
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 15 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1114582
This article is part of the Research TopicProtocols to Success: All the Shots you Took Running up that HillView all 6 articles
 Chenicheri Kizhakkeveettil Keerthana1,2
Chenicheri Kizhakkeveettil Keerthana1,2 Tennyson Prakash Rayginia1,2†
Tennyson Prakash Rayginia1,2† Sadiq Chembothumparambil Shifana1†
Sadiq Chembothumparambil Shifana1† Nikhil Ponnoor Anto3
Nikhil Ponnoor Anto3 Kalishwaralal Kalimuthu1
Kalishwaralal Kalimuthu1 Noah Isakov3
Noah Isakov3 Ruby John Anto1*
Ruby John Anto1*Adenosine monophosphate-activated protein kinase (AMPK) is a key metabolic sensor that is pivotal for the maintenance of cellular energy homeostasis. AMPK contributes to diverse metabolic and physiological effects besides its fundamental role in glucose and lipid metabolism. Aberrancy in AMPK signaling is one of the determining factors which lead to the development of chronic diseases such as obesity, inflammation, diabetes, and cancer. The activation of AMPK and its downstream signaling cascades orchestrate dynamic changes in the tumor cellular bioenergetics. It is well documented that AMPK possesses a suppressor role in the context of tumor development and progression by modulating the inflammatory and metabolic pathways. In addition, AMPK plays a central role in potentiating the phenotypic and functional reprogramming of various classes of immune cells which reside in the tumor microenvironment (TME). Furthermore, AMPK-mediated inflammatory responses facilitate the recruitment of certain types of immune cells to the TME, which impedes the development, progression, and metastasis of cancer. Thus, AMPK appears to play an important role in the regulation of anti-tumor immune response by regulating the metabolic plasticity of various immune cells. AMPK effectuates the metabolic modulation of anti-tumor immunity via nutrient regulation in the TME and by virtue of its molecular crosstalk with major immune checkpoints. Several studies including that from our lab emphasize on the role of AMPK in regulating the anticancer effects of several phytochemicals, which are potential anticancer drug candidates. The scope of this review encompasses the significance of the AMPK signaling in cancer metabolism and its influence on the key drivers of immune responses within the TME, with a special emphasis on the potential use of phytochemicals to target AMPK and combat cancer by modulating the tumor metabolism.
Cancer metabolism relies on the fundamental principle of metabolic reprogramming which leads to the malignant transformation of normal cells into cancer cells. The acquisition of these metabolic alterations is regarded as one of the hallmarks of cancer. Metabolic reprogramming not only affects the biological activity of tumor cells, but it also regulates the differentiation and function of various immune cell populations. Previous literature suggests that the metabolic status of immune cells is crucial for the functional plasticity of the immune cells (1, 2). Over the past few decades, there has been a growing appreciation of the complex molecular crosstalk between the components of the TME and the cellular metabolic pathways. Besides, there is a growing body of evidence suggesting the chemoresistance and malignant progression of tumors to be a manifestation of the metabolic reprogramming that occurs in the immune cells present within the tumor microenvironment (2).
Adenosine monophosphate (AMP)-activated protein kinase (AMPK), is an evolutionarily conserved serine/threonine kinase and the primary regulator of cellular energy homeostasis. AMPK functions as a key metabolic sensor and is actively involved in all planes of energy metabolism and mitochondrial biogenesis (3). AMPK is activated in response to depletion in cellular energy levels resulting from conditions, such as hypoxia and nutrient starvation (4). Upon activation, it promotes ATP production and thus maintains cellular energy homeostasis.
The metabolic stress induced in cells as a result of increase in the cellular AMP : ATP ratio, due to decreased ATP production, and high levels of intracellular AMP, consequently, leads to the activation of AMPK. The resulting allosteric changes promote the phosphorylation of threonine 172(Thr172) within the activation loop of the AMPK catalytic α-subunit (5). This occurs following the binding of AMP/ADP to the γ-regulatory subunit of AMPK. The phosphorylation is mediated by upstream kinases, including liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase (CaMKKβ). Although allosteric activation is triggered only by the binding of AMP, the other two complementary effects can be mimicked by ADP (6). AMPK can sense even subtle changes in AMP concentrations (7–9). The binding of AMP to AMPK inhibits the dephosphorylation of the α-subunit of AMPK and helps maintaining AMPK in its activated state (10). The activation of AMPK in response to energy stress, further, leads to the concomitant inhibition of anabolic pathways and promotion of catabolic pathways that are down-stream of AMPK (11). However, under energy-replete conditions, enzymes such as TGFβ-activated kinase 1 (TAK1); protein phosphatase 2A (PP2A); protein phosphatase 2C (PP2C) and Mg2+/Mn2+-dependent protein phosphatase 1E (PPM1E) render AMPK inactive by maintaining it in an unphosphorylated state (12). A schematic representation highlighting the mechanism of AMPK activation and inactivation is depicted in Figure 1.
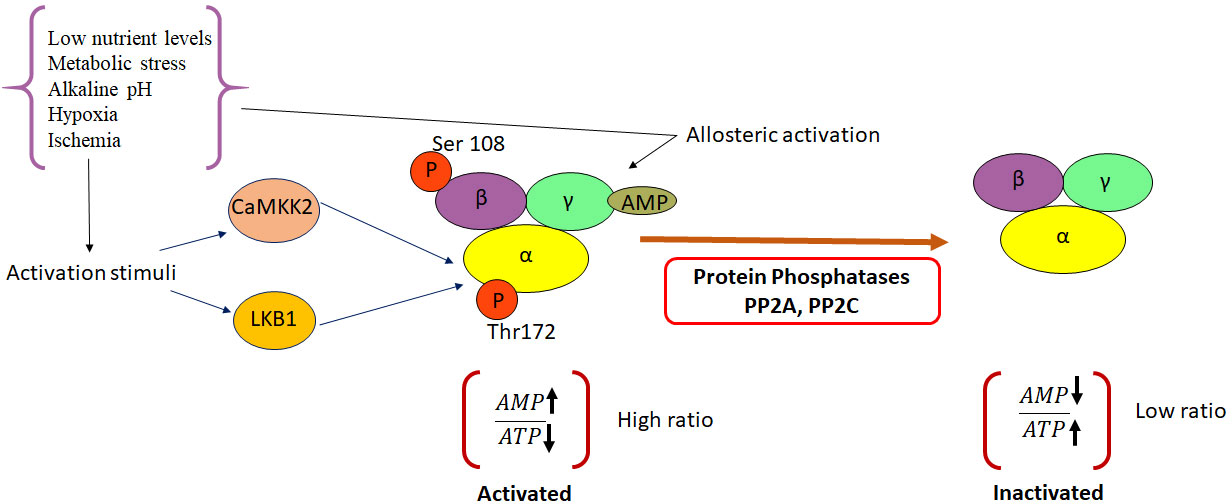
Figure 1 Molecular mechanisms leading to activation and inactivation of AMPK. AMPK is phosphorylated by the two upstream kinases, LKB1 and CAMKKβ in response to various activation stimuli. Protein phosphatases PP2A and C cause de-phosphorylation of AMPK and render it inactive. CAMKK2, Ca2+/Calmodulin-dependent protein kinase kinase 2; LKB1, Liver kinase B1; AMP, Adenosine mono phosphate; PP2A, protein phosphatase 2; PP2C, protein phosphatase 2C; ATP-Adenosine triphosphate.
Given the central role of AMPK in metabolism and the plethora of down-stream metabolic processes that are regulated by this kinase, AMPK has attracted global attention as a potential target for treating metabolic diseases such as obesity, Type 2 diabetes and cancer. Literature review suggests the de-regulation of cellular metabolism to be one of the key drivers of tumorigenesis and cancer progression (13). Besides, recent reports state that AMPK plays a central role in tumor cellular bioenergetics and in evoking an anti-tumor immune response, owing to its molecular crosstalk with various key players of the tumor microenvironment (TME). This review focuses on the role of AMPK in cancer metabolism and AMPK-mediated immunomodulation of the TME, highlighting AMPK as a therapeutic target in cancer patients. Furthermore, we discuss the use of plant-derived anti-cancer agents as activators of AMPK to combat cancer by modulating the tumor metabolism.
Structurally, the protein kinase AMPK is a heterotrimeric αβγ complex, which comprises of a catalytic α-subunit in association with β and γ regulatory subunits (11, 14). The complex structure of the γ -subunit, along with the non-catalytic C-terminal fragments of α and β subunits, in S. pombe was first reported by Townley (3).The β-subunit consists of a glycogen-binding domain (GBD), while the γ-subunit contains Bateman domains responsible for nucleotide binding. AMPK γ-subunit contains four tandem repeats of sequence motifs known as cystathionine-b-synthase (CBS). In AMPK subunits, these repeats assemble to a disc-like shape with one repeat in each quadrant, generating four nucleotide-binding sites. Of these, AMP, ADP, and ATP bind in competition at sites 1 and 3, while site 2 remains vacant. AMP is permanently bound to site 4 (7, 15).
Even though all eukaryotes have homologs of these proteins, the number of subunit genes vary between organisms (11). The aspartate residue (Asp139) in the catalytic loop functions as the base for catalysis, while mutation of aspartate to alanine renders AMPK catalytically inactive. AMPK belongs to a class of protein kinases that are basophilic and hence require the presence of basic residues in the N-terminal sequence, adjacent to the site of phosphorylation on target proteins (16).
