 Yehao Cong
Yehao Cong Yi Wang
Yi Wang Tao Yuan
Tao Yuan Zheng Zhang
Zheng Zhang Jianxun Ge
Jianxun Ge Qi Meng
Qi Meng Ziqing Li
Ziqing Li Shui Sun
Shui Sun- 1Department of Joint Surgery, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, China
- 2Orthopaedic Research Laboratory, Medical Science and Technology Innovation Center, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, Shandong, China
- 3Department of Joint Surgery, Shandong Provincial Hospital, Shandong University, Jinan, Shandong, China
Aseptic loosening (AL) is the most common complication of total joint arthroplasty (TJA). Both local inflammatory response and subsequent osteolysis around the prosthesis are the fundamental causes of disease pathology. As the earliest change of cell behavior, polarizations of macrophages play an essential role in the pathogenesis of AL, including regulating inflammatory responses and related pathological bone remodeling. The direction of macrophage polarization is closely dependent on the microenvironment of the periprosthetic tissue. When the classically activated macrophages (M1) are characterized by the augmented ability to produce proinflammatory cytokines, the primary functions of alternatively activated macrophages (M2) are related to inflammatory relief and tissue repair. Yet, both M1 macrophages and M2 macrophages are involved in the occurrence and development of AL, and a comprehensive understanding of polarized behaviors and inducing factors would help in identifying specific therapies. In recent years, studies have witnessed novel discoveries regarding the role of macrophages in AL pathology, the shifts between polarized phenotype during disease progression, as well as local mediators and signaling pathways responsible for regulations in macrophages and subsequent osteoclasts (OCs). In this review, we summarize recent progress on macrophage polarization and related mechanisms during the development of AL and discuss new findings and concepts in the context of existing work.
1 Introduction
Total joint arthroplasty (TJA) is an extensive and successful surgical therapy, which has been used in the treatment of severely traumatic or arthritic joint diseases. It helps in relieving arthralgia, rebuilding locomotor function, and improving living quality (1). Even so, the long-term survival of joint prosthesis reduces over time, which leads to implant failure, reduced locomotor ability, and heavy financial burden.
The main reason for the failure of TJA is that the joint interface continuously produces debris particles which induce a complex inflammatory response and leads to osteolysis and aseptic loosening (AL) (2–4). At present, the major pathogenesis of AL is the production of wear debris due to the mechanical strength of joints and biological interactions over time. On one hand, the debris induces functional changes in macrophages and promotes the release of cytokines to regulate the immune microenvironment in the bone-implant interface. On the other hand, by further affecting the bone remodeling process, the debris increases bone resorption, eventually resulting in AL (3–6).
Macrophages, the major population of tissue-resident mononuclear phagocytes, are a critical class of cells that have an effect on bacterial recognition and elimination, as well as in the process of innate and adaptive immunity (7). Macrophages have a variety of functions, exhibiting different phenotypes based on practical conditions and responding to microenvironmental signals. Two major phenotypes of macrophages include classically activated macrophages (M1) and alternatively activated macrophages (M2) (8–10). M1 macrophages have proinflammatory properties and are involved in initiating and maintaining the inflammatory state, whereas M2 macrophages have anti-inflammatory properties and take part in tissue homeostasis and repair (8–13).
During the development of inflammation, the polarized state of macrophage is in a dynamic equilibrium. In this regard, macrophages can distinctively adapt to the microenvironment, respectively (9). For example, the increased count of proinflammatory M1 macrophages induced by pathological stimulus leads to periprosthetic osteolysis, whereas anti-inflammatory M2 is favorable to shape an immunomodulatory environment towards osseointegration (14, 15). Therefore, it is essential to figure out the polarization of macrophages and associated regulatory mechanisms during the pathogenesis of AL.
At present, the understanding of macrophages is still limited to some extent. For the past few years, advancing awareness of the impact of macrophage polarization on the pathogenesis of AL has been recognized. This article reviewed interactions between the various receptors, ligands, signal transductions, and other factors related to functional changes of macrophage around the prosthesis. In addition, it cited the research conclusions and reviews regarding other macrophage-related inflammatory regulations, and also emphasized the importance of the functional changes and regulatory mechanisms of macrophages in the bone-implant interface microenvironment.
2 Overview of aseptic loosening
2.1 Clinical features
Harris et al. (16) described a phenomenon of extensive bone resorption leading to loosening without infection in four patients after receiving hip arthroplasty surgery, and this is the first detailed description of AL (16). In definition, AL can be generally described as a failure of the fixation of one or more prosthetic components without any infection (17). It may probably originate from inadequate initial fixation, mechanical loss of fixation over time, or biological loss of fixation, all of which are the leading causes of particles-induced osteolysis around the prosthesis (4, 17). Joint pain is the typical symptom of patients with AL, which always become worse when the affected joint carries out physical activity or bears weight. Impaired gait and restricted range of motion are often discovered in the physical exam of these patients (17).
As one of the major reasons for the failure of artificial joint implants, AL is the main cause of revision surgery (18–20). Due to the high complication rate, the requirement for complex technology, and the heavy economic burden brought by revision surgery (18), extensive studies have focused on the pathogenesis of AL in order to develop diagnostic and therapeutic avenues with more sensitivity and efficacy. The majority of published works reported that AL is mainly caused by wear particles-induced periprosthetic osteolysis (PPOL) (2–4).
2.2 Pathogenesis
Loosening of the prosthesis is a very complex process, involving many mechanical and biological aspects (21, 22). The main biological factor is the biological response of cells to a variety of wear particles (21), for example, wear particles can promote macrophage polarization to M1, and release proinflammatory cytokines and chemokines (23, 24). These cellular responses and subsequent activities are determined by many factors, such as the physical and chemical properties of the material, including the size, morphology, and composition of the material (23, 25–29). Moreover, the presence of endotoxin can also affect these cellular responses and activities (30–32). From the aspects of the disease host, patient-related risk factors, such as age, sex, obesity, smoking, and genetic variation, also play a role in AL pathogenesis (33–38). However, comorbidities affect the occurrence of AL even larger. Patients with hemophilia are reported to have a higher risk of AL (39). Elevated inflammatory activity will increase the risk of loosening after TJA in patients with rheumatoid arthritis (RA), thus the indication of arthroplasty for RA patients should be more strictly controlled (40).
2.3 Pathological feature
The chronic inflammation at the bone-implant interface, accompanied by osteolytic destruction in the surrounding bone, is the major pathological feature of AL (5, 6, 41, 42). Based on histopathological findings, there are numerous infiltrating CD68-positive mononuclear/macrophages, foreign body giant cells (sometimes organized as foreign body granulomas), and wear particles in the periprosthetic connective tissue (42, 43). Scattered fibroblasts and T cells can be observed in the surrounding area of infiltrating macrophage (42). In addition, endotoxin contamination is also present around the prosthesis (44, 45). Regarding the cytokines, there is a significant increase in the expression of proinflammatory factors in the tissue, including interleukin-1β (IL-1β), IL-2, IL-8, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) (41). It was found that in the mice skull implanted with titanium (Ti) particles, macrophages polarized into M1 macrophages in the early phase of the inflammatory responses, and partial tissue restoration was observed in the resolution of inflammation after 6 to 8 weeks (46). However, long-term chronic inflammation eventually leads to osteolytic destruction (6). Studies on the various polarization phenotypes of macrophages may help in further explaining the pathogenesis of AL.
3 Overview of macrophages in aseptic loosening
3.1 Origin of macrophages and wear particles
Monocytes/macrophages originally come from the hematopoietic stem cell (HSC) in the bone marrow and subsequently enter the peripheral blood. In responding to the local inflammation, circulating monocytes leave the bloodstream and mobilize into the local tissues. Upon stimulation by several growth factors, proinflammatory cytokines, or microbial products, circulating monocytes further differentiate into macrophages (11, 12). In addition to that, resident tissue macrophages recruited from the bone marrow are necessary drivers of inflammatory and tissue regenerative responses (47). The initial recruitment of inflammatory cells results from chemotactic factors produced by macrophages (48). When circulating macrophage or monocyte recruitment or activation is disrupted, the early inflammatory response is often diminished (49). Conclusively, the number of tissue-resident macrophages can increase exponentially, including locally proliferating macrophages and monocytes recruited from the bone marrow (50–52).
Due to the detection of ultra-high molecular weight polyethylene (UHMWPE) and various kinds of high-density material debris in tissue samples from patients, submicron-sized wear particles are usually considered potential causes of AL pathogenesis (53). During disease progress, macrophages play a crucial role in recognizing wear particles and releasing a large number of proinflammatory cytokines and chemokines, such as IL-1β, TNF-α, IL-6 (15, 23, 24, 26, 54–58). Diverse cytokines individually modulate the function of cells located at the interface between the prosthesis and the surrounding bone, and collectively affect other cells through diverse signaling mechanisms, ultimately leading to particles-induced inflammatory osteolysis. Detailed interactions between cytokines and cells are to be reviewed in section 4.
3.2 PAMPs and subclinical infection
Common pathogen-associated molecular patterns (PAMPs) include lipopolysaccharide (LPS) and lipoteichoic acid (LTA). LPS is a typical endotoxin and a major component of the outer cell wall of Gram-negative bacteria, whereas LTA is a cell wall polymer discovered in Gram-positive bacteria (59, 60). PAMPs regulate macrophage polarization through toll-like receptors (TLRs) and further promote the release of cytokines (61–63).
Previous studies have reported that some active bacteria or its structural components could be found in the tissues surrounding the loosened implant, even in the absence of any clinical or microbial evidence of infection (44, 45). In fact, subclinical infection is difficult to identify, and the probable cause of this is the growth pattern of biofilm. By firmly anchoring to the surface of the implanted prosthesis, biofilm may have protected inside microorganisms that are being infected from elimination, therefore it is reasonable to suspect these bacterial biofilms anchoring on the surface of the loosened implant as the latent source of endotoxin (45, 64). In addition, PAMPs may also originate from bacterial colonies residing in the gastrointestinal tract, the oral cavity, or even the wounds in the skin, where these bacteria and PAMPs are occasionally transferred to the circulating blood, in turn reaching the implant (65, 66). Evidenced by an in vivo experiment based on mouse balloon models, endotoxin in blood circulation could adhere to Ti particles and consequently induce macrophage aggregation (67).
3.3 Functional changes of macrophages
Macrophages are activated and alter their functions to defend against infections and present antigens to other immune cells, thereby regulating the immune responses (Figure 1) (68).
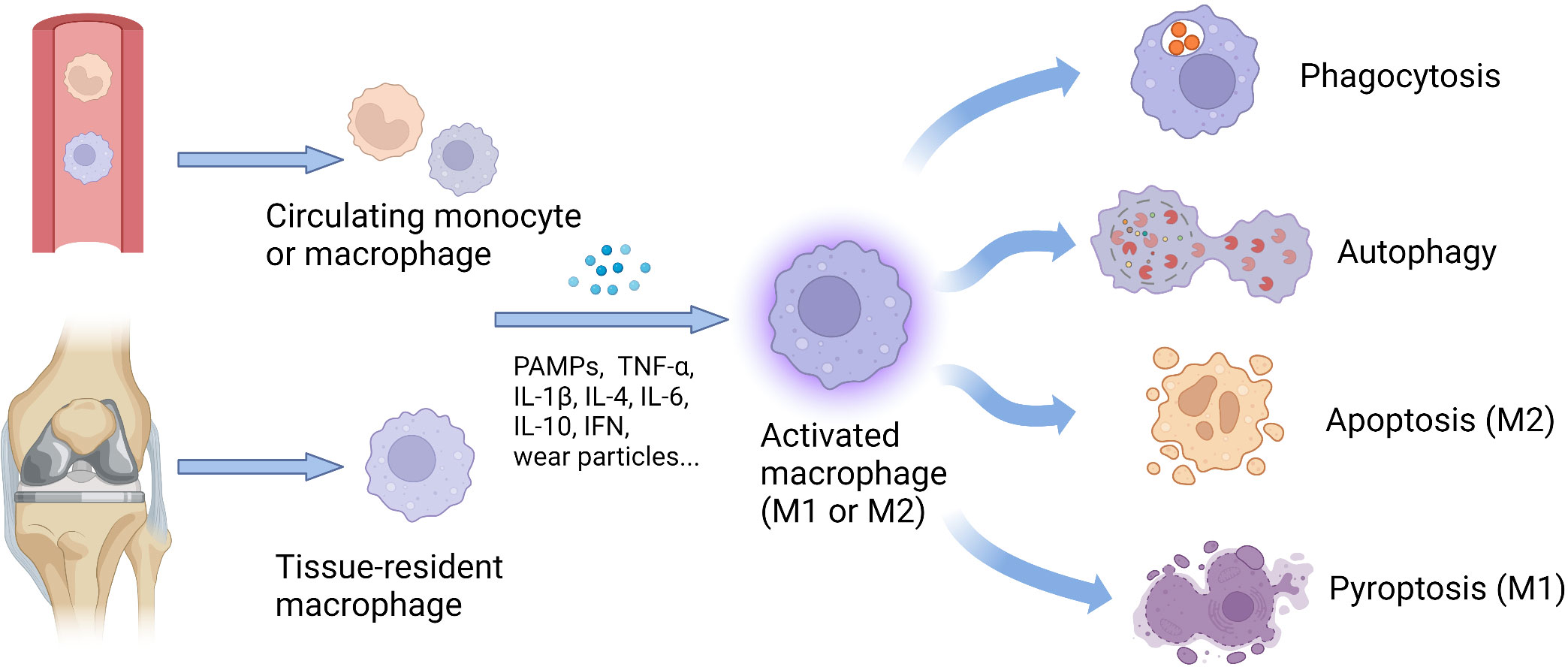
Figure 1 Origin and functional changes of macrophages. The number of tissue-resident macrophages can increase exponentially, including locally proliferating macrophages and monocytes recruited from the circulating peripheral blood. Stimulated by various factors, such as cytokines, PAMPs, and wear particles, the macrophages with different origins could be activated and further polarize into M1 or M2 phenotype. According to cellular phenotypes, activated macrophages show diverse functions, including phagocytosis, autophagy, apoptosis, and pyroptosis, to defend against infections and present antigens to other immune cells, thereby regulating the immune responses. IFN, interferon; IL, interleukin; M1, classically activated macrophages; M2, alternatively activated macrophages; PAMPs, pathogen-associated molecular patterns; TNF-α, tumor necrosis factor-alpha.
