
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 January 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1118449
This article is part of the Research Topic Plasticity of monocytes/macrophages: phenotypic changes during disease progression View all 17 articles
 Zhun Xiao1†
Zhun Xiao1† Minghao Liu1†
Minghao Liu1† Fangming Yang1Guangwei Liu1Jiangkai Liu1Wenxia Zhao1Suping Ma1*
Fangming Yang1Guangwei Liu1Jiangkai Liu1Wenxia Zhao1Suping Ma1* Zhongping Duan2*
Zhongping Duan2*Non-alcoholic fatty liver disease (NAFLD) has now become the leading chronic liver disease worldwide with lifestyle changes. This may lead to NAFLD becoming the leading cause of end-stage liver disease in the future. To date, there are still no effective therapeutic drugs for NAFLD. An in-depth exploration of the pathogenesis of NAFLD can help to provide a basis for new therapeutic agents or strategies. As the most important immune cells of the liver, macrophages play an important role in the occurrence and development of liver inflammation and are expected to become effective targets for NAFLD treatment. Programmed cell death (PCD) of macrophages plays a regulatory role in phenotypic transformation, and there is also a certain connection between different types of PCD. However, how PCD regulates macrophage polarization has still not been systematically elucidated. Based on the role of lipid metabolic reprogramming in macrophage polarization, PCD may alter the phenotype by regulating lipid metabolism. We reviewed the effects of macrophages on inflammation in NAFLD and changes in their lipid metabolism, as well as the relationship between different types of PCD and lipid metabolism in macrophages. Furthermore, interactions between different types of PCD and potential therapeutic agents targeting of macrophages PCD are also explored.
Non-alcoholic fatty liver disease (NAFLD) is currently the most common liver disease, and affects approximately one-third of the world’s population (1). According to the severity and the pathological phase, NAFLD can be divided into non-alcoholic fatty liver (NAFL), non-alcoholic fat hepatitis (NASH), liver fibrosis and cirrhosis. Although NAFL has no clinically significant, there is evidence suggests that approximately 25% of patients with NAFL progress to NASH (2). The presence of NASH promotes the progression of liver pathology and increases the incidence of adverse outcomes compared to NAFL. In recent years, the global proportion of NAFLD-associated hepatocellular carcinoma (HCC) has increased year by year and may gradually become the main cause of HCC (3). Therefore, it is important to find effective therapeutic agents to block the pathological progression of NAFLD, especially NASH. The transition from NAFL to NASH is the result of a complex multifactorial effect, which involves a complex liver cell population (both parenchymal and non-parenchymal cells) as well as pathological signals from visceral fat and intestine. The pathogenesis of NAFLD was considered to be the “two-hit hypothesis” (4). Lipid accumulation in hepatocytes represents the “first hit”, while other factors such as oxidative stress are referred to as the “second hit”. However, recent studies have suggested that NAFLD progression may be influenced by various factors such as environment, metabolism, gut microbiota, and genetic factors (5, 6). The simultaneous changes of insulin resistance, genetic and epigenetic factors, mitochondrial dysfunction, endoplasmic reticulum stress, microbiota, chronic low-grade inflammation, etc. led to the progress of NAFLD, which was named “multiple parallel hits hypothesis” (7).
The main sources of lipid deposition in the liver include adipose tissue lipolysis, hepatic de novo lipogenesis (DNL), and diet, with the former accounting for the majority (8). Excess fatty acids are taken up and intracellularly transported in the liver by hepatocytes, macrophages, and other liver cells. When excess free fatty acids (FFAs) exceed the antioxidant capacity of the body, inflammation occurs. These mechanisms have been well summarized in previous reviews (9, 10). Macrophages are an important component of innate immunity. Macrophages in the liver mainly include tissue-resident Kupffer cells (KCs), monocyte-derived macrophages (MoMFs) and subcapsular macrophages discovered in recent years (11). Subcapsular macrophages play a major role in the defense against infectious agents from the abdominal cavity and are not the topic of this article. KCs are the most abundant tissue-resident macrophages in the mammalian body, accounting for 80-90% of all tissue-resident macrophages (12). KCs are mainly localized in the reticuloendothelial system and sense risk factors from the intestine and adipose tissue as well as multiple signals from the liver microenvironment. KCs constitute the hepatic immune homeostasis and alert when the balance is disturbed. When inflammation is induced, numerous monocytes are recruited to the liver (13). This may contribute to the chronic low-grade inflammation in NAFLD. Considering the important impact of macrophages on hepatic inflammation and their ability to process lipids, they play an important role in the pathological progression of NAFLD. Programmed cell death (PCD) of macrophages is closely associated with the development of inflammation (14). This paper mainly focuses on the effects of lipid metabolism and PCD on the phenotype of macrophages in NAFLD and the relationship between them. The possibility of targeting PCD of macrophages in the treatment of NAFLD has also been explored.
Inflammation is a major pathological factor in the progression of NAFLD, and hepatocyte death is one of the crucial triggers of liver inflammation (15). Macrophages play an important role in the inflammatory response in NAFLD due to their ability to clear pathogens and recruit circulating inflammatory cells (2). Typically, macrophages can be classified into two phenotypes, the classically activated M1 type and the alternatively activated M2 type (Figure 1). M1 macrophages are induced by lipopolysaccharide (LPS) and Th1 cytokines such as interferon-γ (IFN-γ) and granulocyte-macrophage colony-stimulating factor (GM-CSF) alone or in combination to secrete pro-inflammatory factors such as interleukin 1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α), while M2 macrophages are induced by Th2 cytokines such as IL-4 and IL-13 to secrete anti-inflammatory factors such as IL-10 and transforming growth factor-β (TGF-β) (16). The balance of M1 and M2 macrophages is an important determinant of the pathological changes in the liver under inflammatory conditions. However, the M1 and M2 classification cannot describe macrophages accurately due to the heterogeneity and functional diversity of macrophages. More specific markers of different macrophage phenotypes are needed.
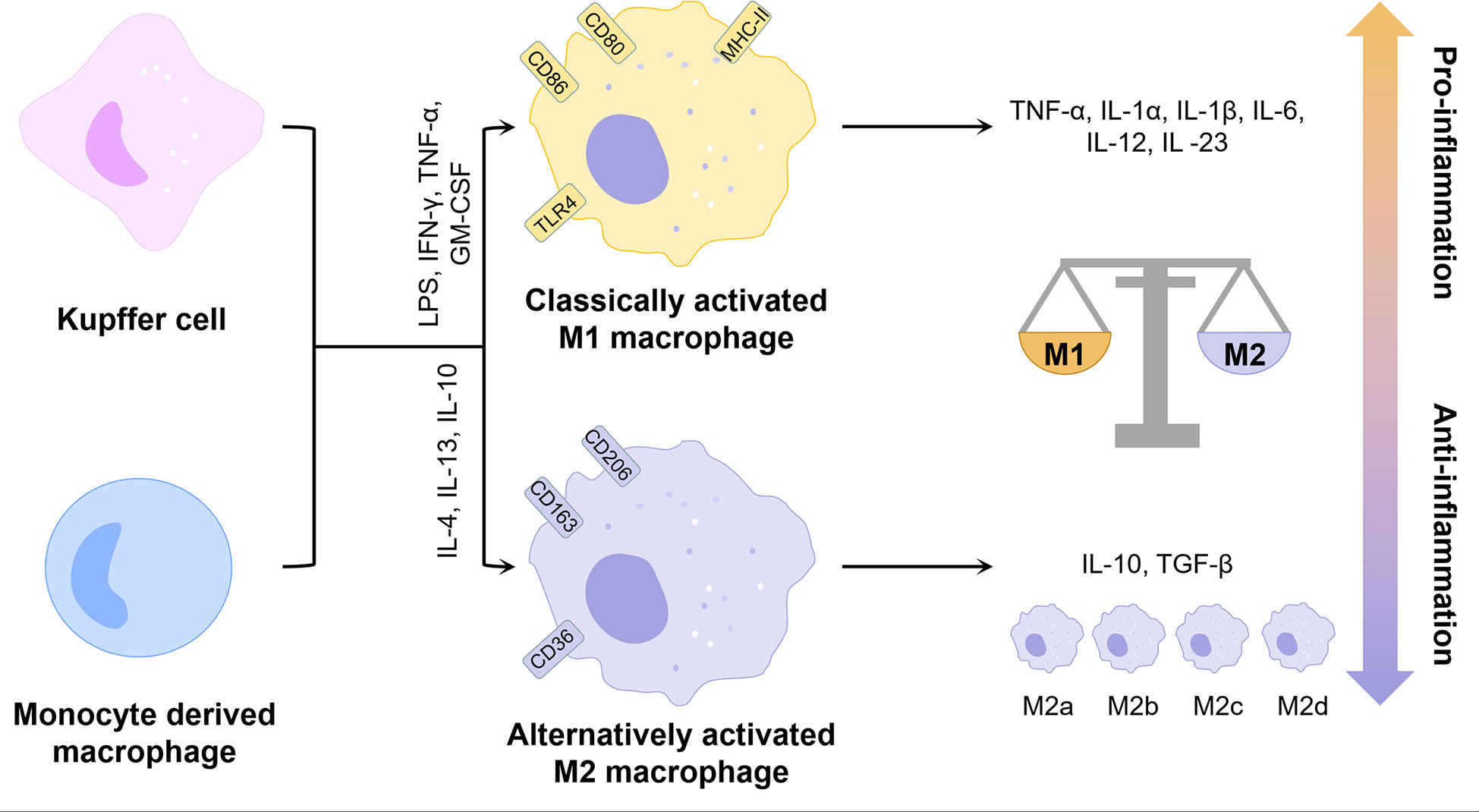
Figure 1 Phenotype and function of liver macrophages. KCs and monocyte-derived macrophages can be polarized by different factors into two distinct phenotypes, classically activated M1 and alternatively activated M2. M2 macrophages can be further classified into multiple phenotypes. Overall, M1 exhibits pro-inflammatory properties and M2 exhibits anti-inflammatory properties. The balance between the two determines the direction of liver inflammation. GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; MHC-II, major histocompatibility complex class IITGF-β, transforming growth factor-β; TLR4, toll-like receptor 4; TNF-α, tumor necrosis factor-α.
KCs are liver-resident macrophages and account for 15% of all liver cells (17). It is now clear that KCs originate from yolk-sac-derived erythro-myeloid progenitors expressing colony stimulating factor 1 receptor (CSF1R) (18). In the context of NAFLD, KCs are the major source of cytokines and chemokines (19). For example, KCs promote steatosis and insulin resistance by secreting IL-1β to downregulate peroxisome proliferative activated receptor α (PPARα) expression in hepatocytes (20). A previous study has demonstrated that consumption of KCs attenuates high fat or high sucrose diet-induced NASH in rats (21). On the one hand, portal vein-derived LPS can bind to toll-like receptor 4 (TLR4) on the surface of KCs to induce their polarization toward M1 pro-inflammatory phenotype and enhance pro-inflammatory cytokines including monocyte chemoattractant protein-1 (MCP1, also known as C-C motif chemokine ligand 2, CCL2), TNF-α and IL-6 expression via yes-associated Protein (YAP) (22). On the other hand, KCs can also promote hepatocyte apoptosis and inflammatory progression by producing TNF, TNF-related apoptosis-inducing ligand (TRAIL) and factor associated suicide (Fas) ligand through phagocytosis of apoptotic bodies (23). In addition, lipotoxic hepatocytes can release mitochondrial DNA (mtDNA) to induce nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) dependent inflammation by binding to transmembrane protein 173 (TMEM173 or STING) on the surface of KCs (24), while KCs themselves can activate the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome via mtDNA released from mitochondria thereby promoting the progression of NASH (25).
MoMFs are recruited to the liver under inflammatory conditions and transformed to different phenotypes in response to stimulation by complex cytokines. Recent studies have confirmed that lipotoxic hepatocytes can secrete extracellular vesicles rich in MicroRNA 192-5p (26), C-X-C Motif Chemokine Ligand 10 (CXCL10) (27), ceramide (28) to promote M1 polarization of MoMFs and secrete pro-inflammatory cytokines. Meanwhile, extracellular vesicles can also promote the recruitment of MoMFs through an integrin β1 (ITGβ1)-dependent pathway (29). Except for CXCL10, various chemokines including CCL2 played important roles in the infiltration of MoMFs and M1 polarization, while inhibition or knockdown of CXCL10 and CCL2 showed inhibition of liver macrophage infiltration and improvement of inflammation in the mouse model of NASH (30, 31). Lymphocyte antigen 6C (Ly6C) is a marker of mouse monocytes, which divides circulating monocytes into two major subpopulations, Ly6Chi and Ly6Clo (32). The Ly6Clo subgroup shows anti-inflammatory properties, while the Ly6Chi subgroup exhibits pro-inflammatory properties and constitutes the main pathological mechanism of NASH progression. Ly6Chi MoMFs can be converted to Ly6Clo MoMFs after the clearance of apoptotic hepatocytes (33). A dual C-C Motif Chemokine Receptor 2 (CCR2)/CCR5 antagonist, cenicriviroc (CVC), demonstrated improvement in NASH-related liver fibrosis and inhibition of steatohepatitis progression in a completed phase II clinical trial enrolling 289 patients with NASH (34). A study in a mouse model of NASH showed that CVC inhibited Ly6Chi MoMFs infiltration and fibrosis progression, but had no direct effect on macrophage polarization (35).
Due to the immune tolerance characteristic of the liver, KCs are mainly involved in maintaining inflammatory homeostasis, while MoMFs play a major role in acute and chronic liver inflammation (32). Under homeostatic conditions, the hepatic macrophage population contains only a small amount of MoMFs. KCs could directly inhibit the inflammatory response of MoMFs by secreting miR-690-containing exosomes, whereas miR-690 of KCs showed low expression during the progression of NASH (36), which might promote the recruitment and pro-inflammatory transformation of MoMFs. In addition, activated KCs can promote the recruitment of Ly6Chi MoMFs by secreting CCL2 (31). KCs proliferate for self-renewal in the liver, but the mechanism of renewal is impaired during NASH. When KCs are depleted, some Ly6Chi MoMFs are even able to convert to KCs to replenish the hepatic KCs pool under specific factors (37). However, compared with embryo-derived KCs, monocyte-derived KCs do not effectively promote hepatic triglyceride storage and exhibit pro-inflammatory properties in the liver, thereby exacerbating liver injury in NASH (38).
