 Youri Bertrand
Youri Bertrand Alba Sánchez-Montalvo
Alba Sánchez-Montalvo Valérie Hox
Valérie Hox Antoine Froidure1,4
Antoine Froidure1,4
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 March 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1117749
This article is part of the Research Topic Immunoglobulin A in Infection and Inflammation View all 8 articles
Immunoglobulin A (IgA) is the most abundant Ig in mucosae where it plays key roles in host defense against pathogens and in mucosal immunoregulation. Whereas intense research has established the different roles of secretory IgA in the gut, its function has been much less studied in the lung. This review will first summarize the state-of-the-art knowledge on the distribution and phenotype of IgA+ B cells in the human lung in both homeostasis and disease. Second, it will analyze the studies looking at cellular and molecular mechanisms of homing and priming of IgA+ B cells in the lung, notably following immunization. Lastly, published data on observations related to IgA and IgA+ B cells in lung and airway disease such as asthma, cystic fibrosis, idiopathic pulmonary fibrosis, or chronic rhinosinusitis, will be discussed. Collectively it provides the state-of-the-art of our current understanding of the biology of IgA-producing cells in the airways and identifies gaps that future research should address in order to improve mucosal protection against lung infections and chronic inflammatory diseases.
IgA is the most secreted antibody isotype in the human body, mainly at mucosal surfaces where it plays protective roles such as immune exclusion. There are two subclasses of this immunoglobulin, namely IgA1 and IgA2. Compared to IgA1, IgA2 has a shorter hinge region and is therefore more resistant to pathogen-derived bacterial proteases (1, 2). There is only 10 to 20% of IgA2 in the blood (3), whereas this proportion increases in the lung mucosa, with approximately 25 to 30% of IgA2 in bronchoalveolar fluid (3, 4). In addition, monomeric IgA is predominant in serum in contrast with mucosal sites where there is mostly dimeric IgA (d-IgA) (80%) (2, 5). In the bronchus, epithelial cells express the polymeric Ig receptor (pIgR) which binds IgA and can be internalized to transport it to the lumen. The transcytosis ends with the cleavage of the C-terminal part of pIgR, the secretory component (SC), which is bound to d-IgA (6). The complex consisting of d-IgA and SC is called secretory IgA (S-IgA) and its main role is to prevent pathogens in the mucosal lumen from entering the body, which is called the immune exclusion (7–9). Beside pIgR, IgA can interact with cells through different receptors such as IgA Fc receptor I (FcαRI or CD89) or the transferrin receptor 1 (CD71) (10, 11). CD89 is the main one in human, expressed by myeloid cells: neutrophils, eosinophils, monocytes, macrophages and dendritic cells (11). Mice do not express CD89 (but retained to Fcα/μR, for both IgA and IgM), leading to the development of transgenic mice expressing the human CD89 to enable taking this interaction into account whilst studying the role of IgA in vivo, e.g. in (11)IgA nephropathy (12–14), cancer (15) or the inflammatory role of serum IgA (16).
IgA-producing B cells in the mucosa were mostly studied in the gut and much less in the airways, leading to gaps in knowledge for the later. This review aims to describe the different subsets of IgA+ B cells in the airways during homeostasis and their changes and roles following immunization and in disease such as IgA deficiency or chronic airway/lung diseases. It also summarizes the mechanisms underlying activation and homing of IgA+ B cells in the lung, as compared to the gut.
Currently identified B-cell subclasses have been mainly studied in blood and usually express CD45, CD19 and CD20. Naïve (IgD+/IgM+), transitional, memory unswitched (CD27+ in humans and IgM+), memory switched (CD27+, IgE+, IgG+ or IgA+) and antibody-secreting B cells (CD20-, CD38+ and CD138+) also known as plasma cells (PCs) are the main types of conventional (B2) B cells (17, 18). The majority (60-70%) of circulating/blood B cells are naive, expressing both IgD and IgM while IgA+ memory B cells (MBCs) represent ˜ 10% of B cells in the peripheral blood (19) (Figure 1). They are circulating and can relocate to secondary lymphoid organs (SLOs) such as the lymph nodes (LNs) or tertiary lymphoid organs (TLOs). In the lung, the mucosa associated lymphoid tissue is known as the inducible bronchus associated lymphoid tissue (iBALT) (20). iBALT is a lymphoid aggregate that is usually rarely detectable in the lung at homeostasis in mouse or in man, but can develop following infections or during chronic inflammation (20, 21).
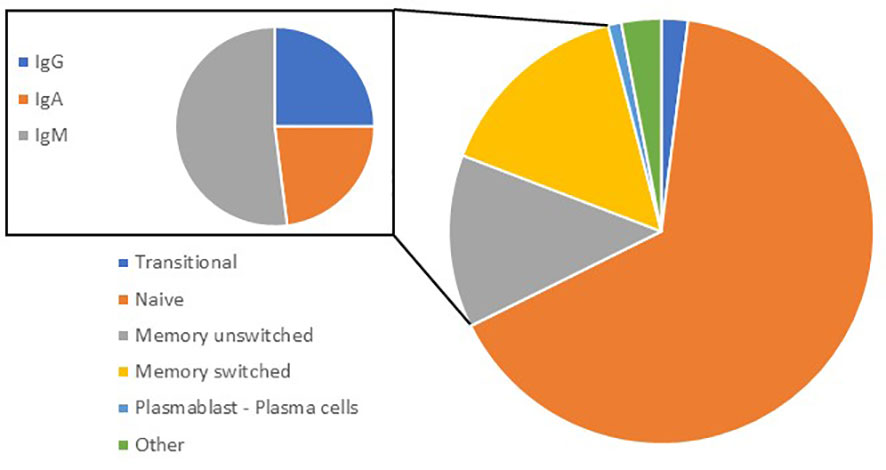
Figure 1 Proportion of B-cell subsets in blood. In peripheral blood, transitional (CD24+ CD38+) represent 2.4%; naive (CD27-; IgD+) 65%; memory unswitched (CD27+; IgD+) 15%; memory switched (CD27+; IgD-) 13; plasmablasts/plasma cells (CD24-; CD38+) 1%; and others 2.5%. Out of memory switched B cells (inset), 23% are IgG+, 21% are IgA+: 21%, and 52% are IgM+ (IgM+ only or IgM+/IgD+).
Although circulating B cells are present in the lung, other specific subtypes are represented in this organ. A subset of tissue-resident memory B cells (BRMs) has been identified in the lung in 2012 (22) and was described as the main producer of IgA in the respiratory system (23). This subset of B cells has mostly been studied in mice; they were identified by iv injection of labelled anti-CD45 antibodies followed by sorting and FACS analysis of B cells (24). In this study, non-circulating cells represented almost half of B cells in the lung (24, 25). Even though a specific marker for this subset was not established (26), mouse BRMs express CD69 and CD103 (22, 24, 26, 27) (Table 1). A larger proportion of BRMs are switched to IgA as compared to circulating blood B cells (24). Their presence and ability to stay (and not recirculate) in the lung is probably due to an increased expression of CXCR3, a receptor to the CXCL11 chemokine (23, 24). In addition, CXCR3 KO mice do not have BRMs and display an impaired production of IgA in the lung (23). BRMs cells have been described in different locations in the lung; after immunization against influenza virus, BRMs were observed in iBALT (28, 29) while alveolar BRMs were also reported after a similar immunization to influenza (29) or Streptococcus pneumoniae (29, 30) (Figure 2). Barker et al. also identified a putative subset of BRMs in the lungs from human donors, where most memory B cells (CD27+) expressed CD69 while naive B cells (IgD+) did not (30). Moreover, most of these CD69+ B cells were class-switched and did not express CD38, indicating that they were not yet differentiated into plasma cells (30).

Table 1 Comparison of IgA+ B cells in airways and in the gut.
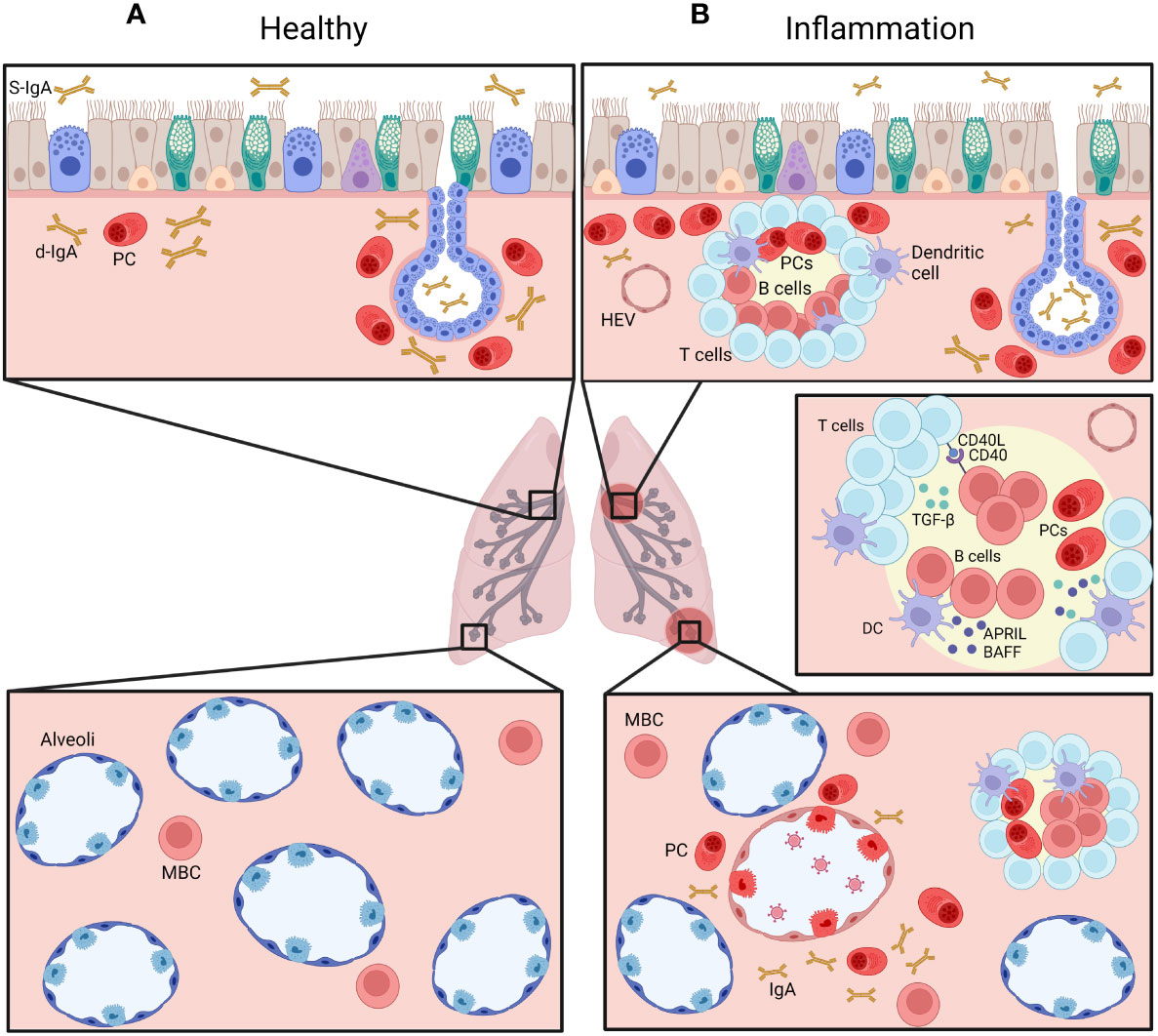
Figure 2 Distribution of IgA+ B cells in the airways, at homeostasis (A) and during chronic inflammatory condition (B). Plasma cells (PC) are normally mainly found beneath the airway epithelium and in submucosal glands (upper panels), representing a pro-IgA niche, and secrete d-IgA, which is transported through pIgR-mediated routing to the mucosal lumen. IgA+ memory B cells are also found in the alveolar septa (lower panels). During chronic inflammation, accumulation of IgA+ PCs is observed, along increased numbers of IgA+ B cells within lymphoid follicles, following cognate interactions with T cells as well as innate signaling from dendritic cells (DC). Created with BioRender.com.
Plasma cells (PC) are the terminal differentiation stage of the B-cell lineage that secretes antibodies. They differentiate from GC B cells or memory B cells and are CD27+, CD38+ and CD138+ but CD20- (18). In the mucosa, IgA is the most represented isotype (70% to 90%) (31, 32), most of them (around 80%) being located in the gut, especially in the gut-associated lymphoid tissue (GALT) (32, 33). A recent research of 3 control subjects suggested that IgA1 is the predominant subclass among IgA PCs in healthy lungs (two thirds of IgA+ PCs) (34), while IgA2 increases during the late stage of COVID infection (34).