3.3.1 Proinflammatory and anti-inflammatory functions
Macrophages amplify the inflammatory process by releasing proinflammatory factors to remove pathogens or other foreign bodies (9, 47). During tissue injury, the local cells which are infected by pathogens and undergo necrosis or pyroptosis could release PAMPs or damage-associated molecular patterns (DAMPs) that activate inflammatory signaling in macrophages and other resident cell populations. Activated cells recruit neutrophils, monocytes, and other inflammation-related cells into the tissue by a release series of cytokines. Once the acute injury is under control, macrophages supply nutrition to the tissues where they are located by decomposing remnants and secreting growth factors and mediators, exerting their function effectively in inhibiting inflammation (47). Some macrophages, characterized by producing of growth factors, including platelet-derived growth factor (PDGF), insulin-like growth factor 1 (IGF-1), and vascular endothelial growth factor-α (VEGF-α), are associated with tissue repair and help in promoting cell proliferation and vascular development and thus alleviating local hypoxia that occurs after injury (47). They also produce soluble mediators, such as transforming growth factor-β1 (TGF-β1) that induce local and recruited fibroblasts to differentiate into myofibroblasts, thereby synthesizing extracellular matrix components and promoting wound closure (69). In the final phase of tissue repairment, monocytes and macrophages present an anti-inflammatory phenotype (47, 70). It was found that these macrophages responded to inhibitory mediators such as IL-10 in the local microenvironment, eventually leading to relieving inflammation (71, 72).
Phagocytosis mainly belongs to the function of M1 macrophages, which goes along with the proinflammatory process, although M2 macrophages also demonstrate a weak function in phagocytosis (73, 74). Stimulated by the serum from Behçet’s disease (BD) patients, monocyte-derived macrophages (MDMs) could differentiate into M1 macrophages with enhanced phagocytic capacity (75). Subsequently, M1 macrophages display an enhanced capacity for the elimination of pathogens, which largely results from their increased production of superoxide, NO, and their derivatives (76). M2 MDMs also demonstrate the ability to phagocytose escherichia coli and cancer cells. Further research found that the phagocytosis in M2 macrophages mainly owes to their surface markers, such as CD14, CD206, and CD163 (77). However, compared with M0 and M2 macrophages, LPS-treated M1 macrophages exhibited an obviously higher ability for phagocytic activity (78).
3.3.2 Autophagy
Both autophagy and phagocytosis in macrophages are lysosomal-dependent catabolic processes, by which cells can engulf and deliver cargo to the lysosomes for digestion via forming transient vesicular structures (autophagosome and phagosome) (79). Acting as scavenger cells, macrophages could phagocytize cellular debris, invading pathogens, and other apoptotic cells (80, 81). Phagocytizing dead cells in macrophages contribute to diverse immune and inflammatory signals that could also trigger intracellular autophagy in macrophages (82, 83). Other studies have revealed more relations between autophagy and phagocytosis. Autophagy promotes phagocytosis and the clearance of pathogens via the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in macrophages (84). As a novel function for autophagy proteins, the LC3-associated phagocytosis pathway (LAP) is closely associated with phagocytosis in macrophages, which exerts its role in blocking proinflammatory signals upon phagocytosis of dying cells and preventing the presentation of autoantigen to other cells (85, 86). Interestingly, autophagy-deficient macrophages may boost phagocytosis through increased scavenger receptor expression (87).
Regarding the functional changes in activated macrophages, vitamin D could restore anti-inflammatory M2 macrophages in an autophagy-dependent manner (88). Similarly, ubiquitin-specific protease 19 (USP19) could inhibit inflammatory responses and promote M2 polarization by increasing autophagy flux (89). Controversially, another study reported that inhibition of autophagy could drive macrophages to the M2 phenotype (90). In AL, a recent study revealed that by activating LAP, aluminum oxide nanoscale particles (Al-n) attenuated the macrophage M1 polarization and inhibited the secretion of inflammatory factors, leading to the prevention of the AL pathogenesis induced by particles in vivo (26). Although autophagy has been shown to be involved in the regulation of macrophage polarization, evidence regarding the regulatory mechanisms is underdeveloped.
3.3.3 Apoptosis
Apoptosis is the sequential death of cells via the mechanisms called programmed cell death (PCD) (91), which is characterized by morphological changes in the cellular structures together with a series of enzyme-dependent biochemical processing (92). In the resolution phase of inflammation, the infiltrating leukocytes execute the acute innate response and undergo apoptosis, subsequently cleared by phagocytic macrophages. In this course, macrophages undergo reprogramming from inflammatory to anti-inflammatory, leading to the relief of inflammation (93).
Efficient clearance of early apoptotic cells requires macrophages polarizing into the M2c phenotype (94). The capacity of M2 macrophages to uptake apoptotic cells depends on several necessary molecules, such as Mer tyrosine kinase, Axl receptor tyrosine kinase, growth arrest-specific 6 (Gas-6) (94, 95). In addition, the macrophage itself could also undergo apoptosis induced by wear particles (96). Consequently, the increase in macrophage apoptosis limits the proinflammatory function of macrophages (97, 98), seemly to be another important mechanism regarding the delayed inflammatory response in AL. The caspase-3, a key mediator related to apoptosis, was detected in periprosthetic tissues in a mouse osteolysis model induced by UHMWPE particles (99, 100). Furthermore, apoptosis is associated with phenotypic changes in macrophages. Transcription factor Zhx2 deficiency could enrich the expression of M2 phenotype markers and as well promote the apoptosis of macrophages (101). Likewise, through the inhibition of M2-specific gene expression and apoptotic cell death, Delta-like-ligand 4 (DLL4) may prevent macrophage from polarizing into the M2 phenotype (102).
3.3.4 Pyroptosis
Different from apoptosis, pyroptosis is a proinflammatory type of PCD mediated by the gasdermin family, which usually leads to cell swelling and rapid rupture of plasma membranes, as well as the release of immunogenic cell contents, thereby exaggerating inflammatory status (103). In the aspect of the immune response, pyroptosis induced by the activation of pattern recognition receptors (PRRs) can stimulate inflammatory responses, independent of its effect in promoting cytokine induction (104). Pyroptosis is undoubtedly related to macrophage polarization. On one hand, by secreting exosomal cathepsin S, M1 macrophages induce pyroptosis in pancreatic acinar cells via the caspase1-mediated classical pyrolysis pathway, resulting in inflammation and pancreatic tissue damage (105). On the other hand, exosomal Mir-30D-5p of polymorphonuclear neutrophils (PMNs) is reported to induce M1 polarization by upregulating TNF-α, IL-1β, IL-6 and triggering pyroptosis in macrophages, leading to sepsis-associated acute lung injury (106).
Moreover, some mutual signals could mediate both M1 polarization and pyroptosis in macrophage, such as the caspase-1/GSDMD signaling pathway (107) and METTL3/MALAT1/PTBP1/USP8/TAK1 axis (108). The overexpression of brain and muscle Arnt-like protein 1 (BMAL1) also reduces the production of inflammasomes and pyroptosis in macrophages, as well as decreases the proportion of M1 phenotype via the TLR2/NF-κB pathway (109). A recent study has found that macrophages can activate the NLRP3 inflammasome and initiate subsequent pyroptosis to affect AL pathogenesis in mice model of cobalt-chromium-molybdenum (CoCrMo) alloy particles-induced osteolysis (110). As a result, wear particles not only induce M1 polarization and production of proinflammatory cytokines, but also boost inflammation by increasing pyroptosis in macrophages and inducing local tissue impairment.
3.4 Bone remodeling and osteoclastogenesis
Bone remodeling is a dynamic and balanced process maintained by osteoblasts (OBs) and osteoclasts (OCs), which is deeply affected by the receptor activator of nuclear factor NF-κB ligand/osteoprotegerin (RANKL/OPG) ratio. Disruption of this homeostasis leads to severe skeletal disorders (111). OBs are a type of bone-forming cells that are derived from bone marrow mesenchymal stem cells (BMSCs) and could respond to anabolic factors, such as bone morphogenic proteins (BMPs) (112). On the other hand, OCs are the unique cells known to absorb bone at or near the bone surface, which originate from bone marrow-derived monocytes/macrophages (BMMs) (113, 114).
Osteoclastogenesis is a complex process involving events of proliferation, differentiation, cell fusion, and multinucleation (111). The primary osteoclastogenic factor is RANKL which triggers a complex network of signaling pathways including NF-κB and mitogen-activated protein kinases (MAPK), via the receptor RANK on OC progenitors. By further activating the nuclear factor of activated T cells c1 (NFATc1), the master transcriptional factor of osteoclastogenesis, the fate of OC progenitors is decided by the controlling of key osteoclastogenic genes, such as tartrate-resistant acid phosphatase (TRAP) and cathepsin K (CTSK) (113–115). Calcineurin, a powerful mediator of transcriptional activity of NFATc1, is regulated by cytosolic calcium (Ca2+) downstream of the TEC kinases and phospholipase Cγ (PLCγ), all of which are governed by both RANK and immunoreceptor tyrosine-based activation motif (ITAM)-based signaling. In addition, sustained intracellular Ca2+ oscillations are required for OC formation and functions, which will be introduced in the following section 5.5.3 (116). On the opposite side, OPG, as a decoy receptor, blocks RANKL binding to its cellular receptor RANK. An active OC is a highly polarized cell with a distinctive cytoskeletal organization and has the ability to create the sealing zone which is a site of tight membrane apposition to the bone surface, thereby executing its function as a bone-resorbing machine (116).
4 Polarization and phenotype of macrophages
Two major phenotypes of macrophages in response to environmental stimuli are M1 and M2. Usually, the phenotype of macrophages can be transformed by reprogramming (117, 118). Moreover, a recent study based on a spectrum of activated human macrophages revealed that there are continuous intermediate phenotypes between two opposite terminal phenotypes (119). Under such background, researchers use the term “polarization” to define the preference pattern of gene expression and protein synthesis in macrophages after different stimuli (120). “Naive” M0 macrophages, the prototype of M1 and M2 macrophages, are characterized by the expression of CD11b and F4/80, and emerge from committed myeloid progenitors in the presence of macrophage colony-stimulating factor (M-CSF). Although lacking the expression of antigen-presenting molecules (MHC-II) and co-stimulatory molecules (B7), M0 macrophages could readily phagocytose cellular debris or pathogens (78, 121).
The polarization to M1 macrophages has two main inducible sources, either microbial products or the cytokines secreted by TH1 lymphocytes. By recognizing pathogens and presenting antigens to T lymphocytes, M1 macrophages play a critical role in triggering adaptive immunity in the body. During immunization, M1 macrophages produce high amounts of proinflammatory factors (IL-1 β, IL-6, TNF-α, IL-12, and IL-23, among others), which in turn promote Th1 proinflammatory response (8, 10, 12, 122, 123). In contrast, M2 macrophages are mainly responsible for inflammatory relief and tissular repairs. Discovered in the early 1990s, the polarization to M2 macrophages is related to IL-4, IL-10, or IL-13 which are produced by innate and adaptive immune cells, such as mast cells, basophils, and Th2 lymphocytes (9, 122–124). By producing multiple growth factors and cytokines, such as TGF-β, IGF-1, PDGF, VEGF, IL-8, and endothelial growth factor (EGF), M2 macrophages are aided in the suppression of local inflammation and thus beneficial for tissue repairs (8, 10, 12, 122).
4.1 M1 macrophage in AL
4.1.1 Stimulus from wear particles and endotoxin
In the bone-implant interface, macrophage polarized to the M1 phenotype is mainly stimulated by wear particles and endotoxin (55). Upon continuous stimulation, M1 macrophages cause tissue damage by strengthening local inflammation via the secretion of TNF-α, IL-1β, IL-6, and IL-8 and the reduction of IL-10 expression (15, 23, 24, 26, 54–58). Besides, other cytokines such as IFN-β (15) and various chemokines, including chemokine (C-X-C motif) ligand 9 (CXCL9), CXCL10, and CXCL11 (56) secreted by M1 macrophages also play a role in AL pathogenesis. These biological reactions are largely affected by the size and type of wear particles (23, 26). Endotoxin also contributes to macrophage differentiation into the M1 phenotype by stimulating TLRs on the cell surface. Particles with PAMPs adhering, such as LTA and LPS, could induce more production of the proinflammatory cytokine than those without endotoxin (30, 32). On the contrary, the removal of endotoxins from particles significantly reduces the cellular activity of macrophages (30) and inhibits OC differentiation (31).
4.1.2 Surface receptor and DAMPs
It is well-established that both polyethylene (PE) and Ti particles can promote the expression of TLRs and various proinflammatory factors in macrophages (43, 125). A recent in vivo experiment also confirmed that alloy particles could induce significantly higher numbers of TLR-1, -4, and -6 positive cells in the synovial layer of joints (126). In the downstream of TLRs, NLRP3, ASC, caspase-1, and TNF-α and IL-1β were found to be positive through the colocalization with CD68 in the tissues around the revised prosthesis (43). The NF-κB, MAPK, and TAK1 pathways are involved in mediating signaling transduction downstream of TLRs (61–63, 127–129). In addition to TLRs, macrophages can engulf PMMA debris through macrophage receptors with collagenous structure (MARCO), which is a key pathogenic factor in promoting the phagocytosis of polymethyl methacrylate (PMMA) debris in aging macrophages (130).
As the main alternative hypothesis to PAMPs, DAMPs also activate TLRs during AL. The term “DAMPs” refer to self-molecules released by dying or damaged cells, which are defined as endogenous danger molecules due to they could activate the innate immune system by interacting with pattern recognition receptors (PRRs) (131). DAMPs are recognized by various membrane-bound receptors, including PRRs and non-PRRs, and also by intracellular sensors, notably through inflammasome (132, 133). Through binding to specific receptors, DAMPs activate the inflammatory process and recruit immune cells like neutrophils and monocytes. Therefore, after the clearance of DAMPs, the recruited leukocytes will change from a proinflammatory into a reparative program (133). Cobalt alloy particles could induce macrophage-associated inflammatory responses and bone loss through DAMPs rather than activated TLR4, due to the partially absent of metal-binding histidines in TLR4 (134). However, it has also been thought that the DAMPs generated in response to particles are insufficient to activate TLR2 or TLR4 in these cells (32).
Although DAMPs contribute to the host’s immune defense, they also promote pathological inflammatory responses. The DAMPs, such as high-mobility group box 1 (HMGB1), S100 proteins, and heat shock proteins (HSPs), are commonly known as regulatory molecules of inflammatory responses (131). HMGB1 is an ancient DNA-binding nucleoprotein. It can be passively released from dying cells or actively secreted by monocytes, macrophages, and myeloid dendritic cells (135, 136). HMGB1 could induce M1 polarization via TLR2, TLR4, and RAGE/NF−κB signaling pathways, leading to LPS−induced acute lung injury (124), meanwhile, it significantly yielded the expression of the M1 marker inducible nitric oxide synthase (iNOS) while decreasing the M2 marker IL-10 in macrophages (137). In vitro, HMGB1 silencing down-regulated the secretion of inflammatory cytokines in macrophages, which cannot be reversed by the exogenous HMGB1 (138). Interestingly, HMGB1 could also trigger M2 macrophage polarization via the TLR2/NOX2/autophagy axis (139). In the same aspect, loss of HMGB1 in macrophages can increase the differentiation of proinflammatory macrophages and enhance inflammatory response under specific conditions, not the otherwise (140). In this regard, HMGB1 may induce distinct macrophage phenotypes probably due to different redox isoforms (141). Besides HMGB1, the expression level of several members of HSPs is closely related to distinct stages of polarization in macrophages (142). Compared with unpolarized macrophages, a significant up-regulation of members of the HSP70 family (HSPA2 and HSPA8), as well as the HSP90 family (HSP90AA1) can be observed in M1 macrophages. On the other hand, changes in HSP expression were also observed in macrophages during the M2 polarization, although with only five transcripts being significantly modulated. Among them, DNAJB5, HSPA13, HSPBAP1 were upregulated, whereas HSPH1 and HSPB1 were down-regulated (142).
