 Indiwari Gopallawa
Indiwari Gopallawa Ruchika Dehinwal
Ruchika Dehinwal Vaibhav Bhatia3
Vaibhav Bhatia3 Vikramsingh Gujar
Vikramsingh Gujar Narendra Chirmule
Narendra Chirmule
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1119564
Lungs are important respiratory organs primarily involved in gas exchange. Lungs interact directly with the environment and their primary function is affected by several inflammatory responses caused by allergens, inflammatory mediators, and pathogens, eventually leading to disease. The immune architecture of the lung consists of an extensive network of innate immune cells, which induce adaptive immune responses based on the nature of the pathogen(s). The balance of immune responses is critical for maintaining immune homeostasis in the lung. Infection by pathogens and physical or genetic dysregulation of immune homeostasis result in inflammatory diseases. These responses culminate in the production of a plethora of cytokines such as TSLP, IL-9, IL-25, and IL-33, which have been implicated in the pathogenesis of several inflammatory and autoimmune diseases. Shifting the balance of Th1, Th2, Th9, and Th17 responses have been the targets of therapeutic interventions in the treatment of these diseases. Here, we have briefly reviewed the innate and adaptive i3mmune responses in the lung. Genetic and environmental factors, and infection are the major causes of dysregulation of various functions of the lung. We have elaborated on the impact of inflammatory and infectious diseases, advances in therapies, and drug delivery devices on this critical organ. Finally, we have provided a comprehensive compilation of different inflammatory and infectious diseases of the lungs and commented on the pros and cons of different inhalation devices for the management of lung diseases. The review is intended to provide a summary of the immunology of the lung, with an emphasis on drug and device development.
Lungs are important respiratory organs involved in providing oxygen to the body. Every day, the lungs interact directly with a myriad of different-sized particles from the environment. Most of these particles are filtered through the mucosal barriers present in the upper respiratory tract. These barriers thus efficiently protect the body from pathogens, allergens, and various foreign bodies. Large particles are removed by physical barriers and forces such as coughing and sneezing, triggered by the muscular-nervous system. Smaller particles are entrapped, degraded, and processed by epithelial and a battery of cells in the mucosal lining of the trachea. Interactions of these external substances with specific Toll-like receptors present on the inner epithelium of the respiratory tract and phagocytosis of these substances activate the innate immune system (1). This system includes the induction of anti-microbial peptides, chemokines, and cytokines. These mediators of the innate immune system subsequently recruit and activate a complex array of adaptive immune responses, which have been classified based on the kind of cytokines into Th1, Th2, Th9, and Th17 responses. The precise regulation, i.e., activation, persistence, and termination of these responses involve activation of transcription factors through a variety of signal transduction pathways.
In this review, we have summarized various aspects of pulmonary immune homeostasis mediated through regulated activation of innate and adaptive immune responses. We have elaborated on the pathogenesis of inflammatory autoimmune diseases and infectious diseases. Further, we have highlighted the advances in novel therapies and drug delivery devices that can help in developing drugs to treat lung diseases.
Homeostasis is a state of balance. Homeostasis within the lung is maintained by harmonious coordination of different immune pathways, which includes the active participation of alveolar epithelial cells (AECs) and macrophages (Figure 1). AEC I and AEC II make up most of the mature alveolar epithelium and AEC I covers almost 99% of the lung surface area which is crucial for gas exchange and protection against a myriad of inhaled external particles. Alveolar epithelium is developed by organized processes that begin in the ventral foregut endoderm during development. Interactions between the foregut epithelium and its associated mesenchyme promote the formation of a functioning respiratory system and its compartmentalization along the proximal-distal axis (2, 3). The lung epithelium is a physical barrier between the lumen and underlying submucosa, protecting the lung tissue from bacteria, viruses, allergies, and other harmful substances. Besides acting as a physical barrier, AECs are crucial in the lung’s immune response. Although both AEC I and AEC II are essential in airway defense mechanisms, AEC II is more immunologically active (4). AECs produce several immune factors such as cytokines and chemokines that are responsible for immune cell activation and differentiation and act as antigen-presenting cells for specific T cells (5–7). Other cells, such as macrophages, also participate in the normal physiological functions of the lung and are involved in some acute and chronic diseases (8, 9). There are two types of lower respiratory tract macrophages: alveolar macrophages (AMs) in the alveoli and interstitial macrophages (IMs) in the interstitium. Both kinds of macrophages produce robust responses to a wide range of stimuli. AMs are innately suppressive and play an essential role in maintaining immunological homeostasis and host defense.
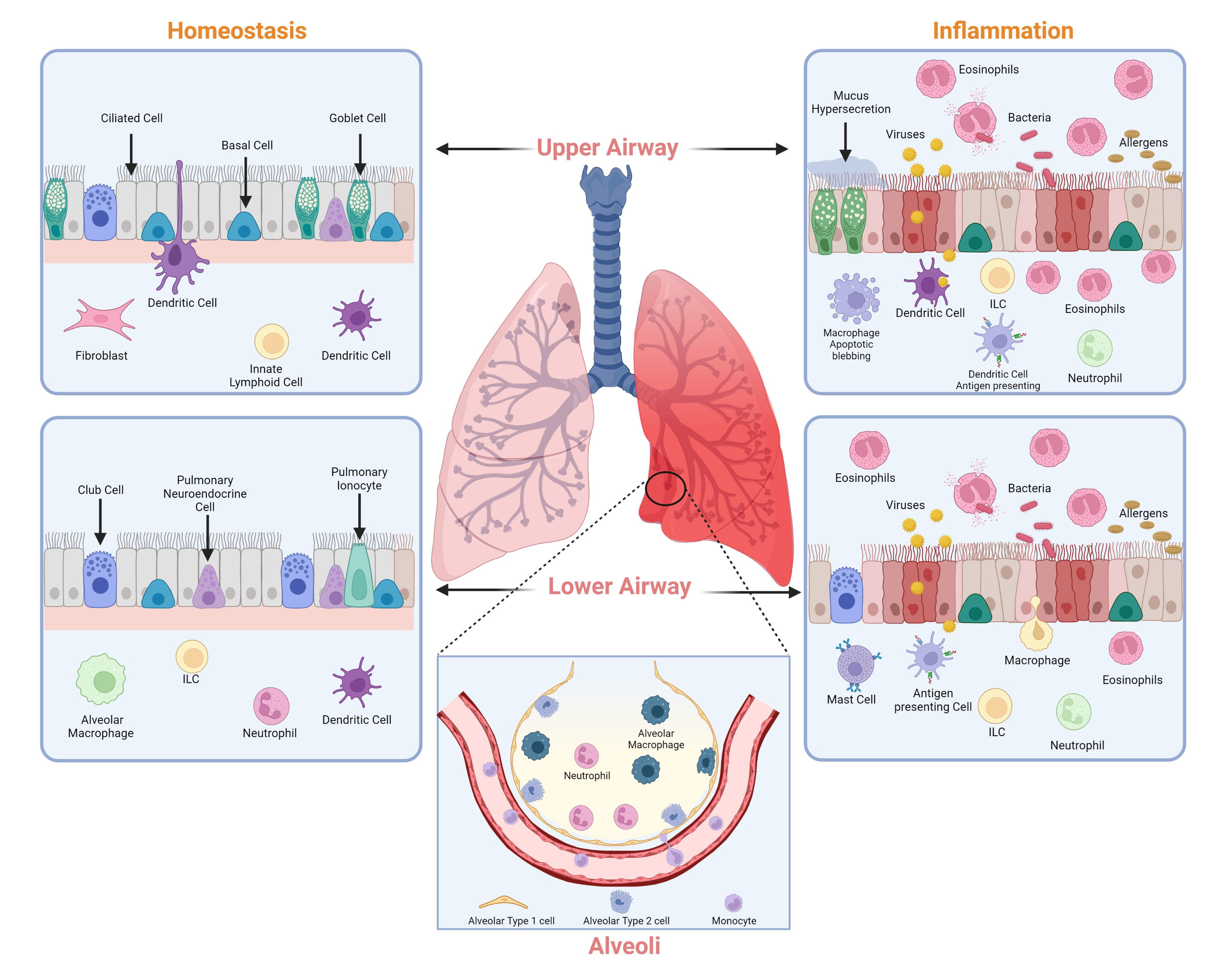
Figure 1 Major cell types of the lung. Cell types that contribute to the maintenance of lung homeostasis (left) and lung inflammation (right) in the upper and lower airways. Image created with Biorender.com.
In contrast to AMs, IMs have a regulatory function within the lung tissue (10). While AMs are regulated through non-specific defenses such as phagocytosis, tumor necrosis factor-alpha (TNF-α), and interferon gamma (IFN- γ), IMs have a higher proclivity for releasing cytokines related to the adaptive immune response (11). Overall, both AECs and macrophages work together to maintain the decorum in the lung under normal conditions and contribute to regulating the immune system.
A detailed understanding of the pathogenesis of inflammatory diseases and lung infections has given insights into the multifactorial components of the immune system in regulating lung functions. We have reviewed these two conditions and listed the immunological pathways that contribute to the immunopathogenesis of various inflammatory diseases and infections. There are several learnings of immunological pathways from these diverse pathologies that have yielded novel therapies. We have outlined the advances in delivery devices for therapeutic interventions that have been developed to treat lung diseases.
Every day, the respiratory system, processes 10,000–12,000 L of air. Due to this, the lung epithelium is exposed to several particulates mixed with a complex mixture of gases (12). While most of the air includes safe environmental components, sometimes, the inhaled air contains harmful microbes and allergens. The conducting airways and gas exchange surfaces must deal with such hazardous substances and quickly react to protect the host. To achieve protection, the respiratory system must coordinate with the pulmonary immune system to protect the host from a plethora of threats. Less virulent microbes are generally obliterated by primary defenses like mucociliary clearance and alveolar macrophages in the respiratory tract. On the contrary, more virulent microbes and a large number of microbes need to be eliminated by inflammatory processes initiated by the innate immune system (13). The pulmonary immune system is composed of innate and adaptive immune systems. The innate immune system is the first line of defense against pathogens and sterile threats. Innate protection is primarily provided at the cellular level by coordinated actions of the airway and alveolar epithelial cells, resident macrophages, recruited neutrophils, and monocytes, which respond rapidly to inhaled materials (Table 1). The initial checkpoint is provided by epithelial cells, dendritic cells, and alveolar macrophages during antigen infiltration, which induces a proinflammatory downstream immune response. The innate defense in the respiratory tract is coordinated by several components such as lysozymes, lactoferrin, secretory leukoprotease inhibitor, proinflammatory cytokines, chemokines along with antimicrobial secretory IgAs (sIgA), defensins and cathelicidins (29–31) The intercellular communication between the airway and the immune cells is facilitated by lipid mediators, complement factors, chemo-attractants, and chemokines. The biological function of chemokines is relayed by different cytokines, including several interleukins and thymic stromal lymphopoietin (TSLP) (32, 33). These substances, secreted by local airway cells, generate customized immunological responses.
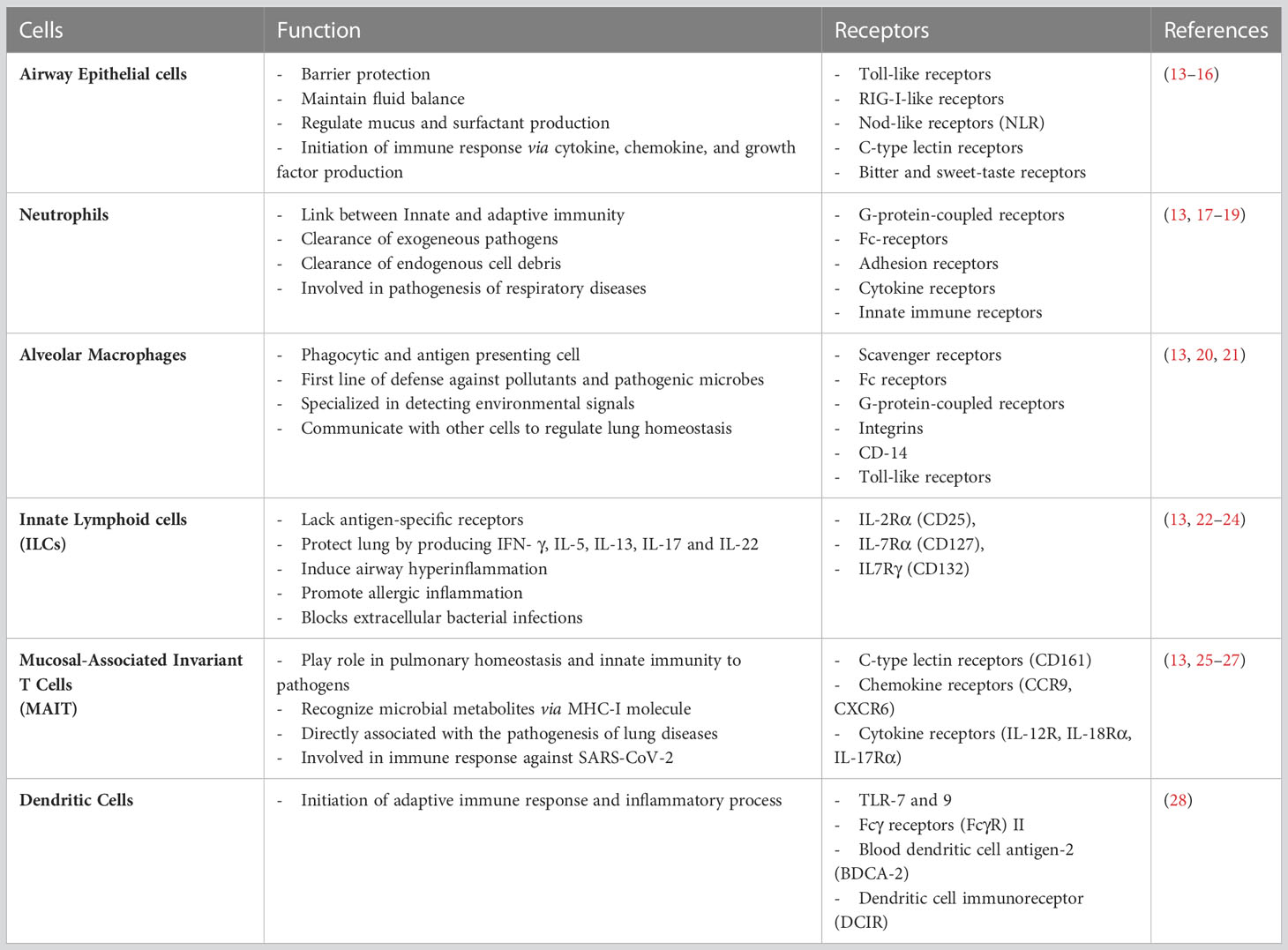
Table 1 Cells involved in primary Immune response of the lung.