Following the sequencing of the mammalian AMPK, it was found to be a homolog of the Saccharomyces cerevisiae protein kinase SNF1, which was previously identified as a regulator of gene transcription in response to glucose starvation (3, 16–19). The three alternative subunits of AMPK can form up to 12 distinct isoforms (14). There are two α subunit isoforms in mammalian AMPK and one in S.cervisea, which has an N-terminal kinase domain followed by an autoinhibitory region, plus a C-terminal domain involved in complex formation. β-subunits have two isoforms in mammalian AMPK and three in S.cervisea. Those subunits contain a canonical Ser/Thr kinase domain (KD), an adenine nucleotide sensor segment, termed as linker, an autoinhibitory domain (AID) and a subunit-interacting C-terminal domain (CTD), the latter of which contains the serine/threonine rich activation loop containing phosphorylation sites for various downstream proteins (11, 20–22). There are three γ-subunit isoforms in mammalian AMPK and one in S.cervisea. The γ- subunits have four copies of CBS motifs preceding unrelated N-terminal extensions of varying lengths. The CBS motifs function in pairs to form a discrete structural unit called a Bateman domain which are the regulatory binding sites for AMP and ATP (11, 16).
The structural and functional modules of the three AMPK subunits share a high degree of amino acid similarity. The α1- and α2-subunits share a sequence identity of 77%, which increases to 90% for the protein kinase module and 61% in the C-terminus. The α-subunits share 60% sequence identity with the kinase domain of sucrose non-fermenting 1 (SNF1) and 46% identity with the entire subunit (16). The CBM motifs of β1 and β2 retain ~80% identity, while the CBS modules across the three γ isoforms share ~60% conservation. Some of the AMPK subunits are known to be alternately spliced in different tissues allowing for further regulation and complexity (5, 16). This basic architecture is conserved in the S. cerevisiae as well as in mammalian homologs (3). Figure 2 is a graphical representation of the domain structure and the isoforms of AMPK.
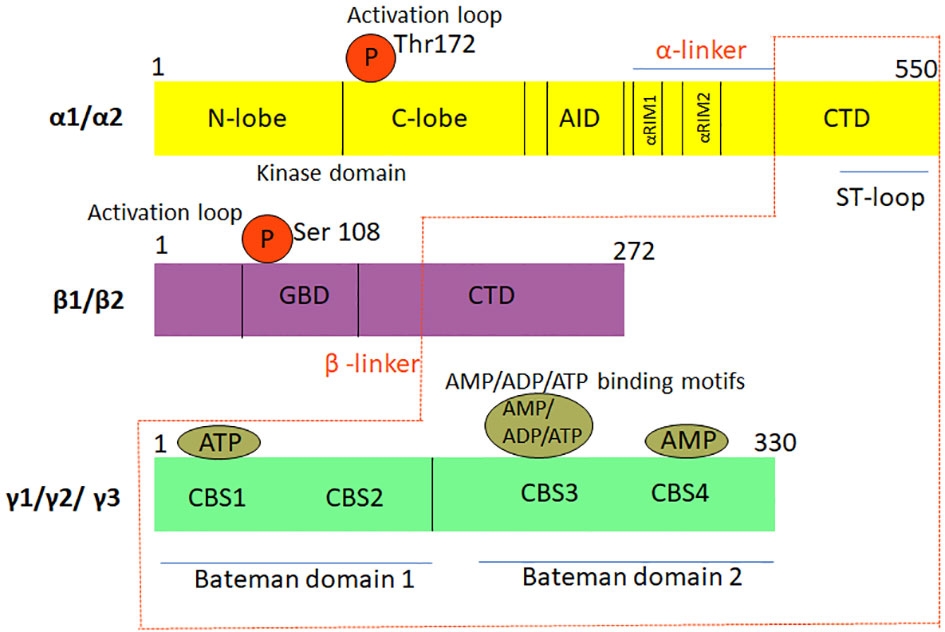
Figure 2 Graphical representation of the domain structure and isoforms of AMPK. The three sub-units, namely, αβγ, which constitute the heterotrimeric complex structure of AMPK have different isoforms. While the α and β sub-units have two isoforms each, the γ sub-unit has three isoforms. Thr, threonine; Ser, Serine; AID, Autoinhibitory domain; ST-loop, Stem loop; CTD, C-terminal domain; αRIM1/2, α-subunit motifs; α/β linker, regulatory linker region; GBD, GTPase protein binding domain; CBS1 domain, Cystathionine beta synthase1 domain; CBS2 domain, Cystathionine beta synthase2 domain; CBS3 domain, Cystathionine beta synthase3 domain; CBS4 domain, Cystathionine beta synthase 4 domain AMP, Adenosine monophosphate; ADP, Adenosine diphosphate; ATP, Adenosine triphosphate.
AMPK activity governs a plethora of metabolic and physiological processes. This kinase is dysregulated in various chronic diseases, such as diabetes, neurodegenerative and neuromuscular diseases, cardiovascular diseases, and cancer (23). The following section substantiates some of the key findings on the physiological roles of AMPK under normal and disease conditions:
Being a multi-substrate serine/threonine kinase, AMPK provides binding sites for an array of regulatory nucleotides. The primary physiological manifestation of activated AMPK is to redirect metabolism towards increased catabolism and decreased anabolism through the phosphorylation of key proteins in response to metabolic stress. This metabolic reprogramming is initiated in order to ensure the replenishment of intracellular ATP levels back to normal (12, 13).Studies using AMPKα1 and α2 knockout mouse models by Violett et al., revealed that glucose homoeostasis was unaltered in AMPKα1−/− mice whereas, high plasma glucose levels and low plasma insulin concentrations were observed in AMPKα2−/− mice although the insulin secretion was not altered in both types of mice. Furthermore, the team also reported that the AMPKα2 catalytic subunit modulates the activity of the autonomous nervous system in vivo (24).
Accumulation of glucose, fatty acids, and amino acids have been reported to suppress AMPK and eventually lead to insulin resistance, while stimulation of AMPK activity improved blood glucose levels, suggesting that development of AMPK activators might potentially function as anti-diabetic drugs (25–27). Metabolic inflexibility and insulin resistance present in Type 2 diabetes mellitus and obesity develop as a result of dysfunctional mitochondria in the muscles (28, 29).
Activation of AMPK in skeletal muscles induces peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) and up-regulates mitochondrial genes (30). These observations suggest that AMPK activators might have a beneficial effect in combating metabolic disorders. Studies also reveal a negative correlation between inflammation and AMPK. The proinflammatory cytokine, tumor necrosis factor α (TNFα), suppresses the phosphorylation of AMPK and increases the expression of protein phosphatase 2C (PP2C) in the skeletal muscles thereby inducing insulin resistance (31).
Interestingly, AMPK has been reported to play a key role in the development of Alzheimer’s disease (AD), a progressive neurodegenerative disease. One of the hallmark features of AD includes the aberrations in β-amyloid metabolism. Previous studies have revealed that AMPK activation is closely linked with the aberrant processing of β-amyloid protein precursor (AβPP). Further, AMPK signaling controls β-amyloid metabolism via the suppression of Glycogen synthase kinase 3β (GSK-3β) (32). Another independent study indicates that AMPK hyperphosphorylation occurs in the brains of mice that have experimental AD, as well as human patients suffering from AD (33, 34). Further studies demonstrated that resveratrol lowers extracellular accumulation of β-amyloid peptides through the activation of AMPK. This study also demonstrated that the inhibition of mammalian target of rapamycin complex 1 (mTORC1) mediated by the activation of AMPK induces autophagy and lysosomal degradation of the β-amyloid peptide (35).
AMPK is also activated under conditions of cardiac ischemia and studies indicate that the intrinsic activation of AMPK protects the heart from injuries induced due to the ischemia (36, 37). Treatment with AMPK activators such as 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) and metformin brought about a significant decrease in contractile dysfunction, apoptosis, and fibrosis in canine heart failure models (38).