4.1.3 Particles-induced inflammation conduce to osteolysis
Recent studies have found that Ti, CoCrMo particles, and LPS could strongly induce inflammatory responses in macrophages, significantly increasing the production of TNF-α and IL-1β, rising RANKL/OPG ratio, and enhancing the OC activity (143–146). The current paradigm holds the view that the induction of OC differentiation by inflammatory cytokines is indirectly yielded under RANKL stimulation (147). Proinflammatory cytokines, such as IL-1β, TNF-α, IL-6, soluble IL-6 receptor, and IL-17, could all increase the production of RANKL from OBs. The effects of proinflammatory cytokines could be balanced by anti-inflammatory cytokines, such as IL-4 and IL-13, which could inhibit RANKL expression (147). In addition, a variety of wear particles could conduce to osteolysis via elevating M1 polarization (15, 24, 57, 58), since M1 macrophages are an important source of TNF-α production (11).
Although the concrete mechanism of TNF-α on OC differentiation is not fully understood, TNF-α signals promote OC differentiation by upregulating several proinflammatory target genes through the activation and nuclear translocation of NF-κB. One such target is RANK, which increases OC activity by mediating RANKL signaling (148). Interestingly, TNF-α produced by LPS/TLR4 signals can regulate OC generation in LPS-treated macrophages through the activation of RANKL signaling, whereas TNF-α in a RAW264.7 cells-based experiment demonstrates it may act as an autocrine/paracrine factor in promoting osteoclastogenesis, independent of RANKL signaling (149). In addition, the co-culture experiments of macrophages and MSCs (or OBs) revealed that PE and Ti particles could inhibit OB function, meanwhile promoting M1 polarization and osteoclastogenesis (150–152), proposing another mechanism for inflammation-induced osteolysis.
4.2 M2 macrophage in AL
4.2.1 Subtypes of M2 macrophage
The classical M1/M2 system was based on experiments in vitro with different stimulation approaches. Subsequent studies revealed that the process of macrophage activation and polarization is much more complex and needed an additional subdivision of the M2 population (153). As a result, the M2 macrophages is further classified into four subtypes: alternative activated macrophages (M2a), type 2 macrophages (M2b), deactivated macrophages (M2c), and M2-like macrophages (M2d) (154). Among them, applied stimuli and the achieved transcriptional changes correspond to particular subtypes: 1) M2a macrophage induced by IL-4 or IL-13 is a profibrotic phenotype, 2) M2b macrophage is stimulated by immune complexes combined with Toll-like receptor or IL-1 receptor agonists, and 3) M2c macrophage is exposure to IL-10, TGF-β, or glucocorticoids, 4) M2d, activated by adenosines or IL-6 (154, 155). For the pathological study, although M2a/c macrophage is found to be beneficial in early inflammatory stages, they have been uncovered to impair tissue remodeling (156). Similarly, M2b macrophages have been thought to be relieving immune responses with minor damage to local tissue (157).
4.2.2 M2 macrophage conduces to inflammatory relief
M2 macrophage has the effect of alleviating inflammation induced by wear particles, through increased expression of IL-10 and decreasing expression of IL-6 and TNF-α (24, 57, 58). In the presence of large amounts of TLR2 ligands, the anti-inflammatory activity of M2 macrophage is inhibited but without evident changes in cell surface markers (158). On the contrary, inhibition of TLR4 caused a shift from inflammatory M1 macrophages toward M2-dominant macrophages (159), suggesting that TLRs act as a switch that plays a role in the regulation of inflammation. In the downstream of TLRs, interleukin-1 receptor-associated kinase (IRAK) -m has been identified as an inhibitor of TLR signaling (160). One study found that knockdown of IRAK-m promoted M1 polarization and inhibited M2 polarization during the mycobacterium tuberculosis infection (161). But the discovery of IRAK-m-mediated local immunosuppression in periprosthetic tissues hints that macrophages have a self-protective mechanism, wherein the wear debris-mediated stimulation is inhibited to prevent overproduction of NK-κB-dependent proinflammatory cytokines and thus suppressing the deleterious host response in AL (162). However, the drawback is that the induction of IRAK-m overexpression triggered by wear debris also appears to conduce to the inhibition of LPS-induced TLR signaling, which leads to low-level biofilm-associated infection and chronic inflammation (162). Therefore, activation of IRAK-m is somehow affected by the local immune environment.
In addition, M2 polarization is regulated by cytokines in the surrounding, especially IL-10 and IL-4. IL-10 is an important anti-inflammatory cytokine (163). Recent studies have verified that IL-10 treatment significantly reduces iNOS expression and promotes CD163 overexpression, suggesting that IL-10 treatment could reprogram bone marrow-derived macrophages (BMDMs) to an M2 phenotype and regulate the process of M1/M2 polarization (164). In vivo experiments revealed that the extra addition of IL-10 partially reversed the inhibitory effect of PE particles on bone ingrowth (165). In the aspect of gene regulation, IL-10 not only decreased the activation of signal transducer and activator of transcription 1 (STAT1), NF-κB p65, and c-Jun N-terminal kinase 1 (JNK1) genes but also increased the expression of STAT3 (164). In addition to IL-10, IL-4 also has the ability to switch the M1 phenotype induced by Ti particles into the M2 phenotype (55, 166). Compared with non-activated macrophages, upon IL-4 stimulation, the shift from M1 to the anti-inflammatory M2 phenotype is more thorough (55).
4.2.3 M2 Macrophage in bone remodeling
Macrophages stimulated by cytokines, such as IL-4 and IL-13, have been confirmed to prevent the OC differentiation and inhibit the function of mature OCs (11, 147). In addition, M2 macrophages may promote OB differentiation by producing cytokines that are critical for osteogenesis, including BMP-2, TGF-β, and IGF-1 (11, 167, 168). Also, by interacting with MSCs, M2 macrophages create an anti-inflammatory environment that is conducive to osseointegration (14). A recent study revealed that preconditioning of murine MSCs with IFN-γ and IL-1β highly significant reduction of CD86 and iNOS protein in macrophages under M1 inducers (LPS + IFN-γ) and diminished TNF-α secretion (169). Additionally, CD86 and iNOS protein expression as well as NO and IL-10 secretion were markedly increased under M2a inducers (IL-4) (169). On the other hand, under the stimulation of IL-4, reduced expression of CD86 and iNOS, as well as increased secretion of nitric oxide (NO) and IL-10 could be discovered in macrophages (169). The secretion of IL-10 could be attributed to the phenotype of M2b macrophages which are generally suggested to be the main subtype of macrophages for inflammatory relief (170). In addition, macrophages stimulated by a conditioned medium from preconditioned MSCs (pre-MSC-CM) may display an overall increased phagocytic capacity (171, 172). Also, M2a macrophages are found to undergo reprogramming to an M2b/M2c phenotype after treatment with the pre-MSC-CM (171, 172), indicating an influence of MSCs behavior on the induction of macrophage polarization.
5 Regulatory mechanisms of macrophages in AL
Several signaling pathways are involved in the process of macrophage polarization (8, 173–179). Various ligands, receptors, transcription factors, and other factors cooperate closely to ensure the precise regulatory capacity of macrophages (8). New research has found that several cytokines can influence M1/M2 polarization through NF-κB, MAPK, and JAK/STAT pathways which are involved in AL (55, 166, 180). Furthermore, depending on the amounts and ratios, molecules in the microenvironment can antagonize or synergistically act, thus in favor of certain macrophage phenotypes (181). Some regulatory mechanisms related to macrophage polarization in the microenvironment of the bone-implant interface are described below (Figure 2).
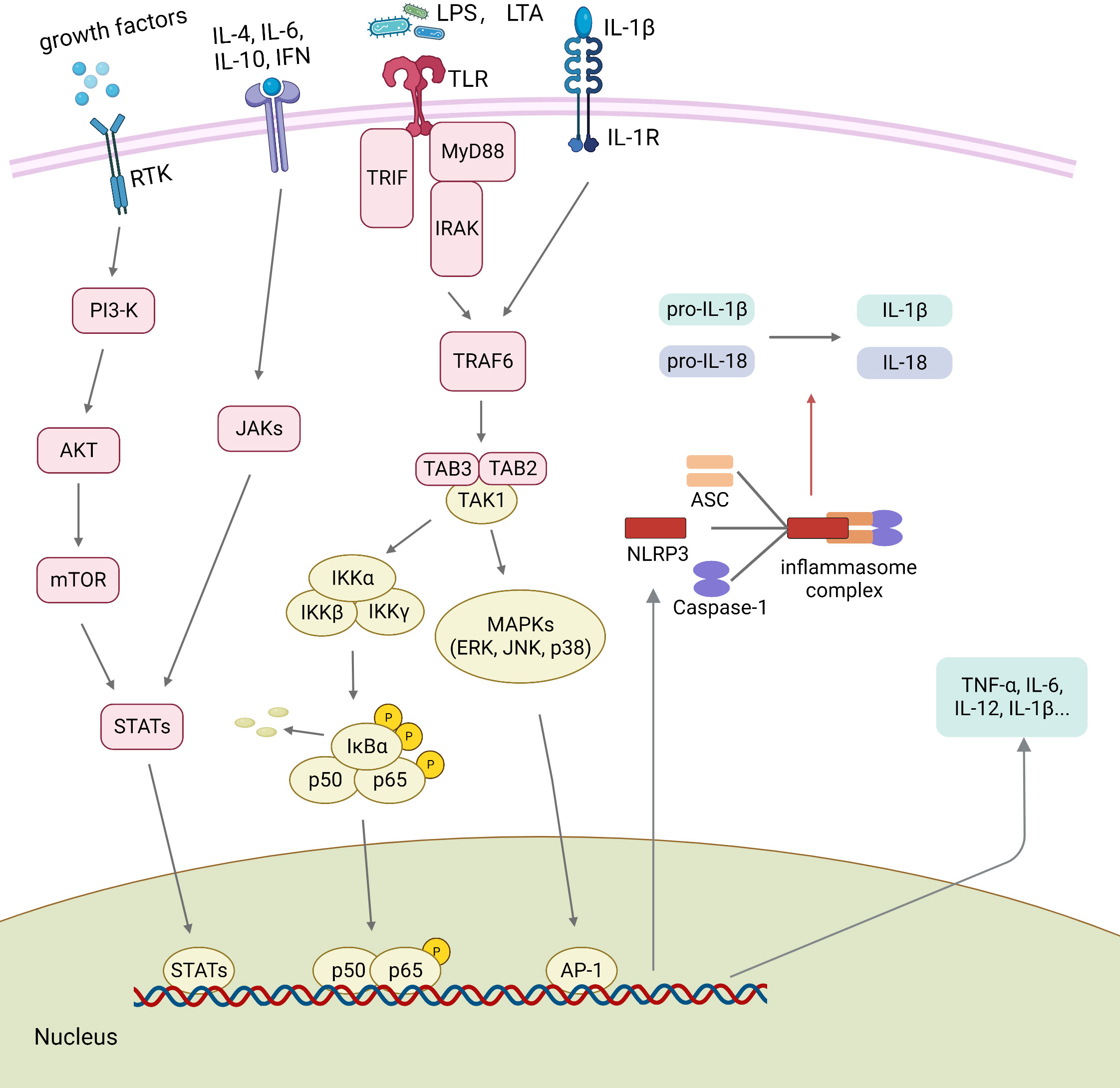
Figure 2 Polarization-related signaling transduction in macrophages. The binding of growth factors or cytokines to their receptors activates STATs through JAKs or PI3K/AKT pathway signaling. Activated TLRs and IL-1βR trigger downstream signaling, including the NF-κB and the MAPK signaling pathways. Several transcription factors, such as AP-1, STATs, and p65, are regulated by the upstream signaling pathways and further promote the assembly of NLRP3 inflammasome. Then, the activated NLRP3 inflammasome promotes the maturation of pro-IL-1β and pro-IL-18. ASC, apoptosis-associated speck-like protein containing a CARD; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; IRAK, Interleukin-1 receptor-associated kinase; LTA, lipoteichoic acid; MyD88, myeloid differentiation primary response protein 88; NLRP3 NOD-like receptor family pyrin domain containing 3; PAMPs, pathogen-associated molecular patterns; RTK, Receptor Tyrosine Kinases; STATs, signal transducers and activators of transcription; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; TRIF, TIR-domain-containing adapter-inducing interferon-beta.
5.1 Toll-like receptors
5.1.1 Overview of toll-like receptors
TLRs have been intensively studied in innate immunity regarding the recognition of PAMPs (182). It has been reported that TLR2 or TLR4 can recruit MyD88 and further bind to the IRAK to form a signal complex called Myddosome. The Myddosome-complex recruits the ubiquitin ligase TNF receptor-associated factor 6 (TRAF6), which triggers the TAK1 kinase signaling cascade pathway and ultimately leads to NF-κB nuclear translocation through the phosphorylation and activation of IκB kinase α/β (IKKα/β) (63, 183, 184).
5.1.2 Toll-like receptors in macrophage polarization
TLRs are closely related to macrophage polarization (63). HMGB1 is an important mediator that induces M1 polarization through the activation of absent in melanoma 2 (AIM2) inflammasome, TLR2/4 and RAGE/NF-κB signaling pathways in macrophages (124). In addition, LPS can activate downstream TAK1, NF-κB, and MAPK signals through the TLR4/MyD88 pathway, which increases the expression of cyclooxygenase-2 (COX-2) and iNOS, promotes M1 polarization and leads to the up-regulated expression of cytokines TNF-α, IL-1β, IL-6, and PGE2 (61, 63). The particles with LTA adhering could increase the expression of proinflammatory factors by the activation of TLR2/NF-κB and MAPK pathways (32, 185). Previous studies have shown that a variety of TLRs, such as TLR2, which participate in local inflammatory and immune responses, are present in periprosthetic tissues after the revision of total hip arthroplasty (rTHA) (43).
5.1.3 NLRP3 inflammasome
NLRP3 inflammasome widely exists in macrophages, granulocytes, antigen-presenting cells (APC), and other immune cells. It consists of the NLRP3 (sensor protein), ASC (adapter protein), and caspase-1 (186). The initiation of NLRP3 inflammasome could be induced by multiple inflammatory stimuli, such as PAMPs. As the core protein of the NLRP3 inflammasome complex, NLRP3 senses endogenous DAMPs and microbial ligands. The production of ROS, low level of intracellular potassium (K+), and the release of lysosomal protease into the cytoplasm are all upstream mechanisms that trigger the activation of NLRP3 inflammasome in several conditions (187–189). NLRP3 inflammasome which is activated by pathologically stimulated macrophages could regulate the production of IL-18, IL-1β, and cleavage of caspase-1 (186, 188, 189).
