 Kana Unuma1*
Kana Unuma1* Dan Tomomasa2Kosuke Noma3Kouhei Yamamoto4Taka-aki Matsuyama5Yohsuke Makino6
Dan Tomomasa2Kosuke Noma3Kouhei Yamamoto4Taka-aki Matsuyama5Yohsuke Makino6 Atsushi Hijikata7Shuheng Wen1
Atsushi Hijikata7Shuheng Wen1 Tsutomu Ogata8Nobuhiko Okamoto9
Tsutomu Ogata8Nobuhiko Okamoto9 Satoshi Okada3Kenichi Ohashi4Koichi Uemura1
Satoshi Okada3Kenichi Ohashi4Koichi Uemura1 Hirokazu Kanegane10
Hirokazu Kanegane10- 1Department of Forensic Medicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 2Department of Pediatrics and Developmental Biology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 3Department of Pediatrics, Hiroshima University, Graduate School of Biomedical and Health Sciences, Hiroshima, Japan
- 4Department of Comprehensive Pathology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 5Department of Legal Medicine, Showa University School of Medicine, Tokyo, Japan
- 6Department of Forensic Medicine, The University of Tokyo, Tokyo, Japan
- 7Department of Life Sciences, Tokyo University of Pharmacy and Life Sciences, Hachioji, Tokyo, Japan
- 8Department of Pediatrics, Hamamatsu University School of Medicine, Hamamatsu, Japan
- 9Department of Medical Genetics, Osaka Women’s and Children’s Hospital, Izumi, Osaka, Japan
- 10Department of Child Health and Development, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
Herein, we report a child with COVID-19 and seemingly no underlying disease, who died suddenly. The autopsy revealed severe anemia and thrombocytopenia, splenomegaly, hypercytokinemia, and a rare ectopic congenital coronary origin. Immunohistochemical analysis demonstrated that the patient had acute lymphoblastic leukemia of the B-cell precursor phenotype (BCP-ALL). The complex cardiac and hematological abnormalities suggested the presence of an underlying disease; therefore, we performed whole-exome sequencing (WES). WES revealed a leucine-zipper-like transcription regulator 1 (LZTR1) variant, indicating Noonan syndrome (NS). Therefore, we concluded that the patient had underlying NS along with coronary artery malformation and that COVID-19 infection may have triggered the sudden cardiac death due to increased cardiac load caused by high fever and dehydration. In addition, multiple organ failure due to hypercytokinemia probably contributed to the patient’s death. This case would be of interest to pathologists and pediatricians because of the limited number of NS patients with LZTR1 variants; the complex combination of an LZTR1 variant, BCP-ALL, and COVID-19; and a rare pattern of the anomalous origin of the coronary artery. Thus, we highlight the significance of molecular autopsy and the application of WES with conventional diagnostic methods.
1 Introduction
Noonan syndrome (NS) is caused by pathogenic variants of genes encoding components of the RAS/MAPK signaling pathway (1), including PTPN11, SOS1, and RAF1 (2). Recently, a leucine-zipper-like transcription regulator 1 (LZTR1) variant was found to be associated with NS using whole-exome sequencing (WES) (1). In 2021, it was reported that the prevalence rate of LZTR1 variants in patients with NS was 4%–6% (2, 3), which was less than 50 cases (4).
The clinical characteristics of patients with NS harboring LZTR1 variants are similar to those with other NS genotypes, including epicanthal folds, low-set ears, blepharoptosis, webbed neck, pectus excavatum-carinatum, cryptorchidism, short stature, intellectual disability, and cardiac anomalies (5). Meanwhile, abnormalities in stature, cardiac function, and neurodevelopment are considerably different between these groups of patients (6). For example, typical characteristics of children with NS include short stature related to growth hormone deficiency; however, only four cases of growth hormone deficiency have been reported in patients with NS harboring LZTR1 variants (4).
Additionally, patients with NS typically present cardiovascular anomalies, with pulmonary stenosis, hypertrophic cardiomyopathy, and atrial septal defects—the most prevalent (3, 7). Correspondingly, 79.4% (27/34) of patients with NS harboring LZTR1 variants reported in 2019 had heart disease, the most frequent being hypertrophic cardiomyopathy (71.4%) (2, 8). In a study of eight Japanese patients with NS harboring LZTR1 variants, one patient (c. 742G>A, p.Gly248Arg, a variant in Kelch 4 domain) was diagnosed to have an anomalous origin of the coronary artery with peripheral pulmonary stenosis by echocardiography (8).