AMPK activation has been extensively investigated as a potential therapeutic target in combating different types of cancer. AMPK phenotypically functions as a tumor suppressor by resisting pro-tumorigenic metabolic processes and directly inducing cell-cycle arrest in cancer cells. Cyclooxygenase 2 (COX-2) plays an important role in cancer stemness. Interestingly, AMPK also acts as a COX-2 inhibitor in cancers of the breast and colon (39). AMPK activation is critical in alleviating metabolic and energetic stresses associated with tumor progression. Besides, AMPK activation triggers the onset of multiple cell death mechanisms. It influences the cell cycle checkpoints, autophagy, mitophagy, and apoptosis. AMPK promotes autophagy and mitophagy by activating UNC-51-like kinase 1 (ULK1) and death-associated protein 1 (DAP1) respectively (40, 41). AMPK initiates the apoptotic program via the activation of p53, p21, and p27. It manifests cell cycle arrest via the inhibition of HUR and the concomitant activation of Cyclins A, B1, and D1 (42). Previous reports indicate that metformin, an AMPK activator down-regulates c-MYC in an AMPK-dependent manner in breast cancer cell lines (43).There are several other reports highlighting the significance of AMPK in cancer chemoprevention (44–50). Studies using MT 63–78, a specific and potent direct AMPK activator have revealed that AMPK activation induces mitotic arrest and apoptosis in androgen-sensitive and castration-resistant prostate cancer via mTORC1 blockade and the suppression of de novo lipogenesis (51). Previous studies have documented that treatment of hepatocellular carcinoma cells with the AMPK activators, AICAR and metformin, significantly inhibited their proliferation, and induced cell cycle arrest at the G1-S phase (52). Zou et al. demonstrated that metformin-induced activation of AMPK down-regulated the expression of segment polarity protein dishevelled homolog3 (DVL3), a key oncoprotein that activates the Wnt/β-catenin signaling pathway (53). Down-regulation of DVL3 reduced the levels of β-catenin and its downstream targets, cyclin D1 and c-Myc, resulting in suppression of cell proliferation. AMPK activation has also been reported to induce p53-dependent apoptotic effects in breast cancer cells (9),and inhibit the metastatic potential of melanoma cells by modulating the ERK signaling pathway and reducing the levels of the COX-2 protein (54). It also induces autophagic and apoptotic cell death through AMPK/JNK signaling (55).Vara-Ciruelos et al. have demonstrated the tumor suppressor role of AMPK-α1 by utilizing T-cell-specific knock-outs of Pten and Prkaa1 gene encoding AMPK-α1. In these models, absence of Pten and Prkaa1genes promoted lymphoma development at an early age and the tumors were significantly more aggressive (56).Previous literature also highlights the impacts of AMPK activation in augmenting the chemosensitizing efficacy of natural and synthetic compounds in combination with conventional chemotherapeutics (57–64). A list of various pharmacological activators of AMPK and their implication in different types of cancer is included in Table 1.
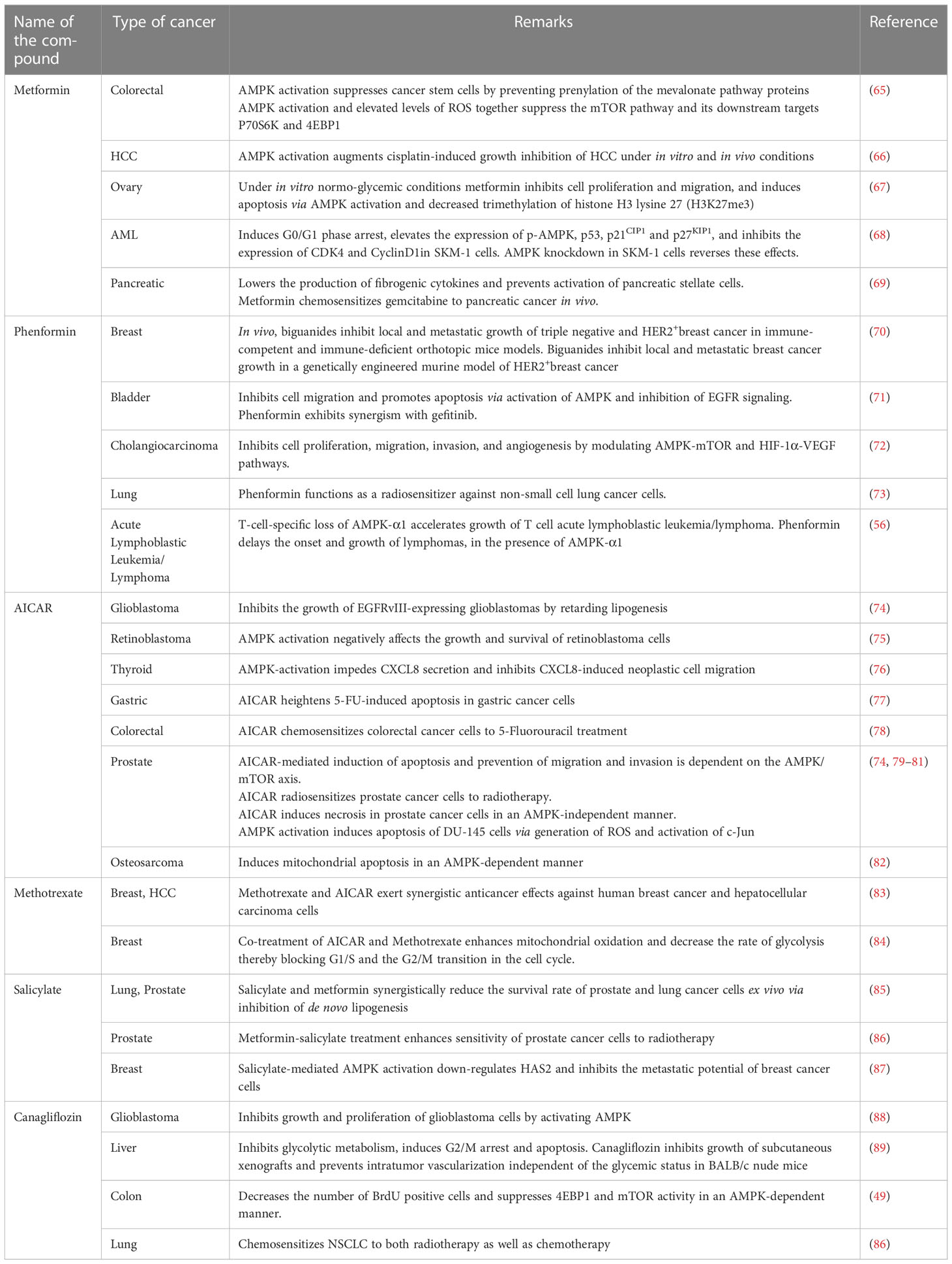
Table 1 Pre-clinical evidences of the anti-cancer effects of pharmacological activation of AMPK.
Cancer cells require an unquenched and persistent supply of energy and nutrients to aid their survival and rapid rates of proliferation. There is an ever-growing need for the uninterrupted functioning of anabolic pathways for promoting cell growth in cancer cells. This includes pathways involved in the synthesis of glucose, fatty acids, phospholipids, protein, and ribosomal RNA. Cancer cells undergo metabolic reprogramming in order to meet the increased metabolic needs and ensure continuous growth and proliferation (90, 91). One of the characteristic traits of cancer cells is aerobic glycolysis, also referred to as the “Warburg effect” wherein, cancer cells rely on cytosolic glycolysis over mitochondrial oxidative phosphorylation irrespective of oxygen supply (92). This selective preference for glycolysis despite its low ATP productivity is for procuring intermediates for anabolic processes, including biosynthesis of glycogen, amino acids, nucleic acids, and lipids as well as for ensuring the increased stability of the mitochondrial membrane in the cancer cells (93).In addition to the changes in the glycolytic phenotype, tumor cells also manifest mitochondrial energy reprogramming to achieve metabolic plasticity. Recent studies attribute tumor aggressiveness and chemoresistance to the hybrid glycolysis/OXPHOS (oxidative phosphorylation) phenotype of cancer cells. Aberration in tumor glucose metabolism and elevated rates of glycolysis contribute to intrinsic/acquired resistance to routinely used anticancer drugs. Thus, the regulation of tumor metabolism would be a promising therapeutic strategy irrespective of the cancer type (94). Activation of AMPK counteracts tumor progression through negative regulation of the Warburg effect of tumor cells (95).AMPK plays a crucial role in regulating carbohydrate, lipid and protein metabolic pathways (96). AMPK quintessentially halts all of these pathways, thereby depriving the cancer cells of energy and nutrients (97). Hence, AMPK functions as a metabolic tumor suppressor by modulating energy levels, enforcing metabolic checkpoints, and inhibiting cell growth (98).
AMPK influences glucose transporter (GLUT)-mediated trafficking of glucose across the plasma membrane. In addition, it promotes cellular glucose uptake by enhancing the mRNA expression of the genes encoding GLUT4 and hexokinase 2 and by mediating the translocation of GLUT4-containing intracellular vesicles across the plasma membrane. Previous studies have reported the role of AMPK in GLUT1/4 vesicle trafficking via the activation of thioredoxin-interacting protein (TXNIP) and Tre-2/Bub2/Cdc16 (TBC) domain family member 1 (TBC1D1) respectively (99). Once glucose is internalized, it is converted to glucose-6-phosphate by the action of hexokinases. Glucose-6-phosphate is further channelized to glycolysis, glycogen synthesis and pentose phosphate pathway. Of all the glucose metabolism pathways, AMPK plays an indispensable role in directly inhibiting aerobic glycolysis and glycogen synthesis (100). AMPK stimulates glycolytic flux through direct phosphorylation of PFKFB2 and PFKFB3 isoforms of the enzyme 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase (101). AMPK phosphorylation also culminates in the inhibition of gluconeogenesis-related enzymes such as glucose-6-phosphatase(G6Pase) and phosphoenol pyruvate carboxykinase (PEPCK) (102). The activation of AMPK impedes glycogen synthesis and activates the rate of glycogen breakdown by promoting the phosphorylation of glycogen phosphorylase (GP).Notably, AMPK activation also enhances insulin sensitivity, inhibits hepatic glucose production in the liver, stimulates glucose uptake in the skeletal muscles, and weakens proinflammatory changes (103).