Macrophages require a lot of energy to maintain their function in inflammation. Metabolic reprogramming has an important role in regulating the function of immune cells, and macrophages of different phenotypes usually exhibit different metabolic profiles (39). Macrophages possess lipid-processing function and play an important role in lipid metabolism. Intracellular lipid metabolism involves a series of complex enzymatic reactions (Figure 2). FFAs taken up by fatty acid transport proteins (e.g. CD36) and fatty acid binding proteins (FABPs) are transformed to acyl-coenzyme A (acyl-CoA) by the action of acyl-CoA synthase (ACS). Acyl-CoA can not only participate in the synthesis of triglycerides, but also converted to acyl-carnitine by carnitine palmitoyltransferase-1 (CPT1). Subsequently, acyl-carnitine is transported into the mitochondria via carnitine-acylcarnitine translocase (CACT) and CPT2 for fatty acid oxidation (FAO) and increases the production of nicotinamide adenine dinucleotide (NADH)/1,5-dihydroflavin adenine dinucleotide (FADH2) thereby promoting oxidative phosphorylation (OXPHOS). The acetyl-CoA produced during this process participates in the TCA cycle and ultimately promotes the production of citrate. Glucose can be converted to pyruvate through glycolysis and then also participate in the TCA cycle through conversion to acetyl-CoA (40). Citrate can be converted back to acetyl-CoA by the action of ATP-citrate lyase (ACLY). Acetyl-CoA is not only involved in de novo lipogenesis (DNL), but also in cholesterol synthesis. The excess fatty acids are used for the synthesis of triglycerides and other complex lipids (41), while cholesterol is transported out of the cell under the regulation of ATP-binding cassette sub-family A member 1 (ABCA1) and ATP-binding cassette sub-family G member 1 (ABCG1) (42).
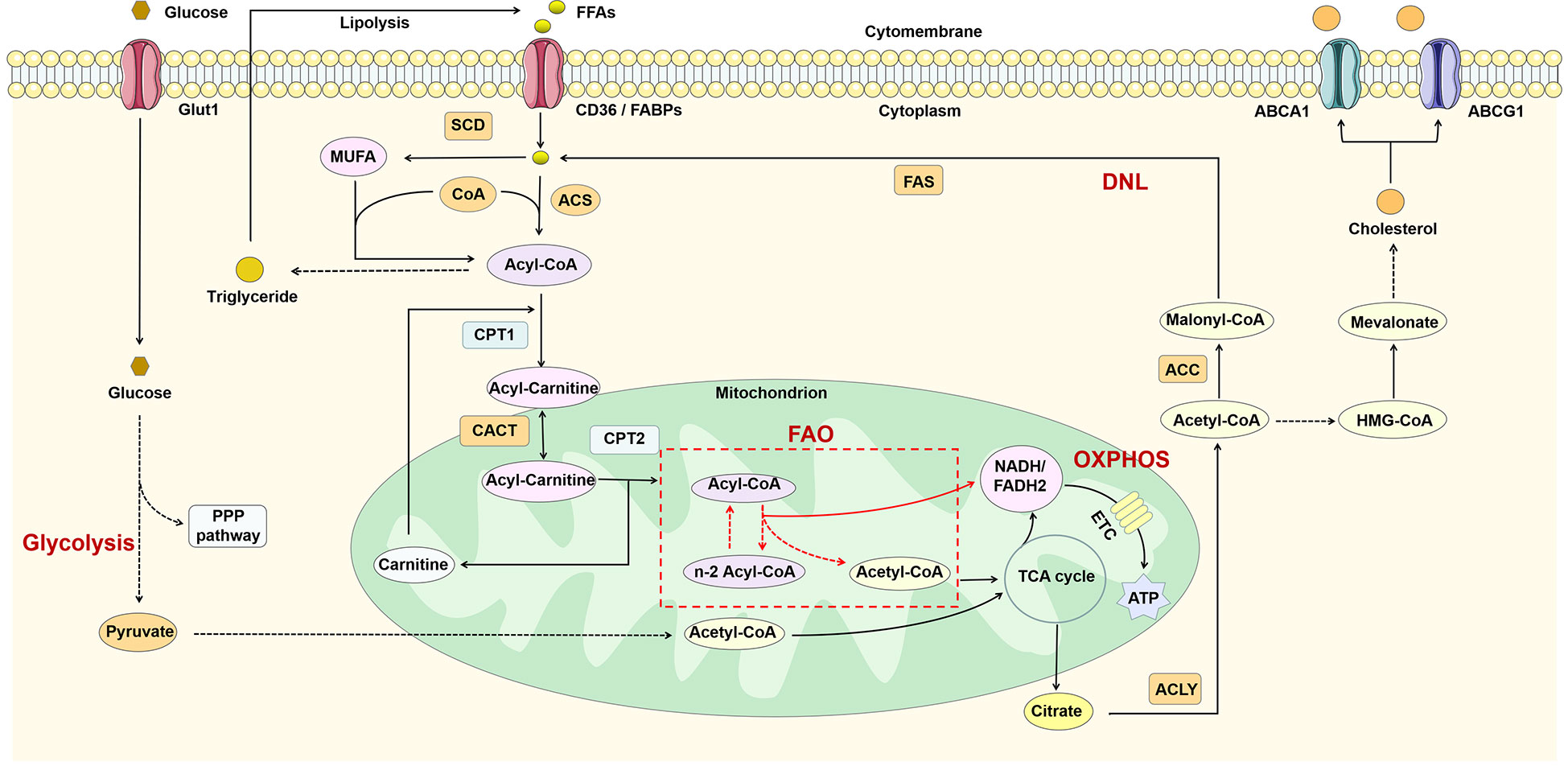
Figure 2 Lipid metabolism in macrophages. FFAs are catabolized to acyl-CoA by the action of ACS and thus participate in triglyceride synthesis and FAO. FAO can participate in the TCA cycle by producing acetyl-CoA and also promote OXPHOS by increasing NADH/FADH2 production. In addition, glucose can also produce acetyl-CoA through glycolysis. The citrate generated by the TCA cycle is reconverted to acetyl-CoA in the cytoplasm by ACL, thus participating in DNL and cholesterol synthesis. ABCA1, ATP-binding cassette sub-family A member 1; ABCG1, ATP-binding cassette sub-family G member 1; ACC, acetyl-CoA carboxylase; ACLY, ATP-citrate lyase; ACS, acyl-CoA synthetase; CACT, carnitine-acylcarnitine translocase; CoA, coenzyme A; CPT1, carnitine palmitoyl transferase 1; DNL, de novo lipogenesis; ETC, electron transport chain; FAO, fatty acid oxidation; FAS, fatty acid synthase; FFAs, free fatty acids; Glut1, glucose transporterisoform 1; MUFA, monounsaturated fatty acid; OXPHOS, oxidative phosphorylation; PPP pathway, pentose phosphate pathway; SCD, stearoyl-coenzyme A desaturase; TCA, tricarboxylic acid.
In M1 macrophages, increased glycolysis not only provides ATP more rapidly, but also promotes the TCA cycle and acetyl-CoA production (43). Sterol regulatory element-binding proteins (SREBPs) and fatty acid synthase (FAS), both key regulators of fatty acid synthesis, not only promote lipid biosynthesis in macrophages, but have also been shown to be critical for the induction of M1 polarization of macrophages (44–46). Different from M1, M2 macrophages possess an intact TCA cycle and enhanced mitochondrial OXPHOS, which depends on fatty acid uptake and FAO (43). CPT1 is involved in the production and mitochondrial transport of fatty acid-derived acyl-carnitine. Inhibition of CPT1 blocks mitochondrial FAO and has been shown to inhibit IL-4-mediated M2 polarization of macrophages (47). Thus, classical LPS/IFN-γ-activated pro-inflammatory M1 macrophages in vitro exhibit enhanced glucose uptake and anaerobic glycolysis, whereas anti-inflammatory M2 macrophages induced by IL-4/IL-13 exhibit enhanced FAO and OXPHOS (Figure 3). Lipid metabolism in M1 macrophages favors lipid synthesis and proinflammatory factor expression, whereas M2 macrophages receive their energy supply through FAO (48, 49). It has been shown that inhibition of FAO inhibits M2 polarization in macrophages (50), while LPS in combination with IFNγ inhibits IL-4-induced M2 repolarization by suppressing OXPHOS in macrophages (51). Due to the high plasticity, macrophages can be phenotypically “repolarized” or “reprogrammed” when induced by the corresponding signals (16). Thus, metabolic regulation may be central to the functional plasticity of macrophages, and FAO plays an essential role in inflammatory and metabolism-mediated phenotypic changes in immune cells.
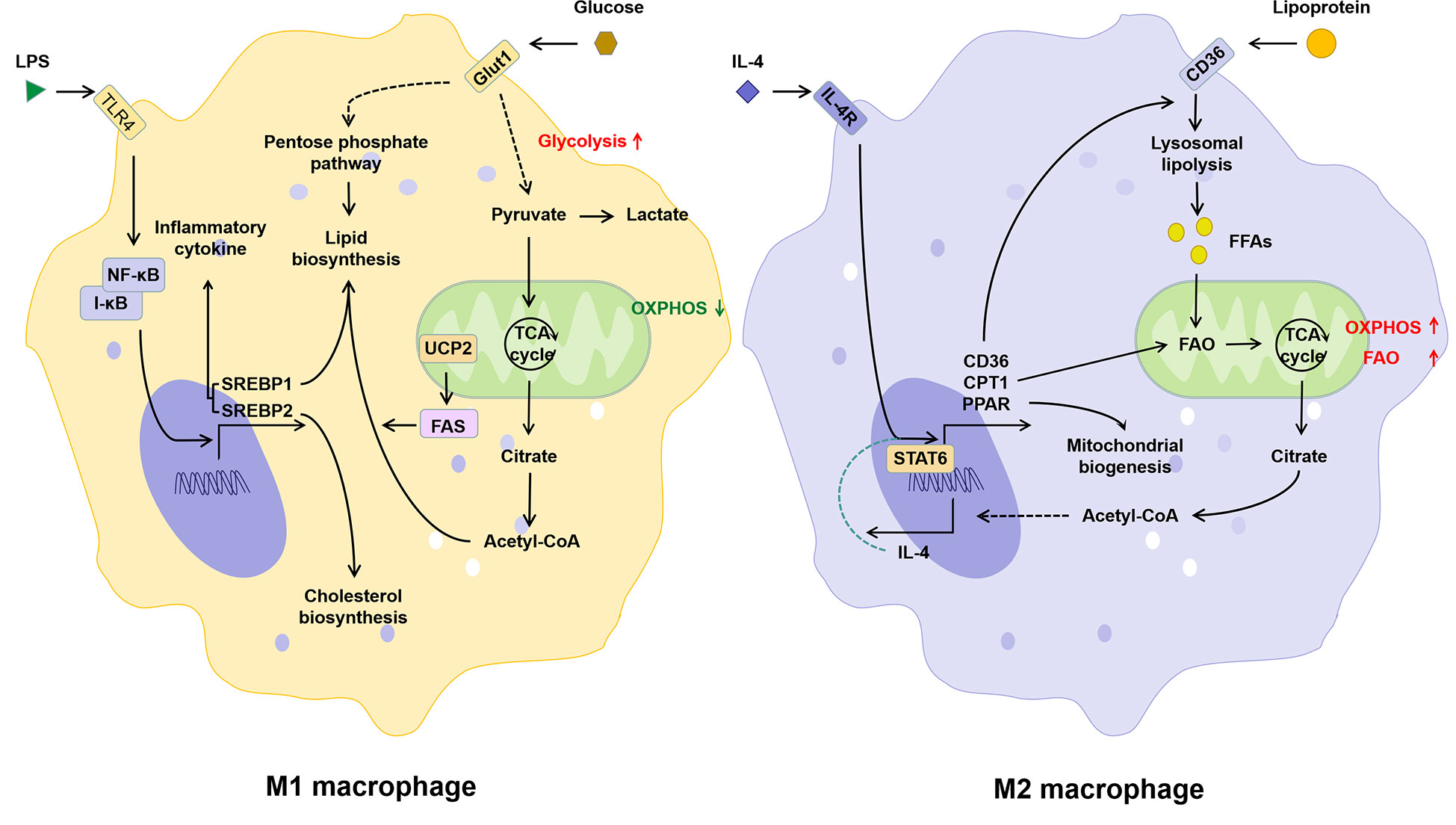
Figure 3 The relationship between lipid metabolism and phenotype of macrophages. Activation of the TLR signaling pathway induced by LPS promotes M1 polarization of macrophages, which is manifested by increased glucose uptake, glycolysis, lipid biosynthesis and decreased OXPHOS. The IL-4 induced M2 polarization of macrophages is manifested as the increase in lipid intake, FAO, and OXPHOS. IL-4, interleukin-4; IL-4R, interleukin-4 receptor; I-κB, inhibitor of NF-κB; LPS, lipopolysaccharide; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PPAR, peroxisome proliferative activated receptor; SREBP, sterol-regulatory element binding protein; STAT6, signal transducer and activator of transcription 6; TLR4, toll-like receptor 4; UCP2, uncoupling protein 2.
The liver is an important organ of lipid metabolism. Based on the close relationship between macrophage lipid metabolism and inflammation, dysregulated lipid metabolism in NAFLD may promote the progression of inflammation by affecting macrophage function. Modulation of lipid metabolism may have potential therapeutic implications for the progression of inflammation in NAFLD by reshaping the M1/M2 balance. Therefore, it is necessary to further clarify the changes in lipid metabolism of macrophages in NAFLD. KCs express receptors such as MSR1, CD36, and TIM4, which recognize and remove membrane lipid components of apoptotic cells and circulating oxidized low-density lipoproteins (ox-LDL). Subsequently, these lipids are degraded to FFAs and cholesterol by lysosomal acid lipase (LAL), which is involved in HDL synthesis (52). This function of KCs is highly conserved in several species (53), and their expression of genes related to lipid metabolism are more abundant than in other tissue-resident macrophages (54). Given the role of KCs in hepatic immune homeostasis, they may maintain specific functions, particularly tissue-specific functions, dependent on certain metabolites or nutrients (55). Imbalance of lipid homeostasis may lead to pro-inflammatory polarization of KCs thereby inducing inflammation in NAFLD. Improvement of NASH by KCs elimination provides evidence for its driving effect on early NASH (20). CD11c+ macrophages may be an important subset driving hepatocyte death-induced inflammation and fibrosis, which promotes disease progression from steatosis to NASH, while KCs are a major source of CD11c+ macrophages (56). Macrophage scavenger receptor 1 (MSR1, CD204) mediates lipid uptake and accumulation in KCs and correlates with the degree of steatosis and steatohepatitis in patients with NAFLD. In a fatty acid-rich environment, MSR1 induces a pro-inflammatory response through the JNK signaling pathway, and its blockade inhibits lipid accumulation in KCs, thereby suppressing their pro-inflammatory polarization and the release of cytokines such as TNF-α (57). Similarly, the knockdown of myeloid forkhead box O1 (FoxO1) induced a shift in macrophage polarization from a pro-inflammatory M1 to an anti-inflammatory M2 phenotype and reduced liver macrophage infiltration in a mouse model of high-fat diet-induced NASH (58). At least for now, it seems that lipid overload may promote M1 polarization of KCs and MoMFs through MSR1 and FoxO1, respectively.
