, and VHL, have been applied in PROTAC strategy (91). PROTAC is a ternary complex that links a POI ligand to an E3 ligase via an optimal linker (92, 93). PROTACs have been reported to target several important signaling pathways in TME and improve antitumor therapy (94). Peptide-based PROTAC of FOXM1 inhibited the expression of PD-L1 and glucose transporter GLUT1 and attenuated carcinogenesis (95). FOXM1-PROTAC mediated degradation of FOXM1 protein in tumor cells and suppressed viability, migration and invasion in several types of tumor cells. In HepG2 and MDA-MB-231 tumor cell xenograft mice, FOXM1-PROTAC retarded tumor growth (95). Moreover, FOXM1-PROTAC reduced the expression of PD-L1, indicating that PROTACs might be used for targeting PD-L1 degradation to improve the immunotherapy in human cancer.
One group developed a new PROTAC molecule 21a that enhanced PD-L1 protein degradation in several types of tumor cells, suggesting that compound 21a might be an alternative way for immunotherapy in cancer patients (96). Another group designed a new resorcinol diphenyl ether-based PROTAC, compound P22, that impaired the interaction between PD-1 and PD-L1 and reactivated the immunity. P22 compound decreased the protein levels of PD-L1 via lysosome-mediated degradation and affected immune functions (97). Degradation of BET protein by the PROTAC technology induced death receptor 5 (DR5)-involved immunogenic cell death (ICD) in colorectal cancer cells, leading to colorectal cancer progression and enhancement of anti-PD-1 antibody blockade (98). Targeting SHP2, a protein tyrosine phosphatase, by PROTACs is useful for cancer immunotherapy partly via regulation of several signaling pathways, such as PD-1/PD-L1, PI3K/AKT, RAS/ERK, JAK/STAT pathways (99). Moreover, nano-PROTACs targeting SHP2 inactivated the CD47/SIRPα and PD-1/PD-L1 pathways, contributing to reinvigoration of T cells and macrophages as well as promotion of antitumor immune response (100). Recently, peptide-PROTACs targeting