The conversion of acetyl-CoA to malonyl-CoA is the rate-limiting step in the de novo synthesis of fatty acids. AMPK was initially identified as a kinase that phosphorylates and inhibits acetyl CoA carboxylase (ACC), the enzyme involved in the conversion of acetyl-CoA to malonyl-CoA (104). AMPK activation leads to the inhibitory phosphorylation of sterol regulatory element-binding protein 1c (SREBP1c). SREBP1cis responsible for enhancing the production of lipogenic enzymes, such as, acetyl-CoA carboxylase 1 (ACC1) and fatty acid (FA) synthase. Further, AMPK negatively regulates the de novo synthesis of cholesterol and triglycerides, and promotes β-oxidation of fatty acids. Additionally, AMPK negatively regulates the first committed step in triglyceride (TG) synthesis that is catalyzed by the enzyme, glycerol-3-phosphate acyltransferase (GPAT). AMPK- mediated inhibition of cholesterol synthesis is induced by phosphorylation of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (HMGCR). Both HMGCR and hormone-sensitive lipase (HSL) enzymes, which are involved in the synthesis of cholesterol and prevention of lipolysis, respectively, are inhibited as a consequence of AMPK activation (105). The role of AMPK in accelerating lipid catabolism is equally important. AMPK facilitates transportation of fatty acids into the mitochondria, where they are subjected to β-oxidation by the enzyme carnitine palmitoyltransferase-1 (CPT-1) (106, 107). Activation of AMPK accelerates the oxidation of fatty acids by inhibiting malonyl Co A and heightens the activity of CPT-1 enzyme (108).
AMPK exercises its inhibitory effects on protein synthesis mainly through the inhibition of mTOR (12). mTOR is a serine/threonine protein kinase that regulates two distinct down-stream catalytic protein complexes, namely, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (109). mTORC1 plays an important role in the growth of cancer cells and in cell division. The major down-stream targets of mTOR that are involved in the translation machinery include p70S6 kinase (p70S6K) and 4E-binding protein (4E-BP). It is well known that eIF4G is a binding site on eIF4E, which is essential for translation initiation (110). However, when 4E-BP is phosphorylated by mTOR, it can no longer bind eIF4E. Various stress factors inhibit mTORC1 activity, which results in the phosphorylation of 4E-BP and in turn, prevention of the translation initiation factor eIF4E (111). AMPK indirectly causes blockade of cap-dependent protein translation by inhibiting mTORC1 expression. This is achieved by phosphorylating tuberous sclerosis complex 2 (TSC2) and regulatory- associated protein of mTOR (RPTOR). Upon inhibition of mTORC1, 4EBP1 gets activated and a simultaneous decrease in the levels of p70S6K is observed. Consequently, the initiation of cap-dependent translation of proteins is halted. Besides, AMPK-mediated activation of eukaryotic elongation factor 2 kinase(eEF2K)also blocks protein synthesis (112). Furthermore, AMPK retards ribosomal RNA synthesis via the inhibitory phosphorylation of transcription initiation factor 1A (TIF-1A). The translational elongation is blocked by AMPK through the phosphorylation of eEF2K, which inhibits eEF2.
Mitochondrial biogenesis replaces fresh mitochondria once the damaged mitochondria are eliminated via autophagic degradation. The process of mitochondrial biogenesis is vital for energy production and cellular response under nutrient-limiting conditions. Interestingly, the substrates for mitochondrial metabolism are generated as part of the autophagic clearance program. Previous research suggests that AMPK regulates mitochondrial biogenesis by regulating peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α) (23). It is one of the cofactors which facilitate transcription of mitochondrial genes. AMPK influences PGC1α either by phosphorylating it directly or deacetylating it in a Sirtuin (silent mating type information regulation 2 homolog) 1(SIRT1)-dependent manner. Besides, AMPK-dependent increase in NAD+/NADH ratio or high levels of nicotinamide phosphoribosyl transferase(NAMPT) expression also lead to an augmented expression of PGC1α (113). AMPK-mediated mitochondrial biogenesis also accelerates the rates of ATP production. This is achieved by increasing the replication of mitochondrial DNA as well as the expression of many nuclear-encoded mitochondrial proteins via the SIRT 1/PGC-1α axis (114). Shen et al., have reported that the steroid hormone, ouabain, induces the simultaneous activation of AMPK and Src pathways in A549 and MCF7 cells and inhibits the mitochondrial OXPHOS in the cancer cells (115).
Figure 3 summarizes the effects of AMPK on the modulation of cancer cell metabolism, which collectively contributes to cell death, cell growth-arrest, inhibition of tumorigenesis and tumor progression.
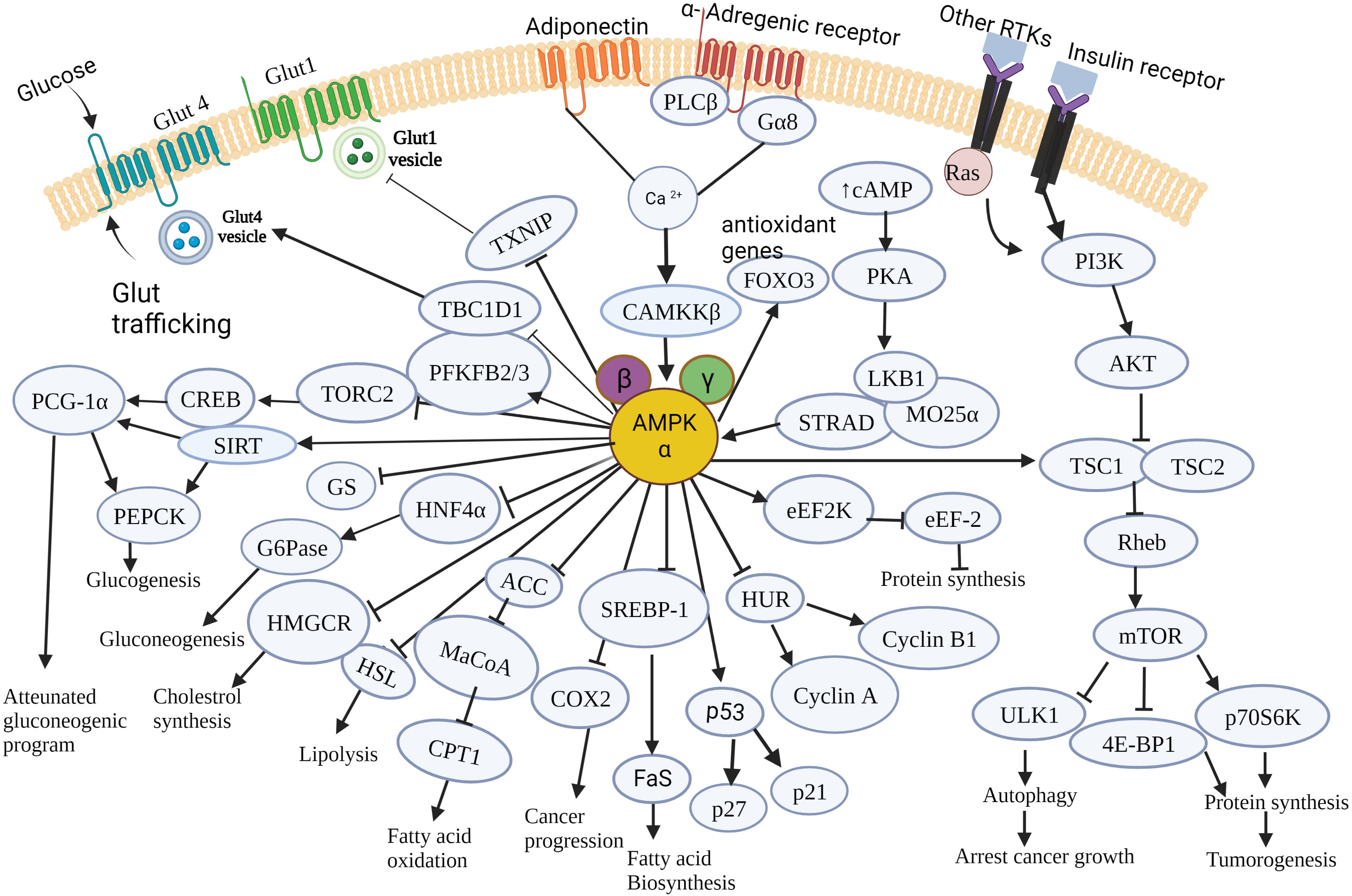
Figure 3 Schematic illustration of the major molecular targets of AMPK and the down-stream signaling events orchestrated by AMPK in cancer cell metabolism. AMPK controls various metabolic pathways that are critical to tumorigenesis and cancer progression. It regulates major cellular events governing cancer cell growth, survival and proliferation and pathways involved in glucose, lipid and protein metabolism. AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; Glut4, Glucose transporter type 4; Glut1, Glucose transporter type1; RTK, Receptor tyrosine kinase; PLC-β, Phospholipase C- β; G6Pase, Glucose -6-phosphatase; HMGCR, Hydroxymethyl glutaryl-CoA reductase; HSL, Hormone sensitive lipase; cAMP, Cyclic adenosine 3’,5’-monophosphate; PI3K, Phosphoinositide 3-kinase; Akt, A protein-serine/threonine kinase; CAMKKβ, Ca2+/Calmodulin-dependent protein kinase kinase 2; PGC1α, PPARG coactivator 1 α; HNF4α, hepatocyte nuclear factor 4 alpha; ACC, acetyl-CoA carboxylase; MaCoA, malonyl CoA; CPT1, Carnitine palmitoyl transferase 1; TSC1, Tuberous sclerosis complex 1; TSC2, tuberous sclerosis complex 2; Rheb – Ras homolog enriched in brain; SREBP1, Sterol regulatory element – binding protein 1; COX2, Cyclooxygenase-2; FaS, Fatty acid synthase; ULK1, unc-51-like kinase 1;p70S6K,70-kDa ribosomal protein S6 kinase; 4EBP1, eIF4E-binding protein 1;HuR,Human antigen R;LKB1,Liver kinase B1; eEF2K,eukaryotic elongation factor 2 kinase; eEF2K, eukaryotic elongation factor 2; PFKFB2/3,6-phosphofructo-2-kinase/fructose -2,6 –biphosphatase 2/3; TBC1D1- tre-2/USP6,BUB2,cdc16 domain family member 1;TXNIP, Thioredoxin interacting protein; CREB, cAMP response element - binding protein; SIRT, Sirtuin; PEPCK, Phosphoenol pyruvate carboxykinase; TORC2, target of rapamycin complex 2; STRAD, STE20-related adaptor protein; MO25α, Mouse protein 25 α; FOXO3, Forkhead box family transcription factor 3; PKA, cAMP-activated protein kinase; GS, Glycogen synthase; Ras, rat sarcoma viral oncogene.