The respiratory adaptive immune system consists of cellular components like T lymphocytes, B lymphocytes, and antigen-presenting cells, along with soluble mediators like cytokines, chemokines, lactoferrin, defensins and sIgAs (34). Following the mechanisms of the innate immune system, the lungs are further protected by the mechanisms of the adaptive immune system against various external and internal threats such as pathogens entering alveoli and microorganisms inside lung cells. The most common immune cell types in the lungs are macrophages and dendritic cells, which are innate cells, and CD4+ and CD8+T lymphocytes, constitute adaptive immune cells (14). Most lung macrophages are AMs and are essential for infection resistance. However, they have been linked to dysregulations in lung inflammation and fibrosis (8). T cells are primed and activated in lymphoid organs before migrating to the lung to respond to respiratory infections. They survive as memory T cells following pathogen clearance. The lung was previously found to have prominent memory T cell populations, which were first assumed to constitute a migratory surveillance population. However, a bulk of lung T cells are now known to survive as noncirculating tissue-resident memory T cells (TRMs). Pools of memory T cells are created throughout a respiratory viral infection and remain for the span of the animal’s life. These cells feature different cytokine production profiles, minimal need for co-stimulation, reduced sensitivity to apoptosis, and persist at high frequency compared to their naïve progenitors. The spleen and draining lymph nodes are secondary lymphoid organs with a significant number of memory cells (15). The functions of T cells especially CD4+ T lymphocytes and how cytokines produced by these T cells play a role in lung diseases such as IPF, COPD, and asthma are described in the section below.
Inflammation is defined as the response of the immune system to injury (35). Inflammation is favorable to the host under normal circumstances. However, an abnormal immune response(s) in the airway can lead to chronic inflammatory responses, contributing to a variety of lung diseases. In this review, we have summarized the role of CD4+ T cells and AEC apoptosis which is a prominent observation in lung biopsies of patients with idiopathic pulmonary fibrosis (IPF). Further, we discuss the role of inflammation in chronic obstructive pulmonary disease (COPD) and asthma. The role of inflammation in IPF is considered somewhat controversial and reviewed extensively elsewhere (36). Additionally, we summarize currently available FDA-approved drugs for IPF, asthma, and COPD (Table 2).
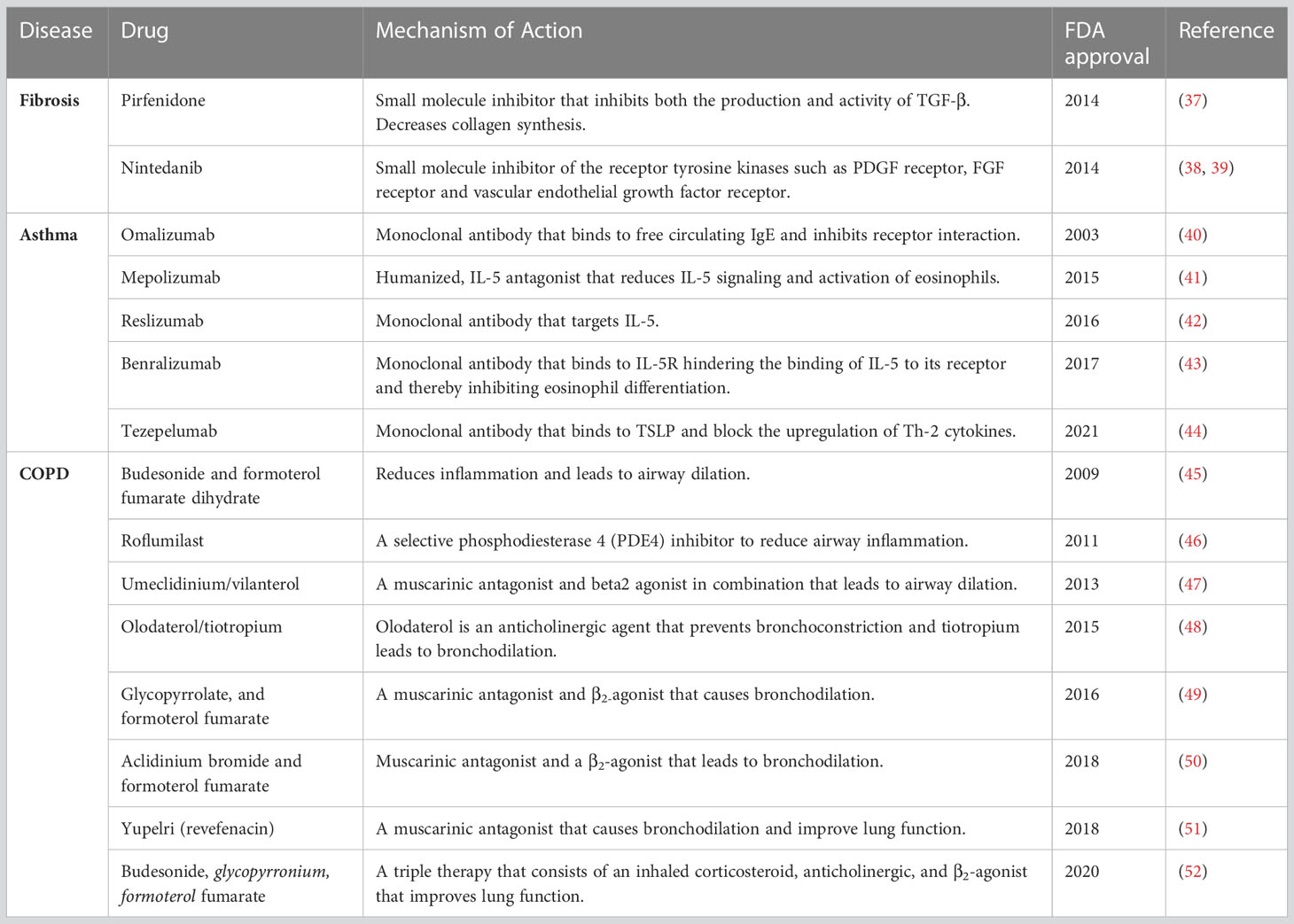
Table 2 Summarizes FDA approved drugs to treat fibrosis, Asthma, and COPD.
Fibrosis is a highly dynamic process, and it can affect any organ including the skin, lungs, liver, and kidney. IPF is defined as an excessive deposition of extracellular matrix (ECM) proteins in the lung tissue, which makes the lungs unable to transport oxygen into the bloodstream efficiently and often leads to lung failure. Although the etiology of IPF is unknown, several risk factors such as cigarette smoking, occupational factors, genetic polymorphisms, and environmental exposure can contribute to the development of IPF. IPF is a clinically heterogeneous disease with an overall prognosis being very poor; typically, it has a median survival of 3–4 years (35, 53). Repetitive injury to the alveolar epithelium, failure to reepithelization, increased AEC apoptosis, and induction of growth factors and profibrotic cytokines such as transforming growth factor-β1 (TGF-β1), TNF-α, platelet-derived growth factor, and connective tissue growth factor (CTGF) are implicated in the progression of IPF (54). In addition to AEC apoptosis, fibroblast differentiation, accumulation of myofibroblasts, and excessive collagen deposition in the alveolar space promote the progression of fibrosis (55, 56).
CD4+ T cells in IPF: One of the main challenges in lung fibrosis is that the exact molecular and cellular mechanisms underlying the disease remain largely unknown. However, damage to the airway epithelium (apoptosis) is considered as an early pathogenic event, with an irregular repair process by epithelial-mesenchymal interactions, rather than an inflammatory response (57). Although over the several decades the role of inflammation has been questioned in fibrosis (58), data from IPF patients and experimental animal models have shown a prominent role of T helper cell type-2 (Th2) cells in IPF (59). It has been shown that each of the major Th2 cytokines such as IL-4, IL-5, and IL-13 contribute to the regulation of tissue remodeling and subsequent fibrosis. In-vitro studies have shown that stimulation with IL-4 increased collagen production in murine fibroblasts (60). Moreover, IL-4 concentrations were increased in the bronchoalveolar lavage fluids from patients with idiopathic pulmonary fibrosis (61). Along similar lines, IL-4 has been shown to induce periostin which may play a role in fibrosis (62).
IL-13 is another Th-2 cytokine that has been shown to play a key role in lung fibrosis and increased expression of IL-13 was repeatedly reported in IPF. Overexpression of IL-13 augmented subepithelial airway fibrosis in the mouse lung (63). Studies done in animal models have demonstrated that treating with anti-IL-13 antibodies significantly decreased aspergillus fumigatus-induced collagen levels in the lung (64). In a different study, it was demonstrated that IL-13 deficient mice were unaffected by radiation-induced fibrosis compared to wild-type mice (65).
In addition to IL-4 and IL-13, it has been observed that IL-5 can also play a role in tissue remodeling in several diseases including pulmonary fibrosis (66). It was demonstrated that IL-5 levels were increased in the lungs of bleomycin-treated mice (67). Further, it was shown that anti-IL-5 antibody caused a significant reduction in lung eosinophils and fibrosis in a bleomycin-induced pulmonary fibrosis mouse model (68). Given the role of IL-4, IL-5, and IL-13, it is crucial to identify other cytokines and receptors that may contribute to the aberrant cellular mechanisms in the IPF lung.
AEC apoptosis in IPF: The respiratory epithelium is a physical and functional barrier, shielding the underlying lung microenvironment. It plays a crucial role in the clearance of pathogens and harmful bacteria (69). Apoptosis, or programmed cell death, is a vital biological process in the maintenance of tissue homeostasis (70). However, in recent years, seminal works have supported the notion that excessive apoptosis is linked to diseases such as IPF, emphysema, and acute lung injury (71). It has been shown in a variety of animal models that blockade of AEC apoptosis reduces subsequent fibrosis (72). A broad-spectrum caspase inhibitor has been shown to decrease caspase-1 and caspase-3 activity, reducing the number of apoptotic cells. This led to a reduction in lung collagen in a bleomycin-induced mouse model (73). At a cellular and molecular level, there are many signaling pathways and factors that contribute to AEC apoptosis, such as the angiotensin (ANG) system. Several studies have demonstrated that manipulation of the ANG system can control apoptosis in the alveolar epithelium (74). Therefore, protecting AECs from apoptosis and sustaining their function might be an effective therapeutic strategy to decrease lung injury.
Current treatment strategies for IPF: Recent research contains a wealth of information on cellular and molecular signaling mechanisms in IPF, and we now have a better understanding of its pathophysiology. Specifically, IPF is no longer considered a pro-inflammatory disorder, but rather it is the result of a fibroproliferative environment and aberrant wound healing mechanisms (75) Currently, two antifibrotic therapies are available for IPF patients: pirfenidone and nintedanib. Pirfenidone has been recognized for its antioxidant and antifibrotic effects (76) is a tyrosine kinase inhibitor that inhibits fibroblast proliferation and differentiation and improves lung function (77). Given the heterogeneity of IPF, targeting multiple signaling pathways and multiple cell types may offer novel therapeutics to patients.
COPD is a disease with high morbidity and mortality rates. It is characterized by chronic inflammation that leads to poorly reversible airway obstruction (78). The most common risk factor for the development of COPD is cigarette smoke. However, other harmful environmental factors such as exposure to air pollutants might contribute to COPD. Patients with COPD have significant levels of circulating inflammatory markers, demonstrating the presence of systemic inflammation (79). Persistent airway inflammation in COPD contributes to airway remodeling, loss of small airways, and emphysema (80). Emphysema is a major pathological change of COPD, which is characterized by the enlargement of alveolar airspaces due to the destruction of the alveolar epithelium (81).
Inflammation in COPD: The pathogenesis of COPD is multifaceted and the underlying cellular and molecular mechanisms remain poorly understood. In COPD, the inflammatory response involves both innate and adaptive immunity. The predominant form of COPD is neutrophilic inflammation, which correlates with the rate of decline in lung function (82, 83). In addition to the well-known augmented influx of neutrophils in COPD, aberrant clearance of neutrophils possibly contributes to the dysregulated inflammation in COPD (84). Aberrant clearance of neutrophils might be a result of defective phagocytic responses in alveolar macrophages. It was demonstrated that cigarette smoke could hinder the clearance of neutrophils, inducing pro-inflammatory cytokines such as TNF-α (85). According to a recent study by Borges et al., the administration of TNF-α weakened the removal of apoptotic cells from the lungs in a mouse model of LPS-induced neutrophilic inflammation (86). Even though neutrophil-associated inflammation is the most common inflammatory phenotype in COPD, 10%–40% of COPD patients demonstrate increased eosinophilic inflammation (87). Additionally, it was shown that airway β-defensin-1 levels were significantly elevated in the sputum of COPD patients (88) whereas secretory IgA (sIgA) levels were markedly reduced due to altered epithelial cell differentiation in the airways of COPD patients (89) Following exposure to cigarette smoke, pollutants, and microbes, alarmins such as TSLP, IL-33, and IL-25 are released from the epithelium. Following T-cell differentiation, Th-2 cells produce IL-4, IL-5, and IL-13, all of which contribute to an inflammatory environment (Figure 2) (90). Induction of IL-4 can lead to an increase in total IgE levels in eosinophilic COPD, which might contribute to a decline in lung function (91).

