
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1118610
This article is part of the Research TopicBiomarkers and Immunotherapy for Genitourinary TumorsView all 30 articles
 Giuseppe Schepisi1*
Giuseppe Schepisi1* Caterina Gianni1
Caterina Gianni1 Maria Concetta Cursano1Valentina Gallà2Cecilia Menna1
Maria Concetta Cursano1Valentina Gallà2Cecilia Menna1 Chiara Casadei1
Chiara Casadei1 Sara Bleve1
Sara Bleve1 Cristian Lolli1
Cristian Lolli1 Giovanni Martinelli1Giovanni Rosti1
Giovanni Martinelli1Giovanni Rosti1 Ugo De Giorgi1
Ugo De Giorgi1Germ cell tumors (GCTs) represent a heterogeneous neoplasm family affecting gonads and rarely occurring in extragonadal areas. Most of patients have a good prognosis, often even in the presence of metastatic disease; however, in almost 15% of cases, tumor relapse and platinum resistance are the main challenges. Thus, novel treatment strategies with both improved antineoplastic activity and minor treatment-related adverse events compared with platinum are really expected. In this context, the development and the high activity demonstrated by immune checkpoint inhibitors in solid tumors and, subsequently, the interesting results obtained from the use of chimeric antigen receptor (CAR-) T cell therapy in hematological tumors, have stimulated research in this direction also in GCTs. In this article, we will analyze the molecular mechanisms underlying the immune action in the development of GCTs, and we will report the data from the studies that tested the new immunotherapeutic approaches in these neoplasms.
Germ cell tumors (GCTs) represent a heterogeneous neoplasm family affecting gonads and rarely occurring in extragonadal areas, such as retroperitoneum, mediastinum and pineal gland (1–3).
GCT is the most common neoplasm affecting young males between 15 to 44 years of age. The vast majority of patients have a good prognosis with high cure rates even in the presence of metastatic disease (4). However, in a small percentage of cases, GCTs deserve a poor prognosis with tumor relapse and resistance to platinum-based chemotherapy, and are treated with aggressive approaches including high-dose chemotherapy with support of hematopoietic progenitor cells (5–7). In addition, because of young age at diagnosis and high cure rate, many patients experience long-term survival related problems, including physical and psychosocial issues, most of which related to previous antitumor treatments, in particular chemotherapy (8–10). Thus, novel treatment strategies with both improved antineoplastic activity and minor treatment-related adverse events compared with platinum are really expected (11, 12). In this context, the development and the high activity demonstrated by immune checkpoint inhibitors in solid tumors and, subsequently, the interesting results obtained from the use of chimeric antigen receptor (CAR-)T cell therapy in hematological tumors, have stimulated research in this direction also in solid tumors, and more recently in GCTs (13). In this article, we will analyze the molecular mechanisms underlying the immune action in the development of GCTs, and we will report the data from the studies that tested the new immunotherapeutic approaches in these neoplasms.
GCTs represent a highly curable neoplasm, but almost 15% of patients experienced recurrence; the exact mechanism of platinum resistance is not fully understood, but it is believed to have a multifactorial origin (14). One reason could be hided into relationship between GCT cells and surrounding tumor microenvironment (TME), which is currently under investigation.
Stromal cells and the extracellular matrix (ECM) can promote neoplastic proliferation and inhibit apoptosis mechanisms. In turn, cancer cells can influence TME activity (15, 16 Indeed, the latter is not constitutionally a protumoral environment, but surrounding conditions may transform an immune TME into a immune suppressive status and viceversa (17). In testis, microenvironment has an critical role during both developmental process and neoplastic transformation (18). The testis structure is divided into seminiferous tubules and interstitium. The testicular interstitial zone is composed of fibrocytes, androgen-producing Leydig cells, and immune cells, including lymphocytes, macrophages, mastocytes, natural killer, and dendritic cells (19–21).
Testis represents an immunologically privileged organ in mammals, because of its immune TME-mediate protection against autoimmune attack and its deficitary response to antigens. This “prerogative” is probably also involved in spermatogenesis and steroidogenesis mechanisms (22). In literature, spontaneous GCT regression cases are rarely reported, probably due to both the patient’s immune TME and the alteration in tumor vascularization (23). Moreover, GCT patients have been described to activate specific CD8+ and CD4+T cell-mediated immune responses against cancer/testis antigens. T cells are strongly present only in conjunction with the expression of these antigens, while they are much less numerous after treatment (24). However, it is currently unclear whether immunological privilege is implicated in GCT development (25). Several studies have been conducted in order to describe any difference in the immune response in case of GCT or GCT in situ: it has been reported that, comparing GCT with non-oncological testis diseases or in normal testis, T cells are ubiquitarious, whereas B cells and dendritic cells have been detected in GCT samples. Furthermore, pro-inflammatory cytokines, such as IL-6, IL-1β TNF-α), anti-inflammatory cytokines (TGF-β1), Th1-related cytokines (IFN-γ and IL-2), and chemokines (CXCL-13, CXCL-10, and CCL-5) were reported as strongly expressed only in GCTs (26). Notwithstanding, Hvarness et al. did not report any active immune surveillance in GCTs, with similar immune cell concentration in both GCT and normal testis samples (27). Recently, Skowron et al. analyzed the role of cross talk between GCT cells and TME, and demonstrated that this interaction stimulated the expression of ECM proteins, such as collagen I/IV and fibronectin, which in turn altered its structure, leading to a pro-tumoral TME. In those conditions, the researchers observed much more effective migration and adhesion properties in GCT cells as well as enhanced platinum resistance. The latter study suggests that targeting ECM (28) could become a novel therapeutic option, especially in relapsed GCT patients.
Among solid tumors, GCT represent an example of neoplasm without any significant mutational burden, as confirmed by The Cancer Genome Atlas (TCGA) (29). Several genome wide studies has been conducted in GCTs: in 4–31% of seminomas, and up to 14% of non-seminomas, driver mutations were detected in three genes (KRAS, NRAS and KIT) (30–33). Since their low incidence in GCT, a single universal mutation could not explain tumorigenesis in testis. So, it has been suggested a polygenic influence in GCT genesis and proliferation, involving an interaction among several susceptibility genes (up to 50 risk loci has been detected to date) (34). A recent study conducted in 137 GCT patients confirmed low mutation in the three known mutated genes (KIT, KRAS, and NRAS) and reported a frequency of 0.5 mutations per megabase (29). Similarly, to testicular GCTs, primary mediastinal GCTs also have low mutational burden, present mutations in RAS pathways, and since they are exclusively non-seminomatous tumors, KIT mutations are rare. However, unlike testicular GCTs, primary mediastinal tumors exhibit a higher percentage of p53 mutations (35).
GCT subtypes represent the developmental steps from embryonic stem cells toward more differentiated cells to somatic tissues. The mapping of GCT DNA-methylation status (methylome) clearly correlates with the state of differentiation in the GCT histotypes: Seminomas are typically unmethylated or severely hypomethylated tumors, Embryonal carcinomas demonstrate low to intermediate levels of global DNA methylation; well-differentiated yolk sac tumors and teratomas show high levels of DNA methylation (36). Therefore, this histological variability correlates both with the tumoral epigenetic heterogeneity, and with the epigenetic landscape of healthy tissues: in fact, hypermethylated pattern has been reported in differentiated somatic cells as well (37–39). Non-CpG methylation, acetylation, and methylation of histones are also involved in GCT development, but they are scarcely understood to date, instead of the microRNA (miR) signaling, which improved our knowledge about GCT molecular biology. The overexpression of pluripotency markers such as NANOG, OCT3/4 or a tissue stem cell factor KIT and its ligand are correlated with the unique GCT germline origin (40–42). Indeed, their expression has been linked to epigenetic regulation with both DNA methylation and histone acetylation (43–46).