PD-1/PD-L1 degradation were designed and exhibited high potential activity to degrade PD-1/PD-L1 in tumor cells, which caused tumor cell death (101). PROTACs targeting hematopoietic progenitor kinase1 (HPK1) regulated T cell function and potentiated the efficacy of CAR-T cell-based immunotherapies (102). Cotton et al. developed antibody-based PROTACs (AbTACs) to disrupt PD-L1 protein stability. AbTACs recruited RNF43 E3 ligase and induced the lysosomal degradation of PD-L1 (103). These reports decipher that PROTACs are novel compounds for targeting PD-1/PD-L1 or other critical factors in immunosuppressive pathways to improve the efficacy of immunotherapy.
In conclusion, the E3 ligases govern the ubiquitination and degradation of PD-1/PD-L1 in the TME (Table 1). Targeting E3 ligases might be a potential strategy for promoting antitumor immunity in human cancer (Figure 1). Several Strategies have been proposed for developing PD-1 inhibitors, including PROTACs (104). It is important to note several issues for readers to fully understand the role of E3 ubiquitin ligases to regulate TME and PD-1/PD-L1 in immunotherapy. Some E3 ubiquitin ligases regulated specific substrates, not PD-1/PD-L1 proteins, to influence TME in tumor cells. FBXL8 downregulation increased accumulation of CCND2 and IRF5 and reduced the cancer-promoting chemokines, modulated TME, leading to repressing tumor metastasis in breast cancer (105). COP1 E3 ligase knockdown reduced chemokine secretion and macrophage infiltration, increased immune checkpoint blockade efficacy, promoted tumor suppressive immunity in the TME. COP1 acted as the E3 ligase to induce polyubiquitination and degradation of the C/ebpδ, resulting in activation of macrophage chemoattractant genes (106).
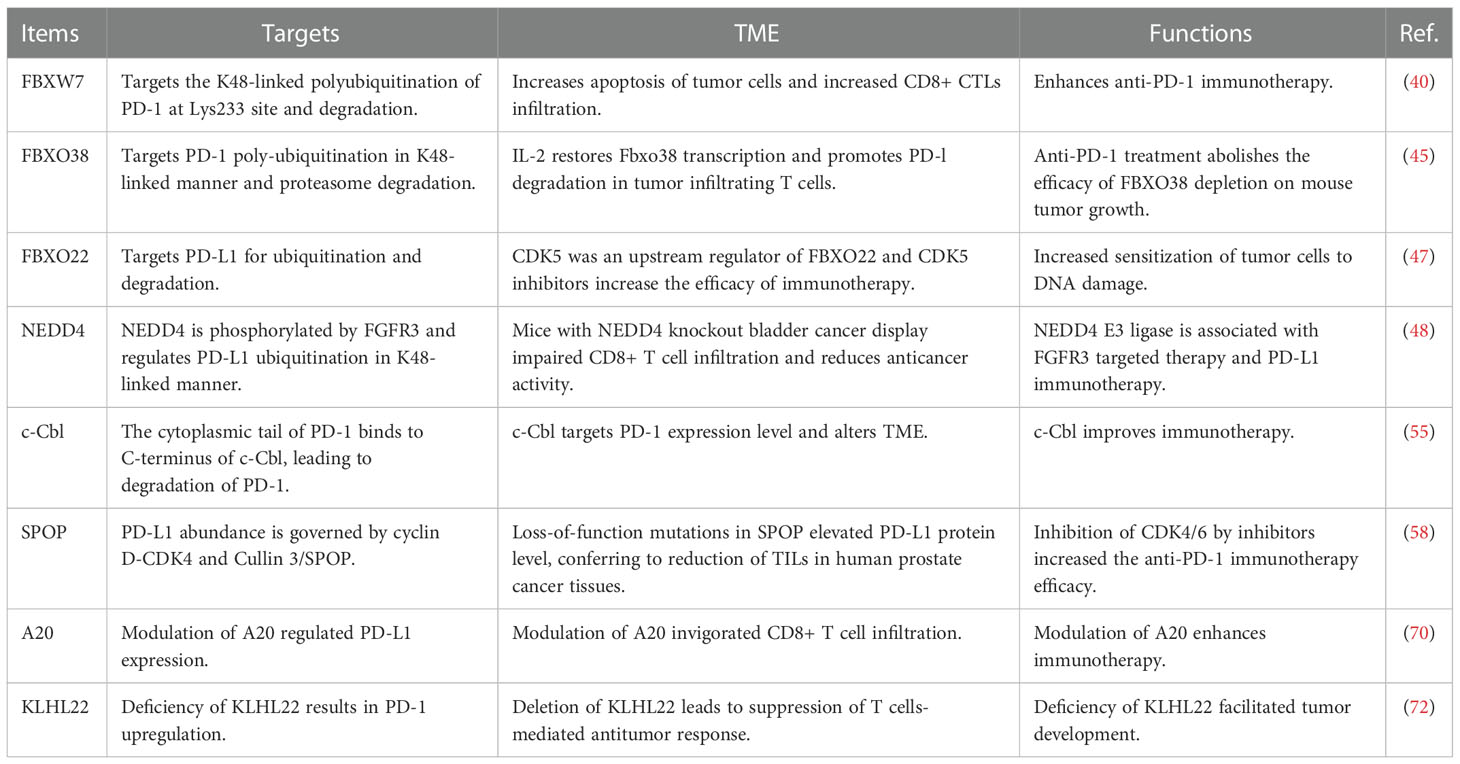
Table 1 The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels.
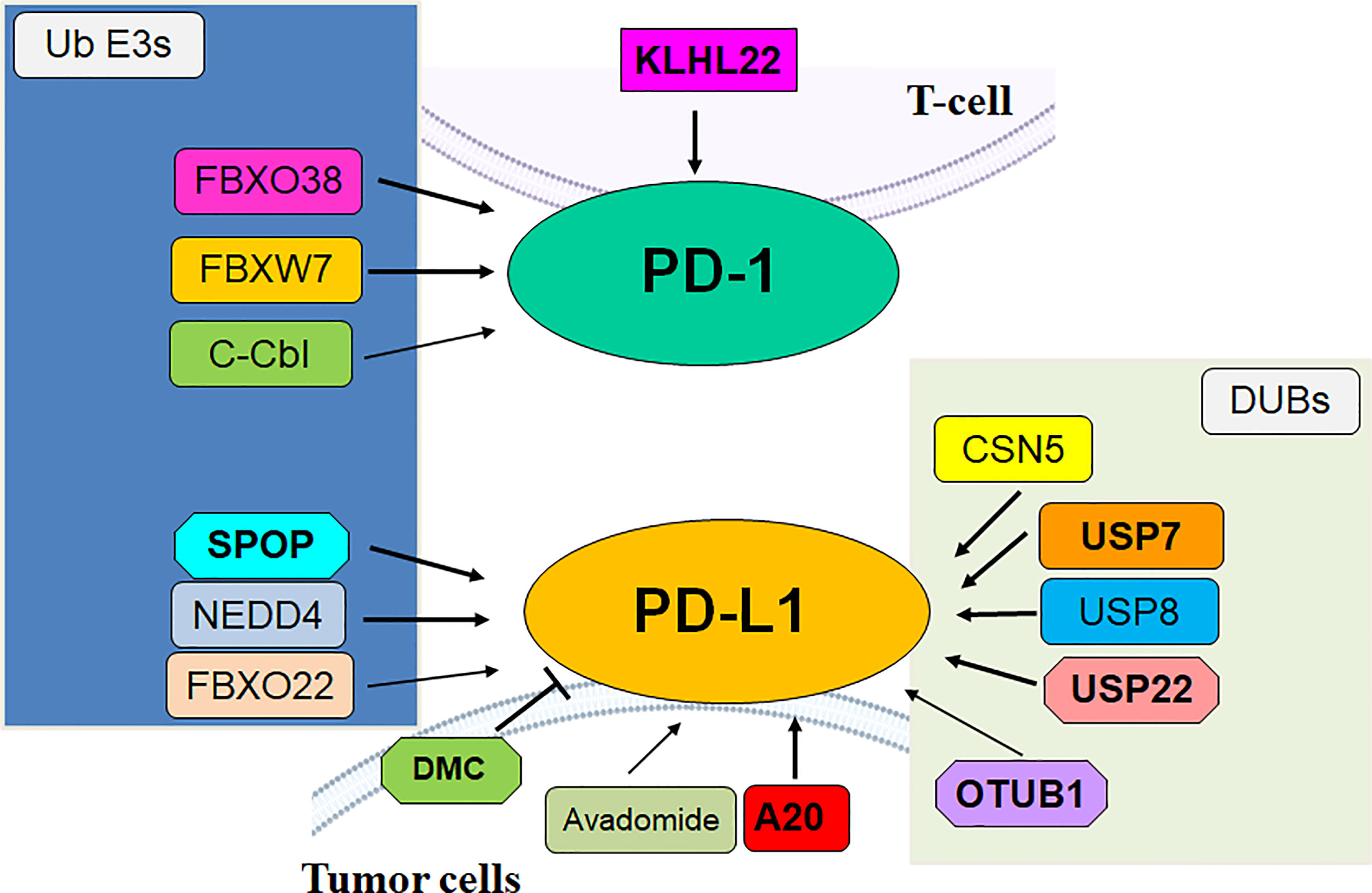
Figure 1 The role of E3 ubiquitin ligases and DUBs in regulation of PD-1/PD-L1 in cancer.