Figure 2 The role of the alarmins in the lung epithelium. Exposure to allergens, viruses, and bacteria releases cytokines from the lung epithelium that can activate dendritic cells (DC) and innate lymphoid cells (ILC-2) that belong to the innate immune system. DCs differentiate naïve T cells into Th2 cells that can secrete IL-4, IL-13, and IL-5. These cytokines play a role in airway remodeling. Similarly, ILC-2s secrete Th2 cytokines. Image created with BioRender.com.
Current treatment strategies for COPD: Although current therapies for COPD have significantly improved the management of the disease, novel therapeutic approaches are still urgently needed, especially for suppressing inflammation in small airways (92). Cigarette smoke is one of the major factors for COPD and smoking cessation has so far been shown to diminish disease progression. Currently, several methods of nicotine replacement therapies are in use. However, the efficacy of these therapies remain to be further investigated (93). A variety of other interventions can lower the exacerbations in COPD, including influenza vaccination (94), anti-cholinergic bronchodilators (95), long-acting β2-agonists (LABAs) (96), and inhaled corticosteroids (ICSs). Further research is needed to understand the cell signaling mechanisms of COPD so that biomarkers can be identified and new therapies can be developed (92).
Asthma is a chronic inflammatory disease characterized by airway obstruction, excessive mucus production, and airway hyperresponsiveness (AHR). Asthma affects over 300 million people worldwide (97). The prevalence of asthma is increasing in many parts of the world, contributing to a substantial global economic burden. Clinically, asthma patients show episodes of cough, chest tightness, wheeze, and shortness of breath (55). There is currently no cure for asthma, but treatment can help ameliorate symptoms and improve the quality of life. Currently, the therapeutic challenge is to generate better inhalation technologies to improve the delivery of anti-inflammatory agents to the lung parenchyma (98). It was interesting to note that polymorphisms in antimicrobial proteins such as β-defensins can contribute to the severity of asthma. In fact, Baines et al., demonstrated that airway β-defensin-1 was elevated in severe asthma patients regardless of the inflammatory phenotype and may act as an effective biomarker and potential therapeutic target (88). Recent seminal work indicates that asthma is generally associated with Th2 immune responses, which is like other conditions such as atopic dermatitis. In this section of the review, we discuss the Th2 immune responses in asthma. Further, we discuss the available treatments and their limitations for asthma.
Th2 immune responses in asthma: The pathophysiology of asthma is complex and involves various host-environment interactions and inflammatory responses occurring at various levels. There are two subtypes of asthma: allergic and non-allergic. Allergic asthma is the most common form of disease that affects many individuals. Allergic asthma may get triggered in response to house dust mites, animal dander, mold, and pollens, whereas non-allergic asthma might be the result of respiratory viral infections, cold air, and tobacco smoke leading to chronic airway inflammation (99). Inflammation of the airways is a key component that leads to airway remodeling and damage. Th2 cytokines, including interleukin (IL)-5, IL-4, and IL-13 play a significant role in initiating the inflammatory cascade in asthma (100). A recent study showed that blockade of Th2-cell-mediated responses reduced symptoms for many patients, thereby demonstrating the central role of Th2 cells in the pathophysiology of asthma (101).
Eosinophils are bilobed leukocytes that are present in low numbers in healthy individuals. However, during asthma, Th2 cytokines such as IL-5 can induce prolonged eosinophil survival, activation, and trafficking to the site of injury (102). Recruitment of eosinophils to the site of injury results in an induction of cytokines and growth factors such as tumor growth factor b (TGF-β), leading to airway remodeling (103). An increased number of eosinophils have been observed in bronchoalveolar lavage (BAL) fluid and bronchial tissue in asthma patients (104). Further, elevated eosinophil counts correlate with disease severity, suggesting that these cells may play a significant role in asthma pathogenesis (104). IL-4 is a major cytokine in the development of inflammation in asthma. It is associated with the induction of isotype switching and secretion of IgE by B cells (105). IgE production, in turn, triggers the release of inflammatory cytokines, such as histamine, from mast cells that causes contraction of the smooth muscle in the airways, edema, and increased mucus secretion (106). Additionally, another crucial activity of IL-4 in stimulating inflammation is the induction of vascular cell adhesion molecule (VCAM)-1 on vascular endothelium in the asthmatic lung (107). Through the interaction of VCAM-1 and integrins on leukocytes, T lymphocytes, monocytes, and eosinophils can migrate to the injury site (108). Aberrant production of IL-4 due to genetic mutations or hyperresponsiveness to this cytokine might further contribute to the severity of asthma (109). Another cytokine that contributes to the pathophysiology of asthma is IL-13, a pleiotropic Th2 cytokine that contributes to goblet cell differentiation, activation of fibroblasts, and increased airway hyperresponsiveness (110). Further, IL-13 is believed to be a central regulator of mucus hypersecretion and fibrosis, leading to airway remodeling (111).
Most recently, a Th9 subtype of CD4+ T cells has emerged. These cells produce IL-9 a pleiotropic cytokine that may play role in inflammatory responses in asthma (83). IL-9 is secreted not only by CD4+ T cells but also by a variety of other T cells such as mast cells, and innate lymphoid cells (ILC-2) cells (112). It has been shown that in the presence of IL-4 and TGF-β, naïve CD4+ T cells develop into Th9 cells. In allergic diseases IL-9 has been shown to induce IgE production by B cells, activate mast cells, and play a pivotal role in mucus secretion (113, 114).
Role of thymic stromal lymphopoietin in asthma: Our understanding of the airway epithelium in driving asthma pathogenesis has progressed extensively in recent years. In response to allergic and non-allergic triggers, airway epithelial cells release cytokines, known as “alarmins,” such as TSLP, IL-33, and IL-25 (Figure 2), which stimulate downstream inflammatory responses (115). More recently, TSLP has emerged as an important factor in the pathogenesis of asthma, and increased levels of TSLP have been detected in asthma patients (116). TSLP can be induced by proinflammatory cytokines such as IL-1, TNF-α, bacterial and viral infections, Toll-like receptors, and allergens. The nuclear factor-kappa-B (NF-kB) pathway plays an important role in the mechanism of TSLP activation (117). Once TSLP is upregulated, it binds to its receptor TSLPR (118). A recent study showed that IL-7Rα binds to the TSLP/TSLPR complex with high affinity (119), and this ternary complex initiates a signaling cascade that activates STAT5 transcriptional factors (120). TSLP can interact with dendritic cells (DCs) to enhance antigen presentation to induce CD4+ cells (121). TSLP-activated mature DCs can then migrate to lymph nodes to initiate adaptive responses. Further, during non-allergic asthma, TSLP activates innate lymphoid cells (ILC-2) to produce Th2-like cytokines (121). According to a recent study, the blockade of TSLP reduces allergen-induced inflammation in animal models (122).
Current treatment options for severe asthma: Multiple phenotypes of severe asthma have been identified, showing the significance of tailored treatment options to meet individual needs. Most patients with asthma attain decent control of the disease with the use of inhaled corticosteroids and bronchodilators (123) However, many patients with asthma, especially in developing countries, remain unattended and suffer from poor quality of life (124). Recently, monoclonal antibodies have emerged as a new treatment option for uncontrolled asthma, targeting different signaling molecules in the pathways causing airway inflammation. Omalizumab is the first FDA-approved drug to treat severe asthma (125). It binds to IgE and prevents binding to its receptor on mast cells (125). Mepolizumab is a fully-humanized anti–IL-5 antibody that binds to IL-5 with high affinity to prevent IL-5 from binding to IL-5 receptors on eosinophils (126). Although many treatment options are available for patients with asthma, not all people with severe disease can benefit from the same biological drug, and therefore, an appropriate biological drug must be carefully selected for each patient. Although management of asthma varies among individuals, it’s crucial to protect patients with asthma from infectious lung diseases caused by bacteria and viruses. People with asthma are more susceptible to developing pneumonia due to previous lung damage. The section below explains several lung infectious diseases such as bronchitis, pneumonia, and disease pathogenesis.
The etiology of lung infections generally involves the acquisition of a pathogen, its dissemination, and invasion of the underlying respiratory tract tissue. Respiratory tract infections are classified based on their symptomatology and anatomic involvement. Upper respiratory tract infections are usually benign, transitory, and self-limiting. These are caused by viruses, bacteria, mycoplasma, and fungi (Table 3), mostly resulting in the common cold, epiglottitis, laryngitis, pharyngitis (sore throat), or sinusitis (sinus infection). On the contrary, lower respiratory tract infections are predominantly caused by bacteria, mycoplasma, viruses, and sometimes fungi (Table 3). These result in bronchitis, bronchiolitis, and pneumonia, which can be severe to fatal.
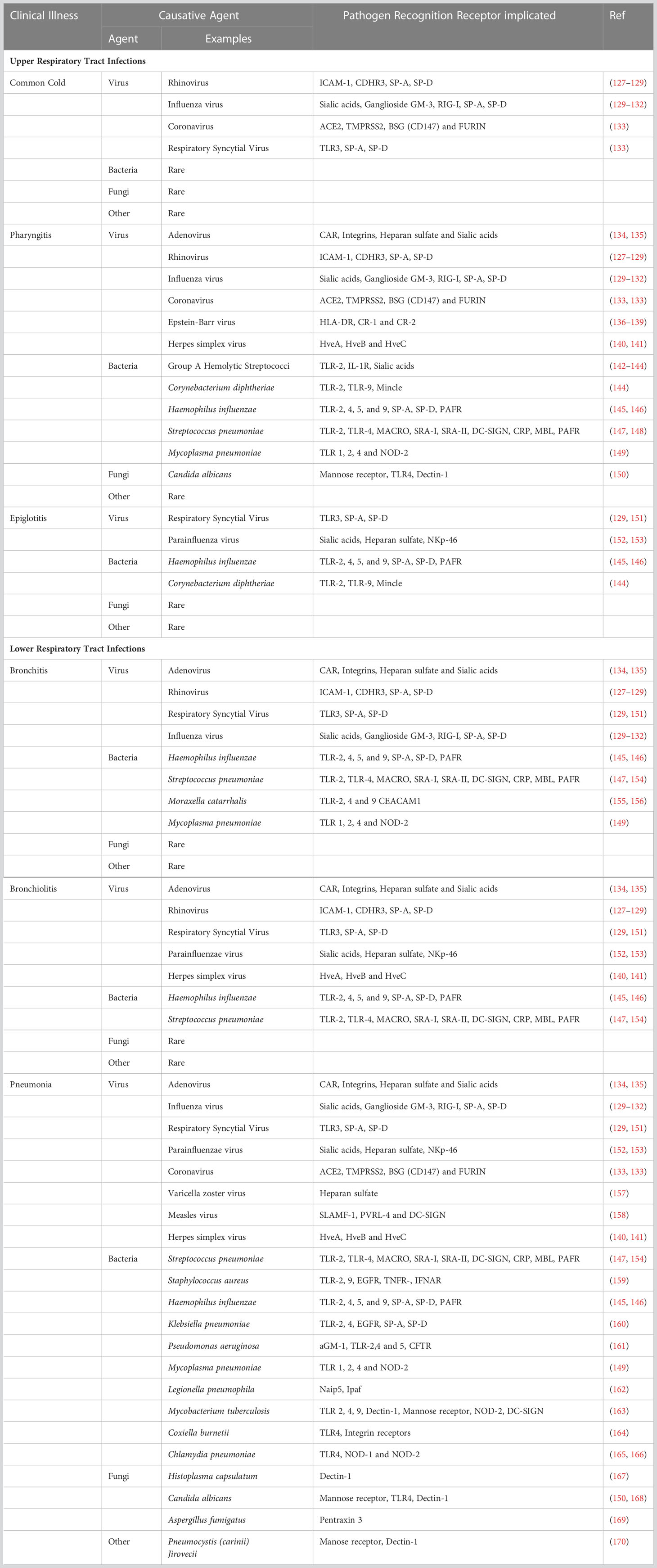
Table 3 Infectious diseases of the Lung.
Etiology: Bronchitis and bronchiolitis involve inflammation of the bronchial tree. Bronchitis is usually preceded by an upper respiratory tract infection and can be caused by a combination of environmental factors and bacterial infections by pathogens such as Haemophilus influenzae and Streptococcus pneumoniae. Chronic bronchitis is classically defined as persistent cough and sputum production with periodic exacerbation (171). Bronchiolitis is a viral respiratory disease of infants and is caused primarily by respiratory syncytial virus (RSV), influenza viruses, or rhinovirus. Bronchiolitis is a mild, self-limited infection in most children but may sometimes progress to respiratory failure in infants (172).
Pathogenesis and clinical manifestations: When the bronchial tree is infected, inflammation ensues, and the mucosa becomes hyperemic and edematous. Bronchial secretion increases in the mucosa and mucociliary function of the epithelium reduces. This reduces lower respiratory volume and leads to increased dyspnea, tachypnea, large amounts of sputum, tightness in the chest, fatigue, and confusion (173). The extent of damage to the mucosa can depend on the organisms(s) involved, bacterial toxins secreted, and other metabolic products produced (174). Infants with bronchiolitis suffer from inflammation in the airways and necrosis of the respiratory epithelium. Bronchial and bronchiolar walls thicken in bronchiolitis, and an exudate made up of necrotic material and respiratory secretions narrow the bronchial lumen, leading to airway obstruction. Clinical manifestations include fever, deepening of cough, dyspnea, tachypnea, wheezing, or actual lack of breath. Areas of air-pockets can develop, which may eventually result in respiratory failure and death (172, 174).
Microbiological diagnosis: Cough in the absence of fever, tachycardia, and tachypnea suggests bronchitis; however, bacteriological examination and culture of purulent respiratory secretions must be done for confirmation (175). Rapid diagnostic tests can be used to detect several pathogens linked to acute bronchitis; however, not all rapid tests are widely available and cost-effective. Aspirations of nasopharyngeal secretions or swabs from infants with bronchiolitis are collected as samples and serologic tests are done to identify the virus (176).