On the other hand, studies have also found that NLRP3 inflammasome participates in macrophage polarization (89, 175, 190, 191). In a co-culture experiment, force-pre-treated human periodontal ligament cells (hPDLCs) promote M1 polarization and increase the secretion of IL-1β in macrophages via the activation of NLRP3 inflammasome (191). On the contrary, by inhibiting the activation of NLRP3 inflammasome, metformin induces M2 polarization in macrophages and promotes wound healing in rat dorsal skin (175). Moreover, USP19 could directly promote M2 polarization by suppressing the activation of NLRP3 inflammasome to interferon regulatory factor-4 (IRF-4) (89). Bruton’s tyrosine kinase (BTK) is a key factor in TLR4-related pathways, which could activate NF-κB signaling and promote p65 phosphorylation, ASC oligomerization, and caspase-1 activation (192). It has been recently found that BTK could promote TiAl6V4 alloy particles (TiPs)-induced inflammation in BMDMs by positively regulating NF-κB activation, NLRP3 inflammasome formation, and M1 polarization (193). On the contrary, NLRP3 silencing attenuates the promotive effect of conditioned exosomes on M1 polarization in a particles-induced osteolysis model (194).
5.1.4 Syk
Syk is a key component of TLRs in recognizing PAMPs and activating immune responses (195–199). Moreover, Syk plays a critical role in LPS-induced M1 polarization (200). In monocytes, pharmacological inhibition of Syk prevents LPS-induced TLR4 phosphorylation (199), suggesting that Syk may be involved in tyrosine phosphorylation of the TIR domain in TLR4. Recently, PMMA and HA particles were confirmed to augment the expression of CD86, and secretion of cytokines, such as TNF-α and IL-6, in a Syk- and MAPK-dependent (phosphorylation of ERK and p38) manner (56). Also, Syk is reported to be closely associated with the NLRP3 inflammasome-mediated proinflammatory cytokine release (201). In addition, Syk could control NLRP3 activation and IL-1β synthesis in macrophages in response to fungal infections (202).
5.2 NF-κB Signaling Pathway
5.2.1 Overview of NF-κB signaling
In the resting state, NF-κB dimers are sequestered in the cytoplasm in an inactive state by the IκB family of proteins, including IκBα, IκBβ, IκBϵ, and the NF-κB precursors, p105 and p100. The IκB kinase (IKK) complex consists of kinases IKKα, IKKβ, and IKKγ. Upon receiving the activation signals, IκB protein is phosphorylated by IKK complexes, leading to proteasomal degradation of IκB. The released NF-κB dimers are then translocating into the nucleus, where they can bind to specific sites on DNA to regulate gene transcription (75).
5.2.2 NF-κB signaling in macrophage polarization
LPS-induced M1 polarization is dependent on NF-κB p65 activation, and the treatment with IKKβ inhibitors reduces the mRNA expression of M1 markers in macrophages (203). Therefore, IKK inhibitors reduce LPS-induced expressions of IL-1, IL-6, IL-10, TNF-α, and IFN (204). Recent studies have revealed that diverse wear particles (e.g., Ti, TiPs, HA, PE, PMMA) could induce the macrophage to release proinflammatory cytokines and chemokines, which are accomplished by the activation of NF-κB signaling pathways and associated macrophage polarization (15, 26, 54, 205). Qiu et al. found that stimulation of LPS or Ti particles could up-regulate p-IKKβ, p-IκBα, and p-p65 and significantly increase the translocation of p65 into the nucleus in BMDMs. Meanwhile, these particles-induced inflammatory infiltrations increase the number of OCs (TRAP-positive cells) and thereby decrease bone mineral density, eventually leading to PPOL (15). Similarly, Gao et al. found that PMMA particles could up-regulate iNOS and decrease the production of Arginase-1 (Arg-1) and IL-10 by the activation of p65 nuclear translocation in macrophages (54).
5.2.3 NF-κB signaling in osteoclastogenesis
Under the same settings, NF-κB can be activated by RANKL or LPS to augment particles-induced bone loss via the enhancement of osteoclastogenesis (206, 207). Upstream stimuli-triggered NF-κB signaling could act on transcription factors (c-fos and NFATc1) and regulate the expression of osteoclastogenesis-related genes, thus promoting OC differentiation and functions (206, 208, 209).
5.3 MAPK signaling pathway
5.3.1 Overview of MAPK signaling
The MAPK signaling pathway takes part in cell differentiation, proliferation, and apoptosis (210). MAPK pathways are organized into three-tiered cascades consisting of three factors: MAPK, MAPK kinase, and MAPKK kinase. During the phosphorelay process, MAPKKKs which are serine/threonine protein kinases, phosphorylate and activate MAPKKs, and then dually phosphorylate the threonine and tyrosine residues of the conserved TXY motif that belongs to the activation loop of MAPKs, including extracellular signal-regulated kinase (ERK), JNK, and p38 (210, 211).
5.3.2 MAPK signaling in macrophage polarization
MAPK signaling pathway is closely associated with macrophage polarization (61, 212–214). The activation of the MAPK signaling pathway promotes polarization of M1 macrophages, expression of iNOS, and down-regulation of CD206, thereby mediating the secretion of cytokines, such as TNF-α, IL-1β, and IL-6 (214). On the other hand, inhibiting the phosphorylation of JNK, ERK, and p38 in MAPK pathways may switch repolarized M1 to reprogrammed M2 phenotype (213, 214). Studies elucidate that the MAPK signaling pathway is located downstream of TLRs, wherein LPS could activate the MAPK pathway through TLR4/MyD88 pathway to induce M1 polarization and participate in the inflammatory response (61, 62, 215). Further, PMMA and hydroxyapatite (HA) particles are reported to result in M1 polarization by increasing phosphorylation of ERK and p38 (56). Similar to NF-κB signaling, MAPK also mediates wear particles-induced OC differentiation and the following osteolysis (206).
5.4 JAK/STAT Signaling Pathway
5.4.1 Overview of JAK/STAT signaling
STAT proteins are potent cytoplasmic transcription factors wherein several family members have participated in macrophage polarization and OC formation, including STAT1, STAT3, and STAT6 (216, 217). Growing studies reveal the involvement of the JAK/STAT pathway in regulating multiple biological events, such as innate and adaptive immunity, cell growth and differentiation, and programmed cell death (217). In the aspect of functioning, phosphorylated STATs promote monomeric dimerization through their SH2 domains and further translocation into the nucleus, where STATs regulate the transcription of target genes (216, 217).
5.4.2 JAK/STAT signaling in macrophage polarization
Several studies have revealed the mediating role of STATs in macrophage polarization. PARP14 silencing or RBM4 knockdown can promote IFN-γ-induced signaling transduction via STAT1 activation, which leads to M1 polarization in macrophages (218, 219). Also, STAT1/6 pathway mediates M1/M2 polarization in macrophages after physalin D stimulation (220). By phosphorylating STAT6, protocatechuic acid (PCA) decreases the activation of NF-κB signaling and gives a bias towards M2 polarization over M1 polarization in macrophages (173). Conclusively, an obvious antagonism between STAT1 and STAT6 has been described to promote M1 and M2 cell polarization, respectively (220). In addition, the activation of JAK/STAT3 signaling facilitates macrophage transformation to M2c polarization (180). In an AL mouse model, compared with the pure stimulation of UHMWPE particles, the extra addition of IL-10 significantly decreases iNOS-positive cells and increases CD163-positive cells via reducing the transcription of STAT1, NF-κB p65, and JNK1, and promoting the expression of STAT6 (164).
5.4.3 JAK/STAT signaling in osteoclastogenesis
Compared with STAT1 and STAT6, STAT3 is closely related to osteoclastogenesis. Therefore, inhibition of STAT3 can negatively affect RANKL-mediated OC formation and functions (221). An in vivo experiment demonstrated that treatment with TiPs pellet could promote the expression of STAT3 and production of RANKL in OBs, thereby stimulating OCs formation in particles-induced osteolysis models, whereas the activation of STAT3 also mediates nano-particles-induced IL-6-dependent inflammatory response in OBs (222).
5.5 Calcium (Ca2+) signaling
5.5.1 Overview of Ca2+ signaling
The centration of Ca2+ in the cytoplasm ([Ca2+]c) is 20, 000 times lower than that outside the cell. This is achieved by the Ca2+-related transportation and exchange under the dependence on specific proteins. When activated by a variety of external stimuli, cells respond by an increase in the [Ca2+]c and trigger downstream signaling, in the form of Ca2+ spikes or oscillations (223). Ca2+ signaling participates in various biological processes, resulting from a complex switch between the activation and inactivation of Ca2+-permeable channels (224). The excitability of Ca2+ signaling is determined by intracellular Ca2+ oscillations, which are attributed by the extracellular Ca2+ influx, the effect of ITAM/PLCγ/IP3 signaling on the release of Ca2+ from ER, and the capacity of collecting Ca2+ from the cytosol by SERCA. In addition, depending on the depletion of ER Ca2+ storage, extracellular Ca2+ entry could also be accomplished by the activation of STIM-mediated TRPC channels and Orai1 channels, which is the so-called SOCE mechanism (225, 226) (Figure 3).
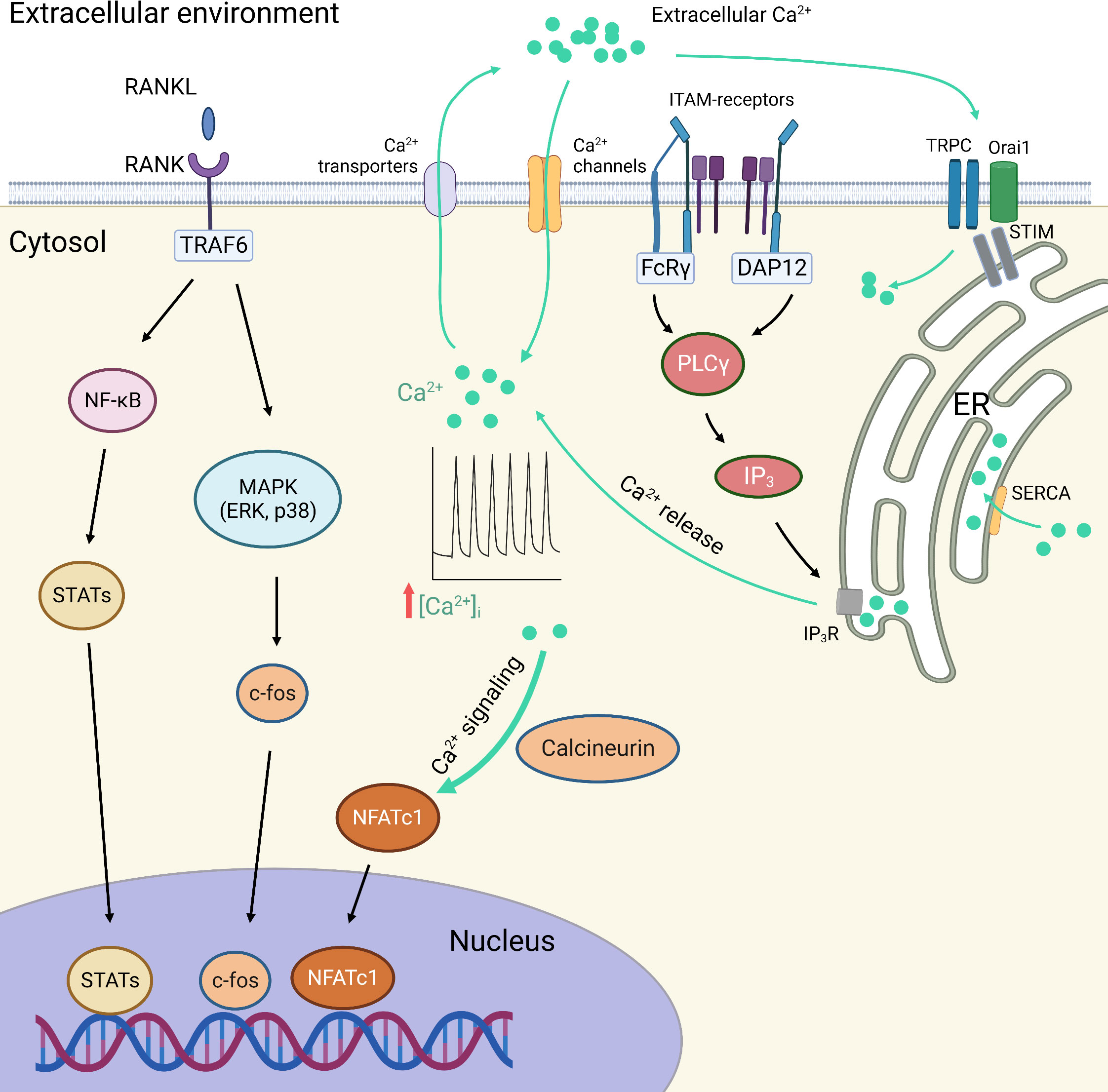
Figure 3 RANKL and calcium (Ca2+) signaling in macrophage polarization and osteoclastogenesis. The transcriptional factors responsible for macrophage polarization and osteoclastogenesis are regulated by the NF-κB and MAPK signaling pathways, as well as the Ca2+ signaling pathways. The excitability of Ca2+ signaling is determined by intracellular Ca2+ oscillations, which are attributed by the extracellular Ca2+ influx, the effect of ITAM/PLCγ/IP3 signaling on the release of Ca2+ from ER, and the capacity of collecting Ca2+ from the cytosol by SERCA. In addition, depending on the depletion of ER Ca2+ storage, extracellular Ca2+ entry could also be accomplished by the activation of STIM-mediated TRPC channels and Orai1 channels, which is the so-called SOCE mechanism. Ca2+, calcium ions; DAP12, DNAX-activation protein of 12 kDa; ER, endoplasmic reticulum; FcRγ, Fc receptor gamma-chain; IP3, inositol 1,4,5-trisphosphate; IP3R, inositol 1,4,5-trisphosphate receptor; ITAM, immunoreceptor tyrosine-based activation motif; MAPK, mitogen-activated protein kinases; NFATc1, nuclear factor of activated T cells c1; PLCγ, phospholipase C-gamma; RANK, receptor activator of nuclear factor-kappa B; RANKL, receptor activator of nuclear factor-kappa B ligand; SERCA, Sarco/endoplasmic reticulum Ca(2+)-ATPase; SOCE, store-operated Ca2+ entry; STATs, signal transducers and activators of transcription; STIM, stromal interaction molecule; TRAF6, tumor necrosis factor receptor-associated factor 6; TRPC channels, transient receptor potential canonical channels.