In GCT, carcinoembryonic antigen claudin 6 (CLDN6), a tight junction associated membrane protein may represent an ideal chimeric antigen receptor (CAR) antigen because of its extracellular loop that can be targeted by T cells; moreover, it is silenced during organogenesis, thus is not expressed in healthy cells but only in various cancer cells, including GCT: indeed CLDN6 is expressed in approximately 93% of GCT (47).
Since the first new checkpoint inhibitors were approved in the oncology field, researchers have simultaneously begun to evaluate new immunological markers in different tumor histologies, including GCTs. Among these novel immune-related biomarkers, programmed-death receptor axis, including (i.e. PD-1 and its ligand PD-L1) was tested in GCT also; one immunohistochemistry study conducted by Fankhauser et al. confirmed its activation: indeed, a frequent PD-L1 expression in 479 GCT tissue samples was reported, regardless of the histological subtype (73% of seminoma and 64% of non-seminoma patients, respectively) (48). Analyzing data from TCGA database, a surrogate signature of “T-cell inflamed genes” was demonstrated in 47% GCT samples (49). Lobo et al. analyzed both CTLA4 and PD-L1 expression in GCTs: albeit they found high rates of CTLA-4 and PD-L1 expression in GCTs (96.3% and 85.5%, respectively), no significant correlations were demonstrated either between CTLA-4 expression and the GCT characteristics, such as IGCCCG grouping, rete testis or lymphovascular invasion, staging, nor between CTLA-4 intensity and recurrence-free survival (RFS) (p = 0.934). Instead, they demonstrated a PD-L1 expression in 24.9% of samples, with no significant differences between seminomas and non-seminomas, although PD-L1 resulted more frequent in choriocarcinomas than in teratomas. Curiously, they did not found any differences in terms of RFS among PD-1 positive and negative cases (50).
Another study applied a multiplicative quick score to evaluate PD-L1 expression in a semi-quantitative manner, demonstrating a correlation between scores and clinical outcome: in fact, significantly better PFS (HR=0.40; P =0.008) and OS (HR=0.43; P =0.040) were reported in GCT patients low PD-L1 expression levels (51). The predictive role of PD-L1 expression was also confirmed by Chovanec et al. The authors evaluated PD-L1 expression on tumor infiltrating lymphocytes (TIL) – whose prognostic role was previously demonstrated by Bols et al. (52) – and demonstrated that high PD-L1 expression on TIL was correlated with a significantly better prognosis than cases with lower levels (53). The same correlation was reported by Boldrini et al. (54) in the development of childhood malignant extracranial GCTs. Cheng et al. reported that PD-L1–positive TILs are detected in 85,9% of seminomas, 91% of embryonal carcinomas, 60% of yolk sac tumors 54,5% of choriocarcinomas, and 35.7% of teratomas (55). Shah et al. detected T-cell–inflamed TME, which is inversely correlated with AFP levels, more frequently in seminomas than in other GCT (49). Analyzing immune cells other than lymphocytes, similar differences in PD1 expression between seminomas and non-seminomas has been highlighted by other studies. Sadigh et al. detected PD-L1+ tumor-associated macrophages (TAM) rather in seminomas than non seminomas (56). Analyzing 22 types of immune TME, Song et al. demonstrated high expression of CD8+ T-cells, macrophages, and dendritic cells in GCTs compared with normal samples (57). Siska et al. reported an activated CD3+ T-cell infiltration, PD-L1 hyperexpression, and implemented PD-1/PD-L1 spatial interaction in seminomas; this characteristics were correlated with the better prognosis of this histotype, whereas high macrophage and neutrophil gene signatures were more frequently shown in nonseminomas. In both cases, decreased T-cell and NK-cell signatures,and elevated Treg, neutrophil, mast cell, and macrophage signatures were reported in advanced stage of GCTs (58).
More recently, in a Polish study conducted in a 180 GCT patient cohort,a correlation among 1) lower expression of immune checkpoint proteins V-domain Ig suppressor of T cell activation (VISTA) and PD-L1 on TME, 2) elevated inflammatory marker platelet-to-lymphocyte ratio and 3) higher risk of events was reported, suggesting an involvement of both local and systemic anti-tumor immune response in GCTs (59).
In spite of this, PD-L1 expression level does not seem to be predictive of response to immune-checkpoint inhibitors. This uncertainty in PD-L1 predicting response is quite common in several tumors. In fact, using PD-1/PD-L1 inhibitors, a significant response was more often reported in cases with PD-L1 expression, but some responses in PD-L1 negative tumors were described as well (60). To date, in GCT patients, PD-L1 expression in tumor and TIL correlates with an abundant immunogenic microenvironment but not with immunotherapy response. Perhaps, this incongruence could be only due to our incomplete knowledge on immune machinery.
A comprehensive molecular characterization conducted by Shen et al. did not demonstrated a significant neoantigen signal in GCT, so the disappointing results of immune check-point inhibitors in GCT could be partly due to very low mutational burden (29).
Two other studies evaluated the role of a systemic-immune infiltration index (SII), a marker of proinflammatory microenvironment, which is obtained from total neutrophils count, lymphocytes, and platelets. The first one evaluated several markers (i.e., low albumin and hemoglobin, high leukocytes, neutrophils, CRP, neutrophils to lymphocyte ratio, and SII) and demonstrated their correlation with poor prognosis in GCT (61). The second study confirmed a correlation between higher SII levels and poor prognosis in two independent GCT patient cohorts. The authors also evaluated the combined prognostic value of SII and PD-L1 expression on TIL, and reported a better prognosis in cases with low SII and high PD-L1 on TIL (62). Both the research group demonstrated the prognostic significance of SII regardless of the standard IGCCCG risk criteria (61, 62). Recently, these results were confirmed by Ribnikar et al. (63). Interestingly, poor prognosis in cases with high SII levels could be the prove that proinflammatory mechanisms stimulated by an aggressive tumor microenvironment represent the effect of an unsuccessful fight of the human immune system against the tumor progression (36). More recently, another study confirmed the prognostic role of SII in GCTs treated with high-dose chemotherapy (64).
Regarding other TME elements, Tumor Associated Fibroblasts (TAFs) have been shown to stimulate proliferation and metastasis in several tumor types (65–68). Indeed, TAFs secrete several soluble factors, which promote ECM to produce further soluble factors, including VEGF, HGF, TGFb, IL6, CXCL12, and CCL2 (69), which are involved in tumor proliferation. Moreover, GCT cells shown a significant miR-125b expression, which stimulates secretion of tumor-derived chemokines, such as CSF1 and CX3CL1, which in turn increase TAM recruitment (70). Other soluble factors, in particular IL-8, can lead to an increase in NF-kB and ABCB1, which are responsible of reduced Cisplatin sensitivity: this mechanism, described in gastric cancer cells, could be applicable to GCTs as well (71). It is therefore interesting to note that precisely the destruction of tumor cells mediated by platinum-based chemotherapy could at the same time stimulate the secretion of protumoral factors in the tumor stroma (72).
Another study evaluated the prognostic role of proinflammatory cytokines, like as IFN-α2, IL-2Rα, or IL-16, demonstrating their correlation with high risk clinical characteristics and poor survival in GCT (73). Another interesting cytokine is IL13RA2, strongly expressed in normal testicular cells and currently studied as a potential CAR target against other tumor types, such as glioblastoma multiforme (74).
Moreover, some researchers evaluated the prognostic role of β-1,4-galactosyl transferase-I (B4GALT1) in GCT. BAGALT1 is an enzyme involved in interaction and adhesion of immune cells; its role in disease control in stage I non-small cell lung cancer patients was reported by Lu et al. (75). Nilius et al. demonstrated that high B4GALT1 expression in peripheral T cells represents a marker of lower risk of relapse in GCT patients underwent salvage high-dose chemotherapy and peripheral stem cell transplant. The authors suggest that activated peripheral T cells may be crucial in cancer control. In fact, lectin stimulation of mononuclear cells with Concanavalin A determined a B4GALT1 upregulation from CD4+ T cells, which was correlated with IL-10 hyperexpression. The latter was in turn correlated with better outcome in GCT patients (76).