Prevention and treatment: With only a few exceptions, viral infections are treated with symptomatic care. Children with mild-to-moderate symptoms can be treated with nasal saline, antipyretics, and a cool-mist humidifier. On the contrary, children with severe respiratory distress should be admitted to the hospital and carefully monitored. The use of corticosteroids, bronchodilators, or prophylactic measures may benefit children with chronic bronchitis. RSV infections in infants may be treated with ribavirin, and influenza type A viruses may be treated with amantadine and rimantadine (175).
Pneumonia is an inflammation of the lung parenchyma, caused by several factors such as environmental contaminants, autoimmune diseases, and infection. Anatomically, pneumonia can be classified as lobar pneumonia (affecting the entire lobe) or bronchopneumonia (affecting patches of the lobe). Further, it can be etiologically classified as community-acquired pneumonia (CAP, infection in a previously healthy individual) or hospital-acquired pneumonia (infection in a hospitalized individual within 48 hours of admission).
Etiology: Bacterial pneumonias- S. pneumoniae is the most common cause of CAP and a leading cause of illness in children below 2 years of age, the elderly, and immunocompromised individuals (177). Pneumonias caused by other Streptococci are uncommon. Streptococcus pyogenes pneumonia is often associated with hemorrhagic pneumonitis and empyema. Pneumonia due to H. influenzae (usually nontypable) and Klebsiella pneumoniae are more common among patients over 50 years old with chronic obstructive lung disease or alcoholism, whereas pneumonia due to Mycoplasma pneumoniae and Chlamydophila pneumoniae are more common in school-aged children. Other less common agents causing pneumonias include Francisella tularensis, Legionella pneumophila, Yersinia pestis, and Neisseria meningitidis (178). Mycobacterium tuberculosis (Mtb) causes pulmonary pneumonia; however, the incidence of tuberculosis is low in industrialized countries. Nonetheless, it remains to be a significant public health problem in the United States.
Viral pneumonias- Pneumonias by RSV and Adenoviruses are rare in the healthy population. Pneumonia caused by influenza viruses is still a cause of high mortality in the elderly and in patients with underlying diseases. A serious complication of influenza is a secondary bacterial pneumonia which can be fatal in infants.
Other pneumonias- Actinomyces and Nocardia spp can cause pneumonitis. Aspergillus and Candida spp cause pneumonias in severely ill or immunosuppressed patients and neonates, whereas Pneumocystis carinii causes life-threatening pneumonia in patients with HIV.
Pathogenesis and clinical manifestations: The onset of pneumonia is quite abrupt; it develops with a shaking chill, fever, malaise, cough, and dyspnoea. Left untreated, this toxic illness can progress to acute respiratory failure, septic shock, multiorgan failure, and death within several days from onset (179). Respiratory failure due to pneumonia occurs when inflammatory exudate fills the alveoli and reduces its normal functional capacity. The persistence of pulmonary artery blood flow to the consolidated lung airspace during pneumonia leads to an intrapulmonary shunt that can also result in respiratory failure (180).
Microbiological diagnosis: A clinical examination of sputum is done to identify the infectious agent. Cultures of sputum, blood, and pleural fluid help in identifying the causative agent. The sputum quellung test can be used to identify S. pneumoniae, H. influenzae, and K. pneumoniae by serotype. In addition, rapid diagnostic tests based on PCR, ELISA, or other DNA probes can be used to identify other causative agents of pneumonia. Viral infections are usually diagnosed by serological testing for related antigens or antibodies in pulmonary secretions.
Prevention and treatment: Clinical diagnosis of the pathogen causing pneumonia is of utmost importance to deploy effective treatment options. Amoxicillin or amoxicillin clavulanate is the most common first-line antimicrobial agent used for CAP (181). In children suspected of having influenza and moderate-to-severe CAP, early antiviral therapy has been proven to be very helpful (182). Pneumococcal vaccines are available for high-risk populations; and along with yearly influenza vaccinations have been proven to reduce pneumonia burden and hospitalizations in children and adults (176). However, due to the presence of allelic variations amongst the bacterial serotypes, the current vaccines do not provide protection against all serotypes (183, 184). Since transmission occurs by droplets or contact, good hand washing, and good personal hygiene are the most important measures to prevent the spread of pneumonia.
COVID-19 is a viral infection caused by coronavirus SARS-CoV-2 that can progress to severe acute respiratory syndrome with pneumonia and acute respiratory distress syndrome (185). In 2019 the disease spread rapidly and became a pandemic affecting more than 600 million people worldwide. Histologically, COVID-19 shows diffuse alveolar damage corresponding to the phase of the disease (acute to fibrotic) and can be divided into 3 main injury patterns: epithelial, vascular and fibrotic affecting the upper respiratory tract in mild disease and bilateral lobes of the lung in more severe disease. The etiology of the disease includes attachment of SARS-CoV-2 virus to target cells via angiotensin converting enzyme-2 (ACE-2) receptors that promotes release of viral RNA into the host cell, initiating replication and further dissemination (186). Clinical features of the disease include fever, dry cough, shortness of breath, fatigue, myalgias, nausea or headache and weakness that may further develop into pneumonia, ARDS or death as the pathology worsens (187).
The pathophysiology involves diffuse or spotty changes in lung parenchyma that turn edematous due to congestion, punctuate hemorrhages, and hemorrhagic necrosis, particularly at the peripheral edges of the pulmonary lobe. Bronchi and bronchioles may show chronic inflammation with marked thickening of bronchial mucosa due to edema. The peripheral or central pulmonary arterial vessels are filled with platelet-fibrin thrombi and inflammatory cells and may result in fibrinoid necrosis, wall thickening, luminal stenosis, or occlusion, leading to the development of pulmonary hypertension in later stages in some critical patients (188). The clinical diagnosis is based on detection of viral RNA by RT-PCR in Nasopharyngeal swabs or sputum samples. The treatment guidelines recommend home management for patients with mild symptoms and hospital care for patients with severe disease requiring oxygenation support. New age mRNA-based vaccines along with viral vector based or whole inactivated viral vaccines are already being prescribed as a strategy to manage viral transmission (189).
To treat lung-related pathologies, the delivery of drugs and vaccines through the nasal and upper respiratory pathway is required. The ongoing COVID-19 pandemic involves a virus that causes significant damage to the lung. There have been several therapies and vaccines that treat and prevent disease progression; however, there is very limited development in delivery to the lung. In the following section, we have reviewed various formulations, devices, and human factors that contribute to the development of upper respiratory tract therapies. We have further compared the requirements for reaching the nasal mucosa, upper respiratory, and mid respiratory tract with those for deep lower respiratory tract.
Drug delivery through the respiratory route is a major strategy to treat asthma and various lung diseases. The devices for delivery of aerosols to treat respiratory diseases have been improvised over several decades from the classical models of nebulizers and pressurized metered-dose inhalers to a myriad of inhaler designs and devices, including dry powder inhalers, soft mist inhalers, and smart nebulizers. Educating and training on the proper use of these devices, determining the suitability of the device type and design according to the personal ease and convenience of the patient, and close monitoring of patient recovery following the use of appropriate inhaler device and the type of formulation are crucial to derive maximum health benefits from the administration of medications using these devices. Delivery of drugs directly to the airways of the respiratory tract and lungs can help reduce the dosage of medication required and improve the effectiveness of the therapy as compared to oral administration of drugs or systemic administration into the bloodstream of the patient.
The effective treatment of respiratory disorders using inhalation devices is closely associated with the formulations available for different types of diseases and the efficiency of the inhaler to aerosolize and deliver the drug to the appropriate location in the respiratory tract. For example, albuterol and ipratropium are bronchodilators used to treat COPD. These are available as liquid formulations (solutions) that can be conveniently aerosolized and delivered to the patient using nebulizers and pressurized metered-dose inhalers (pMDI) (190–193). Three processes govern the deposition of aerosol particles in the respiratory tract: inertial impaction, sedimentation, and diffusion. For most of the inhaled medications, the principal mechanisms of aerosol deposition are impaction and sedimentation (194, 195). Particles of size 3-7 µm get deposited by impaction predominantly in the oropharynx and the larger conduits of the respiratory tract. This impaction takes place particularly at the bifurcation of airways and sites, posing an obstruction to free flow of air, whereas the smallest aerosol particles (2-3 µm) can reach the terminal bronchioles and alveoli (194–196). Aerosol particle sedimentation is influenced by several factors including gravity, dimensions of air conduits, and breath hold time. The fine-particle fraction (FPF) of the inhaled drug that contains <5 µm aerosol particles is most crucial in bringing about desired clinical effects. Inhalers differ widely in respect to the fine-particle fraction, ranging from 10%–50% for pMDI, 12%–35% for dry powder inhalers (DPI), to 30%–50% for the soft mist inhalers (SMI).
The physicochemical and pharmacokinetic properties of drugs, such as half-life, molecular weight, solubility, lipophilicity, charge, and protein binding are important in determining their clearance from the lungs (8). Lipid-soluble molecules can traverse the respiratory tract epithelium by passive transport, whereas hydrophilic molecules travel through extracellular conduits. Medications administered by inhalation are deliberately rendered lipophilic to allow slow dissipation from the lungs and thus exert their effect over a longer duration.
The choice and suitability of an aerosol delivery device for a particular patient depends on the recommendations of the physician, available types of formulations, and compliance with the personal ease and comfort of the patient (Table 4 and Figure 3).
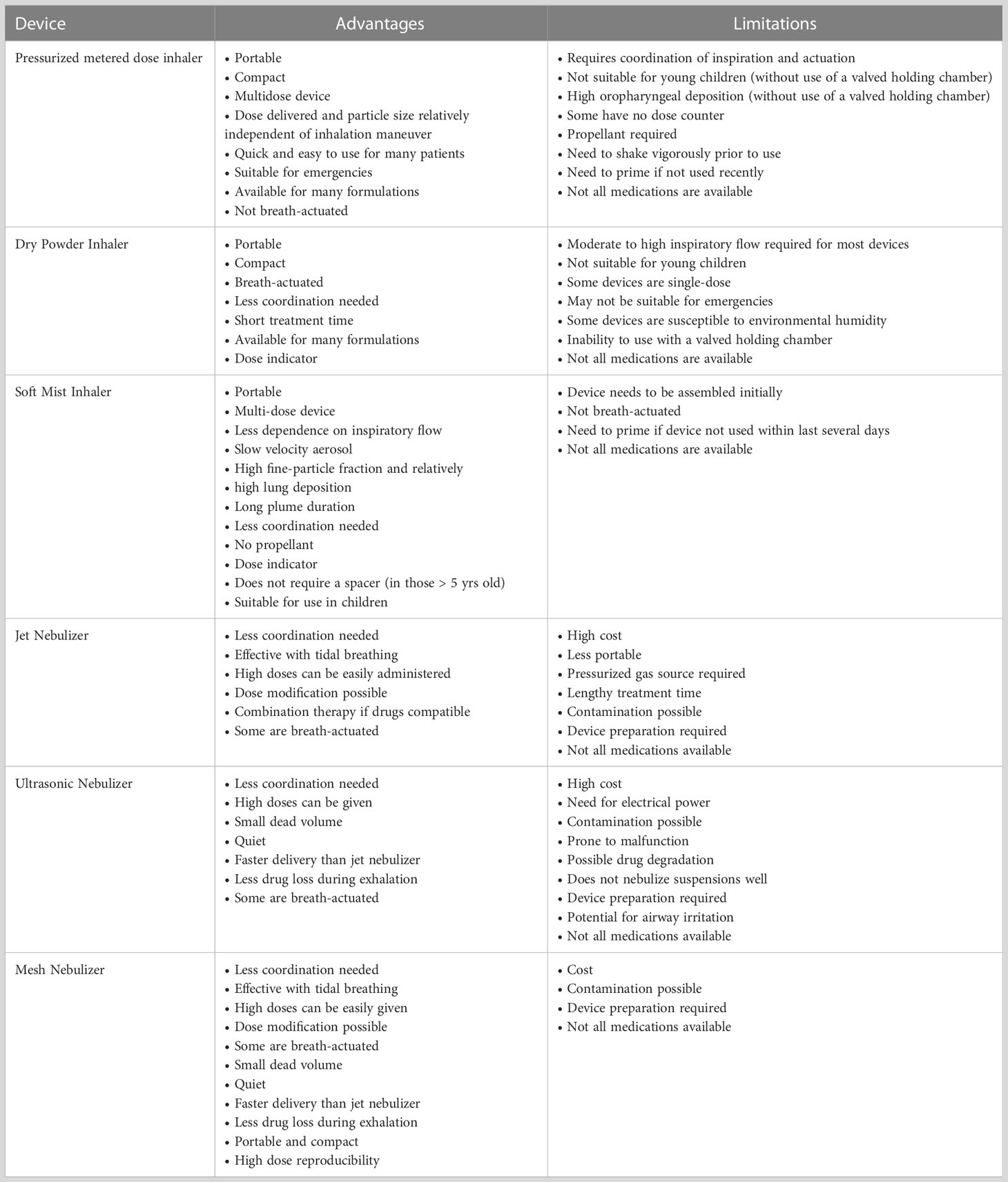
Table 4 Comparison of inhalers.