As most IgA+ PCs are located in the gut, IgA-secreting PCs are also present in the murine lung. Twenty days after primary influenza virus infection or intranasal vaccination, IgA+ PCs derived from BRMs can be observed in the submucosal areas, beneath the bronchial epithelium (23, 29). Interestingly, IgA+ PCs are already detectable 4 days post-secondary infection, next to the alveoli instead of more proximal airways (29). IgA+ PCs have also been described in the lung after challenging ovalbumin (OVA)-sensitized mice (35).
A unique subset of IgA+, long-lived PCs (IgA+ LLPC), able to survive for years, was identified in the bone marrow (36, 37). Further studies led to identify that bone marrow IgA+ PCs were Ly6ahi TIGIT- and had a particularly high capacity to produce antibodies and a BCR being highly mutated due to SHM in the GC (38). It was thus suggested that IgA+ LLPC are differentiating in the mucosa before recirculating and relocating to the bone marrow. Interestingly, IgA+ LLPC were also observed in the gut, 80% of LLPCs being located in GALT (33, 39). Gut and bone marrow LLPCs share a long-lived phenotype (B220-), the same antibody repertoire and somatic hypermutation rate (40). The gut predominance as source of those cells could be explained by the fact that the gut represents the main source of overall IgA+ B cells. In addition, more attention has been given to gut-derived rather than lung-derived LLPCs (e.g., IgE+ LLPCs). However, one study showed that a subset of IgA+ LLPCs was induced upon intranasal vaccination or local allergen challenge (ovalbumin) that was more efficient than systemic sensitization (35, 41). Thus, the condition of induction and exact role of lung-derived IgA+ LLPCs remain unclear and should be further studied especially in human. (Table 1).
In blood as well as in tissue, there are several subsets of B cells that can be classified as Bregs because of their regulatory role; these include CD24+/CD27+ with both a memory and regulatory phenotype; CD24+/CD38+ immature cells, CD27+/CD38+ plasmablasts as well as a subset of CD25+/CD71+ B cells and CD43+ B1 cells. Bregs may modulate inflammation, notably via cytokine secretion of IL-10, IL-35 or TGF-β (42). A recent study discovered a subset of blood-derived IgA+ Bregs that produce IL-10 also expressing PD-L1 (43). Interestingly, whereas those IgA+ Bregs expressed IL-10, PD-L1 and Fas-L, they did not express the classical Breg markers, suggesting that IgA+ Bregs may represent a new subset (Table 1). Another study identified a subset of circulating IgA-secreting Bregs in mice that was induced following intranasal exposure to a fusion protein made of flagellin (FlaA) and Bet v1 allergen (44). They showed production of both IgG and IgA by those Bregs. Whereas those IgA antibodies could not bind to the triggering antigen, it was demonstrated that IgA+ Bregs could inhibit Th2 inflammation, opening therapeutic perspectives in allergy (44).
B1 cells have been discovered in mice as the main subset of B cells found in the pleural and peritoneal cavities (45). Considered as part of the innate immune system, B1 cells are developed at neonatal stages and have the ability to secrete antibodies prior to any infection or antigen exposure (45, 46). B1 cells from these cavities are characterized by their expression of CD19, surface IgM (high), surface IgD (low), CD43 and CD11b (47, 48). This population can be further divided into two main phenotypes, namely B1a cells (CD5+) and B1b cells (CD5-) (49). In the pleural cavity, the majority of B1 cells (75%) are corresponding to the B1a phenotype (48) but those cells may trans-differentiate to B1b cells after stimulation by pathogens (48, 50). Both B1 subsets have the ability to produce “natural antibodies” (NAbs) (49), which can be IgM but also IgG3 or IgA isotype. NAbs can be defined as non-specific antibodies synthesized prior to immunization (51, 52). They are cross-reactive and detect a large spectrum of antigens with low affinity, including self-antigens (51, 53) and a hypothesis even suggests that NAbs rise following exposure to self-antigens (53). Because of their cross-reactivity, NAbs secreted in the mucosa can interact with potential pathogens even during the first encounter with the microorganism and play a key role in the innate immune response to infections (54–56). For instance, anti-GAL NAbs are observed in serum since childhood and are able to bind to several microorganisms (57) and anti-phosphorylcholine NAbs recognize Gram-positive bacteria (46). In addition, it has been shown that their low affinity autoreactivity can play a helpful, homeostatic role in, notably with anti-phosphoryl NAbs that can bind to apoptotic cell membranes (46, 58).
Although there is no consensus yet for specific markers of B1 cells in the human body (52, 53), some “B1-like cells” have been described and characterized as CD27+, CD43+, CD70- in peripheral blood and in the umbilical cord (46, 53, 59). A review on the ontogeny of human B1 cells highlighted that this subset represents 1% of B cells in the adult blood, this number decreasing with age (60). When looking specifically at IgA-producing B1 cells, a population expressing CD27 and CD43 was described in blood (61, 62). Moreover, a particular phenotype of IgA+ innate lymphocytes named “natural helpers” was observed in both gut and lung (63–66). Researchers discovered that those natural helpers were producing cytokines similar to Th2 such as IL-5, IL-6 and IL-13 (64, 65). In the lung, these natural helpers have been only characterized in mouse models, with a potential role in allergic asthma (65). (Table 1).
B cells circulating in the bloodstream can enter tissues, either in lymph nodes (LN) or in mucosa associated lymphoid tissue (MALT) (33, 67, 68). Although the generic mechanisms beyond the homing to those locations are similar, some specific axis are also involved. The homing of B cells has been well described in the gut; in contrast, there are many differences with the iBALT. The iBALT is rarely observed in healthy individuals, their induction requiring the presence of inflammatory signals (20, 69). In response to IL-17, different stromal cells present at the early stage of iBALT formation, such as follicular dendritic cells, can recruit B cells (70, 71). This recruitment starts in high endothelial venules (HEVs), at the border of iBALT, where B cells circulating in the bloodstream can interact with endothelial cells to home into this tissue. Several phenotypes of circulating B cells (naïve, transitional, memory unswitched, memory switched) express L-selectin (CD62-L), a receptor to peripheral lymph node addressin (PNAd) that is a signalling molecule on the membrane of endothelial cells in iBALT (67). This leads to the attraction of B cells to the walls of HEVs, allowing the adhesion of B cells to the endothelium via α4β1 integrins interacting with V-CAM1 (67). This is an important difference compared to the homing into GALT, where the adhesion is driven by the binding of α4β7 integrins to Mad-CAM1 (67). Subsequently, the CXCL13 chemokine secreted by DCs in the central zone will attract B cells through their CCR5 receptor, leading to the formation of a B-cell follicle in the iBALT (20, 70). It has been described that in some iBALT structures, fibroblast-like cells could alternatively guide B cells via the CXCL12-CCR4 axis (71).
The development of mucosal vaccines is a real challenge, with homing of B cells to the lung being an important element of this research. Back in 2004, it was proven that after intranasal inoculation of SARS-CoV virus, mice developed pulmonary lymphoid follicles along with upregulation of the chemokines CXCL9, CXCL10 and their receptor CXCR3 (72). Similar results were observed when mice were intranasally inoculated with Bordetella bronchiseptica (73). Moreover, transgenic mice lacking CXCR3 had fewer lymphocytes in lung tissue and displayed a delayed clearance of the bacteria (73). More recently a murine model of intranasal immunization with Influenza virus demonstrated an increase of B cells in the lung as well as higher levels of CXCL11 and IgA (22, 23). The studies showed that the IgA-producing B cells in those murine models were also expressing CXCR3 and that this receptor was a key for local IgA production, as CXCR3-/- mice had fewer IgA+ cells in the lung (22, 23). A study conducted on delta-inulin, a potential adjuvant, showed that pulmonary immunization against Influenza coupled with this adjuvant led to a 4-fold increase of the number of CXCR3+ class-switched memory B cells in the lung as well as an increase of local IgA and IgG production (74). When re-challenged by a lethal exposure to Influenza, mice that did not receive the adjuvant during the immunization suffered from weight loss and poor survival after 8 to 9 days while mice who received the adjuvant were fully protected (74). Collectively these studies in murine models suggest that the homing of B cells via CXCR3 pathway is key to the immune response to infection and could be targeted for improving the development of mucosal vaccines.
It is known that a class-switch recombination (CSR) to IgA can happen via T-cell dependent or independent pathways (33, 75–77). Since 1989, transforming growth factor β1 (TGF-β1) has been identified as the main cytokine driving CSR of naive B cells to IgA by a T-cell dependent pathway (78–80) (Figure 2). In the lymph nodes of the lungs or the gut, more specifically in the germinal center (GC), naive B cells can be activated when their BCR binds to their cognate antigen. Whenever this event happens, the expression of CCR7 will increase at the expense of the expression of CXCR5. Thus, those B cells will be attracted by CCL21, at the periphery of the GC, where CD4+ Th cells reside (77, 81). Th cells can secrete active TGF-β1, which binds to TGF-β receptor (TGF-βR) and activates its serine/threonine kinase activity (75, 82), leading to the phosphorylation and activation of SMAD2 and SMAD3 and allowing them to interact with SMAD4 and RunX3 to promote the expression of Iα and Cα, which are mandatory for the initiation of CSR (75, 80, 83). In addition, CD40 ligand (CD40L), a protein from the tumor necrosis factor (TNF) family also expressed by Th cells, is mandatory for the T-cell dependent pathway (84) (Figure 3). CD40L can bind to CD40, a transmembrane protein of B cells which will induce the recruitment of TNF receptor associated factor (TRAF) followed by activation of the NF-κb kinase inhibitor (IKK) (75, 85), inducing its phosphorylation and subsequently its ubiquitination to allow NF-κB to play its transcription factor activity and promote the transcription of AICDA (75, 85, 86). Other factors can synergize with CD40L, such as IL-4 which is able to activate the transcription factor STAT6 which also promotes AICDA transcription (86). The process of T-cell dependent CSR toward IgA takes a week (75) and can lead to different endpoints; B cells can return to the follicle, proliferate and undergo somatic hypermutation (SHM), or recirculate in the periphery after differentiation into MBCs or ASCs (77).
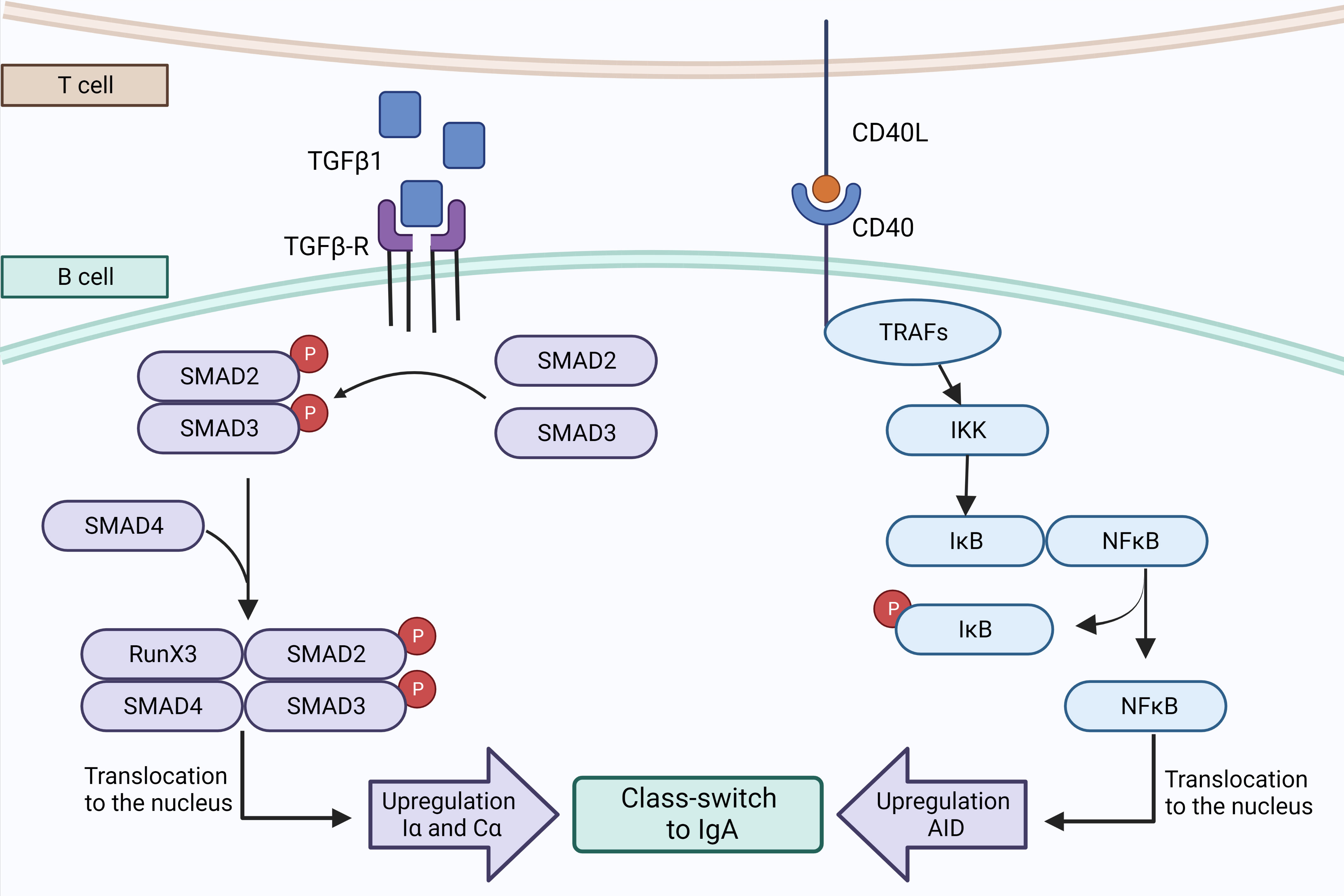
Figure 3 T cell-dependent pathway to IgA synthesis. Th cells may secrete active TGFβ1 which binds to TGFβ-R, triggering the phosphorylation and dimerization of Smad2 and Smad3 that are interacting with Smad4 and RunX3 to promote the transcription of Iα and Cα. The second signal consists of CD40 that interacts with CD40L expressed by T cells, to recruit a complex of TRAFs protein that activates IκB kinase. The phosphorylation of IκB by IKK unleashes NFκB and allows its translocation to the nucleus where it promotes the transcription of AID and CSR to IgA. Created with BioRender.com.
