 Minhan Yi
Minhan Yi Jiaxin Li
Jiaxin Li Shijie Jian2
Shijie Jian2 Yuan Zhang
Yuan Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 28 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1119315
This article is part of the Research TopicBeyond the boundaries of inflammation in Atypical ParkinsonismView all 4 articles
Background: The dysfunction of immune system and inflammation contribute to the Parkinson’s disease (PD) pathogenesis. Cytokines, oxidative stress, neurotoxin and metabolism associated enzymes participate in neuroinflammation in PD and the genes involved in them have been reported to be associated with the risk of PD. In our study, we performed a quantitative and causal analysis of the relationship between inflammatory genes and PD risk.
Methods: Standard process was performed for quantitative analysis. Allele model (AM) was used as primary outcome analysis and dominant model (DM) and recessive model (RM) were applied to do the secondary analysis. Then, for those genes significantly associated with the risk of PD, we used the published GWAS summary statistics for Mendelian Randomization (MR) to test the causal analysis between them.
Results: We included 36 variants in 18 genes for final pooled analysis. As a result, IL-6 rs1800795, TNF-α rs1799964, PON1 rs854560, CYP2D6 rs3892097, HLA-DRB rs660895, BST1 rs11931532, CCDC62 rs12817488 polymorphisms were associated with the risk of PD statistically with the ORs ranged from 0.66 to 3.19 while variants in IL-1α, IL-1β, IL-10, MnSOD, NFE2L2, CYP2E1, NOS1, NAT2, ABCB1, HFE and MTHFR were not related to the risk of PD. Besides, we observed that increasing ADP-ribosyl cyclase (coded by BST1) had causal effect on higher PD risk (OR[95%CI] =1.16[1.10-1.22]) while PON1(coded by PON1) shown probably protective effect on PD risk (OR[95%CI] =0.81[0.66-0.99]).
Conclusion: Several polymorphisms from inflammatory genes of IL-6, TNF-α, PON1, CYP2D6, HLA-DRB, BST1, CCDC62 were statistically associated with the susceptibility of PD, and with evidence of causal relationships for ADP-ribosyl cyclase and PON1 on PD risk, which may help understand the mechanisms and pathways underlying PD pathogenesis.
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases, the main risk factors for which are genetic background, environmental variables, aging, and their interactions (1). Its typical pathological changes include the formation of α-synuclein (α-syn) positive inclusion bodies in neurons and axons (Lewy bodies and Lewy neurites) and the loss of dopaminergic neurons (1). Resting tremor, stiffness, bradykinesia, and other clinical symptoms of PD are brought on by the increasing weakening of dopaminergic neurons in the substantia nigra (2). Currently, a great amount of clinical and genetic evidences has revealed that inflammation and immune system malfunction are related to the development of PD (3, 4).
According to some theories, both central and peripheral inflammation begin to manifest in the prodromal stage of PD and remain as the condition worsens (4). The origin of inflammation arises from the central nervous system (CNS), where resting microglia are activated by α-syn, triggering an inflammatory cascade response that leads to the death of dopaminergic neurons (3, 5). Particularly, the activated microglia can release pro-inflammatory cytokines such as interleukins (ILs) and tumor necrosis factor-α (TNF-α), which eventually produce damage to dopaminergic neurons (6, 7). To make matters worse, immune cells from the peripheral circulation infiltrate the brain parenchyma through the compromised blood-brain barrier (BBB) and trigger immune responses via several pathways (8–10). Meanwhile, higher levels of inflammatory factors released by immune cells, such as IL-6, IL-1β, and TNF-α are also found in peripheral blood of PD patients, indicating the occurrence of peripheral inflammation (11, 12). However, it is important to note that the activation of peripheral inflammatory is nonspecific and can be evaluated using some generalized markers like neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) (13). The discordant central inflammatory response is enhanced concurrently with peripheral immune system activation, which may be a factor exacerbating the neurodegeneration (4).
Besides, autoimmunity and the impairment in resolving inflammation also participate in the PD-related inflammation response and promote the development of PD (10, 14, 15). There are a high number of infiltrating T cells in the ventral midbrain of PD patients, which are autoreactive and can recognize disease-altered self-proteins (e.g., α-syn) as foreign antigens through histocompatibility complex (MHC) molecules and drive helper and cytotoxic T cell responses (10, 15). The alleles and haplotypes of MHC class II genes, like HLA-DRB, has been extensively studied in its association with the risk of PD (8, 16, 17). Physiologically, a carefully regulated immune network is involved in mitigating the progression of inflammation to reduce the tissue damage it causes (14, 18). The balance between effector T cells and regulatory T cells in circulation and some specialized pro-resolving lipid mediators in CNS contribute to the resolution of neuroinflammation and the maintenance of immune homeostasis (18, 19). Accelerating the resolution of early neuroinflammation induced by α-syn could prevent the damage of dopaminergic neurons and the onset of PD (20).
Furthermore, due to mitochondrial dysfunction, inflammation can also be triggered by oxidative stress, which is a significant factor in neurodegeneration (21). Oxidative stress and inflammation interact to produce excitotoxicity, neuronal degeneration, and axonal damage, all of which are eventually significant contributors to the development of PD (22). Studies have shown that Nrf2, nitric oxide synthase (NOS), manganese superoxide dismutase (MnSOD), cytochrome P450s (CYPs), hemochromatosis (HFE) and methylenetetrahydrofolate reductase (MTHFR) participate in the development and progress of PD through pathways related to the oxidative stress, including mitochondrial dysfunction, DNA damage, nerve cell apoptosis, and neuroinflammation (23–27).
Overall, the innate and adaptive immune systems play critical roles in the neuroinflammatory process in PD, including oxidative stress, activation and infiltration of immune cells, and the production of inflammatory mediators (3, 21, 28). Single nucleotide polymorphisms (SNPs) of immunological and inflammatory genes can influence the risk of PD by influencing the immune system and inflammatory response since PD is directly tied to genetics. Genetic factors in PD converge on immune function and inflammation through the activation of immune cells and the release of inflammatory mediators (29). Changes in the inflammatory genes may make a person more vulnerable to the formation of oxidative stress and the activation of the neuroimmune system, both of which can result in the death of dopaminergic neurons (30, 31). Researches on inflammatory polymorphic locus identified by genome-wide association studies (GWAS) study have also exemplified the significance of neuroinflammation in the pathogenesis of PD (32, 33).
Although previous studies have shown a close relationship between inflammation and PD, few studies have investigated the causality between them. Mendelian Randomization (MR) is a reliable genetic epidemiology method, which uses genetic variants as instrumental variable (IV) to assure whether causality exists between exposure and outcome, maybe a powerful tool to explore the causality between inflammation and PD (34). Bottigliengo D et al. investigated the causal role of inflammation on PD by conducting MR analysis (35). They included C-reactive protein (CRP), IL-6, IL-1 receptor antagonist and TNF-α in a two-sample MR analysis and suggested the pro-inflammatory activity of IL-6 could be a determinant of prodromal PD. Nevertheless, other than this study, no other articles have been reported on the causal relationship between inflammation and PD.
Therefore, to reach a comprehensive and updated conclusion, we performed a quantitative and causal analysis to explore the role of inflammatory genes in PD risk in order to bring new understanding of the mechanisms and pathways underlying PD pathogenesis and may provide the theoretical basis for finding the potential biomarkers and implementing anti-inflammatory and immunological treatment in PD. In addition to the genes included in the existing studies, we collected the original researches related to inflammation-related genes and PD as much as possible. Based on the function of genes, we divided them into five groups: genes of cytokines, genes involved in the oxidative stress, genes of neurotoxin-associated enzymes, genes of metabolism-associated enzymes and inflammatory polymorphic locus identified by GWAS study (Figure 1). Our study would be an important supplement on the topic of PD’s genetic susceptibility and also provide idea related to its mechanism and treatment.
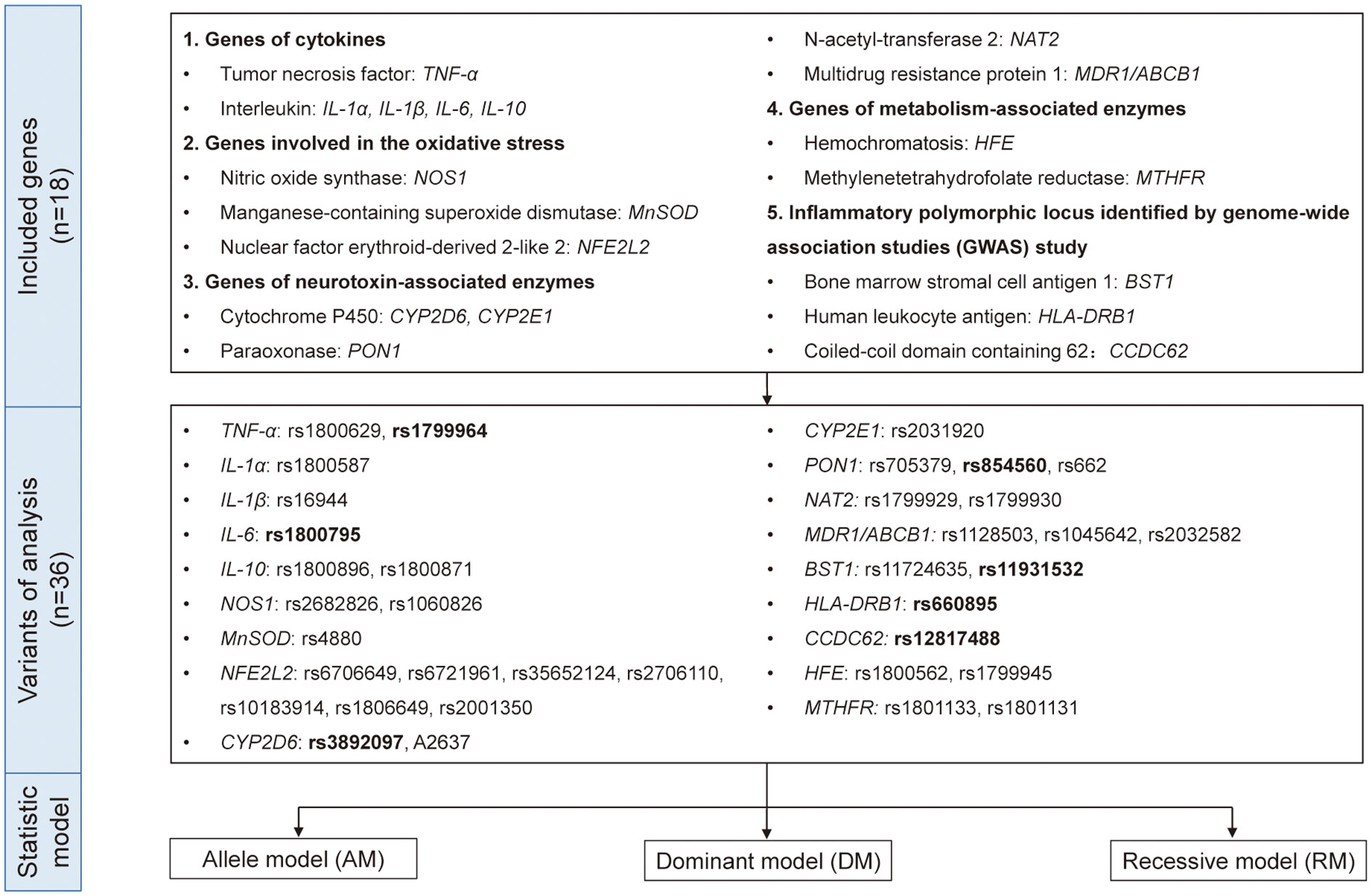
Figure 1 The genes, variants, and data analysis models included in the quantitative analysis. Eighteen genes with 36 variants from five different functional types were included in this study. We used allele model (AM), dominant model (DM), and recessive model (RM) for quantitative analysis and the variants in bold indicated p<0.05 in either model.
Researchers independently retrieved and screened literature, and the inconsistent views were discussed with the third party. Key words were “Parkinson’s disease”, “Parkinso*”, “variants”, “genetic”, “specific genes” (TNF-α, IL-6, IL-1α, IL-1β, IL-10, NOS1, MnSOD, NFE2L2, CYP2D6, PON1, CYP2E1, NAT2, ABCB1, BST1, HLA-DRB, CCDC62, HFE, MTHFR involved in five different inflammation-related group (genes of cytokines, genes involved in the oxidative stress, genes of neurotoxin-associated enzymes, genes of metabolism-associated enzymes and inflammatory polymorphic locus identified by GWAS study) in PD). The detailed searching strategy was listed in Supplementary Table 1.
Inclusion criteria using PICOS (participants, interventions, comparators, outcomes, and studies approach) were applied to screen articles:
Participants: the PD diagnosis from each researched cohort was according to widely accepted criteria (36, 37).