Despite the accumulated observations supporting the tumor suppressing role of AMPK, there exist conflicting views on the pro-tumorigenic and pro-neoplastic features of AMPK. The tumor-suppressive function of AMPK might be overpowered by intracellular stress or oncogenic signals in malignant cancer cells (116).For instance, previous reports suggest that the activation of AMPK as a result of stress induced by glucose depletion or hypoxia enables tumor cells to become more resistant to metabolic stress (113, 117–119). Besides, AMPK-mediated autophagy confers a pro-survival advantage to cancer cells. Autophagy promotes cell growth and survival by providing metabolic substrates for biosynthesis, thus, fulfilling the metabolic demands of proliferating cancer cells. Besides, autophagy is also responsible for inducing chemoresistance in cancer cells (120, 121). There are also reports suggesting that although mTORC1inhibition prevents protein synthesis and cell proliferation, mTORC2 may activate the PI3K-Akt signaling pathway and promote tumor survival (106). A study by Laderoute et al, reports that AMPK promotes the growth of orthotopic tumors excised from breast cancer cells but does not affect the proliferation or survival of these cells in vitro. Experiments utilizing [13C] glucose tracers indicated that AMPK supports tumor glucose metabolism by combining the glycolytic and non-oxidative pentose phosphate cycles (122). Studies on astrocytoma murine models reveal that AMPK is an inducer of tumor cell proliferation. Furthermore, an elevated level of activated AMPK was found in human glioblastoma, in which inhibition of AMPK resulted in a significant decrease in the tumor cell growth rate (123). These results suggest that AMPK is not only involved in the regulation of ATP levels in cancer cells, but also in the regulation of cell replication. AMPK is required for increased mitochondrial biogenesis in response to glucose limitation. Studies by Chaube et al. suggest that under glucose-limiting conditions, cancer cells achieve metabolic homeostasis and adapt to metabolic stress via the activation of AMPK-p38-PGC-1α axis (124).Additional reports suggest that under conditions of nutrient starvation, AMPK may exhibit pro-tumorigenic effects and aid tumor survival, whereas in the presence of sufficient nutrients, AMPK exhibits tumor suppressing effects. The aforementioned data indicates that the pro- or anti-tumorigenic roles of AMPK are likely to be dependent on the levels of the nutrients in the TME (125). Notably, another independent study involving a pan-cancer analysis using multi-omics approach reveals that genetic as well as transcriptional aberrations in AMPK signaling elicits tissue-dependent pro- or anti-tumor impacts across major cancer types (126).
Some of the recent studies throw light on the anti-cancer effects of pharmacological inhibitors of AMPK. Dorsomorphin or Compound C, the only known AMPK-specific inhibitor augmented the anti-cancer effects of Aspirin against HER-2-positive breast cancer in an AMPK-independent manner, by regulating lipid metabolism mediated by c-myc (127). Another study identified BAY-3827 as a novel AMPK inhibitor. The compound also inhibited ribosomal 6 kinase (RSK) family proteins.BAY-3827 displayed excellent anti-proliferative effects against androgen-dependent prostate cancer cell lines by blocking HMGCR, fatty acid synthase (FASN) and PFKFB2, all of which are strongly up-regulated by androgen treatment (128). Thus, it would be safe to say that AMPK is a potential molecular target for cancer therapy employing chemoprevention and chemosensitization approaches, irrespective of its individual pro- or anti-tumorigenic/neoplastic effects.
The TME comprises of a heterogeneous collection of infiltrating and resident host immune cells, secreted factors, blood and lymph vessels, fibroblasts, endothelial cells, extracellular vesicles, and extracellular matrix. The stromal components together with the tumor cells are collectively referred to as the tumor microenvironment and its composition varies across different types of tumors (129). The TME facilitates cell survival and proliferation and promotes angiogenesis, local invasion, and metastasis of cancer (130–132). It includes cellular components of adaptive immunity, namely, T lymphocytes, dendritic cells (DC), and B lymphocytes, as well as those of innate immunity, including, macrophages, polymorphonuclear leukocytes, and natural killer (NK) cells, all of which exhibit context-dependent pro- or anti-tumorigenic functions (133). The metabolic changes occurring at the TME dictates the phenotypic and functional reprogramming of these immune cells. Immunotherapeutic drugs function by targeting specific components of the TME and shifting them from a pro-tumorigenic to an anti-tumorigenic phenotype (134). Owing to its diverse functions in regulating fundamental cellular activities, AMPK is instrumental in tumor metabolic transformation and controlling the metabolic plasticity of various immune cell types within the TME, which in turn potentiates an anti-tumor immune response (132).
T cell activation in the TME is governed by various metabolic pathways such as, aerobic glycolysis, amino acid metabolism, glutaminolysis, and de novo fatty acid synthesis. The LKB1-AMPK signaling pathway stimulates catabolic pathways and ATP generation which in turn, facilitate metabolic reprogramming in T cells. AMPK enhances glutaminolysis and mitochondrial OXPHOS which aids in T cell survival. AMPK activation promotes fatty acid oxidation (FAO). The AMPK-mTOR axis is responsible for the generation of memory T cells (Tmem)and adaptation of effector T cells (Teff) to nutritional stress (135). The inhibition of glycolysis and concomitant activation of FAO and oxidative phosphorylation directs regulatory T cells (Tregs) to undergo metabolic reprogramming which culminates in Tregs-mediated immunosuppression and tumor progression. On the contrary, cytotoxic CD8+ T cells play a crucial role in tumor immunosurveillance and elicit anti-tumor immune response by secreting interferon (IFN)-γ and granzyme B (GZB) (136).Rao et al., have demonstrated that AMPK regulates protein phosphatase activity, which controls survival and function of CD8+ T cells, thereby enhancing their role in tumor immunosurveillance (95). Besides, AMPK is indispensable for the sustained long-term proliferation of T cells and the survival of effector/memory T cell populations (137). In particular, AMPK promotes the accumulation of effector/memory T cells in competitive homeostatic proliferation settings (138). AMPK is found to be activated in CD8+ T cells during primary immune response. It also helps effector T cells (Teff) in their metabolic adaptation in response to reduced glucose availability. Teff cells actively engage in glutamine-dependent oxidative phosphorylation (OXPHOS) to maintain ATP concentrations and cell viability under low glucose conditions. Further, AMPKα1-deficient Teff cells display reduced mitochondrial bioenergetics and cellular ATP in response to glucose limitation (139). Notably, cytotoxic T cells (CD8+ T cells) detect abnormal tumor antigens expressed on cancer cells and target them for destruction. AMPKα-1-mediated inhibition of mTORC1 activity in cytotoxic T lymphocytes (CTLs) is required for CD8+ T-cell memory (140). Previous studies have revealed that inactivation of both AMPKα1 and α2 coupled with Kirsten rat sarcoma virus (KRAS) activation promotes tumorigenesis and decreases the infiltration of CD8+/CD4+ T cells (141). Braverman et al., have reported that increasing AMPK activity orchestrates oxidative metabolism, proliferation, and in vitro recovery of human CD4+ T cells. They also suggest AMPK as a potential candidate for improving the yield of more functional T cells for CAR-T cell therapy (142). The metabolic fitness of T cells is pivotal for antitumor immunity. A recent report suggests that AMPK is essential for the sustained long-term proliferation of T cells and the survival of effector/memory T cell sub-populations (143). However, under conditions of restricted nutritional supply, T cells acquire an exhausted phenotype inside the immune-suppressive tumor milieu. T cell exhaustion culminates in the loss of effector activities and changes in T cell signaling and is characterized by reduced rates of glycolysis and OXPHOS and the onset of mitochondrial dysfunction (135).