5.5.2 Ca2+ signaling in macrophage polarization
Ca2+ signaling is related to behavior changes in macrophages, such as polarization and phagocytosis, largely depending on Ca2+ uptake in mitochondria (227). Many studies have implicated that distinct Ca2+ entry channels determine the IFN-induced M1 polarization or IL-4-induced M2 polarization. Naive or M2 macrophages exhibit a robust Ca2+ entry that is dependent on the activity of Orai1 channels (228). As a result, blockade of Ca2+ entry inhibits NF-κB/STAT1 or STAT6 signaling events and consequently lowers cytokine production that is essential for M1 or M2 polarization in macrophages (228). In detail, Ca2+ influx facilitates M1 polarization, enabling the high productivity of proinflammatory mediators, such as cytokines and chemokines (229). Transient receptor canonical ion channel 1 (TRPC1)-mediated calcium entry seems to play a crucial role in M1 polarization, due to a non-selective TRPC1 current is found in macrophages with M1 phenotype (228, 230). Therefore, knockdown or blockade of the Kir2.1 channel significantly suppresses M1 polarization and promotes M2 polarization (231). Of note, a study found that transient receptor potential vanilloid 1 (TRPV1)-induced Ca2+ influx could promote the phosphorylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), but leads to the inhibition of M1 polarization (232), bringing uncertainty in elucidating regulatory mechanism between Ca2+ signaling and macrophage polarization.
5.5.3 Ca2+ signaling in osteoclastogenesis
Except for macrophage polarization, Ca2+ oscillations are the well-known mechanism in triggering RANKL-induced osteoclastogenesis and bone resorption (233, 234). The increase in the [Ca2+]c is a fundamental process for mediating OC biology, involving in OC proliferation, differentiation, and resorptive function. At the molecular level, cytosolic Ca2+ binds to calmodulin and subsequently activates calcineurin, leading to the activation of NFATc1 which is required for OC differentiation (235). Therefore, by blocking the Ca2+ entry channels, inhibition of the activity of Ca2+/calmodulin‐dependent protein kinase IV (CaMKIV) and calcineurin will lead to a reduction in the nuclear translocation of c-Fos and NFATc1, ultimately resulting in the suppression of osteoclastogenesis (236). Similarly, directly interfering with intracellular Ca2+ oscillations would also have the same negative impact on osteoclastogenesis (237). More content associated with Ca2+ oscillations in OC biology has been reviewed in the article by Okada H, et al. (234).
6 Future directions
Functional changes of macrophages reflect the intricate and constant regulation of the local environment by the network of cells and cytokines. While macrophage phenotypes have been roughly classified into representative M1 and M2 subtypes, investigating the concrete mechanism for the shift between proinflammatory phenotype and anti-inflammatory state is still challenging, especially in identifying responsible genes and proteins. In this regard, Ca2+ signaling based on different stimuli shows bidirectional effects on both macrophage polarization and osteoclast activation, which may be worth further investigation. While it seems clear that the behavior changes of macrophages, such as phagocytosis, pyroptosis, and apoptosis, are closely related to AL pathology, the triggered conditions and signaling events involved have not been fully elucidated. While PAMPs and DAMPs contribute to the response of macrophages to wear particles via TLRs, the main reason for the associated activation of TLRs is still controversial. Likewise, DAMPs affect macrophage programming is also unclear. Therefore, the role of PAMPs and DAMPs in AL still needs to be further elucidated. In addition, the local environment in periprosthetic tissue is complex due to the diversity of involved immune cells, such as dendritic cells and lymphocytes, thus highlighting the importance of intercellular communications via different signals. To address unresolved issues, applying CRISPR/Cas9 technology which enables accurate and efficient genome editing, would help in effectively elucidating the underlying mechanisms of macrophages in AL (238).
For the therapeutic strategy of AL, we believe the relief of inflammation and inhibition of bone resorption are keys to success. Through an in-depth study into the mechanisms of interaction between cytokines and macrophages, we may instruct a more accurate regulation of the inflammatory process in order to maintain a balance between immune defense and tissue homeostasis.
Author contributions
YC mainly contributed to the topic determination, literature selection, and draft written. YW (leading), TY, ZZ, JG, and QM screened detailed information from the literature and helped in the manuscript discussion. SS and ZL approved the direction of the topic, revised the manuscript, and supervised the work.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 82100936 and 82272485) and Taishan Scholars Foundation of Shandong Province (no. tsqnz20221170).
Acknowledgments
Draft of Figures 1–3 was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Price AJ, Alvand A, Troelsen A, Katz JN, Hooper G, Gray A, et al. Knee replacement. Lancet (London England). (2018) 392(10158):1672–82. doi: 10.1016/S0140-6736(18)32344-4
2. Jiang Y, Jia T, Wooley PH, Yang SY. Current research in the pathogenesis of aseptic implant loosening associated with particulate wear debris. Acta orthopaedica Belgica. (2013) 79(1):1–9.
3. Zhang L, Haddouti EM, Welle K, Burger C, Wirtz DC, Schildberg FA, et al. The effects of biomaterial implant Wear debris on osteoblasts. Front Cell Dev Biol (2020) 8:352. doi: 10.3389/fcell.2020.00352
4. Abu-Amer Y, Darwech I, Clohisy JC. Aseptic loosening of total joint replacements: Mechanisms underlying osteolysis and potential therapies. Arthritis Res Ther (2007) 9(Suppl 1):S6. doi: 10.1186/ar2170
5. Hodges NA, Sussman EM, Stegemann JP. Aseptic and septic prosthetic joint loosening: Impact of biomaterial wear on immune cell function, inflammation, and infection. Biomaterials (2021) 278:121127. doi: 10.1016/j.biomaterials.2021.121127
6. Goodman SB, Gallo J. Periprosthetic osteolysis: Mechanisms, prevention and treatment. J Clin Med (2019) 8(12). doi: 10.3390/jcm8122091
7. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci (2017) 19(1). doi: 10.3390/ijms19010092
8. Juhas U, Ryba-Stanisławowska M, Szargiej P, Myśliwska J. Different pathways of macrophage activation and polarization. Postepy higieny i medycyny doswiadczalnej (2015) 69:496–502. doi: 10.5604/17322693.1150133
9. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
10. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology (2018) 154(2):186–95. doi: 10.1111/imm.12910
11. Muñoz J, Akhavan NS, Mullins AP, Arjmandi BH. Macrophage polarization and osteoporosis: A review. Nutrients (2020) 12(10). doi: 10.3390/nu12102999
12. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
13. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
14. Liu W, Li J, Cheng M, Wang Q, Yeung KWK, Chu PK, et al. Zinc-modified sulfonated polyetheretherketone surface with immunomodulatory function for guiding cell fate and bone regeneration. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2018) 5(10):1800749. doi: 10.1002/advs.201800749
15. Qiu J, Peng P, Xin M, Wen Z, Chen Z, Lin S, et al. ZBTB20-mediated titanium particle-induced peri-implant osteolysis by promoting macrophage inflammatory responses. Biomaterials science. (2020) 8(11):3147–63. doi: 10.1039/D0BM00147C
16. Harris WH, Schiller AL, Scholler JM, Freiberg RA, Scott R. Extensive localized bone resorption in the femur following total hip replacement. J Bone Joint Surg Am volume. (1976) 58(5):612–8. doi: 10.2106/00004623-197658050-00005
17. Anil U, Singh V, Schwarzkopf R. Diagnosis and detection of subtle aseptic loosening in total hip arthroplasty. J arthroplasty. (2022) 37(8):1494–500. doi: 10.1016/j.arth.2022.02.060
18. Delanois RE, Mistry JB, Gwam CU, Mohamed NS, Choksi US, Mont MA. Current epidemiology of revision total knee arthroplasty in the united states. J arthroplasty. (2017) 32(9):2663–8. doi: 10.1016/j.arth.2017.03.066
19. Tarazi JM, Chen Z, Scuderi GR, Mont MA. The epidemiology of revision total knee arthroplasty. J knee surgery. (2021) 34(13):1396–401. doi: 10.1055/s-0041-1735282
20. Schwartz AM, Farley KX, Guild GN, Bradbury TL Jr. Projections and Epidemiology of Revision Hip and Knee Arthroplasty in the United States to 2030. J Arthroplasty (2020) 35(6S):S79–85. doi: 10.1016/j.arth.2020.02.030
21. Sundfeldt M, Carlsson LV, Johansson CB, Thomsen P, Gretzer C. Aseptic loosening, not only a question of wear: A review of different theories. Acta orthopaedica. (2006) 77(2):177–97. doi: 10.1080/17453670610045902
22. Wei J, Cai X, Wang Y, Chen H, Zhang B. [Mechanisms, prevention, and treatments of prosthetic aseptic loosening]. Zhongguo xiu fu chong jian wai ke za zhi = Zhongguo xiufu chongjian waike zazhi = Chin J reparative reconstructive Surg (2010) 24(3):296–300.
23. Schoenenberger AD, Schipanski A, Malheiro V, Kucki M, Snedeker JG, Wick P, et al. Macrophage polarization by titanium dioxide (TiO(2)) particles: Size matters. ACS biomaterials Sci engineering. (2016) 2(6):908–19. doi: 10.1021/acsbiomaterials.6b00006
24. Li B, Hu Y, Zhao Y, Cheng M, Qin H, Cheng T, et al. Curcumin attenuates titanium particle-induced inflammation by regulating macrophage polarization In vitro and in vivo. Front Immunol (2017) 8:55. doi: 10.3389/fimmu.2017.00055
25. Li C, Jiang C, Peng M, Li T, Yang Z, Liu Z, et al. Proinflammatory and osteolysis-inducing effects of 3D printing Ti6Al4V particles in vitro and in vivo. RSC Adv (2018) 8(4):2229–39. doi: 10.1039/C7RA12677H
26. Hu X, Xu L, Fu X, Huang J, Ji P, Zhang Z, et al. The TiO(2)-μ implant residual is more toxic than the Al(2)O(3)-n implant residual via blocking LAP and inducing macrophage polarization. Nanoscale (2021) 13(19):8976–90. doi: 10.1039/D1NR00696G
27. Li J, Li Y, Peng X, Li B, Qin H, Chen Y. In vivo analysis of the effects of CoCrMo and Ti particles on inflammatory responses and osteolysis. RSC advances. (2018) 8(10):5151–7. doi: 10.1039/C7RA12325F
28. Liu GY, Xu YS, Jiang WL, Leng NN, Chen JM. [Role and mechanism of endoplasmic reticulum stress response induced by wear particles in osteolysis]. Zhongguo gu shang = China J orthopaedics traumatology (2020) 33(12):1148–56. doi: 10.12200/j.issn.1003-0034.2020.12.012
29. Longhofer LK, Chong A, Strong NM, Wooley PH, Yang SY. Specific material effects of wear-particle-induced inflammation and osteolysis at the bone-implant interface: A rat model. J orthopaedic translation. (2017) 8:5–11. doi: 10.1016/j.jot.2016.06.026
30. Bechtel CP, Gebhart JJ, Tatro JM, Kiss-Toth E, Wilkinson JM, Greenfield EM. Particle-induced osteolysis is mediated by TIRAP/Mal in vitro and in vivo: Dependence on adherent pathogen-associated molecular patterns. J Bone Joint Surg Am volume (2016) 98(4):285–94. doi: 10.2106/JBJS.O.00736
31. Bi Y, Seabold JM, Kaar SG, Ragab AA, Goldberg VM, Anderson JM, et al. Adherent endotoxin on orthopedic wear particles stimulates cytokine production and osteoclast differentiation. J Bone mineral research: Off J Am Soc Bone Mineral Res (2001) 16(11):2082–91. doi: 10.1359/jbmr.2001.16.11.2082
32. Greenfield EM, Beidelschies MA, Tatro JM, Goldberg VM, Hise AG. Bacterial pathogen-associated molecular patterns stimulate biological activity of orthopaedic wear particles by activating cognate toll-like receptors. J Biol Chem (2010) 285(42):32378–84. doi: 10.1074/jbc.M110.136895
33. Hagman DS, Granade CM, Smith LS, Yakkanti MR, Malkani AL. Results of cemented posterior-stabilized total knee arthroplasty in obese patients with an average 10-year follow-up. J arthroplasty. (2020) 35(8):2097–100. doi: 10.1016/j.arth.2020.04.010
34. Koks S, Wood DJ, Reimann E, Awiszus F, Lohmann CH, Bertrand J, et al. The genetic variations associated with time to aseptic loosening after total joint arthroplasty. J arthroplasty. (2020) 35(4):981–8. doi: 10.1016/j.arth.2019.11.004
35. Lee SW, Kim WY, Song JH, Kim JH, Lee HH. Factors affecting periprosthetic bone loss after hip arthroplasty. Hip pelvis. (2021) 33(2):53–61. doi: 10.5371/hp.2021.33.2.53
36. Levent A, Suero EM, Gehrke T, Citak M. Risk factors for aseptic loosening after total knee arthroplasty with a rotating-hinge implant: A case-control study. J Bone Joint Surg Am volume. (2021) 103(6):517–23. doi: 10.2106/JBJS.20.00788
37. Mavčič B, Antolič V, Dolžan V. Association of NLRP3 and CARD8 inflammasome polymorphisms with aseptic loosening after primary total hip arthroplasty. J orthopaedic research: Off Publ Orthopaedic Res Soc (2020) 38(2):417–21. doi: 10.1002/jor.24474
38. Schiffner E, Latz D, Thelen S, Grassmann JP, Karbowski A, Windolf J, et al. Aseptic loosening after THA and TKA - do gender, tobacco use and BMI have an impact on implant survival time? J orthopaedics (2019) 16(3):269–72. doi: 10.1016/j.jor.2019.03.018
39. Fenelon C, Murphy EP, Fahey EJ, Murphy RP, O'Connell NM, Queally JM. Total knee arthroplasty in hemophilia: Survivorship and outcomes-a systematic review and meta-analysis. J arthroplasty. (2022) 37(3):581–92.e1. doi: 10.1016/j.arth.2021.10.015