Growth differentiation factor 15 (GDF15) facilitated tumor immunosuppression via interaction with CD48 on Treg cells and downregulation of E3 ligase STUB1, leading to accumulation of FOXP3 protein in liver cancer (107). UBR5 E3 ligase augmented immunosuppressive macrophage and modulated the TME and facilitated tumor growth and metastasis in ovarian cancer via maintaining β-catenin signaling (108). NEDD4 E3 ligase suppressed T-cell-induced antitumor immunity via targeting the immune checkpoint GITR for degradation in melanoma (109). Depletion of RNF43 E3 ligase remodeled the TIME and facilitated Kras-induced oncogenesis in pancreatic cancer (110). TRAF6 E3 ligase reshaped TME by increasing the immunosuppressive functions of MDSCs via targeting K63-linked ubiquitination and phosphorylation of STAT3 (111). Knockdown of E3 ubiquitin ligase Cbl-b increased CAR T-cell effects and blocked CD8+ T-cell exhaustion, upregulated the expression of IFN-γ and TNFα, and accelerated tumor cell killing (112). Calponin 1 can bind with PDLIM7 and protect its disruption by the NEDD4-1, resulting in activation of ROCK1/MLC pathway. Calponin 1 elevated matrix stiffness in CAF and enhance 5-Fu chemoresistance by activation of YAP in gastric cancer (113).
Senescent stromal cells develop epiregulin (EREG), a member of the EGF family. EREG, a ligand of EGFR, can regulate EGFR-induced oncogenesis. EREG inhibits cellular sensitivity to TKIs treatment (114). High expression of EREG was associated with tumor stage, metastasis and survival in human cancer patients. DNA-damaging agents (DDAs), such as bleomycin, mitoxantrone, and doxorubicin, induced the expression of EREG in stromal cells (115). Stromal EREG levels were linked to adverse clinical outcomes. Moreover, EREG upregulation was due to activation of NF-κB signaling pathway. Furthermore, stromal EREG changed recipient tumor cell phenotypes in prostate cancer cells (115). MARCHF4 (membrane associated ring-CH-type finger 4), a member of E3 ubiquitin protein ligase, promoted viability of prostate cancer cells after mitoxantrone treatment. Overexpression of MARCHF4 induced EMT in prostate cancer cells. MARCHF4 overexpression caused chemotherapeutic agent resistance in prostate cancer cells. Targeting EREG in the damaged TME enhanced treatment efficacy in mice (115). Stromal cell-derived EREG-mediated drug resistance was partly due to MARCHF4 upregulation in recipient tumor cells (115).
HDM201, an inhibitor of MDM2 E3 ligase, has been reported to increase the numbers of dendritic cells in mice (116). Moreover, HDM201 elevated the CD8+/Treg ration and promoted the numbers of Tbet+Eomes+CD8+ T cells in tumor, and this phenotype alteration was abrogated by p53 depletion in tumor cells. Anti-PD-1/PD-L1 therapy in combination with HDM201 enhanced tumor regressions (116). Notably, the function of HDM201 in tumorigenesis is dependent on induction of antitumor immunity. Suppression of MDM2 by its inhibitors stimulated adaptive immunity, which can be promoted by inactivation of PD-1/PD-L1 pathway in cancer patients with p53 wild-type tumors (116). Therefore, inhibition of MDM2 E3 ligase increased anti-PD-1/PD-L1 therapeutic efficacy via regulation of immune and stromal microenvironment in p53 wild-type cancer patients. Trim35 E3 ligase influenced the TIME and reduced the DLBCL progression via targeting a regulator of circadian rhythmicity CLOCK for degradation and modulating NK cell infiltration (117). Galectin-9 augmented an immunosuppression in TME via accelerating TRIM29-mediated degradation of STING in human cancers (118). FBW7-induced degradation of ZEB2 was reported to associate with EMT and TME, resulting in enhancement of colorectal CSCs and drug resistance (119). Depletion of stromal hedgehog signaling Smoothened promoted proliferation of pancreatic cancer via initiating RNF5-induced degradation of PTEN and subsequent activation of AKT (120). In addition, UBR5 E3 ligase accelerated tumor growth and metastasis via regulation of apoptosis, necrosis, EMT and angiogenesis in TNBC (121). Besides, ubiquitination of PD-1/PD-L1, its phosphorylation, glycosylation, palmitoylation, and acetylation have been reported (28, 122). Most studies focused on PD-1 and PD-L1 regulations, whereas PD-L2 regulation by E3 ligases was largely unclear. It is pivotal to determine the PTM regulatory mechanism of PD-L2 in cancer immunotherapy. Taken together, targeting E3 ubiquitin ligases to modulate the PD-1/PD-L1 protein levels might be a promising approach to improve immunotherapeutic effects in cancer patients.