T cell-independent pathways allow B cells to switch to IgA faster and can be mediated by dendritic cells (DCs) (75, 76, 87) (Figure 4). It has been shown that lung DCs are able to induce a local CSR toward IgA with a better efficiency than spleen DCs (88). The main cytokines secreted by DCs that induce IgA switching are B-cell activating factor (BAFF, or BLyS) and A proliferation-inducing ligand (APRIL) (76), two cytokines of the TNF superfamily (89). Both are ligands to the B-cell maturation antigen (BCMA) and transmembrane activator et CALM interactor (TACI) (90), whereas BAFF may also bind a selective BAFF-receptor (BAFF-R) (90).
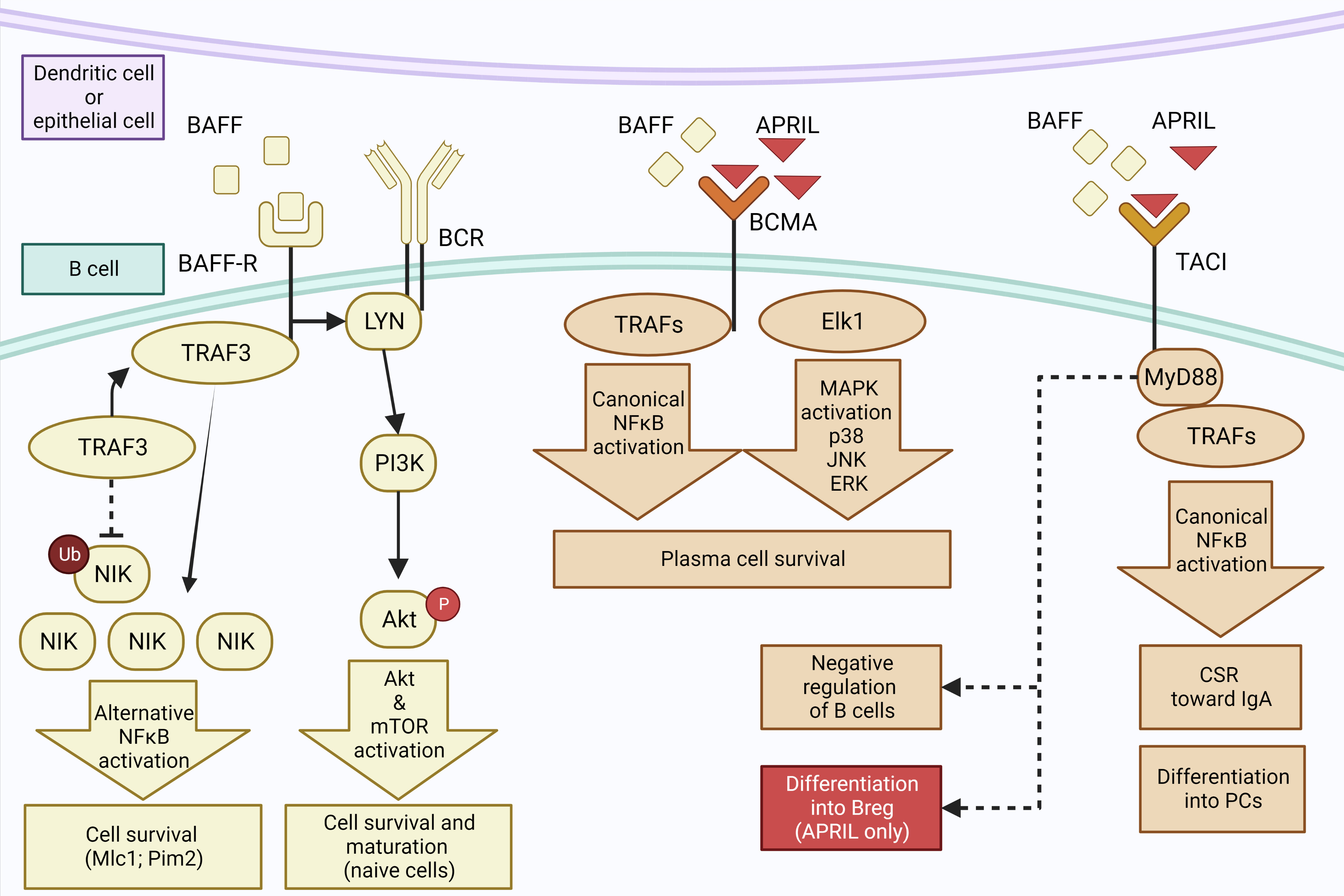
Figure 4 T cell-independent pathway to IgA synthesis. Epithelial and dendritic cells may secrete BAFF that binds to BAFF-R, inducing an interaction between BAFF-R and TRAF3 which induces ubiquitinylation of NIK. Accumulation of NIK leads to activation of NFkB that promotes B-cell survival. BAFF-R also interacts with BCR and Lyn, activating the PI3 kinase and Akt/mTOR pathways to promote survival and maturation of naive B cells. Secondly, BCMA receptor can also bind BAFF as well as APRIL, inducing the recruitment of TRAFs and the activation of NFkB as well as of Elk1 and MAP Kinase pathways, promoting the survival of plasma cells. Third, ligation of TACI by BAFF or APRIL activates the canonical NFkB pathway via its interaction with MyD88, leading to the differentiation of B cells into PCs and class switch to IgA. It may also induce a negative regulation of B cells through an unknown mechanism, or the differentiation of B cells into Bregs when the binding by APRIL occurs in the absence of BAFF. Created with BioRender.com.
TACI receptor has slightly stronger affinity for BAFF than APRIL (91) but it is not able to bind all forms of those molecules; only oligomeric forms of APRIL or BAFF can bind to TACI (92). Interestingly, TACI is able to induce survival signal but also to negatively regulate B cells (92). The pathways behind these opposite activities are not fully understood but TACI can interact with MyD88, an activator of NF-κB (43, 93) as well as with TRAF3 that inhibits the NF-κB pathway (77). The interaction between TACI and BAFF and APRIL can induce the CSR to IgA but also the differentiation into PCs by promoting the expression of Prdm1 (93), which encodes Blimp-1 that is a key transcription factor promoting XPB1 and inducing a secretory phenotype (94).
It has been reported that ligation of TACI by APRIL in the absence of BAFF, also induces CSR to IgA (5). Although the mechanisms of CSR driven by TACI remain unclear, TACI could interact with the MyD88 protein adaptor in order to activate NF-κB, inducing CSR by promoting the transcription of activation induced cytidine deaminase (AICDA) or CH genes which are crucial for CSR (93). In addition, a recent in-vitro study showed that APRIL stimulation of TACI, coupled with IL-21 and in the absence of BAFF or TGF-β, induces the differentiation of B cells isolated from PBMC to a subset of Bregs expressing IgA and IL-10 (43). This study also suggested that this specific subset of IgA+ Bregs could play a protective role against experimental autoimmune encephalomyelitis or arthritis (43). A similar cell phenotype was observed in mice after immunization against an allergen, and induction of IgA+ Bregs in this context was also dependent on MyD88 and NFkB activation (44). Signalling pathways underlying the differentiation of B cells into IgA+ Bregs could open the way for new therapeutic strategies in allergic and inflammatory airway diseases.
BCMA is the second TNF receptor able to bind both BAFF and APRIL, with a stronger affinity for the later (95). This receptor plays a key role in the survival of PCs, a defect of BCMA or its ligand resulting in reduced number of PCs in the bone marrow (96). The signalling pathways downstream of BCMA remain unclear but it is clear that it cannot interact with Myd88 (97)and that they lead to survival signalling pathways such as the classical NF-κB, Elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase pathways (97, 98). BAFF can also bind with a strong affinity to BAFF-R, which is present on all B-cell subsets (99). This will induce the activation of both the canonical and alternative NF-kB pathways, leading to the induction of survival signals for B cells. In addition, ligation between BAFF and BAFF-R causes the activation of phosphatidyl-inositol 3 kinase (PI3K), which activates the downstream signalling pathway of Akt and also induces survival of B cells.
Retinoic acid (RA), a widely studied molecule derived from vitamin A, can interact with specific nuclear receptors expressed in various organs (100). In the lung, RA plays an important role for prenatal lung organogenesis (100) and supports post-injury alveolar regeneration in the adult lung (101). In addition to interactions with epithelial or mesenchymal cells, it also contributes to maintenance of mucosal immunity (102–105). Thus, DCs can synthesize RA which interacts with B cells via their RA-receptor (106). It has been demonstrated in vitro that RA induces a class-switch to IgA in B cells from human tonsils (106) and murine spleen by interacting with RA-receptors on B cells (107). In addition, RA may accelerate the differentiation process from B cells to PCs (108), further increasing the production of IgA antibodies. This is collectively supported by the fact that a defect in vitamin A results in impaired generation of IgA+ B cells (109).
Although selective IgA deficiency (IgAD) is the most prevalent immune deficiency, affecting 1 out of 100 to 1000 individuals and that most IgAD subjects are asymptomatic, this defect increases the risk of upper and lower respiratory tract infections (110). Studies in mice showed that although naive IgA+/+ and IgA-/- mice have the same susceptibility against influenza infection (111–113), whenever they were exposed to influenza virus 5 weeks after vaccination without adjuvant, the survival of IgA -/- mice was decreased compared to controls, suggesting that IgA could provide postvaccinal protection (113). This statement is reinforced by the fact that local immunization against influenza induces the secretion of IgA by PCs as well as BRMs in submucosal areas of the airways and offers a better protection against infection than systemic immunization, which does not affect IgA levels (23). It is however known that IgA-/- mice display abnormalities in generating other isotypes such as IgE or IgG (114). Nevertheless, intranasal vaccination that promotes S-IgA responses has gained interest, notably following COVID pandemics.
Chronic obstructive pulmonary disease (COPD) is a common respiratory disease and the third cause of death worldwide since 2019 (115). It is associated with tobacco exposure and characterized by emphysema and obstruction of small airways (116). Lymphoid aggregates are developing near the distal airways of COPD patients (117) and our group described that IgA+ B cells in those areas were the only ones that upregulate during disease, mostly in severe patients (118). An accumulation of IgA1 was also observed in the subepithelial area (119), consistently with an increased synthesis combined with the lack of transport to the lumen through the pIgR which is downregulated in COPD (120). In addition, IgA autoantibodies against cytokeratins 18 and 19, expressed by epithelial cells, are increased in the plasma from COPD patients (121). Activation of B cells in this disease could be mediated by T cells, and more specifically by IL-21-secreting Th17 cells within lymphoid follicles (118). It is also possible that airway epithelial cells contribute to activate B cells as primary bronchial epithelial cells from COPD patients provide signals (such as IL-6) to B cells to promote their IgA synthesis, possibly through TACI upregulation (119, 122).