40. Böhler C, Weimann P, Alasti F, Smolen JS, Windhager R, Aletaha D. Rheumatoid arthritis disease activity and the risk of aseptic arthroplasty loosening. Semin Arthritis rheumatism. (2020) 50(2):245–51. doi: 10.1016/j.semarthrit.2019.07.011
41. Christiansen RJ, Münch HJ, Bonefeld CM, Thyssen JP, Sloth JJ, Geisler C, et al. Cytokine profile in patients with aseptic loosening of total hip replacements and its relation to metal release and metal allergy. J Clin Med (2019) 8(8). doi: 10.3390/jcm8081259
42. Pajarinen J, Cenni E, Savarino L, Gomez-Barrena E, Tamaki Y, Takagi M, et al. Profile of toll-like receptor-positive cells in septic and aseptic loosening of total hip arthroplasty implants. J BioMed Mater Res A. (2010) 94(1):84–92. doi: 10.1002/jbm.a.32674
43. Naganuma Y, Takakubo Y, Hirayama T, Tamaki Y, Pajarinen J, Sasaki K, et al. Lipoteichoic acid modulates inflammatory response in macrophages after phagocytosis of titanium particles through toll-like receptor 2 cascade and inflammasomes. J BioMed Mater Res A. (2016) 104(2):435–44. doi: 10.1002/jbm.a.35581
44. Nalepka JL, Lee MJ, Kraay MJ, Marcus RE, Goldberg VM, Chen X, et al. Lipopolysaccharide found in aseptic loosening of patients with inflammatory arthritis. Clin orthopaedics related Res (2006) 451:229–35. doi: 10.1097/01.blo.0000224050.94248.38
45. Wasko MK, Goodman SB. Emperor's new clothes: Is particle disease really infected particle disease? J orthopaedic research: Off Publ Orthopaedic Res Soc (2016) 34(9):1497–504. doi: 10.1002/jor.23292
46. Eger M, Hiram-Bab S, Liron T, Sterer N, Carmi Y, Kohavi D, et al. Mechanism and prevention of titanium particle-induced inflammation and osteolysis. Front Immunol (2018) 9:2963. doi: 10.3389/fimmu.2018.02963
47. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol (2017) 79:593–617. doi: 10.1146/annurev-physiol-022516-034356
48. Wynn TA, Barron L. Macrophages: Master regulators of inflammation and fibrosis. Semin liver disease. (2010) 30(3):245–57. doi: 10.1055/s-0030-1255354
49. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest (2005) 115(1):56–65. doi: 10.1172/JCI200522675
50. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med (2013) 210(11):2477–91. doi: 10.1084/jem.20121999
51. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Sci (New York NY). (2011) 332(6035):1284–8. doi: 10.1126/science.1204351
52. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi: 10.1038/nri3073
53. Stratton-Powell AA, Williams S, Tipper JL, Redmond AC, Brockett CL. Mixed material wear particle isolation from periprosthetic tissue surrounding total joint replacements. J Biomed materials Res Part B Appl biomaterials (2022) 110(10):2276–89. doi: 10.1002/jbm.b.35076
54. Gao XR, Ge J, Li WY, Zhou WC, Xu L, Geng DQ. NF-κB/let-7f-5p/IL-10 pathway involves in wear particle-induced osteolysis by inducing M1 macrophage polarization. Cell Cycle (Georgetown Tex). (2018) 17(17):2134–45. doi: 10.1080/15384101.2018.1515549
55. Rao AJ, Gibon E, Ma T, Yao Z, Smith RL, Goodman SB. Revision joint replacement, wear particles, and macrophage polarization. Acta biomaterialia. (2012) 8(7):2815–23. doi: 10.1016/j.actbio.2012.03.042
56. Mahon OR, O'Hanlon S, Cunningham CC, McCarthy GM, Hobbs C, Nicolosi V, et al. Orthopaedic implant materials drive M1 macrophage polarization in a spleen tyrosine kinase- and mitogen-activated protein kinase-dependent manner. Acta biomaterialia. (2018) 65:426–35. doi: 10.1016/j.actbio.2017.10.041
57. Liu YW, An SB, Yang T, Xiao YJ, Wang L, Hu YH. Protection effect of curcumin for macrophage-involved polyethylene Wear particle-induced inflammatory osteolysis by increasing the cholesterol efflux. Med Sci monitor: Int Med J Exp Clin Res (2019) 25:10–20. doi: 10.12659/MSM.914197
58. Yan Z, Tian X, Zhu J, Lu Z, Yu L, Zhang D, et al. Metformin suppresses UHMWPE particle-induced osteolysis in the mouse calvaria by promoting polarization of macrophages to an anti-inflammatory phenotype. Mol Med (Cambridge Mass). (2018) 24(1):20. doi: 10.1186/s10020-018-0013-x
59. Percy MG, Gründling A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu Rev Microbiol (2014) 68:81–100. doi: 10.1146/annurev-micro-091213-112949
60. Venkataranganayaka Abhilasha K, Kedihithlu Marathe G. Bacterial lipoproteins in sepsis. Immunobiology (2021) 226(5):152128. doi: 10.1016/j.imbio.2021.152128
61. Byun EB, Sung NY, Park JN, Yang MS, Park SH, Byun EH. Gamma-irradiated resveratrol negatively regulates LPS-induced MAPK and NF-κB signaling through TLR4 in macrophages. Int immunopharmacol (2015) 25(2):249–59. doi: 10.1016/j.intimp.2015.02.015
62. Wu TT, Tai YT, Cherng YG, Chen TG, Lin CJ, Chen TL, et al. GATA-2 transduces LPS-induced il-1β gene expression in macrophages via a toll-like receptor 4/MD88/MAPK-dependent mechanism. PloS One (2013) 8(8):e72404. doi: 10.1371/journal.pone.0072404
63. Jang HM, Kang GD, Van Le TK, Lim SM, Jang DS, Kim DH. 4-methoxylonchocarpin attenuates inflammation by inhibiting lipopolysaccharide binding to toll-like receptor of macrophages and M1 macrophage polarization. Int immunopharmacol (2017) 45:90–7. doi: 10.1016/j.intimp.2017.02.003
64. Neut D, van Horn JR, van Kooten TG, van der Mei HC, Busscher HJ. Detection of biomaterial-associated infections in orthopaedic joint implants. Clin orthopaedics related Res (2003) 413):261–8. doi: 10.1097/01.blo.0000073345.50837.84
65. Friedlander AH. Oral cavity staphylococci are a potential source of prosthetic joint infection. Clin Infect diseases: an Off Publ Infect Dis Soc America (2010) 50(12):1682–3. doi: 10.1086/653003
66. Greenfield EM. Do genetic susceptibility, toll-like receptors, and pathogen-associated molecular patterns modulate the effects of wear? Clin orthopaedics related Res (2014) 472(12):3709–17. doi: 10.1007/s11999-014-3786-4
67. Wang G, Zhang P, Zhao J. Endotoxin contributes to artificial loosening of prostheses induced by titanium particles. Med Sci monitor: Int Med J Exp Clin Res (2018) 24:7001–6. doi: 10.12659/MSM.910039
68. Plüddemann A, Mukhopadhyay S, Gordon S. Innate immunity to intracellular pathogens: Macrophage receptors and responses to microbial entry. Immunol Rev (2011) 240(1):11–24. doi: 10.1111/j.1600-065X.2010.00989.x
69. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug discovery. (2012) 11(10):790–811. doi: 10.1038/nrd3810
70. Ramachandran P, Iredale JP, Fallowfield JA. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin liver disease. (2015) 35(2):119–31. doi: 10.1055/s-0035-1550057
71. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity (2014) 40(5):720–33. doi: 10.1016/j.immuni.2014.03.012
72. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity (2014) 40(5):706–19. doi: 10.1016/j.immuni.2014.03.011
73. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Advanced materials (Deerfield Beach Fla). (2020) 32(40):e2002054. doi: 10.1002/adma.202002054
74. Al Haq AT, Tseng HY, Chen LM, Wang CC, Hsu HL. Targeting prooxidant MnSOD effect inhibits triple-negative breast cancer (TNBC) progression and M2 macrophage functions under the oncogenic stress. Cell Death Dis (2022) 13(1):49. doi: 10.1038/s41419-021-04486-x
75. Wu X, Wang Z, Shi J, Yu X, Li C, Liu J, et al. Macrophage polarization toward M1 phenotype through NF-κB signaling in patients with behçet's disease. Arthritis Res Ther (2022) 24(1):249. doi: 10.1186/s13075-022-02938-z
76. Canton J. Phagosome maturation in polarized macrophages. J leukocyte Biol (2014) 96(5):729–38. doi: 10.1189/jlb.1MR0114-021R
77. Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci Rep (2019) 9(1):1925. doi: 10.1038/s41598-018-38127-9
78. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol (2015) 53(5):676–88. doi: 10.1165/rcmb.2015-0012OC
79. Li G, Sherchan P, Tang Z, Tang J. Autophagy & phagocytosis in neurological disorders and their possible cross-talk. Curr neuropharmacol (2021) 19(11):1912–24. doi: 10.2174/1570159X19666210407150632
80. Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol (2002) 14(1):123–8. doi: 10.1016/S0952-7915(01)00307-7
81. Gordon S. Phagocytosis: An immunobiologic process. Immunity (2016) 44(3):463–75. doi: 10.1016/j.immuni.2016.02.026
82. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev (2016) 273(1):357–70. doi: 10.1111/imr.12453
83. Lee JW, Nam H, Kim LE, Jeon Y, Min H, Ha S, et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy (2019) 15(5):753–70. doi: 10.1080/15548627.2018.1556946
84. Xu SL, Lin Y, Zhu XZ, Liu D, Tong ML, Liu LL, et al. Autophagy promotes phagocytosis and clearance of treponema pallidum via the NLRP3 inflammasome in macrophages. J Eur Acad Dermatol Venereol (2020) 34(9):2111–9. doi: 10.1111/jdv.16463
85. Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-associated phagocytosis and inflammation. J Mol Biol (2017) 429(23):3561–76. doi: 10.1016/j.jmb.2017.08.012
86. Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci (2019) 132(5). doi: 10.1242/jcs.222984
87. Bonilla DL, Bhattacharya A, Sha Y, Xu Y, Xiang Q, Kan A, et al. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity (2013) 39(3):537–47. doi: 10.1016/j.immuni.2013.08.026
88. Das LM, Binko AM, Traylor ZP, Peng H, Lu KQ. Vitamin d improves sunburns by increasing autophagy in M2 macrophages. Autophagy (2019) 15(5):813–26. doi: 10.1080/15548627.2019.1569298
89. Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol (2021) 18(10):2431–42. doi: 10.1038/s41423-020-00567-7
90. Chang CP, Su YC, Lee PH, Lei HY. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy (2013) 9(4):619–21. doi: 10.4161/auto.23546
91. D'Arcy MS. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int (2019) 43(6):582–92. doi: 10.1002/cbin.11137
92. Elmore S. Apoptosis: A review of programmed cell death. Toxicol pathol (2007) 35(4):495–516. doi: 10.1080/01926230701320337
93. Maimon N, Zamir ZZ, Kalkar P, Zeytuni-Timor O, Schif-Zuck S, Larisch S, et al. The pro-apoptotic ARTS protein induces neutrophil apoptosis, efferocytosis, and macrophage reprogramming to promote resolution of inflammation. Apoptosis: an Int J programmed Cell Death (2020) 25(7-8):558–73. doi: 10.1007/s10495-020-01615-3
94. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol (Baltimore Md: 1950). (2012) 189(7):3508–20. doi: 10.4049/jimmunol.1200662
95. Nadella V, Wang Z, Johnson TS, Griffin M, Devitt A. Transglutaminase 2 interacts with syndecan-4 and CD44 at the surface of human macrophages to promote removal of apoptotic cells. Biochim Biophys Acta (2015) 1853(1):201–12. doi: 10.1016/j.bbamcr.2014.09.020
96. Catelas I, Petit A, Zukor DJ, Marchand R, Yahia L, Huk OL. Induction of macrophage apoptosis by ceramic and polyethylene particles in vitro. Biomaterials (1999) 20(7):625–30. doi: 10.1016/S0142-9612(98)00214-2
97. Wang Q, Ye C, Sun S, Li R, Shi X, Wang S, et al. Curcumin attenuates collagen-induced rat arthritis via anti-inflammatory and apoptotic effects. Int immunopharmacol (2019) 72:292–300. doi: 10.1016/j.intimp.2019.04.027
98. Schumacher MA, Hedl M, Abraham C, Bernard JK, Lozano PR, Hsieh JJ, et al. ErbB4 signaling stimulates pro-inflammatory macrophage apoptosis and limits colonic inflammation. Cell Death Dis (2017) 8(2):e2622. doi: 10.1038/cddis.2017.42
99. Landgraeber S, Putz S, Schlattjan M, Bechmann LP, Totsch M, Grabellus F, et al. Adiponectin attenuates osteolysis in aseptic loosening of total hip replacements. Acta biomaterialia. (2014) 10(1):384–93. doi: 10.1016/j.actbio.2013.08.031
100. He J, Wang Y, Xu LH, Qiao J, Ouyang DY, He XH. Cucurbitacin IIa induces caspase-3-dependent apoptosis and enhances autophagy in lipopolysaccharide-stimulated RAW 264.7 macrophages. Int Immunopharmacol (2013) 16(1):27–34. doi: 10.1016/j.intimp.2013.03.013
101. Erbilgin A, Seldin MM, Wu X, Mehrabian M, Zhou Z, Qi H, et al. Transcription factor Zhx2 deficiency reduces atherosclerosis and promotes macrophage apoptosis in mice. Arteriosclerosis thrombosis Vasc Biol (2018) 38(9):2016–27. doi: 10.1161/ATVBAHA.118.311266
102. Pagie S, Gérard N, Charreau B. Notch signaling triggered via the ligand DLL4 impedes M2 macrophage differentiation and promotes their apoptosis. Cell communication Signaling (2018) 16(1):4. doi: 10.1186/s12964-017-0214-x
103. Frank D, Vince JE. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death differentiation. (2019) 26(1):99–114. doi: 10.1038/s41418-018-0212-6
104. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
105. Xia W, Lu Z, Chen W, Zhou J, Zhao Y. Excess fatty acids induce pancreatic acinar cell pyroptosis through macrophage M1 polarization. BMC Gastroenterol. (2022) 22(1):72. doi: 10.1186/s12876-022-02146-8
106. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care (London England). (2021) 25(1):356. doi: 10.1186/s13054-021-03775-3
107. Li N, Chen J, Geng C, Wang X, Wang Y, Sun N, et al. Myoglobin promotes macrophage polarization to M1 type and pyroptosis via the RIG-I/Caspase1/GSDMD signaling pathway in CS-AKI. Cell Death discovery. (2022) 8(1):90. doi: 10.1038/s41420-022-00894-w
108. Shu B, Zhou YX, Li H, Zhang RZ, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death discovery. (2021) 7(1):368. doi: 10.1038/s41420-021-00756-x
109. Chen L, Yu C, Xu W, Xiong Y, Cheng P, Lin Z, et al. Dual-targeted nanodiscs revealing the cross-talk between osteogenic differentiation of mesenchymal stem cells and macrophages. ACS nano (2023) 17(3):3153–67. doi: 10.1021/acsnano.2c12440