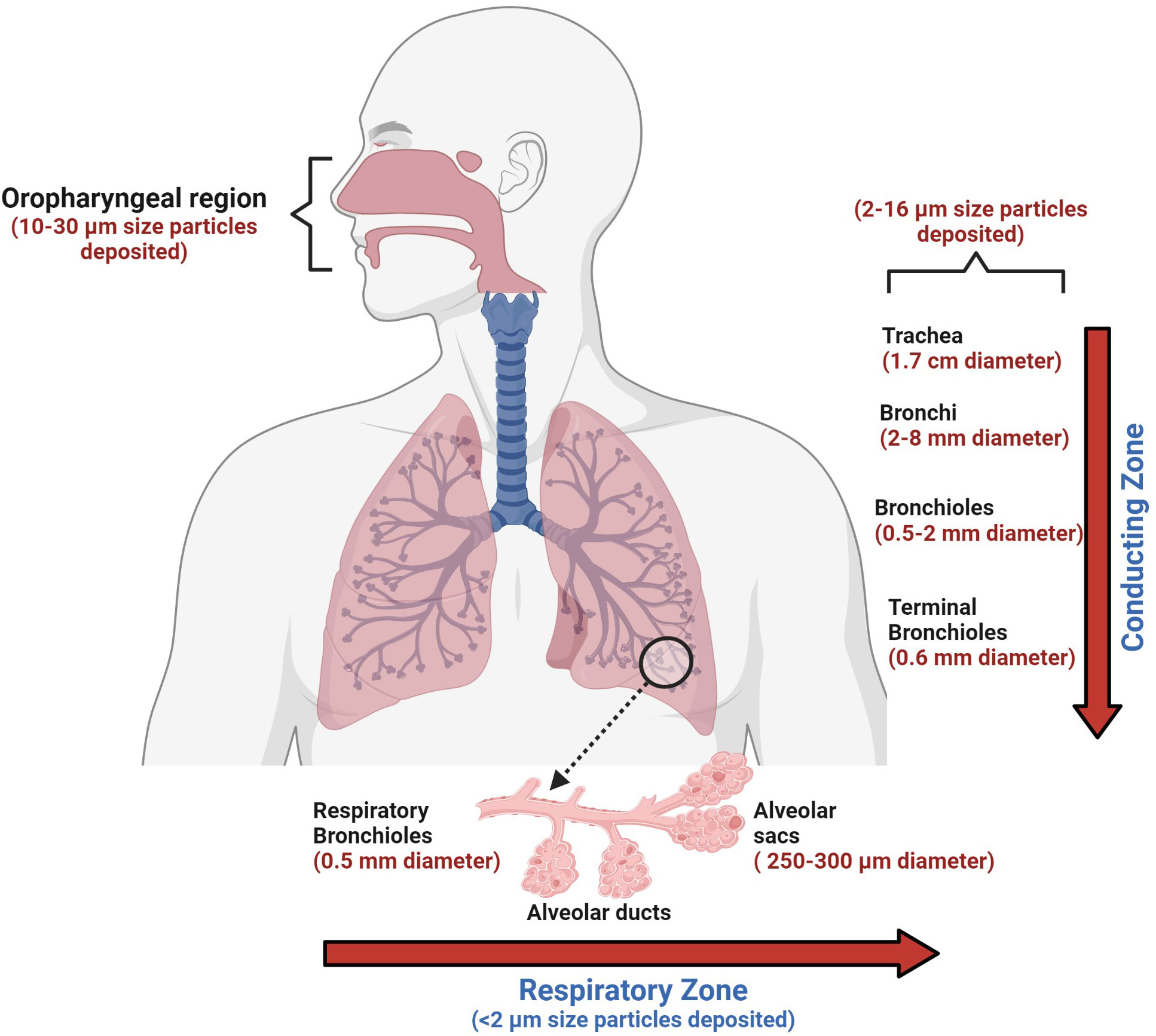
Figure 3 The human conducting and respiratory zones. The conducting and respiratory zones include the nasal cavity, pharynx, larynx, trachea, bronchi, bronchioles, and alveolar sacs. The air-conducting zones become smaller and smaller as the airways branch out into smaller tubes. Trachea, a 10 cm long structure with the largest diameter, is made of cartilage rings that provide structural support to the upper respiratory region. As the trachea branches into bronchi and further into smaller bronchioles, the inner epithelial lining also changes from thicker, taller, and ciliated cells to a thinner epithelium which has shorter and non-ciliated cells. The thinner lining promotes the exchange of inhaled air into the bloodstream in the alveolar sacs. Orchestrated movement of cilia in the tracheal region, faster air currents, and mucus efficiently trap particles larger than 10 microns, contrary to particles less than 2 microns. Particles that are less than 2 microns have higher chances to reach alveoli, which can lead to impaired gas exchange, which may result in serious health consequences.
pMDIs were the foremost among the extensively used inhaler devices (190, 191, 197) and continue to be popular to date due to ready availability, reasonably low cost, multidose drug administration, small size, easy portability, and the availability of many compatible formulations. The majority of the patients with asthma or COPD are administered with bronchodilators (albuterol/ipratropium) using pMDIs. Inhaled drugs for the treatment of obstructive lung disorders are widely available for use with pMDI, either singly or in combination. The formulation used in PMDIs may be a solution, suspension, or co-suspension. In addition, it contains other ingredients like propellants, co-solvents, and surfactants (for example, lecithin or oleic acid) (198). Sometimes, pMDIs show a problem of flocculation of drug crystals, leading to sedimentation. Particle crystal agglomeration can be minimized using surfactants. A novel strategy, known as co-suspension technology, helps to ameliorate this problem. It involves attaching phospholipid particles to the drug molecules to render a more uniform drug suspension, enabling the formulation of drug combinations and relying less on the need for shaking before use (199). pMDIs are not suitable for inhaled drug administration to young children without the use of a valved holding chamber. Moreover, administration using pMDIs results in high deposition of the drug in the oropharynx with relatively less amount reaching the finer airways in the lungs. The requirement of a propellant and the need to shake vigorously before use are some other concerns pertaining to the use of pMDIs.
Nebulizers are the earliest among the modern methods of delivering aerosols to the lungs for respiratory drug delivery. Nebulizers are routinely used in hospitals and home settings. Several newer nebulization technologies have enabled more efficient use of nebulizers as drug delivery devices. A jet nebulizer harnesses a stream of compressed gas to generate droplets from the drug solution or suspension (200). Some conventional jet nebulizer designs (like non-vented jet nebulizers) result in substantial loss of the aerosolized drug into the room air. This problem has been circumvented by the development of mesh nebulizers and ultrasonic nebulizers. Mesh nebulizers can produce droplets of uniform size without imposing high mechanical stress on the drug solution, they are associated with relatively less temperature rise during the nebulization process (201), and they can administer higher drug doses leading to faster patient recovery (202), they do not require baffles, thus minimizing residual losses, and they can nebulize a myriad of solutions and suspensions. These devices operate using batteries or a direct power supply, eliminating the requirement of an external gas flow. However, mesh nebulizers have some issues associated with them. They are costlier than jet nebulizers. The apertures in the mesh are prone to clog, and the high surface area of the mesh (owing to the presence of numerous apertures) requires careful cleaning (202). Examples of FDA-approved drugs for delivery using mesh nebulizers include glycopyrrolate (a long-acting muscarinic antagonist for COPD) and aztreonam (an antibiotic for cystic fibrosis).
Ultrasonic nebulizers are electrically powered devices in which a drug solution is subjected to ultrasonic waves of high frequency, resulting in an aerosol generation. Portable, small-sized ultrasonic nebulizers are available in the market for the administration of bronchodilators. They are more expensive than jet nebulizers but are comparable to mesh nebulizers in this regard. Treprostinil (available for inhalation under the brand name Tyvaso) has been successfully used to treat patients with pulmonary arterial hypertension using an ultrasonic nebulizer (203). A major drawback of ultrasonic nebulizers is the possible drug inactivation due to ultrasonic waves. Moreover, they are not efficient in nebulizing a suspension (204).
Soft mist inhalers (SMIs) (Respimat, Boehringer Ingelheim, Ingelheim am Rhein, Germany) were originally available for the administration of ipratropium/albuterol, which later became available for other bronchodilators (205, 206). A compressed spring within the device pushes the drug through a membrane, emitting an aerosol (205). It is not necessary to shake before using the device. It is a portable, multi-dose device that generates aerosols with a high fine-particle fraction, resulting in substantial deposition of drugs in the lungs. In comparison to pMDIs, Respimat discharges aerosols slower (205, 206) and over a longer duration (-1.2 s as compared to < 0.5 s from pMDIs) which may reduce the coordination required for actuation and inspiration, thus improving the likelihood for greater lung deposition. Further, it does not require a propellant and is suitable for use in children.
Dry powder inhalers (DPIs) are excellent alternatives to pMDIs as they help to circumvent many limitations associated with pMDIs such as low lung deposition of drugs and the need for propellants. In 1967, Fisons (Ipswich, UK) launched the Spinhaler® device. Thereafter, striking advances have been made in DPI design. and a wide variety of medications are available as dry powder formulations, making them extremely useful to patients with respiratory tract diseases. Some of the commercially available DPI designs are Genuair, Breezhaler, Podhaler, Dreamboat, Conix, Twincer, Twincaps, and Staccato. They are compact and easily portable and can help impart short treatment times. The TOBI® Podhaler® (Novartis, Basel, Switzerland) holds merit over jet nebulizers for the treatment of cystic fibrosis. Administration of Tobramycin Inhalation Solution (TIS™) (Novartis, Basel, Switzerland) using a jet-nebulizer posed several problems. For example, the device requires long inhalation times cleaning of the nebulizer is required after every use, which increases the chances of lung infection from devices that are not cleaned properly (207). Moreover, nebulizers are rather voluminous and require a jet of compressed air to generate aerosols. Further, TIS should be refrigerated during storage (208), and the product may undergo degradation before use. Considering these factors, the TOBI Podhaler is safer and more convenient for cystic fibrosis patients.
However, there are some demerits of DPIs. Due to inter-particulate forces like electrostatic interactions and van der Waals forces (209), micronized powders are adhesive/cohesive and spontaneously form agglomerates. These agglomerates must be deagglomerated prior to or during the processes of aerosolization and inhalation to produce a substantial fraction of particles within the range of 1–5 µm in size. These particles can reach small tubules and alveolar spaces in the lungs. In passive DPIs, the energy required to overcome the above-mentioned inter-particulate forces is derived from the patient’s inspiratory flow (210, 211), whereas, in active DPIs, energy is harnessed via other sources. The merit of a passive DPI is that it averts the need to synchronize the actuation and inspiration maneuver by the patient. However, this requires a pronounced inspiratory effort on the part of the patient, which might be difficult for the elderly and children, and those suffering from COPD or asthma (212). Because the rheological properties of micronized powders are often poor, in most formulations, drug particles are blended with larger carrier particles (30–90 μm in size) such as lactose to facilitate deagglomeration and powder flow (213). The performance of DPIs is susceptible to environmental humidity as dry powder formulations might absorb moisture from the environment. Further, these devices are not amicable to use with valved holding chambers.
A wide variety of inhaler devices are commercially available today; however, knowledge of correct device use, device maintenance, and stringent compliance to the instructions provided by the medical practitioner are pivotal in achieving maximum and timely therapeutic benefits. Newer models of inhalers are constantly on the horizon to make them more user-friendly and convenient. Effective communication and coordination among doctors, nurses, and patients and being updated with the latest developments in the field of inhaler device designs and applications can help in the effective and timely implementation of treatment regimens.
Exosomes are naturally occurring nanoparticles present in most bodily fluids such as serum, plasma, urine, breast milk, saliva, ascites, and synovial and amniotic fluids. These extracellular vesicles are like bacterial outer membrane vesicles and vary in size from 20 microns to 200, assisting in cell-cell communication and intercellular transport (17, 214). The exosomes contain exogenous biomolecules such as mRNA, proteins, DNA, miRNA, lipids and conserved transmembrane proteins such as CD81, CD63, and CD9, which are used as biomarkers for identifying exosomes. As the expression of these transmembrane proteins is based on cell type, they are responsible for delivering the cargo by identifying the specific recipient cells (17, 18). Several studies have demonstrated the advantages and ability of these nanocarriers over synthetic liposomes for drug delivery (18, 19). Current bioengineering techniques permit modification of exosome surface to enhance cargo loading, allowing superior drug delivery to targeted cell types. Even though biological nanoparticles have been investigated as drug delivery vehicles for several diseases, using exosomes for treating pulmonary disorders is still in its early phase. Recently Kim et al. developed an engineered exosome with therapeutic peptides and curcumin, which can be delivered into the lungs by inhalation. These engineered exosomes significantly reduced proinflammatory cytokines and inhibited inflammation (19). In another study by Popowski et al., lung-derived exosomes were used as inhaled nanoparticle delivery systems, which delivered mRNA and protein drugs. As these exosomes are delivered by inhalation and are naturally derived, the pulmonary bioavailability of the encapsulated drugs increases significantly (22). The drug must reach the deep lung tissue for treating lower respiratory diseases, and the pulmonary distribution should be optimal. Drug-carrying exosomes fulfill all these criteria and are naturally optimized molecules that can be delivered to the targeted site using currently available techniques. However, further investigation and clinical studies need to be directed to completely understand the long-term health effects of these biological nanoparticles. Further, more studies are required to understand their full potential as drug delivery systems.
The normal functioning of the lung requires complex interactions of many different cell types embedded in a precise architecture of the lung. Normal human lungs are constantly exposed to a plethora of inflammatory molecules, allergens, pollutants, and microbes from the environment and can induce cough, shortness of breath, chest pain, and fever. The nature of the immune responses to these foreign entities involves a coordinated action of the innate and adaptive immunity, which enables a balanced response and minimize lung pathology.
In this review we have described various aspects of lung homeostasis and innate and adaptive responses required to maintain a balanced state. We reviewed lung inflammatory responses and infectious diseases contributing to lung disease pathogenesis with a brief description of COVID-19 lung pathology. Further, we reviewed currently available therapies to treat lung diseases and devices used to deliver drugs including exosomes, directed to the airways of the respiratory tract. The review provides a guide to lung immunology expanding on the interplay of different aspects of the immune system, inflammation and lung associated infectious diseases and how various strategies are used for lung pathology treatment. It aims to provide a technical insight into the current landscape of drugs and devices used to treat various lung diseases and discusses their limitations to initiate a colloquy about future targets of therapeutic interventions.
IG: Wrote the Lung Inflammation Section; RD: Wrote the Infectious Diseases Section; VB: Wrote the Therapy and Devices Section; VG: Wrote the Lung Immunology and Exosomes as drug delivery Section; NC: Conceived the concept and directed the writing. All authors contributed to the article and approved the submitted version. IG, RD and VB contributed equally to the manuscript.