Nuclear receptors, including PPARs and liver X receptor (LXR), are ligand-dependent transcription factors and involved in the regulation of lipid and glucose metabolism genes and inflammation-regulated genes (59). This provides another link between macrophage lipid metabolism and inflammation. The three isoforms of PPARs, PPARα, β/δ, and γ, play different but complementary regulatory roles in lipid metabolism and inflammation in the liver (48). The current studies have confirmed the promotion and necessity of PPARγ and PPARβ/δ on M2 polarization of macrophages (60, 61). And LXR can also induce the anti-inflammatory phenotype of MoMFs by inhibiting TLR2, TLR4 and TLR9 and related pathways (62). Interestingly, although LXR was expressed in both hepatocytes and macrophages, an LXRα agonist, DMHCA, selectively activated LXRα in macrophages (63), suggesting the feasibility of targeting LXR in macrophages. In addition, retinoic acid-related orphan nuclear receptor alpha (RORα) is also thought to regulate M2 polarization in macrophages in NAFLD (64). However, the significance of RORα-specific deletion in macrophages for NASH progression remains controversial (65). In addition to regulating lipid metabolism and inflammation, nuclear receptors can also act as redox sensors to sense metabolic stress and thus prevent oxidative damage (66). Due to the important role of oxidative stress in promoting lipid metabolism disorders and inflammation, targeting nuclear receptors may improve NASH progression by regulating macrophage phenotype through antioxidant, anti-inflammatory, and regulating lipid metabolism.
Although the current study confirms the importance of lipid metabolism reprogramming on the pro-inflammatory phenotype of macrophages, changes in the phenotype and lipid metabolism of hepatic macrophage subsets need to be further explored. A recent study showed that depletion of a subset of CD206hi ESAM+ KCs or silencing of their fatty acid transporter protein CD36 reversed obesity and steatosis in mice (67). CD36 and CD206 are both commonly considered to be M2 markers (68). These conflicting results further demonstrate the complexity of macrophage function. Furthermore, the role of lipid metabolism in macrophage polarization remains controversial, although it is now generally accepted that glycolysis defines M1 macrophages and FAO defines M2 (43). Glycolysis, OXPHOS and FAO may determine phenotypic shifts through complex interactions rather than a single pathway, which remains to be elucidated.
Cells may die from accidental cell death (ACD) or regulatory cell death (RCD). RCD is a strictly regulated form of cell death induced by complex molecular mechanisms, and it is also known as PCD when it occurs in the absence of external environmental interference (69). PCD plays an important role in host defense against pathogens and maintenance of body homeostasis, while its over-activation or tolerance leads to the development of disease. Several types of PCD have been identified including autophagy, apoptosis, necroptosis, pyroptosis, and ferroptosis (Figure 4). We focused on these types of PCD in macrophages in the NASH phase of NAFLD.
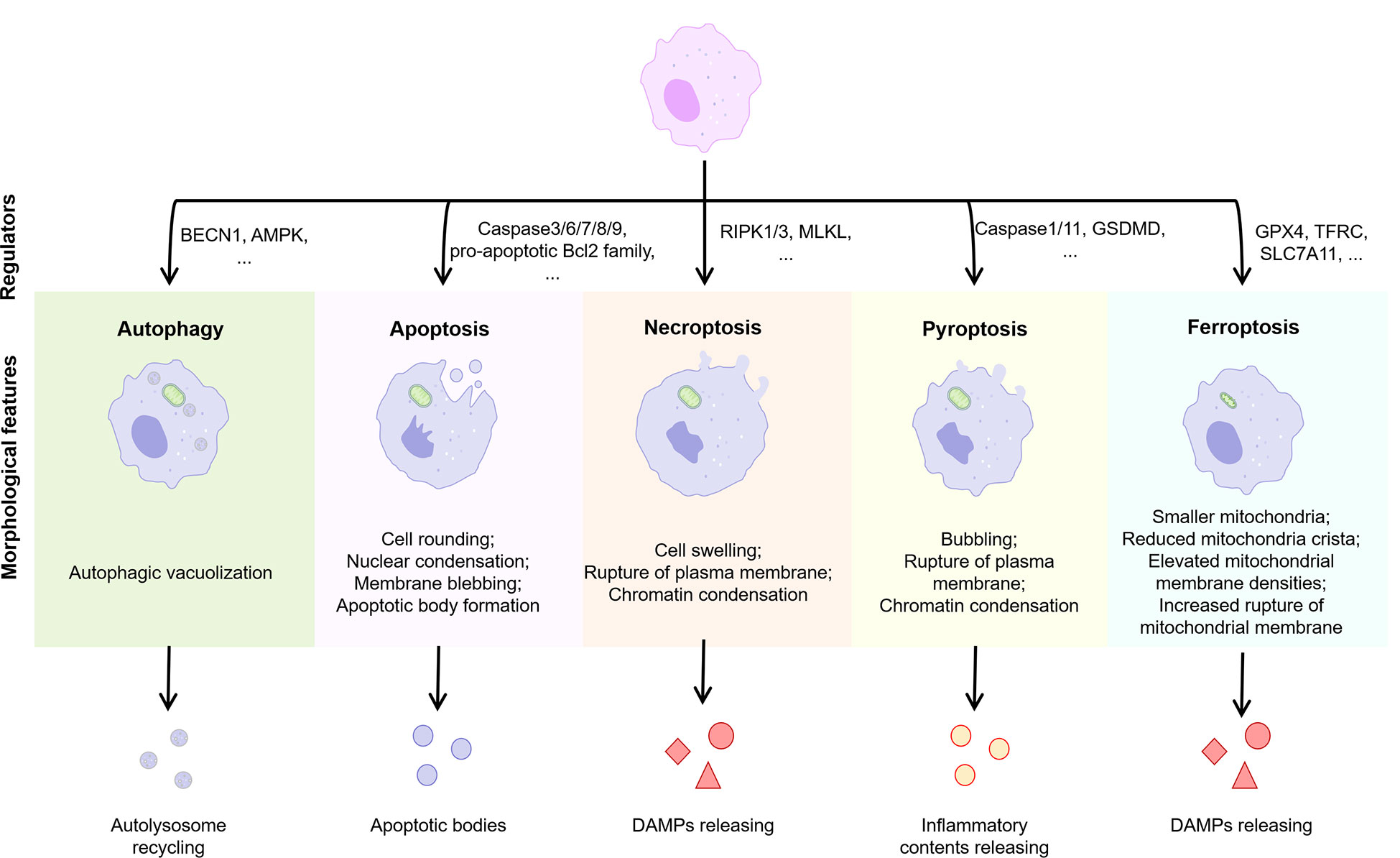
Figure 4 Features of different types of PCD. Normal cells are induced to different forms of death by different stimuli or signals, including autophagy, apoptosis, necroptosis, pyroptosis, and ferroptosis. Different PCD types exhibit different morphological features and play an important regulatory role in the progression of inflammation. BECN1, beclin 1; AMPK, adenosine 5’-monophosphate (AMP)-activated protein kinase; RIPK, receptor-interacting protein kinase; MLKL, mixed lineage kinase domain-like; GSDMD, gasderminD; DAMPs, damage associated molecular patterns; GPX4, glutathione peroxidases 4; TFRC, transferrin receptor; SLC7A11, solute carrier family 7 member 11.
Autophagy is a catabolic process that degrades damaged organelles and abnormally accumulated proteins via lysosomes (70). Autophagy can be classified as macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy according to the pathways of cytoplasmic materials into lysosomes (71). In addition, autophagy can be further classified into selective and non-selective autophagy according to the specificity of degradation substrates. Physiological autophagy is essential for maintaining cellular homeostasis, and its regulation of macrophage function is widely recognized (72). It has been demonstrated that autophagy is impaired in human livers with steatosis or NASH and that promoting autophagy inhibits the pro-inflammatory activation of human macrophage cell lines (73). Typically, autophagy of macrophages in most NAFLD-related studies refers to macroautophagy, the lack of which amplifies hepatic steatosis and/or liver injury through overactivation of the innate immune response. Macrophage-specific deletion of macroautophagy-dependent autophagy-associated proteins such as autophagy protein 5 (ATG5) was shown to increase IL1α and IL1β secretion thereby exacerbating liver inflammation and fibrosis in mice (74). It is suggested that autophagy contributes to the down-regulation of macrophage-induced inflammatory responses, whereas insufficient autophagy may lead to macrophage polarization toward pro-inflammatory M1. Dysbiosis of intestinal flora and increased intestinal permeability in NAFLD patients lead to elevated serum LPS levels, which induce increased secretion of pro-inflammatory factors and reactive oxygen species (ROS) through binding to TLR4, a pattern recognition receptor on the membrane surface of macrophages (75). High concentrations of ROS activated the oxidative stress regulatory switch nuclear factor erythroid 2-related factor 2 (Nrf2) and further induced transcription of antioxidant genes and sequestosome 1 (SQSTM1, also known as p62), thereby inducing p62-dependent selective autophagy in macrophages (76). In contrast, autophagy-deficient hepatic macrophages promoted liver inflammation and fibrosis by enhancing the mitochondrial ROS (mtROS)/NF-κB/IL-1α/β pathway (77).
As a cellular energy sensor, adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) can promote macrophage M2 polarization through mitochondrial autophagy (78). However, both free fatty acids and hyperglycemia can lead to autophagy deficiency through the inhibition of AMPK, thereby inhibiting macrophage M2 polarization (79, 80). A study based on liver-specific SQSTM1 knockout mice showed that SQSTM1 induces hepatocyte autophagy by promoting the interaction between AMPK and unc-51 Like Autophagy Activating Kinase 1 (ULK1) and further activates the kelch-1ike ECH- associated protein 1 (Keap1)-Nrf2 signaling pathway to protect the liver from lipotoxicity in mice (81). As an important regulator of nutrient perception, growth, and metabolism, the mammalian target of rapamycin complex (mTORC) phosphorylates ULK1 to inhibit the interaction between AMPK and ULK1 (82), exhibiting a regulatory role in autophagy in contrast to SQSTM1. In addition, AMPK can also inhibit mTORC1 function by phosphorylating Raptor in the mTORC1 complex (83). These studies suggest an antagonism between AMPK and mTORC1 on the regulatory function of autophagy. However, previous studies have demonstrated that SQSTM1 also interacts with Raptor to activate mTORC1 (84). Persistent deficiency of mTORC1 in macrophages has also been shown to inhibit their M2 polarization by inducing lysosomal dysfunction (85). The role of mTORC1 in the regulation of macrophage autophagy and phenotype remains puzzling.
Lipid deposition in the liver impairs local oxygen homeostasis, followed by tissue hypoxia-induced adaptive responses that ultimately affect the homeostasis of hepatic lipid metabolism (86). Since oxygen is transported through the blood, hypoxia means impaired blood microcirculation. Therefore, hypoxia and nutritional disorders occur simultaneously. Nutrient depletion caused by hypoxia activates AMPK and simultaneously inactivates mTORC1 thereby inducing autophagy to maintain cell survival. As a key regulator of hypoxia, Hypoxia-inducible factor 1 subunit alpha (HIF-1α) can broadly regulate the expression of hypoxia-inducible genes and the activation of various signaling pathways (87). A study based on methionine and choline-deficient L-amino acid diet (MCD)-fed mice and NASH patients showed that HIF-1α expression in hepatic macrophages was induced by palmitic acid, thereby reducing autophagic flux and targeting inflammasomes to increase IL-1β production (88). Although HIF-1α and AMPK/mTORC1 regulate macrophage autophagy and phenotypic transformation as important links of hypoxia and energy regulation, respectively, their interactions are still not systematically elucidated.
Currently, studies on HIF-1α and AMPK regulation of lipid metabolism in NAFLD are mainly carried out in hepatocytes (89, 90). The mechanisms by which lipid metabolism reprogramming in macrophages regulates autophagy are still poorly understood. Monoacylglycerol lipase (MAGL), the rate-limiting enzyme in the degradation of monoacylglycerols, not only degrades triacylglycerols to free fatty acids and glycerol, but also metabolizes endogenous cannabinoid receptor ligand 2-arachidonyl acid glycerol to arachidonic acid (91). During chronic liver injury, MAGL inhibited macrophage autophagy to promote inflammation and fibrosis, whereas inhibition of MAGL reduced the number of Ly-6Chi macrophages and increased the number of Ly-6Clo macrophages in an autophagy-dependent manner (92). In high-fat diet-induced obese mice, MAGL deletion also exhibited improved inflammation and insulin resistance in adipose tissue and reduced triglyceride levels in the liver (93). This provides evidence for a link between macrophage autophagy and lipid metabolism reprogramming. In addition, macrophage autophagy reduces programmed death ligand 1 (PD-L1) expression and thus inhibits hepatocellular carcinogenesis, while defective autophagy induces its immunosuppressive phenotype and thus promotes hepatocellular carcinoma progression (94). Given the increased incidence of NAFLD-associated HCC, regulation of macrophage autophagy may be valuable in reducing NAFLD-associated HCC.
Apoptosis is a Caspase-dependent cell death that includes intrinsic pathways induced by DNA damage, ROS accumulation, and endoplasmic reticulum stress (also known as the mitochondrial pathway) as well as exogenous apoptotic pathways initiated by the binding of other cellular soluble or cell surface ligands including TNF, FasL, or TRAIL to death receptors (95). The Caspase family is a family of evolutionarily conserved cysteine-dependent endonucleases that are primarily involved in cell death and inflammatory responses. Caspases involved in apoptosis are divided into two main categories: initiating Caspases (Caspases-2, 8, 9 and 10) and effector Caspases (Caspases-3, 6 and 7) (96). Current studies have amply demonstrated that Caspases-3, 6, 7, 8, and 9 promote the progression of NASH (97). Among them, Caspases-8 is mainly involved in the exogenous apoptotic pathway, while Caspases-9 is mainly involved in the endogenous apoptotic pathway. Increased positivity of the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay in liver tissue of NASH patients demonstrates the involvement of apoptosis in the progression of NASH (98). Most of the current studies on apoptosis have been conducted on hepatocytes and few on macrophages. A study based on a high-fat diet-induced mouse model of NAFLD and primary KCs demonstrated that IL-4-activated M2 KCs could release IL-10 to promote apoptosis of M1 KCs thereby reducing liver inflammation and hepatocyte injury in NAFLD (99). This study suggests an intrinsic regulatory mechanism for the balance of M1 and M2 macrophages, i.e., M2 macrophages induce protective apoptosis of M1 macrophages and their dysfunction leads to the accumulation of M1 macrophages in the liver and the progression of inflammation.
Endoplasmic reticulum stress is significantly associated with lipotoxicity and NASH (100). C/EBP homologous protein (CHOP) is a transcription factor downstream of protein kinase RNA-activated-like ER kinase. CHOP is induced by endoplasmic reticulum stress and its deletion prevents apoptosis induced by endoplasmic reticulum stress (101). As endoplasmic reticulum stress can induce TRAIL receptors and activate Caspase-8 (102), this may string endogenous apoptotic and exogenous apoptotic pathways together. Early studies demonstrated that CHOP deficiency promotes macrophage resistance to lipotoxicity and the progression of inflammation in NAFLD (103). Therefore, CHOP may have the effect of inducing apoptosis of M1 macrophages and thus regulating hepatic macrophage homeostasis to protect the liver from steatohepatitis. The role of LXRα in promoting M2 polarization in hepatic macrophages has been discussed above. Paradoxically, a peritoneal macrophage-based study demonstrated that LXRα can inhibit the CHOP pathway induced by endoplasmic reticulum stress and thus inhibit macrophage apoptosis (104). The relationship between CHOP and LXRα in M1 macrophages in the liver obviously needs to be further explored.