110. Wu YL, Zhang CH, Teng Y, Pan Y, Liu NC, Liu PX, et al. Propionate and butyrate attenuate macrophage pyroptosis and osteoclastogenesis induced by CoCrMo alloy particles. Military Med Res (2022) 9(1):46. doi: 10.1186/s40779-022-00404-0
111. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature (2003) 423(6937):337–42. doi: 10.1038/nature01658
112. Amarasekara DS, Kim S, Rho J. Regulation of osteoblast differentiation by cytokine networks. Int J Mol Sci (2021) 22(6). doi: 10.3390/ijms22062851
113. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet (2003) 4(8):638–49. doi: 10.1038/nrg1122
114. Park JH, Lee NK, Lee SY. Current understanding of RANK signaling in osteoclast differentiation and maturation. Molecules Cells (2017) 40(10):706–13. doi: 10.14348/molcells.2017.0225
115. Cui Z, Feng C, Chen J, Wang Y, Meng Q, Zhao S, et al. Network pharmacology deciphers the action of bioactive polypeptide in attenuating inflammatory osteolysis via the suppression of oxidative stress and restoration of bone remodeling balance. Oxid Med Cell longevity. (2022) 2022:4913534. doi: 10.1155/2022/4913534
116. Veis DJ, O'Brien CA. Osteoclasts, master sculptors of bone. Annu Rev Pathol (2022) 24(18):257–81. doi: 10.1146/annurev-pathmechdis-031521-040919
117. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in cryptococcus neoformans infection. mBio (2013) 4(3):e00264–13. doi: 10.1128/mBio.00264-13
118. Cai Y, Wen J, Ma S, Mai Z, Zhan Q, Wang Y, et al. Huang-Lian-Jie-Du decoction attenuates atherosclerosis and increases plaque stability in high-fat diet-induced ApoE(-/-) mice by inhibiting M1 macrophage polarization and promoting M2 macrophage polarization. Front Physiol (2021) 12:666449. doi: 10.3389/fphys.2021.666449
119. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
120. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
121. Wang C, Yu X, Cao Q, Wang Y, Zheng G, Tan TK, et al. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol (2013) 14:6. doi: 10.1186/1471-2172-14-6
122. Porta C, Riboldi E, Ippolito A, Sica A. Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol (2015) 27(4):237–48. doi: 10.1016/j.smim.2015.10.003
123. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
124. Wang J, Li R, Peng Z, Hu B, Rao X, Li J. HMGB1 participates in LPS−induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF−κB signaling pathways. Int J Mol Med (2020) 45(1):61–80. doi: 10.3892/ijmm.2019.4402
125. Kandahari AM, Yang X, Laroche KA, Dighe AS, Pan D, Cui Q. A review of UHMWPE wear-induced osteolysis: The role for early detection of the immune response. Bone Res (2016) 4:16014. doi: 10.1038/boneres.2016.14
126. Cheng X, Jansson V, Kretzer JP, Bader R, Utzschneider S, Paulus AC. The expression levels of toll-like receptors after metallic particle and ion exposition in the synovium of a murine model. J Clin Med (2021) 10(16). doi: 10.3390/jcm10163489
127. Zhang C, Li C, Li S, Qin L, Luo M, Fu G, et al. Small heterodimer partner negatively regulates TLR4 signaling pathway of titanium particles-induced osteolysis in mice. J Biomed nanotechnol (2018) 14(3):609–18. doi: 10.1166/jbn.2018.2533
128. Vargas-Hernández O, Ventura-Gallegos JL, Ventura-Ayala ML, Torres M, Zentella A, Pedraza-Sánchez S. THP-1 cells increase TNF-α production upon LPS + soluble human IgG co-stimulation supporting evidence for TLR4 and fcγ receptors crosstalk. Cell Immunol (2020) 355:104146. doi: 10.1016/j.cellimm.2020.104146
129. Yuan J, Lin F, Chen L, Chen W, Pan X, Bai Y, et al. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology (2022) 30(2):487–98. doi: 10.1007/s10787-022-00942-y
130. Yamada C, Beron-Pelusso C, Algazzaz N, Heidari A, Luz D, Rawas-Qalaji M, et al. Age-dependent effect between MARCO and TLR4 on PMMA particle phagocytosis by macrophages. J Cell Mol Med (2019) 23(8):5827–31. doi: 10.1111/jcmm.14494
131. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune network. (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
132. Galli G, Vacher P, Ryffel B, Blanco P, Legembre P. Fas/CD95 signaling pathway in damage-associated molecular pattern (DAMP)-sensing receptors. Cells (2022) 11(9). doi: 10.3390/cells11091438
133. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
134. Samelko L, Landgraeber S, McAllister K, Jacobs J, Hallab NJ. Cobalt alloy implant debris induces inflammation and bone loss primarily through danger signaling, not TLR4 activation: Implications for DAMP-ening implant related inflammation. PloS One (2016) 11(7):e0160141. doi: 10.1371/journal.pone.0160141
135. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi: 10.1038/nature00858
136. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature (2009) 462(7269):99–103. doi: 10.1038/nature08512
137. Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol (2015) 308(1):F69–75. doi: 10.1152/ajprenal.00484.2014
138. Jiang Y, Chen R, Shao X, Ji X, Lu H, Zhou S, et al. HMGB1 silencing in macrophages prevented their functional skewing and ameliorated EAM development: Nuclear HMGB1 may be a checkpoint molecule of macrophage reprogramming. Int immunopharmacol (2018) 56:277–84. doi: 10.1016/j.intimp.2018.01.013
139. Shiau DJ, Kuo WT, Davuluri GVN, Shieh CC, Tsai PJ, Chen CC, et al. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci Rep (2020) 10(1):13582. doi: 10.1038/s41598-020-70137-4
140. Yang X, Zhang B, Yu P, Liu M, Zhang C, Su E, et al. HMGB1 in macrophage nucleus protects against pressure overload induced cardiac remodeling via regulation of macrophage differentiation and inflammatory response. Biochem Biophys Res Commun (2022) 611:91–8. doi: 10.1016/j.bbrc.2022.04.053
141. Qu H, Heinbäck R, Salo H, Ewing E, Espinosa A, Aulin C, et al. Transcriptomic profiling reveals that HMGB1 induces macrophage polarization different from classical M1. Biomolecules (2022) 12(6). doi: 10.3390/biom12060779
142. Fagone P, Di Rosa M, Palumbo M, De Gregorio C, Nicoletti F, Malaguarnera L. Modulation of heat shock proteins during macrophage differentiation. Inflammation research: Off J Eur Histamine Res Soc [et al]. (2012) 61(10):1131–9. doi: 10.1007/s00011-012-0506-y
143. Geng T, Sun S, Chen X, Wang B, Guo H, Zhang S, et al. Strontium ranelate reduces the progression of titanium particle-induced osteolysis by increasing the ratio of osteoprotegerin to receptor activator of nuclear factor-κB ligand in vivo. Mol Med Rep (2018) 17(3):3829–36. doi: 10.3892/mmr.2017.8292
144. Hu S, Xue Y, He J, Chen C, Sun J, Jin Y, et al. Irisin recouples osteogenesis and osteoclastogenesis to protect wear-particle-induced osteolysis by suppressing oxidative stress and RANKL production. Biomaterials science. (2021) 9(17):5791–801. doi: 10.1039/D1BM00563D
145. Liao L, Lin Y, Liu Q, Zhang Z, Hong Y, Ni J, et al. Cepharanthine ameliorates titanium particle-induced osteolysis by inhibiting osteoclastogenesis and modulating OPG/RANKL ratio in a murine model. Biochem Biophys Res Commun (2019) 517(3):407–12. doi: 10.1016/j.bbrc.2019.07.115
146. Wang Q, Ge G, Liang X, Bai J, Wang W, Zhang W, et al. Punicalagin ameliorates wear-particle-induced inflammatory bone destruction by bi-directional regulation of osteoblastic formation and osteoclastic resorption. Biomaterials science. (2020) 8(18):5157–71. doi: 10.1039/D0BM00718H
147. Souza PP, Lerner UH. The role of cytokines in inflammatory bone loss. Immunol investigations. (2013) 42(7):555–622. doi: 10.3109/08820139.2013.822766
148. Luo G, Li F, Li X, Wang ZG, Zhang B. TNF−α and RANKL promote osteoclastogenesis by upregulating RANK via the NF−κB pathway. Mol Med Rep (2018) 17(5):6605–11. doi: 10.3892/mmr.2018.8698
149. AlQranei MS, Senbanjo LT, Aljohani H, Hamza T, Chellaiah MA. Lipopolysaccharide- TLR-4 axis regulates osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol (2021) 22(1):23. doi: 10.1186/s12865-021-00409-9
150. Wang L, Wang Q, Wang W, Ge G, Xu N, Zheng D, et al. Harmine alleviates titanium particle-induced inflammatory bone destruction by immunomodulatory effect on the macrophage polarization and subsequent osteogenic differentiation. Front Immunol (2021) 12:657687. doi: 10.3389/fimmu.2021.657687
151. Zhu K, Yang C, Dai H, Li J, Liu W, Luo Y, et al. Crocin inhibits titanium particle-induced inflammation and promotes osteogenesis by regulating macrophage polarization. Int immunopharmacol (2019) 76:105865. doi: 10.1016/j.intimp.2019.105865
152. Lin T, Kohno Y, Huang JF, Romero-Lopez M, Pajarinen J, Maruyama M, et al. NFκB sensing IL-4 secreting mesenchymal stem cells mitigate the proinflammatory response of macrophages exposed to polyethylene wear particles. J BioMed Mater Res A. (2018) 106(10):2744–52. doi: 10.1002/jbm.a.36504
153. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
154. Huang X, Li Y, Fu M, Xin HB. Polarizing macrophages in vitro. Methods Mol Biol (Clifton NJ) (2018) 1784:119–26. doi: 10.1007/978-1-4939-7837-3_12
155. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J pathol (2015) 185(10):2596–606. doi: 10.1016/j.ajpath.2015.06.001
156. Lin CW, Chen CC, Huang WY, Chen YY, Chen ST, Chou HW, et al. Restoring Prohealing/Remodeling-Associated M2a/c Macrophages Using ON101 Accelerates Diabetic Wound Healing. JID Innov (2020) 2(5):100138. doi: 10.1016/j.xjidi.2022.100138
157. Yue Y, Yang X, Feng K, Wang L, Hou J, Mei B, et al. M2b macrophages reduce early reperfusion injury after myocardial ischemia in mice: A predominant role of inhibiting apoptosis via A20. Int J Cardiol (2017) 245:228–35. doi: 10.1016/j.ijcard.2017.07.085
158. Quero L, Hanser E, Manigold T, Tiaden AN, Kyburz D. TLR2 stimulation impairs anti-inflammatory activity of M2-like macrophages, generating a chimeric M1/M2 phenotype. Arthritis Res Ther (2017) 19(1):245. doi: 10.1186/s13075-017-1447-1
159. Abdollahi E, Keyhanfar F, Delbandi AA, Falak R, Hajimiresmaiel SJ, Shafiei M. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-κB activation in human endothelial cells and differentiated macrophages. Eur J Pharmacol (2022) 918:174715. doi: 10.1016/j.ejphar.2021.174715
160. Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr., Medzhitov R, Flavell RA. IRAK-m is a negative regulator of toll-like receptor signaling. Cell (2002) 110(2):191–202. doi: 10.1016/S0092-8674(02)00827-9
161. Shen P, Li Q, Ma J, Tian M, Hong F, Zhai X, et al. IRAK-m alters the polarity of macrophages to facilitate the survival of mycobacterium tuberculosis. BMC Microbiol (2017) 17(1):185. doi: 10.1186/s12866-017-1095-2
162. Zhang Y, Yu S, Xiao J, Hou C, Li Z, Zhang Z, et al. Wear Particles promote endotoxin tolerance in macrophages by inducing interleukin-1 receptor-associated kinase-m expression. J BioMed Mater Res A. (2013) 101(3):733–9. doi: 10.1002/jbm.a.34375
163. Saraiva M, Vieira P, O'Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020) 217(1). doi: 10.1084/jem.20190418
164. Jiang J, Jia T, Gong W, Ning B, Wooley PH, Yang SY. Macrophage polarization in IL-10 treatment of particle-induced inflammation and osteolysis. Am J pathol (2016) 186(1):57–66. doi: 10.1016/j.ajpath.2015.09.006
165. Goodman S, Trindade M, Ma T, Lee M, Wang N, Ikenou T, et al. Modulation of bone ingrowth and tissue differentiation by local infusion of interleukin-10 in the presence of ultra-high molecular weight polyethylene (UHMWPE) wear particles. J BioMed Mater Res A. (2003) 65(1):43–50. doi: 10.1002/jbm.a.10279
166. Antonios JK, Yao Z, Li C, Rao AJ, Goodman SB. Macrophage polarization in response to wear particles in vitro. Cell Mol Immunol (2013) 10(6):471–82. doi: 10.1038/cmi.2013.39
167. Gong L, Zhao Y, Zhang Y, Ruan Z. The macrophage polarization regulates MSC osteoblast differentiation in vitro. Ann Clin Lab Sci (2016) 46(1):65–71.
168. Zhang Y, Böse T, Unger RE, Jansen JA, Kirkpatrick CJ, van den Beucken J. Macrophage type modulates osteogenic differentiation of adipose tissue MSCs. Cell Tissue Res (2017) 369(2):273–86. doi: 10.1007/s00441-017-2598-8
169. Philipp D, Suhr L, Wahlers T, Choi YH, Paunel-Görgülü A. Preconditioning of bone marrow-derived mesenchymal stem cells highly strengthens their potential to promote IL-6-dependent M2b polarization. Stem Cell Res Ther (2018) 9(1):286. doi: 10.1186/s13287-018-1039-2
170. MacKenzie KF, Clark K, Naqvi S, McGuire VA, Nöehren G, Kristariyanto Y, et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase a-SIK-CRTC3 pathway. J Immunol (Baltimore Md: 1950). (2013) 190(2):565–77. doi: 10.4049/jimmunol.1202462
171. Holthaus M, Santhakumar N, Wahlers T, Paunel-Görgülü A. The secretome of preconditioned mesenchymal stem cells drives polarization and reprogramming of M2a macrophages toward an IL-10-Producing phenotype. Int J Mol Sci (2022) 23(8). doi: 10.3390/ijms23084104
172. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators inflammation. (2015) 2015:816460. doi: 10.1155/2015/816460
173. Liu Y, Wang X, Pang J, Zhang H, Luo J, Qian X, et al. Attenuation of atherosclerosis by protocatechuic acid via inhibition of M1 and promotion of M2 macrophage polarization. J Agric Food Chem (2019) 67(3):807–18. doi: 10.1021/acs.jafc.8b05719
174. Li Z, Zhu X, Xu R, Wang Y, Hu R, Xu W. Deacylcynaropicrin inhibits RANKL-induced osteoclastogenesis by inhibiting NF-κB and MAPK and promoting M2 polarization of macrophages. Front Pharmacol (2019) 10:599. doi: 10.3389/fphar.2019.00599
175. Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Trans Res (2019) 11(2):655–68.