BH and TC wrote the manuscript. HZ and JL made the table and figures. PW and GS edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AML, acute myeloid leukemia; AMPK, adenosine 5’-monophosphate-activated protein kinase; BET, Bromodomain and extra-terminal domain; β-TrCP, beta-transducin repeats-containing protein; CAFs, cancer-associated fibroblasts; CAR, Chimeric antigen receptor; c-Cbl, casitas B lymphoma; CSC, cancer stem cell; CDK, cyclin-dependent kinase; CLL, chronic lymphocytic leukemia; CTL, cytotoxic T lymphocytes; DLBCL, diffuse large B-cell lymphoma; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; GSK3β, glycogen synthase kinase 3β; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HECT, homologous to E6AP C terminus; FBW7, F-box and WD repeat domain containing 7; FGFR3, fibroblast growth factor receptor 3; MDSCs, Myeloid-derived suppressor cells; mPGES-1, microsomal prostaglandin E synthase-1; NEDD4, neuronally expressed developmentally downregulated 4; NSCLC, non-small cell lung cancer; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; PROTACs, proteolysis targeting chimeras; PPARγ, peroxisome proliferator-activated receptor gamma; RING, really interesting new gene; RBP, RING-in-between-RING; SCF, Skp1-Cullin1-F-box protein; SHP2, Src homology 2 domain-containing phosphatase 2; STAT3, signal transducer and activator of transcription 3; TIME, tumor immune microenvironment; TME, tumor microenvironment; TNBC, triple-negative breast cancer; TILs, tumor-infiltrating lymphocytes; TRAF6, Tumor necrosis factor receptor-associated factor 6.
1. Joyce JA, Fearon DT. T Cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348:74–80. doi: 10.1126/science.aaa6204
2. Pantel K, Alix-Panabieres C. Tumour microenvironment: informing on minimal residual disease in solid tumours. Nat Rev Clin Oncol (2017) 14:325–6. doi: 10.1038/nrclinonc.2017.53
3. Kao KC, Vilbois S, Tsai CH, Ho PC. Metabolic communication in the tumour-immune microenvironment. Nat Cell Biol (2022) 24:1574–83. doi: 10.1038/s41556-022-01002-x
4. Grisaru-Tal S, Rothenberg ME, Munitz A. Eosinophil-lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat Immunol (2022) 23:1309–16. doi: 10.1038/s41590-022-01291-2
5. Petroni G, Buque A, Coussens LM, Galluzzi L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat Rev Drug Discovery (2022) 21:440–62. doi: 10.1038/s41573-022-00415-5
6. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi: 10.1038/ni.2703
7. Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell (2020) 78:1019–33. doi: 10.1016/j.molcel.2020.05.034
8. Labrie M, Brugge JS, Mills GB, Zervantonakis IK. Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat Rev Cancer (2022) 22:323–39. doi: 10.1038/s41568-022-00454-5
9. Liu J, Chen T, Li S, Liu W, Wang P, Shang G. Targeting matrix metalloproteinases by E3 ubiquitin ligases as a way to regulate the tumor microenvironment for cancer therapy. Semin Cancer Biol (2022) 86:259–68. doi: 10.1016/j.semcancer.2022.06.004
10. Zhang W, Li S, Li C, Li T, Huang Y. Remodeling tumor microenvironment with natural products to overcome drug resistance. Front Immunol (2022) 13:1051998. doi: 10.3389/fimmu.2022.1051998
11. Kirchhammer N, Trefny MP, Auf der Maur P, Laubli H, Zippelius A. Combination cancer immunotherapies: Emerging treatment strategies adapted to the tumor microenvironment. Sci Transl Med (2022) 14:eabo3605. doi: 10.1126/scitranslmed.abo3605
12. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
13. Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol (2018) 19:59–70. doi: 10.1038/nrm.2017.83
14. Mowen KA, David M. Unconventional post-translational modifications in immunological signaling. Nat Immunol (2014) 15:512–20. doi: 10.1038/ni.2873
15. Cheng J, Guo J, Wang Z, North BJ, Tao K, Dai X, et al. Functional analysis of cullin 3 E3 ligases in tumorigenesis. Biochim Biophys Acta Rev Cancer (2018) 1869:11–28. doi: 10.1016/j.bbcan.2017.11.001
16. Wang W, Liu W, Chen Q, Yuan Y, Wang P. Targeting CSC-related transcription factors by E3 ubiquitin ligases for cancer therapy. Semin Cancer Biol (2022) 87:84–97. doi: 10.1016/j.semcancer.2022.11.002
17. Wang P, Dai X, Jiang W, Li Y, Wei W. RBR E3 ubiquitin ligases in tumorigenesis. Semin Cancer Biol (2020) 67:131–44. doi: 10.1016/j.semcancer.2020.05.002
18. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol (2021) 18:345–62. doi: 10.1038/s41571-021-00473-5
19. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8:239–45. doi: 10.1038/ni1443
20. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14:1212–8. doi: 10.1038/ni.2762
21. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol (2022) 19:775–90. doi: 10.1038/s41571-022-00689-z
22. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12030738