Interventions: genetic sequencing of variants in inflammation-related genes of interest were performed by PCR-based methods or other accepted methods;
Controls: controls were neither having PD nor other neurological diseases.
Outcomes: available data to calculate the number of carriers and non-carriers of the gene variants.
Studies approach: original studies provided sufficient data to do pooled analysis.
Exclusion criteria including: 1) neurological diseases not PD or without control groups; 2) not original studies including editorial, review, systematic review etc.; 3) functional studies using animal or cell models; 4) studies not having sufficient data to calculate odd ratio (OR) and 95% confidence interval (CI) in all models.
Then, authors independently extracted the detailed information from the included studies. The data extraction table were as follows: first author, publication year, ethnics, number of allele carriers in cases or controls, number of cases, number of controls, number of genotype carriers in cases, number of genotypes in controls. The Newcastle-Ottawa Scale (NOS) scores were used to evaluate the quality of the included articles. If there was any disagreement on data extraction, a third researcher was asked to make a decision.
Revman 5.3 software was used to calculate pooled OR and 95%CI. Three models were applied to do the association analyses: allele model (AM, indicated “a” distribution between case group and control group), dominant model (DM, indicated “aa + Aa” distribution between case group and control group), and recessive model (RM, indicated “aa” distribution between case group and control group). “A” represented wild type allele, “a” represented mutated allele. P <0.05 was considered statistically significant.
The I2 and Q test were performed to analyze the heterogeneity. If I2> 50, the random-effect model was used, otherwise if I2 ≤ 50, the fix-effect model was applied. The publication bias was measured by the symmetry of funnel plot. If the plot was in a symmetrical shape, no publication bias was shown. Otherwise, publication bias was observed. Sensitivity analysis was performed by sequentially removing one article at a time.
For the genes with statistically significant results in the quantitative analysis, we further explored the causality between proteins they encode and the risk of PD by conducting a two-sample MR analysis. After searching the GWAS data of the included genes, we evaluated the causal associations between corresponding proteins [ADP-ribosyl cyclase (coded by BST1) and PON1 (coded by PON1)] and PD in two directions (Figure 2).
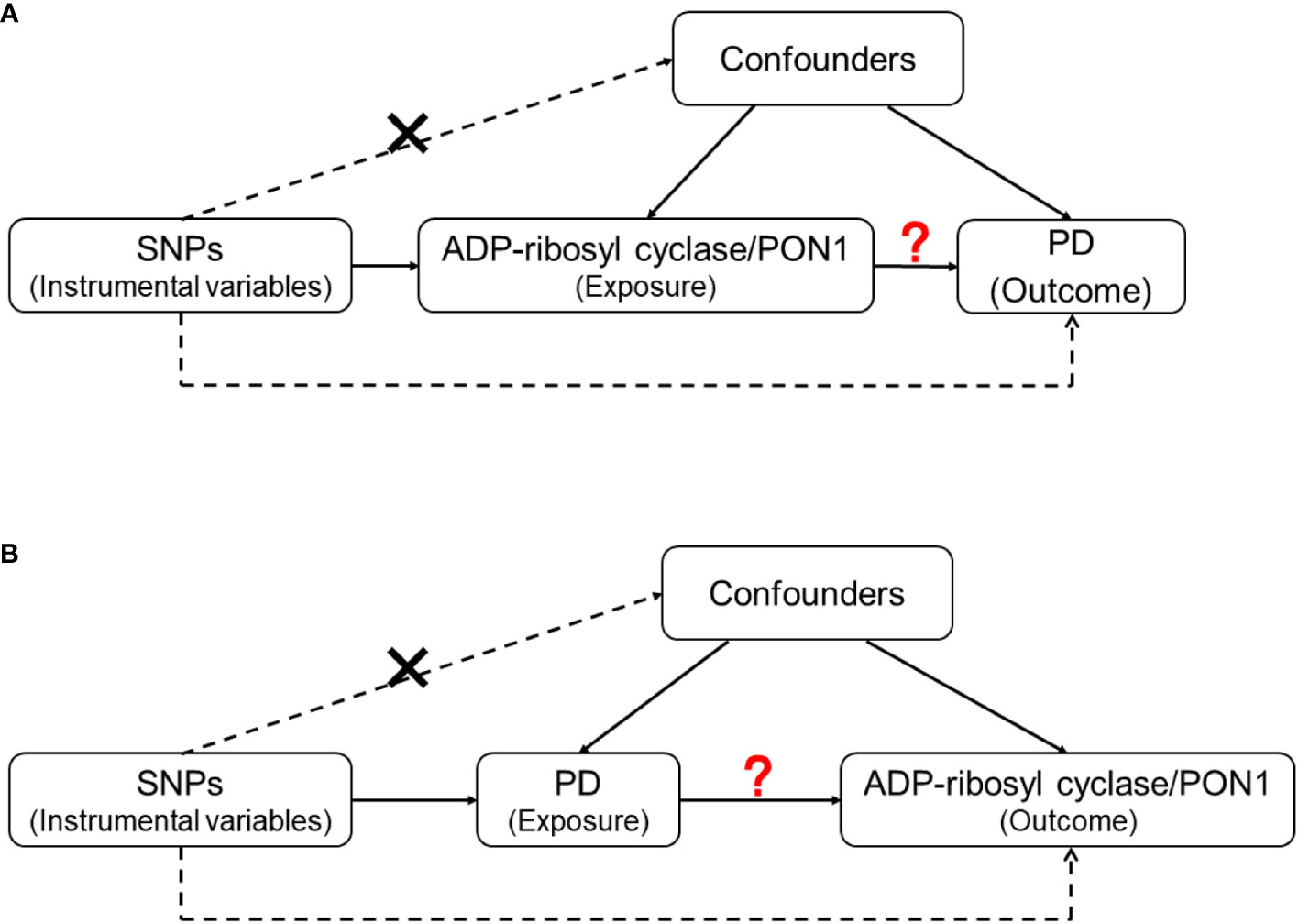
Figure 2 The design of Mendelian Randomization (MR) analysis to assess causality between PD and proteins coded by inflammatory genes. (A) SNPs independently associated with ADP-ribosyl cyclase (coded by BST1) and PON1(coded by PON1) from GWAS summary statistic were used as instrumental variables to explore the causal effect of ADP-ribosyl cyclase and PON1 on PD. (B) SNPs independently associated with PD from GWAS summary statistic were used as instrumental variables to explore the causal effect of PD on ADP-ribosyl cyclase and PON1 respectively. In addition to the association assumption, another two assumptions of MR include: (1) SNPs are not associated with the confounders of exposure and outcome; (2) there is no feasible pathway between the genetic variations and outcome other than through exposure.
We used a published GWAS summary statistics from International Parkinson Disease Genomics Consortium (IPDGC) Study that contained 482,730 individuals with 37,688 PD from Europe (38). ADP-ribosyl cyclase GWAS summary statistics were obtained from a German cohort included 997 European (39). In this study, the plasma ADP-ribosyl cyclase levels of participants were quantified by proteomics measurements using the SOMAscan platform. Besides, we also include the PON1 GWAS summary statistics from the Milieu Intérieur cohort which contained 400 participants from Europe (40). The level of PON1 in plasm were quantified by protein immunoassay. The specific information was summarized in Table 1.
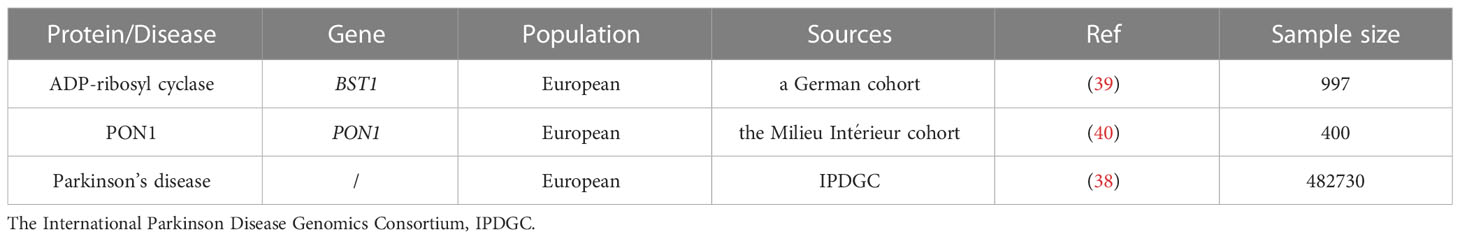
Table 1 Summary of genome-wide association study (GWAS) datasets for MR analysis.
There are three assumptions for instrumental variable (IV) selection in two-sample MR analysis: (1) the selected genetic variants are associated with the exposure; (2) the used IV variants are not associated with the confounders of exposure and outcome; (3) there is no feasible pathway between the genetic variations and outcome other than through exposure (41). In detail, when using ADP-ribosyl cyclase, PON1 and PD as exposure, we selected associated variants with p < 5 × 10−8 (38–40). Then, the independently associated variants were included as IV with the criteria of r2 < 0.1 within distance of 1000kb.
We used the method of inverse-variance weighting (IVW) (42) and Mendelian randomization-pleiotropy residual sum and outlier (MR-PRESSO) (43) as the primary outcomes that assumed that all SNPs are valid instrument variables. In sensitivity analyses, we used MR Egger (44) and Weighted median (45) to correct for any potential violations of the assumptions. These methods are performed as they operate in different ways and rely on different assumptions for valid inferences to assess the reliability of MR analysis. Besides, heterogeneity was analyzed by Cochran’s Q-test of IVW and MR Egger, and pleiotropy was tested by the intercept of MR Egger analysis. When heterogeneity was detected for associated relationships, we used the RadialMR package to remove outliers and applied above analysis again (46).
As can be seen from the flowchart (Figure 1 and Supplementary Figure 1), articles were retrieved for each research gene separately using three databases (PubMed, Embase and Web of Science database). By removing overlapping articles, reading title/abstract and full-text screening. Final original articles were included for pooled analysis by different genes separately. The detailed information of included original articles and genotypes distributions were presented in Table 2 and Supplementary Tables 2, 3. Thirty-six variants in 18 genes associated with inflammatory mechanisms in PD were involved. The results of quantitative analysis were presented in Table 3. The functions of these genes were classified by five groups: genes of cytokines, genes involved in the oxidative stress, genes of neurotoxin-associated enzymes, genes of metabolism-associated enzymes and inflammatory polymorphic locus identified by GWAS study.
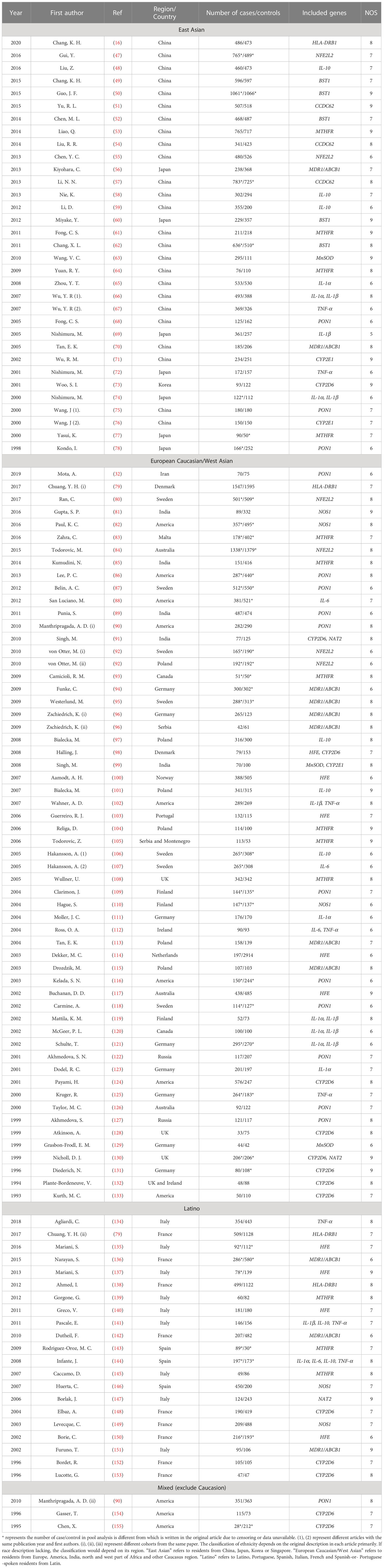
Table 2 The characteristics of all included publications for quantitative analysis.