Patients with NS have an increased risk of hematological abnormalities (9–11); particularly, transient myeloproliferative disorder is observed in approximately 10% of pediatric patients (12). Juvenile myelomonocytic leukemia is another common hematological malignancy; approximately 90% of patients with NS and myelomonocytic leukemia have mutually exclusive PTPN11, NRAS, KRAS, NF1, or CBL variants (12). However, reports on association between LZTR1 variants and hematological abnormalities are scarce.
Herein, we present a child who might have died due to COVID-19. A molecular autopsy of the patient revealed LZTR1 variant-associated NS with a complex combination of acute lymphoblastic leukemia of the B-cell precursor (BCP-ALL) phenotype, and a rare ectopic congenital coronary origin.
2 Case history and symptoms at presentation
A 5-year-old boy, with no known medical history, developed a high fever (37.8°C) 6 days before his death. The patient was not administered any medication as the SARS-CoV-2 antigen test result was negative. On day 3 following fever onset, the patient’s body temperature dropped to below 37°C; however, his parents noticed that his face was abnormally pale. The fever (39.2°C) relapsed on day 5 after the initial episode. On day 6, the patient could barely drink or eat. When the patient’s mother tried to put him to bed, he moaned and convulsed with eyes open; the patient was rushed to the hospital emergency room via an ambulance. He went into cardiac arrest in the ambulance and died in the hospital despite the best efforts of cardiopulmonary resuscitation. The child’s blood analysis revealed severe anemia, thrombocytopenia, and hypercytokinemia, suggesting hemophagocytic lymphohistiocytosis (HLH). The detailed laboratory findings are summarized in Table 1.

Table 1 Representative laboratory findings of the patient’s blood analysis.
3 Autopsy findings
An autopsy was performed on the boy within 34 h after his death. Physical examination revealed the following: height, 108 cm; weight, 19.2 kg. No injuries were observed. The child was not prenatally diagnosed with any disease. The computed tomography scan showed no obvious acute or chronic fractures; furthermore, there were no major findings that pointed to a definite cause of death. The patient’s polymerase chain reaction test result was positive for SARS-CoV-2.