Patients with cystic fibrosis (CF) are particularly susceptible to airway and lung infections. CF is a genetic autosomal recessive disease due to a mutation on the gene encoding for thecystic fibrosis transmembrane conductance regulator (CFTR) protein, a ion channel for chloride anions (123), leading to dehydration of the airway mucus that becomes sticky and promotes respiratory infections with opportunistic pathogens such as Pseudomonas aeruginosa (Pa) (124). In CF lungs, lymphoid aggregates that include IgA+ PCs (CD138+) were observed (124). Similarly, increased levels of IgA can be observed in serum and bronchoalveolar lavage (BAL) from CF patients compared to healthy controls (125). In addition, the increase in IgA in serum and airway secretions was correlated with chronic infection by Pa (126–128). These results were similar to murine models, where it was also demonstrated that Pa infection leads to an upregulation of pIgR in both control and CF mice as well as an increased production of IgA, leading to an increase of BAL S-IgA (126). It was also reported that BAFF mediates the induction of IgA following Pa infection in mice (129), whereas depletion of BAFF increases the susceptibility to Pa of CF mice. Thus, it seems that Pa infection induces a specific IgA immune response that is associated in CF with a global upregulation of the IgA/pIgR system, which relate notably to BAFF and IL-17 signalling. This response is not associated with the eradication/clearance of the pathogen but rather accompanies chronic infection.
In addition, IgA can also represent a biomarker of infection or disease severity in CF. It is long known that CF patients display elevated levels of autoantibodies, among which IgA autoantibodies, in serum and sputum (130, 131). It can be already observed in pediatric cohorts but the levels of some specific antibodies increase with the age (132–134). The most studied IgA autoantibodies in CF are antineutrophil cytoplasm antibodies (ANCA) and more specifically the one targeting bactericidal/permeability-increasing protein (BPI) (132, 135–138). Neutrophils are major immune cells present in CF airways, putatively playing protective roles against pathogens (139) while also exacerbating airway damage, e.g. by releasing neutrophil elastase that degrades peribronchial tissue (139). In addition, dead neutrophils are the main source of extracellular DNA that further thickens the mucus of CF patients (140). A meta-analysis on the topic indicates that around 50% of the patients had elevated levels of BPI-ANCA IgA (137). They also demonstrated that the levels of BPI-ANCA were associated with increased prevalence of P. aeruginosa infection, increased response to P. aeruginosa infection and decreased lung function (137). IgA autoantibodies directed against double-stranded DNA were also observed both in serum and sputum from CF patients but only the systemic one was associated with a worse outcome (140). Finally, IgA autoantibodies against PAD-4 were identified, also in serum and sputum secretions, correlating with disease severity (141). Furthermore, it was reported that serum levels of IgG BPI-ANCA correlated those of antibodies against P.aeruginosa (132)whereas these IgG autoantibodies are not present in the airways where only IgA BPI-ANCA are observed, despite that IgG antibodies against P.aeruginosa are also present, suggesting different pathways underlying local/airway vs systemic autoimmune responses (132). In another work in children with CF, an increase in the number of B cells was reported in blood compared to BAL, also supporting this hypothesis (142). Finally, a study on the response to P.aeruginosa infection in children demonstrated differences between systemic and local responses (143), all children with CF -whether they had chronic, acute, or even no history of P. aeruginosa infection- displaying higher BAL levels of IgA against P. aeruginosa than did control children. On the other hand, children with no history of P. aeruginosa infection had similar levels of serum specific IgA as control children (143). Thus, whereas IgA may play a protective role against infection, it is likely that it also play roles during autoimmune responses in the lung. Whereas IgA autoantibodies are well documented in CF, the presence of autoreactive B cells and their location or biology has been less studied. The relationship between a specific subset of natural killer T cells and IgG autoreactive B cells in the lung was explored in a murine model, showing that the number of germinal centers autoreactive IgG+ cells in the lung was inversely correlated to this T cell subset (144). To our knowledge, this has not been evaluated with IgA autoreactive B cells, probably mostly due to difficulties in isolating or detecting antigen specific B cells particularly in tissues where a method has however been developed using biotinylated specific antigens revealed by avidin-cyanin 3 (145).
Finally, the IgA transport system seems also altered in this disease (116, 146), with upregulation of pIgR expression allowing increased transcytosis of IgA into mucosal secretions (118, 137). despite an intrinsic inhibitory effect of the mutated CFTR on pIgR expression in cells or animals (126, 146).. When CFTR-mutated mice were infected with P.aeruginosa, pIgR expression was upregulated, probably through the IL-17 pathway, indicating that infection could restore and even upregulate the IgA/pIgR system in this disease (126).
In asthma, an association was recently described between disease severity and increased blood IgA+ B cells in a cohort of 154 patients and 28 healthy individuals (147). Using flow cytometry, this study showed that more severe asthma was associated with a decrease of circulating naïve and transitional B cells and an increase of IgA+ B cells compared to controls as well as compared to patients with mild asthma (147). It was also described that BAFF levels were increased in lung tissue and BAL (but not in serum) in human asthma (148, 149). In ovalbumin (OVA)-induced allergic experimental asthma (95), it was shown that those mice had increased IgA levels in both serum and BAL (150). Interestingly, eosinophils, which are major effector cells of allergic asthma, have a high expression of the myeloid IgA-receptor FCαR1 in mice (150), as observed in allergic patients (151). S-IgA can readily induce the degranulation of eosinophils (152, 153), suggesting that IgA could contribute to the pathogenesis of mucosal inflammation in asthma (150). Furthermore, inhibition of CD40-CD40L or OX40-OX40L interactions in mice results in lower total and OVA-specific IgA levels in lung tissue, BAL and serum, which could suggest that this increase is at least partly due to a T–cell-dependent mechanism (150).
IgA and IgA+ B cells could also play a role in both autoimmunity and fibrogenesis in interstitial lung disease. In particular, idiopathic pulmonary fibrosis (IPF), a deadly pathology characterized by the destruction of the architecture of the distal lung with an accumulation of collagen due to the hyperactivation of fibroblasts (154, 155). Recent data suggest that B cells and IgA could play a role in this pathology. First, lymphoid aggregates were observed in IPF lungs (156, 157) and elevated concentration of IgA antibodies in serum from patients with IPF has been associated with a decreased survival (158). IPF patients also display increased circulating IgA autoantibodies compared to healthy individuals (159, 160), which correlate with the number of TLOs in the lung (160). In addition, increased numbers of PCs and transitional B cells were present in the IPF lung tissue, and TLOs were strongly stained for IgA (159). Moreover, an increase of CXCL13 was observed in blood as well as in the lung tissue from IPF patients, particularly in TLOs and near the fibroblast foci (161). Furthermore, B cells in TLOs express CXCR5 (161) but not Ki67 (157), suggesting that they could mainly be recruited from blood through a CXCL13-CXCR5 axis. It is known that TGF-β is increased in IPF tissue (162), probably produced by Th cells in TLOs (159), leading to the hypothesis that CSR to IgA could be induced locally. The precise role of IgA+ B cells in IPF remains however unclear and should be further studied.
Few data exist on the role of IgA and IgA-producing B cells in sinonasal diseases (163). In patients with selective IgAD, 48-78% display recurrent upper airway infections, including rhinitis and rhinosinusitis (164–166), likely due to the major role of IgA against viral infections (167). Upper airway infections with viruses such as rhinovirus (168), influenza (169) and SARS-CoV2 (170) lead to an increase of S-IgA in the nasal lavage, which plays a protective role in man (169) and mice (126, 127). Additional evidence emerged from studies in endurance athletes, strenuous exercise decrease upper airway S-IgA production that correlates to the increased rate of upper respiratory tract infections (171–173).
In patients with allergic rhinitis (AR), nasal allergen challenge induces local IgA production (153), and specific nasal IgA responses have been reported for different allergens, including house dust mites (174), grass (175), ragweed (176, 177), birch pollen (178, 179), and red cedar (180) with antigen-specific IgA levels correlating with nasal symptoms (180). However, patients with IgAD are more likely to develop AR (165, 166) and AR patients have lower total IgA levels in the serum as well as in their nasal tissue compared to healthy controls (181, 182). One study showed that intranasal administration of ragweed-specific IgA protected against allergic inflammation in sensitized mice (183), and that the production of allergen-specific IgA in neonatal mice prevented the development of cockroach allergy (183). Several studies also reported that successful allergen immunotherapyis associated with allergen-specific IgA responses (184–191) along increased IgA+ B cells, PCs, and Bregs that correlate with the clinical benefit (192, 193).
In chronic rhinosinusitis (CRS) patients it has been reported that 16.7% has low levels of serum IgA, with 6.2% matching the definition of selective IgAD (194). Similarly CRS is more frequently reported in individuals with IgAD (up to 78%) compared to individuals with normal IgA levels (166). In patients suffering from CRS with nasal polyps (CRSwNP), increased numbers of B cells and PCs are found in polyp tissue (195, 196) and these PCs abundantly produce Igs including IgA. It has been reported that these B cells infiltrating NPs are contributing to organize TLOs (196–198), suggesting a unique B cell activation environment within NPs that is distinct from classic GC-mediated mechanisms (199). Several studies also reported overexpression BAFF in patients with CRSwNP (200–203) and of local IgA+ PCs (204), IgA1+ B cells serving as precursors for IgA2+ B cells (205). One recent paper also suggested the possibility of local conversion of antigen-specific IgA to IgE (206), but this needs to be validated. Increased S-IgA levels were observed in CRSwNP compared to healthy controls in one study (204), but not in another (207). The latter study also showed increased tissue IgA in CRSwNP with paradoxically reduced IgA antibody levels to S. aureus enterotoxin B (207), a powerful pro-Th2 superantigen. Elevated levels of IgA autoantibodies to double-stranded DNA and basement membrane proteins have also been observed in CRSwNP (208).
In the lung, IgA-producing B cells and IgA+ PCs are found in the airways, close to the surface epithelium and around submucosal glands, as well as in the lung parenchyma. Following its transport across the epithelium through pIgR-mediated trafficking, IgA antibodies protect the conducting airways via immune exclusion of inhaled pathogens and antigens. During disease state such as infections or COPD, iBALT can develop and recruits B cells by producing the chemokine CXCL13 that attracts the CCR5-expressing B cells. T cells that are present in iBALT can induce class-switching to IgA, through TGF-β and concomitant interaction between CD40L and CD40. Dendritic cells may also induce IgA synthesis in the lung by secreting APRIL and/or BAFF that bind to three different receptors and lead to the activation of NF-κB and Akt pathways that underlie survival and maturation of naïve B-cells. Interestingly, TACI activation is closely related to class-switching to IgA and differentiation into PCs.
Lung-tissue resident memory B cells were identified in the lung, but within airways and alveoli. Upon antigen rechallenge, those resident cells can move near the new infection site via CXCL13-CCR5 interaction, where they differentiate into PCs to secrete protective antibodies. This host response was not observed upon systemic immunization, suggesting that mucosal vaccines could represent a specific route for prevention against respiratory infections. In addition, a subset of Bregs that produce IgA was discovered following activation by APRIL in the absence of BAFF, or following activation by a specific fusion protein containing an allergen. Importantly, IgA+ Bregs produce IL-10 and are potent regulators of allergic inflammatory responses in mice. In contrast, IgA and IgA+ B cells could also contribute to chronic inflammation, possibly through autoimmune mechanisms. iBALT from COPD and IPF patients include IgA+ B cells, a feature that correlates with increased levels of circulating IgA autoantibodies directed against KRT18 and KRT19 (in COPD) or citrullinated proteins (in IPF). In the upper airways, IgA+ B cells may also play different roles, as patients with IgA deficiency are more susceptible to develop allergic rhinitis or CRS on the one hand, while in CSRwNP IgA autoantibodies against double stranded DNA could be detrimental on the other.
There remain many gaps in knowledge regarding IgA immunity in the lungs. Some of them are the characterization of B1-like and LLPC subsets, the selective pro-IgA signalling operating in the lung (including molecular signalling, e.g. behind TACI), as well as the regulation by IgA of airway/lung microbiome. Most importantly, future research should also address the functional role of IgA+ B cells that are present in several inflammatory diseases in the airways, to determine whether, how, and under which circumstances they exert protective and/or deleterious roles.
YB drafted the manuscript. AS and VH wrote the upper airway chapter. AF revised the lung chapters. CP revised the manuscript. All authors contributed to the article and approved the submitted version.