Hinsch et al. evaluated the immunohistochemic expression of T Cell immunoreceptor with Ig and ITIM domains (TIGIT) in 78 seminoma samples, and reported frequent expression of this immune checkpoint receptor, albeit with high variability in the relative prevalence of TIGIT+ and PD-1+ cells (77).
Two other promising therapeutic targets in immunotherapy are T-cell immunoglobulin and mucin domain-3 (TIM-3) and lymphocyte activation gene-3 (LAG-3): the first one is involved in T-cell exhaustion, which in turn could determine a failure of PD-1 monotherapy blockade or adaptive resistance to anti-PD-1 agents (78, 79). The second one is involved in immune homeostasis through an inhibition of T cell activation and cytokine secretion. In addition, higher LAG3 expression on TILs was correlated with higher PD-L1 expression (80). In spite of this, LAG3 and TIM3 expression in GCT cells were not higher than in nearby normal cells (81).
Recently, mismatch-repair (MMR) deficiency has been significantly related to PD-L1 expression, in different tumor types, including GCT (50). This deficiency makes the tumor more immune sensitive, and more prone to express higher levels of PD-L1. In GCTs, a correlation between MMR-deficiency, microsatellite instability (MSI) and platinum resistance was reported by Honecker et al. (82). More recently, the correlation between low MMR proteins expression and lower platinum sensitivity was confirmed in GCTs (83).
The potential immunotherapeutic strategy in GCT was investigated in several case reports and small patient cohorts (Figure 1).
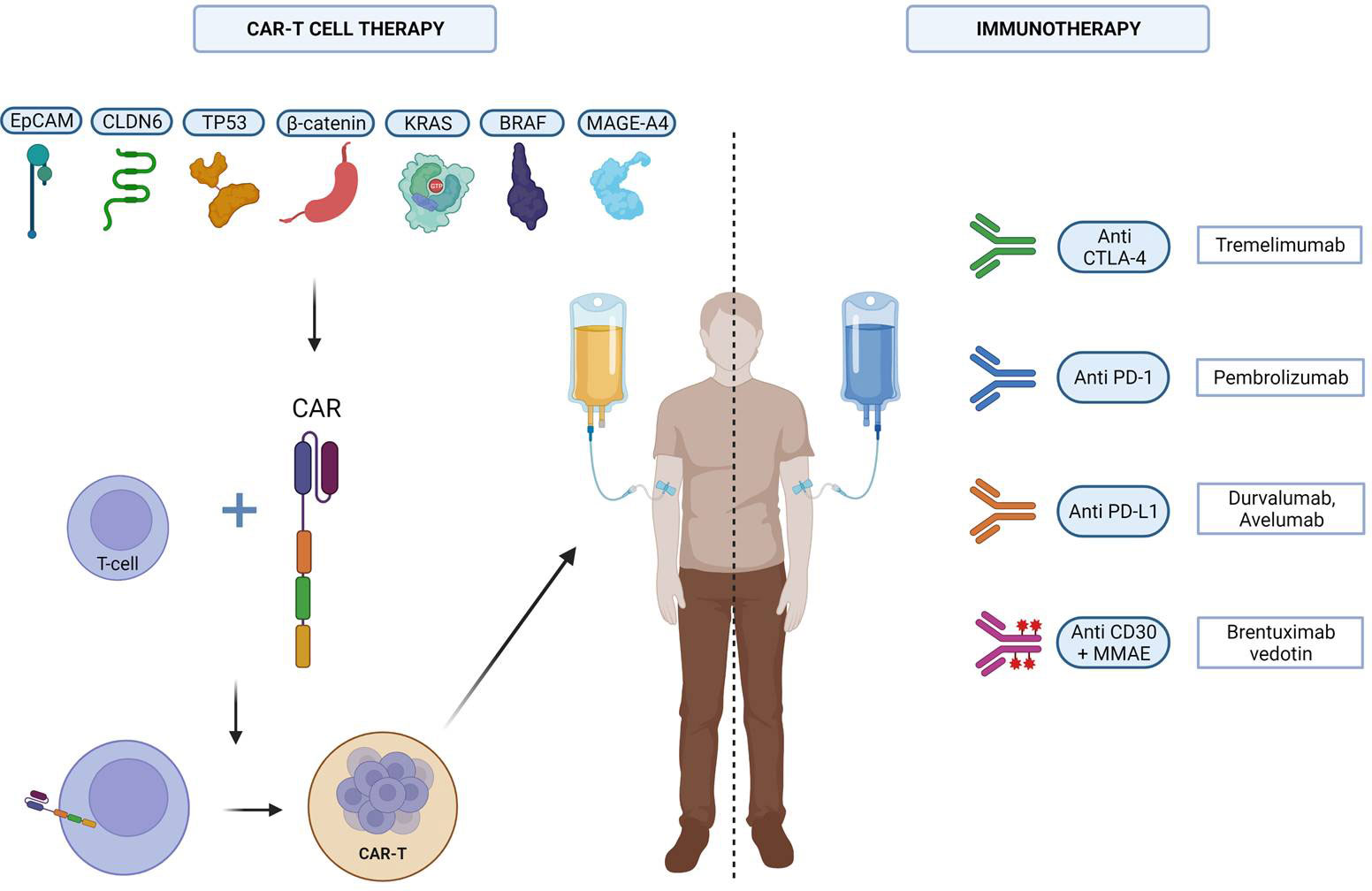
Figure 1 The different immunotherapeutic strategies tested against GCTs. Created with Biorender.
Most of the studies testing immunotherapeutic strategies against GCTs are based on PD-L1 checkpoint inhibitors (84, 85).
One trial reported a 33% of tumor volume regression based on RECIST version 1.1 and a 49% regression based on immune-related response criteria in a small cohort of embryonal cell carcinoma underwent a single dose of anti-PD-1 immune therapy (49). Zschäbitz et al. evaluated a series of seven platinum-refractory GCTs underwent high-dose chemotherapy and stem cell transplantation, and subsequently treated with anti-PD1 Nivolumab or Pembrolizumab. Four of them experienced rapid tumor progression and died after single-dose of immunotherapeutic drug. Only one PR was shown in one of the other three enrolled patients, but they underwent concomitant etoposide (86). Chi et al. reported a durable (radiographic and beta-HCG) response to Nivolumab in a pretreated poor risk metastatic choriocarcinoma (87). In another case report, one patient with choriocarinoma was treated with Pembrolizumab, but he experienced a rapid PD, thus immunotherapy was prematurely stopped (88). A phase II, single-arm trial (NCT02499952) tested anti-PD1 Pembrolizumab in 12 platinum-refractory GCT patients, enrolled regardless of PD-L1 expression. No OR was reported, with only SD in two patients for approximately 7 months and 5 months, respectively (89). Another phase II clinical trial (NCT02721732) was conducted in a small cohort of 12 GCT patients (10 men, 2 women) treated with Pembrolizumab. The drug was well tolerated. A SD was reported in 3 patients, but no OR was shown. The median PFS was 2.4 months and the median OS was 10.6 months (90).
Overall, these studies demonstrated a very limited antitumor activity of anti-PD-1 inhibitor monotherapy in GCTs (Table 1). Other researchers tested anti-PD-L1 inhibitors.
