 Cuicui Wang1
Cuicui Wang1 Xiaolin Cui
Xiaolin Cui Shuang Wu
Shuang Wu Zhiming Lu
Zhiming Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1117172
This article is part of the Research Topic The Emerging Role of the Neuroimmune Axis as a Therapeutic Target in Alzheimer's Disease View all 7 articles
Alzheimer’s disease (AD) is defined as a severe chronic degenerative neurological disease in human. The pathogenic mechanism of AD has been convincingly elucidated by the “amyloid cascade hypothesis” with the main focus of the pathological accretion of β-amyloid (Aβ) peptides outside the cell. However, increasing evidence suggests that this hypothesis is weak in explaining the pathogenesis of AD. Neuroinflammation is crucial in the development of AD, which is proven by the elevated levels of inflammatory markers and the identification of AD risk genes relevant to the innate immune function. Here, we summarize the effects of microglia-mediated neuroinflammation on AD, focusing on the temporal and spatial changes in microglial phenotype, the interactions among microglia, Aβ, tau, and neurons, and the prospects and recent advances in neuroinflammation as a diagnostic and therapeutic target of AD.
Alzheimer’s disease (AD) is the most common type of dementia, with its case number projected to be nearly doubled in Europe and tripled worldwide by 2050 (1). The brain atrophy, characterized by the accumulation of amyloid plaques and neuronal fibrillary tangles as well as the loss of neurons and synapses, are considered as the main pathological feature of AD. The “amyloid cascade hypothesis” is currently the most popular molecular mechanism regulating the AD pathogenesis, indicating that the misfolding and aggregation of β-amyloid (Aβ) result in a linear cascade of AD pathology. However, the addition of Aβ to the brains of healthy humans or mice does not cause the development of AD. Similarly, removal of Aβ from the brains of AD patients could not improve the medical conditions, suggesting that the “amyloid cascade hypothesis” alone can no longer fully explain the development of AD (2, 3). To date, it is well-known that neuroinflammation is responsible for the pathogenesis of AD. A total of more than 40 loci are confirmed as the target genes related to the late-onset AD (LOAD), with many related genes concentrated in glial cells. In particular, microglia have become a central topic in AD research (4). Microglia are natural immune cells in the central nervous system (CNS), participating in nerve development by phagocytosis and clearance of damaged neurons and synapses (5, 6). However, once activated to show the proinflammatory phenotype, microglia produce deleterious substances in patients with AD. This review focuses on neuroinflammation, microglial phenotype, and their interrelations with AD pathology, showing significant prospects of neuroinflammation and microglia as diagnostic and therapeutic strategies during the therapy of AD.
AD is a neurodegenerative disease characterized by gradual deterioration of cognitive ability, with notably gradual death of neurons and often severe impairment of cognitive ability in memory and learning. It is well known that AD is a prolonged neurological disease with its development divided into three stages, i.e., preclinical stage, mild cognitive impairment (MCI) stage, and demented stage (7, 8). In about 20 years ago, the “amyloid cascade hypothesis” was proposed to explain the pathogenesis of AD based on the formation of abnormal Aβ plaques in different regions of the brain, suggesting that the abnormal accumulation of Aβ outside the cells was the main cause of the cascade effect leading to neuronal injury (9, 10). However, the accuracy of Aβ as a biomarker for AD is seriously challenged because Aβ is found in approximately 30% of cognitively normal elderly people (11). Similarly, the results of Aβ clearance as a treatment for AD patients are not satisfactory, indicating that the “amyloid cascade hypothesis” is unable to fully explain the molecular mechanism of AD pathogenesis (12, 13). Furthermore, another significant pathological change in AD is the intracellular aggregation of neuronal fibrillary tangles formed by tau aggregation (14). Moreover, several alternative hypotheses, i.e., the oxidative stress hypothesis, the cholinergic hypothesis, and the neuroinflammation hypothesis, are also proposed to provide significant insights into the pathogenesis of AD (15–17). Among these hypotheses, the neuroinflammation has been shown to be involved in the entire process of AD. Growing evidence suggests that persistent glial cell-mediated neuroinflammation is the primary cause for both neurodegenerative processes and cognitive deficits in AD patients. Inflammation is not only a passive outcome but also a contributor to the occurrence of AD, attracting increasing attention from medical scholars to further explore the important functions of neuroinflammation in the development of AD (18).
Neuroinflammation is a reaction of inflammation in the CNS due to pathological damage occurring in the peripheral or CNS, resulting in the production of proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, IL-8, and tumor necrosis factor (TNF), chemokines, complement cytokines as well as some small molecule messengers, e.g., prostaglandins, nitric oxide (NO), and reactive oxygen species (ROS) (19). Generally, the acute inflammation in the brain exists as a defense against damage to the nervous system, infection, and other stimuli. However, with prolonged inflammation, the acute inflammation gradually becomes chronic to stimulate the nervous system. The chronic inflammation could continue to release various inflammatory cytokines, resulting in a proinflammatory response that is consistently stronger than the anti-inflammatory response, thus aggravating the inflammation, ultimately damaging the neurons and causing various pathological changes in the body (20). Endogenous bioactive lipids regulate a large number of cellular and molecular processes related to health and disease, especially during inflammation. All major endogenous bioactive lipids, i.e., eicosanoids, specialized pro-catabolic lipid mediators, lysophospholipids, and endocannabinoids, are closely associated with chronic inflammation (21). Although these molecules are originally described as neuromodulators/neurotransmitters, they have been characterized as important regulators of glial function (22) and will in turn disrupt tissue homeostasis to cause chronic damage, such as AD (23). Given the higher expression and production of inflammatory cytokines in microglia than those of other glial cells, microglia are considered the main sources of brain inflammation. Furthermore, the microglia have attracted increasing attention from the medical scholars due to their complex phenotypes. Moreover, microglia are also closely related to the loss of synapses, tau phosphorylation, and poor memory, making these biological processes increasingly the centers of the study of neuroinflammation in AD (24).
Microglia are considered as the resident macrophages of the CNS, originated from mesoderm and migrating to the CNS during its development (25). On the one hand, microglia are known with an essential protective effect in the physiological processes of the CNS and are considered an important protection to maintain the homeostasis, constituting the frontline defense of the innate immune system and regulating the number of neurons in the CNS. During the body development, microglia promote neuronal survival and differentiation as well as circuit formation through brain-derived neurotrophic factor signaling; microglia also participate in synaptic formation related to learning. For example, studies have shown that CX3CR1 in microglia is essential for the survival of layer V neurons, while the microglia-derived IGF1 is a trophic factor for maintaining the neuronal survival, whereas the neurotrophic factors released by microglia could induce programmed cell death and maintain body stability (26). On the other hand, persistently activated microglia could contribute to the pathogenesis of neurodegenerative disease. For example, studies have shown that overactivated microglia could produce proinflammatory cytokines and chemokines at high levels, leading to neuronal dysfunction (27). Meanwhile, the abnormal activation of the complement-microglia axis is involved in the synaptic loss in the early stages of AD, which is closely related to the cognitive performance. Studies have shown that the inhibition of microglia complement receptors in a mouse model could reduce the degree of early synaptic loss (28, 29), while the microglia-mediated synaptic pruning may be the underlying mechanism of synaptic loss and memory impairment induced by long-term alcohol exposure (30).