AS is supported by an Aspirant fellowship of the Fonds National de la Recherche Scientifique, Belgium. (FRC), Brussels, Belgium. CP is postdoctoral specialist of the FNRS (grant 1.R016.20), Belgium. VH is supported by clinicien-chercheur mandates of (grant FNRS 1R20221F), Belgium.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
APRIL, A proliferation-inducing ligand; AR, Allergic rhinitis; BAFF, B cell activating factor; BAFF-R, B cell activating-factor receptor; BCMA, B cell maturation antigen; Breg, Regulatory B cell; BRM, Tissue-resident memory B cells; CF, Cystic fibrosis; COPD, Chronic obstructive pulmonary disease; CRS, Chronic rhinosinusitis; CRSsNP, Chronic rhinosinusitis without nasal polyps; CRSwNP, Chronic rhinosinusitis with nasal polyps; CSR, Class switch recombination; DC, Dendritic cell; d-IgA, Dimeric IgA; GALT, Gut-associated lymphoid tissue; GC, Germinal center; HEV, High endothelium venule; iBALT, inducible bronchus-associated lymphoid tissue; IgAD, Selective IgA deficiency; IPF, Idiopathic pulmonary fibrosis; LLPC, Long-lived plasma cell; MALT, Mucosae-associated lymphoid tissue; MBC, Memory B cells; Nabs, Natural antibodies; NP, Nasal polyps; OVA, Ovalbumine; Pa, Pseudomonas aeruginosa; PC, Plasma cells; pIgR, Polymeric immunoglobulin receptor; RA, Retinoic acid; SC, secretory component; s-IgA, Secretory IgA; TACI, Transmembrane activation and CALM interactor; TLO, Tertiary lymphoid organ; TNF, Tumor necrosis factor; TRAF, TNF receptor associated factor
1. Mistry D, Stockley RA. IgA1 protease. Int J Biochem Cell Biol (2006) 38(8):1244–8. doi: 10.1016/j.biocel.2005.10.005
2. Woof JM, Russell MW. Structure and function relationships in IgA. Mucosal Immunol (2011) 4(6):590–7. doi: 10.1038/mi.2011.39
4. Burnett D, Crocker J, Stockley RA. Cells containing IgA subclasses in bronchi of subjects with and without chronic obstructive lung disease. J Clin Pathol (1987) 40(10):1217–20. doi: 10.1136/jcp.40.10.1217
5. Pilette C, Ouadrhiri Y, Godding V, Vaerman JP, Sibille Y. Lung mucosal immunity: immunoglobulin-a revisited. Eur Respir J (2001) 18(3):571–88. doi: 10.1183/09031936.01.00228801
6. Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol (2011) 4(6):598–602. doi: 10.1038/mi.2011.37
7. Sun K, Johansen FE, Eckmann L, Metzger DW. An important role for polymeric ig receptor-mediated transport of IgA in protection against streptococcus pneumoniae nasopharyngeal carriage. J Immunol (2004) 173(7):4576–81. doi: 10.4049/jimmunol.173.7.4576
8. Tjärnlund A, Rodríguez A, Cardona PJ, Guirado E, Ivanyi J, Singh M, et al. Polymeric IgR knockout mice are more susceptible to mycobacterial infections in the respiratory tract than wild-type mice. Int Immunol (2006) 18(5):807–16. doi: 10.1093/intimm/dxl017
9. Li Y, Jin L, Chen T. The effects of secretory IgA in the mucosal immune system. BioMed Res Int (2020) 2020:2032057. doi: 10.1155/2020/2032057
10. Mkaddem SB, Christou I, Rossato E, Berthelot L, Lehuen A, Monteiro RC. IgA, IgA receptors, and their anti-inflammatory properties. Curr Top Microbiol Immunol (2014) 382:221–35. doi: 10.1007/978-3-319-07911-0_10
11. Monteiro RC, van de Winkel JGJ. IgA fc receptors. Annu Rev Immunol (2003) 21(1):177–204. doi: 10.1146/annurev.immunol.21.120601.141011
12. Monteiro RC, Suzuki Y. Are there animal models of IgA nephropathy? Semin Immunopathol (2021) 43(5):639–48. doi: 10.1007/s00281-021-00878-5
13. Suzuki H, Suzuki Y. Murine models of human IgA nephropathy. Semin Nephrol (2018) 38(5):513–20. doi: 10.1016/j.semnephrol.2018.05.021
14. Xu L, Li B, Huang M, Xie K, Li D, Li Y, et al. Critical role of kupffer cell CD89 expression in experimental IgA nephropathy. PloS One (2016) 11(7):e0159426. doi: 10.1371/journal.pone.0159426
15. Boross P, Lohse S, Nederend M, Jansen JHM, van Tetering G, Dechant M, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med (2013) 5(8):1213–26. doi: 10.1002/emmm.201201929
16. Bos A, Aleyd E, van der Steen LPE, Winter PJ, Heemskerk N, Pouw SM, et al. Anti-FcαRI monoclonal antibodies resolve IgA autoantibody-mediated disease. Front Immunol (2022) 13:732977. doi: 10.3389/fimmu.2022.732977
17. Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for b cell subpopulations from infancy to adulthood. Clin Exp Immunol (2010) 162(2):271–9. doi: 10.1111/j.1365-2249.2010.04206.x
18. Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, et al. Circulating human b and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica (2010) 95(6):1016–20. doi: 10.3324/haematol.2009.018689
19. Allman D, Pillai S. Peripheral b cell subsets. Curr Opin Immunol (2008) 20(2):149–57. doi: 10.1016/j.coi.2008.03.014
20. Silva-Sanchez A, Randall TD. Role of iBALT in respiratory immunity. Curr Top Microbiol Immunol (2020) 426:21–43. doi: 10.1007/82_2019_191
21. Tschernig T, Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology (2000) 68(1):1–8. doi: 10.1159/000028109
22. Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, et al. Memory b cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc Natl Acad Sci U S A (2012) 109(7):2485–90. doi: 10.1073/pnas.1115369109
23. Oh JE, Song E, Moriyama M, Wong P, Zhang S, Jiang R, et al. Intranasal priming induces local lung-resident b cell populations that secrete protective mucosal antiviral IgA. Sci Immunol (2021) 6(66):eabj5129. doi: 10.1126/sciimmunol.abj5129
24. Fan L, Wu Q, Kang S, Yang B, Wu C. The phenotypic and functional study of tissue b cells in respiratory system provided important information for diseases and development of vaccines. J Cell Mol Med (2021) 25(5):2621–32. doi: 10.1111/jcmm.16278
25. Lee CM, Oh JE. Resident memory b cells in barrier tissues. Front Immunol (2022) 13:953088. doi: 10.3389/fimmu.2022.953088
26. Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, et al. The establishment of resident memory b cells in the lung requires local antigen encounter. Nat Immunol (2019) 20(1):97–108. doi: 10.1038/s41590-018-0260-6
27. Allie SR, Randall TD. Resident memory b cells. Viral Immunol (2020) 33(4):282–93. doi: 10.1089/vim.2019.0141
28. Tan HX, Juno JA, Esterbauer R, Kelly HG, Wragg KM, Konstandopoulos P, et al. Lung-resident memory b cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci Immunol (2022) 7(67):eabf5314. doi: 10.1126/sciimmunol.abf5314
29. MacLean AJ, Richmond N, Koneva L, Attar M, Medina CAP, Thornton EE, et al. Secondary influenza challenge triggers resident memory b cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity (2022) 55(4):718–733.e8. doi: 10.1016/j.immuni.2022.03.003
30. Barker KA, Etesami NS, Shenoy AT, Arafa EI, Lyon de Ana C, Smith NM, et al. Lung-resident memory b cells protect against bacterial pneumonia. J Clin Invest (2021) 131(11):e141810. doi: 10.1172/JCI141810
31. Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, et al. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood (2009) 113(11):2461–9. doi: 10.1182/blood-2008-04-153544
32. Gommerman JL, Rojas OL, Fritz JH. Re-thinking the functions of IgA(+) plasma cells. Gut Microbes (2014) 5(5):652–62. doi: 10.4161/19490976.2014.969977
33. Brandtzaeg P, Johansen FE. Mucosal b cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev (2005) 206(1):32–63. doi: 10.1111/j.0105-2896.2005.00283.x
34. Ferreira-Gomes M, Kruglov A, Durek P, Heinrich F, Tizian C, Heinz GA, et al. SARS-CoV-2 in severe COVID-19 induces a TGF-β-dominated chronic immune response that does not target itself. Nat Commun (2021) 12(1):1961. doi: 10.1038/s41467-021-22210-3
35. Luger EO, Fokuhl V, Wegmann M, Abram M, Tillack K, Achatz G, et al. Induction of long-lived allergen-specific plasma cells by mucosal allergen challenge. J Allergy Clin Immunol (2009) 124(4):819–26. doi: 10.1016/j.jaci.2009.06.047
36. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature (1997) 388(6638):133–4. doi: 10.1038/40540
37. Höfer T, Muehlinghaus G, Moser K, Yoshida T, E. Mei H, Hebel K, et al. Adaptation of humoral memory. Immunol Rev (2006) 211(1):295–302. doi: 10.1111/j.0105-2896.2006.00380.x
38. Liu X, Yao J, Zhao Y, Wang J, Qi H. Heterogeneous plasma cells and long-lived subsets in response to immunization, autoantigen and microbiota. Nat Immunol (2022) 23(11):1564–76. doi: 10.1038/s41590-022-01345-5
39. Lightman SM, Utley A, Lee KP. Survival of long-lived plasma cells (LLPC): Piecing together the puzzle. Front Immunol (2019) 10:965. doi: 10.3389/fimmu.2019.00965
40. Wilmore JR, Gaudette BT, Gómez Atria D, Rosenthal RL, Reiser SK, Meng W, et al. IgA plasma cells are long-lived residents of gut and bone marrow that express isotype- and tissue-specific gene expression patterns. Front Immunol (2021) 12:791095. doi: 10.3389/fimmu.2021.791095
41. Hassan AO, Shrihari S, Gorman MJ, Ying B, Yaun D, Raju S, et al. An intranasal vaccine durably protects against SARS-CoV-2 variants in mice. Cell Rep (2021) 36(4):109452. doi: 10.1016/j.celrep.2021.109452
42. Rosser EC, Mauri C. Regulatory b cells: origin, phenotype, and function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
43. Fehres CM, van Uden NO, Yeremenko NG, Fernandez L, Franco Salinas G, van Duivenvoorde LM, et al. APRIL induces a novel subset of IgA+ regulatory b cells that suppress inflammation via expression of IL-10 and PD-L1. Front Immunol (2019) 10:1368. doi: 10.3389/fimmu.2019.01368
44. Goretzki A, Lin YJ, Meier C, Dorn B, Wolfheimer S, Jamin A, et al. Stimulation of naïve b cells with a fusion protein consisting of FlaA and bet v 1 induces regulatory b cells ex vivo. Allergy (2022). doi: 10.1111/all.15542
45. Meyer-Bahlburg A. B-1 cells as a source of IgA. Ann New York Acad Sci (2015) 1362(1):122–31. doi: 10.1111/nyas.12801
46. Rothstein TL, Griffin DO, Holodick NE, Quach TD, Kaku H. Human b-1 cells take the stage. Ann N Y Acad Sci (2013) 1285:97–114. doi: 10.1111/nyas.12137
47. Ghosn EEB, Yang Y, Tung J, Herzenberg LA, Herzenberg LA. CD11b expression distinguishes sequential stages of peritoneal b-1 development. Proc Natl Acad Sci U S A (2008) 105(13):5195–200. doi: 10.1073/pnas.0712350105