Tumor-associated macrophages (TAMs) are present in abundance across various types of malignant tumors and promote tumor angiogenesis, extravasation and metastasis (144).Macrophages are responsible for driving anti-tumor immunity via phagocytosis and antigen presentation. Macrophages exist in two distinct phenotypes namely, the ‘classically’ activated, tumoricidal phenotype, M1φ, and the ‘alternatively’ activated pro-tumorigenic phenotype, M2φ (145).Some documented results indicate that AMPK inhibition leads to an up-surge in LPS-induced macrophage inflammatory function (141). Previous reports also state that AMPK prevents the polarization of monocyte-derived macrophages towards the M2φ subtype (146).Chiang et al., reported that metformin-mediated AMPK activation suppresses the skewing of macrophage towards M2φ subtype in breast cancer cells (147). The metabolic control of macrophage polarization relies partially upon glycolysis. A shift towards increased glycolytic rates leads to the activation of M1φ and is governed by the activation of mTOR through the Akt-HIF-1α pathway. Conversely, M2φ selectively utilize FAO, instead of aerobic glycolysis, to meet the energy requirements of OXPHOS. While HIF-1α and NF-κB favour the M1φ, PGC1β, peroxisome proliferator-activated receptors (PPAR) and STAT6 skew the balance towards M2φ. AMPK influences M1/M2φ polar mitochondrial biogenesis of macrophages by deacetylating proteins such as, SIRT1 with NAD+, and suppressing HIF-1α and NF-κB (1).
Previous reports also suggest that AMPK is involved in the molecular crosstalk between macrophages and cytokines. For instance, studies on the effect of macrophage stimulation using anti-inflammatory cytokines causes rapid phosphorylation of AMPK as opposed to the inactivation of AMPK upon the stimulation using pro-inflammatory LPS stimulus. AMPK directs signaling pathways in macrophages in a manner that suppresses pro-inflammatory responses and promotes macrophage polarization to an anti-inflammatory functional phenotype (146). AMPKα silencing elevated the mRNA expression levels of LPS-induced TNF-α, IL-6, and cyclooxygenase-2. Similarly, transfection of dominant negative AMPKα1 gene increased TNF-α and IL-6 expression levels and down-regulated IL-10 expression in macrophages, upon LPS stimulation (147).Another recent study revealed that AMPK inactivation potentiates the development of LPS-induced inflammatory injury (148). In the TME, AMPK has been reported to suppress the secretion of various pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, interleukins-1/2/8, MCP-1, IFN-γ and chemokines such as RANTES, CCL 1/2/5/10/11 (148).Previous studies indicate that stimulation of wild type macrophages using anti-inflammatory cytokine,IL-10 results in rapid activation of AMPK, phosphoinositide 3-kinase(PI3K) and mTORC1 (149).AMPK is known to inhibit the functions of pro-inflammatory molecules such as IL-1 β. Previous reports suggest that AMPK activation inhibited IL-1-stimulated CXCL10 secretion, via the down-regulation of the MKK4/JNK and IKK/IκBα/NF-κB signaling axis (150).
Myeloid-derived suppressor cells (MDSC) are an immunosuppressive class of immune cells that are pathologically activated in various tumor types. MDSCs promote tumor progression and enhance tumor cell survival, angiogenesis, invasion, metastasis and production of immunosuppressive cytokines such as IL-10 and TGF-β. Recent studies indicate that splenic MDSCs impede T cell response in a ROS-dependent manner, whereas, tumor-infiltrating MDSCs inhibit anti-CD3/28-stimulated response through nitric oxide (NO) production and secretion of Arginase1 (Arg1). MDSCs mediate the addition of nitrate groups to chemokines thereby, blocking CD8+ T cells from entering into the tumor site (151). AMPK block the expansion and activation of MDSCs. It curtails the function of MDSCs via inhibition of JAK-STAT, NF-κB, C/EBPβ, CHOP, and HIF-1α pathways that are crucial for the development and migration of MDSCs (152–154).
The influence of AMPK on various immune checkpoints is yet another remarkable event in the alteration of the TME (116). Immune checkpoint molecules are co-stimulatory cell surface receptors that are expressed on the surface of several immune cells, which bind with their corresponding ligand molecules and in turn, prevent an immune attack against self-antigens. However, cancer cells use this feature of immune checkpoints to evade immune attacks. This confers a survival advantage to the cancer cells. Immunotherapy drugs aim at inhibiting these immune checkpoints rendering cancer cells susceptible to immune attacks (155).Blocking of the programmed cell death 1 (PD-1) receptor is currently being used as a first-line treatment option against lung cancer. A recent report indicates that GSK3β-mediated inhibition of glycogen synthase down-regulatesPD-1 expression levels on CD8+ T cells in B16F10, murine melanoma cells (136).Several reports also suggest the role of AMPK as an immune checkpoint inhibitor. AMPK activation causes phosphorylation of PD-L1 on Ser283 and disrupts its interaction with chemokine like factor (CKLF)-like MARVEL transmembrane domain containing 4 (CMTM4), which leads to the degradation of PD-1 ligand (PD-L1). AMPK also potentiates ER-associated degradation of PD-L1 (156, 157). Dai et al., have reported that, in syngeneic mouse tumor models, AMPK agonists enhance the efficacy of anti-CTLA-4 immunotherapy and improve the overall survival rate (156). Besides, reports also suggest the potential use of AMPK activators in combination with anti-VEGF/PD-1 agents as a dual-targeted therapy against ovarian cancer (116). A recent report by Pokhrel et al. states that AMPK drives anti-tumor immunity by blocking PD-1 expression via the HMGCR/p38 MAPK/GSK3β signaling pathway (136). They also report on the synergic antitumor effect of AMPK activators with anti-PD-1 antibodies, anti-CTLA-4 antibodies, or HMGCR inhibitors in murine tumor models (158). Another study has documented the AMPK-mediated blockade of PD-1 through a reduction of tumor hypoxia (159). Taken together, these findings highlight the central role of AMPK in the inhibition of major immune checkpoints. The current knowledgebase can be further expanded by evaluating the therapeutic efficacy of immunotherapy drugs in combination with AMPK inhibitors against multiple cancer types. Figure 4 represents AMPK-mediated modulation of various components of the TME and the signalling pathways associated with it.
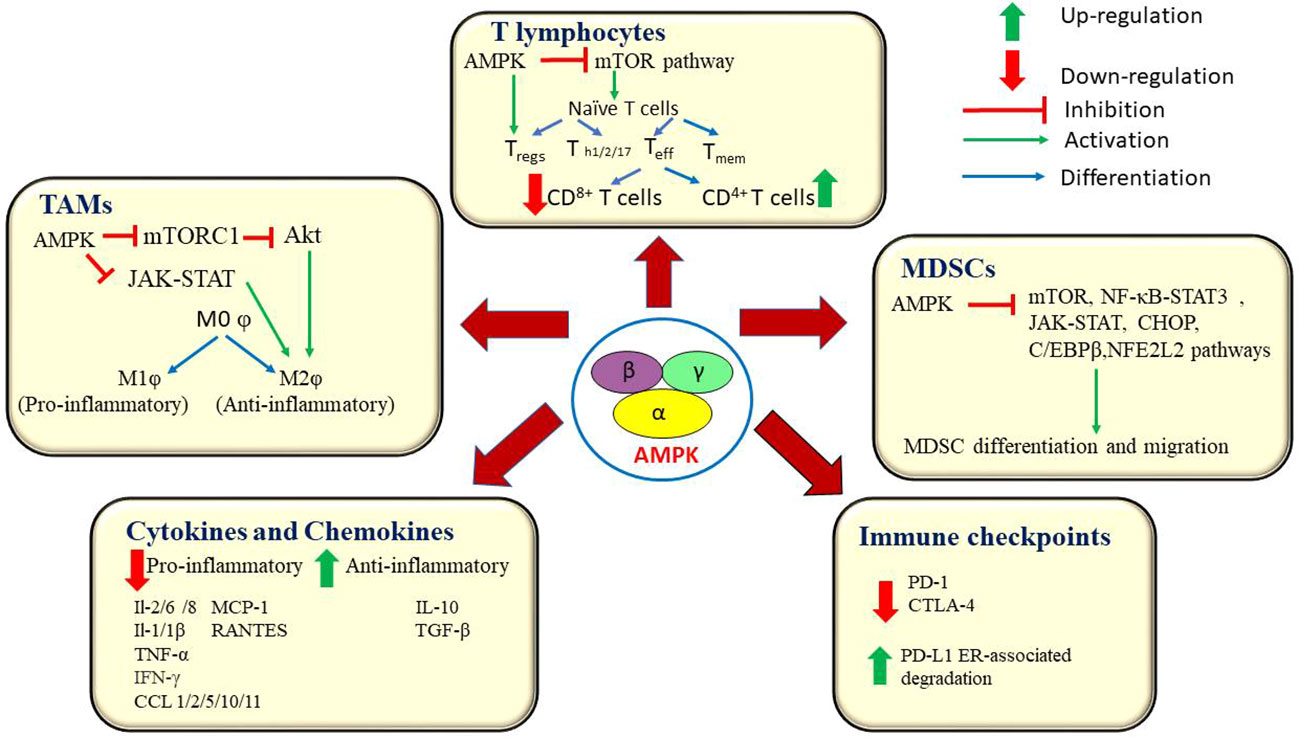
Figure 4 AMPK orchestrates an anti-tumor immune response by interacting with key players at the tumor microenvironment. AMPK influences the functions of T lymphocytes, macrophages and myeloid-derived suppressor cells. AMPK down-regulates the production of pro-inflammatory cytokines and chemokines and blocks the immune checkpoint molecules, PD-1 and CTLA-4. AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; Akt, A protein-serine/threonine kinase; mTORC1, mammalian target of rapamycin complex 1; Teff, Effector T cells; Tmem, Memory T cells; Treg, Regulatory T cells; Th1/2/17, Helper T cells1/2/17; PD-1, programmed cell death 1; IL-2/6/8/1/1β, Interleukin 2/6/8/1/1β; TNF α, Tumor necrosis factor α; IFN α, interferon gamma; PDL1, Programmed cell death ligand 1; CTLA-4, Cytotoxic T-lymphocyte – associated antigen 4; MCP-1, Monocyte chemoattractant protein -1; RANTES, Regulated upon activation, normal T cell Expressed, and Secreted; TGF-β, Transforming growth factor; MDSCs, Myeloid – derived suppressor cells; NF-κB, Nuclear factor kappa B; JAK/STAT, Janus kinase/signal transducers and activators of transcription; C/EBPR, CCAAT/enhancer – binding protein beta; CCL1/2/5/10/11, Chemokine (C-C motif) ligand 1/2/5/10/11; CHOP, C/EBP homologous protein; NFE2L2, Nuclear factor erythroid-derived 2-like 2; MOφ, non – activated macrophage; M1φ, pro-inflammatory macrophage; M2φ, anti-inflammatory macrophage.