The suppressor of cytokine signaling (SOCS) family is a class of proteins with negative feedback regulation on cytokine signaling pathways, including eight members of SOCS1 to SOCS7 and cytokine-inducible SH2-containing protein (CIS) (105). Currently, SOCS1, SOCS2 and SOCS3 are the most studied in macrophages. IL-4-induces SOCS2 expression, IFN-γ induces SOCS3 expression, while SOCS1 can be induced by both (106). Differently, induction of SOCS1 by IL-4 is signal transducer and activator of transcription 6 (STAT6) dependent, whereas induction of SOCS1 by IFN-γ is STAT1 dependent. At the same time, IL-4 induced SOCS1 can inhibit the expression of STAT6 and form a negative feedback signal. A recent study showed that SOCS2 expression in macrophages was negatively correlated with the degree of NASH (107). The study also found that SOCS2 plays a role in inhibiting inflammation and apoptosis via NF-κB and inflammasome signaling pathway in macrophages during NASH. In addition, SOCS1 and SOCS3 have been shown to inhibit the exogenous apoptotic pathway in other cell lines such as renal tubular epithelial cells and prostate cancer cells, respectively (108, 109). Overall, SOCS2 may promote M2 polarization and inhibit apoptosis in macrophages, while SOCS1 and SOCS3 may promote M1 polarization and inhibit apoptosis. The three are induced by different signals to inhibit exogenous apoptosis in macrophages of the corresponding phenotype thereby regulating the progression of NASH. Notably, both SOCS1 and SOCS3 have been shown to have inhibitory effects on FAO (110–112). Although these studies were not performed in macrophages, the mechanisms all involved inhibition of the Janus kinase (JAK)/STAT pathway. Different from SOCS1 and SOCS3, the effect of SOCS2 on lipid metabolism has received rarely attention. Although it has been shown to prevent hepatic steatosis due to high-fat diet (113), some studies suggest that it does not promote the increase of FAO in the liver (114). In conclusion, it remains to be clarified whether the SOCS family is involved in the regulation of macrophage polarization and apoptosis by inhibiting FAO through the JAK/STAT pathway.
Necroptosis is a caspase-independent necrotic cell death program regulated by receptor interacting serine/threonine kinase 1 (RIPK1) and RIPK3. It can be triggered by extracellular stimuli that activate inflammation and cell death. Various innate immune signaling pathways such as TNFR, TLR and interferon receptors (IFNRs) can induce the binding of RIPK1 and RIPK3, which then leads to phosphorylation and translocation of mixed lineage kinase domain-like pseudokinase (MLKL) to the cell membrane, ultimately leading to necroptosis and release of damage-associated molecular patterns (DAMPs) (115). Precisely, RIPK1 induces apoptosis rather than necroptosis when Caspase-8 is present. When Caspase-8 is inhibited, deubiquitinated RIPK1 does not bind to complex 2, but to RIPK3, forming a necrosome complex and subsequently recruiting and activating MLKL (116). Thus, inhibition of Caspase-8 is as important as activation of RIPK3 for necroptosis. In contrast, RIPK1, although involved in the induction of necroptosis, may not be necessary for necroptosis. Depletion of nicotinamide adenine dinucleotide (NAD+) is sufficient to trigger necroptosis in a RIPK3- and MLKL-dependent manner (117), which provides a non-classical necroptotic pathway.
Necroptosis plays an important role in macrophage polarization and inflammation. A study based on aged mice showed that aging led to increased necroptosis in liver macrophages and release of pro-inflammatory factors including TNFα, IL-6 and IL-1β, while necrostatin-1s, a necroptosis inhibitor, significantly reduced M1 macrophages and improved inflammation (118). This study suggests that necroptosis promotes M1 polarization of macrophages. O-acetylglucosamine glycosylation modification (O-GlcNAcylation) is a specific glycosylation modification of intracellular proteins that can affect the localization, function and stability of substrate proteins (119). LPS-activated M1 macrophages exhibit attenuated hexose biosynthetic pathway and protein O-GlcNAcylation, while O-GlcNAc transferase (OGT) mediated O-GlcNAcyclization of RIPK3 prevented the hetero interaction and homo interaction of RIPK3-RIPK1, thereby inhibiting macrophage necroptosis (120). A study based on patients with NASH and choline-deficient L-amino acid-defined diet (CDAA)-induced NAFLD models in mice showed that hepatic RIPK3 correlated with NAFLD severity in humans and mice and RIPK3 deficiency ameliorated CDAA-induced inflammation, fibrosis and carcinogenesis in mice (121). Even though RIPK3 has now emerged as one of the promising targets for the treatment of NASH, the mechanism by which necroptosis regulates macrophage polarization has not been systematically elucidated. Studies based on the RIP3-deficient mouse model demonstrated that RIPK3 promotes the TLR4-NF-κB pathway via Rho-associated coiled-coil-containing protein kinase (ROCK)1 and thereby induces M1 polarization of macrophages in the liver (122). The effect of necroptosis on M1 macrophage activation and pro-inflammation may also be paracrine-related. Interestingly, M1 but not M2 macrophages exhibited higher RIPK3 and MLKL expression when BMDMs were intervened with necroptosis inducers (123). In contrast, another study expressed a different result. Blockade of TAK1, the RIPK1 inhibitor, induced more intense necroptosis in M2 but not M1 peripheral blood monocyte-derived macrophages in the context of Caspase inhibition (124). The mechanisms underlying these different results are still unclear. Necroptosis may be involved in both depletion of M2 macrophages and activation of M1 to promote the progression of inflammation in NASH.
There may be a correlation between the necroptosis of macrophages and their lipid metabolism reprogramming. RIPK3 was downregulated in macrophages in HCC and promoted FAO via the ROS-Caspase1-PPAR pathway, which induced the reprogramming of fatty acid metabolism and ultimately induced M2 polarization in tumor-associated macrophages (125). An atherosclerosis-based study found that MLKL deficiency exacerbated lipid accumulation despite reducing the occurrence of necroptosis in macrophages (126). Another study based on the high-fat diet-induced NAFLD model in mice also showed that RIPK3 deficiency inhibited inflammation while exacerbating hepatic steatosis (127). These studies suggest that the inhibition of necroptosis may promote increased lipid uptake or de novo synthesis of fatty acids. Therefore, the relationship between necroptosis and the reprogramming of lipid metabolism in macrophages remains to be further clarified.
Pyroptosis is a form of PCD mediated by inflammasome activation, which is manifested by continuous cellular distension until the cell membrane ruptures, thereby releasing cellular contents to activate an intense inflammatory response. Classical pyroptosis is regulated by the inflammasome composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), pro-Caspase-1 and gasdermin D (GSDMD) (128). Various exogenous and endogenous signals including LPS and ATP can induce inflammasome formation followed by activation of Caspase-1. Activated Caspase-1 cleaves pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18, while cleaving the pyroptotic substrate GSDMD and forming membrane pores to induce pyroptosis and releasing IL-1β and IL-18 (129). The non-classical pyroptosis pathways are cytoplasmic LPS-mediated activation of Caspase-4/5/11 and cleavage by GSDMD (130). Xu et al. (131) showed that GSDMD and its fragment GSDMD-N protein expression, which induce pyroptosis, were significantly increased in liver tissues of NAFLD/NASH patients and correlated with NAFLD activity score and fibrosis. In contrast, MCD-fed GSDMD-/- mice were free from steatohepatitis and fibrosis, demonstrating the role of GSDMD-mediated pyroptosis in promoting NASH. In addition to GSDMD, most members of the gasdermin family can also induce pyroptosis (132), but their role in NAFLD still needs further evaluation.
Most studies on pyroptosis have focused on hepatocytes, but inflammasomes are mainly expressed in immune cells, especially macrophages (133). As an important regulator of anti-inflammation and antioxidant, Nrf2 is downregulated in the liver of NASH patients (134). Macrophage-specific Nrf2 knockdown promotes ROS and IL-1β production via a YAP-NLRP3-dependent manner thereby exacerbating NASH progression (135), suggesting the promotion of macrophage pyroptosis on NASH progression. Meanwhile, the gasdermin family may also induce mtROS release by targeting the mitochondrial membrane, thereby triggering NLRP3 inflammasome activation (136, 137). These studies suggest that pyroptosis may promote NASH progression by inducing ROS release and thus amplifying the cascade of pyroptosis and inflammation. Given the regulation of pyroptosis by the NLRP3 inflammasome, inhibition of the NLRP3 inflammasome may attenuate the inflammatory response of liver tissue by inhibiting macrophage pyroptosis (138). NLRP3 blockade also shows improvement in liver inflammation and fibrosis in atherogenic diet-fed foz/foz mice with NASH (139). In addition to the NLRP3 inflammasome, other inflammasome complexes, such as the NLR family CARD domain containing 4 (NLRC4) inflammasome, can also be involved in NAFLD inflammatory progression by promoting macrophage pyroptosis (140). In conclusion, inflammasome activation induces macrophage pyroptosis on the one hand and mediates macrophage polarization on the other. This provides evidence for a close relationship between liver macrophage pyroptosis and pro-inflammatory polarization in NASH.
As mentioned above, macrophage polarization is regulated by disorders of lipid metabolism. Previous studies have demonstrated that lipids released from dead hepatocytes in NASH activate macrophages to overexpress NLRP3 inflammasome and Caspase-1 (141). Therefore, macrophage pyroptosis may be associated with lipid metabolism. Bile acids are endogenous ligands for nuclear receptors that regulate lipid and energy metabolism (142). As a bile acid receptor, G protein-coupled bile acid receptor 1 (GPBAR1, also known as TGR5)-mediated bile acid signaling plays a key role in integrating glucose, lipid and energy metabolism (143). TGR5 activates PPARα and PPAR-γ coactivator 1 alpha (PGC-1α) to increase mitochondrial oxidative phosphorylation and energy metabolism and inhibit NF-κB-mediated pro-inflammatory cytokine production (144, 145), which is important for the metabolic reprogramming of M2 macrophages. Shi et al. (146) found that TGR5 expression was significantly reduced in the liver tissue of NASH patients and mouse models, while TGR5 knockdown exacerbated liver injury and inflammation and promoted macrophage M1 polarization in mice. Mechanistically, TGR5 signaling inhibits NLRP3-mediated macrophage M1 polarization thereby ameliorating hepatic steatosis and inflammation. Although the available evidence suggests an association between macrophage pyroptosis and lipid metabolism, the regulatory mechanisms are still poorly understood.
Ferroptosis is a form of iron-dependent cell death mediated by lipid peroxidation, whose main biochemical features are iron deposition and lipid peroxidation (147). Both increased iron uptake and decreased iron excretion may lead to iron overload, which in turn leads to excessive ROS production and lipid peroxidation through Fenton reaction and enzymatic oxygenation, subsequently triggering ferroptosis. The liver is one of the most important organs for iron storage and metabolism. Due to the abnormal lipid deposition in the liver of NASH patients, this may promote the development of ferroptosis. The correlation between disease progression and liver iron overload in NAFLD patients has been demonstrated (148). A bioinformatics study showed that the grading of liver steatosis was associated with 8 iron death-related genes including ACSL3, ACSL4, AKR1C1, AKR1C2, CS, FADS2, GSS and PGD (149). Another study also showed that the expression of SLC11A2, CP, SLC40A1, and ACSL5 was downregulated in the livers of NASH patients compared to healthy livers, while the expression of FTL, FTH1, ACSL4, and ACSL6 was upregulated (150). Actually, targeted ferroptosis has been shown to improve inflammation in both MCD and choline-deficient, ethionine-supplemented (CDE) diet-induced NASH models in mice (151, 152). Current studies on the role of ferroptosis in NAFLD progression have focused on hepatocytes and HSCs (153). Since liver iron is mainly distributed in hepatocytes and reticuloendothelial system (macrophages), iron deposition in macrophages may play a role in NASH. An earlier multicenter study that included 849 patients with NAFLD has also demonstrated that iron deposition in macrophages is associated with severe NASH and advanced liver histological features (154).
Previous studies have confirmed that iron chelation in M1 macrophages may contribute to the development of chronic inflammation, while iron export from M2 macrophages may promote the growth of adjacent cells in the microenvironment (155), suggesting that macrophage polarization is associated with altered iron metabolism. A study based on BMDMs showed that iron overload increased the levels of M1 products (e.g. IL-6, TNF-α and IL-1β), promoting their polarization to the M1 type while exacerbating steatohepatitis and liver fibrosis (156). This study also showed that iron overload inhibited M2 polarization in BMDMs in the presence of IL-4. This differential performance may be associated with higher expression of Hamp and ferritin heavy chain (FTH)/ferritin light chain (FTL) and lower expression of ferroportin (FPN1, also known as SLC40A1) and iron regulatory proteins 1/2 (IRP1/2) in M1 macrophages compared to M2 (157). Moreover, the stronger antioxidant capacity of M1 macrophages enhanced their resistance to iron overload (158). Thus, under the same conditions, M2 macrophages may be induced to die, while the M1 type survives. As iron may be involved in the regulation of energy production and amino acid catabolism, the regulation of iron metabolism in polarized macrophages may alter the macrophage phenotype. For example, anti-inflammatory M2 macrophages in the tumor microenvironment can be converted to pro-inflammatory M1 macrophages via ferroptosis (159, 160). However, whether macrophages have this property in NASH remains to be elucidated. Notably, a study based on human monocytic leukemia THP-1 cell-derived macrophages showed that macrophages exhibit M2 polarization rather than M1 in response to chronic iron overload (161), but this may be related to metabolic changes in tumor-associated macrophages.
There is also a connection between ferroptosis and lipid metabolism, which has been well summarized in a recent review (162). Ferroptosis is characterized by iron-dependent peroxidation with phospholipids containing polyunsaturated fatty acyl (PUFA) chains as substrates. Monounsaturated fatty acids (MUFAs), as inhibitor of iron death (163), can be synthesized de novo in cells and participate in membrane lipid composition. Lipid metabolism may control the composition of membrane lipid by regulating the balance of PUFAs and MUFAs.Interestingly, sterol-regulatory element binding protein 1 (SREBP-1), which regulates MUFAs synthesis, is upregulated in M1 macrophages. One possible explanation is that ferroptosis signaling induces M1 polarization in macrophages, while reprogramming of lipid metabolism increases their resistance to ferroptosis. Similarly, not only fatty acid β-oxidation is increased in M2 macrophages, but also lipid transport proteins such as CD36 are upregulated. Given that fatty acid β-oxidation reduces the accumulation of PUFAs and thus inhibits lipid peroxidation (164), and that CD36-mediated lipid uptake increases susceptibility to ferroptosis (165), the ferroptosis-mediated shift in macrophage phenotype may ultimately depend on the disruption of the balance between PUFAs and MUFAs.