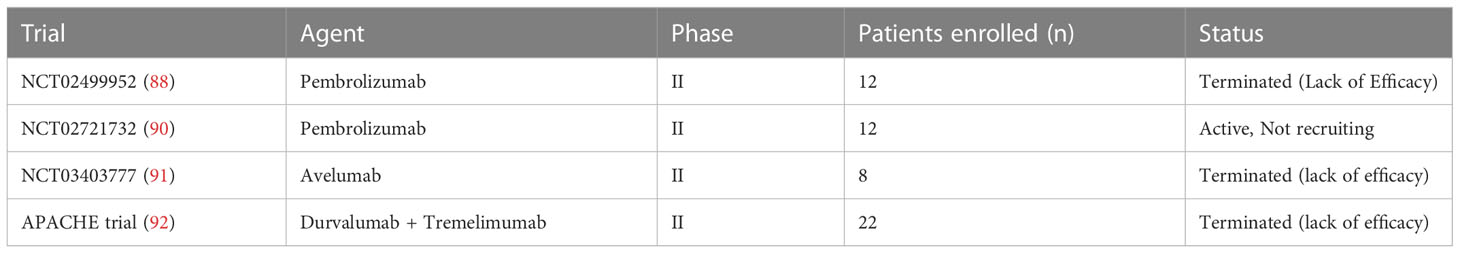
Table 1 Trials presenting anti-PD-1 inhibitor in GCTs.
A phase II clinical trial tested the PD-L1 inhibitor Avelumab in eight patients with relapsed/refractory GCT. Avelumab was well tolerated but with a limited activity in this small patient cohort (91). In another open label, phase II clinical trial, the PD-L1 inhibitor Durvalumab, alone or in combination with the anti-CTLA4 inhibitor Tremelimumab, was administered in a cohort of 22 GCT patients (11 underwent Durvalumab alone and 11 combination of both treatments). However, 72.7% of patient cohort treated with Durvalumab monotherapy experienced rapid PD, thus that arm was prematurely closed. One case of PR and one of SD was reported in the combination arm. PD-L1 expression was not correlated with tumor response (92).
The poor results obtained with anti-PD1/PD-L1 inhibitors prompted researchers to focus on new immunological targets, for example on conjugated antibodies, such as brentuximab vedotin. This is an anti-CD30 antibody conjugate comprising a chimeric antibody bound to cell-surface antigen CD30 covalently conjugated to the cytotoxic antitubulin agent monomethylauristatin E. In a phase II study (NCT01461538), the researchers enrolled seven relapsed/refractory CD30-positive GCT patients, which underwent brentuximab-vedotin every 3 weeks. The authors reported two OR an one CR, which persisted for more than 4 years after four treatment cycles. In the other patient, a PR was demonstrated after 2 cycles, but after the fourth one, this patient experienced a rapid PD (93). In another study, Brentuximab Vedotin was administered in a cohort of 24 CD30-positive GCT patients. Eleven of them experienced a serum tumor markers reduction, whereas 11.1% of them reported a 3-month PFS and 85.7% of them 6-month OS. In a case report, a combination of Brentuximab Vedotin and Pembrolizumab in a highly pretreated patients with GCT led to a CR but at the cost of severe toxicities (grade 3 immune-mediated hepatitis, grade 3 polyneuropathy) (94).
CAR-T cells are genetically engineered T cells, which display antigen-specific receptors on its external cell membrane. They are composed of four domains: (1) on the external extremity, a single-chain antibody fragment (scFV) also known as the antigen-binding domain; (2) a hinge region, which links scFV with the (3) transmembrane region; and (4) an intracellular region, which comprises the signal transduction part of the TCR, linked with one or two costimulatory domains (95).
Compared to other types of immunotherapy, CARs provide the following advantages: first of all, the immune mechanism of action depends on a surface–antigen interaction. Thus it is not MHC restricted, and that allows to use CAR-T cell strategy also in tumors without significant MHC expression (96). Moreover, The low antigen affinity in TCRs can determine off-target toxicities (97). In addition, CAR-T cells also provide T-cell lytic property (98).
To date, compared with haematological neoplasms, the road to using CAR-T cell therapy approach in solid tumors is much more complex; this is due to various problems, the most important being intra-tumor heterogeneity and TME-mediated protumoral activity (99). Indeed, no CAR-T cell therapy is approved in solid tumors.
Recently, four early-phase studies presented at ESMO Congress 2022 could represent a step forward in the development of CAR-T cell therapy into solid tumors (Table 2). Different approaches were tested: in two studies CAR-T cell therapies (100, 101) were used, a vaccine-targeted therapy in a third trial (102) and a T-cell receptor T-cell therapy in the fourth one (103), respectively. Among them, only one enrolled GCT patients (100).
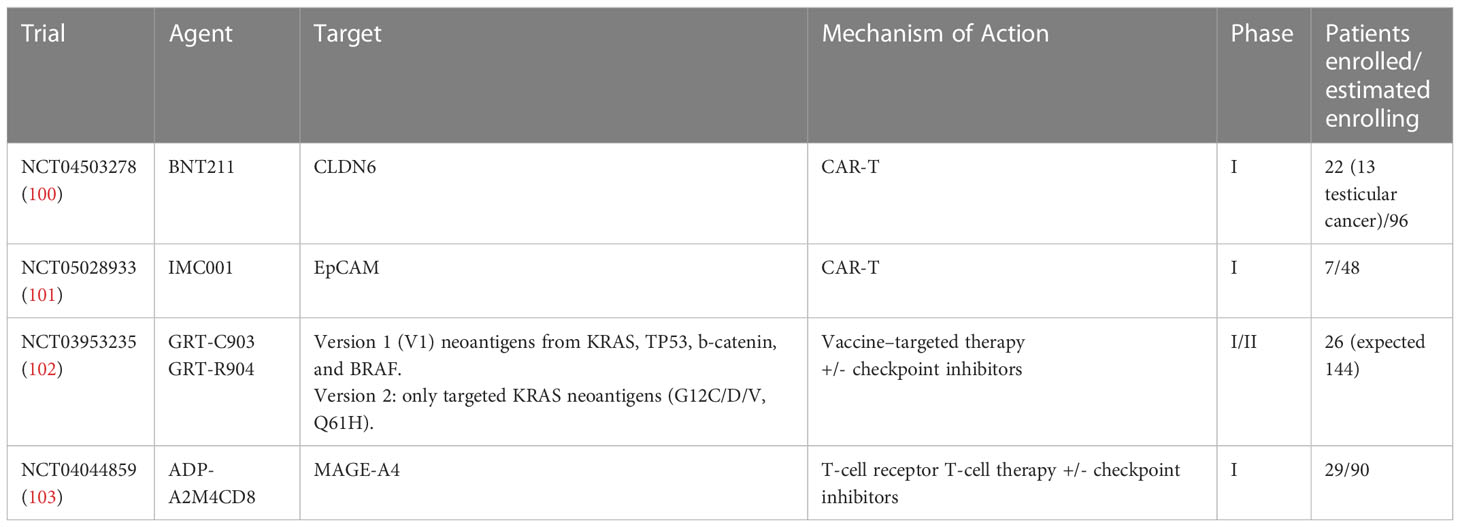
Table 2 Ongoing clinical trials with adaptive cell therapies on solid tumors presented at ESMO 2022.