Even though the aforementioned chemical activators of AMPK show promising anti-cancer effects in the pre-clinical studies, there are drawbacks which prohibit their direct translation into the clinics. For instance, the synthetic biguanides, metformin and phenformin, were proposed to be used for the clinical management of type 2 diabetes in the 1950s. However, the clinical use of phenformin was withdrawn in the 1970s owing to a rare, but fatal condition induced by the drug known as ‘lactic acidosis’ (6). Mitochondrial impairment and ATP depletion leads to an acceleration of the glycolytic flux. Subsequently, there is an increased glucose uptake and excessive lactate generation. The excess lactate escapes into the circulation instead of undergoing further oxidation, giving rise to lactic acidosis (160). Although, metformin is safer than phenformin, it does induce lactic acidosis when used for long term. Incidences of such drug-induced toxicity and adverse side effects, and chemoresistance caused by conventional chemotherapeutic drugs limit the therapeutic efficacy of chemotherapy. The concept of chemoprevention and chemosensitization through dietary intervention has evolved to abate these detrimental effects. Over the past few decades, several studies have enumerated the immense therapeutic efficacy and pharmacological safety of various phytochemicals, highlighting them as suitable anti-cancer agents. Notably, many of these phytochemicals exert anti-cancer effects via the activation of AMPK, which results in increased apoptosis and inhibition of cell proliferation (161). Some of the phytochemicals that are well-known activators of AMPK include resveratrol, quercetin, berberine, ginsenoside, curcumin, epigallocatechin gallate(EGCG), theaflavin, hispidulin and galegine (6, 162). Recently, we have reported that Uttroside B, a novel saponin identified in our lab exhibits excellent anti-HCC effect via the up-regulation of AMPK and the concomitant down-regulation of mTOR (163). Taken together, substitution of phytochemical activators instead of chemical activators of AMPK would be a promising and pharmacologically safer strategy in treating different types of cancers. Table 2 enlists some of the major phytochemicals which generate anti-cancer effects via AMPK activation.
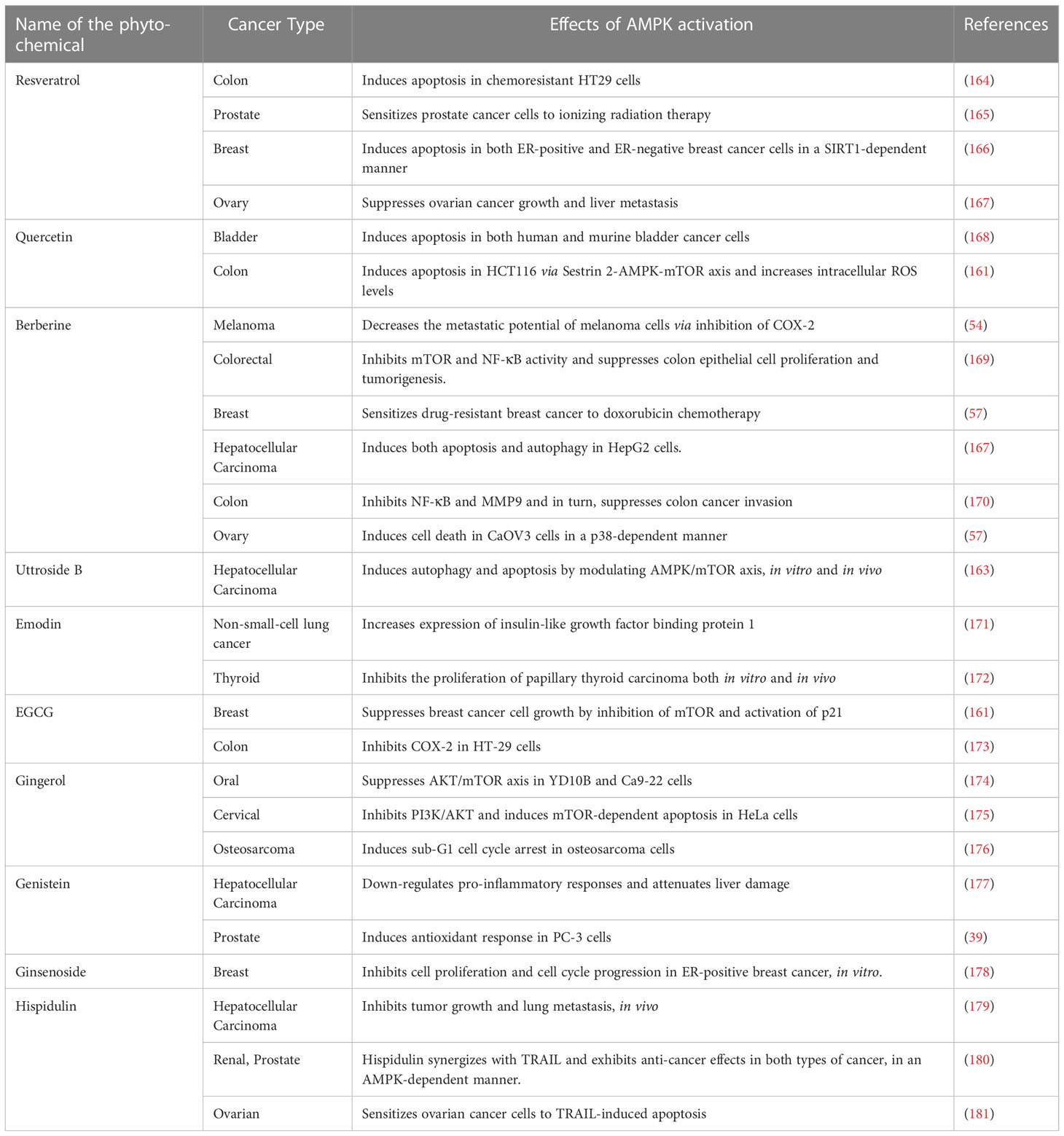
Table 2 The implications of AMPK activation in various cancer types mediated by some of the major plant-derived anti-cancer agents.
Cancer cells being aberrant variants of normal cells, mimic normal cells to gain a survival advantage and escape cell death machinery and immune surveillance. This makes the eradication of cancer cells from the body a very challenging prospect. The unique metabolic features in the TME and cancer cells foster each other, resulting in sustained cell proliferation, tumor progression, and metastasis of cancer. Therefore, identifying molecules that act as focal points connecting cancer metabolism and anti-tumor immune response would be a promising strategy to combat cancer. In this scenario, AMPK acts as the nexus between cellular energy homeostasis, tumor bioenergetics, and anti-tumor immunity. Thus, AMPK would be a suitable candidate for targeted cancer therapy.
There are numerous pre-clinical evidences stating the dual nature of AMPK in cancer. However, the controversy over the positive and negative regulatory effects of AMPK in the context of cancer cannot be resolved without substantial clinical evidence (126). The research pertaining to the clinical outcomes of differential regulation of AMPK signaling in cancer is still in a nascent stage. Thus, it is quintessential to explore the causes and effects of the contradicting roles of AMPK in cancer progression using human tissue samples and PDX models of different types of cancer before drawing any conclusions. Future research aimed at elucidating the role of tumor type, tissue site and nutrient status of various cancer types in deciding the pro- or anti-tumorigenic functions of AMPK and the consequent clinical implication in patients is warranted.
Previous literature suggests that AMPK influences the differentiation and function of T cells and macrophages. A recent study revealed that AMPKα1 promotes mitochondrial homeostasis and persistence of B Cell memory although it hampers primary antibody responses (182). However, the literature on the role of AMPK in regulating the B cell metabolism and functions of B lymphocytes and humoral immunity is limited (1). Hence, a mechanistic evaluation of AMPK-mediated regulation of humoral immunity in various cancer types will facilitate in expanding the current knowledge.