48. Yenson V, Baumgarth N. Purification and immune phenotyping of b-1 cells from body cavities of mice. Methods Mol Biol (2021) 2270:27–45. doi: 10.1007/978-1-0716-1237-8_2
49. Baumgarth N. The double life of a b-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol (2011) 11(1):34–46. doi: 10.1038/nri2901
50. Savage HP, Kläsener K, Smith FL, Luo Z, Reth M, Baumgarth N. TLR induces reorganization of the IgM-BCR complex regulating murine b-1 cell responses to infections. eLife (2019) 8:e46997. doi: 10.7554/eLife.46997.015
51. Holodick NE, Rodríguez-Zhurbenko N, Hernández AM. Defining natural antibodies. Front Immunol (2017) 8:872. doi: 10.3389/fimmu.2017.00872
52. Panda S, Ding JL. Natural antibodies bridge innate and adaptive immunity. J Immunol (2015) 194(1):13–20. doi: 10.4049/jimmunol.1400844
53. Maddur MS, Lacroix-Desmazes S, Dimitrov JD, Kazatchkine MD, Bayry J, Kaveri SV. Natural antibodies: From first-line defense against pathogens to perpetual immune homeostasis. Clinic Rev Allerg Immunol (2020) 58(2):213–28. doi: 10.1007/s12016-019-08746-9
54. Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. marginal zone and B1 b cells as part of a ‘natural immune memory’. Immunol Rev (2000) 175:70–9. doi: 10.1111/j.1600-065X.2000.imr017515.x
55. Hayakawa K, Hardy RR. Development and function of b-1 cells. Curr Opin Immunol (2000) 12(3):346–54. doi: 10.1016/S0952-7915(00)00098-4
56. Baldan A, Gonen A, Choung C, Que X, Marquart TJ, Hernandez I, et al. ABCG1 is required for pulmonary b-1 b cell and natural antibody homeostasis. J Immunol (2014) 193(11):5637–48. doi: 10.4049/jimmunol.1400606
57. Hamanova M, Chmelikova M, Nentwich I, Thon V, Lokaj J. Anti-gal IgM, IgA and IgG natural antibodies in childhood. Immunol Lett (2015) 164(1):40–3. doi: 10.1016/j.imlet.2015.02.001
58. Shaw PX, Goodyear C, Chang MK, Witztum JL, Silverman GJ. The autoreactivity of anti-phosphorylcholine antibodies for atherosclerosis-associated neo-antigens and apoptotic cells. J Immunol (2003) 170(12):6151–7. doi: 10.4049/jimmunol.170.12.6151
59. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J Exp Med (2011) 208(1):67–80. doi: 10.1084/jem.20101499
60. Kageyama Y, Katayama N. Ontogeny of human B1 cells. Int J Hematol (2020) 111(5):628–33. doi: 10.1007/s12185-019-02775-y
61. Verbinnen B, Covens K, Moens L, Meyts I, Bossuyt X. Human CD20+CD43+CD27+CD5– b cells generate antibodies to capsular polysaccharides of streptococcus pneumoniae. J Allergy Clin Immunol (2012) 130(1):272–5. doi: 10.1016/j.jaci.2012.04.040
62. Wiest M, Upchurch K, Hasan MM, Cardenas J, Lanier B, Millard M, et al. Phenotypic and functional alterations of regulatory b cell subsets in adult allergic asthma patients. Clin Exp Allergy (2019) 49(9):1214–24. doi: 10.1111/cea.13439
63. Koyasu S, Moro K. Th2-type innate immune responses mediated by natural helper cells. Ann New York Acad Sci (2013) 1283(1):43–9. doi: 10.1111/nyas.12106
64. Koyasu S, Moro K. Innate Th2-type immune responses and the natural helper cell, a newly identified lymphocyte population. Curr Opin Allergy Clin Immunol (2011) 11(2):109–14. doi: 10.1097/ACI.0b013e3283448808
65. Halim TYF, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity (2012) 36(3):451–63. doi: 10.1016/j.immuni.2011.12.020
66. Koyasu S, Moro K. Natural ‘helper’ cells in the lung: good or bad help? Immunity (2012) 36(3):317–9. doi: 10.1016/j.immuni.2012.03.001
67. Xu B, Wagner N, Pham LN, Magno V, Shan Z, Butcher EC, et al. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by l-selectin/PNAd, α4β1 Integrin/VCAM-1, and LFA-1 adhesion pathways. J Exp Med (2003) 197(10):1255–67. doi: 10.1084/jem.20010685
68. Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol (2003) 3(10):822–9. doi: 10.1038/nri1203
69. Randall TD. Bronchus-associated lymphoid tissue (BALT): Structure and function. Adv Immunol (2010) 107:187. doi: 10.1016/B978-0-12-381300-8.00007-1
70. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12(7):639–46. doi: 10.1038/ni.2053
71. Fleige H, Ravens S, Moschovakis GL, Bölter J, Willenzon S, Sutter G, et al. IL-17-induced CXCL12 recruits b cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med (2014) 211(4):643–51. doi: 10.1084/jem.20131737
72. Glass WG, Subbarao K, Murphy B, Murphy PM. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J Immunol (2004) 173(6):4030–9. doi: 10.4049/jimmunol.173.6.4030
73. Widney DP, Hu Y, Foreman-Wykert AK, Bui KC, Nguyen TT, Lu B, et al. CXCR3 and its ligands participate in the host response to bordetella bronchiseptica infection of the mouse respiratory tract but are not required for clearance of bacteria from the lung. Infect Immun (2005) 73(1):485–93. doi: 10.1128/IAI.73.1.485-493.2005
74. Tomar J, Patil HP, Bracho G, Tonnis WF, Frijlink HW, Petrovsky N, et al. Advax augments b and T cell responses upon influenza vaccination via the respiratory tract and enables complete protection of mice against lethal influenza virus challenge. J Control Release (2018) 288:199–211. doi: 10.1016/j.jconrel.2018.09.006
75. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol (2008) 8(6):421–34. doi: 10.1038/nri2322
76. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3(9):822–9. doi: 10.1038/ni829
77. Kato A, Hulse KE, Tan BK, Schleimer RP. B-lymphocyte lineage cells and the respiratory system. J Allergy Clin Immunol (2013) 131(4):933–57. doi: 10.1016/j.jaci.2013.02.023
78. Sonoda E, Matsumoto R, Hitoshi Y, Ishii T, Sugimoto M, Araki S, et al. Transforming growth factor beta induces IgA production and acts additively with interleukin 5 for IgA production. J Exp Med (1989) 170(4):1415–20. doi: 10.1084/jem.170.4.1415
79. Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine b lymphocytes. J Exp Med (1989) 170(3):1039–44. doi: 10.1084/jem.170.3.1039
80. Stavnezer J, Kang J. The surprising discovery that TGFβ specifically induces the IgA class switch. J Immunol (2009) 182(1):5–7. doi: 10.4049/jimmunol.182.1.5
81. Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Förster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines b-cell position. Nature (2002) 416(6876):94–9. doi: 10.1038/416094a
82. Rubtsov YP, Rudensky AY. TGFβ signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol (2007) 7(6):443–53. doi: 10.1038/nri2095
83. Park SR, Lee JH, Kim PH. Smad3 and Smad4 mediate transforming growth factor-β1-induced IgA expression in murine b lymphocytes. Eur J Immunol (2001) 31(6):1706–15. doi: 10.1002/1521-4141(200106)31:6<1706::AID-IMMU1706>3.0.CO;2-Z
84. Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A (2001) 98(22):12614–9. doi: 10.1073/pnas.221456898
85. Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol (2005) 5(6):435–45. doi: 10.1038/nri1629
86. Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol (2004) 16(3):395–404. doi: 10.1093/intimm/dxh042
87. Fagarasan S, Honjo T. T-Independent immune response: New aspects of b cell biology. Science (2000) 290(5489):89–92. doi: 10.1126/science.290.5489.89
88. Naito T, Suda T, Suzuki K, Nakamura Y, Inui N, Sato J, et al. Lung dendritic cells have a potent capability to induce production of immunoglobulin a. Am J Respir Cell Mol Biol (2008) 38(2):161–7. doi: 10.1165/rcmb.2007-0237OC
89. Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr Opin Immunol (2005) 17(3):282–9. doi: 10.1016/j.coi.2005.04.005
90. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-r mediate isotype switching in b cells. J Exp Med (2005) 201(1):35–9. doi: 10.1084/jem.20032000
91. Day ES, Cachero TG, Qian F, Sun Y, Wen D, Pelletier M, et al. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry (2005) 44(6):1919–31. doi: 10.1021/bi048227k
92. Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-r, is solely activated by oligomeric BAFF and APRIL to support survival of activated b cells and plasmablasts. Blood (2008) 111(3):1004–12. doi: 10.1182/blood-2007-09-110874
93. He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating b cells through the adaptor MyD88. Nat Immunol (2010) 11(9):836–45. doi: 10.1038/ni.1914
94. Tsuji S, Cortesão C, Bram RJ, Platt JL, Cascalho M. TACI deficiency impairs sustained blimp-1 expression in b cells decreasing long-lived plasma cells in the bone marrow. Blood (2011) 118(22):5832–9. doi: 10.1182/blood-2011-05-353961
95. Li L, Jiang Y, Su L, Feng D, Wei J, Sun J. Study on the mechanism of selective interaction of BR3 and BCMA with BAFF and APRIL. Protein Pept Lett (2020) 27(11):1114–23. doi: 10.2174/0929866527666200413101757
96. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
97. Cornelis R, Chang HD, Radbruch A. Keeping up with the stress of antibody production: BAFF and APRIL maintain memory plasma cells. Curr Opin Immunol (2021) 71:97–102. doi: 10.1016/j.coi.2021.06.012
98. Hatzoglou A, Roussel J, Bourgeade MF, Rogier E, Madry C, Inoue J, et al. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-κB, elk-1, c-jun n-terminal kinase, and p38 mitogen-activated protein Kinase1. J Immunol (2000) 165(3):1322–30. doi: 10.4049/jimmunol.165.3.1322
99. Khan WN. B cell receptor and BAFF receptor signaling regulation of b cell homeostasis. J Immunol (2009) 183(6):3561–7. doi: 10.4049/jimmunol.0800933
100. Fernandes-Silva H, Araújo-Silva H, Correia-Pinto J, Moura RS. Retinoic acid: A key regulator of lung development. Biomolecules (2020) 10(1):152. doi: 10.3390/biom10010152
101. Hind M, Gilthorpe A, Stinchcombe S, Maden M. Retinoid induction of alveolar regeneration: from mice to man? Thorax (2009) 64(5):451–7. doi: 10.1136/thx.2008.105437
102. Timoneda J, Rodríguez-Fernández L, Zaragozá R, Marín MP, Cabezuelo MT, Torres L, et al. Vitamin a deficiency and the lung. Nutrients (2018) 10(9):1132. doi: 10.3390/nu10091132
103. Sirisinha S. The pleiotropic role of vitamin a in regulating mucosal immunity. Asian Pac J Allergy Immunol (2015) 33(2):71–89.