An autopsy revealed splenomegaly (140 g, normal range: 45–50 g) and hepatomegaly (810 g, normal range: 550–600 g). The heart weighed 122 g, which is within the normal range for a 5-year-old boy. We did not find an anomalous positioning, abnormal chamber arrangement, significant ventricular wall thickening, or chamber dilatation. However, an acute angle take-off of the left coronary artery (LCA) from the non-coronary cusp (NCC) was observed (Figure 1A). The left main trunk (LMT) passed through a long course along the Valsalva sinus wall (Figure 1B), and a histological section of the LMT revealed an eccentric intimal fibrous thickening indicative of approximately 50% stenosis (Figure 1C). No other macro/microscopic anomaly including COVID-19 pneumonia was found.
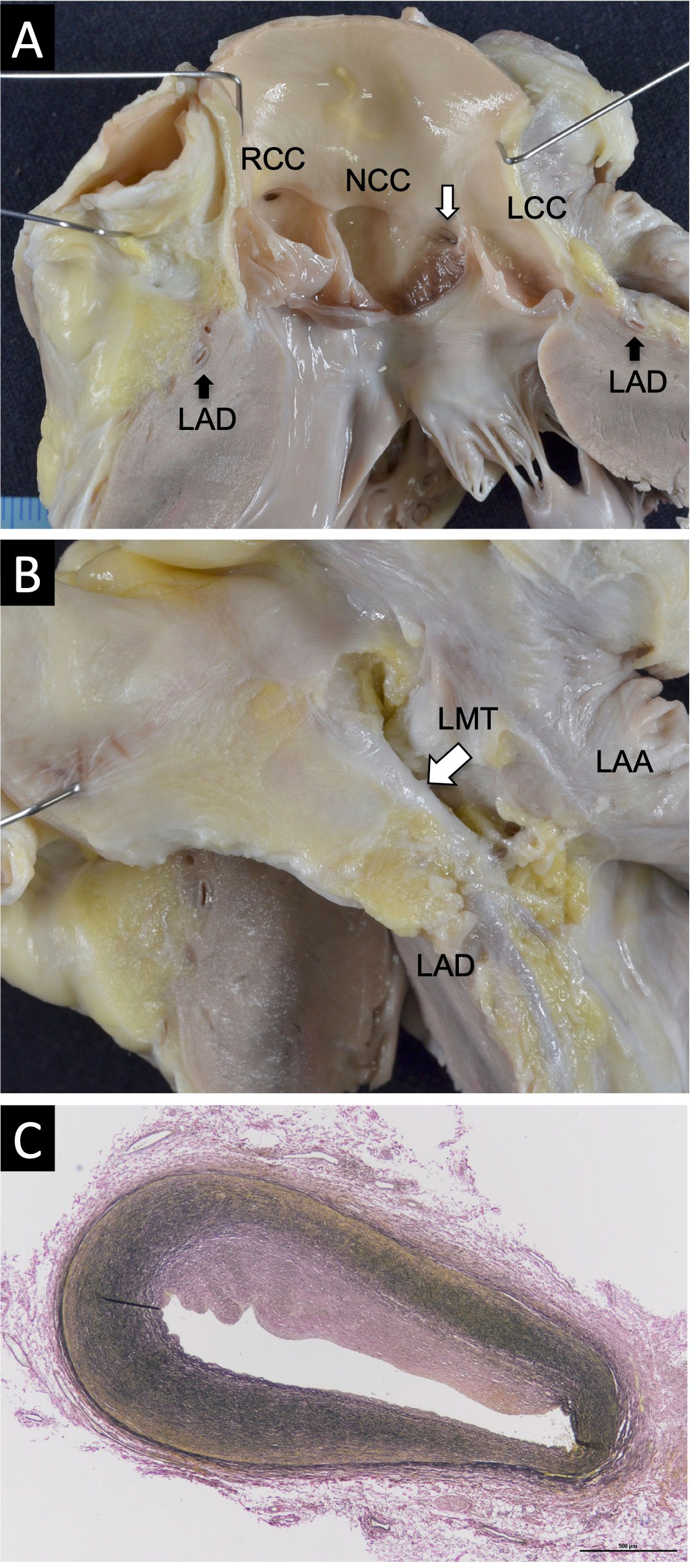
Figure 1 Macroscopic and microscopic pathological findings of the heart. (A) An acute angle take-off of the LCA from the NCC. (B) The LMT passing through a long course along the wall of Valsalva sinus. (C) Masson’s trichrome staining of the LMT revealed an eccentric intimal fibrous thickening indicating approximately 50% stenosis (magnification: ×10).
The child was diagnosed to have severe anemia and thrombocytopenia; therefore, further pathological examination was performed. Hematoxylin and eosin staining of the liver revealed diffused lymphoblast proliferation around the Glisson’s capsule (Figure 2A). Immunostaining of the liver using standard avidin–biotin immunohistochemical techniques showed positive staining of the cell cytoplasm and cytomembrane for CD34 (a hematopoietic stem cell marker; Figure 2B), TdT (a marker of precursor lymphoid cells containing B and T cells; Figure 2C), and CD79a (a pan-B-cell marker; Figure 2D). These findings were consistent with a pre-B-cell phenotype. The lymphoblasts also infiltrated the lung, spleen, kidney, and pancreas (Supplementary Figure 1).