IG is an employee of AstraZeneca and may hold stock ownership and/or stock options or interests in the company. NC is employed by SymphonyTech Biologics and VB is employed by Lamark.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, et al. Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol (2013) 45:2568–73. doi: 10.1016/j.biocel.2013.08.009
3. Rock JR, Hogan BL. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu Rev Cell Dev Biol (2011) 27:493–512. doi: 10.1146/annurev-cellbio-100109-104040
4. Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res (2001) 2:33–46. doi: 10.1186/rr36
5. Debbabi H, Ghosh S, Kamath AB, Alt J, Demello DE, Dunsmore S, et al. Primary type II alveolar epithelial cells present microbial antigens to antigen-specific CD4+ T cells. Am J Physiol Lung Cell Mol Physiol (2005) 289:L274–9. doi: 10.1152/ajplung.00004.2005
6. Lo B, Hansen S, Evans K, Heath JK, Wright JR. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. J Immunol (2008) 180:881–8. doi: 10.4049/jimmunol.180.2.881
7. Pechkovsky DV, Goldmann T, Ludwig C, Prasse A, Vollmer E, Muller-Quernheim J, et al. CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir Res (2005) 6:75. doi: 10.1186/1465-9921-6-75
8. Shi T, Denney L, An H, Ho LP, Zheng Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J Leukoc Biol (2021) 110:107–14. doi: 10.1002/JLB.3RU0720-418R
9. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front Immunol (2021) 12:753940. doi: 10.3389/fimmu.2021.753940
10. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
11. Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest (2009) 119:3723–38. doi: 10.1172/JCI39717
12. Riches DWH, Martin TR. Overview of innate lung immunity and inflammation. Methods Mol Biol (2018) 1809:17–30. doi: 10.1007/978-1-4939-8570-8_2
13. Hartl D, Tirouvanziam R, Laval J, Greene CM, Habiel D, Sharma L, et al. Innate immunity of the lung: From basic mechanisms to translational medicine. J Innate Immun (2018) 10:487–501. doi: 10.1159/000487057
14. Greene CM, Hiemstra PS. Innate immunity of the lung. J Innate Immun (2020) 12:1–3. doi: 10.1159/000504621
15. Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc (2005) 2:403–11. doi: 10.1513/pats.200508-090JS
16. Ardain A, Marakalala MJ, Leslie A. Tissue-resident innate immunity in the lung. Immunology (2020) 159:245–56. doi: 10.1111/imm.13143
17. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
18. Futosi K, Fodor S, Mocsai A. Reprint of neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17:1185–97. doi: 10.1016/j.intimp.2013.11.010
19. Liu J, Pang Z, Wang G, Guan X, Fang K, Wang Z, et al. Advanced role of neutrophils in common respiratory diseases. J Immunol Res (2017) 2017:6710278. doi: 10.1155/2017/6710278
20. Monick MM, Hunninghake GW. Activation of second messenger pathways in alveolar macrophages by endotoxin. Eur Respir J (2002) 20:210–22. doi: 10.1183/09031936.02.00252001
21. Waters CM, Roan E, Navajas D. Mechanobiology in lung epithelial cells: measurements, perturbations, and responses. Compr Physiol (2012) 2:1–29. doi: 10.1002/cphy.c100090
22. Bi J, Cui L, Yu G, Yang X, Chen Y, Wan X. NK cells alleviate lung inflammation by negatively regulating group 2 innate lymphoid cells. J Immunol (2017) 198:3336–44. doi: 10.4049/jimmunol.1601830
23. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
24. Panda SK, Colonna M. Innate lymphoid cells in mucosal immunity. Front Immunol (2019) 10:861. doi: 10.3389/fimmu.2019.00861
25. Eckle SB, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HE, Reantragoon R, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J Exp Med (2014) 211:1585–600. doi: 10.1084/jem.20140484
26. Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med (2013) 210:2305–20. doi: 10.1084/jem.20130958
27. Wen X, Zhang X, Nian S, Wei G, Guo X, Yu H, et al. Title of article: Mucosal-associated invariant T cells in lung diseases. Int Immunopharmacol (2021) 94:107485. doi: 10.1016/j.intimp.2021.107485
28. Hossain MK, Wall KA. Use of dendritic cell receptors as targets for enhancing anti-cancer immune responses. Cancers (Basel) (2019) 11(3):418. doi: 10.3390/cancers11030418
29. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol (2011) 45:189–201. doi: 10.1165/rcmb.2011-0011RT
30. Schutte BC, McCray PB Jr. [beta]-defensins in lung host defense. Annu Rev Physiol (2002) 64:709–48. doi: 10.1146/annurev.physiol.64.081501.134340
31. Cole AM, Waring AJ. The role of defensins in lung biology and therapy. Am J Respir Med (2002) 1:249–59. doi: 10.1007/BF03256616
32. Gauvreau GM, Sehmi R, Ambrose CS, Griffiths JM. Thymic stromal lymphopoietin: its role and potential as a therapeutic target in asthma. Expert Opin Ther Targets (2020) 24:777–92. doi: 10.1080/14728222.2020.1783242
33. Parnes JR, Molfino NA, Colice G, Martin U, Corren J, Menzies-Gow A. Targeting TSLP in asthma. J Asthma Allergy (2022) 15:749–65. doi: 10.2147/JAA.S275039
34. Curtis JL. Cell-mediated adaptive immune defense of the lungs. Proc Am Thorac Soc (2005) 2:412–6. doi: 10.1513/pats.200507-070JS
35. Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res (2010) 690:24–39. doi: 10.1016/j.mrfmmm.2009.09.005
36. Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal (2008) 10:287–301. doi: 10.1089/ars.2007.1897
37. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
38. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J (2015) 45:1434–45. doi: 10.1183/09031936.00174914
39. Fala L. Ofev (Nintedanib): First tyrosine kinase inhibitor approved for the treatment of patients with idiopathic pulmonary fibrosis. Am Health Drug Benefits (2015) 8:101–4.
40. Kaplan AP, Giménez-Arnau AM, Saini SS. Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy (2017) 72:519–33. doi: 10.1111/all.13083
41. Fala L. Nucala (Mepolizumab): First IL-5 antagonist monoclonal antibody FDA approved for maintenance treatment of patients with severe asthma. Am Health Drug Benefits (2016) 9:106–10.
42. Máspero J. Reslizumab in the treatment of inadequately controlled asthma in adults and adolescents with elevated blood eosinophils: clinical trial evidence and future prospects. Ther Adv Respir Dis (2017) 11:311–25. doi: 10.1177/1753465817717134
43. Dávila González I, Moreno Benítez F, Quirce S. Benralizumab: A new approach for the treatment of severe eosinophilic asthma. J Investig Allergol Clin Immunol (2019) 29:84–93. doi: 10.18176/jiaci.0385
44. Dorey-Stein ZL, Shenoy KV. Tezepelumab as an emerging therapeutic option for the treatment of severe asthma: Evidence to date. Drug Des Devel Ther (2021) 15:331–8. doi: 10.2147/DDDT.S250825
45. Spoelstra FM, Postma DS, Hovenga H, Noordhoek JA, Kauffman HF. Additive anti-inflammatory effect of formoterol and budesonide on human lung fibroblasts. Thorax (2002) 57:237–41. doi: 10.1136/thorax.57.3.237
46. Baye J. Roflumilast (daliresp): a novel phosphodiesterase-4 inhibitor for the treatment of severe chronic obstructive pulmonary disease. Pharmacy and Therapeutics (2012) 37:149–61. P t.
47. Babu KS, Morjaria JB. Umeclidinium in chronic obstructive pulmonary disease: latest evidence and place in therapy. Ther Adv Chronic Dis (2017) 8:81–91. doi: 10.1177/2040622317700822
48. Mosley JF 2nd, Smith LL, Dutton BN. Tiotropium Bromide/Olodaterol (Stiolto respimat): Once-daily combination therapy for the maintenance of COPD. Pharmacy and Therapeutics (2016) 41:97–102. P t.
49. Al-Salama ZT, Frampton JE. Glycopyrronium/Formoterol: A review in COPD. Drugs (2019) 79:1455–66. doi: 10.1007/s40265-019-01186-x
50. D’Urzo AD, Singh D, Donohue JF, Chapman KR, Wise RA. Aclidinium bromide/formoterol fumarate as a treatment for COPD: an update. Expert Rev Respir Med (2021) 15:1093–106. doi: 10.1080/17476348.2021.1920403
51. Li F, Yang J. Revefenacin for the treatment of chronic obstructive pulmonary disease. Expert Rev Clin Pharmacol (2019) 12:293–8. doi: 10.1080/17512433.2019.1587292
52. Ferguson GT, Rabe KF, Martinez FJ, Fabbri LM, Wang C, Ichinose M, et al. Triple therapy with budesonide/glycopyrrolate/formoterol fumarate with co-suspension delivery technology versus dual therapies in chronic obstructive pulmonary disease (KRONOS): a double-blind, parallel-group, multicentre, phase 3 randomised controlled trial. Lancet Respir Med (2018) 6:747–58. doi: 10.1016/S2213-2600(18)30327-8
53. Sauleda J, Núñez B, Sala E, Soriano JB. Idiopathic pulmonary fibrosis: Epidemiology, natural history, phenotypes. Med Sci (Basel) (2018) 6(4):110. doi: 10.3390/medsci6040110
54. Fujiwara K, Kobayashi T, Fujimoto H, Nakahara H, D'Alessandro-Gabazza CN, Hinneh JA, et al. Inhibition of cell apoptosis and amelioration of pulmonary fibrosis by thrombomodulin. Am J Pathol (2017) 187:2312–22. doi: 10.1016/j.ajpath.2017.06.013
55. Mims JW. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol (2015) 5 Suppl 1:S2–6. doi: 10.1002/alr.21609
56. Ashley SL, Sisson TH, Wheaton AK, Kim KK, Wilke CA, Ajayi IO, et al. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol (2016) 54:482–92. doi: 10.1165/rcmb.2015-0148OC
57. Passalacqua G, Mincarini M, Colombo D, Troisi G, Ferrari M, Bagnasco D, et al. IL-13 and idiopathic pulmonary fibrosis: Possible links and new therapeutic strategies. Pulm Pharmacol Ther (2017) 45:95–100. doi: 10.1016/j.pupt.2017.05.007
58. Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair (2012) 5:11. doi: 10.1186/1755-1536-5-11
59. Lo Re S, Lison D, Huaux F. CD4+ T lymphocytes in lung fibrosis: diverse subsets, diverse functions. J Leukoc Biol (2013) 93:499–510. doi: 10.1189/jlb.0512261
60. Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol (1994) 152:3606–14. doi: 10.4049/jimmunol.152.7.3606
61. Wijsenbeek MS, Kool M, Cottin V. Targeting interleukin-13 in idiopathic pulmonary fibrosis: from promising path to dead end. Eur Respir J (2018) 52:1802111. doi: 10.1183/13993003.02111-2018
62. Okamoto M, Hoshino T, Kitasato Y, Sakazaki Y, Kawayama T, Fujimoto K, et al. Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur Respir J (2011) 37:1119–27. doi: 10.1183/09031936.00059810
63. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest (1999) 103:779–88. doi: 10.1172/JCI5909
64. Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel SL, Hogaboam CM. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol (2001) 166:5219–24. doi: 10.4049/jimmunol.166.8.5219
65. Chung SI, Horton JA, Ramalingam TR, White AO, Chung EJ, Hudak KE, et al. IL-13 is a therapeutic target in radiation lung injury. Sci Rep (2016) 6:39714. doi: 10.1038/srep39714
66. Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol (2004) 4:583–94. doi: 10.1038/nri1412
67. She YX, Yu QY, Tang XX. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discovery (2021) 7:52. doi: 10.1038/s41420-021-00437-9
68. Gharaee-Kermani M, McGarry B, Lukacs N, Huffnagle G, Egan RW, Phan SH. The role of IL-5 in bleomycin-induced pulmonary fibrosis. J Leukoc Biol (1998) 64:657–66. doi: 10.1002/jlb.64.5.657
69. Chuquimia OD, Petursdottir DH, Periolo N, Fernández C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect Immun (2013) 81:381–9. doi: 10.1128/IAI.00950-12
70. Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J (2008) 32:1631–8. doi: 10.1183/09031936.00176807
71. Gopallawa I, Uhal BD. Molecular and cellular mechanisms of the inhibitory effects of ACE-2/ANG1-7/Mas axis on lung injury. Curr Top Pharmacol (2014) 18:71–80.
72. Budinger GR, Mutlu GM, Eisenbart J, Fuller AC, Bellmeyer AA, Baker CM, et al. Proapoptotic bid is required for pulmonary fibrosis. Proc Natl Acad Sci U.S.A. (2006) 103:4604–9.