In the rapidly changing environment of brain, precise differentiation of microglial phenotypes is essential to explore the pathological development of AD. The phenotypic characters of microglia could be divided into resting phase (M0) and activated phases (M1 and M2) (31, 32) (Figure 1). In the resting microglia, cytosol remains in a fixed position. The two-photon imaging of the cerebral cortex reveals that microglia at the resting phase are still functioning as a sensitive surveillance, with their cytosols and protrusions continuously monitoring changes in the microenvironment of the brain parenchyma (33). The cytosols of microglia at this phase are mostly highly branched, i.e., these cells are known as multipolar or branched microglia, which are particularly sensitive to the homeostasis disturbance of the internal brain environment. In AD, the brain homeostasis is impaired, accompanied by changes in both brain microenvironment and glial cell characteristics, while the microglia mainly show the activated anti-inflammatory phenotype M2, featured by the release of anti-inflammatory cytokines, including transforming growth factor-β (TGF-β), IL-4, IL-10, and IL-13, and enhanced phagocytosis (34). The persistent inflammation transforms microglia to the activated proinflammatory phenotype M1, which promotes inflammation and leads to increased concentrations of proinflammatory cytokines, including TNF-α, IL-4, IL-6, IL-12, and IL-18, accompanied by impaired phagocytosis (35), while the microglial cell body is enlarged, the processes become shorter, and the outline of microglial cells is changed to round (33). These polarized states differ in triggering stimuli, phenotypic markers, and expression of secretory mediators (Figure 1). In AD, the neuroinflammation is increased with the disease progression. Specifically, the neuroinflammation level reaches the first peak in the early development of AD, attributed to the initial anti-inflammatory response, and then a second peak during the transition from stage MCI to stage dementia, suggesting the change of microglia to a proinflammatory phenotype (36, 37). A longitudinal study revealed that microglial activation may attempt to repair damage during the initial stages of AD, while in LOAD, microglia could be harmful, producing proinflammatory molecules to cause neuronal damage (36). For example, microglia initially phagocytose apoptotic cells through Anexelekto (Axl) and Mer anti-inflammatory receptors (38), whereas the chronic microglial activation could lead to severe pathological changes and neurological complications. It is worth noting that although the M1/M2 nomenclature is rather useful in experimental paradigms, it has also been fiercely discussed, especially in microglia: in particular, M1- and M2-like phenotypes can be considered two extremes of a rather blurred distribution, rather than 2 distinct populations (39). Some authors strongly avoided this dichotomy, whereas others acknowledged that they still use it even with its limitations for lack of a better term (40). Dark microglial (DM) is a recently described phenotype, also known as “dystrophic” microglia, which was shown to precede the spread of Tau pathology (41, 42). Its dark appearance is detected under the high spatial resolution transmission electron microscopy (43). In the APP/PS1 mouse model, black microglia with complete and clear abnormal chromatin and dark appearance are observed. Compared with C57BL/6 mice, the density of black microglia in APP/PS1 mice is significantly increased and concentrated near both Aβ plaques and dystrophic neurites (44). The molecular mechanisms underlying the functions of different types of microglia remain to be explored.
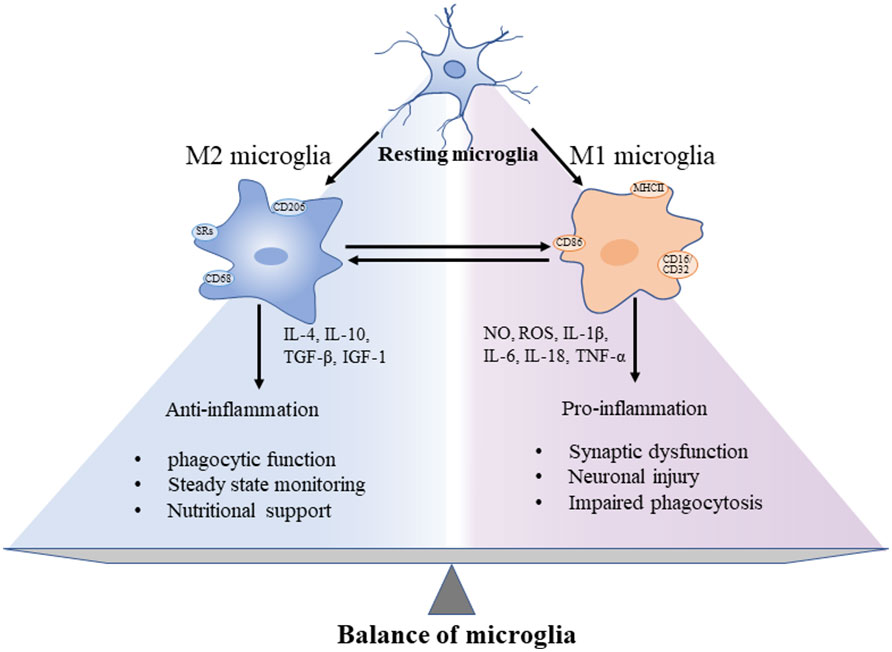
Figure 1 Different functions of microglia showing the “two-sided” characteristics of microglia and the balance between pro- and anti-inflammation in the progression of CNS neuroinflammation. These microglial phenotypes display different cell surface receptors and secreted factors, among which, by secreting anti-inflammatory cytokines and trophic factors to promote phagocytosis, maintain homeostasis, and nourish nerves. In addition, the surface tightening was activated to cause both synaptic dysfunction and neuronal damage.
Compared to microglia at steady state, hundreds of genes markedly up- and down-regulated in the early or late-response microglia have been revealed by the single-cell differential expression (SCDE) software packages. For example, a subpopulation of immune response-related genes, encoding the chemokines cytokine ligand 3 (CCL3) and cytokine ligand 4 (CCL4) as well as CXC chemokine ligand 16 (CXCL16) and macrophage migration inhibitory factor (MIF), are up-regulated in microglia subpopulations 1 week following p25 induction, while the expressions of genes encoding proteins such as Axl and apolipoprotein E (Apoe) were elevated in most late-responding cells except in 1-week CK-p25 cells (45). Previous studies revealed significant activation of microglia during MCI phase of rising load, followed by a decrease in microglia activation levels as Aβ load approached the AD levels, showing a positive correlation between increased inflammation levels and tau load when tau tangles are formed in Aβ-positive patients with mild cognitive impairment with prodromal AD (46). Moreover, microglia in brains of Braak stages V and VI undergo more noteworthy morphological changes compared to those from brains in earlier stages. The main manifestations include shortened or reduced branches of microglia, cytoplasmic fragmentation, and globular formation (47, 48).