Due to the complex signal environment in the body, there is significant crosstalk between different types of PCD (Figure 5). As one of the main inducing pathways of exogenous apoptosis, the TNF signaling pathway also induces necroptosis. The difference is that necroptosis is RIPK3-dependent MLKL activation, while apoptosis is manifested as activation of Caspase-8. A study based on the mouse NASH model induced by MCD showed that Caspase-8 could balance the over-activation of RIPK3-dependent necroptosis, suggesting the mutual inhibition of RIPK3 and Caspase-8 (166). However, the study was performed based on hepatocytes rather than macrophages. Increased autophagy inhibits necroptosis by upregulating ATG16L1 (167) and inhibits apoptosis by inhibiting Caspase-8 activity (168). MLKL, another key regulator of necrotic apoptosis, has been demonstrated to participate in autophagy inhibition in a RIPK3-independent manner in FFC diet (high in fat, fructose and cholesterol) induced NASH mice and palmitic acid treated primary mouse hepatocytes (169). These studies suggest that Caspase-8 is a key node in balancing apoptosis and necroptosis, while MLKL may be an essential node in balancing autophagy and necroptosis. In addition to Caspase-8, Caspase-6 has also been shown to be involved in the interaction between autophagy and apoptosis. As an important participant in autophagy, AMPK can also inhibit apoptosis by phosphorylating Caspase-6 to inhibit its function (170), suggesting an antagonistic mechanism between autophagy and endogenous apoptosis. Notably, the relationship between autophagy and necroptosis is not merely antagonistic. For example, a recent study showed that RIPK3 can directly bind and activate AMPK (171). Considering the mutual inhibition of AMPK and mTOR to regulate autophagic signaling in the downstream, this may be an important link in the balance of autophagy and necroptosis. As an upstream regulator of RIPK3, RIPK1 regulates apoptosis and necroptosis through Caspase-8 and RIPK3, respectively (115). Caspase-8 has been shown to cleave GSDMD to induce pyroptosis (172). These studies all illustrate the complex interaction network among pyroptosis, apoptosis and necroptosis. In experimental and clinical NASH, RIPK1 is phosphorylated and activated mainly in liver macrophages, especially in BMDMs (173). As mentioned above, palmitic acid-induced a decrease in autophagic flux of macrophages (88). However, palmitic acid also induced the activation of RIPK1 (173). Thus, fatty acid-induced inflammatory activation of macrophages was accompanied by an inhibition of autophagy and an increase in apoptosis, necroptosis, and pyroptosis, while the predominant PCD type may be associated with different inducing factors and cytokine expression.
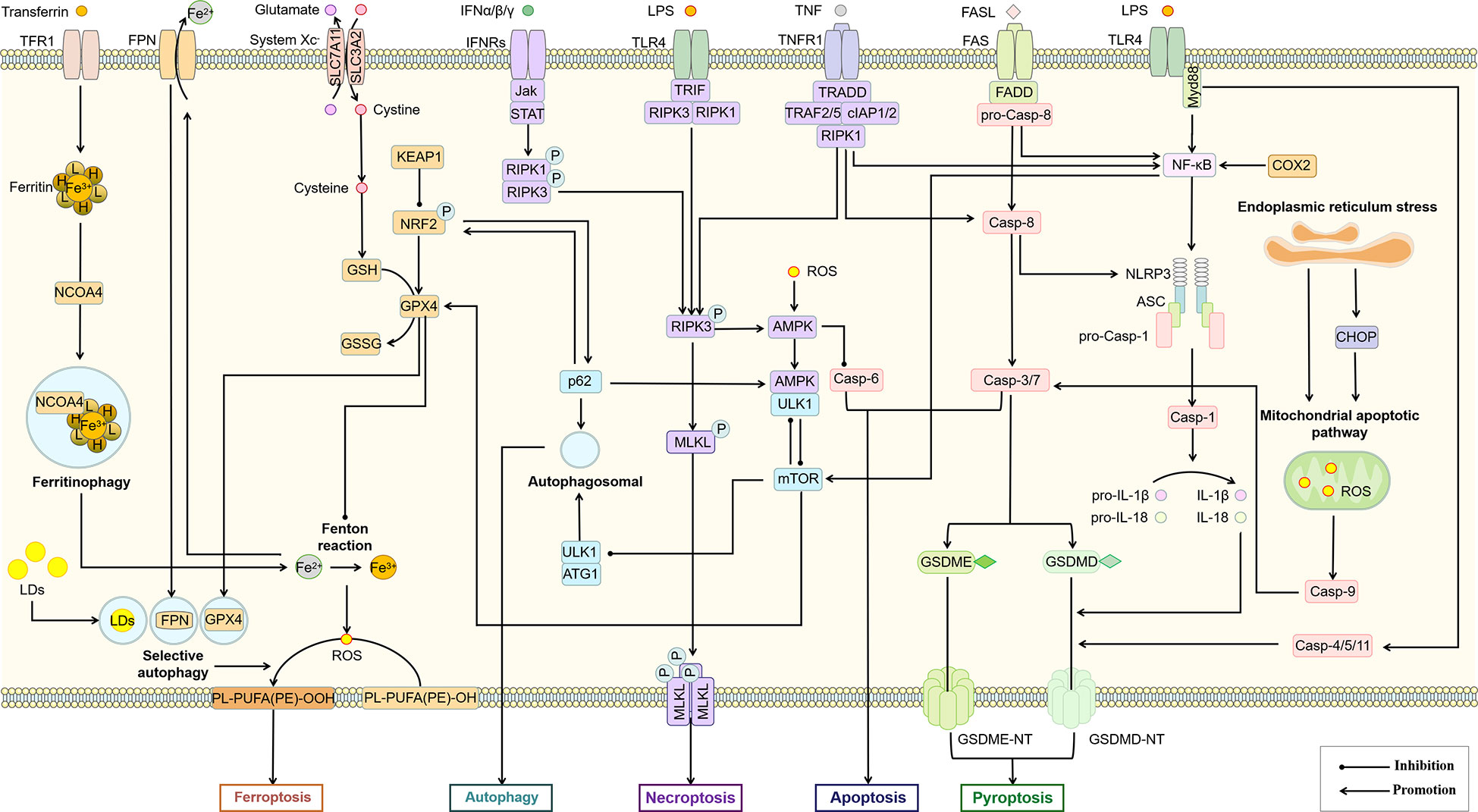
Figure 5 Association of different types of PCD in macrophages. The interaction between different PCD constitutes a complex regulatory network for survival or death of macrophages. AMPK, mTOR, Casp-8, RIPK3, Bcl-2 and p62 may be important nodes in the interaction of autophagy, apoptosis, necroptosis, pyroptosis and ferroptosis in this cell death network. ASC, apoptosis-associated speck-like protein; Casp, Caspase; CHOP, C/EBP homologous protein; cIAP, cellular inhibitor of apoptosis protein; COX-2, cyclooxygenase-2; FADD, Fas-associating protein with a novel death domain; FASL, Fas Ligand; FPN, ferroportin; GSDME, gasdermin E; GSH, glutathione; GSSG, glutathiol; IFN, interferon; Jak, janus kinase; KEAP1, kelch-1ike ECH-associated protein 1; LDs, lipid droplets; mTOR, mammalian target of rapamycin; Myd88, myeloid differentiation primary response 88; NCOA4, nuclear receptor coactivator 4; NLRP3, NOD-like receptor thermal protein domain associated protein 3; NRF2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; SLC3A2, solute carrier family 3 member 2; TFR1, transferrin receptor 1; TNFR1, tumor necrosis factor receptor 1; TRADD, TNFR1-associated death domain protein; TRAF, TNF-receptor associated factor.
Activation of the NLRP3 inflammasome is not only the initiating link of classical scorch death, but also leads to other types of PCD, including apoptosis, necroptosis, and ferroptosis (132). Prostaglandin-endoperoxide synthase 2 (PTGS2), one of the markers of iron death, regulates the synthesis of cyclooxygenase-2 (COX-2). However, COX-2 increases pro-IL-1β and NLRP3 expression through NF-κB activation and mediates NLRP3 inflammasome activation by enhancing Caspase-1 activation through promoting mitochondrial damage and ROS production (174), which may lead to an increase in pyroptosis. As a major member of the antioxidant system and an important regulator of ferroptosis, glutathione peroxidases 4 (GPX4) also showed the function of inhibiting macrophage pyroptosis (175). Therefore, pyroptosis and ferroptosis may promote each other and thus regulate macrophage pro-inflammatory polarization. The relationship between selective autophagy and ferroptosis has also been widely demonstrated (176). Autophagy promotes ferroptosis through selective degradation of ferritin (177), GPX4 (178), SLC40A1 (179), aryl hydrocarbon receptor nuclear translocator-like (ARNTL) (180), and lipid droplets (181). As a widely recognized inhibitor of autophagy, mTORC1 has also been shown to inhibit ferroptosis by regulating GPX4 synthesis (182). Since DAMPs including proteoglycan decorin (DCN) secreted by ferroptotic cells can bind to advanced glycosylation end-product-specific receptor (AGER) on macrophages and further trigger the production of pro-inflammatory cytokines in an NF-κB-dependent manner. Macrophage-selective autophagy and ferroptosis may promote each other and induce the formation of the NASH inflammatory microenvironment (183). Interestingly, although both pyroptosis and selective autophagy can promote the occurrence of ferroptosis, they do not promote each other. The inhibition of macrophage autophagy has been previously mentioned to be associated with increased pyroptosis. The inhibition of macrophage autophagy has been previously mentioned to be associated with increased pyroptosis. A recent study also confirmed that Tim-4, a phosphatidylserine (PS) receptor, activates liver kinase B1 (LKB1)/AMPKα-mediated autophagy to inhibit NLRP3 inflammasome activation, thereby improving the release of IL-1β and IL-18 from macrophages (184), suggesting that Tim-4+ macrophages may inhibit the onset of pyroptosis through autophagy. On the other hand, peritoneal Tim-4 macrophages could inhibit CD8+ T proliferation (185), while activated CD8+ T cells could release granzyme B to induce increased macrophage pyroptosis and promote NAFLD progression (186). Thus, Tim-4-mediated macrophage autophagy not only directly inhibits pyroptosis, but also indirectly inhibits macrophage pyroptosis by suppressing CD8+ T cell activation, suggesting an antagonistic relationship between autophagy and pyroptosis.
It is now generally accepted that the progression of NAFLD is caused by liver lipotoxicity. Possible mediators of lipotoxicity include free cholesterol, saturated free fatty acids, diacylglycerol, lysophosphatidylcholine, sphingolipids, and ceramides (187). Lipotoxic mediators not only induce damage and death of hepatocytes thereby recruiting macrophages, but also directly induce M1 polarization of macrophages. Mitochondria serve as important sites of energy metabolism and regulate liver lipid metabolism and oxidative stress. Changes in mitochondrial metabolism and physiology may underlie the corresponding phenotypes of macrophage activation induced by various signals, including alterations in oxidative metabolism, mitochondrial membrane potential and tricarboxylic acid cycle, as well as the release of mtROS and mtDNA and alterations in mitochondrial ultrastructure (188). Excess ROS attacks biological membranes leading to lipid peroxidation, which not only direct damages phospholipids but also acts as a cell death signal to induce PCD. Numerous studies have confirmed that mitochondrial ROS can induce a variety of PCD in macrophages including pyroptosis (189), autophagy (190), apoptosis (191), necroptosis (192), and ferroptosis (193), suggesting that macrophage polarization and death are closely related to disturbed energy metabolism and oxidative stress. However, it is poorly understood that how dysregulated lipid metabolism in the complex in vivo environment leads to different types of PCD in macrophages. Mitochondria play an important role as the energy center in different types of PCD (164), and the development of single cell omics and mitochondriomics may provide valuable information. This aspect is still poorly understood and requires continuous and intensive research.
As drivers of hepatic steatosis, inflammation, fibrosis and important players in hepatic lipid metabolism, macrophages are attractive therapeutic targets for the treatment of NAFLD. The main strategies currently used to target macrophages include inhibition of monocyte infiltration and inhibition of pro-inflammatory macrophage polarization (194). The improvement of liver inflammation by inhibition of MoMFs infiltration has been well supported by evidence in preclinical studies. A randomized, double-blind, multinational phase 2b study showed that canicriviroc, a dual chemokine receptor CCR2/CCR5 inhibitor, doubled the proportion of patients with at least 1 stage of fibrosis improvement after 1 year despite no improvement in liver inflammation (34). The nuclear receptor family mediates anti-inflammatory polarization of macrophages, thus providing a link between inflammation and lipid metabolism and may be a promising target for NAFLD treatment. Drugs targeting nuclear receptors for the treatment of NAFLD including pan-PPAR agonist (Lanifibranor), PPAR-α/δ agonists (elafibranor), PPAR-α/γ agonist (Saroglitazar) and FXR agonist (Obecholic acid) have been well summarised (48). These drugs may have the effect of modulating both lipid metabolism and phenotypes of macrophages, but more evidence is needed.
PCD is involved in the regulation of the pro-inflammatory polarization of macrophages. Although apoptosis does not induce intense inflammation, apoptosis inhibition has also been considered a therapeutic strategy for NAFLD. As mentioned above, the Caspase family plays an important role in M1 polarization and apoptosis of macrophages. A double-blind, placebo-controlled clinical trial demonstrated that 28 days of treatment with the pan-Caspase inhibitor emricasan significantly reduced ALT and Caspase-3/7 activation in patients with NAFLD (195). However, another clinical study showed that 72 weeks of emricasan treatment did not improve liver histology in patients with NASH fibrosis and may have worsened fibrosis and ballooning (196). Similarly, apoptosis signal-regulated kinase 1 (ASK1) promotes the mitochondrial apoptotic pathway. A multicenter phase 2 clinical trial showed that 24 weeks of treatment with Selonsertib, an ASK1 inhibitor, had no effect on histological inflammation or ballooning despite a reduction in liver fibrosis (197). As the pro-inflammatory properties of hepatocytes and hepatic stellate cells are also regulated by PCD, it is difficult to identify which type or types of liver cells are targeted by these drugs in vivo. Multiple types of PCD may act in combination to induce the pro-inflammatory polarization of macrophages. These targeted drugs may have induced other types of PCD and thus failed to improve liver inflammation. Inhibition of Caspases, particularly Caspase-8, may lead to a bias towards necroptosis. Necroptosis of monocytes induced by LPS and pan-Caspase inhibitors increased CXCL1/2, TNF-α and IL-6 expression, whereas inhibition of RIPK3 resulted in a decrease in CXCL1 and CXCL2 and an increase in TNF-α (127). This result suggests that inhibition of a single type of PCD does not resolve inflammation completely. In addition, some natural drugs and their active ingredients have extremely strong anti-inflammation and anti-oxidation capabilities (198), which help regulate the death of macrophages, and are also potential therapeutic drugs for NAFLD. Licochalcone B (LicoB), a main component of the traditional medicinal herb licorice, is a specific inhibitor of the NLRP3 inflammasome which directly binds to never in mitosis A-related kinase 7 (NEK7) and inhibits the interaction between NLRP3 and NEK7 (199). Glycyrrhetinic acid, another active ingredient of licorice, can also improve the damaged autophagy flux and reduce the excessive production of inflammatory cytokines such as TNF-α, IL-6 and IL-1β by regulating the STAT3-HIF-1 pathway of macrophages (200). Curcumin and berberine, two of the most studied natural products for the treatment of NAFLD, have shown positive results in several clinical trials (201, 202). Mechanistic studies have also demonstrated the effect of both on macrophage polarization (203, 204). However, whether regulation of PCD is involved remains unclear.