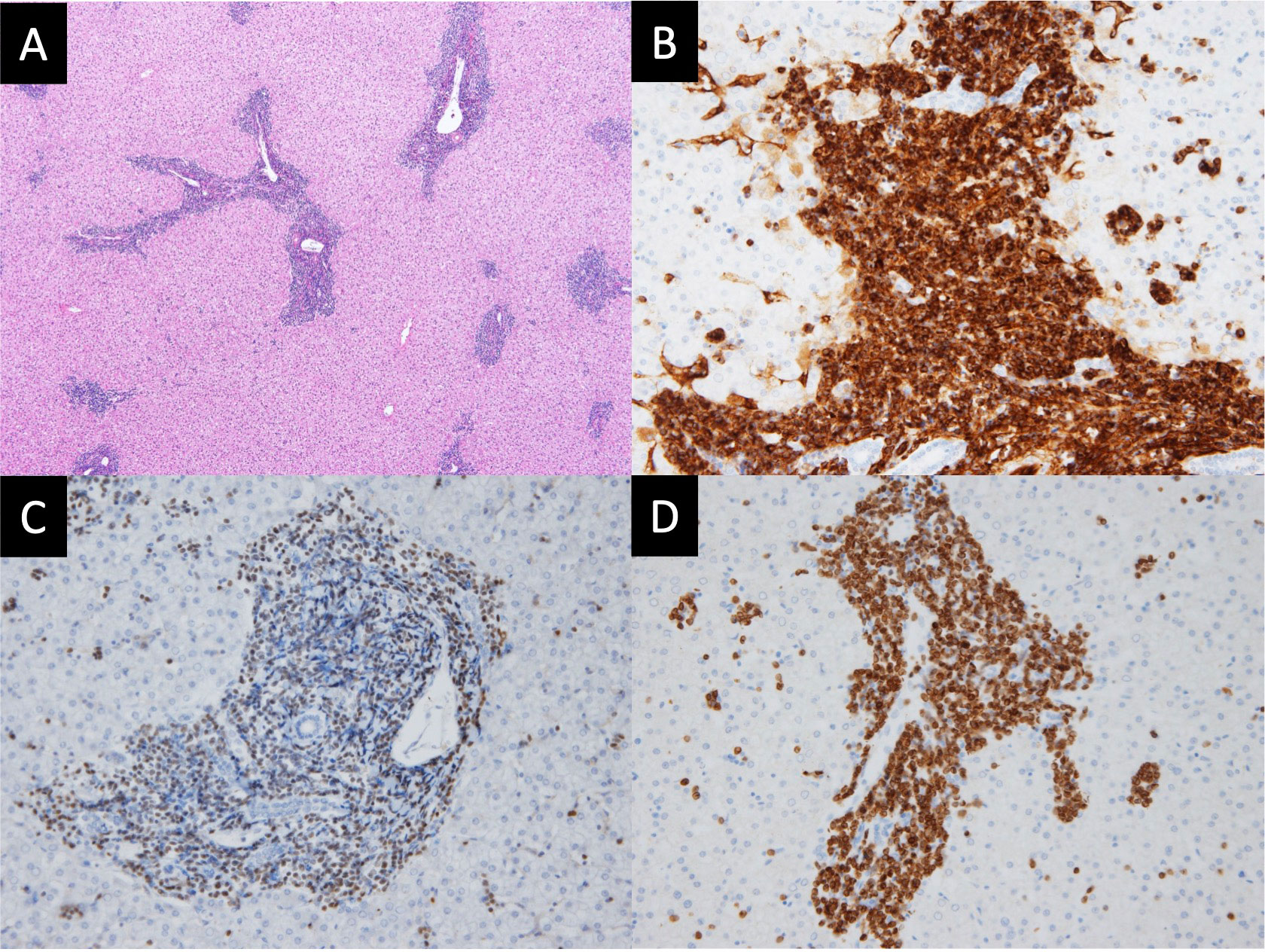
Figure 2 Histopathology and immunohistochemistry of the liver. (A) Hematoxylin and eosin (HE) staining depict diffused proliferation of lymphoblasts around Glisson’s capsule (magnification: ×200). (B) CD34 staining is positive in the cytoplasm and cytomembrane (magnification: ×400). (C) TdT staining is positive in the nucleus (magnification: ×400). (D) CD79a staining is positive in the cytoplasm and cytomembrane (magnification: ×400). These findings of the liver are consistent with the diagnosis of B-cell lymphoblastic lymphoma.