Analysis of plaque-associated microglia and those not associated with plaques in the TgCRND mouse model of AD showed that the K1 current in microglia around plaques is significantly increased with evident morphological changes, while microglia not associated with plaques showed only slight changes (48, 49). Furthermore, microglia acquired in plaque-rich regions are found to show different transcriptional features than those of microglia derived from non-plaque regions (50, 51).
Microglia have shown different transcriptional responses to Aβ peptide aggregates in vitro or in vivo, and even to exogenous oligomer Aβ (oAβ) or fibrillar Aβ (fAβ) (52), with the expressions of RNAs related to cell cycle and phagocytosis increased by oAβ and fAβ, respectively, and they responded differently depending on the timing of the stimulus. Furthermore, research has shown that in mouse models of AD, the microglial proliferation is increased linearly with the development of the disease, whereas the Aβ load reached a plateau in the early clinical stages of the disease (53, 54), suggesting that microgliosis at this stage is associated with the tau tangles.
Although the results of postmortem of microglia in the brains of cognitively normal participants have shown similar levels of gene expression profiles to those of mice, the genes that regulate microglia during aging are only partially overlapping between humans and mice (55). This variation could partially explain the frequently failed translation of anti-inflammatory strategies into clinical practice. As a microglial surface receptor, TREM2 has been found to have inconsistent effects in vitro and in vivo, with its expression regulated by inflammation (56). Studies have shown that the TREM2 expression is decreased during acute inflammatory responses in vitro (57) and is increased in both AD patients (58) and mouse models of Aβ and Tau pathology (59). Therefore, it is necessary to distinguish between acute and chronic microglial activations in order to elucidate the molecular mechanisms underlying the TREM2 expression. It is essential to detect microglia and respond to neural deformation signals. The in vivo studies have shown that TREM2 is widely involved in the uptake of Aβ by microglia, whereas the in vitro studies have shown inconsistent results (56, 57).
Studies have shown that the microglial phenotype regulation depends primarily on the relationship between pattern recognition receptors (PRRs) and molecules released from the surrounding cells (60). In AD, these PRRs could sense Aβ, pathogen-associated molecular patterns (PAMPs), or other damage-associated molecular patterns (DAMPs) to influence the microglial phenotype. Microglia activation is characterized by enhanced responsiveness to various inflammatory factors or injury stimuli. After the microglia perceive the DAMPs and PAMPs through toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), or other PRRs, the signaling through PRRs could trigger an inflammatory response and proinflammatory cytokine secretion (Table 1). Generally, these activated microglia are called disease-associated microglia (DAM) (73–77). Furthermore, the Aβ binding particularly to TLR4 has been shown to activate microglia (63, 78). The main receptors expressed by activated microglia include scavenger receptors (SRs), TLRs, triggering receptors in myeloid cells 2 (TREM-2), CD14, CD47, Fcγ receptors (FcγRs), CD200 receptor (CD200R) and receptor for advanced glycation end products (RAGE). Specifically, CD14, TLR2, and TLR4 are required for fAβ-stimulated microglial activation (79), though the molecular mechanisms regulating this microglial activation remain unclear. However, this sustained activation of microglia is adverse because the extended activation of TLR2 and TLR4 in microglia could induce the secretion of Aβ (80). TREM2 signaling appears to be required for the maintenance of microglial metabolism, while the deficiency of TREM2 leads to impaired microglial survival, migration and, phagocytosis (81). In addition, the mild activation of TREM2 has shown anti-inflammatory function to inhibit TLR4 receptor-induced proinflammatory response (82–84). However, the role of TREM2 in the inflammatory process is still controversial. For example, the injection of AD-Tau into the brain of mice with amyloidosis to induce Aβ-dependent Tau deposition showed that TREM2 consistently activated microglia and aggravated Tau pathology as well as neurodystrophy (85). Furthermore, other Aβ peptides, fibrils, and APPS are also potent activators of microglia (86). In AD pathology, the PRRs bind to different types of Aβ with different affinities to initiate the activation of glial cells, causing accumulation of more microglia and then increasing the density of microglia (87). Moreover, Aβ could activate the NF-KB-dependent pathways to bind to the surface of microglia to activate extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways, ultimately triggering the proinflammatory gene expression (88, 89). An autopsy study of the temporal neocortex in 15 control subjects without dementia and 91 AD patients has indicated that in the postmortem brain of AD patients, even after the Aβ plaque growth ends, the density of microglia is increased linearly and positively correlated with NFTs burden, which is consistently observed in the non-amnesic AD variant of primary progressive aphasia (54, 90). Inconsistent with other studies, the activation of Wnt-3a in primary microglia could promote microglia into the proinflammatory state of AD (91–93). Furthermore, the exogenous interfering factors, e.g., lipopolysaccharides (LPS), injected intraperitoneally in 5×FAD mice and APP23 mice are subsequently observed in the microglial hyperresponsiveness around the dense core plaque (94). Due to its significant biological importance, the activation of microglia caused by inflammation which is induced by overactivated microglia has attracted increasing attention to explore the molecular mechanisms regulating the microglial phenotypic variations.

Table 1 Microglia receptors associated with Alzheimer’s disease (AD).
In the 1990s, microglia were proved to have evident interactions with Aβ and tau (Figure 2). Although their interconnections have been extensively investigated, the molecular mechanisms underlying their interconnections remain unclear. The glial cells and neurons interact in a synchronous manner to facilitate the occurrence and development of AD. The microglia influence the neuronal activity by direct physical contact or by releasing paracrine signal (94). Studies have shown that the overproduction of Aβ by neurons could stimulate the NF-κB pathway in astrocytes to trigger an increase in the extracellular expression of complement C3 and then to reverse to an adverse effect on both neurons and microglia, leading to neuronal damage and microglia activation (95, 96). Inconsistently, the activated microglia induce the A1 neurotoxic astrocytes to cause the loss of the ability of A1 astrocytes to foster neuronal survival, growth, synaptic initiation and phagocytosis, and induce the neuronal and oligodendrocyte death (97). Notably, microglia are recruited to neurons 7 days before their elimination with the neuronal loss reduced (98). In transgenic mice, microglia rapidly respond to plaque formation through the process of expansion, and then migrate to plaques and accumulate around amyloid plaques. The size of plaques varies with microglia volume, i.e., larger plaques are generally surrounded by larger microglia (99). In young APPswe/PS1d9xYFP transgenic mice, a dynamic plaque-forming process is detected in 24 hours and microglia are activated and recruited to the area in 1-2 days after the appearance of the new plaque, which is followed by progressive neuropathy (100). Among all types of Aβ peptides, the microglia induced by small oligomers of Aβ have shown stronger neurotoxicity relative to the relatively large oligomers, ultimately causing more severe neuronal death (101). Furthermore, the in vivo imaging has revealed that microglia could function as transporters to help Aβ propagate in unaffected tissues (102). Moreover, the chronically activated microglia around Aβ plaques could release inflammatory factors while engulfing plaques, leading to excessive activation of microglia and ultimately aggravating the disease (103).