Given the possible relationship between PCD-regulated macrophage polarization and lipid metabolism, drugs that regulate lipid metabolism may also improve NAFLD by modulating PCD of macrophages and thereby inhibiting the pro-inflammatory polarization. For example, Ezetimibe blocks the NLRP3 inflammasome-IL-1β pathway in macrophages in an autophagy dependent manner, and regulates the interaction between hepatocytes and macrophages through extracellular vesicles (73). In addition, sodium dependent glucose transporters 2 (SGLT2) inhibitors not only control blood glucose by inhibiting the reabsorption of glucose by the proximal tubules of the kidney, but also show regulatory effects on lipid metabolism, such as lipid synthesis and FAO (205). Empagliflozin, one of the SGLT2 inhibitors, also shows the role of regulating the AMPK/mTOR signal pathway to enhance autophagy of liver macrophages in T2DM mouse models with NAFLD (206). The therapeutic strategy of targeting lipid metabolism and PCD for the treatment of NAFLD is gradually being emphasized, and the research progress and clinical trials of inhibitors of the relevant targets are well summarized in a recent review (207). However, it is unclear whether these inhibitors target hepatic macrophages. In Table 1, we briefly summarize potential small molecule drugs that may improve NAFLD by regulating PCD of macrophages. The safety and efficacy of these small molecule drugs still need to be supported by more clinical evidence.
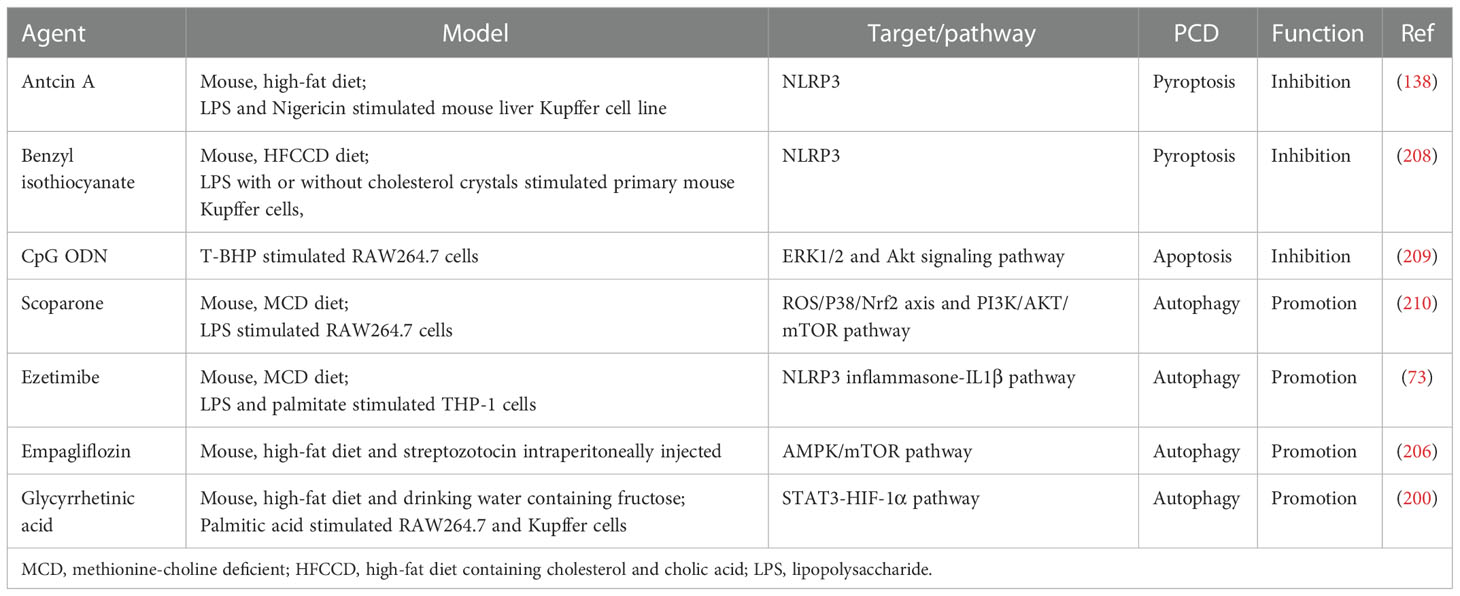
Table 1 Potential small molecule drugs that regulate macrophage death to improve NAFLD.
Lipid metabolism disorders are an important factor in the development of NAFLD. Lipid deposition is not the main inducement of cell damage, but it makes cells more vulnerable to the influence of internal and external environments and aggravates cell damage (211). Once the fuse is ignited, disordered lipid metabolism can rapidly exacerbate the hepatic inflammatory cascade. Compared to other liver disease, NAFLD is more likely to be susceptible to severe damage from lipid peroxidation. This may be part of the reason that NAFLD can progress to HCC without the stage of liver cirrhosis. As an important regulator of hepatic inflammatory homeostasis, the M1/M2 imbalance in macrophages leads to the development and progression of inflammation. Based on the important role and huge number of macrophages in liver immune cells, targeting macrophages is of great significance to improve the development and progression of inflammation in NAFLD. Lipids, as key metabolites in macrophage polarization, are closely associated with macrophage function. Conventional opinion suggests that M1 macrophages are dependent on glycolysis for energy while M2 macrophages are dependent on FAO. However, this view has been challenged by some data in recent years, which demonstrate the complexity of macrophage metabolism. Therefore, it remains difficult to answer whether intervention in the lipid metabolic reprogramming of macrophages can improve NASH, and the metabolic profile of different phenotypes of macrophages still needs to be further clarified.
PCD is closely related to the polarization of macrophages. Compared with M2 macrophages, M1 macrophages may be more tolerant to various types of PCD, which leads to its survival in inflammation. At present, most studies targeting macrophages to treat NAFLD only focus on different types of PCD. As mentioned above, various types of PCD crosstalk with each other, which makes it difficult to obtain satisfactory results by blocking a single type of PCD. Other types of PCD can continue to promote the progress of inflammation as a complementary or alternative way. Therefore, elucidating the relationship between different types of PCD and the main regulatory factors will help to effectively regulate the proinflammatory polarization of macrophages. Regulation of macrophage polarization by targeting key regulators of specific macrophage populations to inhibit the pro-inflammatory PCD may be a promising therapeutic strategy for NAFLD. Most studies only provided preliminary evidence for the correlation between PCD and lipid metabolism of macrophages. Based on the regulation of lipid metabolism reprogramming on macrophage polarization, exploring the relationship between PCD and lipid metabolism may help to clarify how PCD regulates the phenotypic transformation of macrophages, and provide a basis for the strategy of targeting macrophages in the treatment of NAFLD.
A suitable animal model is important for mechanistic studies and pre-clinical evaluation of drugs. The pathology of NAFLD is extremely complex. Animal models of NAFLD, whether induced by high-fat, MCD, CDAA diets or specific gene deletions, are only partially reflect the characteristics of NAFLD in humans. This may have led to frustration in clinical trials of numerous drugs that performed well in pre-clinical studies. In addition, the majority of pre-clinical studies were conducted on mice. Species differences lead to inconsistent expression of phenotypic, inflammation, and metabolism-related genes in human and mouse macrophages. The elucidation of the epigenetic and metabolic characteristics of human macrophages is particularly important for the translation from pre-clinical studies to clinical applications. Further exploration of the links between various types of PCD in macrophages and the links between PCD and lipid metabolism may help to identify specific markers of macrophages. This will not only contribute to the development of drugs targeting macrophages for the treatment of NAFLD, but will also be important for the non-invasive diagnosis and assessment of the degree of liver inflammation and disease progression.
ZX wrote the manuscript. ML involved with project concept. FY, GL, JL and WZ performed data collection. SM and ZD revised the manuscript and were responsible for final approval. All authors contributed to the article and approved the submitted version.
This work is supported by National Natural Science Foundation of China (No. 82104772, 81904154, 82205208), China Postdoctoral Science Foundation (No. 2022M721067), The Science and Technology Research Program of Henan Province (No. 222102310502), Key Research and Promotion Project of Henan Province (No. 202102310168), Special Project of Traditional Chinese Medicine Scientific Research of Henan Province (No. 2018JDZX004) and Traditional Chinese Medicine Science Research Project of Henan Province (No. 2022ZY2008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol (2018) 68:268–79. doi: 10.1016/j.jhep.2017.09.003
2. Orci LA, Kreutzfeldt M, Goossens N, Rubbia-Brandt L, Slits F, Hammad K, et al. Tolerogenic properties of liver macrophages in non-alcoholic steatohepatitis. Liver Int (2020) 40:609–21. doi: 10.1111/liv.14336
3. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
4. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein e knockout mice. Hepatology (2008) 48:770–81. doi: 10.1002/hep.22423
5. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
6. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
7. Caturano A, Acierno C, Nevola R, Pafundi PC, Galiero R, Rinaldi L, et al. Non-alcoholic fatty liver disease: From pathogenesis to clinical impact. Processes (Basel) (2021) 9:135. doi: 10.3390/pr9010135
8. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest (2005) 115:1343–51. doi: 10.1172/JCI23621
9. Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol (2018) 13:321–50. doi: 10.1146/annurev-pathol-020117-043617
10. Pierantonelli I, Svegliati-Baroni G. Nonalcoholic fatty liver disease: Basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation (2019) 103:e1–13. doi: 10.1097/TP.0000000000002480
11. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity (2017) 47:374–88. doi: 10.1016/j.immuni.2017.07.018
12. Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of kupffer cells in normal rat liver. Hepatology (1986) 6:718–22. doi: 10.1002/hep.1840060430
13. Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A, et al. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol (2017) 322:74–83. doi: 10.1016/j.cellimm.2017.10.006
14. Robinson N, Ganesan R, Hegedus C, Kovacs K, Kufer TA, Virag L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol (2019) 26:101239. doi: 10.1016/j.redox.2019.101239
15. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut (2018) 67:963–72. doi: 10.1136/gutjnl-2017-315691
16. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
17. Mirshafiee V, Sun B, Chang CH, Liao YP, Jiang W, Jiang J, et al. Toxicological profiling of metal oxide nanoparticles in liver context reveals pyroptosis in kupffer cells and macrophages versus apoptosis in hepatocytes. ACS Nano (2018) 12:3836–52. doi: 10.1021/acsnano.8b01086
18. Gomez PE, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
19. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
20. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology (2010) 51:511–22. doi: 10.1002/hep.23337
21. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes (2010) 59:347–57. doi: 10.2337/db09-0016
22. Song K, Kwon H, Han C, Chen W, Zhang J, Ma W, et al. Yes-associated protein in kupffer cells enhances the production of proinflammatory cytokines and promotes the development of nonalcoholic steatohepatitis. Hepatology (2020) 72:72–87. doi: 10.1002/hep.30990
23. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology (2003) 38:1188–98. doi: 10.1053/jhep.2003.50472
24. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest (2019) 129:546–55. doi: 10.1172/JCI121842
25. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse kupffer cells through mitochondrial DNA release. Cell Immunol (2018) 332:111–20. doi: 10.1016/j.cellimm.2018.08.006
26. Liu XL, Pan Q, Cao HX, Xin FZ, Zhao ZH, Yang RX, et al. Lipotoxic hepatocyte-derived exosomal MicroRNA 192-5p activates macrophages through Rictor/Akt/Forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology (2020) 72:454–69. doi: 10.1002/hep.31050
27. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of c-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology (2016) 63:731–44. doi: 10.1002/hep.28252
28. Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY, et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology (2020) 159:1487–503. doi: 10.1053/j.gastro.2020.06.031
29. Guo Q, Furuta K, Lucien F, Gutierrez SL, Hirsova P, Krishnan A, et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol (2019) 71:1193–205. doi: 10.1016/j.jhep.2019.07.019
30. Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, et al. CXCL10-mediates macrophage, but not other innate immune cells-associated inflammation in murine nonalcoholic steatohepatitis. Sci Rep (2016) 6:28786. doi: 10.1038/srep28786
31. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
32. Guillot A, Tacke F. Liver macrophages: Old dogmas and new insights. Hepatol Commun (2019) 3:730–43. doi: 10.1002/hep4.1356
33. Wang H, Mehal W, Nagy LE, Rotman Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell Mol Immunol (2021) 18:73–91. doi: 10.1038/s41423-020-00579-3
34. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67:1754–67. doi: 10.1002/hep.29477
35. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology (2018) 67:1270–83. doi: 10.1002/hep.29544
36. Gao H, Jin Z, Bandyopadhyay G, Cunha ERK, Liu X, Zhao H, et al. MiR-690 treatment causes decreased fibrosis and steatosis and restores specific kupffer cell functions in NASH. Cell Metab (2022) 34:978–90. doi: 10.1016/j.cmet.2022.05.008
37. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated kupffer cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
38. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gelineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity (2020) 53:627–40. doi: 10.1016/j.immuni.2020.06.003
39. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
40. Yan J, Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol (2020) 30:979–89. doi: 10.1016/j.tcb.2020.09.006
41. Song Z, Xiaoli AM, Yang F. Regulation and metabolic significance of De novo lipogenesis in adipose tissues. Nutrients (2018) 10:1383. doi: 10.3390/nu10101383
42. Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol (2010) 30:139–43. doi: 10.1161/ATVBAHA.108.179283
43. Batista-Gonzalez A, Vidal R, Criollo A, Carreno LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol (2019) 10:2993. doi: 10.3389/fimmu.2019.02993
44. Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab (2017) 25:412–27. doi: 10.1016/j.cmet.2016.11.009
45. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity (2018) 49:842–56. doi: 10.1016/j.immuni.2018.08.021
46. Carroll RG, Zaslona Z, Galvan-Pena S, Koppe EL, Sevin DC, Angiari S, et al. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J Biol Chem (2018) 293:5509–21. doi: 10.1074/jbc.RA118.001921
47. Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab (2015) 308:E756–69. doi: 10.1152/ajpendo.00362.2014
48. Puengel T, Liu H, Guillot A, Heymann F, Tacke F, Peiseler M. Nuclear receptors linking metabolism, inflammation, and fibrosis in nonalcoholic fatty liver disease. Int J Mol Sci (2022) 23:2668. doi: 10.3390/ijms23052668
49. Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol (2016) 17:216–17. doi: 10.1038/ni.3366
50. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15:846–55. doi: 10.1038/ni.2956
51. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
52. Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity (2022) 55:1515–29. doi: 10.1016/j.immuni.2022.08.002
53. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell (2022) 185:379–96. doi: 10.1016/j.cell.2021.12.018