HLH or the related inborn errors of immunity (IEI) were initially considered, and the child was screened for T-cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles (KRECs). Moreover, tests for autoantibodies against type I interferon (IFN) were performed. The normal levels of TRECs and KRECs (965 and 817 copies/μg DNA, respectively) indicated no T-cell or B-cell immunodeficiencies. Moreover, the absence of autoantibodies against type I IFN suggested no COVID-19-associated IEI (13). The complex cardiac and hematological abnormalities suggested the presence of an underlying disease; therefore, we performed WES. Briefly, genomic DNA was fragmented using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA). Exonic sequences were enriched using xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA) and SureSelect XT HS Reagents (Agilent Technologies, Santa Clara, CA, USA). The captured fragments were purified and sequenced on DNBSEQ-G400RS (MGI Tech, Shenzhen, China) using paired-end reads. WES revealed a heterozygous LZTR1 variant (c.1234C>T, p.Arg412Cys). Sanger sequencing revealed this variant in the nail, heart, brain, and spleen tissues, indicating that it is a germline variant (Supplementary Figure 2). Since the LZTR1 variant is positive for PS1, PM2, PP2, and PP3, both in silico analysis and evaluation under the ACMG guideline indicate that it is likely pathogenic (Supplementary Figure 3). We have also analyzed the model structure of LZTR1 using AlphaFoldDB (Supplementary Figure 4). In the model structure, R412 is located on the loop of the Six-bladed beta-propeller domain, presumably forming an intermolecular interaction site, and its side chain forms hydrogen bonds with N410 and D86. Structural stability assessment of the R412C mutant using FoldX showed no significant change (-0.23 kcal/mol), suggesting that the mutation does not contribute significantly to structural stability (Supplementary Figure 4). However, it does alter the hydrogen bonding network of the loop structure, which may affect the molecular interactions. This variant has been reported in a few cases of NS (14, 15). Skilled geneticists identified mild facial features, such as broad forehead, blepharoptosis, epicanthal folds, hypertelorism, a short nose, and thick lips. However, no signs of a webbed neck or short stature were noted. Thus, the final diagnosis was NS associated with cryptorchidism, BCP-ALL, and a coronary malformation.
Additional blood analyses data are shown in Table 1. The results of split-surface general bacterial cultures of blood, cerebrospinal fluid, and lung were negative. The analysis results of throat swab fluid were negative for respiratory syncytial virus, adenovirus, and antigens of group A Streptococcus, influenza A, influenza B, and human metapneumovirus. Liquid chromatography–mass spectrometry revealed low caffeine concentrations in blood. No other drug was detected.
4 Discussion
In this study, the autopsy of the patient with COVID-19 revealed BCP-ALL and an anomalous origin of the LCA, and WES showed NS with an LZTR1 variant. The severity of NS was relatively mild and, thus, NS was not suspected during the patient’s lifetime. Rapid cardiac arrest could have been caused by a COVID-19-related fever and fluid imbalance such as dehydration combined with coronary artery anomaly, which may cause fatal arrhythmias and relative ischemia. LCA arising from NCC, which was observed in our patient, is considered one of the rarest forms of coronary defects (16), detected in 0.02% (36 out of 174,262) of cases. Among the reported cases, 18 (50%) have been symptomatic, including 11 (31%) cases of sudden cardiac death.
Saji et al. (17) reported the death of a 13-year-old girl after long-distance running. Similar to our patient, this patient had LCA with marked intimal thickening. Other studies have also reported children who died suddenly after rigorous physical exercise, and the LCA of these patients also originated from the commissure between the NCC and left coronary cusp, with a slit-like orifice (18, 19). The origin of the LCA in these cases was similar to that observed in our patient, supporting our assumption of the mode of death described above.
Approximately 85% of childhood ALL cases are of B-cell precursor origin, whereas 15% originate from T cells (20). However, patients with NS frequently develop juvenile myelomonocytic leukemia and seldom ALL (21). Only a few cases of LZTR1 variants in patients with NS and ALL are known. Chinton et al. (2) described a 2-year-old female NS/ALL patient with a variant (p. Gly248Arg) in the Kelch 4 domain. In a study of American patients with NS harboring LZTR1 variants (n = 23), two (c. 2220-17C>A, p. Arg210* and c. 1678G>C, p. Glu563Gln) patients were identified to have ALL. One of the two patients developed ALL at 5 years of age, which progressed to acute myeloid leukemia at 7 years of age; the patient died 2 years later. The other patient developed ALL at 3 years of age and remained in remission (5). In addition, Lztr1 deficiency has been linked to B-cell malignancies in CD19+B220+CD43+ immature B cells in mice (22). Thus, ALL development in our patient might be related to NS. However, the LZTR1 variants associated with ALL are rare and need to be researched further.
Molecular autopsy refers to DNA-based identification of the cause of death. In recent large-scale studies of sudden death of young patients, molecular autopsies were able to uncover a likely or plausible cause of death in 12.6%–28% of cases (23, 24). Comprehensive molecular autopsy, similar to that performed on our patient, has the potential to provide more accurate information by identifying genetic causes of unexpected sudden death. Since WES-based molecular autopsy does not lie in the identification of variants but in determining their predicted pathogenicity, care must be taken not to erroneously determine ambiguous variants as pathogens (24). It should be noted that next-generation sequencing (NGS)-based target gene panel sequencing is useful for identifying the causative gene in a clinically suspected patient without accidental findings. However, WES can identify the causative gene even in patients without clinical diagnosis (25).