73. Kuwano K, Kunitake R, Maeyama T, Hagimoto N, Kawasaki M, Matsuba T, et al. Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am J Physiol Lung Cell Mol Physiol (2001) 280:L316–25. doi: 10.1152/ajplung.2001.280.2.L316
74. Gopallawa I, Uhal BD. Angiotensin-(1-7)/mas inhibits apoptosis in alveolar epithelial cells through upregulation of MAP kinase phosphatase-2. Am J Physiol Lung Cell Mol Physiol (2016) 310:L240–8. doi: 10.1152/ajplung.00187.2015
75. Rafii R, Juarez MM, Albertson TE, Chan AL. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis (2013) 5:48–73.
76. Somogyi V, Chaudhuri N, Torrisi SE, Kahn N, Müller V, Kreuter M. The therapy of idiopathic pulmonary fibrosis: what is next? Eur Respir Rev (2019) 28:190021. doi: 10.1183/16000617.0021-2019
77. Roach K, Castells E, Dixon K, Mason S, Elliott G, Marshall H, et al. Evaluation of pirfenidone and nintedanib in a human lung model of fibrogenesis. Front Pharmacol (2021) 12. doi: 10.3389/fphar.2021.679388
78. King PT. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin Transl Med (2015) 4:68. doi: 10.1186/s40169-015-0068-z
79. Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Rev Evidence Thorax (2010) 65:930–6. doi: 10.1136/thx.2009.130260
80. Siva R, Green RH, Brightling CE, Shelley M, Hargadon B, McKenna S, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J (2007) 29:906–13. doi: 10.1183/09031936.00146306
81. Tsutsumi A, Chubachi S, Irie H, Sasaki M, Yamada Y, Sugiura H, et al. Characteristics of chronic obstructive pulmonary disease patients with robust progression of emphysematous change. Sci Rep (2021) 11:9548. doi: 10.1038/s41598-021-87724-8
82. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax (2021) 76:188–95. doi: 10.1136/thoraxjnl-2020-215167
83. Badolati I, Sverremark-Ekström E, van der Heiden M. Th9 cells in allergic diseases: A role for the microbiota? Scand J Immunol (2020) 91:e12857. doi: 10.1111/sji.12857
84. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol (2013) 48:531–9. doi: 10.1165/rcmb.2012-0492TR
85. Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am J Respir Crit Care Med (2009) 179:1011–21. doi: 10.1164/rccm.200807-1148OC
86. Borges VM, Vandivier RW, McPhillips KA, Kench JA, Morimoto K, Groshong SD, et al. TNFalpha inhibits apoptotic cell clearance in the lung, exacerbating acute inflammation. Am J Physiol Lung Cell Mol Physiol (2009) 297:L586–95. doi: 10.1152/ajplung.90569.2008
87. George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis (2016) 7:34–51. doi: 10.1177/2040622315609251
88. Baines KJ, Wright TK, Simpson JL, McDonald VM, Wood LG, Parsons KS, et al. Airway β-Defensin-1 protein is elevated in COPD and severe asthma. Mediators Inflammation (2015) 2015:407271. doi: 10.1155/2015/407271
89. Polosukhin VV, Cates JM, Lawson WE, Zaynagetdinov R, Milstone AP, Massion PP, et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2011) 184:317–27. doi: 10.1164/rccm.201010-1629OC
90. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J (2019) 54:1900651. doi: 10.1183/13993003.00651-2019
91. Lommatzsch M, Speer T, Herr C, Jörres RA, Watz H, Müller A, et al. IgE is associated with exacerbations and lung function decline in COPD. Respir Res (2022) 23:1. XXXC.s.g. for the. doi: 10.1186/s12931-021-01847-0
92. Barnes PJ, Stockley RA. COPD: current therapeutic interventions and future approaches. Eur Respir J (2005) 25:1084–106. doi: 10.1183/09031936.05.00139104
93. Wagena EJ, van der Meer RM, Ostelo RJ, Jacobs JE, van Schayck CP. The efficacy of smoking cessation strategies in people with chronic obstructive pulmonary disease: results from a systematic review. Respir Med (2004) 98:805–15. doi: 10.1016/j.rmed.2004.06.001
94. Poole P, Chacko EE, Wood-Baker R, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Systematic Rev (2006) 6. doi: 10.1002/14651858.CD002733.pub2
95. Niewoehner DE, Rice K, Cote C, Paulson D, Cooper JAD, Korducki L, et al. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator. Ann Internal Med (2005) 143:317–26. doi: 10.7326/0003-4819-143-5-200509060-00007
96. Bateman ED, Rabe KF, Calverley PMA, Goehring UM, Brose M, Bredenbröker D, et al. Roflumilast with long-acting β2-agonists for COPD: influence of exacerbation history. Eur Respir J (2011) 38:553–60. doi: 10.1183/09031936.00178710
97. Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers (2015) 1:15025. doi: 10.1038/nrdp.2015.25
98. Tulic MK, Christodoulopoulos P, Hamid Q. Small airway inflammation in asthma. Respir Res (2001) 2:333. doi: 10.1186/rr83
99. Mukherjee AB, Zhang Z. Allergic asthma: Influence of genetic and environmental factors. J Biol Chem (2011) 286:32883–9. doi: 10.1074/jbc.R110.197046
100. Leόn B. T Cells in allergic asthma: Key players beyond the Th2 pathway. Curr Allergy Asthma Rep (2017) 17:43. doi: 10.1007/s11882-017-0714-1
101. Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM. Treatment of allergic asthma: Modulation of Th2 cells and their responses. Respir Res (2011) 12:114. doi: 10.1186/1465-9921-12-114
102. Doran E, Cai F, Holweg CTJ, Wong K, Brumm J, Arron JR. Interleukin-13 in asthma and other eosinophilic disorders. Front Med (2017) 4. doi: 10.3389/fmed.2017.00139
103. Walford HH, Doherty TA. Diagnosis and management of eosinophilic asthma: a US perspective. J Asthma Allergy (2014) 7:53–65.
104. Bousquet J, Chanez P, Lacoste JY, Barnéon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. New Engl J Med (1990) 323:1033–9. doi: 10.1056/NEJM199010113231505
105. Baos S, Calzada D, Cremades-Jimeno L, Sastre J, Picado C, Quiralte J, et al. Nonallergic asthma and its severity: Biomarkers for its discrimination in peripheral samples. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01416
106. Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol (1999) 162:6233–7. doi: 10.4049/jimmunol.162.10.6233
107. Moser R, Fehr J, Bruijnzeel PL. IL-4 controls the selective endothelium-driven transmigration of eosinophils from allergic individuals. J Immunol (1992) 149:1432–8. doi: 10.4049/jimmunol.149.4.1432
108. Steinke JW, Borish L. Th2 cytokines and asthma. interleukin-4: its role in the pathogenesis of asthma, and targeting it for asthma treatment with interleukin-4 receptor antagonists. Respir Res (2001) 2:66–70. doi: 10.1186/rr40
109. Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol (2000) 105:1063–70. doi: 10.1067/mai.2000.107604
110. Corren J. Role of interleukin-13 in asthma. Curr Allergy Asthma Rep (2013) 13:415–20. doi: 10.1007/s11882-013-0373-9
111. Rael EL, Lockey RF. Interleukin-13 signaling and its role in asthma. World Allergy Organ J (2011) 4:54–64. doi: 10.1097/WOX.0b013e31821188e0
112. Chen CY, Lee JB, Liu B, Ohta S, Wang PY, Kartashov AV, et al. Induction of interleukin-9-Producing mucosal mast cells promotes susceptibility to IgE-mediated experimental food allergy. Immunity (2015) 43:788–802. doi: 10.1016/j.immuni.2015.08.020
113. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol (2015) 136:433–40.e1. doi: 10.1016/j.jaci.2015.01.021
114. Dong Q, Louahed J, Vink A, Sullivan CD, Messler CJ, Zhou Y, et al. IL-9 induces chemokine expression in lung epithelial cells and baseline airway eosinophilia in transgenic mice. Eur J Immunol (1999) 29:2130–9. doi: 10.1002/(SICI)1521-4141(199907)29:07<2130::AID-IMMU2130>3.0.CO;2-S
115. Porsbjerg CM, Sverrild A, Lloyd CM, Menzies-Gow AN, Bel EH. Anti-alarmins in asthma: targeting the airway epithelium with next-generation biologics. Eur Respir J (2020) 56:2000260. doi: 10.1183/13993003.00260-2020
116. West EE, Kashyap M, Leonard WJ. TSLP: A key regulator of asthma pathogenesis. Drug Discovery Today Dis Mech (2012) 9. doi: 10.1016/j.ddmec.2012.09.003
117. Lee HC, Headley MB, Iseki M, Ikuta K, Ziegler SF. Cutting edge: Inhibition of NF-kappaB-mediated TSLP expression by retinoid X receptor. J Immunol (2008) 181:5189–93. doi: 10.4049/jimmunol.181.8.5189
118. Zhong J, Sharma J, Raju R, Palapetta SM, Prasad TS, Huang TC, et al. TSLP signaling pathway map: a platform for analysis of TSLP-mediated signaling. Database (Oxford) (2014) 2014:bau007. doi: 10.1093/database/bau007
119. Mizutani N, Sae-Wong C, Kangsanant S, Nabe T, Yoshino S. Thymic stromal lymphopoietin-induced interleukin-17A is involved in the development of IgE-mediated atopic dermatitis-like skin lesions in mice. Immunology (2015) 146:568–81. doi: 10.1111/imm.12528
120. Rochman Y, Kashyap M, Robinson GW, Sakamoto K, Gomez-Rodriguez J, Wagner KU, et al. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc Natl Acad Sci U.S.A. (2010) 107:19455–60.
121. Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med (2005) 202:1213–23. doi: 10.1084/jem.20051135
122. Cheng DT, Ma C, Niewoehner J, Dahl M, Tsai A, Zhang J, et al. Thymic stromal lymphopoietin receptor blockade reduces allergic inflammation in a cynomolgus monkey model of asthma. J Allergy Clin Immunol (2013) 132:455–62. doi: 10.1016/j.jaci.2013.05.011
123. Wendell SG, Fan H, Zhang C. G Protein–coupled receptors in asthma therapy: Pharmacology and drug action. Pharmacol Rev (2020) 72:1–49. doi: 10.1124/pr.118.016899
124. Al-Hajjaj MS. Bronchial asthma in developing countries: a major social and economic burden. Ann Thorac Med (2008) 3:39–40. doi: 10.4103/1817-1737.39633
125. McCracken JL, Tripple JW, Calhoun WJ. Biologic therapy in the management of asthma. Curr Opin Allergy Clin Immunol (2016) 16:375–82. doi: 10.1097/ACI.0000000000000284
126. Quirt J, Hildebrand KJ, Mazza J, Noya F, Kim H. Asthma. Allergy Asthma Clin Immunol (2018) 14:50–0. doi: 10.1186/s13223-018-0279-0
127. Marlin SD, Rothlein R. Intercellular adhesion molecules: ICAM-1, ICAM-2 and ICAM-3. In: Delves PJ, editor. Encyclopedia of immunology (Second edition). Oxford: Elsevier (1998). p. 1409–12.
128. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus c binding and replication. Proc Natl Acad Sci U.S.A. (2015) 112:5485–90.
129. Watson A, Madsen J, Clark HW, SP-A and SP-D. Dual functioning immune molecules with antiviral and immunomodulatory properties. Front Immunol (2020) 11:622598. doi: 10.3389/fimmu.2020.622598
130. Vrijens P, Noppen S, Boogaerts T, Vanstreels E, Ronca R, Chiodelli P, et al. Influenza virus entry via the GM3 ganglioside-mediated platelet-derived growth factor receptor β signalling pathway. J Gen Virol (2019) 100:583–601. doi: 10.1099/jgv.0.001235
131. Fujioka Y, Nishide S, Ose T, Suzuki T, Kato I, Fukuhara H, et al. A sialylated voltage-dependent Ca(2+) channel binds hemagglutinin and mediates influenza a virus entry into mammalian cells. Cell Host Microbe (2018) 23:809–818.e5. doi: 10.1016/j.chom.2018.04.015
132. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441:101–5. doi: 10.1038/nature04734
133. Matusiak M, Schürch CM. Expression of SARS-CoV-2 entry receptors in the respiratory tract of healthy individuals, smokers and asthmatics. Respir Res (2020) 21:252. doi: 10.1186/s12931-020-01521-x
134. Kunz AN, Ottolini M. The role of adenovirus in respiratory tract infections. Curr Infect Dis Rep (2010) 12:81–7. doi: 10.1007/s11908-010-0084-5
135. Zhang Y, Bergelson JM. Adenovirus receptors. J Virol (2005) 79:12125–31. doi: 10.1128/JVI.79.19.12125-12131.2005
136. Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, et al. Epstein-Barr Virus uses HLA class II as a cofactor for infection of b lymphocytes. J Virol (1997) 71:4657–62. doi: 10.1128/jvi.71.6.4657-4662.1997
137. Ogembo JG, Kannan L, Ghiran I, Nicholson-Weller A, Finberg RW, Tsokos GC, et al. Human complement receptor type 1/CD35 is an Epstein-Barr virus receptor. Cell Rep (2013) 3:371–85. doi: 10.1016/j.celrep.2013.01.023
138. Tanner J, Whang Y, Sample J, Sears A, Kieff E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J Virol (1988) 62:4452–64. doi: 10.1128/jvi.62.12.4452-4464.1988
139. Simoons-Smit AM, Kraan EM, Beishuizen A, Strack van Schijndel RJ, Vandenbroucke-Grauls CM. Herpes simplex virus type 1 and respiratory disease in critically-ill patients: Real pathogen or innocent bystander? Clin Microbiol Infect (2006) 12:1050–9. doi: 10.1111/j.1469-0691.2006.01475.x
140. Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, et al. Herpes simplex virus glycoprotein d can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol (1998) 72:7064–74. doi: 10.1128/JVI.72.9.7064-7074.1998
141. Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, et al. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology (1998) 246:179–89. doi: 10.1006/viro.1998.9218
142. Fieber C, Kovarik P. Responses of innate immune cells to group a streptococcus. Front Cell Infect Microbiol (2014) 4:140. doi: 10.3389/fcimb.2014.00140
143. LaRock DL, Russell R, Johnson AF, Wilde S, LaRock CN. Group a streptococcus infection of the nasopharynx requires proinflammatory signaling through the interleukin-1 receptor. Infect Immun (2020) 88(10):e00356–20. doi: 10.1128/IAI.00356-20
144. Ott L, Möller J, Burkovski A. Interactions between the re-emerging pathogen corynebacterium diphtheriae and host cells. Int J Mol Sci (2022) 23(6):3298. doi: 10.3390/ijms23063298
145. King P. Haemophilus influenzae and the lung (Haemophilus and the lung). Clin Transl Med (2012) 1:10. doi: 10.1186/2001-1326-1-10
146. Clementi CF, Murphy TF. Non-typeable haemophilus influenzae invasion and persistence in the human respiratory tract. Front Cell Infect Microbiol (2011) 1:1. doi: 10.3389/fcimb.2011.00001
147. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med (2008) 358:716–27. doi: 10.1056/NEJMra074111
148. Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J Biol Chem (2007) 282:28700–8. doi: 10.1074/jbc.M703576200
149. Benedetti F, Curreli S, Zella D. Mycoplasmas-host interaction: Mechanisms of inflammation and association with cellular transformation. Microorganisms (2020) 8(9):1351. doi: 10.3390/microorganisms8091351
150. Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of candida albicans by the innate immune system. Nat Rev Microbiol (2008) 6:67–78. doi: 10.1038/nrmicro1815
151. Groskreutz DJ, Monick MM, Powers LS, Yarovinsky TO, Look DC, Hunninghake GW. Respiratory syncytial virus induces TLR3 protein and protein kinase r, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J Immunol (2006) 176:1733–40. doi: 10.4049/jimmunol.176.3.1733
152. Bose S, Banerjee AK. Role of heparan sulfate in human parainfluenza virus type 3 infection. Virology (2002) 298:73–83. doi: 10.1006/viro.2002.1484
153. Duev-Cohen A, Isaacson B, Berhani O, Charpak-Amikam Y, Friedman N, Drori Y, et al. Altered NKp46 recognition and elimination of influenza b viruses. Viruses (2020) 13(1):34. doi: 10.3390/v13010034
154. Jang AY, Seo HS, Lin S, Chung GH, Kim HW, Lim S, et al. Molecular characterization of pneumococcal surface protein K, a potential pneumococcal vaccine antigen. Virulence (2017) 8:875–90. doi: 10.1080/21505594.2016.1278334
155. de Vries SP, Bootsma HJ, Hays JP, Hermans PW. Molecular aspects of moraxella catarrhalis pathogenesis. Microbiol Mol Biol Rev (2009) 73:389–406. doi: 10.1128/MMBR.00007-09
156. Hassan F, Ren D, Zhang W, Merkel TJ, Gu XX. Moraxella catarrhalis activates murine macrophages through multiple toll like receptors and has reduced clearance in lungs from TLR4 mutant mice. PloS One (2012) 7:e37610. doi: 10.1371/journal.pone.0037610
157. Jacquet A, Haumont M, Chellun D, Massaer M, Tufaro F, Bollen A, et al. The varicella zoster virus glycoprotein b (gB) plays a role in virus binding to cell surface heparan sulfate proteoglycans. Virus Res (1998) 53:197–207. doi: 10.1016/S0168-1702(97)00149-4
158. Lin LT, Richardson CD. The host cell receptors for measles virus and their interaction with the viral hemagglutinin (H) protein. Viruses (2016) 8(9):250. doi: 10.3390/v8090250
159. Parker D, Prince A. Immunopathogenesis of staphylococcus aureus pulmonary infection. Semin Immunopathol (2012) 34:281–97. doi: 10.1007/s00281-011-0291-7
160. Bengoechea JA, Sa Pessoa J. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol Rev (2019) 43:123–44. doi: 10.1093/femsre/fuy043
161. Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med (2005) 171:1209–23. doi: 10.1164/rccm.200408-1044SO
162. Coers J, Vance RE, Fontana MF, Dietrich WF. Restriction of legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol (2007) 9:2344–57. doi: 10.1111/j.1462-5822.2007.00963.x
163. Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of mycobacterium tuberculosis. Clin Dev Immunol (2011) 2011):405310.