BNT211-01 trial is a phase I first-in-human trial which tested BNT211, i.e. a CAR-T cell targeting CLDN6 both as monotherapy and in combination with a CLDN6-encoding CAR-T cell-amplifying RNA vaccine (CARVac) in patients with CLDN6-positive relapsed/refractory solid tumors (100). Twenty-two patients with GCT (n=13), ovarian (n=4), endometrial (n=1), fallopian tube (n=1), gastric cancer (n=1), sarcoma (n=1), and unknown primary (n=1) underwent CAR-T cell therapy at two different dose levels. 1 complete response (CR), 6 partial response (PR), 7 stable disease (SD) and 5 progressive disease (PD) were reported, with an overall response rate (ORR) of 33% (7/21) and a disease control rate (DCR) of 67% (14/21). Even more encouraging responses were observed in GCT cohort, with an ORR of 57% and a DCR of 85% (1 CR, 3 PR, 2 SD). A CR, in terms of both negative PET-CT scan and tumor markers was reported. Moreover, an abundant persistence of CAR-T cells was observed for >100 days, and in some cases for >200 days. Two patients had dose-limiting toxicities, including pancytopenia after lymphodepletion for the CAR T-cell monotherapy cohort, and hemophagocytic lymphohistiocytosis for the combination cohort, respectively. To date, the generation of CLDN6-based CAR-T cells has been switched to an automated process and dose escalation is ongoing (100).
In solid tumors, currently the most common immunotherapy is immune checkpoint blockade, which maintain the activity of T cells through the linkage with CTLA4 or PD1/PD-L1 molecules (104). The clinical response to these antibodies is generally more significant in tumors that carry a high mutational burden, albeit the response varies among single cases (105). CAR T-cell therapy is a novel fascinating strategy for non-responders to immune checkpoint inhibitors and for less immunogenic neoplasms, and demonstrated sensational results in some hematological tumors (106, 107). Curiously, in GCTs the current situation is the opposite: indeed, studies testing immune checkpoint inhibitors have led to disappointing findings, whereas CAR-T cell therapy showed promising results in a cohort of 13 GCT patients. This guarantees further investigation with a larger sample size. Should the role of CAR-T cell therapy in GCTs be confirmed, this would represent a significant step forward in this category of patients who are so young, for whom only chemotherapy is currently approved. Furthermore, young age should represent an element in favor of good tolerance to treatment, regardless of any known side effects related to CAR-T cell therapy (95). Furthermore, a better understanding of the mechanisms of activation of the immune system, including epigenetic influence in immune checkpoint expression (108), could also lead to a reconsideration of therapy with immune checkpoint inhibitors against GCTs.
Conceptualization: GS and UD. Methodology: GS, CG, UD. Validation: UD. Writing original draft preparation: GS. Writing—review and editing: UD, CG and GS. Supervision: UD, CG, MC, CC, SB, GM, GR. All authors contributed to the article and approved the submitted version.
This work was partly supported thanks to the contribution of Ricerca Corrente by the Italian Ministry of Health within the research line L2 (Innovative therapies, phase I-III clinical trials).
Figure 1 has been created with Biorender.com (https://biorender.com/).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Scholz M, Zehender M, Thalmann GN, Borner M, Thöni H, Studer UE. Extragonadal retroperitoneal germ cell tumor: Evidence of origin in the testis. Ann Oncol (2002) 13(1):121–4. doi: 10.1093/annonc/mdf003
2. Gori S, Porrozzi S, Roila F, Gatta G, De Giorgi U, Marangolo M. Germ cell tumours of the testis. Crit Rev Oncol Hematol (2005) 53(2):141–64. doi: 10.1016/j.critrevonc.2004.05.006
3. Fizazi K, Oldenburg J, Dunant A, Chen I, Salvioni R, Hartmann JT, et al. Assessing prognosis and optimizing treatment in patients with postchemotherapy viable nonseminomatous germ-cell tumors (NSGCT): Results of the SCR2 international study. Ann Oncol (2008) 19(2):259–64. doi: 10.1093/annonc/mdm472
4. Oldenburg J, Berney DM, Bokemeyer C, Climent MA, Daugaard G, Gietema JA, et al. Testicular seminoma and non-seminoma: ESMO-EURACAN clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol (2022) 33(4):362–75. doi: 10.1016/j.annonc.2022.01.002
5. De Giorgi U, Rosti G, Slavin S, Yaniv I, Harousseau JL, Ladenstein R, et al. Salvage high-dose chemotherapy for children with extragonadal germ-cell tumours. Br J Cancer (2005) 93(4):412–7. doi: 10.1038/sj.bjc.6602724
6. Feldman DR, Lorch A, Kramar A, Albany C, Einhorn LH, Giannatempo P, et al. Brain metastases in patients with germ cell tumors: Prognostic factors and treatment options–an analysis from the global germ cell cancer group. J Clin Oncol (2016) 34(4):345–51. doi: 10.1200/JCO.2015.62.7000
7. De Giorgi U, Demirer T, Wandt H, Taverna C, Siegert W, Bornhauser M, et al. Second-line high-dose chemotherapy in patients with mediastinal and retroperitoneal primary non-seminomatous germ cell tumors: The EBMT experience. Ann Oncol (2005) 16(1):146–51. doi: 10.1093/annonc/mdi017
8. Curreri SA, Fung C, Beard CJ. Secondary malignant neoplasms in testicular cancer survivors. Urol Oncol (2015) 33(9):392–8. doi: 10.1016/j.urolonc.2015.05.002
9. Ostrowski KA, Walsh TJ. Infertility with testicular cancer. Urol Clin North Am (2015) 42(3):409–20. doi: 10.1016/j.ucl.2015.05.003
10. Schepisi G, De Padova S, De Lisi D, Casadei C, Meggiolaro E, Ruffilli F, et al. Psychosocial issues in long-term survivors of testicular cancer. Front Endocrinol (Lausanne) (2019) 10:113. doi: 10.3389/fendo.2019.00113
11. Conti M, Tazzari V, Baccini C, Pertici G, Serino LP, De Giorgi U. Anticancer drug delivery with nanoparticles. In Vivo (2006) 20(6A):697–701.
12. De Giorgi U, Rosti G, Aieta M, Testore F, Burattini L, Fornarini G, et al. Phase II study of oxaliplatin and gemcitabine salvage chemotherapy in patients with cisplatin-refractory nonseminomatous germ cell tumor. Eur Urol (2006) 50(5):1032–8. doi: 10.1016/j.eururo.2006.05.011
13. Schepisi G, Cursano MC, Casadei C, Menna C, Altavilla A, Lolli C, et al. CAR-T cell therapy: A potential new strategy against prostate cancer. J Immunother Cancer (2019) 7(1):258. doi: 10.1186/s40425-019-0741-7
14. Skowron MA, Oing C, Bremmer F, Ströbel P, Murray MJ, Coleman N, et al. The developmental origin of cancers defines basic principles of cisplatin resistance. Cancer Lett (2021) 519:199–210. doi: 10.1016/j.canlet.2021.07.037
15. Milia-Argeiti E, Mourah S, Vallée B, Huet E, Karamanos NK, Theocharis AD, et al. EMMPRIN/CD147-encriched membrane vesicles released from malignant human testicular germ cells increase MMP production through tumor-stroma interaction. Biochim Biophys Acta (2014) 1840(8):2581–8. doi: 10.1016/j.bbagen.2014.02.026