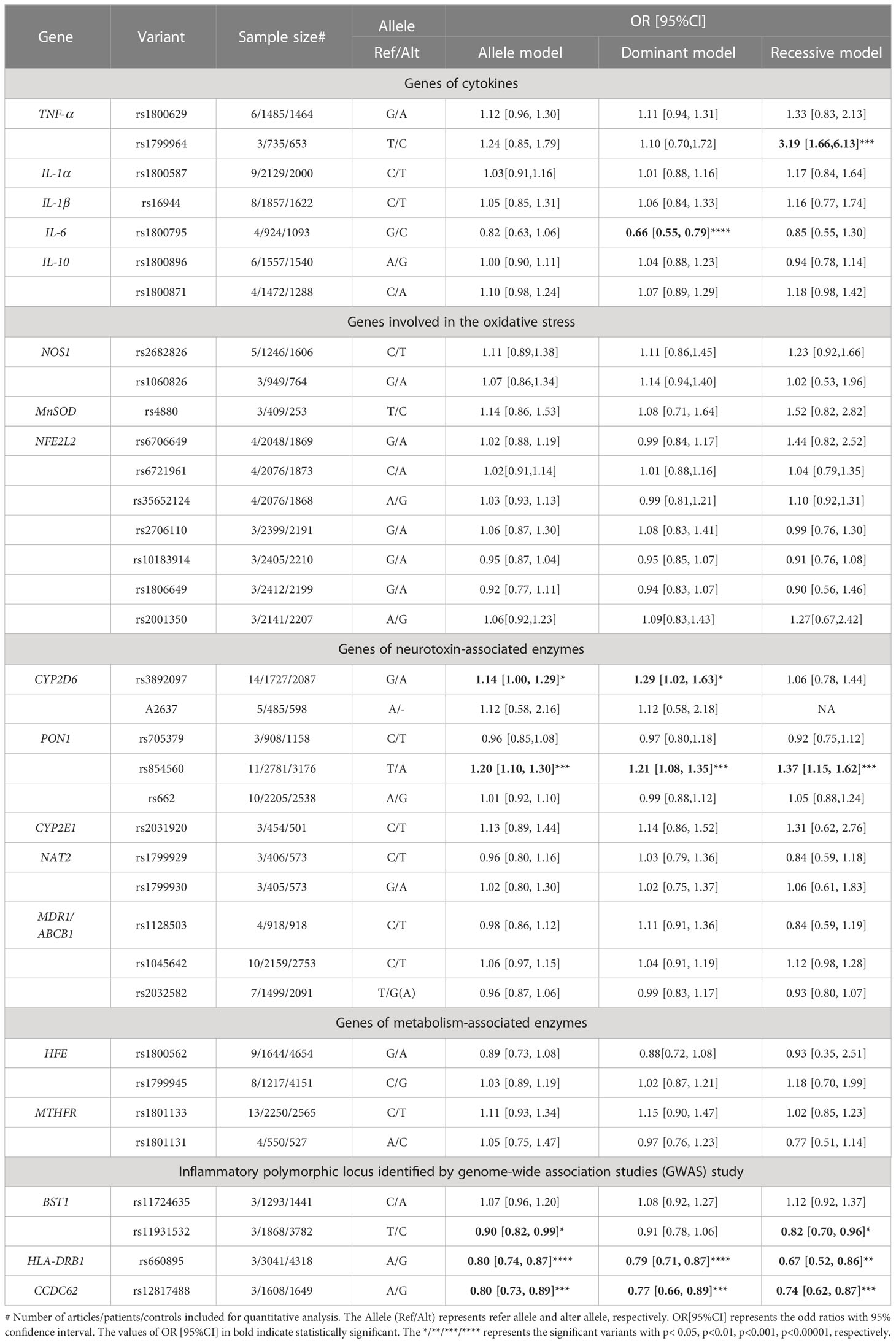
Table 3 The results of quantitative analysis for the association between included variants and the risk of PD in different models.
Seven variants in five genes (TNF-α, IL-6, IL-1α, IL-1β, IL-10) were included in the pooled analysis. In the results of DM and RM models, rs1799964 of TNF-a (RM: OR[95%CI] = 3.19[1.66,6.13], p=0.0005) polymorphism was positively associated with PD risk. In contrary, rs1800795 of IL-6 (DM: OR[95%CI] = 0.66 [0.55, 0.79], p<0.00001) polymorphism was negatively associated with PD risk. About variants in IL-1α (rs1800587), IL-1β(rs16944), IL-10 (rs1800871, rs1800896), all three models (AM, DM, RM) showed these variants were not associated with the risk of PD.
Ten variants in three genes (NOS1, MnSOD, NFE2L2) were included for quantitative analysis. We failed to identify the association between NFE2L2 rs6706649, rs6721961, rs35652124, rs2706110, rs10183914, rs1806649, rs2001350, NOS1 rs2682826, rs1060826, MnSOD rs4880 and PD risk in all three models (AM, DM, RM).
Eleven variants in five genes (CYP2D6, PON1, CYP2E1, NAT2, ABCB1/MDR) were included in the AM for quantitative analysis. CYP2D6 rs3892097 (OR[95%CI] =1.14[1.00-1.29], p=0.04) variant was positively associated with PD risk on 1727 PD cases and 2087 controls. PON1 rs854560 (OR[95%CI] =1.20 [1.10, 1.30], p<0.0001) variant was also positively associated with PD risk in AM on 2781 PD cases and 3176 controls.
In the secondary analysis, by including 2781 PD cases and 3176 controls in DM and RM, PON1 rs854560 variant was positively associated with PD risks (DM: OR[95%CI] =1.21 [1.08-1.35], p=0.0007; RM: OR[95%CI] =1.37[1.15-1.62], p= 0.0003). The significant result was also replicated by DM in CYP2D6 variant rs3892097 (OR[95%CI] =1.29[1.02-1.63], p=0.04). Variant in CYP2E1 (rs2031920) was not associated with PD risk. Variants in NAT2 (rs1799929, rs1799930) or ABCB1 (rs1128503, rs1045642, rs2032582) were not associated with PD risk either.
Four variants in two genes (HFE, MTHFR) were included for quantitative analysis. We failed to identify the association between HFE rs1800562, rs1799945, MTHFR rs1801133, rs1801131 and PD risk in all three models (AM, DM, RM).
We included four variants in three genes (BST1, HLA-DRB, CCDC62) in the pooled analysis. BST1 rs11931532 was negatively related to PD risk in pooled analysis on 1868 PD patients and 3782 controls (AM: OR[95%CI] =0.90[0.82-0.99], p=0.02). In the further analysis, the significant results were also presented in RM about BST1 rs11931532 (OR[95%CI] =0.82[0.70-0.96], p=0.01). By including 3041 PD cases and 4318 controls, we found that HLA-DRB rs660895 was associated with PD risk negatively in all models (AM: OR[95%CI] = 0.80 [0.74, 0.87], p <0.00001; DM: OR[95%CI]= 0.79 [0.71, 0.87], p<0.00001; RM: OR[95%CI]=0.67[0.52-0.86], p=0.002). CCDC62 rs12817488 was also associated with decreased PD risk in quantitative analysis on 1608 PD cases and 1649 controls in all models (AM: OR[95%CI] = 0.80 [0.73, 0.89], p <0.0001; DM : OR[95%CI]=0.77[0.66-0.89], p=0.0005; RM OR[95%CI]=0.74[0.62-0.87], p=0.0003).
Funnel plots of almost all quantitative analysis were symmetric, indicating that there was no publication bias (Supplementary Figures 2, 3). We conducted the sensitivity analysis by comparing the changes in pooled p value, OR and 95%CI after deleting each article at a time in turn (Supplementary Table 4). After removing Ross, O. A. et al. (112) in IL-6 rs1800795, Hague, S. et al. (110) in NOS1 rs1060826, Todorovic, M. et al. (84) in NFE2L2 rs2001350 or Funke, C. et al. (94) in MDR1 rs1128503, the pooled p value or OR changed significantly. This could be due to large or small sample sizes, or differences in genotype distribution on account of ethnicity and region compared with other studies.
When using ADP-ribosyl cyclase (coded by BST1) as exposure and PD as outcome, the primary outcome of IVW model showed that an increased level of ADP-ribosyl cyclase was causally associated with the higher risk of PD (OR[95%CI] = 1.08 [1.01, 1.16], p =0.02) (Figure 3A). The result was almost significant in MR-PRESSO model (OR[95%CI] = 1.08 [1.01, 1.16], p =0.07) (Figure 3A). The results of MR Egger and Weighted median were shown in Supplementary Table 5. Since there were high heterogeneity for above analysis (Supplementary Table 6), we identified and removed those outliers of SNPs. We confirmed that there was no obvious heterogeneity in all models (Supplementary Table 6). And the causality become stronger in both IVW (OR[95%CI] = 1.16 [1.10, 1.22], p =1.64×10-7] and MR-PRESSO (OR[95%CI] = 1.16 [1.14, 1.17], p =1.73×10-3] models (Figure 3A).
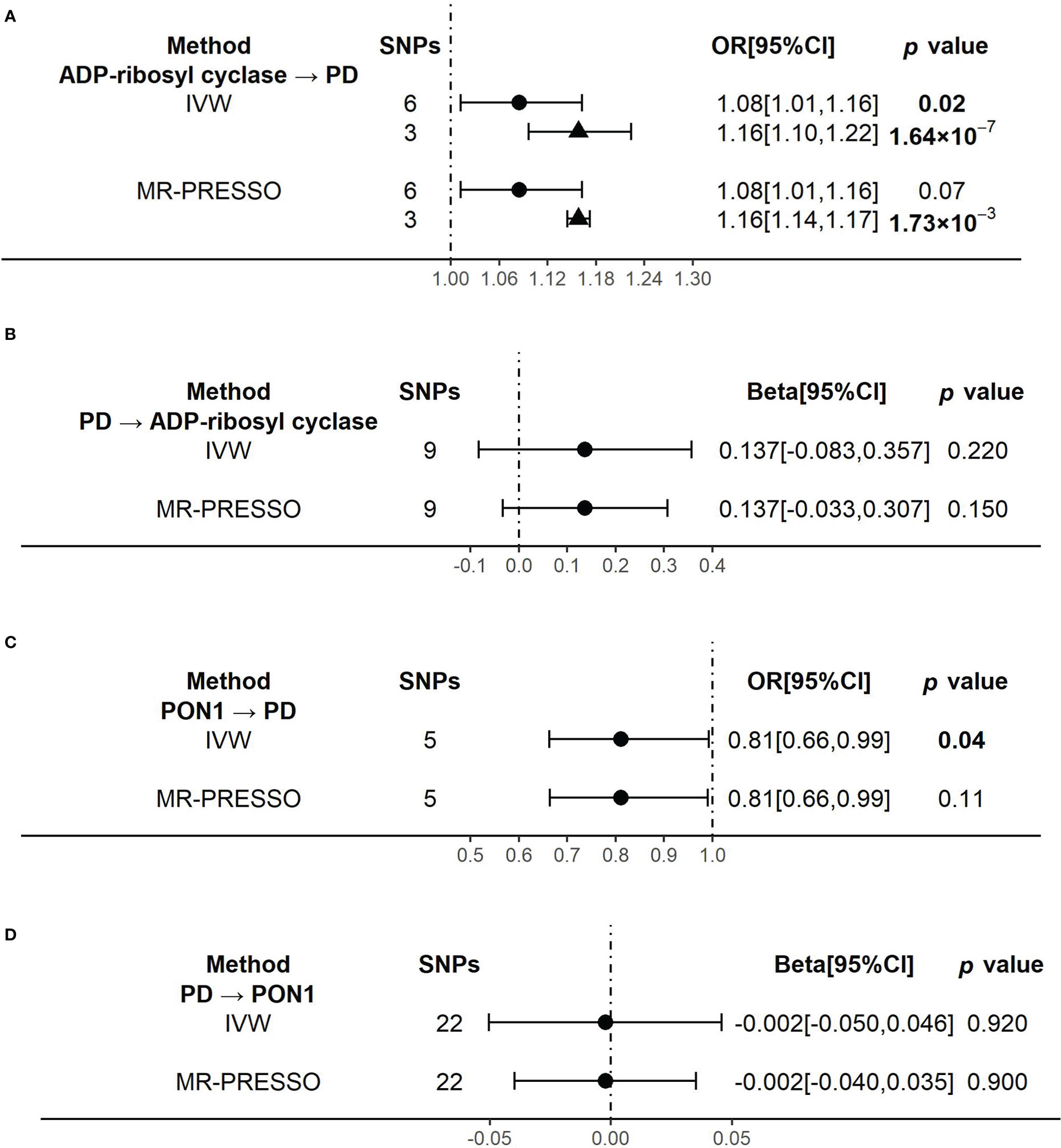
Figure 3 The results of the causal effects between ADP-ribosyl cyclase, PON1 and Parkinson’s disease by Mendelian Randomization analysis (A) The causal effect of ADP-ribosyl cyclase on PD. (B) The causal effect of PD on ADP-ribosyl cyclase. (C) The causal effect of PON1 on PD. (D) The causal effect of PD on PON1.. 95% confidence interval, 95% CI; inverse-variance weighted, IVW; Mendelian randomization-pleiotropy residual sum and outlier, MR-PRESSO; Parkinson’s disease, PD. Circulars illustrate the results from primary instrumental variables (IVs) and triangles mean the results from outliers removed IVs.