164. Marrie TJ. Coxiella burnetii pneumonia. Eur Respir J (2003) 21:713–9. doi: 10.1183/09031936.03.00099703
165. van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. Molecular pathogenesis of the obligate intracellular bacterium coxiella burnetii. Nat Rev Microbiol (2013) 11:561–73. doi: 10.1038/nrmicro3049
166. Wissel H, Schulz C, Koehne P, Richter E, Maass M, Rüdiger M. Chlamydophila pneumoniae induces expression of toll-like receptor 4 and release of TNF-alpha and MIP-2 via an NF-kappaB pathway in rat type II pneumocytes. Respir Res (2005) 6:51. doi: 10.1186/1465-9921-6-51
167. Mittal J, Ponce MG, Gendlina I, Nosanchuk JD. Histoplasma capsulatum: Mechanisms for pathogenesis. Curr Top Microbiol Immunol (2019) 422:157–91.
168. Li Z, Lu G, Meng G. Pathogenic fungal infection in the lung. Front Immunol (2019) 10:1524. doi: 10.3389/fimmu.2019.01524
169. Garlanda C, Hirsch E, Bozza S, Salustri A, Acetis M, Nota R, et al. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature (2002) 420:182–6. doi: 10.1038/nature01195
170. Steele C, Marrero L, Swain S, Harmsen AG, Zheng M, Brown GD, et al. Alveolar macrophage-mediated killing of pneumocystis carinii f. sp. muris involves molecular recognition by the dectin-1 beta-glucan receptor. J Exp Med (2003) 198:1677–88. doi: 10.1084/jem.20030932
171. Rubinstein E, Carbon C, Rangaraj M, Ignacio Santos J, Thys JP, Veyssier P. Lower respiratory tract infections: etiology, current treatment, and experience with fluoroquinolones. Clin Microbiol Infect (1998) 4 Suppl 2:S42–s50. doi: 10.1111/j.1469-0691.1998.tb00693.x
173. Wenzel RP, Fowler AA 3rd. Clinical practice. acute bronchitis. N Engl J Med (2006) 355:2125–30. doi: 10.1056/NEJMcp061493
174. Dasaraju PV, Liu C. Infections of the respiratory system. In: Baron S, editor. Medical microbiology. Galveston (TX: University of Texas Medical Branch at Galveston (1996).
175. Brunton S, Carmichael BP, Colgan R, Feeney AS, Fendrick AM, Quintiliani R, et al. Acute exacerbation of chronic bronchitis: a primary care consensus guideline. Am J Manag Care (2004) 10:689–96.
176. Wright P, Cutts FT. Generic protocol to examine the incidence of lower respiratory infection due to respiratory syncytial virus in children less than five years of age. (Geneva, Switzerland:WHO) (2000).
177. Saxena S, Khan N, Dehinwal R, Kumar A, Sehgal D. Conserved surface accessible nucleoside ABC transporter component SP0845 is essential for pneumococcal virulence and confers protection in vivo. PloS One (2015) 10:e0118154. doi: 10.1371/journal.pone.0118154
179. van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet (2009) 374:1543–56. doi: 10.1016/S0140-6736(09)61114-4
180. Light RB. Pulmonary pathophysiology of pneumococcal pneumonia. Semin Respir Infect (1999) 14:218–26.
181. Clifford V, Tebruegge M, Vandeleur M, Curtis N. Question 3: can pneumonia caused by penicillin-resistant streptococcus pneumoniae be treated with penicillin? Arch Dis Child (2010) 95:73–7.
182. Bradley JS, Byington CL, Shah SS, Alverson B, Carter ER, Harrison C, et al. Executive summary: the management of community-acquired pneumonia in infants and children older than 3 months of age: clinical practice guidelines by the pediatric infectious diseases society and the infectious diseases society of America. Clin Infect Dis (2011) 53:617–30. doi: 10.1093/cid/cir625
183. Kim GL, Seon SH, Rhee DK. Pneumonia and streptococcus pneumoniae vaccine. Arch Pharm Res (2017) 40:885–93. doi: 10.1007/s12272-017-0933-y
184. Wu Y, Hu Q, Dehinwal R, Rakov AV, Grams N, Clemens EC, et al. The not so good, the bad and the ugly: Differential bacterial adhesion and invasion mediated by salmonella PagN allelic variants. Microorganisms (2020) 8(4):489. doi: 10.3390/microorganisms8040489
185. Lamers MM, Haagmans BL. SARS-CoV-2 pathogenesis. Nat Rev Microbiol (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
186. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol (2020) 94(7). doi: 10.1128/JVI.00127-20
187. Reuter M, Rigó M, Formazin M, Liebers F, Latza U, Castell S, et al. Authors' response: Occupation and SARS-CoV-2 infection risk among workers during the first pandemic wave in Germany: potential for bias. Scand J Work Environ Health (2022) 48:588–90. doi: 10.5271/sjweh.4061
188. Pandey P, Agarwal S, Rajkumar. Lung pathology in COVID-19: A systematic review. Int J Appl Basic Med Res (2020) 10:226–33. doi: 10.4103/ijabmr.IJABMR_381_20
189. Tregoning JS, Flight KE, Higham SL, Wang Z, Pierce BF. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat Rev Immunol (2021) 21:626–36. doi: 10.1038/s41577-021-00592-1
190. Hess DR. Aerosol delivery devices in the treatment of asthma. Respir Care (2008) 53:699–723. discussion 723-5.
191. Dolovich MB, Ahrens RC, Hess DR, Anderson P, Dhand R, Rau JL, et al. Device selection and outcomes of aerosol therapy: Evidence-based guidelines: American college of chest Physicians/American college of asthma, allergy, and immunology. Chest (2005) 127:335–71. doi: 10.1378/chest.127.1.335
192. Kim IK, Saville AL, Sikes KL, Corcoran TE. Heliox-driven albuterol nebulization for asthma exacerbations: an overview. Respir Care (2006) 51:613–8.
193. Berlinski A, Willis JR. Albuterol delivery by 4 different nebulizers placed in 4 different positions in a pediatric ventilator in vitro model. Respir Care (2013) 58:1124–33. doi: 10.4187/respcare.02074
194. Laube BL, Janssens HM, de Jongh FH, Devadason SG, Dhand R, Diot P, et al. What the pulmonary specialist should know about the new inhalation therapies. Eur Respir J (2011) 37:1308–31. doi: 10.1183/09031936.00166410
195. Olsson B. BE, Bongstrom L, Edsba¨cker S, Eirefelt S, Ekelund K, Gustavsson L, et al. Pulmonary drug metabolism, clearance, and absorption. New York: Springer (2011).
196. Labiris NR, Dolovich MB. Pulmonary drug delivery. part I: physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol (2003) 56:588–99. doi: 10.1046/j.1365-2125.2003.01892.x
197. Stein SW, Thiel CG. The history of therapeutic aerosols: A chronological review. J Aerosol Med Pulm Drug Delivery (2017) 30:20–41. doi: 10.1089/jamp.2016.1297
198. Hou S. WJ, Li X, Shu H. Practical, regulatory and clinical considerations for development of inhalation drug products. Asian J Pharm Sci (2015) 10:490–500. doi: 10.1016/j.ajps.2015.08.008
199. Ferguson GT, Hickey AJ, Dwivedi S. Co-Suspension delivery technology in pressurized metered-dose inhalers for multi-drug dosing in the treatment of respiratory diseases. Respir Med (2018) 134:16–23. doi: 10.1016/j.rmed.2017.09.012
200. Martin AR, Finlay WH. Nebulizers for drug delivery to the lungs. Expert Opin Drug Delivery (2015) 12:889–900. doi: 10.1517/17425247.2015.995087
201. Lass JS, Sant A, Knoch M. New advances in aerosolised drug delivery: vibrating membrane nebuliser technology. Expert Opin Drug Delivery (2006) 3:693–702. doi: 10.1517/17425247.3.5.693
203. Hill NS, Preston IR, Roberts KE. Inhaled therapies for pulmonary hypertension. Respir Care (2015) 60:794–802. discussion 802-5. doi: 10.4187/respcare.03927
204. Nikander K, Turpeinen M, Wollmer P. The conventional ultrasonic nebulizer proved inefficient in nebulizing a suspension. J Aerosol Med (1999) 12:47–53. doi: 10.1089/jam.1999.12.47
205. Dalby RN, Eicher J, Zierenberg B. Development of respimat(®) soft mist™ inhaler and its clinical utility in respiratory disorders. Med Devices (Auckl) (2011) 4:145–55.
206. Pitcairn G, Reader S, Pavia D, Newman S. Deposition of corticosteroid aerosol in the human lung by respimat soft mist inhaler compared to deposition by metered dose inhaler or by turbuhaler dry powder inhaler. J Aerosol Med (2005) 18:264–72. doi: 10.1089/jam.2005.18.264
207. Blau H, Mussaffi H, Mei Zahav M, Prais D, Livne M, Czitron BM, et al. Microbial contamination of nebulizers in the home treatment of cystic fibrosis. Child Care Health Dev (2007) 33:491–5. doi: 10.1111/j.1365-2214.2006.00669.x
208. Vandevanter DR, Geller DE. Tobramycin administered by the TOBI(®) podhaler(®) for persons with cystic fibrosis: a review. Med Devices (Auckl) (2011) 4:179–88. doi: 10.2147/MDER.S16360
210. Mahler DA. Peak inspiratory flow rate as a criterion for dry powder inhaler use in chronic obstructive pulmonary disease. Ann Am Thorac Soc (2017) 14:1103–7. doi: 10.1513/AnnalsATS.201702-156PS
211. Ghosh S, Ohar JA, Drummond MB. Peak inspiratory flow rate in chronic obstructive pulmonary disease: Implications for dry powder inhalers. J Aerosol Med Pulm Drug Delivery (2017) 30:381–7. doi: 10.1089/jamp.2017.1416
212. Tiddens HA, Geller DE, Challoner P, Speirs RJ, Kesser KC, Overbeek SE, et al. Effect of dry powder inhaler resistance on the inspiratory flow rates and volumes of cystic fibrosis patients of six years and older. J Aerosol Med (2006) 19:456–65. doi: 10.1089/jam.2006.19.456
213. Timsina M.P. MGP, Marriott C, Ganderton D, Yianneskis M. Drug delivery to the respiratory tract using dry powder inhalers. Int J Pharm (1994) 101:1–13. doi: 10.1016/0378-5173(94)90070-1
Keywords: lung, immunity, inflammation, invasion, intervention
Citation: Gopallawa I, Dehinwal R, Bhatia V, Gujar V and Chirmule N (2023) A four-part guide to lung immunology: Invasion, inflammation, immunity, and intervention. Front. Immunol. 14:1119564. doi: 10.3389/fimmu.2023.1119564
Received: 09 December 2022; Accepted: 09 March 2023;
Published: 31 March 2023.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Charles Pilette, Université Catholique de Louvain, BelgiumCopyright © 2023 Gopallawa, Dehinwal, Bhatia, Gujar and Chirmule. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Narendra Chirmule, Y2hpcm11bGVAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.