16. Kozlova N, Grossman JE, Iwanicki MP, Muranen T. The interplay of the extracellular matrix and stromal cells as a drug target in stroma-rich cancers. Trends Pharmacol Sci (2020) 41(3):183–98. doi: 10.1016/j.tips.2020.01.001
17. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–5. doi: 10.1016/j.cub.2020.06.081
18. Müller MR, Skowron MA, Albers P, Nettersheim D. Molecular and epigenetic pathogenesis of germ cell tumors. Asian J Urol (2021) 8(2):144–54. doi: 10.1016/j.ajur.2020.05.009
19. Wang M, Fijak M, Hossain H, Markmann M, Nüsing RM, Lochnit G, et al. Characterization of the micro-environment of the testis that shapes the phenotype and function of testicular macrophages. J Immunol (2017) 198(11):4327–40. doi: 10.4049/jimmunol.1700162
20. Meinhardt A, Wang M, Schulz C, Bhushan S. Microenvironmental signals govern the cellular identity of testicular macrophages. J Leukoc Biol (2018) 104(4):757–66. doi: 10.1002/JLB.3MR0318-086RR
21. Qu N, Ogawa Y, Kuramasu M, Nagahori K, Sakabe K, Itoh M. Immunological microenvironment in the testis. Reprod Med Biol (2019) 19(1):24–31. doi: 10.1002/rmb2.12293
22. Fijak M, Meinhardt A. The testis in immune privilege. Immunol Rev (2006) 213:66–81. doi: 10.1111/j.1600-065X.2006.00438.x
23. Dorantes-Heredia R, Motola-Kuba D, Murphy-Sanchez C, Izquierdo-Tolosa CD, Ruiz-Morales JM. Spontaneous regression as a ‘burned-out’ non-seminomatous testicular germ cell tumor: A case report and literature review. J Surg Case Rep (2019) 2019(1):rjy358. doi: 10.1093/jscr/rjy358
24. Pearce H, Hutton P, Chaudhri S, Porfiri E, Patel P, Viney R, et al. Spontaneous CD4+ and CD8+ T-cell responses directed against cancer testis antigens are present in the peripheral blood of testicular cancer patients. Eur J Immunol (2017) 47(7):1232–42. doi: 10.1002/eji.201646898
25. Kalavska K, Schmidtova S, Chovanec M, Mego M. Immunotherapy in testicular germ cell tumors. Front Oncol (2020) 10:573977. doi: 10.3389/fonc.2020.573977
26. Klein B, Haggeney T, Fietz D, Indumathy S, Loveland KL, Hedger M, et al. Specific immune cell and cytokine characteristics of human testicular germ cell neoplasia. Hum Reprod (2016) 31(10):2192–202. doi: 10.1093/humrep/dew211
27. Hvarness T, Nielsen JE, Almstrup K, Skakkebaek NE, Rajpert-De Meyts E, Claesson MH. Phenotypic characterisation of immune cell infiltrates in testicular germ cell neoplasia. J Reprod Immunol (2013) 100(2):135–45. doi: 10.1016/j.jri.2013.10.005
28. Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol Cancer Ther (2018) 17(6):1147–55. doi: 10.1158/1535-7163.MCT-17-0646
29. Shen H, Shih J, Hollern DP, Wang L, Bowlby R, Tickoo SK, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep (2018) 23(11):3392–406. doi: 10.1016/j.celrep.2018.05.039
30. McIntyre A, Summersgill B, Grygalewicz B, Gillis AJ, Stoop J, van Gurp RJ, et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res (2005) 65(18):8085–9. doi: 10.1158/0008-5472.CAN-05-0471
31. Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, et al. KIT mutations are common in testicular seminomas. Am J Pathol (2004) 164(1):305–13. doi: 10.1016/S0002-9440(10)63120-3
32. Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun (2015) 6:5973. doi: 10.1038/ncomms6973
33. Cheng L, Lyu B, Roth LM. Perspectives on testicular germ cell neoplasms. Hum Pathol (2017) 59:10–25. doi: 10.1016/j.humpath.2016.08.002
34. Litchfield K, Levy M, Orlando G, Loveday C, Law PJ, Migliorini G, et al. Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nat Genet (2017) 49(7):1133–40. doi: 10.1038/ng.3896
35. Urbini M, Schepisi G, Bleve S, Virga A, Gianni C, Gurioli G, et al. Primary mediastinal and testicular germ cell tumors in adolescents and adults: A comparison of genomic alterations and clinical implications. Cancers (Basel) (2021) 13(20):5223. doi: 10.3390/cancers13205223
36. Chovanec M, Albany C, Mego M, Montironi R, Cimadamore A, Cheng L. Emerging prognostic biomarkers in testicular germ cell tumors: Looking beyond established practice. Front Oncol (2018) 8:571. doi: 10.3389/fonc.2018.00571
37. Kremenskoy M, Kremenska Y, Ohgane J, Hattori N, Tanaka S, Hashizume K, et al. Genome-wide analysis of DNA methylation status of CpG islands in embryoid bodies, teratomas, and fetuses. Biochem Biophys Res Commun (2003) 311(4):884–90. doi: 10.1016/j.bbrc.2003.10.078
38. Fukushima S, Yamashita S, Kobayashi H, Takami H, Fukuoka K, Nakamura T, et al. Genome-wide methylation profiles in primary intracranial germ cell tumors indicate a primordial germ cell origin for germinomas. Acta Neuropathol (2017) 133(3):445–62. doi: 10.1007/s00401-017-1673-2
39. Liu J, Shi H, Li X, Chen G, Larsson C, Lui WO. MiR−223−3p regulates cell growth and apoptosis via FBXW7 suggesting an oncogenic role in human testicular germ cell tumors. Int J Oncol (2017) 50(2):356–64. doi: 10.3892/ijo.2016.3807
40. Looijenga LH, Stoop H, de Leeuw HP, de Gouveia Brazao CA, Gillis AJ, van Roozendaal KE, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res (2003) 63(9):2244–50.
41. Cheng L, Sung MT, Cossu-Rocca P, Jones TD, MacLennan GT, De Jong J, et al. OCT4: Biological functions and clinical applications as a marker of germ cell neoplasia. J Pathol (2007) 211(1):1–9. doi: 10.1002/path.2105
42. Mirabello L, Kratz C, Savage S, Greene M. Promoter methylation of candidate genes associated with familial testicular cancer. Int J Mol Epidemiol Genet (2012) 3(3):213–27.
43. Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell (2007) 18(5):1543–53. doi: 10.1091/mbc.e07-01-0029
44. Almstrup K, Nielsen JE, Mlynarska O, Jansen MT, Jørgensen A, Skakkebæk NE, et al. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer (2010) 103(8):1269–76. doi: 10.1038/sj.bjc.6605880
45. Nettersheim D, Biermann K, Gillis AJ, Steger K, Looijenga LH, Schorle H. NANOG promoter methylation and expression correlation during normal and malignant human germ cell development. Epigenetics (2011) 6(1):114–22. doi: 10.4161/epi.6.1.13433
46. Villasante A, Piazzolla D, Li H, Gomez-Lopez G, Djabali M, Serrano M. Epigenetic regulation of nanog expression by Ezh2 in pluripotent stem cells. Cell Cycle (2011) 10(9):1488–98. doi: 10.4161/cc.10.9.15658
47. Zhang C, Guo C, Li Y, Liu K, Zhao Q, Ouyang L. Identification of claudin-6 as a molecular biomarker in pan-cancer through multiple omics integrative analysis. Front Cell Dev Biol (2021) 9:726656. doi: 10.3389/fcell.2021.726656
48. Fankhauser CD, Curioni-Fontecedro A, Allmann V, Beyer J, Tischler V, Sulser T, et al. Frequent PD-L1 expression in testicular germ cell tumors. Br J Cancer (2015) 113(3):411–3. doi: 10.1038/bjc.2015.244
49. Shah S, Ward JE, Bao R, Hall CR, Brockstein BE, Luke JJ. Clinical response of a patient to anti-PD-1 immunotherapy and the immune landscape of testicular germ cell tumors. Cancer Immunol Res (2016) 4(11):903–9. doi: 10.1158/2326-6066.CIR-16-0087