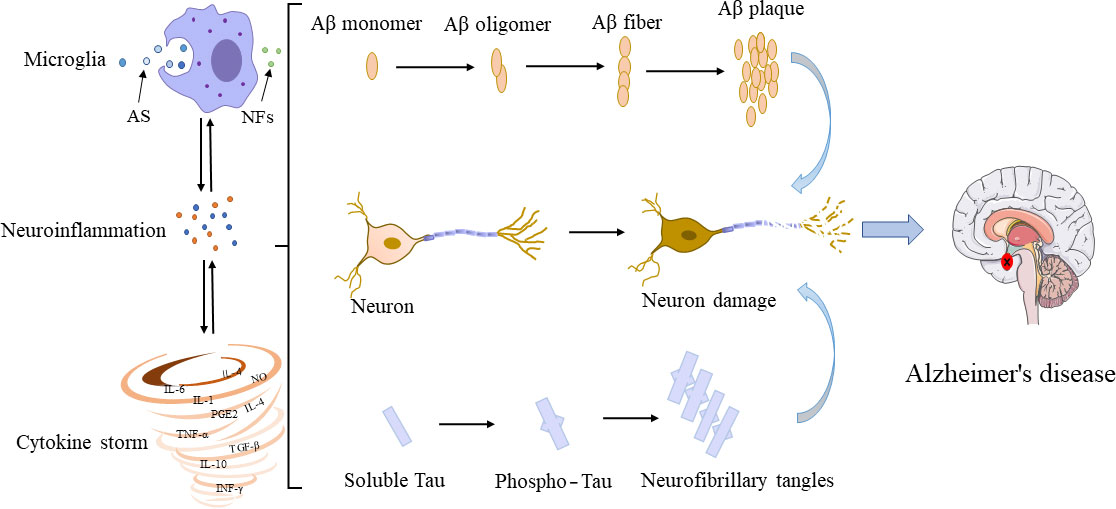
Figure 2 Interconnections among microglia, Aβ, tau, and neurons. In their early developmental stages, microglia enhance the phagocytic capacity and generate phagocytoma abnormal material, such as cell debris and misfolded proteins. Subsequently, M2 microglial cells are promoted to release proinflammatory mediators, leading to an inflammatory storm. In this process, the amyloid monomers gradually accumulated to form amyloid plaques. Meanwhile, free tau proteins are phosphorylated and aggregated in neurons to form neurofibrillary tangles. The accumulation of Aβ plaques and the formation of neurofibrillary tangles could positively feedback to microglia to promote the activation of microglia, leading to impaired ability to phagosome foreign objects. AS, abnormal substances (i.e., cell debris and misfolded protein); NFs, neurotrophic factors.
The relationships between Aβ and neuroinflammation have been extensively studied to provide support of the “amyloid cascade hypothesis,” while studies on the relationships between microglia and tau are sparse. Typically, tau mediates microtubule stability and aggregation by interacting with tubulin; in AD, tau is transformed into an insoluble form once it is dissociated from microtubules, further resulting in the formation of neurofibrillary tangles to increase the levels of reactive microglia surrounding tau (104, 105). The snRNA-seq analysis of nuclei has revealed different expression profiles in the microglia population, with AD2-microglia showing a strong correlation between the abundance and phosphorylated tau burden in the tissue, which are more enriched in tau pathological samples (106). Studies have shown that neurodegenerative microglia (MGnD) overproduce p-tau+ EVs in parallel with both reduction of Aβ plaques and removal of NP tau, which is suggested as a possible mechanistic link for the association between increased Aβ deposition and tau propagation in APP mutant knock-in homozygote (AppNL-G-F) mice (107). However, recent research has indicated that immunosuppressors attenuate tau pathology by inhibiting microglial activation in a mouse model of tau pathology (P301S), providing experimental evidence to link early microglial activation with tau pathology (108). To date, the pathogenic mechanism underlying the microglial promotion of tau deposition remains unclear. It is proved that tau could activate the NF-KB signaling pathway, driving microglia-mediated seeding and spreading of tau, as verified by single-cell RNA-seq analysis (109). Furthermore, studies have shown that the polyglutamine binding protein 1 (PQBP1) in microglia first senses the exogenous tau 3R/4R proteins through direct interaction and then provokes innate immune reactions through triggering the cyclic GMP-AMP synthase (cGAS) in the stimulator of interferon gene (STING) pathway, while PQBP1 is required for sensing the tau-induced NF-KB translocation, NF-κB-dependent transcription of inflammatory genes, brain inflammation, and ultimately cognitive impairment in mice (110). Given the complex interconnections among glial cells, Aβ, tau, and neurons, the communications among these units generate the positive feedback in the inflammatory environment of AD, causing a disordered and self-amplified inflammatory response.
To date, the therapeutic interventions to lower the Aβ level have yielded unsatisfactory results, and even the oral administration of β-site amyloid precursor protein–cleaving enzyme 1 (BACE-1) inhibitors that block Aβ production fails to reduce the cognitive or functional decline in the patients suffering mild to moderate AD (12). Likewise, many clinical trials have not been successful in eliminating various forms of Aβ and tau from the brains of AD patients (13), suggesting that the plaque removal may not be an ideal strategy for treating symptomatic AD, although it could still be a feasible preventive treatment. In contrast, targeting the microglia for therapeutic intervention against neuroinflammation exacerbated by overactivated microglia represents a potentially promising therapeutic strategy for AD. Therefore, it is important to identify the actions of microglia in the process of AD development in order to intervene and treat AD by the following methods: first, suppressing microglial priming in the pre-disease phase; second, limiting the proinflammatory response of microglia; and third, modulating and transforming the phenotypes of microglia to an anti-inflammatory phenotype.
Various factors induce imbalances of the CNS, e.g., aging, systemic inflammation, or stress, leading to the initiation of microglia and causing microglia to show strong susceptibility and responsiveness toward inflammatory stimuli. Therefore, a potential treatment strategy for AD could be to inhibit pre-disease microglial priming. Studies have shown that the middle-aged AD patients with obesity and insulin-resistance are prone to inducing inflammation, while statins exhibit a protective effect against this injury, preventing the initiation of microglia (111, 112). Furthermore, supplementation with folic acid and omega-3 fatty acid has also been shown to intervene the neuroinflammation and reduce the level of inflammation in both the cerebrospinal fluid and blood of individuals with AD (113, 114).