Reversely, when using PD as exposure and ADP-ribosyl cyclase as outcome, the results of all analysis models showed no significant association between them (Figure 3B and Supplementary Table 5). And there was no evidence of heterogeneity in all models (Supplementary Table 6).
When using PON1 (coded by PON1) as exposure and PD as outcome, the primary outcome of IVW model showed that the higher risk of PD was significantly associated with a decreased level of PON1, which indicated PON1 probably had protective effect on PD risk (OR[95%CI] = 0.81 [0.66, 0.99], p =0.04) (Figure 3C). But the causality was weaker in MR-PRESSO model (OR[95%CI] = 0.81 [0.66, 0.99], p =0.11) (Figure 3C). There was no evidence of heterogeneity in the IVW and MR-PRESSO analysis (Supplementary Table 6). The results of MR Egger and Weighted median were shown in Supplementary Table 5.
When we did the reverse analysis, we did not find any significant causality in all the models (Figure 3D and Supplementary Table 5). None of the models showed evidence of heterogeneity (Supplementary Table 6).
The immune system’s dysregulation and inflammatory reactions are now more clearly linked to PD (30). However, due to their reliance on a small number of genes, restricted areas, or territories, and potential for analytical bias, the existing association studies are not sufficiently thorough. After a combination of quantitative analysis and two-sample MR analysis, we found that TNF-α rs1799964, PON1 rs854560 and CYP2D6 rs3892097 were associated with the higher risk of PD while IL-6 rs1800795, HLA-DRB rs660895, BST1 rs11931532 and CCDC62 rs12817488 were related to the lower risk of PD. Besides, we observed that increased plasma level of ADP-ribosyl cyclase (coded by BST1) had causal effect on higher PD risk while PON1(coded by PON1) shown probably protective effect on PD risk. This study may help us to have a deeper understanding of the relationship between the inflammatory variations and PD, and potentially identify biomarkers and create anti-inflammatory and immunological therapy options for PD.
Our study is the most thorough one to date when compared to quantitative analyses on inflammatory genetic variations associated to PD. ZS Ulhaq conducted a meta-analysis in 2020 to clarify the relationship between inflammatory genes and PD. It discovered that while variations in IL-1β, TNF-α were not connected with PD risk, variations in IL-1α, IL-6, IL-8, IL-10, and IL-18 were associated (156). However, we came to the conclusion that TNF-α was linked to an elevated risk of PD from our research. Although these findings were different from what ZS Ulhaq had previously stated, our study included the most recent original publications. Besides, genes of oxidative stress, neurotoxic and metabolism-related enzymes, and inflammatory polymorphism loci discovered by GWAS studies have also been extensively studied in the past and have been shown to impact the risk of PD. These genes were also included in our study. Further, for the genes with statistically significant results in the quantitative analysis, we explore the causality between proteins they encode and the risk of PD by conducting a two-sample MR analysis.
Regarding genes of cytokine specifically, several studies have noted higher levels of TNF-α, ILs, and other pro-inflammatory cytokines are presented in the peripheral blood, cerebrospinal fluid (CSF) of patients with PD and in the striatum of post-mortem brains from patients with PD (157–159).The inflammatory genes encoding these molecules have also been widely studies, though the results might be inconsistent. The gene set-association analysis did not reveal the association between TNF-α, IL-6, IL-8 etc. and PD (144). But Chu et al. considered TNF-α rs1799964, IL-6 rs1800795 and IL-1RA VNTR were shown to be associated with PD risk (160). Our study is a thorough update and addition to the prior studies because it used bigger cohorts. We discovered a favorable correlation between PD and a TNF- α rs1799964. The variation may alter the expression of TNF-α or have an impact on other genes associated with inflammation, contributing to the pathophysiology of PD (161, 162). We also found IL-6 rs1800795 G>C decreased the PD risk, which may lower the level of IL-6 in serum (163). A MR analysis study also demonstrated that the pro-inflammatory activity of the IL-6 could be a determinant of prodromal PD (35).
In addition, PD pathogenesis is highly related to oxidative stress, which can promote the dysfunction of immune system and inflammatory response in turn (23). By altering the detoxification of neurotoxins in PD, metabolizing enzymes such CYPs (CYP2D6, CYP2E1) and paraoxonase (PON1) may impact the likelihood of developing PD. These enzymes’ activity and sensitivity to oxidative damage are strongly related (32, 164). In our quantitative analysis, PON1 rs854560 was positively associated with PD risks in all three models (AM, DM and RM), which was inconsistent with the conclusion of previous meta-analysis conducted by Liu Y. et al. (165). Compared with Liu Y. et al., we included more studies to reach a more reliable conclusion. Besides, we also found CYP2D6 rs3892097 significantly increased the risk of PD, which was consistent with previous meta-analysis conducted by Lu Y. et al. (166). CYP2D6 can catalyze the metabolism of MPTP to toxic 1-methyl-4-phenylpyridinium ion (MPP(+)), which lead to oxidative damage of dopaminergic neurons (167, 168). CYP2D6 rs3892097 may affect the occurrence of PD by changing the metabolic activity of CYP2D6. GWAS has shown that genes including BST1, HLA-DQB1 etc. involved in the “regulation of leucocyte/lymphocyte activity” and “cytokine-mediated signaling” are associated with PD risk (33). HLA-DRA and HLA-DRB alleles encode HLA-DR antigen, acting as regulatory molecule involved in autoimmunity (169). HLA-DRB variants differ in affinity with α-syn antigen epitopes, which influence antigen recognition and subsequent immune response (10). Patients with PD also have a higher expression of MHC class II molecules in peripheral blood mononuclear cells, which is consistent with the inflammatory pattern of PD (170). Therefore, variants in HLA-DRB could alter the risk of PD by regulating the expression of HLA-DRB or its response to α-syn (10, 15). CCDC62/HIP1R loci were identified by the first large-scale meta-analysis of published GWAS in PD (57). These researches are consistent with our research results that the variants in BST1, HLA-DRB, CCDC62 were correlated with PD risk.
Further, we conducted a two-sample MR analysis to investigate the causality between proteins coded by inflammatory genes and the risk of PD and we found increased plasma level of ADP-ribosyl cyclase had causal effect on higher PD risk while PON1 shown probably protective effect on PD risk. Cyclic ADP-ribose (cADPR) is a signal transduction molecule downstream of the dopamine receptors, which is synthesized from β-NAD+ by both cytosolic and membrane-bound forms of ADP-ribosyl cyclase and/or CD38 (171, 172). Higashida, H. et al. indicated that cADPR, as an endogenous inhibitor of mTOR signaling pathway, reduced downstream protein synthesis and thus affected synaptic plasticity of neurons (173). The dysregulation of dopaminergic system is associated with a variety of neurological and psychiatric disorders, including PD (174). Increased ADP-ribosyl cyclase may lead to the disturbance of dopaminergic system by inhibiting mTOR signaling pathway, thus promoting the occurrence of PD. PON1 is an esterase carried by high-density lipoproteins and has antioxidant and anti-inflammatory effects (175). Its detoxification activity is considered to be an important link between environmental exposure to pesticides or pollutants and the risk of neurodegenerative diseases since it is able to hydrolyze active metabolites of organophosphate insecticides (176). The mutation at PON1 rs854560 reduced the scavenging activity of lipoprotein free radicals, which may lead to neuronal damage (176). Thus, a decrease in plasma PON1 level would reduce the capacity of antioxidant and anti-inflammatory and increase the risk of PD. Reversely, our results suggested that the development of PD did not lead to the changes in plasma ADP-ribosyl cyclase and PON1 levels.
Our findings indicated that the onset and progression of PD is closely related to inflammatory response and the disorder of immune system. It would be worth mentioning that PD and atypical parkinsonian syndromes (APS), including multiple system atrophy (MSA) and progressive supranuclear palsy (PSP), have overlapping symptoms that are difficult to make an early diagnose. Nevertheless, PD and APS have different inflammatory patterns, which would be helpful in the differential diagnosis and understanding of the pathogenesis of diseases. Although the occurrence of PSP is also related to the activation of microglia and neuroinflammation, the process may be associated with the accumulation of phosphorylated tau protein, but not α-syn (177). Compared with patients with PD patients, patients with PSP have higher NLR in peripheral blood and significantly increased expression of CRP and microglia-derived cytokines in CSF, including IL-1β, IL-6 and TNF-α (177, 178). MSA is a rapidly progressing neurodegenerative disease characterized by the accumulation of oligodendrocyte inclusions composed of α‐syn (179). Animal model of MSA shows a stronger inflammatory response than PD model (180). Compared with the healthy controls, the PLR in the peripheral blood of MSA patients is significantly increased (13). The NLR and PLR in peripheral blood show no significant difference between patients with MSA and PD (13). However, patients with MSA have higher levels of inflammatory markers in the CSF than patients with PD, including CRP, serum amyloid A, IL-1β, IL-6 and TNF-α, but lower levels of neuroprotective molecules, such as beta nerve growth factor (β-NGF) and Delta and Notch like epidermal growth factor-related receptor (DNER) (178, 181, 182).
Excitingly, the existence of these inflammatory patterns and immune system alterations provides new insights into anti-inflammatory and immunological treatment strategies for neurodegenerative diseases, including PD. Firstly, inhibiting or activating the function of inflammatory genes might delay or halt disease progression during prodrome or prevent disease progression. For example, activation of Nrf2 (the transcription factor of NFE2L2) could alleviate the progression of neurodegenerative diseases by counteracting oxidative stress and inflammation (183). Besides, treatments that have anti-inflammatory factors or enhances anti-inflammatory ability could reduce the occurrence and progression of neurodegenerative diseases. A retrospective cohort study indicated that anti-TNF therapy could effectively reduce the incidence of PD (184).Currently, several clinical trials of anti-inflammatory and immunological therapy for PD are underway, though no effective outcomes are available yet (5). Since the inflammatory cascade has an important impact on the development and progression of neurodegeneration in PD, the initiation of more clinical trials on PD inflammation is rational (5). Furthermore, targeting α-syn with antibodies to slow the transmission and reverse the effects of α-syn pathology is another direction for PD immunotherapy since α-syn plays a key role in the pathogenesis of PD. Monoclonal antibodies against α-syn could inhibit the spread of α-syn, reduce the loss of dopaminergic neurons, and alleviate motor deficits in PD mouse models (185, 186). Clinical trials have shown that PRX002/RG7935, the monoclonal antibody against α-syn, could penetrate BBB and efficiently reduce serum α-syn levels to alleviate the progression of PD (187, 188). So far, the safety and tolerability of the PRX002/RG7935 treatment have been preliminarily verified, and the next phase of clinical trials is needed to explore its effectiveness in the treatment of PD (188).
Nevertheless, it must be admitted that our study has several inescapable limitations. Because we combined all of the reported patients and controls for our quantitative analysis, these cases and controls may not be age or sex matched, which might lead to selection bias. Differences in race might also cause confusion. We were unable to run a subgroup analysis on the variables because of the dearth of data. Furthermore, barely fewer than 5 publications were included in some of our quantitative analysis. To reach a reliable conclusion, further unique investigations are required. Due to the insufficient GWAS data resources, we did not conduct causal analysis for all the proteins encoded by statistically significant genes, only ADP-ribosyl cyclase and PON1 were analyzed.
In conclusion, we included 18 inflammatory genes, including genes encoding cytokines, genes implicated in oxidative stress, genes for neurotoxins and metabolism-related enzymes, and inflammatory polymorphic loci discovered by GWAS analysis that are strongly connected with PD pathogenesis. While variations in IL-1α, IL-1β, IL-10, MnSOD, NFE2L2, CYP2E1, NOS1, NAT2, ABCB1, HFE or MTHFR were not connected to PD risk, we discovered that multiple polymorphisms from IL-6, TNF-α, PON1, CYP2D6, HLA-DRB, BST1, and CCDC62 were statistically correlated with PD risk. Additionally, we indicated the changes in plasm ADP-ribosyl cyclase and PON1 level have causal effects on the risk of PD. Further researches are needed to confirm these findings.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Study design: MY and YZ. Data collection: MY, JL, SJ, BL, LS, ZH, and YZ. Data analysis: MY, JL, SJ, BL, LS, ZH, and YZ. Writing: MY, JL, SJ, BL, ZH, LS, and YZ. Funding: MY and YZ. Administration: MY and YZ. All authors contributed to the article and approved the submitted version.