23. Xiang Z, Li J, Zhang Z, Cen C, Chen W, Jiang B, et al. Comprehensive evaluation of anti-PD-1, anti-PD-L1, anti-CTLA-4 and their combined immunotherapy in clinical trials: A systematic review and meta-analysis. Front Pharmacol (2022) 13:883655. doi: 10.3389/fphar.2022.883655
24. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer (2022) 21:28. doi: 10.1186/s12943-021-01489-2
25. Perez-Ruiz E, Melero I, Kopecka J, Sarmento-Ribeiro AB, Garcia-Aranda M, De Las Rivas J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist Updat (2020) 53:100718. doi: 10.1016/j.drup.2020.100718
26. Zhou X, Sun SC. Targeting ubiquitin signaling for cancer immunotherapy. Signal Transduct Target Ther (2021) 6:16. doi: 10.1038/s41392-020-00421-2
27. Ye P, Chi X, Cha JH, Luo S, Yang G, Yan X, et al. Potential of E3 ubiquitin ligases in cancer immunity: Opportunities and challenges. Cells (2021) 10. doi: 10.3390/cells10123309
28. Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res (2018) 78:6349–53. doi: 10.1158/0008-5472.CAN-18-1892
29. Dai X, Gao Y, Wei W. Post-translational regulations of PD-L1 and PD-1: Mechanisms and opportunities for combined immunotherapy. Semin Cancer Biol (2022) 85:246–52. doi: 10.1016/j.semcancer.2021.04.002
30. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol (2022) 19:287–305. doi: 10.1038/s41571-022-00601-9
31. Yan L, Lin M, Pan S, Assaraf YG, Wang ZW, Zhu X. Emerging roles of f-box proteins in cancer drug resistance. Drug Resist Updat (2020) 49:100673. doi: 10.1016/j.drup.2019.100673
32. Wang Q, Wu L, Cao R, Gao J, Chai D, Qin Y, et al. Fbxo45 promotes the malignant development of esophageal squamous cell carcinoma by targeting GGNBP2 for ubiquitination and degradation. Oncogene (2022) 41:4795–807. doi: 10.1038/s41388-022-02468-7
33. Lin M, Zhang J, Bouamar H, Wang Z, Sun LZ, Zhu X. Fbxo22 promotes cervical cancer progression via targeting p57(Kip2) for ubiquitination and degradation. Cell Death Dis (2022) 13:805. doi: 10.1038/s41419-022-05248-z
34. Wu L, Yu K, Chen K, Zhu X, Yang Z, Wang Q, et al. Fbxo45 facilitates pancreatic carcinoma progression by targeting USP49 for ubiquitination and degradation. Cell Death Dis (2022) 13:231. doi: 10.1038/s41419-022-04675-2
35. Song Y, Lin M, Liu Y, Wang ZW, Zhu X. Emerging role of f-box proteins in the regulation of epithelial-mesenchymal transition and stem cells in human cancers. Stem Cell Res Ther (2019) 10:124. doi: 10.1186/s13287-019-1222-0
36. Lin M, Xu Y, Gao Y, Pan C, Zhu X, Wang ZW. Regulation of f-box proteins by noncoding RNAs in human cancers. Cancer Lett (2019) 466:61–70. doi: 10.1016/j.canlet.2019.09.008
37. Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by f-box proteins. Nat Rev Mol Cell Biol (2013) 14:369–81. doi: 10.1038/nrm3582
38. Cao T, Cui Y, Wang Y, Wu L, Yu K, Chen K, et al. CACNA1C-AS2 inhibits cell proliferation and suppresses cell migration and invasion via targeting FBXO45 and PI3K/AKT/mTOR pathways in glioma. Apoptosis (2022) 27:979–91. doi: 10.1007/s10495-022-01764-7
39. Wang Z, Dai X, Zhong J, Inuzuka H, Wan L, Li X, et al. SCF(beta-TRCP) promotes cell growth by targeting PR-Set7/Set8 for degradation. Nat Commun (2015) 6:10185. doi: 10.1038/ncomms10185
40. Liu J, Wei L, Hu N, Wang D, Ni J, Zhang S, et al. FBW7-mediated ubiquitination and destruction of PD-1 protein primes sensitivity to anti-PD-1 immunotherapy in non-small cell lung cancer. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-005116
41. Wang Z, Liu P, Inuzuka H, Wei W. Roles of f-box proteins in cancer. Nat Rev Cancer (2014) 14:233–47. doi: 10.1038/nrc3700
42. Zhong L, Zhang Y, Li M, Song Y, Liu D, Yang X, et al. E3 ligase FBXW7 restricts M2-like tumor-associated macrophage polarization by targeting c-myc. Aging (Albany NY) (2020) 12:24394–423. doi: 10.18632/aging.202293
43. Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, et al. Triggering Fbw7-mediated proteasomal degradation of c-myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther (2012) 11:1155–65. doi: 10.1158/1535-7163.MCT-12-0066
44. Gstalder C, Liu D, Miao D, Lutterbach B, DeVine AL, Lin C, et al. Inactivation of Fbxw7 impairs dsRNA sensing and confers resistance to PD-1 blockade. Cancer Discovery (2020) 10:1296–311. doi: 10.1158/2159-8290.CD-19-1416
45. Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature (2018) 564:130–5. doi: 10.1038/s41586-018-0756-0
46. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
47. De S, Holvey-Bates EG, Mahen K, Willard B, Stark GR. The ubiquitin E3 ligase FBXO22 degrades PD-L1 and sensitizes cancer cells to DNA damage. Proc Natl Acad Sci U S A (2021) 118(47):e2112674118. doi: 10.1073/pnas.2112674118
48. Jing W, Wang G, Cui Z, Xiong G, Jiang X, Li Y, et al. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-Mediated bladder cancer immune surveillance. Cancer Res (2022) 82:114–29. doi: 10.1158/0008-5472.CAN-21-2362