54. Scott CL, Guilliams M. The role of kupffer cells in hepatic iron and lipid metabolism. J Hepatol (2018) 69:1197–99. doi: 10.1016/j.jhep.2018.02.013
55. Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol (2022) 19:384–408. doi: 10.1038/s41423-021-00791-9
56. Itoh M, Suganami T, Kato H, Kanai S, Shirakawa I, Sakai T, et al. CD11c+ resident macrophages drive hepatocyte death-triggered liver fibrosis in a murine model of nonalcoholic steatohepatitis. JCI Insight (2017) 2:e92902. doi: 10.1172/jci.insight.92902
57. Govaere O, Petersen SK, Martinez-Lopez N, Wouters J, Van Haele M, Mancina RM, et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. J Hepatol (2022) 76:1001–12. doi: 10.1016/j.jhep.2021.12.012
58. Lee S, Usman TO, Yamauchi J, Chhetri G, Wang X, Coudriet GM, et al. Myeloid FoxO1 depletion attenuates hepatic inflammation and prevents nonalcoholic steatohepatitis. J Clin Invest (2022) 132:e154333. doi: 10.1172/JCI154333
59. Dixon ED, Nardo AD, Claudel T, Trauner M. The role of lipid sensing nuclear receptors (PPARs and LXR) and metabolic lipases in obesity, diabetes and NAFLD. Genes (Basel) (2021) 12:645. doi: 10.3390/genes12050645
60. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-gamma activity on kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
61. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J Hepatol (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
62. Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife (2015) 4:e8009. doi: 10.7554/eLife.08009
63. Muse ED, Yu S, Edillor CR, Tao J, Spann NJ, Troutman TD, et al. Cell-specific discrimination of desmosterol and desmosterol mimetics confers selective regulation of LXR and SREBP in macrophages. Proc Natl Acad Sci U.S.A. (2018) 115:E4680–89. doi: 10.1073/pnas.1714518115
64. Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, et al. RORalpha induces KLF4-mediated M2 polarization in the liver macrophages that protect against nonalcoholic steatohepatitis. Cell Rep (2017) 20:124–35. doi: 10.1016/j.celrep.2017.06.017
65. L'Homme L, Sermikli BP, Molendi-Coste O, Fleury S, Quemener S, Le Maitre M, et al. Deletion of the nuclear receptor RORalpha in macrophages does not modify the development of obesity, insulin resistance and NASH. Sci Rep (2020) 10:21095. doi: 10.1038/s41598-020-77858-6
66. Hong T, Chen Y, Li X, Lu Y. The role and mechanism of oxidative stress and nuclear receptors in the development of NAFLD. Oxid Med Cell Longev (2021) 2021:6889533. doi: 10.1155/2021/6889533
67. Bleriot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of kupffer cells regulates metabolism through the expression of CD36. Immunity (2021) 54:2101–16. doi: 10.1016/j.immuni.2021.08.006
68. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: Different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
69. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
70. Gual P, Gilgenkrantz H, Lotersztajn S. Autophagy in chronic liver diseases: the two faces of janus. Am J Physiol Cell Physiol (2017) 312:C263–73. doi: 10.1152/ajpcell.00295.2016
71. Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol (2022). doi: 10.1038/s41580-022-00529-z
72. Wu MY, Lu JH. Autophagy and macrophage functions: Inflammatory response and phagocytosis. Cells (2019) 9:70. doi: 10.3390/cells9010070
73. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy (2017) 13:1767–81. doi: 10.1080/15548627.2017.1356977
74. Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy (2015) 11:1280–92. doi: 10.1080/15548627.2015.1058473
75. Dornas W, Lagente V. Intestinally derived bacterial products stimulate development of nonalcoholic steatohepatitis. Pharmacol Res (2019) 141:418–28. doi: 10.1016/j.phrs.2019.01.026
76. Fukushima H, Yamashina S, Arakawa A, Taniguchi G, Aoyama T, Uchiyama A, et al. Formation of p62-positive inclusion body is associated with macrophage polarization in non-alcoholic fatty liver disease. Hepatol Res (2018) 48:757–67. doi: 10.1111/hepr.13071
77. Sun K, Xu L, Jing Y, Han Z, Chen X, Cai C, et al. Autophagy-deficient kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett (2017) 388:198–207. doi: 10.1016/j.canlet.2016.12.004
78. Tsai ML, Tsai YG, Lin YC, Hsu YL, Chen YT, Tsai MK, et al. IL-25 induced ROS-mediated M2 macrophage polarization via AMPK-associated mitophagy. Int J Mol Sci (2021) 23:3. doi: 10.3390/ijms23010003
79. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12:408–15. doi: 10.1038/ni.2022
80. Wang Q, Wei S, Zhou S, Qiu J, Shi C, Liu R, et al. Hyperglycemia aggravates acute liver injury by promoting liver-resident macrophage NLRP3 inflammasome activation via the inhibition of AMPK/mTOR-mediated autophagy induction. Immunol Cell Biol (2020) 98:54–66. doi: 10.1111/imcb.12297
81. Lee DH, Park JS, Lee YS, Han J, Lee DK, Kwon SW, et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy (2020) 16:1949–73. doi: 10.1080/15548627.2020.1712108
82. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi: 10.1038/ncb2152
83. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
84. Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell (2011) 44:134–46. doi: 10.1016/j.molcel.2011.06.038
85. Liu W, Ye C, Cheng Q, Zhang X, Yao L, Li Q, et al. Macrophage raptor deficiency-induced lysosome dysfunction exacerbates nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol (2019) 7:211–31. doi: 10.1016/j.jcmgh.2018.09.011
86. Lefere S, Van Steenkiste C, Verhelst X, Van Vlierberghe H, Devisscher L, Geerts A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease. Cell Mol Life Sci (2016) 73:3419–31. doi: 10.1007/s00018-016-2222-1
87. Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol (2020) 36:101671. doi: 10.1016/j.redox.2020.101671
88. Wang X, de Carvalho RM, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A, et al. Macrophage-specific hypoxia-inducible factor-1alpha contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology (2019) 69:545–63. doi: 10.1002/hep.30215
89. Fang C, Pan J, Qu N, Lei Y, Han J, Zhang J, et al. The AMPK pathway in fatty liver disease. Front Physiol (2022) 13:970292. doi: 10.3389/fphys.2022.970292
90. Isaza SC, Del PE, Dominguez-Alcon L, Elbouayadi L, Gonzalez-Rodriguez A, Garcia-Monzon C. Hypoxia and non-alcoholic fatty liver disease. Front Med (Lausanne) (2020) 7:578001. doi: 10.3389/fmed.2020.578001
91. Tardelli M. Monoacylglycerol lipase reprograms lipid precursors signaling in liver disease. World J Gastroenterol (2020) 26:3577–85. doi: 10.3748/wjg.v26.i25.3577
92. Habib A, Chokr D, Wan J, Hegde P, Mabire M, Siebert M, et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut (2019) 68:522–32. doi: 10.1136/gutjnl-2018-316137
93. Taschler U, Radner FP, Heier C, Schreiber R, Schweiger M, Schoiswohl G, et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J Biol Chem (2011) 286:17467–77. doi: 10.1074/jbc.M110.215434
94. Deust A, Chobert MN, Demontant V, Gricourt G, Denaes T, Thiolat A, et al. Macrophage autophagy protects against hepatocellular carcinogenesis in mice. Sci Rep (2021) 11:18809. doi: 10.1038/s41598-021-98203-5
95. Izadi M, Ali TA, Pourkarimi E. Over fifty years of life, death, and cannibalism: A historical recollection of apoptosis and autophagy. Int J Mol Sci (2021) 22:12466. doi: 10.3390/ijms222212466
96. Kesavardhana S, Malireddi R, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
97. Shojaie L, Iorga A, Dara L. Cell death in liver diseases: A review. Int J Mol Sci (2020) 21:9682. doi: 10.3390/ijms21249682
98. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology (2003) 125:437–43. doi: 10.1016/s0016-5085(03)00907-7
99. Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 kupffer cells promote M1 kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology (2014) 59:130–42. doi: 10.1002/hep.26607
100. Maiers JL, Malhi H. Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. Semin Liver Dis (2019) 39:235–48. doi: 10.1055/s-0039-1681032
101. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev (1998) 12:982–95. doi: 10.1101/gad.12.7.982
102. Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J (2016) 283:2640–52. doi: 10.1111/febs.13598
103. Malhi H, Kropp EM, Clavo VF, Kobrossi CR, Han J, Mauer AS, et al. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J Biol Chem (2013) 288:18624–42. doi: 10.1074/jbc.M112.442954
104. Che X, Xiao Q, Song W, Zhang H, Sun B, Geng N, et al. Protective functions of liver X receptor alpha in established vulnerable plaques: Involvement of regulating endoplasmic reticulum-mediated macrophage apoptosis and efferocytosis. J Am Heart Assoc (2021) 10:e18455. doi: 10.1161/JAHA.120.018455
105. Yoshimura A, Ito M, Mise-Omata S, Ando M. SOCS: Negative regulators of cytokine signaling for immune tolerance. Int Immunol (2021) 33:711–16. doi: 10.1093/intimm/dxab055
106. Dickensheets H, Vazquez N, Sheikh F, Gingras S, Murray PJ, Ryan JJ, et al. Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun (2007) 8:21–7. doi: 10.1038/sj.gene.6364352
107. Li S, Han S, Jin K, Yu T, Chen H, Zhou X, et al. SOCS2 suppresses inflammation and apoptosis during NASH progression through limiting NF-kappaB activation in macrophages. Int J Biol Sci (2021) 17:4165–75. doi: 10.7150/ijbs.63889
108. Horndasch M, Culig Z. SOCS-3 antagonizes pro-apoptotic effects of TRAIL and resveratrol in prostate cancer cells. Prostate (2011) 71:1357–66. doi: 10.1002/pros.21353
109. Du C, Yao F, Ren Y, Du Y, Wei J, Wu H, et al. SOCS-1 is involved in TNF-alpha-induced mitochondrial dysfunction and apoptosis in renal tubular epithelial cells. Tissue Cell (2017) 49:537–44. doi: 10.1016/j.tice.2017.06.005
110. Luo B, Zou T, Lu N, Chai F, Ye X, Wang Y, et al. Role of suppressor of cytokine signaling 3 in lipid metabolism: Analysis based on a phage-display human liver cDNA library. Biochem Biophys Res Commun (2011) 416:39–44. doi: 10.1016/j.bbrc.2011.10.129
111. Vila L, Roglans N, Alegret M, Sanchez RM, Vazquez-Carrera M, Laguna JC. Suppressor of cytokine signaling-3 (SOCS-3) and a deficit of serine/threonine (Ser/Thr) phosphoproteins involved in leptin transduction mediate the effect of fructose on rat liver lipid metabolism. Hepatology (2008) 48:1506–16. doi: 10.1002/hep.22523
112. Dai Z, Wang H, Jin X, Wang H, He J, Liu M, et al. Depletion of suppressor of cytokine signaling-1a causes hepatic steatosis and insulin resistance in zebrafish. Am J Physiol Endocrinol Metab (2015) 308:E849–59. doi: 10.1152/ajpendo.00540.2014
113. Zadjali F, Santana-Farre R, Vesterlund M, Carow B, Mirecki-Garrido M, Hernandez-Hernandez I, et al. SOCS2 deletion protects against hepatic steatosis but worsens insulin resistance in high-fat-diet-fed mice. FASEB J (2012) 26:3282–91. doi: 10.1096/fj.12-205583
114. LaPensee CR, Lin G, Dent AL, Schwartz J. Deficiency of the transcriptional repressor b cell lymphoma 6 (Bcl6) is accompanied by dysregulated lipid metabolism. PloS One (2014) 9:e97090. doi: 10.1371/journal.pone.0097090
115. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol (2015) 16:689–97. doi: 10.1038/ni.3206
116. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
117. Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M. NAD(+) depletion triggers macrophage necroptosis, a cell death pathway exploited by mycobacterium tuberculosis. Cell Rep (2018) 24:429–40. doi: 10.1016/j.celrep.2018.06.042
118. Mohammed S, Thadathil N, Selvarani R, Nicklas EH, Wang D, Miller BF, et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell (2021) 20:e13512. doi: 10.1111/acel.13512
119. Chang YH, Weng CL, Lin KI. O-GlcNAcylation and its role in the immune system. J BioMed Sci (2020) 27:57. doi: 10.1186/s12929-020-00648-9
120. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting Serine/Threonine-protein kinase 3. Immunity (2019) 50:576–90. doi: 10.1016/j.immuni.2019.01.007
121. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simao AL, Gaspar MM, Majdi A, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut (2021) 70:2359–72. doi: 10.1136/gutjnl-2020-321767
122. Wei S, Zhou H, Wang Q, Zhou S, Li C, Liu R, et al. RIP3 deficiency alleviates liver fibrosis by inhibiting ROCK1-TLR4-NF-kappaB pathway in macrophages. FASEB J (2019) 33:11180–93. doi: 10.1096/fj.201900752R
123. Hao Q, Kundu S, Kleam J, Zhao ZJ, Idell S, Tang H. Enhanced RIPK3 kinase activity-dependent lytic cell death in M1 but not M2 macrophages. Mol Immunol (2021) 129:86–93. doi: 10.1016/j.molimm.2020.11.001
124. Varga Z, Molnar T, Mazlo A, Kovacs R, Jenei V, Kerekes K, et al. Differences in the sensitivity of classically and alternatively activated macrophages to TAK1 inhibitor-induced necroptosis. Cancer Immunol Immunother (2020) 69:2193–207. doi: 10.1007/s00262-020-02623-7
125. Wu L, Zhang X, Zheng L, Zhao H, Yan G, Zhang Q, et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol Res (2020) 8:710–21. doi: 10.1158/2326-6066.CIR-19-0261
126. Rasheed A, Robichaud S, Nguyen MA, Geoffrion M, Wyatt H, Cottee ML, et al. Loss of MLKL (Mixed lineage kinase domain-like protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis. Arterioscler Thromb Vasc Biol (2020) 40:1155–67. doi: 10.1161/ATVBAHA.119.313640
127. Saeed WK, Jun DW, Jang K, Ahn SB, Oh JH, Chae YJ, et al. Mismatched effects of receptor interacting protein kinase-3 on hepatic steatosis and inflammation in non-alcoholic fatty liver disease. World J Gastroenterol (2018) 24:5477–90. doi: 10.3748/wjg.v24.i48.5477
128. Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci (2022) 43:653–68. doi: 10.1016/j.tips.2022.04.003
129. de Carvalho RM, Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol (2022) 17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529
130. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–49. doi: 10.1126/science.1240248
131. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin d plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol (2018) 68:773–82. doi: 10.1016/j.jhep.2017.11.040
132. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
133. Gan C, Cai Q, Tang C, Gao J. Inflammasomes and pyroptosis of liver cells in liver fibrosis. Front Immunol (2022) 13:896473. doi: 10.3389/fimmu.2022.896473
134. Azzimato V, Jager J, Chen P, Morgantini C, Levi L, Barreby E, et al. Liver macrophages inhibit the endogenous antioxidant response in obesity-associated insulin resistance. Sci Transl Med (2020) 12:w9709. doi: 10.1126/scitranslmed.aaw9709
135. Wang P, Ni M, Tian Y, Wang H, Qiu J, You W, et al. Myeloid Nrf2 deficiency aggravates non-alcoholic steatohepatitis progression by regulating YAP-mediated NLRP3 inflammasome signaling. Iscience (2021) 24:102427. doi: 10.1016/j.isci.2021.102427
136. Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga Toxin/Lipopolysaccharide activates caspase-4 and gasdermin d to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep (2018) 25:1525–36. doi: 10.1016/j.celrep.2018.09.071
137. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun (2019) 10:1689. doi: 10.1038/s41467-019-09397-2
138. Ruan S, Han C, Sheng Y, Wang J, Zhou X, Guan Q, et al. Antcin a alleviates pyroptosis and inflammatory response in kupffercells of non-alcoholic fatty liver disease by targeting NLRP3. Int Immunopharmacol (2021) 100:108126. doi: 10.1016/j.intimp.2021.108126
139. Mridha AR, Wree A, Robertson A, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
140. Chen Y, Ma K. NLRC4 inflammasome activation regulated by TNF-alpha promotes inflammatory responses in nonalcoholic fatty liver disease. Biochem Biophys Res Commun (2019) 511:524–30. doi: 10.1016/j.bbrc.2019.02.099
141. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res (2017) 58:1067–79. doi: 10.1194/jlr.M072454
142. Chiang J, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr (2019) 39:175–200. doi: 10.1146/annurev-nutr-082018-124344
143. Chiang J, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol (2020) 318:G554–73. doi: 10.1152/ajpgi.00223.2019
144. Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-x-receptor in lipid and glucose disorders. Trends Pharmacol Sci (2009) 30:570–80. doi: 10.1016/j.tips.2009.08.001
145. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated b cells (NF-kappaB) in mice. Hepatology (2011) 54:1421–32. doi: 10.1002/hep.24525
146. Shi Y, Su W, Zhang L, Shi C, Zhou J, Wang P, et al. TGR5 regulates macrophage inflammation in nonalcoholic steatohepatitis by modulating NLRP3 inflammasome activation. Front Immunol (2020) 11:609060. doi: 10.3389/fimmu.2020.609060
147. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
148. Britton LJ, Subramaniam VN, Crawford DH. Iron and non-alcoholic fatty liver disease. World J Gastroenterol (2016) 22:8112–22. doi: 10.3748/wjg.v22.i36.8112
149. Dai X, Zhang R, Wang B. Contribution of classification based on ferroptosis-related genes to the heterogeneity of MAFLD. BMC Gastroenterol (2022) 22:55. doi: 10.1186/s12876-022-02137-9
150. Day K, Seale LA, Graham RM, Cardoso BR. Selenotranscriptome network in non-alcoholic fatty liver disease. Front Nutr (2021) 8:744825. doi: 10.3389/fnut.2021.744825
151. Li X, Wang TX, Huang X, Li Y, Sun T, Zang S, et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int (2020) 40:1378–94. doi: 10.1111/liv.14428
152. Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis (2019) 10:449. doi: 10.1038/s41419-019-1678-y
153. Wang S, Liu Z, Geng J, Li L, Feng X. An overview of ferroptosis in non-alcoholic fatty liver disease. BioMed Pharmacother (2022) 153:113374. doi: 10.1016/j.biopha.2022.113374
154. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology (2011) 53:448–57. doi: 10.1002/hep.24038
155. Recalcati S, Cairo G. Macrophages and iron: A special relationship. Biomedicines (2021) 9:1585. doi: 10.3390/biomedicines9111585
156. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol (2019) 105:1015–26. doi: 10.1002/JLB.3A0318-108R
157. Marques L, Negre-Salvayre A, Costa L, Canonne-Hergaux F. Iron gene expression profile in atherogenic Mox macrophages. Biochim Biophys Acta (2016) 1862:1137–46. doi: 10.1016/j.bbadis.2016.03.004
158. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
159. Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol (2022) 56:102463. doi: 10.1016/j.redox.2022.102463
160. Li LG, Peng XC, Yu TT, Xu HZ, Han N, Yang XX, et al. Dihydroartemisinin remodels macrophage into an M1 phenotype via ferroptosis-mediated DNA damage. Front Pharmacol (2022) 13:949835. doi: 10.3389/fphar.2022.949835
161. Kao JK, Wang SC, Ho LW, Huang SW, Lee CH, Lee MS, et al. M2-like polarization of THP-1 monocyte-derived macrophages under chronic iron overload. Ann Hematol (2020) 99:431–41. doi: 10.1007/s00277-020-03916-8
162. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell (2022) 82:2215–27. doi: 10.1016/j.molcel.2022.03.022
163. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol (2019) 26:420–32. doi: 10.1016/j.chembiol.2018.11.016
164. Bock FJ, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
165. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33:1001–12. doi: 10.1016/j.cmet.2021.02.015
166. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med (2014) 6:1062–74. doi: 10.15252/emmm.201403856
167. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med (2017) 214:3687–705. doi: 10.1084/jem.20170558
168. Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy (2010) 6:891–900. doi: 10.4161/auto.6.7.13038
169. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S, et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J Hepatol (2020) 73:616–27. doi: 10.1016/j.jhep.2020.03.023
170. Zhao P, Sun X, Chaggan C, Liao Z, In WK, He F, et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science (2020) 367:652–60. doi: 10.1126/science.aay0542
171. Wu W, Wang X, Sun Y, Berleth N, Deitersen J, Schlutermann D, et al. TNF-induced necroptosis initiates early autophagy events via RIPK3-dependent AMPK activation, but inhibits late autophagy. Autophagy (2021) 17:3992–4009. doi: 10.1080/15548627.2021.1899667
172. Gram AM, Booty LM, Bryant CE. Chopping GSDMD: Caspase-8 has joined the team of pyroptosis-mediating caspases. EMBO J (2019) 38:e102065. doi: 10.15252/embj.2019102065
173. Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ (2021) 28:1418–33. doi: 10.1038/s41418-020-00668-w
174. Hua KF, Chou JC, Ka SM, Tasi YL, Chen A, Wu SH, et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1beta production. J Cell Physiol (2015) 230:863–74. doi: 10.1002/jcp.24815
175. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe (2018) 24:97–108. doi: 10.1016/j.chom.2018.05.009
176. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
177. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HR, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12:1425–28. doi: 10.1080/15548627.2016.1187366
178. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U.S.A. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
179. Li J, Liu J, Xu Y, Wu R, Chen X, Song X, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy (2021) 17:3361–74. doi: 10.1080/15548627.2021.1872241
180. Liu J, Yang M, Kang R, Klionsky DJ, Tang D. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy (2019) 15:2033–35. doi: 10.1080/15548627.2019.1659623
181. Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun (2019) 508:997–1003. doi: 10.1016/j.bbrc.2018.12.039
182. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun (2021) 12:1589. doi: 10.1038/s41467-021-21841-w
183. Liu J, Zhu S, Zeng L, Li J, Klionsky DJ, Kroemer G, et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy (2022) 18:2036–49. doi: 10.1080/15548627.2021.2008692
184. Liu W, Bai F, Wang H, Liang Y, Du X, Liu C, et al. Tim-4 inhibits NLRP3 inflammasome via the LKB1/AMPKalpha pathway in macrophages. J Immunol (2019) 203:990–1000. doi: 10.4049/jimmunol.1900117
185. Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell (2021) 39:973–88. doi: 10.1016/j.ccell.2021.05.006
186. Zhang Q, Wang J, Huang F, Yao Y, Xu L. Leptin induces NAFLD progression through infiltrated CD8+ T lymphocytes mediating pyroptotic-like cell death of hepatocytes and macrophages. Dig Liver Dis (2021) 53:598–605. doi: 10.1016/j.dld.2020.10.025
187. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: How metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv Exp Med Biol (2018) 1061:19–44. doi: 10.1007/978-981-10-8684-7_3
188. Wang Y, Li N, Zhang X, Horng T. Mitochondrial metabolism regulates macrophage biology. J Biol Chem (2021) 297:100904. doi: 10.1016/j.jbc.2021.100904
189. Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol (2019) 11:1069–82. doi: 10.1093/jmcb/mjz020
190. Luo Q, Song Y, Kang J, Wu Y, Wu F, Li Y, et al. mtROS-mediated Akt/AMPK/mTOR pathway was involved in copper-induced autophagy and it attenuates copper-induced apoptosis in RAW264.7 mouse monocytes. Redox Biol (2021) 41:101912. doi: 10.1016/j.redox.2021.101912
191. Du S, Li C, Lu Y, Lei X, Zhang Y, Li S, et al. Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics (2019) 9:1878–92. doi: 10.7150/thno.29682
192. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun (2017) 8:14329. doi: 10.1038/ncomms14329
193. Jiang JJ, Zhang GF, Zheng JY, Sun JH, Ding SB. Targeting mitochondrial ROS-mediated ferroptosis by quercetin alleviates high-fat diet-induced hepatic lipotoxicity. Front Pharmacol (2022) 13:876550. doi: 10.3389/fphar.2022.876550
194. Krenkel O, Tacke F. Macrophages in nonalcoholic fatty liver disease: A role model of pathogenic immunometabolism. Semin Liver Dis (2017) 37:189–97. doi: 10.1055/s-0037-1604480
195. Shiffman M, Freilich B, Vuppalanchi R, Watt K, Chan JL, Spada A, et al. Randomised clinical trial: emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment Pharmacol Ther (2019) 49:64–73. doi: 10.1111/apt.15030
196. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol (2020) 72:816–27. doi: 10.1016/j.jhep.2019.11.024
197. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology (2018) 67:549–59. doi: 10.1002/hep.29514
198. Arulselvan P, Fard MT, Tan WS, Gothai S, Fakurazi S, Norhaizan ME, et al. Role of antioxidants and natural products in inflammation. Oxid Med Cell Longev (2016) 2016:5276130. doi: 10.1155/2016/5276130
199. Li Q, Feng H, Wang H, Wang Y, Mou W, Xu G, et al. Licochalcone b specifically inhibits the NLRP3 inflammasome by disrupting NEK7-NLRP3 interaction. EMBO Rep (2022) 23:e53499. doi: 10.15252/embr.202153499
200. Fan Y, Dong W, Wang Y, Zhu S, Chai R, Xu Z, et al. Glycyrrhetinic acid regulates impaired macrophage autophagic flux in the treatment of non-alcoholic fatty liver disease. Front Immunol (2022) 13:959495. doi: 10.3389/fimmu.2022.959495
201. Ngu MH, Norhayati MN, Rosnani Z, Zulkifli MM. Curcumin as adjuvant treatment in patients with non-alcoholic fatty liver (NAFLD) disease: A systematic review and meta-analysis. Complement Ther Med (2022) 68:102843. doi: 10.1016/j.ctim.2022.102843
202. Koperska A, Wesolek A, Moszak M, Szulinska M. Berberine in non-alcoholic fatty liver disease-a review. Nutrients (2022) 14:3459. doi: 10.3390/nu14173459
203. Tong C, Wu H, Gu D, Li Y, Fan Y, Zeng J, et al. Effect of curcumin on the non-alcoholic steatohepatitis via inhibiting the M1 polarization of macrophages. Hum Exp Toxicol (2021) 40:S310–17. doi: 10.1177/09603271211038741
204. Wang Y, Zhou X, Zhao D, Wang X, Gurley EC, Liu R, et al. Berberine inhibits free fatty acid and LPS-induced inflammation via modulating ER stress response in macrophages and hepatocytes. PloS One (2020) 15:e232630. doi: 10.1371/journal.pone.0232630
205. Szekeres Z, Toth K, Szabados E. The effects of SGLT2 inhibitors on lipid metabolism. Metabolites (2021) 11:87. doi: 10.3390/metabo11020087
206. Meng Z, Liu X, Li T, Fang T, Cheng Y, Han L, et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int Immunopharmacol (2021) 94:107492. doi: 10.1016/j.intimp.2021.107492
207. Xu X, Poulsen KL, Wu L, Liu S, Miyata T, Song Q, et al. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct Target Ther (2022) 7:287. doi: 10.1038/s41392-022-01119-3
208. Chen HW, Yen CC, Kuo LL, Lo CW, Huang CS, Chen CC, et al. Benzyl isothiocyanate ameliorates high-fat/cholesterol/cholic acid diet-induced nonalcoholic steatohepatitis through inhibiting cholesterol crystal-activated NLRP3 inflammasome in kupffer cells. Toxicol Appl Pharmacol (2020) 393:114941. doi: 10.1016/j.taap.2020.114941
209. Qu Y, Yang C, Li X, Luo H, Li S, Niu M, et al. CpG-oligodeoxynucleotides alleviate tert-butyl hydroperoxide-induced macrophage apoptosis by regulating mitochondrial function and suppressing ROS production. Oxid Med Cell Longev (2020) 2020:1714352. doi: 10.1155/2020/1714352
210. Liu B, Deng X, Jiang Q, Li G, Zhang J, Zhang N, et al. Scoparone improves hepatic inflammation and autophagy in mice with nonalcoholic steatohepatitis by regulating the ROS/P38/Nrf2 axis and PI3K/AKT/mTOR pathway in macrophages. BioMed Pharmacother (2020) 125:109895. doi: 10.1016/j.biopha.2020.109895
Keywords: non-alcoholic fatty liver disease, inflammation, macrophages, programmed cell death, lipid metabolism
Citation: Xiao Z, Liu M, Yang F, Liu G, Liu J, Zhao W, Ma S and Duan Z (2023) Programmed cell death and lipid metabolism of macrophages in NAFLD. Front. Immunol. 14:1118449. doi: 10.3389/fimmu.2023.1118449
Received: 07 December 2022; Accepted: 06 January 2023;
Published: 18 January 2023.
Edited by:
Chao Yang, Zhejiang University, ChinaReviewed by:
Jingbo Pang, University of Illinois at Chicago, United StatesCopyright © 2023 Xiao, Liu, Yang, Liu, Liu, Zhao, Ma and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suping Ma, bWFzdXBpbmdAMTYzLmNvbQ==; Zhongping Duan, ZHVhbjI1MTdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.