This research was supported by the National Natural Science Foundation of China (No. 82001357), the Hunan Provincial Natural Science Foundation of China (No. 2021JJ80079), the Degree & Postgraduate Education Reform Project of Central South University (No. 2021YJSKSA10), the Undergraduate Education Reform Project of Central South University (No. 2021CG065, No. 2021CG068) and the Research Project of Laboratory Construction and Management of Central South University (No. 202120).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1119315/full#supplementary-material
1. Pang SY, Ho PW, Liu HF, Leung CT, Li L, Chang EES, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of parkinson's disease. Transl Neurodegener (2019) 8:23. doi: 10.1186/s40035-019-0165-9
2. Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet (2004) 363(9423):1783–93. doi: 10.1016/S0140-6736(04)16305-8
3. Tan EK, Chao YX, West A, Chan LL, Poewe W, Jankovic J. Parkinson Disease and the immune system - associations, mechanisms and therapeutics. Nat Rev Neurol (2020) 16(6):303–18. doi: 10.1038/s41582-020-0344-4
4. Joshi N, Singh S. Updates on immunity and inflammation in Parkinson disease pathology. J Neurosci Res (2018) 96(3):379–90. doi: 10.1002/jnr.24185
5. Gundersen V. Parkinson's disease: Can targeting inflammation be an effective neuroprotective strategy? Front Neurosci (2020) 14:580311. doi: 10.3389/fnins.2020.580311
6. Zhang QS, Heng Y, Yuan YH, Chen NH. Pathological alpha-synuclein exacerbates the progression of parkinson's disease through microglial activation. Toxicol Lett (2017) 265:30–7. doi: 10.1016/j.toxlet.2016.11.002
7. Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D, et al. Type-1 interferons contribute to the neuroinflammatory response and disease progression of the mptp mouse model of parkinson's disease. Glia (2016) 64(9):1590–604. doi: 10.1002/glia.23028
8. Pandi S, Chinniah R, Sevak V, Ravi PM, Raju M, Vellaiappan NA, et al. Association of hla-Drb1, Dqa1 and Dqb1 alleles and haplotype in parkinson's disease from south India. Neurosci Lett (2021) 765:136296. doi: 10.1016/j.neulet.2021.136296
9. Williams GP, Schonhoff AM, Jurkuvenaite A, Thome AD, Standaert DG, Harms AS. Targeting of the class ii transactivator attenuates inflammation and neurodegeneration in an alpha-synuclein model of parkinson's disease. J Neuroinflamm (2018) 15(1):244. doi: 10.1186/s12974-018-1286-2
10. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T Cells from patients with parkinson's disease recognize alpha-synuclein peptides. Nature (2017) 546(7660):656–61. doi: 10.1038/nature22815
11. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol (2016) 73(11):1316–24. doi: 10.1001/jamaneurol.2016.2742
12. Kouchaki E, Kakhaki RD, Tamtaji OR, Dadgostar E, Behnam M, Nikoueinejad H, et al. Increased serum levels of tnf-alpha and decreased serum levels of il-27 in patients with Parkinson disease and their correlation with disease severity. Clin Neurol Neurosurg (2018) 166:76–9. doi: 10.1016/j.clineuro.2018.01.022
13. Madetko N, Migda B, Alster P, Turski P, Koziorowski D, Friedman A. Platelet-to-Lymphocyte ratio and neutrophil-tolymphocyte ratio may reflect differences in pd and msa-p neuroinflammation patterns. Neurol Neurochir Pol (2022) 56(2):148–55. doi: 10.5603/PJNNS.a2022.0014
14. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140(6):871–82. doi: 10.1016/j.cell.2010.02.029
15. Garretti F, Agalliu D, Lindestam Arlehamn CS, Sette A, Sulzer D. Autoimmunity in parkinson's disease: The role of alpha-Synuclein-Specific T cells. Front Immunol (2019) 10:303. doi: 10.3389/fimmu.2019.00303
16. Chang KH, Wu YR, Chen YC, Fung HC, Chen CM. Association of genetic variants within hla-Dr region with parkinson's disease in Taiwan. Neurobiol Aging (2020) 87:140 e13– e18. doi: 10.1016/j.neurobiolaging.2019.11.002
17. Sun C, Wei L, Luo F, Li Y, Li J, Zhu F, et al. Hla-Drb1 alleles are associated with the susceptibility to sporadic parkinson's disease in Chinese han population. PloS One (2012) 7(11):e48594. doi: 10.1371/journal.pone.0048594
18. Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment Via the choroid plexus. EMBO J (2014) 33(1):7–22. doi: 10.1002/embj.201386609
19. Tiberi M, Chiurchiu V. Specialized pro-resolving lipid mediators and glial cells: Emerging candidates for brain homeostasis and repair. Front Cell Neurosci (2021) 15:673549. doi: 10.3389/fncel.2021.673549
20. Krashia P, Cordella A, Nobili A, La Barbera L, Federici M, Leuti A, et al. Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of parkinson's disease. Nat Commun (2019) 10(1):3945. doi: 10.1038/s41467-019-11928-w
21. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and Pink1 mitigate sting-induced inflammation. Nature (2018) 561(7722):258–62. doi: 10.1038/s41586-018-0448-9
22. Chiurchiu V, Orlacchio A, Maccarrone M. Is modulation of oxidative stress an answer? the state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid Med Cell Longev (2016) 2016:7909380. doi: 10.1155/2016/7909380
23. von Otter M, Bergstrom P, Quattrone A, De Marco EV, Annesi G, Soderkvist P, et al. Genetic associations of Nrf2-encoding Nfe2l2 variants with parkinson's disease - a multicenter study. BMC Med Genet (2014) 15:131. doi: 10.1186/s12881-014-0131-4
24. Hancock DB, Martin ER, Vance JM, Scott WK. Nitric oxide synthase genes and their interactions with environmental factors in parkinson's disease. Neurogenetics (2008) 9(4):249–62. doi: 10.1007/s10048-008-0137-1
25. Shi H, Deng HX, Gius D, Schumacker PT, Surmeier DJ, Ma YC. Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum Mol Genet (2017) 26(10):1915–26. doi: 10.1093/hmg/ddx100
26. Kim Y, Stahl MC, Huang X, Connor JR. H63d variant of the homeostatic iron regulator (Hfe) gene alters alpha-synuclein expression, aggregation, and toxicity. J Neurochem (2020) 155(2):177–90. doi: 10.1111/jnc.15107
27. Fan X, Zhang L, Li H, Chen G, Qi G, Ma X, et al. Role of homocysteine in the development and progression of parkinson's disease. Ann Clin Transl Neurol (2020) 7(11):2332–8. doi: 10.1002/acn3.51227
28. Yan Z, Yang W, Wei H, Dean MN, Standaert DG, Cutter GR, et al. Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflamm (2021) 8(5):e1036. doi: 10.1212/NXI.0000000000001036
29. Kline EM, Houser MC, Herrick MK, Seibler P, Klein C, West A, et al. Genetic and environmental factors in parkinson's disease converge on immune function and inflammation. Mov Disord (2021) 36(1):25–36. doi: 10.1002/mds.28411
30. Chao Y, Wong SC, Tan EK. Evidence of inflammatory system involvement in parkinson's disease. BioMed Res Int (2014) 2014:308654. doi: 10.1155/2014/308654
31. Karahalil B, Miser Salihoglu E, Elkama A, Orhan G, Saygin E, Yardim Akaydin S. Individual susceptibility has a major impact on strong association between oxidative stress, defence systems and parkinson's disease. Basic Clin Pharmacol Toxicol (2022) 130(1):158–70. doi: 10.1111/bcpt.13659
32. Mota A, Hemati-Dinarvand M, Akbar Taheraghdam A, Reza Nejabati H, Ahmadi R, Ghasemnejad T, et al. Association of Paraoxonse1 (Pon1) genotypes with the activity of Pon1 in patients with parkinson's disease. Acta Neurol Taiwan (2019) 28(3):66–74.
33. Holmans P, Moskvina V, Jones L, Sharma M, International Parkinson's Disease Genomics C, Vedernikov A, et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to parkinson's disease. Hum Mol Genet (2013) 22(5):1039–49. doi: 10.1093/hmg/dds492
34. Smith GD, Ebrahim S. 'Mendelian randomization': Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol (2003) 32(1):1–22. doi: 10.1093/ije/dyg070
35. Bottigliengo D, Foco L, Seibler P, Klein C, Konig IR, Del Greco MF. A mendelian randomization study investigating the causal role of inflammation on parkinson's disease. Brain (2022) 145(10):3444–53. doi: 10.1093/brain/awac193
36. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. Mds clinical diagnostic criteria for parkinson's disease. Mov Disord (2015) 30(12):1591–601. doi: 10.1002/mds.26424
37. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic parkinson's disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry (1992) 55(3):181–4. doi: 10.1136/jnnp.55.3.181
38. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for parkinson's disease: A meta-analysis of genome-wide association studies. Lancet Neurol (2019) 18(12):1091–102. doi: 10.1016/S1474-4422(19)30320-5
39. Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun (2017) 8:14357. doi: 10.1038/ncomms14357
40. Caron B, Patin E, Rotival M, Charbit B, Albert ML, Quintana-Murci L, et al. Integrative genetic and immune cell analysis of plasma proteins in healthy donors identifies novel associations involving primary immune deficiency genes. Genome Med (2022) 14(1):28. doi: 10.1186/s13073-022-01032-y
41. VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology (2014) 25(3):427–35. doi: 10.1097/EDE.0000000000000081
42. Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing mendelian randomization investigations. Wellcome Open Res (2019) 4:186. doi: 10.12688/wellcomeopenres.15555.2
43. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet (2018) 50(5):693–8. doi: 10.1038/s41588-018-0099-7
44. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through egger regression. Int J Epidemiol (2015) 44(2):512–25. doi: 10.1093/ije/dyv080
45. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol (2016) 40(4):304–14. doi: 10.1002/gepi.21965
46. Bowden J, Spiller W, Del Greco MF, Sheehan N, Thompson J, Minelli C, et al. Improving the visualization, interpretation and analysis of two-sample summary data mendelian randomization Via the radial plot and radial regression. Int J Epidemiol (2018) 47(4):1264–78. doi: 10.1093/ije/dyy101
47. Gui Y, Zhang L, Lv W, Zhang W, Zhao J, Hu X. Nfe2l2 variations reduce antioxidant response in patients with Parkinson disease. Oncotarget (2016) 7(10):10756–64. doi: 10.18632/oncotarget.7353
48. Liu Z, Guo J, Wang Y, Li K, Kang J, Wei Y, et al. Lack of association between il-10 and il-18 gene promoter polymorphisms and parkinson's disease with cognitive impairment in a Chinese population. Sci Rep (2016) 6:19021. doi: 10.1038/srep19021
49. Chang KH, Wu YR, Chen YC, Fung HC, Lee-Chen GJ, Chen CM. Stk39, but not Bst1, hla-Dqb1, and Sppl2b polymorphism, is associated with han-Chinese parkinson's disease in Taiwan. Med (Baltimore) (2015) 94(41):e1690. doi: 10.1097/MD.0000000000001690
50. Guo JF, Li K, Yu RL, Sun QY, Wang L, Yao LY, et al. Polygenic determinants of parkinson's disease in a Chinese population. Neurobiol Aging (2015) 36(4):1765 e1– e6. doi: 10.1016/j.neurobiolaging.2014.12.030
51. Yu RL, Guo JF, Wang YQ, Liu ZH, Sun ZF, Su L, et al. The single nucleotide polymorphism Rs12817488 is associated with parkinson's disease in the Chinese population. J Clin Neurosci (2015) 22(6):1002–4. doi: 10.1016/j.jocn.2014.11.024