49. Gott H, Uhl E. FGFR3-TACCs3 fusions and their clinical relevance in human glioblastoma. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23158675
50. Costa R, Carneiro BA, Taxter T, Tavora FA, Kalyan A, Pai SA, et al. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget (2016) 7:55924–38. doi: 10.18632/oncotarget.10482
51. Kacew A, Sweis RF. FGFR3 alterations in the era of immunotherapy for urothelial bladder cancer. Front Immunol (2020) 11:575258. doi: 10.3389/fimmu.2020.575258
52. Wang ZW, Hu X, Ye M, Lin M, Chu M, Shen X. NEDD4 E3 ligase: Functions and mechanism in human cancer. Semin Cancer Biol (2020) 67:92–101. doi: 10.1016/j.semcancer.2020.03.006
53. Ye X, Wang L, Shang B, Wang Z, Wei W. NEDD4: a promising target for cancer therapy. Curr Cancer Drug Targets (2014) 14:549–56. doi: 10.2174/1568009614666140725092430
54. Lyle CL, Belghasem M, Chitalia VC. C-cbl: An important regulator and a target in angiogenesis and tumorigenesis. Cells (2019) 8. doi: 10.3390/cells8050498
55. Lyle C, Richards S, Yasuda K, Napoleon MA, Walker J, Arinze N, et al. C-cbl targets PD-1 in immune cells for proteasomal degradation and modulates colorectal tumor growth. Sci Rep (2019) 9:20257. doi: 10.1038/s41598-019-56208-1
56. Wang Z, Song Y, Ye M, Dai X, Zhu X, Wei W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat Rev Urol (2020) 17:339–50. doi: 10.1038/s41585-020-0314-z
57. Song Y, Xu Y, Pan C, Yan L, Wang ZW, Zhu X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer (2020) 19:2. doi: 10.1186/s12943-019-1124-x
58. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin d-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature (2018) 553:91–5. doi: 10.1038/nature25015
59. Meng F, Su Y, Xu B. Rho-associated protein kinase-dependent moesin phosphorylation is required for PD-L1 stabilization in breast cancer. Mol Oncol (2020) 14:2701–12. doi: 10.1002/1878-0261.12804
60. Zhang H, Xia Y, Wang F, Luo M, Yang K, Liang S, et al. Aldehyde dehydrogenase 2 mediates alcohol-induced colorectal cancer immune escape through stabilizing PD-L1 expression. Adv Sci (Weinh) (2021) 8:2003404. doi: 10.1002/advs.202003404
61. Zhao X, Wei Y, Chu YY, Li Y, Hsu JM, Jiang Z, et al. Phosphorylation and stabilization of PD-L1 by CK2 suppresses dendritic cell function. Cancer Res (2022) 82:2185–95. doi: 10.1158/0008-5472.CAN-21-2300
62. Tang Z, Pilie PG, Geng C, Manyam GC, Yang G, Park S, et al. ATR inhibition induces CDK1-SPOP signaling and enhances anti-PD-L1 cytotoxicity in prostate cancer. Clin Cancer Res (2021) 27:4898–909. doi: 10.1158/1078-0432.CCR-21-1010
63. Ghosh C, Xing Y, Li S, Hoyle RG, Sun M, Li J, et al. Sorting nexin 6 interacts with Cullin3 and regulates programmed death ligand 1 expression. FEBS Lett (2021) 595:2558–69. doi: 10.1002/1873-3468.14191
64. Zhang L, Wang X, Li Y, Han J, Gao X, Li S, et al. C-myb facilitates immune escape of esophageal adenocarcinoma cells through the miR-145-5p/SPOP/PD-L1 axis. Clin Transl Med (2021) 11:e464. doi: 10.1002/ctm2.464
65. Sun W, Cui J, Ge Y, Wang J, Yu Y, Han B, et al. Tumor stem cell-derived exosomal microRNA-17-5p inhibits anti-tumor immunity in colorectal cancer via targeting SPOP and overexpressing PD-L1. Cell Death Discovery (2022) 8:223. doi: 10.1038/s41420-022-00919-4
66. Wu J, Wu Y, Guo Q, Chen S, Wang S, Wu X, et al. SPOP promotes cervical cancer progression by inducing the movement of PD-1 away from PD-L1 in spatial localization. J Transl Med (2022) 20:384. doi: 10.1186/s12967-022-03574-6
67. Dong M, Qian M, Ruan Z. CUL3/SPOP complex prevents immune escape and enhances chemotherapy sensitivity of ovarian cancer cells through degradation of PD-L1 protein. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-005270
68. Gao K, Shi Q, Gu Y, Yang W, He Y, Lv Z, et al. SPOP mutations promote tumor immune escape in endometrial cancer via the IRF1-PD-L1 axis. Cell Death Differ (2022). doi: 10.1038/s41418-022-01097-7
69. Momtazi G, Lambrecht BN, Naranjo JR, Schock BC. Regulators of A20 (TNFAIP3): new drug-able targets in inflammation. Am J Physiol Lung Cell Mol Physiol (2019) 316:L456–69. doi: 10.1152/ajplung.00335.2018
70. Guo W, Ma J, Guo S, Wang H, Wang S, Shi Q, et al. A20 regulates the therapeutic effect of anti-PD-1 immunotherapy in melanoma. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2020-001866
71. Bei Y, Cheng N, Chen T, Shu Y, Yang Y, Yang N, et al. CDK5 inhibition abrogates TNBC stem-cell property and enhances anti-PD-1 therapy. Adv Sci (Weinh) (2020) 7:2001417. doi: 10.1002/advs.202001417
72. Zhou XA, Zhou J, Zhao L, Yu G, Zhan J, Shi C, et al. KLHL22 maintains PD-1 homeostasis and prevents excessive T cell suppression. Proc Natl Acad Sci U S A (2020) 117:28239–50. doi: 10.1073/pnas.2004570117
73. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature (2017) 549:106–10. doi: 10.1038/nature23669
74. Shi C, Wang Y, Wu M, Chen Y, Liu F, Shen Z, et al. Promoting anti-tumor immunity by targeting TMUB1 to modulate PD-L1 polyubiquitination and glycosylation. Nat Commun (2022) 13:6951. doi: 10.1038/s41467-022-34346-x
75. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
76. Wang Y, Sun Q, Mu N, Sun X, Wang Y, Fan S, et al. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun Signal (2020) 18:112. doi: 10.1186/s12964-020-00612-y
77. Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res (2019) 7:1580–90. doi: 10.1158/2326-6066.CIR-18-0910
78. Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B (2021) 11:694–707. doi: 10.1016/j.apsb.2020.11.005
79. Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun (2022) 13:1700. doi: 10.1038/s41467-022-29401-6
80. Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ (2022). doi: 10.1038/s41418-022-01102-z
81. Zhu D, Xu R, Huang X, Tang Z, Tian Y, Zhang J, et al. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ (2021) 28:1773–89. doi: 10.1038/s41418-020-00700-z
82. Yang Z, Xu G, Wang B, Liu Y, Zhang L, Jing T, et al. USP12 downregulation orchestrates a protumourigenic microenvironment and enhances lung tumour resistance to PD-1 blockade. Nat Commun (2021) 12:4852. doi: 10.1038/s41467-021-25032-5
83. Shi D, Wu X, Jian Y, Wang J, Huang C, Mo S, et al. USP14 promotes tryptophan metabolism and immune suppression by stabilizing IDO1 in colorectal cancer. Nat Commun (2022) 13:5644. doi: 10.1038/s41467-022-33285-x
84. Chen Z, Chen Y, Peng L, Wang X, Tang N. 2,5-dimethylcelecoxib improves immune microenvironment of hepatocellular carcinoma by promoting ubiquitination of HBx-induced PD-L1. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2020-001377
85. Ioannou N, Hagner PR, Stokes M, Gandhi AK, Apollonio B, Fanous M, et al. Triggering interferon signaling in T cells with avadomide sensitizes CLL to anti-PD-L1/PD-1 immunotherapy. Blood (2021) 137:216–31. doi: 10.1182/blood.2020006073
86. Fang DD, Tang Q, Kong Y, Wang Q, Gu J, Fang X, et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J Immunother Cancer (2019) 7:327. doi: 10.1186/s40425-019-0750-6
87. Li H, Li CW, Li X, Ding Q, Guo L, Liu S, et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology (2019) 156:1849–1861 e1813. doi: 10.1053/j.gastro.2019.01.252
88. Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-Reticulum-Associated degradation of PD-L1. Mol Cell (2018) 71:606–620 e607. doi: 10.1016/j.molcel.2018.07.030
89. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discovery (2022) 21:181–200. doi: 10.1038/s41573-021-00371-6
90. Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer (2021) 21:638–54. doi: 10.1038/s41568-021-00365-x
91. Wang ZW, Liu Y, Zhu X. PhotoPROTACs: A novel biotechnology for cancer treatment. Trends Cell Biol (2020) 30:749–51. doi: 10.1016/j.tcb.2020.08.003
92. Liu J, Ma J, Liu Y, Xia J, Li Y, Wang ZP, et al. PROTACs: A novel strategy for cancer therapy. Semin Cancer Biol (2020) 67:171–9. doi: 10.1016/j.semcancer.2020.02.006
93. Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat Chem Biol (2019) 15:937–44. doi: 10.1038/s41589-019-0362-y
94. Chatterjee DR, Kapoor S, Jain M, Das R, Chowdhury MG, Shard A. PROTACting the kinome with covalent warheads. Drug Discovery Today (2022) 28:103417. doi: 10.1016/j.drudis.2022.103417
95. Wang K, Dai X, Yu A, Feng C, Liu K, Huang L. Peptide-based PROTAC degrader of FOXM1 suppresses cancer and decreases GLUT1 and PD-L1 expression. J Exp Clin Cancer Res (2022) 41:289. doi: 10.1186/s13046-022-02483-2
96. Wang Y, Zhou Y, Cao S, Sun Y, Dong Z, Li C, et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem (2021) 111:104833. doi: 10.1016/j.bioorg.2021.104833
97. Cheng B, Ren Y, Cao H, Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem (2020) 199:112377. doi: 10.1016/j.ejmech.2020.112377
98. Tong J, Tan X, Risnik D, Gao M, Song X, Ermine K, et al. BET protein degradation triggers DR5-mediated immunogenic cell death to suppress colorectal cancer and potentiate immune checkpoint blockade. Oncogene (2021) 40:6566–78. doi: 10.1038/s41388-021-02041-8
99. Mi D, Li Y, Chen Y. Small-molecule modulators targeting SHP2 for cancer therapy. Anticancer Agents Med Chem (2022). doi: 10.2174/1871520622666220921093052
100. Zhang C, Xu M, He S, Huang J, Xu C, Pu K. Checkpoint nano-PROTACs for activatable cancer photo-immunotherapy. Adv Mater (2022), e2208553. doi: 10.1002/adma.202208553
101. Dai MY, Shi YY, Wang AJ, Liu XL, Liu M, Cai HB. High-potency PD-1/PD-L1 degradation induced by peptide-PROTAC in human cancer cells. Cell Death Dis (2022) 13:924. doi: 10.1038/s41419-022-05375-7