The factor of NF-κB has been implicated in physiological processes related to signaling, cognition, and memory in CNS. This factor could be activated by oxidative stress, neuroinflammation, and other factors, leading to CNS dysfunction in AD patients (115). The production of ROS initiates the enzymatic activities of IKKb to phosphorylate the heterodimer of NF-κB, which is an inhibitor of kappaB (IκB), leading to its degradation via the ubiquitin-proteasome pathway, while the dissociation of IκB from the dimer could initiate the NF-κB influx into the nucleus (116). It is indicated that the activation of NF-κB stimulates the BACE1 expression and Aβ processing, which is probably a novel molecular mechanism regulating the AD progression (117). It has been discovered that inhibition of NF-κB activation by antioxidants, polyphenols, and non-steroidal anti-inflammatory drugs (NSAIDs) is essential to reduce the load of Aβ (118). For example, Tanshinone I is used to inhibit the synthesis and expression of several proinflammatory M1 mediators, significantly suppressing the LPS-induced activation of NF-κB in microglia (119), while Forsythia B regulates the neuroinflammation in APP/PS1 mice, effectively ameliorating cognitive function, reducing the accumulation of Aβ and tau, and attenuating glial cell activation in the hippocampus (120). Similarly, when the NF-kB signaling pathway is inhibited, the release of pathogenic tau by primary microglia could be reduced to rescue the autophagy defect of microglia and reduce the neurotoxic reaction (109).
The NOD-like receptor thermal protein domain associated protein 3 (NLRP3) are highly enriched in microglia comparison to both NLRP1 and NLRP2 (121). Therefore, it is worth further exploring the potential application of NLRP3 in the diagnosis and treatment of AD compared with other types of inflammatory vesicles. Indeed, there is ample evidence to show that the Aβ level in brains of APP/PS1 or TgCRND8 mice could be reduced with the memories modified following the use of small molecular inhibitors of NLRP3 inflammatory vesicles or knockdown (KO) of NLRP3 expression (122, 123). Furthermore, inhibition of NLRP3 inflammasome reduces IL-1β secretion and limits the injury to neighboring tissues, while IL-1β is thought to enhance the expression of s100β protein, which is closely associated with AD (124, 125). For example, benzylisothiocyanate (126), stavudine (127), and artemisinin (128) all have shown therapeutic effects on AD mice by inhibiting NLRP3 activation.
Microglia support the neuronal function by removing toxic damage during the early stages of AD. Interference with the activation of microglia could allow their anti-inflammatory effect to persist for a long time. For example, the activated PPPAR-γ could inhibit the inflammatory response, while in mouse model of AD, treatment with PPAR-γ activators, e.g., pioglitazone and rosiglitazone, could switch microglia to show anti-inflammatory phenotype with phagocytosis (37, 129–131), which is probably related to the phagocytosis of amyloid deposits.
The microglia response to injury suggests the significant potential of microglia used as a diagnostic marker for the development and progression of AD. The release of proinflammatory factors could be detected in the olfactory bulb (OB) of 2-month-old 5×FAD mice, showing significant microglial activation and morphological variations (132), suggesting that microglia-mediated OB neuroinflammation could serve as an early biomarker of AD. Even before the positive β-amyloid pathology is shown, the increased microglial activation is associated with defective NREM rapid-sleep spindle expression in the frontal cortex (24), providing evidence for the earlier diagnosis of AD. The restraint of microglia activation and the associated proinflammatory mediators are diagnostic and therapeutic targets of AD. Because microglia have different phenotypes with varied functions, it will be important to further explore the cause of the microglia activation detected in the initial phase of AD, either the anti-inflammatory M2 phenotype attempting to clear Aβ and protect neurons or an M1 phenotype leading to the neuronal damage. The interventions targeting the proinflammatory phenotype of microglia in the earlier phase of AD could be a potentially efficient strategy to treat AD.
ZL, CW, and XC jointly completed the conception and writing of the review. SZ, XW, and SW completed the picture drawing of the review. YL and LW provided assistance for literature search and manuscript editing. All authors contributed to the article and approved the submitted version.
This work was supported by Key Research and Development Project of Shandong Province (2022CXGC010507).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer's disease. Lancet (2021) 397(10284):1577–90. doi: 10.1016/S0140-6736(20)32205-4
2. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci (2015) 18(6):794–9. doi: 10.1038/nn.4017
3. Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of abeta and cognition in aging and Alzheimer disease. Ann Neurol (2011) 69(1):181–92. doi: 10.1002/ana.22248
4. Arranz AM, De Strooper B. The role of astroglia in alzheimer's disease: pathophysiology and clinical implications. Lancet Neurol (2019) 18(4):406–14. doi: 10.1016/S1474-4422(18)30490-3
5. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
6. Frost JL, Schafer DP. Microglia: Architects of the developing nervous system. Trends Cell Biol (2016) 26(8):587–97. doi: 10.1016/j.tcb.2016.02.006
7. Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer's disease. Lancet (2016) 388(10043):505–17. doi: 10.1016/S0140-6736(15)01124-1
8. 2020 alzheimer's disease facts and figures. Alzheimers Dement (2020) 16(3):319–460. doi: 10.1002/alz.12068
9. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science (1992) 256(5054):184–5. doi: 10.1126/science.1566067
10. Hardy J, Selkoe DJ. The amyloid hypothesis of alzheimer's disease: progress and problems on the road to therapeutics. Science (2002) 297(5580):353–6. doi: 10.1126/science.1072994
11. Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian imaging, biomarkers and lifestyle (AIBL) study of aging. Neurobiol Aging. (2010) 31(8):1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007
12. Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, et al. Randomized trial of verubecestat for mild-to-Moderate alzheimer's disease. N Engl J Med (2018) 378(18):1691–703. doi: 10.1056/NEJMoa1706441
13. Lannfelt L, Relkin NR, Siemers ER. Amyloid-ss-directed immunotherapy for alzheimer's disease. J Intern Med (2014) 275(3):284–95. doi: 10.1111/joim.12168
14. Small SA, Duff K. Linking abeta and tau in late-onset alzheimer's disease: a dual pathway hypothesis. Neuron (2008) 60(4):534–42. doi: 10.1016/j.neuron.2008.11.007
15. Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci (2019) 20(3):148–60. doi: 10.1038/s41583-019-0132-6
16. Wang H, Shen Y, Chuang H, Chiu C, Ye Y, Zhao L. Neuroinflammation in alzheimer's disease: Microglia, molecular participants and therapeutic choices. Curr Alzheimer Res (2019) 16(7):659–74. doi: 10.2174/1567205016666190503151648