176. Li Y, Zhang Z, Xu K, Du S, Gu X, Cao R, et al. Minocycline alleviates peripheral nerve adhesion by promoting regulatory macrophage polarization via the TAK1 and its downstream pathway. Life Sci (2021) 276:119422. doi: 10.1016/j.lfs.2021.119422
177. Yu B, Bai J, Shi J, Shen J, Guo X, Liu Y, et al. MiR-106b inhibition suppresses inflammatory bone destruction of wear debris-induced periprosthetic osteolysis in rats. J Cell Mol Med (2020) 24(13):7490–503. doi: 10.1111/jcmm.15376
178. Lin Y, Zhao JL, Zheng QJ, Jiang X, Tian J, Liang SQ, et al. Notch signaling modulates macrophage polarization and phagocytosis through direct suppression of signal regulatory protein α expression. Front Immunol (2018) 9:1744. doi: 10.3389/fimmu.2018.01744
179. Chen J, Cui Z, Wang Y, Lyu L, Feng C, Feng D, et al. Cyclic polypeptide D7 protects bone marrow mesenchymal cells and promotes chondrogenesis during osteonecrosis of the femoral head via growth differentiation factor 15-mediated redox signaling. Oxid Med Cell longevity. (2022) 2022:3182368. doi: 10.1155/2022/3182368
180. Miki S, Suzuki JI, Takashima M, Ishida M, Kokubo H, Yoshizumi M. S-1-Propenylcysteine promotes IL-10-induced M2c macrophage polarization through prolonged activation of IL-10R/STAT3 signaling. Sci Rep (2021) 11(1):22469. doi: 10.1038/s41598-021-01866-3
181. Mia S, Warnecke A, Zhang XM, Malmström V, Harris RA. An optimized protocol for human M2 macrophages using m-CSF and IL-4/IL-10/TGF-β yields a dominant immunosuppressive phenotype. Scand J Immunol (2014) 79(5):305–14. doi: 10.1111/sji.12162
182. Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
183. Gay NJ, Gangloff M, O'Neill LA. What the myddosome structure tells us about the initiation of innate immunity. Trends Immunol (2011) 32(3):104–9. doi: 10.1016/j.it.2010.12.005
184. Li H, Yoon JH, Won HJ, Ji HS, Yuk HJ, Park KH, et al. Isotrifoliol inhibits pro-inflammatory mediators by suppression of TLR/NF-κB and TLR/MAPK signaling in LPS-induced RAW264.7 cells. Int Immunopharmacol (2017) 45:110–9. doi: 10.1016/j.intimp.2017.01.033
185. Jayakumar T, Yang CM, Yen TL, Hsu CY, Sheu JR, Hsia CW, et al. Anti-inflammatory mechanism of an alkaloid rutaecarpine in LTA-stimulated RAW 264.7 cells: Pivotal role on NF-κB and ERK/p38 signaling molecules. Int J Mol Sci (2022) 23(11). doi: 10.3390/ijms23115889
186. Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol (2019) 10:276. doi: 10.3389/fimmu.2019.00276
187. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
188. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol (Baltimore Md 1950). (2015) 195(4):1685–97. doi: 10.4049/jimmunol.1500509
189. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20(13). doi: 10.3390/ijms20133328
190. Zhang BC, Li Z, Xu W, Xiang CH, Ma YF. Luteolin alleviates NLRP3 inflammasome activation and directs macrophage polarization in lipopolysaccharide-stimulated RAW264.7 cells. Am J Trans Res (2018) 10(1):265–73.
191. Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1β production in inflammatory root resorption. J Clin periodontol (2020) 47(4):451–60. doi: 10.1111/jcpe.13258
192. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6:7360. doi: 10.1038/ncomms8360
193. Lin S, Wen Z, Li S, Chen Z, Li C, Ouyang Z, et al. LncRNA Neat1 promotes the macrophage inflammatory response and acts as a therapeutic target in titanium particle-induced osteolysis. Acta biomaterialia. (2022) 142:345–60. doi: 10.1016/j.actbio.2022.02.007
194. Gao XR, Ge J, Li WY, Zhou WC, Xu L, Geng DQ. miR-34a carried by adipocyte exosomes inhibits the polarization of M1 macrophages in mouse osteolysis model. J BioMed Mater Res A. (2021) 109(6):994–1003. doi: 10.1002/jbm.a.37088
195. Mócsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol (2010) 10(6):387–402. doi: 10.1038/nri2765
196. Ma P, Yue L, Yang H, Fan Y, Bai J, Li S, et al. Chondroprotective and anti-inflammatory effects of amurensin h by regulating TLR4/Syk/NF-κB signals. J Cell Mol Med (2020) 24(2):1958–68. doi: 10.1111/jcmm.14893
197. Slomiany BL, Slomiany A. Helicobacter pylori LPS-induced gastric mucosal spleen tyrosine kinase (Syk) recruitment to TLR4 and activation occurs with the involvement of protein kinase cδ. Inflammopharmacology (2018) 26(3):805–15. doi: 10.1007/s10787-017-0430-4
198. Liu L, Lucas RM, Nanson JD, Li Y, Whitfield J, Curson JEB, et al. The transmembrane adapter SCIMP recruits tyrosine kinase syk to phosphorylate toll-like receptors to mediate selective inflammatory outputs. J Biol Chem (2022) 298(5):101857. doi: 10.1016/j.jbc.2022.101857
199. Chaudhary A, Fresquez TM, Naranjo MJ. Tyrosine kinase syk associates with toll-like receptor 4 and regulates signaling in human monocytic cells. Immunol Cell Biol (2007) 85(3):249–56. doi: 10.1038/sj.icb7100030
200. Li J, Chen YH, Li LZ, Wang F, Song W, Alolga RN, et al. Omics and transgenic analyses reveal that salvianolic acid b exhibits its anti-inflammatory effects through inhibiting the mincle-Syk-Related pathway in macrophages. J Proteome Res (2021) 20(7):3734–48. doi: 10.1021/acs.jproteome.1c00325
201. Malik AF, Hoque R, Ouyang X, Ghani A, Hong E, Khan K, et al. Inflammasome components asc and caspase-1 mediate biomaterial-induced inflammation and foreign body response. Proc Natl Acad Sci United States America. (2011) 108(50):20095–100. doi: 10.1073/pnas.1105152108
202. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature (2009) 459(7245):433–6. doi: 10.1038/nature07965
203. Maehara T, Fujimori K. Contribution of FP receptors in M1 macrophage polarization via IL-10-regulated nuclear translocation of NF-κB p65. Biochim Biophys Acta Mol Cell Biol lipids. (2020) 1865(5):158654. doi: 10.1016/j.bbalip.2020.158654
204. Glushkova OV, Parfenyuk SB, Khrenov MO, Novoselova TV, Lunin SM, Fesenko EE, et al. Inhibitors of TLR-4, NF-κB, and SAPK/JNK signaling reduce the toxic effect of lipopolysaccharide on RAW 264.7 cells. J immunotoxicol (2013) 10(2):133–40. doi: 10.3109/1547691X.2012.700652
205. Guangtao F, Zhenkang W, Zhantao D, Mengyuan L, Qingtian L, Yuanchen M, et al. Icariin alleviates Wear particle-induced periprosthetic osteolysis via down-regulation of the estrogen receptor α-mediated NF-κB signaling pathway in macrophages. Front Pharmacol (2021) 12:746391. doi: 10.3389/fphar.2021.746391
206. Liu Y, Song FM, Ma ST, Moro A, Feng WY, Liao SJ, et al. Vaccarin prevents titanium particle-induced osteolysis and inhibits RANKL-induced osteoclastogenesis by blocking NF-κB and MAPK signaling pathways. J Cell Physiol (2019) 234(8):13832–42. doi: 10.1002/jcp.28063
207. Zhang W, Jiang G, Zhou X, Huang L, Meng J, He B, et al. α-mangostin inhibits LPS-induced bone resorption by restricting osteoclastogenesis via NF-κB and MAPK signaling. Chin Med (2022) 17(1):34. doi: 10.1186/s13020-021-00555-7
208. An S, Han F, Hu Y, Liu Y, Li J, Wang L. Curcumin inhibits polyethylene-induced osteolysis via repressing NF-κB signaling pathway activation. Cell Physiol Biochem Int J Exp Cell physiol biochem Pharmacol (2018) 50(3):1100–12. doi: 10.1159/000494537
209. Li X, Lu Y, Li J, Zhou S, Wang Y, Li L, et al. Photoluminescent carbon dots (PCDs) from sour apple: A biocompatible nanomaterial for preventing UHMWPE wear-particle induced osteolysis via modulating Chemerin/ChemR23 and SIRT1 signaling pathway and its bioimaging application. J nanobiotechnol (2022) 20(1):301. doi: 10.1186/s12951-022-01498-3
210. Lee K, Seo I, Choi MH, Jeong D. Roles of mitogen-activated protein kinases in osteoclast biology. Int J Mol Sci (2018) 19(10). doi: 10.3390/ijms19103004
211. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev (2001) 81(2):807–69. doi: 10.1152/physrev.2001.81.2.807
212. Liu L, Guo H, Song A, Huang J, Zhang Y, Jin S, et al. Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol (2020) 21(1):32. doi: 10.1186/s12865-020-00355-y
213. Zhou F, Mei J, Han X, Li H, Yang S, Wang M, et al. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-κB/MAPK signaling and protecting chondrocytes. Acta Pharm Sin B (2019) 9(5):973–85. doi: 10.1016/j.apsb.2019.01.015
214. Lu J, Zhang H, Pan J, Hu Z, Liu L, Liu Y, et al. Fargesin ameliorates osteoarthritis via macrophage reprogramming by downregulating MAPK and NF-κB pathways. Arthritis Res Ther (2021) 23(1):142. doi: 10.1186/s13075-021-02512-z
215. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine (2008) 42(2):145–51. doi: 10.1016/j.cyto.2008.01.006
216. Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell (2013) 4(3):176–85. doi: 10.1007/s13238-013-2084-3
217. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal transduction targeted Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
218. Huangfu N, Zheng W, Xu Z, Wang S, Wang Y, Cheng J, et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int immunopharmacol (2020) 83:106432. doi: 10.1016/j.intimp.2020.106432
219. Iwata H, Goettsch C, Sharma A, Ricchiuto P, Goh WW, Halu A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun (2016) 7:12849. doi: 10.1038/ncomms12849
220. Ding N, Wang Y, Dou C, Liu F, Guan G, Wei K, et al. Physalin d regulates macrophage M1/M2 polarization via the STAT1/6 pathway. J Cell Physiol (2019) 234(6):8788–96. doi: 10.1002/jcp.27537
221. Hu J, Li X, Chen Y, Han X, Li L, Yang Z, et al. The protective effect of WKYMVm peptide on inflammatory osteolysis through regulating NF-κB and CD9/gp130/STAT3 signalling pathway. J Cell Mol Med (2020) 24(2):1893–905. doi: 10.1111/jcmm.14885
222. Deng Z, Zhang R, Li M, Wang S, Fu G, Jin J, et al. STAT3/IL-6 dependent induction of inflammatory response in osteoblast and osteoclast formation in nanoscale wear particle-induced aseptic prosthesis loosening. Biomaterials science. (2021) 9(4):1291–300. doi: 10.1039/D0BM01256D
223. Islam MS. Calcium signaling: From basic to bedside. Adv Exp Med Biol (2020) 1131:1–6. doi: 10.1007/978-3-030-12457-1_1
224. Dupont G, Combettes L, Bird GS, Putney JW. Calcium oscillations. Cold Spring Harbor Perspect Biol (2011) 3(3). doi: 10.1101/cshperspect.a004226
225. Li Z, Liu T, Gilmore A, Gómez NM, Fu C, Lim J, et al. Regulator of G protein signaling protein 12 (Rgs12) controls mouse osteoblast differentiation via calcium Channel/Oscillation and gαi-ERK signaling. J Bone mineral research: Off J Am Soc Bone Mineral Res (2019) 34(4):752–64. doi: 10.1002/jbmr.3645
226. Di Buduo CA, Abbonante V, Marty C, Moccia F, Rumi E, Pietra D, et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood (2020) 135(2):133–44. doi: 10.1182/blood.2019001103
227. Tedesco S, Scattolini V, Albiero M, Bortolozzi M, Avogaro A, Cignarella A, et al. Mitochondrial calcium uptake is instrumental to alternative macrophage polarization and phagocytic activity. Int J Mol Sci (2019) 20(19). doi: 10.3390/ijms20194966
228. Nascimento Da Conceicao V, Sun Y, Ramachandran K, Chauhan A, Raveendran A, Venkatesan M, et al. Resolving macrophage polarization through distinct Ca(2+) entry channel that maintains intracellular signaling and mitochondrial bioenergetics. iScience (2021) 24(11):103339. doi: 10.1016/j.isci.2021.103339
229. Ji SY, Lee H, Hwangbo H, Hong SH, Cha HJ, Park C, et al. A novel peptide oligomer of bacitracin induces M1 macrophage polarization by facilitating Ca(2+) influx. Nutrients (2020) 12(6). doi: 10.3390/nu12061603
230. Chauhan A, Sun Y, Sukumaran P, Quenum Zangbede FO, Jondle CN, Sharma A, et al. M1 macrophage polarization is dependent on TRPC1-mediated calcium entry. iScience (2018) 8:85–102. doi: 10.1016/j.isci.2018.09.014
231. Chen K, Man Q, Miao J, Xu W, Zheng Y, Zhou X, et al. Kir2.1 channel regulates macrophage polarization via the Ca2+/CaMK II/ERK/NF-κB signaling pathway. J Cell Sci (2022) 135(13). doi: 10.1242/jcs.259544
232. Lv Z, Xu X, Sun Z, Yang YX, Guo H, Li J, et al. TRPV1 alleviates osteoarthritis by inhibiting M1 macrophage polarization via Ca(2+)/CaMKII/Nrf2 signaling pathway. Cell Death Dis (2021) 12(6):504. doi: 10.1038/s41419-021-03792-8
233. Shi Y, Ye L, Shen S, Qian T, Pan Y, Jiang Y, et al. Morin attenuates osteoclast formation and function by suppressing the NF-κB, MAPK and calcium signalling pathways. Phytother Res (2021) 35(10):5694–707. doi: 10.1002/ptr.7229
234. Okada H, Okabe K, Tanaka S. Finely-tuned calcium oscillations in osteoclast differentiation and bone resorption. Int J Mol Sci (2020) 22(1). doi: 10.3390/ijms22010180
235. Kang JY, Kang N, Yang YM, Hong JH, Shin DM. The role of Ca(2+)-NFATc1 signaling and its modulation on osteoclastogenesis. Int J Mol Sci (2020) 21(10). doi: 10.3390/ijms21103646
236. Kim HJ, Lee J, Lee GR, Kim N, Lee HI, Kwon M, et al. Flunarizine inhibits osteoclastogenesis by regulating calcium signaling and promotes osteogenesis. J Cell Physiol (2021) 236(12):8239–52. doi: 10.1002/jcp.30496
237. Okada H, Kajiya H, Omata Y, Matsumoto T, Sato Y, Kobayashi T, et al. CTLA4-ig directly inhibits osteoclastogenesis by interfering with intracellular calcium oscillations in bone marrow macrophages. J Bone mineral research: Off J Am Soc Bone Mineral Res (2019) 34(9):1744–52. doi: 10.1002/jbmr.3754
Keywords: macrophage, polarization, aseptic loosening, arthroplasty, inflammation
Citation: Cong Y, Wang Y, Yuan T, Zhang Z, Ge J, Meng Q, Li Z and Sun S (2023) Macrophages in aseptic loosening: Characteristics, functions, and mechanisms. Front. Immunol. 14:1122057. doi: 10.3389/fimmu.2023.1122057
Received: 12 December 2022; Accepted: 13 February 2023;
Published: 08 March 2023.
Edited by:
Yanan Ma, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Wenjun Liu, Amgen, United StatesXixi Zhang, Dana–Farber Cancer Institute, United States
Xinhe Shan, Penn Medicine, United States
Jingkai Zhou, City of Hope National Medical Center, United States
Copyright © 2023 Cong, Wang, Yuan, Zhang, Ge, Meng, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziqing Li, bGl6aXFpbmdAc2RmbXUuZWR1LmNu; Shui Sun, c3Vuc2h1aUBzZGZtdS5lZHUuY24=
†These authors have contributed equally to this work