104. Hufnagl K, Jensen-Jarolim E. Vitamin a and d in allergy: from experimental animal models and cellular studies to human disease. Allergo J Int (2018) 27(3):72–8. doi: 10.1007/s40629-018-0054-2
105. Stephensen CB, Lietz G. Vitamin a in resistance to and recovery from infection: relevance to SARS-CoV2. Br J Nutr (2021) 126(11):1663–72. doi: 10.1017/S0007114521000246
106. Seo GY, Jang YS, Kim J, Choe J, Han HJ, Lee JM, et al. Retinoic acid acts as a selective human IgA switch factor. Hum Immunol (2014) 75(8):923–9. doi: 10.1016/j.humimm.2014.06.021
107. Seo GY, Jang YS, Kim HA, Lee MR, Park MH, Park SR, et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-β1 to enhance the overall IgA response. J Leukocyte Biol (2013) 94(2):325–35. doi: 10.1189/jlb.0313128
108. Morikawa K, Nonaka M. All-trans-retinoic acid accelerates the differentiation of human b lymphocytes maturing into plasma cells. Int Immunopharmacol (2005) 5(13):1830–8. doi: 10.1016/j.intimp.2005.06.002
109. Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting b cells by intestinal dendritic cells. Science (2006) 314(5802):1157–60. doi: 10.1126/science.1132742
110. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA deficiency: Epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Scand J Immunol (2017) 85(1):3–12. doi: 10.1111/sji.12499
111. Mbawuike IN, Pacheco S, Acuna CL, Switzer KC, Zhang Y, Harriman GR. Mucosal immunity to influenza without IgA: An IgA knockout mouse model. J Immunol (1999) 162(5):2530–7. doi: 10.4049/jimmunol.162.5.2530
112. Zhang Y, Pacheco S, Acuna CL, Switzer KC, Wang Y, Gilmore X, et al. Immunoglobulin a-deficient mice exhibit altered T helper 1-type immune responses but retain mucosal immunity to influenza virus. Immunology (2002) 105(3):286–94. doi: 10.1046/j.0019-2805.2001.01368.x
113. Arulanandam BP, Raeder RH, Nedrud JG, Bucher DJ, Le J, Metzger DW. IgA immunodeficiency leads to inadequate Th cell priming and increased susceptibility to influenza virus infection. J Immunol (2001) 166(1):226–31. doi: 10.4049/jimmunol.166.1.226
114. Arnaboldi PM, Behr MJ, Metzger DW. Mucosal b cell deficiency in IgA–/– mice abrogates the development of allergic lung Inflammation1. J Immunol (2005) 175(2):1276–85. doi: 10.4049/jimmunol.175.2.1276
115. Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet (2020) 396(10258):1204–22. doi: 10.1016/S0140-6736(20)30925-9
116. de Fays C, Carlier FM, Gohy S, Pilette C. Secretory immunoglobulin a immunity in chronic obstructive respiratory diseases. Cells (2022) 11(8):1324. doi: 10.3390/cells11081324
117. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med (2004) 350(26):2645–53. doi: 10.1056/NEJMoa032158
118. Ladjemi MZ, Martin C, Lecocq M, Detry B, Nana FA, Moulin C, et al. Increased IgA expression in lung lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2019) 199(5):592–602. doi: 10.1164/rccm.201802-0352OC
119. Ladjemi MZ, Lecocq M, Weynand B, Bowen H, Gould HJ, Van Snick J, et al. Increased IgA production by b-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur Respir J (2015) 45(4):980–93. doi: 10.1183/09031936.00063914
120. Gohy ST, Detry BR, Lecocq M, Bouzin C, Weynand BA, Amatngalim GD, et al. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease. persistence in the cultured epithelium and role of transforming growth factor-β. Am J Respir Crit Care Med (2014) 190(5):509–21. doi: 10.1164/rccm.201311-1971OC
121. Xiong Y, Gao S, Luo G, Cheng G, Huang W, Jiang R, et al. Increased circulating autoantibodies levels of IgG, IgA, IgM against cytokeratin 18 and cytokeratin 19 in chronic obstructive pulmonary disease. Arch Med Res (2017) 48(1):79–87. doi: 10.1016/j.arcmed.2017.01.007
122. Sato A, Hashiguchi M, Toda E, Iwasaki A, Hachimura S, Kaminogawa S. CD11b+ peyer’s patch dendritic cells secrete IL-6 and induce IgA secretion from naive b cells. J Immunol (2003) 171(7):3684–90. doi: 10.4049/jimmunol.171.7.3684
123. Klimova B, Kuca K, Novotny M, Maresova P. Cystic fibrosis revisited - a review study. Med Chem (2017) 13(2):102–9. doi: 10.2174/1573406412666160608113235
124. Lund-Palau H, Turnbull AR, Bush A, Bardin E, Cameron L, Soren O, et al. Pseudomonas aeruginosa infection in cystic fibrosis: Pathophysiological mechanisms and therapeutic approaches. Expert Rev Respir Med (2016) 10(6):685–97. doi: 10.1080/17476348.2016.1177460
125. Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med (1994) 150(2):448–54. doi: 10.1164/ajrccm.150.2.8049828
126. Collin AM, Lecocq M, Noel S, Detry B, Carlier FM, Aboubakar Nana F, et al. Lung immunoglobulin a immunity dysregulation in cystic fibrosis. EBioMedicine (2020) 60:102974. doi: 10.1016/j.ebiom.2020.102974
127. Pedersen SS, Møller H, Espersen F, Sørensen CH, Jensen T, Høiby N. Mucosal immunity to pseudomonas aemginosa alginate in cystic fibrosis. APMIS (1992) 100(1–6):326–34. doi: 10.1111/j.1699-0463.1992.tb00879.x
128. Aanaes K, Johansen HK, Poulsen SS, Pressler T, Buchwald C, Høiby N. Secretory IgA as a diagnostic tool for pseudomonas aeruginosa respiratory colonization. J Cystic Fibros (2013) 12(1):81–7. doi: 10.1016/j.jcf.2012.07.001
129. Garić D, Tao S, Ahmed E, Youssef M, Kanagaratham C, Shah J, et al. Depletion of BAFF cytokine exacerbates infection in pseudomonas aeruginosa infected mice. J Cyst Fibros (2019) 18(3):349–56. doi: 10.1016/j.jcf.2018.11.015
130. Høiby N, Wiik A. Antibacterial precipitins and autoantibodies in serum of patients with cystic fibrosis. Scand J Respir Dis (1975) 56(1):38–46.
131. Schiøtz PO, Egeskjold EM, Høiby N, Permin H. Autoantibodies in serum and sputum from patients with cystic fibrosis. Acta Pathol Microbiol Scand C (1979) 87(5):319–24.
132. Theprungsirikul J, Skopelja-Gardner S, Meagher RE, Clancy JP, Zemanick ET, Ashare A, et al. Dissociation of systemic and mucosal autoimmunity in cystic fibrosis. J Cyst Fibros (2020) 19(2):196–202. doi: 10.1016/j.jcf.2019.06.006
133. Condino AA, Hoffenberg EJ, Accurso F, Penvari C, Anthony M, Gralla J, et al. Frequency of ASCA seropositivity in children with cystic fibrosis. J Pediatr Gastroenterol Nutr (2005) 41(1):23–6. doi: 10.1097/01.mpg.0000166801.61708.60
134. Hirche TO, Stein J, Hirche H, Hausmann J, Wagner TO, Behrens F, et al. Increased levels of anti-glycan antibodies in patients with cystic fibrosis. Eur J Med Res (2011) 16(9):385–90. doi: 10.1186/2047-783X-16-9-385
135. Hovold G, Lindberg U, Ljungberg JK, Shannon O, Påhlman LI. BPI-ANCA is expressed in the airways of cystic fibrosis patients and correlates to platelet numbers and pseudomonas aeruginosa colonization. Respir Med (2020) 170:105994. doi: 10.1016/j.rmed.2020.105994
136. Lindberg U, Carlsson M, Löfdahl CG, Segelmark M. BPI-ANCA and long-term prognosis among 46 adult CF patients: A prospective 10-year follow-up study. Clin Dev Immunol (2012) 2012:370107. doi: 10.1155/2012/370107
137. Iwuji K, Larumbe-Zabala E, Bijlani S, Nugent K, Kanu A, Manning E, et al. Prevalence of Bactericidal/Permeability-increasing protein autoantibodies in cystic fibrosis patients: Systematic review and meta-analysis. Pediatr Allergy Immunol Pulmonol (2019) 32(2):45–51. doi: 10.1089/ped.2018.0970
138. Lachenal F, Nkana K, Nove-Josserand R, Fabien N, Durieu I. Prevalence and clinical significance of auto-antibodies in adults with cystic fibrosis. Eur Respir J (2009) 34(5):1079–85. doi: 10.1183/09031936.00006009
139. Laval J, Ralhan A, Hartl D. Neutrophils in cystic fibrosis. Biol Chem (2016) 397(6):485–96. doi: 10.1515/hsz-2015-0271
140. Yadav R, Linnemann RW, Kahlenberg JM, Bridges LS, Stecenko AA, Rada B. IgA autoantibodies directed against self DNA are elevated in cystic fibrosis and associated with more severe lung dysfunction. Autoimmunity (2020) 53(8):476–84. doi: 10.1080/08916934.2020.1839890
141. Linnemann RW, Yadav R, Zhang C, Sarr D, Rada B, Stecenko AA. Serum anti-PAD4 autoantibodies are present in cystic fibrosis children and increase with age and lung disease severity. Autoimmunity (2022) 55(2):109–17. doi: 10.1080/08916934.2021.2021193
142. Shanthikumar S, Ranganathan SC, Saffery R, Neeland MR. Mapping pulmonary and systemic inflammation in preschool aged children with cystic fibrosis. Front Immunol (2021) 12:733217. doi: 10.3389/fimmu.2021.733217
143. Moore R, Kyd JM, Carzino R, Armstrong D, Grimwood K, Otczyk DC, et al. Mucosal and systemic antibody responses to potential pseudomonas aeruginosa vaccine protein antigens in young children with cystic fibrosis following colonization and infection. Hum Vaccin Immunother (2013) 9(3):506–14. doi: 10.4161/hv.23226
144. Siegmann N, Worbs D, Effinger F, Bormann T, Gebhardt M, Ulrich M, et al. Invariant natural killer T (iNKT) cells prevent autoimmunity, but induce pulmonary inflammation in cystic fibrosis. Cell Physiol Biochem (2014) 34(1):56–70. doi: 10.1159/000362984
145. Wahren-Herlenius M, Salomonsson S. Detection of antigen specific b-cells in tissues. Methods Mol Med (2007) 136:19–24. doi: 10.1007/978-1-59745-402-5_2
146. Gohy S, Moeremans A, Pilette C, Collin A. Immunoglobulin a mucosal immunity and altered respiratory epithelium in cystic fibrosis. Cells (2021) 10(12):3603. doi: 10.3390/cells10123603
147. Habener A, Grychtol R, Gaedcke S, DeLuca D, Dittrich AM, Happle C, et al. IgA+ memory b-cells are significantly increased in patients with asthma and small airway dysfunction. Eur Respir J (2022) 60(5):2102130. doi: 10.1183/13993003.02130-2021
148. Samitas K, Malmhäll C, Rådinger M, Ramos-Ramirez P, Lu Y, Deák T, et al. Precursor b cells increase in the lung during airway allergic inflammation: A role for b cell-activating factor. PloS One (2016) 11(8):e0161161. doi: 10.1371/journal.pone.0161161
149. Casaro M, Souza VR, Oliveira FA, Ferreira CM. OVA-induced allergic airway inflammation mouse model. Methods Mol Biol (2019) 1916:297–301. doi: 10.1007/978-1-4939-8994-2_28
150. Hong GU, Lim JY, Kim NG, Shin JH, Ro JY. IgE and IgA produced by OX40–OX40L or CD40–CD40L interaction in b cells–mast cells re-activate FcϵRI or FcαRI on mast cells in mouse allergic asthma. Eur J Pharmacol (2015) 754:199–210. doi: 10.1016/j.ejphar.2015.02.023
151. Monteiro RC, Hostoffer RW, Cooper MD, Bonner JR, Gartland GL, Kubagawa H. Definition of immunoglobulin a receptors on eosinophils and their enhanced expression in allergic individuals. J Clin Invest (1993) 92(4):1681–5. doi: 10.1172/JCI116754
152. Abu-Ghazaleh RI, Fujisawa T, Mestecky J, Kyle RA, Gleich GJ. IgA-induced eosinophil degranulation. J Immunol (1989) 142(7):2393–400. doi: 10.4049/jimmunol.142.7.2393
153. Terada N, Terada Y, Shirotori K, Ishikawa K, Togawa K, Konno A. Immunoglobulin as an eosinophil degranulation factor: Change in immunoglobulin level in nasal lavage fluid after antigen challenge. Acta Oto Laryngol (1996) 116(6):863–7. doi: 10.3109/00016489609137942
154. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet (2017) 389(10082):1941–52. doi: 10.1016/S0140-6736(17)30866-8
155. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers (2017) 3:17074. doi: 10.1038/nrdp.2017.74
156. Todd NW, Scheraga RG, Galvin JR, Iacono AT, Britt EJ, Luzina IG, et al. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J Inflammation Res (2013) 6:63–70. doi: 10.2147/JIR.S40673
157. Xue J, Kass DJ, Bon J, Vuga L, Tan J, Csizmadia E, et al. Plasma b lymphocyte stimulator and b cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol (2013) 191(5):2089–95. doi: 10.4049/jimmunol.1203476
158. Ten Klooster L, van Moorsel CHM, Kwakkel-van Erp JM, van Velzen-Blad H, Grutters JC. Immunoglobulin a in serum: An old acquaintance as a new prognostic biomarker in idiopathic pulmonary fibrosis. Clin Exp Immunol (2015) 181(2):357–61. doi: 10.1111/cei.12636