17. Hampel H, Mesulam MM, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, et al. The cholinergic system in the pathophysiology and treatment of alzheimer's disease. Brain (2018) 141(7):1917–33. doi: 10.1093/brain/awy132
18. Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer's disease. Cell (2013) 153(3):707–20. doi: 10.1016/j.cell.2013.03.030
19. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol (2021) 17(3):157–72. doi: 10.1038/s41582-020-00435-y
20. Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, et al. Aggregated tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol (2019) 137(4):599–617. doi: 10.1007/s00401-018-01957-y
21. Leuti A, Fazio D, Fava M, Piccoli A, Oddi S, Maccarrone M. Bioactive lipids, inflammation and chronic diseases. Adv Drug Delivery Rev (2020) 159:133–69. doi: 10.1016/j.addr.2020.06.028
22. Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol (2020) 16(1):9–29. doi: 10.1038/s41582-019-0284-z
23. Scipioni L, Ciaramellano F, Carnicelli V, Leuti A, Lizzi AR, De Dominicis N, et al. Microglial endocannabinoid signalling in AD. Cells (2022) 11(7):1237. doi: 10.3390/cells11071237
24. Mander BA, Dave A, Lui KK, Sprecher KE, Berisha D, Chappel-Farley MG, et al. Inflammation, tau pathology, and synaptic integrity associated with sleep spindles and memory prior to beta-amyloid positivity. Sleep (2022) 45(9):zsac135. doi: 10.1093/sleep/zsac135
25. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience (1990) 39(1):151–70. doi: 10.1016/0306-4522(90)90229-W
26. Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci (2013) 16(5):543–51. doi: 10.1038/nn.3358
27. Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. (2004) 1(1):14. doi: 10.1186/1742-2094-1-14
28. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352(6286):712–6. doi: 10.1126/science.aad8373
29. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature (2016) 534(7608):538–43. doi: 10.1038/nature18283
30. Lan L, Wang H, Zhang X, Shen Q, Li X, He L, et al. Chronic exposure of alcohol triggers microglia-mediated synaptic elimination inducing cognitive impairment. Exp Neurol (2022) 353:114061. doi: 10.1016/j.expneurol.2022.114061
31. Wendimu MY, Hooks SB. Microglia phenotypes in aging and neurodegenerative diseases. Cells (2022) 11(13):2091. doi: 10.3390/cells11132091
32. Colton C, Wilcock DM. Assessing activation states in microglia. CNS neurol Disord Drug targets. (2010) 9(2):174–91. doi: 10.2174/187152710791012053
33. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
34. Udeochu JC, Shea JM, Villeda SA. Microglia communication: Parallels between aging and alzheimer's disease. Clin Exp neuroimmunol (2016) 7(2):114–25. doi: 10.1111/cen3.12307
35. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
36. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in alzheimer's disease trajectory. Brain (2017) 140(3):792–803. doi: 10.1093/brain/aww349
37. Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in alzheimer's disease: a prospective study using 18F-DPA-714 PET imaging. Brain (2016) 139(Pt 4):1252–64. doi: 10.1093/brain/aww017
38. Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature (2016) 532(7598):240–4. doi: 10.1038/nature17630
39. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
40. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron (2022) 110(21):3458–83. doi: 10.1016/j.neuron.2022.10.020
41. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in alzheimer's disease. Acta Neuropathol (2009) 118(4):475–85. doi: 10.1007/s00401-009-0556-6
42. St-Pierre MK, Bordeleau M, Tremblay ME. Visualizing dark microglia. Methods Mol Biol (2019) 2034:97–110. doi: 10.1007/978-1-4939-9658-2_8
43. Bisht K, Sharma KP, Lecours C, Sanchez MG, El Hajj H, Milior G, et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia (2016) 64(5):826–39. doi: 10.1002/glia.22966
44. St-Pierre MK, Carrier M, Gonzalez Ibanez F, Simoncicova E, Wallman MJ, Vallieres L, et al. Ultrastructural characterization of dark microglia during aging in a mouse model of alzheimer's disease pathology and in human post-mortem brain samples. J Neuroinflammation. (2022) 19(1):235. doi: 10.1186/s12974-022-02595-8
45. Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep (2017) 21(2):366–80. doi: 10.1016/j.celrep.2017.09.039
46. Ismail R, Parbo P, Madsen LS, Hansen AK, Hansen KV, Schaldemose JL, et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in alzheimer's disease: a longitudinal PET study. J Neuroinflammation. (2020) 17(1):151. doi: 10.1186/s12974-020-01820-6
47. Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, et al. Soluble phospho-tau from alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathol (2016) 132(6):897–916. doi: 10.1007/s00401-016-1630-5
48. Ekonomou A, Savva GM, Brayne C, Forster G, Francis PT, Johnson M, et al. Stage-specific changes in neurogenic and glial markers in alzheimer's disease. Biol Psychiatry (2015) 77(8):711–9. doi: 10.1016/j.biopsych.2014.05.021
49. Plescher M, Seifert G, Hansen JN, Bedner P, Steinhauser C, Halle A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an alzheimer's disease mouse model. Glia (2018) 66(7):1464–80. doi: 10.1002/glia.23318
50. Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, et al. Transcriptional signature in microglia associated with abeta plaque phagocytosis. Nat Commun (2021) 12(1):3015. doi: 10.1038/s41467-021-23111-1
51. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47(3):566–81 e9. doi: 10.1016/j.immuni.2017.08.008
52. McFarland KN, Ceballos C, Rosario A, Ladd T, Moore B, Golde G, et al. Microglia show differential transcriptomic response to abeta peptide aggregates ex vivo and in vivo. Life Sci Alliance (2021) 4(7):e202101108. doi: 10.26508/lsa.202101108
53. Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of alzheimer's disease not evident in mouse models. Cell Rep (2018) 22(3):832–47. doi: 10.1016/j.celrep.2017.12.066
54. Serrano-Pozo A, Mielke ML, Gomez-Isla T, Betensky RA, Growdon JH, Frosch MP, et al. Reactive glia not only associates with plaques but also parallels tangles in alzheimer's disease. Am J Pathol (2011) 179(3):1373–84. doi: 10.1016/j.ajpath.2011.05.047
55. Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci (2017) 20(8):1162–71. doi: 10.1038/nn.4597
56. Zheng H, Liu CC, Atagi Y, Chen XF, Jia L, Yang L, et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol Aging. (2016) 42:132–41. doi: 10.1016/j.neurobiolaging.2016.03.004
57. Qin Q, Teng Z, Liu C, Li Q, Yin Y, Tang Y. TREM2, microglia, and alzheimer's disease. Mech Ageing Dev (2021) 195:111438. doi: 10.1016/j.mad.2021.111438
58. Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, Walker DG. TREM2 protein expression changes correlate with alzheimer's disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathol (2015) 25(4):469–80. doi: 10.1111/bpa.12190
59. Perez SE, Nadeem M, He B, Miguel JC, Malek-Ahmadi MH, Chen K, et al. Neocortical and hippocampal TREM2 protein levels during the progression of alzheimer's disease. Neurobiol Aging. (2017) 54:133–43. doi: 10.1016/j.neurobiolaging.2017.02.012