52. Chen ML, Lin CH, Lee MJ, Wu RM. Bst1 Rs11724635 interacts with environmental factors to increase the risk of parkinson's disease in a Taiwanese population. Parkinsonism Relat Disord (2014) 20(3):280–3. doi: 10.1016/j.parkreldis.2013.11.009
53. Liao Q, Li NN, Mao XY, Chang XL, Zhao DM, Zhang JH, et al. Mthfr C677t variant reduces risk of sporadic parkinson's disease in ethnic Chinese. Acta Neurol Scand (2014) 130(1):e30–4. doi: 10.1111/ane.12245
54. Liu RR, Zhou LL, Cheng X, Sun MX, Hu YB, Chen SF, et al. Ccdc62 variant Rs12817488 is associated with the risk of parkinson's disease in a han Chinese population. Eur Neurol (2014) 71(1-2):77–83. doi: 10.1159/000354333
55. Chen YC, Wu YR, Wu YC, Lee-Chen GJ, Chen CM. Genetic analysis of Nfe2l2 promoter variation in Taiwanese parkinson's disease. Parkinsonism Relat Disord (2013) 19(2):247–50. doi: 10.1016/j.parkreldis.2012.10.018
56. Kiyohara C, Miyake Y, Koyanagi M, Fujimoto T, Shirasawa S, Tanaka K, et al. Mdr1 C3435t polymorphism and interaction with environmental factors in risk of parkinson's disease: A case-control study in Japan. Drug Metab Pharmacokinet (2013) 28(2):138–43. doi: 10.2133/dmpk.dmpk-12-rg-075
57. Li NN, Tan EK, Chang XL, Mao XY, Zhang JH, Zhao DM, et al. Genetic association study between Stk39 and Ccdc62/Hip1r and parkinson's disease. PloS One (2013) 8(11):e79211. doi: 10.1371/journal.pone.0079211
58. Nie K, Zhang Y, Gan R, Wang L, Zhao J, Huang Z, et al. Polymorphisms in Immune/Inflammatory cytokine genes are related to parkinson's disease with cognitive impairment in the han Chinese population. Neurosci Lett (2013) 541:111–5. doi: 10.1016/j.neulet.2013.02.024
59. Li D, He Q, Li R, Xu X, Chen B, Xie A. Interleukin-10 promoter polymorphisms in Chinese patients with parkinson's disease. Neurosci Lett (2012) 513(2):183–6. doi: 10.1016/j.neulet.2012.02.033
60. Miyake Y, Tanaka K, Fukushima W, Kiyohara C, Sasaki S, Tsuboi Y, et al. Lack of association between Bst1 polymorphisms and sporadic parkinson's disease in a Japanese population. J Neurol Sci (2012) 323(1-2):162–6. doi: 10.1016/j.jns.2012.09.008
61. Fong CS, Shyu HY, Shieh JC, Fu YP, Chin TY, Wang HW, et al. Association of mthfr, mtr, and mtrr polymorphisms with parkinson's disease among ethnic Chinese in Taiwan. Clin Chim Acta (2011) 412(3-4):332–8. doi: 10.1016/j.cca.2010.11.004
62. Chang XL, Mao XY, Li HH, Zhang JH, Li NN, Burgunder JM, et al. Association of gwas loci with pd in China. Am J Med Genet B Neuropsychiatr Genet (2011) 156B(3):334–9. doi: 10.1002/ajmg.b.31167
63. Wang V, Chen SY, Chuang TC, Shan DE, Soong BW, Kao MC. Val-9ala and Ile+58thr polymorphism of mnsod in parkinson's disease. Clin Biochem (2010) 43(12):979–82. doi: 10.1016/j.clinbiochem.2010.05.009
64. Yuan RY, Sheu JJ, Yu JM, Hu CJ, Tseng IJ, Ho CS, et al. Methylenetetrahydrofolate reductase polymorphisms and plasma homocysteine in levodopa-treated and non-treated parkinson's disease patients. J Neurol Sci (2009) 287(1-2):64–8. doi: 10.1016/j.jns.2009.09.007
65. Zhou YT, Yang JF, Zhang YL, Wang XY, Chan P. Protective role of interlekin-1 alpha gene polymorphism in Chinese han population with sporadic parkinson's disease. Neurosci Lett (2008) 445(1):23–5. doi: 10.1016/j.neulet.2008.08.054
66. Wu YR, Chen CM, Hwang JC, Chen ST, Feng IH, Hsu HC, et al. Interleukin-1 alpha polymorphism has influence on late-onset sporadic parkinson's disease in Taiwan. J Neural Transm (Vienna) (2007) 114(9):1173–7. doi: 10.1007/s00702-007-0726-4
67. Wu YR, Feng IH, Lyu RK, Chang KH, Lin YY, Chan H, et al. Tumor necrosis factor-alpha promoter polymorphism is associated with the risk of parkinson's disease. Am J Med Genet B Neuropsychiatr Genet (2007) 144B(3):300–4. doi: 10.1002/ajmg.b.30435
68. Fong CS, Cheng CW, Wu RM. Pesticides exposure and genetic polymorphism of paraoxonase in the susceptibility of parkinson's disease. Acta Neurol Taiwan (2005) 14(2):55–60.
69. Nishimura M, Kuno S, Kaji R, Yasuno K, Kawakami H. Glutathione-S-Transferase-1 and interleukin-1beta gene polymorphisms in Japanese patients with parkinson's disease. Mov Disord (2005) 20(7):901–2. doi: 10.1002/mds.20477
70. Tan EK, Chan DK, Ng PW, Woo J, Teo YY, Tang K, et al. Effect of Mdr1 haplotype on risk of Parkinson disease. Arch Neurol (2005) 62(3):460–4. doi: 10.1001/archneur.62.3.460
71. Wu RM, Cheng CW, Chen KH, Shan DE, Kuo JW, Ho YF, et al. Genetic polymorphism of the Cyp2e1 gene and susceptibility to parkinson's disease in Taiwanese. J Neural Transm (Vienna) (2002) 109(11):1403–14. doi: 10.1007/s00702-002-0721-8
72. Nishimura M, Mizuta I, Mizuta E, Yamasaki S, Ohta M, Kaji R, et al. Tumor necrosis factor gene polymorphisms in patients with sporadic parkinson's disease. Neurosci Lett (2001) 311(1):1–4. doi: 10.1016/s0304-3940(01)02111-5
73. Woo SI, Kim JW, Seo HG, Park CH, Han SH, Kim SH, et al. Cyp2d6*4 polymorphism is not associated with parkinson's disease and has no protective role against alzheimer's disease in the Korean population. Psychiatry Clin Neurosci (2001) 55(4):373–7. doi: 10.1046/j.1440-1819.2001.00877.x
74. Nishimura M, Mizuta I, Mizuta E, Yamasaki S, Ohta M, Kuno S. Influence of interleukin-1beta gene polymorphisms on age-at-Onset of sporadic parkinson's disease. Neurosci Lett (2000) 284(1-2):73–6. doi: 10.1016/s0304-3940(00)00991-5
75. Wang J, Liu Z. No association between paraoxonase 1 (Pon1) gene polymorphisms and susceptibility to parkinson's disease in a Chinese population. Mov Disord (2000) 15(6):1265–7. doi: 10.1002/1531-8257(200011)15:6<1265::aid-mds1034>3.0.co;2-0
76. Wang J, Liu Z, Chan P. Lack of association between cytochrome P450 2e1 gene polymorphisms and parkinson's disease in a Chinese population. Mov Disord (2000) 15(6):1267–9. doi: 10.1002/1531-8257(200011)15:6<1267::aid-mds1035>3.0.co;2-w
77. Yasui K, Kowa H, Nakaso K, Takeshima T, Nakashima K. Plasma homocysteine and mthfr C677t genotype in levodopa-treated patients with pd. Neurology (2000) 55(3):437–40. doi: 10.1212/wnl.55.3.437
78. Kondo I, Yamamoto M. Genetic polymorphism of paraoxonase 1 (Pon1) and susceptibility to parkinson's disease. Brain Res (1998) 806(2):271–3. doi: 10.1016/s0006-8993(98)00586-1
79. Chuang YH, Lee PC, Vlaar T, Mulot C, Loriot MA, Hansen J, et al. Pooled analysis of the hla-Drb1 by smoking interaction in Parkinson disease. Ann Neurol (2017) 82(5):655–64. doi: 10.1002/ana.25065
80. Ran C, Wirdefeldt K, Brodin L, Ramezani M, Westerlund M, Xiang F, et al. Genetic variations and mrna expression of Nrf2 in parkinson's disease. Parkinsons Dis (2017) 2017:4020198. doi: 10.1155/2017/4020198
81. Gupta SP, Kamal R, Mishra SK, Singh MK, Shukla R, Singh MP. Association of polymorphism of neuronal nitric oxide synthase gene with risk to parkinson's disease. Mol Neurobiol (2016) 53(5):3309–14. doi: 10.1007/s12035-015-9274-3
82. Paul KC, Sinsheimer JS, Rhodes SL, Cockburn M, Bronstein J, Ritz B. Organophosphate pesticide exposures, nitric oxide synthase gene variants, and gene-pesticide interactions in a case-control study of parkinson's disease, California (USA). Environ Health Perspect (2016) 124(5):570–7. doi: 10.1289/ehp.1408976
83. Zahra C, Tabone C, Camilleri G, Felice AE, Farrugia R, Bezzina Wettinger S. Genetic causes of parkinson's disease in the Maltese: A study of selected mutations in Lrrk2, mthfr, qdpr and spr. BMC Med Genet (2016) 17(1):65. doi: 10.1186/s12881-016-0327-x
84. Todorovic M, Newman JR, Shan J, Bentley S, Wood SA, Silburn PA, et al. Comprehensive assessment of genetic sequence variants in the antioxidant 'Master regulator' Nrf2 in idiopathic parkinson's disease. PloS One (2015) 10(5):e0128030. doi: 10.1371/journal.pone.0128030
85. Kumudini N, Uma A, Naushad SM, Mridula R, Borgohain R, Kutala VK. Association of seven functional polymorphisms of one-carbon metabolic pathway with total plasma homocysteine levels and susceptibility to parkinson's disease among south indians. Neurosci Lett (2014) 568:1–5. doi: 10.1016/j.neulet.2014.03.044
86. Lee PC, Rhodes SL, Sinsheimer JS, Bronstein J, Ritz B. Functional paraoxonase 1 variants modify the risk of parkinson's disease due to organophosphate exposure. Environ Int (2013) 56:42–7. doi: 10.1016/j.envint.2013.03.004
87. Belin AC, Ran C, Anvret A, Paddock S, Westerlund M, Hakansson A, et al. Association of a protective paraoxonase 1 (Pon1) polymorphism in parkinson's disease. Neurosci Lett (2012) 522(1):30–5. doi: 10.1016/j.neulet.2012.06.007
88. San Luciano M, Ozelius L, Lipton RB, Raymond D, Bressman SB, Saunders-Pullman R. Gender differences in the Il6 -174g>C and Esr2 1730g>a polymorphisms and the risk of parkinson's disease. Neurosci Lett (2012) 506(2):312–6. doi: 10.1016/j.neulet.2011.11.032
89. Punia S, Das M, Behari M, Dihana M, Govindappa ST, Muthane UB, et al. Leads from xenobiotic metabolism genes for parkinson's disease among north indians. Pharmacogenet Genomics (2011) 21(12):790–7. doi: 10.1097/FPC.0b013e32834bcd74
90. Manthripragada AD, Costello S, Cockburn MG, Bronstein JM, Ritz B. Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology (2010) 21(1):87–94. doi: 10.1097/EDE.0b013e3181c15ec6
91. Singh M, Khanna VK, Shukla R, Parmar D. Association of polymorphism in cytochrome P450 2d6 and n-Acetyltransferase-2 with parkinson's disease. Dis Markers (2010) 28(2):87–93. doi: 10.3233/DMA-2010-0688
92. von Otter M, Landgren S, Nilsson S, Celojevic D, Bergstrom P, Hakansson A, et al. Association of Nrf2-encoding Nfe2l2 haplotypes with parkinson's disease. BMC Med Genet (2010) 11:36. doi: 10.1186/1471-2350-11-36
93. Camicioli RM, Bouchard TP, Somerville MJ. Homocysteine is not associated with global motor or cognitive measures in nondemented older parkinson's disease patients. Mov Disord (2009) 24(2):176–82. doi: 10.1002/mds.22227
94. Funke C, Soehn AS, Tomiuk J, Riess O, Berg D. Genetic analysis of coding snps in blood-brain barrier transporter Mdr1 in European parkinson's disease patients. J Neural Transm (Vienna) (2009) 116(4):443–50. doi: 10.1007/s00702-009-0196-y
95. Westerlund M, Belin AC, Anvret A, Hakansson A, Nissbrandt H, Lind C, et al. Association of a polymorphism in the Abcb1 gene with parkinson's disease. Parkinsonism Relat Disord (2009) 15(6):422–4. doi: 10.1016/j.parkreldis.2008.11.010
96. Zschiedrich K, Konig IR, Bruggemann N, Kock N, Kasten M, Leenders KL, et al. Mdr1 variants and risk of Parkinson disease. association with pesticide exposure? J Neurol (2009) 256(1):115–20. doi: 10.1007/s00415-009-0089-x
97. Bialecka M, Klodowska-Duda G, Kurzawski M, Slawek J, Gorzkowska A, Opala G, et al. Interleukin-10 (Il10) and tumor necrosis factor alpha (Tnf) gene polymorphisms in parkinson's disease patients. Parkinsonism Relat Disord (2008) 14(8):636–40. doi: 10.1016/j.parkreldis.2008.02.001