159. Heukels P, van Hulst JAC, van Nimwegen M, Boorsma CE, Melgert BN, von der Thusen JH, et al. Enhanced bruton’s tyrosine kinase in b-cells and autoreactive IgA in patients with idiopathic pulmonary fibrosis. Respir Res (2019) 20(1):232. doi: 10.1186/s12931-019-1195-7
160. Solomon JJ, Matson S, Kelmenson LB, Chung JH, Hobbs SB, Rosas IO, et al. IgA antibodies directed against citrullinated protein antigens are elevated in patients with idiopathic pulmonary fibrosis. Chest (2020) 157(6):1513–21. doi: 10.1016/j.chest.2019.12.005
161. Vuga LJ, Tedrow JR, Pandit KV, Tan J, Kass DJ, Xue J, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2014) 189(8):966–74. doi: 10.1164/rccm.201309-1592OC
162. Khalil N, O’Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, et al. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol (1991) 5(2):155–62. doi: 10.1165/ajrcmb/5.2.155
163. Sánchez Montalvo A, Gohy S, Rombaux P, Pilette C, Hox V. The role of IgA in chronic upper airway disease: Friend or foe? Front Allergy (2022) 3:852546. doi: 10.3389/falgy.2022.852546
164. Yel L. Selective IgA deficiency. J Clin Immunol (2010) 30(1):10–6. doi: 10.1007/s10875-009-9357-x
165. Aytekin C, Tuygun N, Gokce S, Dogu F, Ikinciogullari A. Selective IgA deficiency: Clinical and laboratory features of 118 children in turkey. J Clin Immunol (2012) 32(5):961–6. doi: 10.1007/s10875-012-9702-3
166. Aghamohammadi A, Cheraghi T, Gharagozlou M, Movahedi M, Rezaei N, Yeganeh M. IgA deficiency: Correlation between clinical and immunological phenotypes. J Clin Immunol (2009) 29(1):130–6. doi: 10.1007/s10875-008-9229-9
167. Brandtzaeg P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine (2007) 25(30):5467–84. doi: 10.1016/j.vaccine.2006.12.001
168. Igarashi Y, Skoner D, Doyle W, White M, Fireman P, Kaliner M. Analysis of nasal secretions during experimental rhinovirus upper respiratory infections. J Allergy Clin Immunol (1993) 92(5):722–31. doi: 10.1016/0091-6749(93)90016-9
169. van Riet E, Ainai A, Suzuki T, Hasegawa H. Mucosal IgA responses in influenza virus infections; thoughts for vaccine design. Vaccine (2012) 30(40):5893–900. doi: 10.1016/j.vaccine.2012.04.109
170. Russell MW, Moldoveanu Z, Ogra PL, Mestecky J. Mucosal immunity in COVID-19: A neglected but critical aspect of SARS-CoV-2 infection. Front Immunol (2020) 30:11. doi: 10.3389/fimmu.2020.611337
171. Fahlman MM, Engels HJ. Mucosal IgA and URTI in american college football players: A year longitudinal study. Med Sci Sports Exerc (2005) 37(3):374–80. doi: 10.1249/01.MSS.0000155432.67020.88
172. Gleeson M, Pyne DB. Respiratory inflammation and infections in high-performance athletes. Immunol Cell Bio (2016) 94(2):124–31. doi: 10.1038/icb.2015.100
173. Turner SEG, Loosemore M, Shah A, Kelleher P, Hull JH. Salivary IgA as a potential biomarker in the evaluation of respiratory tract infection risk in athletes. J Allergy Clin Immunol Pract (2021) 9(1):151–9. doi: 10.1016/j.jaip.2020.07.049
174. Nahm D, Kim H, Park H. Elevation of specific immunoglobulin a antibodies to both allergen and bacterial antigen in induced sputum from asthmatics. Eur Respir J (1998) 12(3):540–5. doi: 10.1183/09031936.98.12030540
175. Aghayan-Ugurluoglu R, Ball T, Vrtala S, Schweiger C, Kraft D, Valenta R. Dissociation of allergen-specific IgE and IgA responses in sera and tears of pollen-allergic patients: A study performed with purified recombinant pollen allergens. J Allergy Clin Immunol (2000) 105(4):803–13. doi: 10.1067/mai.2000.104782
176. Peebles RS, Liu MC, Adkinson NF, Lichtenstein LM, Hamilton RG. Ragweed-specific antibodies in bronchoalveolar lavage fluids and serum before and after segmental lung challenge: IgE and IgA associated with eosinophil degranulation. J Allergy Clin Immunol (1998) 101(2):265–73. doi: 10.1016/S0091-6749(98)70392-6
177. Reed CE, Bubak M, Dunnette S, Blomgren J, Pfenning M, Wentz-Murtha P. Ragweed-specific IgA in nasal lavage fluid of ragweed-sensitive allergic rhinitis patients: Increase during the pollen season. Int Arch Allergy Immunol (1991) 94(1–4):275–7. doi: 10.1159/000235382
178. Benson M, Reinholdt J, Cardell LO. Allergen-reactive antibodies are found in nasal fluids from patients with birch pollen-induced intermittent allergic rhinitis, but not in healthy controls. Allergy (2003) 58(5):386–92. doi: 10.1034/j.1398-9995.2003.00113.x
179. Keen C, Johansson S, Reinholdt J, Benson M, Wennergren G. Bet v 1-specific IgA increases during the pollen season but not after a single allergen challenge in children with birch pollen-induced intermittent allergic rhinitis. Pediatr Allergy Immunol (2005) 16(3):209–16. doi: 10.1111/j.1399-3038.2005.00264.x
180. Suzuki M, Yokota M, Ozaki S, Matsumoto T, Nakamura Y. Japanese Cedar pollen–specific IgA in nasal secretions and nasal allergy symptoms. Ann Otol Rhinol Laryngol (2019) 128(4):330–7. doi: 10.1177/0003489418823791
181. Bahceciler NN, Arikan C, Taylor A, Akdis M, Blaser K, Barlan IB. Impact of sublingual immunotherapy on specific antibody levels in asthmatic children allergic to house dust mites. Int Arch Allergy Immunol (2005) 136(3):287–94. doi: 10.1159/000083956
182. Stokes CR, Taylor B, Turner MW. Association of house-dust and grass-pollen allergies with specific iga antibody deficiency. Lancet (1974) 304(7879):485–8. doi: 10.1016/s0140-6736(74)92014-5
183. Schwarze J, Cieslewicz G, Joetham A, Sun LK, Sun WN, Chang TW. Antigen-specific immunoglobulin-a prevents increased airway responsiveness and lung eosinophilia after airway challenge in sensitized mice. Am J Respir Crit Care Med (1998) 158(2):519–25. doi: 10.1164/ajrccm.158.2.9801014
184. Durham SR, Penagos M. Sublingual or subcutaneous immunotherapy for allergic rhinitis? J Allergy Clin Immunol (2016) 349(e10):339–49. doi: 10.1016/j.jaci.2015.12.1298
185. Shamji MH, Larson D, Eifan A, Scadding GW, Qin T, Lawson K. Differential induction of allergen-specific IgA responses following timothy grass subcutaneous and sublingual immunotherapy. J Allergy Clin Immunol (2021) 1071(e11):1061–71. doi: 10.1016/j.jaci.2021.03.030
186. Henriques A, Nunes R, Loureiro G, Martinho A, Pais M, Segorbe-Luís A. Alterations on peripheral blood b cell subsets induced by allergic rhinitis. Inflammation Res (2015) 64(3–4):145–9. doi: 10.1007/s00011-015-0803-3
187. Francis JN, James LK, Paraskevopoulos G, Wong C, Calderon MA, Durham S. Grass pollen immunotherapy: IL-10 induction and suppression of late responses precedes IgG4 inhibitory antibody activity. J Allergy Clin Immunol (2008) 121(5):1120–5. doi: 10.1016/j.jaci.2008.01.072
188. Jutel M, Akdis M, Budak F, Aebischer-Casaulta C, Wrzyszcz M, Blaser K. IL-10 and TGF-β cooperate in the regulatory T cell response to mucosal allergens in normal immunity and specific immunotherapy. Eur J Immunol (2003) 33(5):1205–14. doi: 10.1002/eji.200322919
189. Schmidt-Weber CB, Blaser K. Regulation and role of transforming growth factor-β in immune tolerance induction and inflammation. Curr Opin Immunol (2004) 16(6):709–16. doi: 10.1016/j.coi.2004.09.008
190. Ciprandi G, Amici M, Tosca M, Marseglia G. Serum transforming growth factor-β levels depend on allergen exposure in allergic rhinitis. Int Arch Allergy Immunol (2010) 152(1):66–70. doi: 10.1159/000260085
191. Pilette C, Nouri-Aria KT, Jacobson MR, Wilcock LK, Detry B, Walker SM. Grass pollen immunotherapy induces an allergen-specific IgA2 antibody response associated with mucosal TGF-β expression. J Immunol (2007) 178(7):4658–66. doi: 10.4049/jimmunol.178.7.4658
192. Wirz OF, Głobińska A, Ochsner U, Veen W, Eller E, Christiansen ES. Comparison of regulatory b cells in asthma and allergic rhinitis. Allergy (2019) 74(4):815–8. doi: 10.1111/all.13672
193. Kim AS, Doherty TA, Karta MR, Das S, Baum R, Rosenthal P. Regulatory b cells and T follicular helper cells are reduced in allergic rhinitis. J Allergy Clin Immunol (2016) 1195(e5):1192–5. doi: 10.1016/j.jaci.2016.03.017
194. Chee L, Graham SM, Carothers DG, Ballas ZK. Immune dysfunction in refractory sinusitis in a tertiary care setting. Laryngoscope (2001) 111(2):233–5. doi: 10.1097/00005537-200102000-00008
195. Van Zele T, Claeys S, Gevaert P, Van Maele G, Holtappels G, Van Cauwenberge P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy (2006) 61(11):1280–9. doi: 10.1111/j.1398-9995.2006.01225.x
196. Feldman S, Kasjanski R, Poposki J, Hernandez D, Chen JN, Norton JE. Chronic airway inflammation provides a unique environment for b cell activation and antibody production. Clin Exp Allergy (2017) 47(4):457–66. doi: 10.1111/cea.12878
197. Gevaert P, Holtappels G, Johansson SGO, Cuvelier C, Cauwenberge P, Bachert C. Organization of secondary lymphoid tissue and local IgE formation to staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy: Eur J Allergy Clin Immunol (2005) 60(1):71–9. doi: 10.1111/j.1398-9995.2004.00621.x
198. Lau A, Lester S, Moraitis S, Ou J, Psaltis AJ, McColl S. Tertiary lymphoid organs in recalcitrant chronic rhinosinusitis. J Allergy Clin Immunol (2017) 1373(e6):1371–3. doi: 10.1016/j.jaci.2016.08.052
199. Wang Z, Song J, Wang H, Li J, Xiao Q, Yu Z. Stromal cells and b cells orchestrate ectopic lymphoid tissue formation in nasal polyps. Allergy (2021) 76(5):1416–31. doi: 10.1111/all.14612
200. Dilidaer ZY ZL. X H, J z, l h. increased BAFF expression in nasal polyps is associated with local IgE production, Th2 response and concomitant asthma. Eur Arch Oto Rhino Laryngol (2017) 274(4):1883–90. doi: 10.1007/s00405-016-4435-1
201. Kato A, Peters A, Suh L, Carter R, Harris KE, Chandra R. Evidence of a role for b cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol (2008) 121(6):1385–92. doi: 10.1016/j.jaci.2008.03.002
202. Yoon YH, Jin J, Kwon KR, Kim SH, Rha KS, Kim YM. The role of b cell activating factor (BAFF) expression on pathogenesis of nasal polyp in chronic rhinosinusitis with nasal polyposis. Rhinology (2014) 52(4):390–6. doi: 10.4193/Rhin13.154
203. Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci (2005) 1050(1):34–9. doi: 10.1196/annals.1313.004
204. Tsybikov NN, v. EE, Kuznik BI, v. FE, Magen E. Anticytokine autoantibodies in chronic rhinosinusitis. Allergy Asthma Proc (2015) 36(6):473–80. doi: 10.2500/aap.2015.36.3880
205. Lin M, Du L, Brandtzaeg P, Pan-Hammarstrom A. IgA subclass switch recombination in human mucosal and systemic immune compartments. Mucosal Immunol (2014) 7(3):511–20. doi: 10.1038/mi.2013.68
206. Takeda K, Sakakibara S, Yamashita K, Motooka D, Nakamura S, Hussien MA. Allergic conversion of protective mucosal immunity against nasal bacteria in patients with chronic rhinosinusitis with nasal polyposis. J Allergy Clin Immunol (2019) 1175(e15):1163–75. doi: 10.1016/j.jaci.2018.07.006
207. Hupin C, Rombaux P, Bowen H, Gould H, Lecocq M, Pilette C. Downregulation of polymeric immunoglobulin receptor and secretory IgA antibodies in eosinophilic upper airway diseases. Allergy: Eur J Allergy Clin Immunol (2013) 68(12):1589–97. doi: 10.1111/all.12274
Keywords: IgA+ B cells, lung B cells, lung mucosal immunity, upper airway immunity, airway disease
Citation: Bertrand Y, Sánchez-Montalvo A, Hox V, Froidure A and Pilette C (2023) IgA-producing B cells in lung homeostasis and disease. Front. Immunol. 14:1117749. doi: 10.3389/fimmu.2023.1117749
Received: 06 December 2022; Accepted: 14 February 2023;
Published: 01 March 2023.
Edited by:
Louis Boon, JJP Biologics, PolandReviewed by:
Maria Manuela Rosado, Sapienza University of Rome, ItalyCopyright © 2023 Bertrand, Sánchez-Montalvo, Hox, Froidure and Pilette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Pilette, Y2hhcmxlcy5waWxldHRlQHVjbG91dmFpbi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.