60. Rajesh Y, Kanneganti TD. Innate immune cell death in neuroinflammation and alzheimer's disease. Cells (2022) 11(12):1885. doi: 10.3390/cells11121885
61. McDonald CL, Hennessy E, Rubio-Araiz A, Keogh B, McCormack W, McGuirk P, et al. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of alzheimer's disease. Brain Behav Immun (2016) 58:191–200. doi: 10.1016/j.bbi.2016.07.143
62. Zhou C, Sun X, Hu Y, Song J, Dong S, Kong D, et al. Genomic deletion of TLR2 induces aggravated white matter damage and deteriorated neurobehavioral functions in mouse models of alzheimer's disease. Aging (2019) 11(17):7257–73. doi: 10.18632/aging.102260
63. Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of alzheimer's disease. J Neuroinflammation. (2008) 5:23. doi: 10.1186/1742-2094-5-23
64. Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, et al. TLR4 mutation reduces microglial activation, increases abeta deposits and exacerbates cognitive deficits in a mouse model of alzheimer's disease. J Neuroinflammation. (2011) 8:92. doi: 10.1186/1742-2094-8-92
65. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated caspase-6 activation. Cell Death Differ (2015) 22(10):1676–86. doi: 10.1038/cdd.2015.16
66. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
67. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493(7434):674–8. doi: 10.1038/nature11729
68. Milner MT, Maddugoda M, Gotz J, Burgener SS, Schroder K. The NLRP3 inflammasome triggers sterile neuroinflammation and alzheimer's disease. Curr Opin Immunol (2021) 68:116–24. doi: 10.1016/j.coi.2020.10.011
69. Barczuk J, Siwecka N, Lusa W, Rozpedek-Kaminska W, Kucharska E, Majsterek I. Targeting NLRP3-mediated neuroinflammation in alzheimer's disease treatment. Int J Mol Sci (2022) 23(16):8979. doi: 10.3390/ijms23168979
70. Bravo J, Ribeiro I, Terceiro AF, Andrade EB, Portugal CC, Lopes IM, et al. Neuron-microglia contact-dependent mechanisms attenuate methamphetamine-induced microglia reactivity and enhance neuronal plasticity. Cells (2022) 11(3):355. doi: 10.3390/cells11030355
71. Rabaneda-Lombarte N, Serratosa J, Bove J, Vila M, Saura J, Sola C. The CD200R1 microglial inhibitory receptor as a therapeutic target in the MPTP model of parkinson's disease. J Neuroinflammation. (2021) 18(1):88. doi: 10.1186/s12974-021-02132-z
72. Liu LQ, Liu XR, Zhao JY, Yan F, Wang RL, Wen SH, et al. Brain-selective mild hypothermia promotes long-term white matter integrity after ischemic stroke in mice. CNS Neurosci Ther (2018) 24(12):1275–85. doi: 10.1111/cns.13061
73. Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in alzheimer's disease. Lancet Neurol (2015) 14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5
74. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
75. Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci (2005) 25(36):8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005
76. Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. (2009) 6:1. doi: 10.1186/1742-2094-6-1
77. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol (2014) 258:5–16. doi: 10.1016/j.expneurol.2014.01.001
78. Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in abeta uptake and clearance. Brain (2006) 129(Pt 11):3006–19. doi: 10.1093/brain/awl249
79. Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar a{beta}-stimulated microglial activation. J Neurosci (2009) 29(38):11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009
80. Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, et al. Activation of toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem (2006) 281(6):3651–9. doi: 10.1074/jbc.M508125200
81. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in alzheimer's disease. Cell (2017) 170(4):649–63 e13. doi: 10.1016/j.cell.2017.07.023
82. Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol (Baltimore Md 1950). (2006) 177(4):2051–5. doi: 10.4049/jimmunol.177.4.2051
83. Zhong L, Chen XF, Zhang ZL, Wang Z, Shi XZ, Xu K, et al. DAP12 stabilizes the c-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J Biol Chem (2015) 290(25):15866–77. doi: 10.1074/jbc.M115.645986
84. Zhong L, Zhang ZL, Li X, Liao C, Mou P, Wang T, et al. TREM2/DAP12 complex regulates inflammatory responses in microglia via the JNK signaling pathway. Front Aging Neurosci (2017) 9:204. doi: 10.3389/fnagi.2017.00204
85. Jain N, Lewis CA, Ulrich JD, Holtzman DM. Chronic TREM2 activation exacerbates abeta-associated tau seeding and spreading. J Exp Med (2023) 220(1):e20220654. doi: 10.1084/jem.20220654
86. Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein e. Nature (1997) 388(6645):878–81. doi: 10.1038/42257
87. Islam R, Choudhary H, Rajan R, Vrionis F, Hanafy KA. An overview on microglial origin, distribution, and phenotype in alzheimer's disease. J Cell Physiol (2022). doi: 10.1002/jcp.30829
88. Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in alzheimer's disease. Curr Drug Targets Inflammation Allergy (2005) 4(2):247–56. doi: 10.2174/1568010053586237
89. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and alzheimer's disease. Neurobiol Aging. (2000) 21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X
90. Ohm DT, Fought AJ, Martersteck A, Coventry C, Sridhar J, Gefen T, et al. Accumulation of neurofibrillary tangles and activated microglia is associated with lower neuron densities in the aphasic variant of alzheimer's disease. Brain Pathol (2021) 31(1):189–204. doi: 10.1111/bpa.12902
91. Halleskog C, Schulte G. Pertussis toxin-sensitive heterotrimeric g(alphai/o) proteins mediate WNT/beta-catenin and WNT/ERK1/2 signaling in mouse primary microglia stimulated with purified WNT-3A. Cell Signal (2013) 25(4):822–8. doi: 10.1016/j.cellsig.2012.12.006
92. Dijksterhuis JP, Arthofer E, Marinescu VD, Nelander S, Uhlen M, Ponten F, et al. High levels of WNT-5A in human glioma correlate with increased presence of tumor-associated microglia/monocytes. Exp Cell Res (2015) 339(2):280–8. doi: 10.1016/j.yexcr.2015.10.022
93. Hooper C, Sainz-Fuertes R, Lynham S, Hye A, Killick R, Warley A, et al. Wnt3a induces exosome secretion from primary cultured rat microglia. BMC Neurosci (2012) 13:144. doi: 10.1186/1471-2202-13-144
94. Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, et al. Immune hyperreactivity of abeta plaque-associated microglia in alzheimer's disease. Neurobiol Aging. (2017) 55:115–22. doi: 10.1016/j.neurobiolaging.2017.03.021
95. Lian H, Yang L, Cole A, Sun L, Chiang Angie CA, Fowler Stephanie W, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with alzheimer’s disease. Neuron (2015) 85(1):101–15. doi: 10.1016/j.neuron.2014.11.018
96. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer's disease. J Neurosci (2016) 36(2):577–89. doi: 10.1523/JNEUROSCI.2117-15.2016
97. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541(7638):481–7. doi: 10.1038/nature21029
98. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of alzheimer's disease. Nat Neurosci (2010) 13(4):411–3. doi: 10.1038/nn.2511
99. Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci (2008) 28(16):4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008
100. Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of alzheimer's disease. Nature (2008) 451(7179):720–4. doi: 10.1038/nature06616
101. Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large Soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci (2017) 37(1):152–63. doi: 10.1523/JNEUROSCI.1698-16.2016
102. d'Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezo C, Erny D, et al. Microglia contribute to the propagation of abeta into unaffected brain tissue. Nat Neurosci (2022) 25(1):20–5. doi: 10.1038/s41593-021-00951-0
103. Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid a improves alzheimer's disease-related pathology. Proc Natl Acad Sci U S A. (2013) 110(5):1941–6. doi: 10.1073/pnas.1215165110
104. Althafar ZM. Targeting microglia in alzheimer's disease: From molecular mechanisms to potential therapeutic targets for small molecules. Molecules (2022) 27(13):4124. doi: 10.3390/molecules27134124
105. Azevedo EP, Ledo JH, Barbosa G, Sobrinho M, Diniz L, Fonseca AC, et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis (2013) 4:e789. doi: 10.1038/cddis.2013.325
106. Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, et al. Distinct amyloid-beta and tau-associated microglia profiles in alzheimer's disease. Acta Neuropathol (2021) 141(5):681–96. doi: 10.1007/s00401-021-02263-w
107. Clayton K, Delpech JC, Herron S, Iwahara N, Ericsson M, Saito T, et al. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol Neurodegener. (2021) 16(1):18. doi: 10.1186/s13024-021-00440-9
108. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron (2007) 53(3):337–51. doi: 10.1016/j.neuron.2007.01.010
109. Wang C, Fan L, Khawaja RR, Liu B, Zhan L, Kodama L, et al. Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat Commun (2022) 13(1):1969. doi: 10.1038/s41467-022-29552-6
110. Jin M, Shiwaku H, Tanaka H, Obita T, Ohuchi S, Yoshioka Y, et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat Commun (2021) 12(1):6565. doi: 10.1038/s41467-021-26851-2
111. Dias HK, Brown CL, Polidori MC, Lip GY, Griffiths HR. LDL-lipids from patients with hypercholesterolaemia and alzheimer's disease are inflammatory to microvascular endothelial cells: mitigation by statin intervention. Clin Sci (Lond). (2015) 129(12):1195–206. doi: 10.1042/CS20150351
112. Verdile G, Keane KN, Cruzat VF, Medic S, Sabale M, Rowles J, et al. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and alzheimer's disease. Mediators Inflamm (2015) 2015:105828. doi: 10.1155/2015/105828
113. Chen H, Liu S, Ji L, Wu T, Ji Y, Zhou Y, et al. Folic acid supplementation mitigates alzheimer's disease by reducing inflammation: A randomized controlled trial. Mediators Inflamm (2016) 2016:5912146. doi: 10.1155/2016/5912146
114. Vedin I, Cederholm T, Freund Levi Y, Basun H, Garlind A, Faxén Irving G, et al. Effects of docosahexaenoic acid-rich n-3 fatty acid supplementation on cytokine release from blood mononuclear leukocytes: the OmegAD study. Am J Clin Nutr (2008) 87(6):1616–22. doi: 10.1093/ajcn/87.6.1616
115. Kounatidis I, Ligoxygakis P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol (2012) 2(5):120075. doi: 10.1098/rsob.120075
116. Jones SV, Kounatidis I. Nuclear factor-kappa b and Alzheimer disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol (2017) 8:1805. doi: 10.3389/fimmu.2017.01805
117. Shi Z, Hong Y, Zhang K, Wang J, Zheng L, Zhang Z, et al. BAG-1M co-activates BACE1 transcription through NF-kappaB and accelerates abeta production and memory deficit in alzheimer's disease mouse model. Biochim Biophys Acta Mol Basis Dis (2017) 1863(9):2398–407. doi: 10.1016/j.bbadis.2017.05.014
118. Wentzell J, Kretzschmar D. Alzheimer's disease and tauopathy studies in flies and worms. Neurobiol Dis (2010) 40(1):21–8. doi: 10.1016/j.nbd.2010.03.007
119. Fu Y, Yang J, Wang X, Yang P, Zhao Y, Li K, et al. Herbal compounds play a role in neuroprotection through the inhibition of microglial activation. J Immunol Res (2018) 2018:9348046. doi: 10.1155/2018/9348046
120. Kong F, Jiang X, Wang R, Zhai S, Zhang Y, Wang D. Forsythoside b attenuates memory impairment and neuroinflammation via inhibition on NF-kappaB signaling in alzheimer's disease. J Neuroinflammation. (2020) 17(1):305. doi: 10.1186/s12974-020-01967-2
121. de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. J Cereb Blood Flow Metab (2014) 34(3):369–75. doi: 10.1038/jcbfm.2013.227
122. Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav Immun (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
123. Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of alzheimer's disease. Mol Neurobiol (2018) 55(3):1977–87. doi: 10.1007/s12035-017-0467-9
124. Lambert JC, Ferreira S, Gussekloo J, Christiansen L, Brysbaert G, Slagboom E, et al. Evidence for the association of the S100beta gene with low cognitive performance and dementia in the elderly. Mol Psychiatry (2007) 12(9):870–80. doi: 10.1038/sj.mp.4001974
125. Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, et al. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. (1996) 17(5):761–6. doi: 10.1016/0197-4580(96)00104-2
126. Lee CM, Lee DS, Jung WK, Yoo JS, Yim MJ, Choi YH, et al. Benzyl isothiocyanate inhibits inflammasome activation in e. coli LPS-stimulated BV2 Cells Int J Mol Med (2016) 38(3):912–8. doi: 10.3892/ijmm.2016.2667
127. La Rosa F, Saresella M, Marventano I, Piancone F, Ripamonti E, Al-Daghri N, et al. Stavudine reduces NLRP3 inflammasome activation and modulates amyloid-beta autophagy. J Alzheimers Dis (2019) 72(2):401–12. doi: 10.3233/JAD-181259
128. Shi JQ, Zhang CC, Sun XL, Cheng XX, Wang JB, Zhang YD, et al. Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-kappaB and NLRP3 inflammasome activation. CNS Neurosci Ther (2013) 19(4):262–8. doi: 10.1111/cns.12066
129. Esmaeili MA, Yadav S, Gupta RK, Waggoner GR, Deloach A, Calingasan NY, et al. Preferential PPAR-alpha activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum Mol Genet (2016) 25(2):317–27. doi: 10.1093/hmg/ddv477
130. Escribano L, Simon AM, Gimeno E, Cuadrado-Tejedor M, Lopez de Maturana R, Garcia-Osta A, et al. Rosiglitazone rescues memory impairment in alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology (2010) 35(7):1593–604. doi: 10.1038/npp.2010.32
131. Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci (2012) 32(48):17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012
Keywords: Alzheimer’s disease, neuroinflammation, microglia activation, therapy, β-amyloid
Citation: Wang C, Zong S, Cui X, Wang X, Wu S, Wang L, Liu Y and Lu Z (2023) The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. Immunol. 14:1117172. doi: 10.3389/fimmu.2023.1117172
Received: 06 December 2022; Accepted: 10 February 2023;
Published: 22 February 2023.
Edited by:
Alexandru Movila, Indiana University, United StatesReviewed by:
Pritam Das, Mayo Clinic Florida, United StatesCopyright © 2023 Wang, Zong, Cui, Wang, Wu, Wang, Liu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiming Lu, bHV6aGltaW5nQHNkdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.