102. Si J, Shi X, Sun S, Zou B, Li Y, An D, et al. Hematopoietic progenitor Kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T cell-based immunotherapies. Cancer Cell (2020) 38:551–566 e511. doi: 10.1016/j.ccell.2020.08.001
103. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc (2021) 143:593–8. doi: 10.1021/jacs.0c10008
104. Chen W, Huang Y, Pan W, Xu M, Chen L. Strategies for developing PD-1 inhibitors and future directions. Biochem Pharmacol (2022) 202:115113. doi: 10.1016/j.bcp.2022.115113
105. Chang SC, Hsu W, Su EC, Hung CS and Ding JL. Human FBXL8 is a novel E3 ligase which promotes BRCA metastasis by stimulating pro-tumorigenic cytokines and inhibiting tumor suppressors. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12082210
106. Wang X, Tokheim C, Gu SS, Wang B, Tang Q, Li Y, et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell (2021) 184:5357–5374 e5322. doi: 10.1016/j.cell.2021.09.006
107. Wang Z, He L, Li W, Xu C, Zhang J, Wang D, et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J Immunother Cancer (2021) 9. doi: 10.1136/jitc-2021-002787
108. Song M, Yeku OO, Rafiq S, Purdon T, Dong X, Zhu L, et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat Commun (2020) 11:6298. doi: 10.1038/s41467-020-20140-0
109. Guo Y, Yang L, Lei S, Tan W, Long J. NEDD4 negatively regulates GITR via ubiquitination in immune microenvironment of melanoma. Onco Targets Ther (2019) 12:10629–37. doi: 10.2147/OTT.S212317
110. Hosein AN, Dangol G, Okumura T, Roszik J, Rajapakshe K, Siemann M, et al. Loss of Rnf43 accelerates kras-mediated neoplasia and remodels the tumor immune microenvironment in pancreatic adenocarcinoma. Gastroenterology (2022) 162:1303–1318 e1318. doi: 10.1053/j.gastro.2021.12.273
111. Song G, Zhang Y, Tian J, Ma J, Yin K, Xu H, et al. TRAF6 regulates the immunosuppressive effects of myeloid-derived suppressor cells in tumor-bearing host. Front Immunol (2021) 12:649020. doi: 10.3389/fimmu.2021.649020
112. Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of cbl-b inhibits CD8(+) T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer (2021) 9. doi: 10.1136/jitc-2020-001688
113. Lu Y, Jin Z, Hou J, Wu X, Yu Z, Yao L, et al. Calponin 1 increases cancer-associated fibroblasts-mediated matrix stiffness to promote chemoresistance in gastric cancer. Matrix Biol (2022). S0945-053X(22)00137-8. doi: 10.1016/j.matbio.2022.11.005
114. Cheng WL, Feng PH, Lee KY, Chen KY, Sun WL, Van Hiep N, et al. The role of EREG/EGFR pathway in tumor progression. Int J Mol Sci (2021) 22. doi: 10.3390/ijms222312828
115. Wang C, Long Q, Fu Q, Xu Q, Fu D, Li Y, et al. Targeting epiregulin in the treatment-damaged tumor microenvironment restrains therapeutic resistance. Oncogene (2022) 41:4941–59. doi: 10.1038/s41388-022-02476-7
116. Wang HQ, Mulford IJ, Sharp F, Liang J, Kurtulus S, Trabucco G, et al. Inhibition of MDM2 promotes antitumor responses in p53 wild-type cancer cells through their interaction with the immune and stromal microenvironment. Cancer Res (2021) 81:3079–91. doi: 10.1158/0008-5472.CAN-20-0189
117. Tan X, Cao F, Tang F, Lu C, Yu Q, Feng S, et al. Suppression of DLBCL progression by the E3 ligase Trim35 is mediated by CLOCK degradation and NK cell infiltration. J Immunol Res (2021) 2021:9995869. doi: 10.1155/2021/9995869
118. Zhang CX, Huang DJ, Baloche V, Zhang L, Xu JX, Li BW, et al. Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis (2020) 9:65. doi: 10.1038/s41389-020-00248-0
119. Li N, Babaei-Jadidi R, Lorenzi F, Spencer-Dene B, Clarke P, Domingo E, et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis (2019) 8:13. doi: 10.1038/s41389-019-0125-3
120. Pitarresi JR, Liu X, Avendano A, Thies KA, Sizemore GM, Hammer AM, et al. Disruption of stromal hedgehog signaling initiates RNF5-mediated proteasomal degradation of PTEN and accelerates pancreatic tumor growth. Life Sci Alliance (2018) 1:e201800190. doi: 10.26508/lsa.201800190
121. Liao L, Song M, Li X, Tang L, Zhang T, Zhang L, et al. E3 ubiquitin ligase UBR5 drives the growth and metastasis of triple-negative breast cancer. Cancer Res (2017) 77:2090–101. doi: 10.1158/0008-5472.CAN-16-2409
Keywords: E3 ligases, cancer, therapy, PD-1, PD-L1, TME
Citation: Hou B, Chen T, Zhang H, Li J, Wang P and Shang G (2023) The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front. Immunol. 14:1123244. doi: 10.3389/fimmu.2023.1123244
Received: 13 December 2022; Accepted: 03 January 2023;
Published: 17 January 2023.
Edited by:
Huanfa Yi, Jilin University, ChinaReviewed by:
Yuyong Tan, The second Xiangya Hospital, Central South University, ChinaCopyright © 2023 Hou, Chen, Zhang, Li, Wang and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guanning Shang, c2hhbmdndWFubmluZ0Bob3RtYWlsLmNvbQ==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.