98. Halling J, Petersen MS, Grandjean P, Weihe P, Brosen K. Genetic predisposition to parkinson's disease: Cyp2d6 and hfe in the faroe islands. Pharmacogenet Genomics (2008) 18(3):209–12. doi: 10.1097/FPC.0b013e3282f5106e
99. Singh M, Khan AJ, Shah PP, Shukla R, Khanna VK, Parmar D. Polymorphism in environment responsive genes and association with Parkinson disease. Mol Cell Biochem (2008) 312(1-2):131–8. doi: 10.1007/s11010-008-9728-2
100. Aamodt AH, Stovner LJ, Thorstensen K, Lydersen S, White LR, Aasly JO. Prevalence of haemochromatosis gene mutations in parkinson's disease. J Neurol Neurosurg Psychiatry (2007) 78(3):315–7. doi: 10.1136/jnnp.2006.101352
101. Bialecka M, Klodowska-Duda G, Kurzawski M, Slawek J, Opala G, Bialecki P, et al. Interleukin-10 gene polymorphism in parkinson's disease patients. Arch Med Res (2007) 38(8):858–63. doi: 10.1016/j.arcmed.2007.06.006
102. Wahner AD, Sinsheimer JS, Bronstein JM, Ritz B. Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch Neurol (2007) 64(6):836–40. doi: 10.1001/archneur.64.6.836
103. Guerreiro RJ, Bras JM, Santana I, Januario C, Santiago B, Morgadinho AS, et al. Association of hfe common mutations with parkinson's disease, alzheimer's disease and mild cognitive impairment in a Portuguese cohort. BMC Neurol (2006) 6:24. doi: 10.1186/1471-2377-6-24
104. Religa D, Czyzewski K, Styczynska M, Peplonska B, Lokk J, Chodakowska-Zebrowska M, et al. Hyperhomocysteinemia and methylenetetrahydrofolate reductase polymorphism in patients with parkinson's disease. Neurosci Lett (2006) 404(1-2):56–60. doi: 10.1016/j.neulet.2006.05.040
105. Todorovic Z, Dzoljic E, Novakovic I, Mirkovic D, Stojanovic R, Nesic Z, et al. Homocysteine serum levels and mthfr C677t genotype in patients with parkinson's disease, with and without levodopa therapy. J Neurol Sci (2006) 248(1-2):56–61. doi: 10.1016/j.jns.2006.05.040
106. Hakansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, et al. Investigation of genes coding for inflammatory components in parkinson's disease. Mov Disord (2005) 20(5):569–73. doi: 10.1002/mds.20378
107. Hakansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to parkinson's disease. Am J Med Genet B Neuropsychiatr Genet (2005) 133B(1):88–92. doi: 10.1002/ajmg.b.30136
108. Wullner U, Kolsch H, Linnebank M. Methylenetetrahydrofolate reductase in parkinson's disease. Ann Neurol (2005) 58(6):972–3. doi: 10.1002/ana.20696
109. Clarimon J, Eerola J, Hellstrom O, Tienari PJ, Singleton A. Paraoxonase 1 (Pon1) gene polymorphisms and parkinson's disease in a Finnish population. Neurosci Lett (2004) 367(2):168–70. doi: 10.1016/j.neulet.2004.05.108
110. Hague S, Peuralinna T, Eerola J, Hellstrom O, Tienari PJ, Singleton AB. Confirmation of the protective effect of inos in an independent cohort of Parkinson disease. Neurology (2004) 62(4):635–6. doi: 10.1212/01.wnl.0000110191.38152.29
111. Moller JC, Depboylu C, Kolsch H, Lohmuller F, Bandmann O, Gocke P, et al. Lack of association between the interleukin-1 alpha (-889) polymorphism and early-onset parkinson's disease. Neurosci Lett (2004) 359(3):195–7. doi: 10.1016/j.neulet.2004.01.058
112. Ross OA, O'Neill C, Rea IM, Lynch T, Gosal D, Wallace A, et al. Functional promoter region polymorphism of the proinflammatory chemokine il-8 gene associates with parkinson's disease in the Irish. Hum Immunol (2004) 65(4):340–6. doi: 10.1016/j.humimm.2004.01.015
113. Tan EK, Drozdzik M, Bialecka M, Honczarenko K, Klodowska-Duda G, Teo YY, et al. Analysis of Mdr1 haplotypes in parkinson's disease in a white population. Neurosci Lett (2004) 372(3):240–4. doi: 10.1016/j.neulet.2004.09.046
114. Dekker MC, Giesbergen PC, Njajou OT, van Swieten JC, Hofman A, Breteler MM, et al. Mutations in the hemochromatosis gene (Hfe), parkinson's disease and parkinsonism. Neurosci Lett (2003) 348(2):117–9. doi: 10.1016/s0304-3940(03)00713-4
115. Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z. Polymorphism in the p-glycoprotein drug transporter Mdr1 gene: A possible link between environmental and genetic factors in parkinson's disease. Pharmacogenetics (2003) 13(5):259–63. doi: 10.1097/01.fpc.0000054087.48725.d9
116. Kelada SN, Costa-Mallen P, Checkoway H, Viernes HA, Farin FM, Smith-Weller T, et al. Paraoxonase 1 promoter and coding region polymorphisms in parkinson's disease. J Neurol Neurosurg Psychiatry (2003) 74(4):546–7. doi: 10.1136/jnnp.74.4.546
117. Buchanan DD, Silburn PA, Chalk JB, Le Couteur DG, Mellick GD. The Cys282tyr polymorphism in the hfe gene in Australian parkinson's disease patients. Neurosci Lett (2002) 327(2):91–4. doi: 10.1016/s0304-3940(02)00398-1
118. Carmine A, Buervenich S, Sydow O, Anvret M, Olson L. Further evidence for an association of the paraoxonase 1 (Pon1) met-54 allele with parkinson's disease. Mov Disord (2002) 17(4):764–6. doi: 10.1002/mds.10172
119. Mattila KM, Rinne JO, Lehtimaki T, Roytta M, Ahonen JP, Hurme M. Association of an interleukin 1b gene polymorphism (-511) with parkinson's disease in Finnish patients. J Med Genet (2002) 39(6):400–2. doi: 10.1136/jmg.39.6.400
120. McGeer PL, Yasojima K, McGeer EG. Association of interleukin-1 beta polymorphisms with idiopathic parkinson's disease. Neurosci Lett (2002) 326(1):67–9. doi: 10.1016/s0304-3940(02)00300-2
121. Schulte T, Schols L, Muller T, Woitalla D, Berger K, Kruger R. Polymorphisms in the interleukin-1 alpha and beta genes and the risk for parkinson's disease. Neurosci Lett (2002) 326(1):70–2. doi: 10.1016/s0304-3940(02)00301-4
122. Akhmedova SN, Yakimovsky AK, Schwartz EI. Paraoxonase 1 met–leu 54 polymorphism is associated with parkinson's disease. J Neurol Sci (2001) 184(2):179–82. doi: 10.1016/s0022-510x(01)00439-7
123. Dodel RC, Lohmuller F, Du Y, Eastwood B, Gocke P, Oertel WH, et al. A polymorphism in the intronic region of the il-1alpha gene and the risk for parkinson's disease. Neurology (2001) 56(7):982–3. doi: 10.1212/wnl.56.7.982
124. Payami H, Lee N, Zareparsi S, Gonzales McNeal M, Camicioli R, Bird TD, et al. Parkinson's disease, Cyp2d6 polymorphism, and age. Neurology (2001) 56(10):1363–70. doi: 10.1212/wnl.56.10.1363
125. Kruger R, Hardt C, Tschentscher F, Jackel S, Kuhn W, Muller T, et al. Genetic analysis of immunomodulating factors in sporadic parkinson's disease. J Neural Transm (Vienna) (2000) 107(5):553–62. doi: 10.1007/s007020070078
126. Taylor MC, Le Couteur DG, Mellick GD, Board PG. Paraoxonase polymorphisms, pesticide exposure and parkinson's disease in a Caucasian population. J Neural Transm (Vienna) (2000) 107(8-9):979–83. doi: 10.1007/s007020070046
127. Akhmedova S, Anisimov S, Yakimovsky A, Schwartz E. Gln –> arg 191 polymorphism of paraoxonase and parkinson's disease. Hum Hered (1999) 49(3):178–80. doi: 10.1159/000022868
128. Atkinson A, Singleton AB, Steward A, Ince PG, Perry RH, McKeith IG, et al. Cyp2d6 is associated with parkinson's disease but not with dementia with lewy bodies or alzheimer's disease. Pharmacogenetics (1999) 9(1):31–5. doi: 10.1097/00008571-199902000-00005
129. Grasbon-Frodl EM, Kosel S, Riess O, Muller U, Mehraein P, Graeber MB. Analysis of mitochondrial targeting sequence and coding region polymorphisms of the manganese superoxide dismutase gene in German Parkinson disease patients. Biochem Biophys Res Commun (1999) 255(3):749–52. doi: 10.1006/bbrc.1998.9998
130. Nicholl DJ, Bennett P, Hiller L, Bonifati V, Vanacore N, Fabbrini G, et al. A study of five candidate genes in parkinson's disease and related neurodegenerative disorders. European study group on atypical parkinsonism. Neurology (1999) 53(7):1415–21. doi: 10.1212/wnl.53.7.1415
131. Diederich N, Hilger C, Goetz CG, Keipes M, Hentges F, Vieregge P, et al. Genetic variability of the cyp 2d6 gene is not a risk factor for sporadic parkinson's disease. Ann Neurol (1996) 40(3):463–5. doi: 10.1002/ana.410400319
132. Plante-Bordeneuve V, Davis MB, Maraganore DM, Marsden CD, Harding AE. Debrisoquine hydroxylase gene polymorphism in familial parkinson's disease. J Neurol Neurosurg Psychiatry (1994) 57(8):911–3. doi: 10.1136/jnnp.57.8.911
133. Kurth MC, Kurth JH. Variant cytochrome P450 Cyp2d6 allelic frequencies in parkinson's disease. Am J Med Genet (1993) 48(3):166–8. doi: 10.1002/ajmg.1320480311
134. Agliardi C, Guerini FR, Zanzottera M, Riboldazzi G, Zangaglia R, Bono G, et al. Tnf-alpha -308 g/a and -238 g/a promoter polymorphisms and sporadic parkinson's disease in an Italian cohort. J Neurol Sci (2018) 385:45–8. doi: 10.1016/j.jns.2017.12.011
135. Mariani S, Ventriglia M, Simonelli I, Bucossi S, Siotto M, Donno S, et al. Association between sex, systemic iron variation and probability of parkinson's disease. Int J Neurosci (2016) 126(4):354–60. doi: 10.3109/00207454.2015.1020113
136. Narayan S, Sinsheimer JS, Paul KC, Liew Z, Cockburn M, Bronstein JM, et al. Genetic variability in Abcb1, occupational pesticide exposure, and parkinson's disease. Environ Res (2015) 143(Pt A):98–106. doi: 10.1016/j.envres.2015.08.022
137. Mariani S, Ventriglia M, Simonelli I, Spalletta G, Bucossi S, Siotto M, et al. Effects of hemochromatosis and transferrin gene mutations on peripheral iron dyshomeostasis in mild cognitive impairment and alzheimer's and parkinson's diseases. Front Aging Neurosci (2013) 5:37. doi: 10.3389/fnagi.2013.00037
138. Ahmed I, Tamouza R, Delord M, Krishnamoorthy R, Tzourio C, Mulot C, et al. Association between parkinson's disease and the hla-Drb1 locus. Mov Disord (2012) 27(9):1104–10. doi: 10.1002/mds.25035
139. Gorgone G, Curro M, Ferlazzo N, Parisi G, Parnetti L, Belcastro V, et al. Coenzyme Q10, hyperhomocysteinemia and mthfr C677t polymorphism in levodopa-treated parkinson's disease patients. Neuromolecular Med (2012) 14(1):84–90. doi: 10.1007/s12017-012-8174-1
140. Greco V, De Marco EV, Rocca FE, Annesi F, Civitelli D, Provenzano G, et al. Association study between four polymorphisms in the hfe, tf and tfr genes and parkinson's disease in southern Italy. Neurol Sci (2011) 32(3):525–7. doi: 10.1007/s10072-011-0504-9
141. Pascale E, Passarelli E, Purcaro C, Vestri AR, Fakeri A, Guglielmi R, et al. Lack of association between il-1beta, tnf-alpha, and il-10 gene polymorphisms and sporadic parkinson's disease in an Italian cohort. Acta Neurol Scand (2011) 124(3):176–81. doi: 10.1111/j.1600-0404.2010.01441.x
142. Dutheil F, Beaune P, Tzourio C, Loriot MA, Elbaz A. Interaction between Abcb1 and professional exposure to organochlorine insecticides in Parkinson disease. Arch Neurol (2010) 67(6):739–45. doi: 10.1001/archneurol.2010.101