5 Conclusion
Herein, we presented a case of sudden child death. The death may have resulted from cardiac complications due to NS with a complex combination of BCP-ALL, COVID-19, and a rare pattern of an anomalous origin of the coronary artery. Our case study could be valuable for pathologists and pediatric practitioners as it emphasizes the significance of molecular autopsy. WES or whole-genome sequencing could be used in the diagnosis or even prevention of sudden child mortality.
Data availability statement
The findings of the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
A forensic autopsy was performed on the boy as requested by the public prosecutor. For this type of case report, formal consent is not required. All procedures were performed in accordance with the ethical standards of our institutional research committee and tenets of the 1964 Helsinki Declaration and its later amendments.
Author contributions
Conception and design of the research: KaU, HK. Acquisition of data: KaU, DT, KN, KY, TM. Analysis and interpretation of the data: KaU, TM, AH, YM, SW, TO, NO, SO, KO, KoU, HK. Writing of the manuscript: KaU, HK. Critical revision of the manuscript for intellectual content: TM, YM, HK. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Japan Agency for Medical Research and Development (Grant Number: JP21fk0108436) to SO. The funding body played no role in 1) the design of the study; 2) the collection, analysis, and interpretation of data; 3) the writing of the report; and 4) the decision to submit the manuscript for publication.
Acknowledgments
The authors thank Dr. Yoko Aoki MD, PhD (Tohoku University), Dr. Kenji Kurosawa MD, PhD (Kanagawa Children’s Medical Center), and Maki Taniguchi (Hiroshima University) for diagnosing the patient to have NS. The authors also thank Dr. Hidenori Ohnishi MD, PhD (Gifu University) for providing critical discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1121059/full#supplementary-material
References
1. Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with noonan syndrome. J Med Genet (2015) 52:413–21. doi: 10.1136/jmedgenet-2015-103018
2. Chinton J, Huckstadt V, Mucciolo M, Lepri F, Novelli A, Gravina LP, et al. Providing more evidence on LZTR1 variants in noonan syndrome patients. Am J Med Genet A (2020) 182:409–14. doi: 10.1002/ajmg.a.61445
3. Shehade-Awwad N, Yeshayahu Y, Pinhas-Hamiel O, Katz U. Differences in severity of cardiovascular anomalies in children with noonan syndrome based on the causative gene. Front Pediatr (2022) 10:946071. doi: 10.3389/fped.2022.946071
4. Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC Endocr Disord (2021) 21:2. doi: 10.1186/s12902-020-00666-6
5. Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive noonan syndrome associated with biallelic LZTR1 variants. Genet Med (2018) 20:1175–85. doi: 10.1038/gim.2017.249
6. Güemes M, Martín-Rivada Á, Ortiz-Cabrera NV, Martos-Moreno GÁ, Pozo-Román J, Argente J. LZTR1: Genotype expansion in noonan syndrome. Horm Res Paediatr (2019) 92:269–75. doi: 10.1159/000502741
7. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics (2010) 126:746–59. doi: 10.1542/peds.2009-3207
8. Umeki I, Niihori T, Abe T, Kanno SI, Okamoto N, Mizuno S, et al. Delineation of LZTR1 mutation-positive patients with noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum Genet (2019) 138:21–35. doi: 10.1007/s00439-018-1951-7
9. Bader-Meunier B, Tchernia G, Miélot F, Fontaine JL, Thomas C, Lyonnet S, et al. Occurrence of myeloproliferative disorder in patients with noonan syndrome. J Pediatr (1997) 130:885–9. doi: 10.1016/s0022-3476(97)70273-7
10. Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in noonan, Costello, cardiofaciocutaneous and leopard syndromes. Am J Med Genet C Semin Med Genet (2011) 157C:83–9. doi: 10.1002/ajmg.c.30300
11. Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukaemia and noonan syndrome. J Med Genet (2014) 51:689–97. doi: 10.1136/jmedgenet-2014-102611
12. Niemeyer CM. RAS diseases in children. Haematologica (2014) 99:1653–62. doi: 10.3324/haematol.2014.114595
13. Eto S, Nukui Y, Tsumura M, Nakagama Y, Kashimada K, Mizoguchi Y, et al. Neutralizing type I interferon autoantibodies in Japanese patients with severe COVID-19. J Clin Immunol (2022) 42:1360–70. doi: 10.1007/s10875-022-01308-3
14. Dempsey E, Haworth A, Ive L, Dubis R, Savage H, Serra E, et al. A report on the impact of rapid prenatal exome sequencing on the clinical management of 52 ongoing pregnancies: A retrospective review. BJOG (2021) 128:1012–9. doi: 10.1111/1471-0528.16546
15. Quaio C, Moreira CM, Novo-Filho GM, Sacramento-Bobotis PR, Groenner Penna M, Perazzio SF, et al. Diagnostic power and clinical impact of exome sequencing in a cohort of 500 patients with rare diseases. Am J Med Genet C Semin Med Genet (2020) 184:955–64. doi: 10.1002/ajmg.c.31860
16. Bravo-Jaimes K, Balan P, Garcia-Sayan E. Controversies on the cusp: Anomalous origin of the left coronary artery from the non-coronary cusp. Cureus (2020) 12:e7993. doi: 10.7759/cureus.7993
17. Saji T, Yamamoto K, Hashiguchi R, Matsuo N, Yabe Y. Hypoplastic left coronary artery. in association with occlusive intimal thickening of a coronary artery with ectopic ostium and with atresia of the left coronary ostium. Jpn Heart J (1985) 26:603–12. doi: 10.1536/ihj.26.603
18. Hamamichi Y, Okada E, Ichida F. Anomalous origin of the main stem of the left coronary artery from the non-facing sinus of valsalva associated with sudden death in a young athlete. Cardiol Young (2000) 10:147–9. doi: 10.1017/s1047951100006624
19. Ishikawa T, Otsuka T, Suzuki T. Anomalous origin of the left main coronary artery from the noncoronary sinus of valsalva. Pediatr Cardiol (1990) 11:173–4. doi: 10.1007/BF02238853
20. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet (2008) 371:1030–43. doi: 10.1016/S0140-6736(08)60457-2
21. Hasle H. Malignant diseases in noonan syndrome and related disorders. Horm Res (2009) 72 Suppl 2:8–14. doi: 10.1159/000243773
22. Chen S, Vedula RS, Cuevas-Navarro A, Lu B, Hogg SJ, Wang E, et al. Impaired proteolysis of noncanonical RAS proteins drives clonal hematopoietic transformation. Cancer Discovery (2022) 12:2434–53. doi: 10.1158/2159-8290.CD-21-1631
23. Webster G, Puckelwartz MJ, Pesce LL, Dellefave-Castillo LM, Vanoye CG, Potet F, et al. Genomic autopsy of sudden deaths in young individuals. JAMA Cardiol (2021) 6:1247–56. doi: 10.1001/jamacardio.2021.2789
24. Shanks GW, Tester DJ, Ackerman JP, Simpson MA, Behr ER, White SM, et al. Importance of variant interpretation in whole-exome molecular autopsy: Population-based case series. Circulation (2018) 137:2705–15. doi: 10.1161/CIRCULATIONAHA.117.031053
Keywords: SARS-CoV-2, BCP-ALL, noonan syndrome (NS), sudden child death, autopsy
Citation: Unuma K, Tomomasa D, Noma K, Yamamoto K, Matsuyama T-a, Makino Y, Hijikata A, Wen S, Ogata T, Okamoto N, Okada S, Ohashi K, Uemura K and Kanegane H (2023) Case Report: Molecular autopsy underlie COVID-19-associated sudden, unexplained child mortality. Front. Immunol. 14:1121059. doi: 10.3389/fimmu.2023.1121059
Received: 11 December 2022; Accepted: 31 March 2023;
Published: 18 April 2023.
Edited by:
Sara Sebnem Kilic, Bursa Uludağ University, TürkiyeReviewed by:
Antonio Oliva, Catholic University of the Sacred Heart, ItalyAbhinav Jain, Mayo Clinic, United States
Copyright © 2023 Unuma, Tomomasa, Noma, Yamamoto, Matsuyama, Makino, Hijikata, Wen, Ogata, Okamoto, Okada, Ohashi, Uemura and Kanegane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kana Unuma, dW51bWEubGVnbUB0bWQuYWMuanA=