50. Lobo J, Rodrigues Â, Guimarães R, Cantante M, Lopes P, Maurício J, et al. Detailed characterization of immune cell infiltrate and expression of immune checkpoint molecules PD-L1/CTLA-4 and MMR proteins in testicular germ cell tumors disclose novel disease biomarkers. Cancers (Basel) (2019) 11(10):1535. doi: 10.3390/cancers11101535
51. Cierna Z, Mego M, Miskovska V, Machalekova K, Chovanec M, Svetlovska D, et al. Prognostic value of programmed-Death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann Oncol (2016) 27(2):300–5. doi: 10.1093/annonc/mdv574
52. Bols B, Jensen L, Jensen A, Brendstrup O. Immunopathology of in situ seminoma. Int J Exp Pathol (2000) 81(3):211–7. doi: 10.1046/j.1365-2613.2000.00151.x
53. Chovanec M, Cierna Z, Miskovska V, Machalekova K, Svetlovska D, Kalavska K, et al. Prognostic role of programmed-death ligand 1 (PD-L1) expressing tumor infiltrating lymphocytes in testicular germ cell tumors. Oncotarget (2017) 8(13):21794–805. doi: 10.18632/oncotarget.15585
54. Boldrini. R, De Pasquale MD, Melaiu O, Chierici M, Jurman G, Benedetti MC, et al. Tumor-infiltrating T cells and PD-L1 expression in childhood malignant extracranial germ-cell tumors. Oncoimmunology (20182018) 8(2):e1542245. doi: 10.1080/2162402X.2018.1542245
55. Cheng X, Dai H, Wan N, Moore Y, Vankayalapati R, Dai Z. Interaction of programmed death-1 and programmed death-1 ligand-1 contributes to testicular immune privilege. Transplantation (2009) 87(12):1778–86. doi: 10.1097/TP.0b013e3181a75633
56. Sadigh S, Farahani SJ, Shah A, Vaughn D, Lal P. Differences in PD-L1-Expressing macrophages and immune microenvironment in testicular germ cell tumors. Am J Clin Pathol (2020) 153(3):387–95. doi: 10.1093/ajcp/aqz184
57. Song Y, Qi X, Kang J, Wang X, Ou N, Zhu J, et al. Identification of new biomarkers in immune microenvironment of testicular germ cell tumour. Andrologia (2021) 53(3):e13986. doi: 10.1111/and.13986
58. Siska PJ, Johnpulle RAN, Zhou A, Bordeaux J, Kim JY, Dabbas B, et al. Deep exploration of the immune infiltrate and outcome prediction in testicular cancer by quantitative multiplexed immunohistochemistry and gene expression profiling. Oncoimmunology (2017) 6(4):e1305535. doi: 10.1080/2162402X.2017.1305535
59. Pęksa R, Kunc M, Popęda M, Piątek M, Bieńkowski M, Żok J, et al. Combined assessment of immune checkpoint regulator VISTA on tumor-associated immune cells and platelet-to-Lymphocyte ratio identifies advanced germ cell tumors with higher risk of unfavorable outcomes. Cancers (2021) 13(8):1750. doi: 10.3390/cancers13081750
60. Huang Q, Zhang H, Hai J, Socinski MA, Lim E, Chen H, et al. Impact of PD-L1 expression, driver mutations and clinical characteristics on survival after anti-PD-1/PD-L1 immunotherapy versus chemotherapy in non-Small-Cell lung cancer: A meta-analysis of randomized trials. Oncoimmunology (2018) 7(12):e1396403. doi: 10.1080/2162402X.2017.1396403
61. Fankhauser CD, Sander S, Roth L, Gross O, Eberli D, Sulser T, et al. Systemic inflammatory markers have independent prognostic value in patients with metastatic testicular germ cell tumours undergoing first-line chemotherapy. Br J Cancer (2018) 118(6):825–30. doi: 10.1038/bjc.2017.467
62. Chovanec M, Cierna Z, Miskovska V, Machalekova K, Kalavska K, Rejlekova K, et al. Systemic immune-inflammation index in germ-cell tumours. Br J Cancer (2018) 118(6):831–8. doi: 10.1038/bjc.2017.460
63. Ribnikar D, Stukalin I, Bedard PL, Hamilton RJ, Jewett M, Warde P, et al. The prognostic value of neutrophil-To-Lymphocyte ratio in metastatic testicular cancer. Curr Oncol (2020) 28:107–14. doi: 10.3390/CURRONCOL28010014
64. Cursano MC, Kopf B, Scarpi E, Menna C, Casadei C, Schepisi G, et al. Prognostic role of systemic inflammatory indexes in germ cell tumors treated with high-dose chemotherapy. Front Oncol (2020) 10:1325. doi: 10.3389/fonc.2020.01325
65. Linxweiler J, Hajili T, Körbel C, Berchem C, Zeuschner P, Müller A, et al. Cancer-associated fibroblasts stimulate primary tumor growth and metastatic spread in an orthotopic prostate cancer xenograft model. Sci Rep (2020) 10(1):12575. doi: 10.1038/s41598-020-69424-x
66. Louault K, Li RR, DeClerck. YA. Cancer-associated fibroblasts: Understanding their heterogeneity. Cancers (Basel) (2020) 12(11):3108. doi: 10.3390/cancers12113108
67. Pan X, Zheng L. Epigenetics in modulating immune functions of stromal and ImmuneCells in the tumor microenvironment. Cell Mol Immunol (2020) 17(9):940–53. doi: 10.1038/s41423-020-0505-9
68. Pape J, Magdeldin T, Stamati K, Nyga A, Loizidou M, Emberton M, et al. Cancer-associated fibroblasts mediate cancer progression and remodel the tumouroid stroma. Br J Cancer (2020) 123(7):1178–90. doi: 10.1038/s41416-020-0973-9
69. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
70. Batool. A, Wang. YQ, Hao. XX, Chen. SR, Liu. YX. A MiR-125b/CSF1-CX3CL1/Tumor- associated macrophage recruitment axis controls testicular germ cell tumor growth. Cell Death Dis (2018) 9(10):962. doi: 10.1038/s41419-018-1021-z
71. Zhai J, Shen J, Xie G, Wu J, He M, Gao L, et al. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett (2019) 454:37–43. doi: 10.1016/j.canlet.2019.04.002
72. Skowron MA, Eul K, Stephan A, Ludwig GF, Wakileh GA, Bister A, et al. Profiling the 3D interaction between germ cell tumors and microenvironmental cells at the transcriptome and secretome level. Mol Oncol (2022) 16(17):3107–27. doi: 10.1002/1878-0261.13282
73. Svetlovska D, Miskovska V, Cholujova D, Gronesova P, Cingelova S, Chovanec M, et al. Plasma cytokines correlated with disease characteristics, progression-free survival, and overall survival in testicular germ-cell tumor patients. Clin Genitourin Cancer (2017) 15(3):411–416.e2. doi: 10.1016/j.clgc.2017.01.027
74. Jakobsen MK, Gjerstorff MF. CAR T-cell cancer therapy targeting surface Cancer/Testis antigens. Front Immunol (2020) 11:1568. doi: 10.3389/fimmu.2020.01568
75. Lu Y, Wang L, Liu P, Yang P, You M. Gene-expression signature predicts postoperative recurrence in stage I non-small cell lung cancer patients. PloS One (2012) 7(1):e30880. doi: 10.1371/journal.pone.0030880
76. Nilius V, Killer MC, Timmesfeld N, Schmitt M, Moll R, Lorch A, et al. High β-1,4-Galactosyltransferase-I expression in peripheral T-lymphocytes is associated with a low riskof relapse in germ-cell cancer patients receiving high-dose chemotherapy with autologous stem cell reinfusion. Oncoimmunology (2018) 7(5):e1423169. doi: 10.1080/2162402X.2017.1423169
77. Hinsch A, Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, et al. Expression of the immune checkpoint receptor TIGIT in seminoma. Oncol Lett (2019) 18(2):1497–502. doi: 10.3892/ol.2019.10428
78. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276(1):97–111. doi: 10.1111/imr.12520
79. He Y, Cao J, Zhao. C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther (2018) 11:7005–9. doi: 10.2147/OTT.S170385
80. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9(5-6):176–89. doi: 10.18632/genesandcancer.180