143. Rodriguez-Oroz MC, Lage PM, Sanchez-Mut J, Lamet I, Pagonabarraga J, Toledo JB, et al. Homocysteine and cognitive impairment in parkinson's disease: A biochemical, neuroimaging, and genetic study. Mov Disord (2009) 24(10):1437–44. doi: 10.1002/mds.22522
144. Infante J, Garcia-Gorostiaga I, Sanchez-Juan P, Sanchez-Quintana C, Gurpegui JL, Rodriguez-Rodriguez E, et al. Inflammation-related genes and the risk of parkinson's disease: A multilocus approach. Eur J Neurol (2008) 15(4):431–3. doi: 10.1111/j.1468-1331.2008.02092.x
145. Caccamo D, Gorgone G, Curro M, Parisi G, Di Iorio W, Menichetti C, et al. Effect of mthfr polymorphisms on hyperhomocysteinemia in levodopa-treated parkinsonian patients. Neuromolecular Med (2007) 9(3):249–54. doi: 10.1007/s12017-007-8006-x
146. Huerta C, Sanchez-Ferrero E, Coto E, Blazquez M, Ribacoba R, Guisasola LM, et al. No association between parkinson's disease and three polymorphisms in the enos, nnos, and inos genes. Neurosci Lett (2007) 413(3):202–5. doi: 10.1016/j.neulet.2006.11.044
147. Borlak J, Reamon-Buettner SM. N-acetyltransferase 2 (Nat2) gene polymorphisms in parkinson's disease. BMC Med Genet (2006) 7:30. doi: 10.1186/1471-2350-7-30
148. Elbaz A, Levecque C, Clavel J, Vidal JS, Richard F, Amouyel P, et al. Cyp2d6 polymorphism, pesticide exposure, and parkinson's disease. Ann Neurol (2004) 55(3):430–4. doi: 10.1002/ana.20051
149. Levecque C, Elbaz A, Clavel J, Richard F, Vidal JS, Amouyel P, et al. Association between parkinson's disease and polymorphisms in the nnos and inos genes in a community-based case-control study. Hum Mol Genet (2003) 12(1):79–86. doi: 10.1093/hmg/ddg009
150. Borie C, Gasparini F, Verpillat P, Bonnet AM, Agid Y, Hetet G, et al. Association study between iron-related genes polymorphisms and parkinson's disease. J Neurol (2002) 249(7):801–4. doi: 10.1007/s00415-002-0704-6
151. Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, et al. Expression polymorphism of the blood-brain barrier component p-glycoprotein (Mdr1) in relation to parkinson's disease. Pharmacogenetics (2002) 12(7):529–34. doi: 10.1097/00008571-200210000-00004
152. Bordet R, Broly F, Destee A, Libersa C, Lafitte JJ. Lack of relation between genetic polymorphism of cytochrome p-450iid6 and sporadic idiopathic parkinson's disease. Clin Neuropharmacol (1996) 19(3):213–21. doi: 10.1097/00002826-199619030-00003
153. Lucotte G, Turpin JC, Gerard N, Panserat S, Krishnamoorthy R. Mutation frequencies of the cytochrome Cyp2d6 gene in Parkinson disease patients and in families. Am J Med Genet (1996) 67(4):361–5. doi: 10.1002/(SICI)1096-8628(19960726)67:4<361::AID-AJMG8>3.0.CO;2-P
154. Gasser T, Muller-Myhsok B, Supala A, Zimmer E, Wieditz G, Wszolek ZK, et al. The Cyp2d6b allele is not overrepresented in a population of German patients with idiopathic parkinson's disease. J Neurol Neurosurg Psychiatry (1996) 61(5):518–20. doi: 10.1136/jnnp.61.5.518
155. Chen X, Xia Y, Gresham LS, Molgaard CA, Thomas RG, Galasko D, et al. Apoe and Cyp2d6 polymorphism with and without parkinsonism-dementia complex in the people of chamorro, Guam. Neurology (1996) 47(3):779–84. doi: 10.1212/wnl.47.3.779
156. Ulhaq ZS, Garcia CP. Inflammation-related gene polymorphisms associated with parkinson’s disease: An updated meta-analysis. Egyptian J Med Hum Genet (2020) 21(1):14. doi: 10.1186/s43042-020-00056-6
157. Nilsonne G, Lekander M. Circulating interleukin 6 in Parkinson disease. JAMA Neurol (2017) 74(5):607–8. doi: 10.1001/jamaneurol.2017.0037
158. Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (Il)-1 beta, il-2, il-4, il-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and parkinson's disease. Neurosci Lett (1996) 211(1):13–6. doi: 10.1016/0304-3940(96)12706-3
159. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (Tnf-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett (1994) 165(1-2):208–10. doi: 10.1016/0304-3940(94)90746-3
160. Chu K, Zhou X, Luo BY. Cytokine gene polymorphisms and parkinson's disease: A meta-analysis. Can J Neurol Sci (2012) 39(1):58–64. doi: 10.1017/s0317167100012695
161. Sharma S, Sharma A, Kumar S, Sharma SK, Ghosh B. Association of tnf haplotypes with asthma, serum ige levels, and correlation with serum tnf-alpha levels. Am J Respir Cell Mol Biol (2006) 35(4):488–95. doi: 10.1165/rcmb.2006-0084OC
162. Zhao Y, Du Y, Zhao S, Guo Z. Single-nucleotide polymorphisms of microrna processing machinery genes and risk of colorectal cancer. Onco Targets Ther (2015) 8:421–5. doi: 10.2147/OTT.S78647
163. Olivieri F, Bonafe M, Cavallone L, Giovagnetti S, Marchegiani F, Cardelli M, et al. The -174 C/G locus affects in vitro/in vivo il-6 production during aging. Exp Gerontol (2002) 37(2-3):309–14. doi: 10.1016/s0531-5565(01)00197-8
164. Franco R, Li S, Rodriguez-Rocha H, Burns M, Panayiotidis MI. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to parkinson's disease. Chem Biol Interact (2010) 188(2):289–300. doi: 10.1016/j.cbi.2010.06.003
165. Liu YL, Yang J, Zheng J, Liu DW, Liu T, Wang JM, et al. Paraoxonase 1 polymorphisms L55m and Q192r were not risk factors for parkinson's disease: A huge review and meta-analysis. Gene (2012) 501(2):188–92. doi: 10.1016/j.gene.2012.03.067
166. Lu Y, Peng Q, Zeng Z, Wang J, Deng Y, Xie L, et al. Cyp2d6 phenotypes and parkinson's disease risk: A meta-analysis. J Neurol Sci (2014) 336(1-2):161–8. doi: 10.1016/j.jns.2013.10.030
167. Chattopadhyay M, Chowdhury AR, Feng T, Assenmacher CA, Radaelli E, Guengerich FP, et al. Mitochondrially targeted cytochrome P450 2d6 is involved in monomethylamine-induced neuronal damage in mouse models. J Biol Chem (2019) 294(26):10336–48. doi: 10.1074/jbc.RA119.008848
168. Bajpai P, Sangar MC, Singh S, Tang W, Bansal S, Chowdhury G, et al. Metabolism of 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine by mitochondrion-targeted cytochrome P450 2d6: Implications in Parkinson disease. J Biol Chem (2013) 288(6):4436–51. doi: 10.1074/jbc.M112.402123
169. Klein J, Sato A. The hla system. first of two parts. N Engl J Med (2000) 343(10):702–9. doi: 10.1056/NEJM200009073431006
170. Kannarkat GT, Cook DA, Lee JK, Chang J, Chung J, Sandy E, et al. Common genetic variant association with altered hla expression, synergy with pyrethroid exposure, and risk for parkinson's disease: An observational and case-control study. NPJ Parkinsons Dis (2015) 1:15002–. doi: 10.1038/npjparkd.2015.2
171. Bockaert J, Marin P. Mtor in brain physiology and pathologies. Physiol Rev (2015) 95(4):1157–87. doi: 10.1152/physrev.00038.2014
172. Higashida H, Salmina AB, Olovyannikova RY, Hashii M, Yokoyama S, Koizumi K, et al. Cyclic adp-ribose as a universal calcium signal molecule in the nervous system. Neurochem Int (2007) 51(2-4):192–9. doi: 10.1016/j.neuint.2007.06.023
173. Higashida H, Kamimura SY, Inoue T, Hori O, Islam MS, Lopatina O, et al. Cyclic adp-ribose as an endogenous inhibitor of the mtor pathway downstream of dopamine receptors in the mouse striatum. J Neural Transm (Vienna) (2018) 125(1):17–24. doi: 10.1007/s00702-016-1666-7
174. Nagatsu T. The catecholamine system in health and disease -relation to tyrosine 3-monooxygenase and other catecholamine-synthesizing enzymes. Proc Jpn Acad Ser B Phys Biol Sci (2007) 82(10):388–415. doi: 10.2183/pjab.82.388
175. Menini T, Gugliucci A. Paraoxonase 1 in neurological disorders. Redox Rep (2014) 19(2):49–58. doi: 10.1179/1351000213Y.0000000071
176. Androutsopoulos VP, Kanavouras K, Tsatsakis AM. Role of paraoxonase 1 (Pon1) in organophosphate metabolism: Implications in neurodegenerative diseases. Toxicol Appl Pharmacol (2011) 256(3):418–24. doi: 10.1016/j.taap.2011.08.009
177. Alster P, Madetko N, Koziorowski D, Friedman A. Microglial activation and inflammation as a factor in the pathogenesis of progressive supranuclear palsy (Psp). Front Neurosci (2020) 14:893. doi: 10.3389/fnins.2020.00893
178. Starhof C, Winge K, Heegaard NHH, Skogstrand K, Friis S, Hejl A. Cerebrospinal fluid pro-inflammatory cytokines differentiate parkinsonian syndromes. J Neuroinflamm (2018) 15(1):305. doi: 10.1186/s12974-018-1339-6
179. Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, et al. Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol (1999) 155(4):1241–51. doi: 10.1016/s0002-9440(10)65226-1
180. Hoffmann A, Ettle B, Battis K, Reiprich S, Schlachetzki JCM, Masliah E, et al. Oligodendroglial alpha-Synucleinopathy-Driven neuroinflammation in multiple system atrophy. Brain Pathol (2019) 29(3):380–96. doi: 10.1111/bpa.12678
181. Hall S, Janelidze S, Surova Y, Widner H, Zetterberg H, Hansson O. Cerebrospinal fluid concentrations of inflammatory markers in parkinson's disease and atypical parkinsonian disorders. Sci Rep (2018) 8(1):13276. doi: 10.1038/s41598-018-31517-z
182. Santaella A, Kuiperij HB, van Rumund A, Esselink RAJ, van Gool AJ, Bloem BR, et al. Inflammation biomarker discovery in parkinson's disease and atypical parkinsonisms. BMC Neurol (2020) 20(1):26. doi: 10.1186/s12883-020-1608-8
183. Dinkova-Kostova AT, Copple IM. Advances and challenges in therapeutic targeting of Nrf2. Trends Pharmacol Sci (2023). doi: 10.1016/j.tips.2022.12.003
184. Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol (2018) 75(8):939–46. doi: 10.1001/jamaneurol.2018.0605
185. Lindstrom V, Fagerqvist T, Nordstrom E, Eriksson F, Lord A, Tucker S, et al. Immunotherapy targeting alpha-synuclein protofibrils reduced pathology in (Thy-1)-H[A30p] alpha-synuclein mice. Neurobiol Dis (2014) 69:134–43. doi: 10.1016/j.nbd.2014.05.009
186. Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing c-Terminal-Truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in parkinson's disease-like models. J Neurosci (2014) 34(28):9441–54. doi: 10.1523/JNEUROSCI.5314-13.2014
187. Foltynie T, Langston JW. Therapies to slow, stop, or reverse parkinson's disease. J Parkinsons Dis (2018) 8(s1):S115–S21. doi: 10.3233/JPD-181481
188. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of Prx002/Rg7935, an anti-Alpha-Synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol (2018) 75(10):1206–14. doi: 10.1001/jamaneurol.2018.1487
Keywords: inflammation, Parkinson’s disease, genetics, polymorphism, causal analysis
Citation: Yi M, Li J, Jian S, Li B, Huang Z, Shu L and Zhang Y (2023) Quantitative and causal analysis for inflammatory genes and the risk of Parkinson’s disease. Front. Immunol. 14:1119315. doi: 10.3389/fimmu.2023.1119315
Received: 08 December 2022; Accepted: 08 February 2023;
Published: 28 February 2023.
Edited by:
Iwona Kurkowska-Jastrzebska, Institute of Psychiatry and Neurology (IPiN), PolandReviewed by:
Natalia Madetko-Alster, Department of Neurology, PolandCopyright © 2023 Yi, Li, Jian, Li, Huang, Shu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Zhang, emhhbmd5dWFuOTE5NEBjc3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.