81. Tu L, Guan R, Yang H, Zhou Y, Hong W, Ma L, et al. Assessment of the expression of the immune checkpoint molecules PD-1, CTLA4, TIM-3 and LAG-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int J Cancer (2020) 147(2):423–39. doi: 10.1002/ijc.32785
82. Honecker F, Wermann H, Mayer F, Gillis AJ, Stoop H, van Gurp RJ, et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol (2009) 27(13):2129–36. doi: 10.1200/JCO.2008.18.8623
83. Rudolph C, Melau C, Nielsen JE, Vile Jensen K, Liu D, Pena-Diaz J, et al. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell Oncol (Dordr) (2017) 40(4):341–55. doi: 10.1007/s13402-017-0326-8
84. Kalavska K, Kucerova L, Schmidtova S, Chovanec M, Mego M. Cancer stem cell niche and immune-active tumor microenvironment in testicular germ cell tumors. Adv Exp Med Biol (2020) 1226:111–21. doi: 10.1007/978-3-030-36214-0_9
85. Velado-Eguskiza A, Gomez-Santos L, Badiola I, Sáez FJ, Alonso E. Testicular germ cell tumours and proprotein convertases. Cancers (Basel) (2022) 14(7):1633. doi: 10.3390/cancers14071633
86. Zschäbitz S, Lasitschka F, Hadaschik B, Hofheinz RD, Jentsch-Ullrich K, Grüner M, et al. Response to anti-programmed cell death protein-1 antibodies in men treated for platinum refractory germ cell cancer relapsed after high-dose chemotherapy and stem cell transplantation. Eur J Cancer (2017) 76:1–7. doi: 10.1016/j.ejca.2017.01.033
87. Chi EA, Schweizer MT. Durable response to immune checkpoint blockade in a platinum-refractory patient with nonseminomatous germ cell tumor. Clin Genitourin Cancer (2017) 15(5):e855–7. doi: 10.1016/j.clgc.2017.04.005
88. Loh KP, Fung C. Novel therapies in platinum-refractory metastatic GermCell tumor: A case report with a focus on a PD-1 inhibitor. Rare Tumors (2017) 9(2):6867. doi: 10.4081/rt.2017.6867
89. Adra N, Einhorn LH, Althouse SK, Ammakkanavar. NR, Musapatika D, Albany C, et al. Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: A Hoosier cancer research network study GU14-206. Ann Oncol (2018) 29(1):209–14. doi: 10.1093/annonc/mdx680
90. Tsimberidou AM, Vo HH, Subbiah V, Janku F, Piha-Paul S, Yilmaz B, et al. Pembrolizumab in patients with advanced metastatic germ cell tumors. Oncologist (2021) 26(7):558–e1098. doi: 10.1002/onco.13682
91. Mego M, Svetlovska D, Chovanec M, Rečkova. M, Rejlekova. K, Obertova. J, et al. Phase II study of avelumab in multiple Relapsed/Refractory germ cell cancer. Invest New Drugs (2019) 37(4):748–54. doi: 10.1007/s10637-019-00805-4
92. Necchi A, Giannatempo P, Raggi D, Mariani L, Colecchia M, Farè E, et al. An open-label randomized phase 2 study of durvalumab alone or in combination with tremelimumab in patients with advanced germ cell tumors (APACHE): Results from the first planned interim analysis. Eur Urol (2019) 75(1):201–3. doi: 10.1016/j.eururo.2018.09.010
93. Albany C, Einhorn L, Garbo L, Boyd T, Josephson N, Feldman DR. Treatment of CD30- expressing germ cell tumors and sex cord stromal tumors with brentuximab vedotin: Identification and report of seven cases. Oncologist (2018) 23(3):316–23. doi: 10.1634/theoncologist.2017-0544
94. Mayrhofer K, Strasser-Weippl K, Niedersüß-Beke D. Pembrolizumab plus brentuximab-vedotin in a patient with pretreated metastatic germ cell tumor. Ann Hematol Oncol (2018) 5:1196.
95. Schepisi G, Conteduca V, Casadei C, Gurioli G, Rossi L, Gallà V, et al. Potential application of chimeric antigen receptor (CAR)-T cell therapy in renal cell tumors. Front Oncol (2020) 10:565857. doi: 10.3389/fonc.2020.565857
96. Pantuck M, Palaskas N, Drakaki A. Next generation T-cell therapy for genitourinary malignancies, part a: Introduction and current state of the art. Cancer Treat Res Commun (2018) 17:8–12. doi: 10.1016/j.ctarc.2018.08.001
97. Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, et al. Chimeric antigen receptor engineering: A right step in the evolution of adoptive cellular immunotherapy. Int Rev Immunol (2015) 34(2):154–87. doi: 10.3109/08830185.2015.1018419
98. Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, et al. Engineered T cells for pancreatic cancer treatment. HPB (Oxford) (2011) 13(9):643–50. doi: 10.1111/j.1477-2574.2011.00344.x
99. Martino M, Naso V, Loteta B, Canale FA, Pugliese M, Alati C, et al. Chimeric antigen receptor T-cell therapy: What we expect soon. Int J Mol Sci (2022) 23(21):13332. doi: 10.3390/ijms232113332
100. Mackensen A, Haanen JBAG, Koenecke C, Alsdorf W, Wagner-Drouet E, Heudobler D, et al. LBA38 BNT211-01: A phase I trial to evaluate safety and efficacy of CLDN6 CAR T cells and CLDN6-encoding MRNA vaccine-mediated in vivo expansion in patients with CLDN6 positive advanced solid tumours. Ann Oncol (2022) 33:S1404–5. doi: 10.1016/j.annonc.2022.08.035
101. Fang W, Luo T, Lu Z, Zhang H, Tong C, Wang S, et al. 737MO - EpCAM-targeted CAR-T cell therapy in patients with advanced colorectal and gastric cancers. Ann Oncol (2022) 33:S331–55. doi: 10.1016/j.annonc.2022.07.863
102. Kyi CK, Spira A, Carbone DP, Johnson ML, Henick. BS, Johnson B, et al. 736MO- personalized, off-the-Shelf KRAS neoantigen-specific immunotherapy for the treatment of advanced solid tumors: Clinical benefit associated with decreases in CtDNA (SLATE-KRAS). Ann Oncol (2022) 33:S331–55. doi: 10.1016/j.annonc.2022.07.862
103. Hong DS, Asch A, Calvo E, Zugazagoitia J, Charlson J, Butler MO, et al. 735MO- updated safety and efficacy from SURPASS, the phase I trial of ADP-A2M4CD8, a next-generation autologous T-cell receptor T-cell therapy, in previously treated patients with unresectable or metastatic. Annal Oncol (2022) 33:S331–55. doi: 10.1016/j.annonc.2022.07.861
104. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
105. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
106. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature (2017) 545(7655):423–31. doi: 10.1038/nature22395
107. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/NATURE12477
Keywords: germ cell tumors, GCT, chimeric antigen receptor, CAR-T, immune checkpoint inhibitors, testicular cancer, treatment
Citation: Schepisi G, Gianni C, Cursano MC, Gallà V, Menna C, Casadei C, Bleve S, Lolli C, Martinelli G, Rosti G and De Giorgi U (2023) Immune checkpoint inhibitors and Chimeric Antigen Receptor (CAR)-T cell therapy: Potential treatment options against Testicular Germ Cell Tumors. Front. Immunol. 14:1118610. doi: 10.3389/fimmu.2023.1118610
Received: 07 December 2022; Accepted: 03 February 2023;
Published: 13 February 2023.
Edited by:
Hui Dai, Peking University, ChinaReviewed by:
Chenghua Yang, Second Military Medical University, ChinaCopyright © 2023 Schepisi, Gianni, Cursano, Gallà, Menna, Casadei, Bleve, Lolli, Martinelli, Rosti and De Giorgi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Schepisi, Z2l1c2VwcGUuc2NoZXBpc2lAaXJzdC5lbXIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.