AMPK-mediated disruption of cancer cell metabolism or alteration of the TME components to inhibit cancer progression would provide immense therapeutic benefits (183). These results would aid in the formulation of novel therapeutic regimen involving the activation or inactivation of AMPK, in a context-dependent manner, for mitigating cancer progression. Furthermore, substituting chemical activators/inhibitors of AMPK with their phytochemical functional equivalents may help circumvent the side-effects of chemo drugs without compromising their anti-cancer efficacy.
RA conceptualized the work; CK, TR, SS, KK, and NA collected, analyzed, and interpreted the relevant literature and wrote the manuscript; CK prepared the tables; CK and SS prepared the figures; NA, KK, NI, and RA critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
We thank the Department of Biotechnology, Government of India for the financial support.
The Figure 3 included in this article has been created with BioRender.com. We thank Dr. Ramakrishnan Muthuswamy, Nanjing Forestry University, China, for the help rendered in creating this image. We also thank Ms. Aiswarya US and Ms. Jannet S for their technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kim J. Regulation of immune cell functions by metabolic reprogramming. J Immunol Res (2018) 2018:1–12. doi: 10.1155/2018/8605471
2. Shi R, Tang YQ, Miao H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm (2020) 1(1):47–68. doi: 10.1002/mco2.6
3. Kemp BE, Oakhill JS, Scott JW. AMPK structure and regulation from three angles. Structure (2007) 15(10):1161–3. doi: 10.1016/j.str.2007.09.006
4. Hardie DG, Ross FA, Hawley SA. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13(4):251–62. doi: 10.1038/nrm3311
5. Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. Structure of mammalian AMPK and its regulation by ADP. Nature (2011) 472(7342):230–3. doi: 10.1038/nature09932
6. Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab (2013) 18(4):556–66. doi: 10.1016/j.cmet.2013.08.019
7. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol (2011) 13(9):1016–23. doi: 10.1038/ncb2329
8. Lin S-C, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab (2018) 27(2):299–313. doi: 10.1016/j.cmet.2017.10.009
9. Li X, Wang L, Zhou XE, Ke J, De Waal PW, Gu X, et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res (2015) 25(1):50–66. doi: 10.1038/cr.2014.150
10. Hardie DG. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell Biol (2007) 8(10):774–85. doi: 10.1038/nrm2249
11. Yan Y, Zhou XE, Xu HE, Melcher K. Structure and physiological regulation of AMPK. Int J Mol Sci (2018) 19(11):3534. doi: 10.3390/ijms19113534
12. Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol (2019) 9(7):190099. doi: 10.1098/rsob.190099
13. Hardie DG. AMP-activated protein kinase–an energy sensor that regulates all aspects of cell function. Genes Dev (2011) 25(18):1895–908. doi: 10.1101/gad.17420111
14. Yan Y, Mukherjee S, Harikumar KG, Strutzenberg TS, Zhou XE, Suino-Powell K, et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science (2021) 373(6553):413–9. doi: 10.1126/science.abe7565
15. Hardie DG, Schaffer BE, Brunet A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol (2016) 26(3):190–201. doi: 10.1016/j.tcb.2015.10.013
16. Scott JW, Oakhill JS, van Denderen BJ. AMPK/SNF1 structure: a menage a trois of energy-sensing. Front Bioscience-Landmark (2009) 14(2):596–610. doi: 10.2741/3266
17. Carling D, Aguan K, Woods A, Verhoeven AJ, Beri RK, Brennan CH, et al. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J Biol Chem (1994) 269(15):11442–8. doi: 10.1016/S0021-9258(19)78143-5
18. Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, et al. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem (1994) 269(4):2361–4. doi: 10.1016/S0021-9258(17)41951-X
19. Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem (1994) 269(30):19509–15. doi: 10.1016/S0021-9258(17)32198-1
20. Hurley RL, Barré LK, Wood SD, Anderson KA, Kemp BE, Means AR, et al. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem (2006) 281(48):36662–72. doi: 10.1074/jbc.M606676200
21. Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Phosphorylation by akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem J (2014) 459(2):275–87. doi: 10.1042/BJ20131344
22. Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, et al. Inhibition of AMPK catabolic action by GSK3. Mol Cell (2013) 50(3):407–19. doi: 10.1016/j.molcel.2013.03.022
23. Jeon S-M. Regulation and function of AMPK in physiology and diseases. Exp Mol Med (2016) 48(7):e245–5. doi: 10.1038/emm.2016.81
24. Viollet B, Andreelli F, Jørgensen SB, Perrin C, Flamez D, Mu J, et al. Physiological role of AMP-activated protein kinase (AMPK): Insights from knockout mouse models. Biochem Soc Trans (2003) 31(1):216–9. doi: 10.1042/bst0310216
25. Tremblay F, Brûlé S, Hee Um S, Li Y, Masuda K, Roden M, et al. Identification of IRS-1 ser-1101 as a target of S6K1 in nutrient-and obesity-induced insulin resistance. Proc Natl Acad Sci (2007) 104(35):14056–61. doi: 10.1073/pnas.0706517104
26. Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine-and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes (2010) 59(10):2426–34. doi: 10.2337/db09-1870
27. Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. Nutrient excess in AMPK downregulation and insulin resistance. J endocrinology Diabetes Obes (2013) 1(1):1008.
28. Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J (1999) 13(14):2051–60. doi: 10.1096/fasebj.13.14.2051
29. Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol-Endocrinol And Metab (2000) 279(5):E1039–44. doi: 10.1152/ajpendo.2000.279.5.E1039
30. Jäger S, Handschin C, St.-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci (2007) 104(29):12017–22. doi: 10.1073/pnas.0705070104
31. Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, et al. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab (2006) 4(6):465–74. doi: 10.1016/j.cmet.2006.11.005
32. Cai Z, Li B, Li K, Zhao B. Down-regulation of amyloid-β through AMPK activation by inhibitors of GSK-3β in SH-SY5Y and SH-SY5Y-AβPP695 cells. J Alzheimer's Dis (2012) 29(1):89–98. doi: 10.3233/JAD-2012-111649
33. Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of aβ oligomers through tau phosphorylation. Neuron (2013) 78(1):94–108. doi: 10.1016/j.neuron.2013.02.003
34. Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of alzheimer's disease. J Neurosci (2007) 27(4):824–31. doi: 10.1523/JNEUROSCI.4345-06.2007
35. Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J Biol Chem (2010) 285(12):9100–13. doi: 10.1074/jbc.M109.060061
36. Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol (2000) 10(20):1247–55. doi: 10.1016/S0960-9822(00)00742-9
37. Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest (2004) 114(4):495–503. doi: 10.1172/JCI19297
38. Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation (2009) 119(19):2568–77. doi: 10.1161/CIRCULATIONAHA.108.798561
39. Park CE, Yun H, Lee EB, Min BI, Bae H, Choe W, et al. The antioxidant effects of genistein are associated with AMP-activated protein kinase activation and PTEN induction in prostate cancer cells. J medicinal Food (2010) 13(4):815–20. doi: 10.1089/jmf.2009.1359
40. Naik PP, Mukhopadhyay S, Praharaj PP, Bhol CS, Panigrahi DP, Mahapatra KK, et al. Secretory clusterin promotes oral cancer cell survival via inhibiting apoptosis by activation of autophagy in AMPK/mTOR/ULK1 dependent pathway. Life Sci (2021) 264:118722. doi: 10.1016/j.lfs.2020.118722
41. Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy (2015) 11(8):1341–57. doi: 10.1080/15548627.2015.1061849
42. Cai X, Hu X, Cai B, Wang Q, Li Y, Tan X, et al. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle G1/G0 phase arrest and p21CIP and p27KIP expression and downregulation of cyclin D1 in vitro and in vivo. Oncol Rep (2013) 30(5):2449–57. doi: 10.3892/or.2013.2718
43. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun (2012) 3(1):1–11. doi: 10.1038/ncomms1859
44. Lee JD, Choi MA, Ro SW, Yang WI, Cho AE, Ju HL, et al. Synergic chemoprevention with dietary carbohydrate restriction and supplementation of AMPK-activating phytochemicals: the role of SIRT1. Eur J Cancer Prev (2016) 25(1):54. doi: 10.1097/CEJ.0000000000000141
45. Tong X, Bridgeman BB, Smith KA, Avram MJ, Pelling JC. AMPK-mTOR axis as key target for chemoprevention of UV-induced skin cancer by the bioflavonoid apigenin. Cancer Res (2012) 72(8_Supplement):1587–7. doi: 10.1158/1538-7445.AM2012-1587
46. Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc Natl Acad Sci (2014) 111(8):3020–5. doi: 10.1073/pnas.1318440111
47. Knobloch TJ, Ryan NM, Bruschweiler-Li L, Wang C, Bernier MC, Somogyi A, et al. Metabolic regulation of glycolysis and AMP activated protein kinase pathways during black raspberry-mediated oral cancer chemoprevention. Metabolites (2019) 9(7):140. doi: 10.3390/metabo9070140
48. Wang Y, Ma W, Zheng W. Deguelin, a novel anti-tumorigenic agent targeting apoptosis, cell cycle arrest and anti-angiogenesis for cancer chemoprevention. Mol Clin Oncol (2013) 1(2):215–9. doi: 10.3892/mco.2012.36
49. Shrotriya S, Tyagi A, Deep G, Orlicky DJ, Wisell J, Wang XJ, et al. Grape seed extract and resveratrol prevent 4-nitroquinoline 1-oxide induced oral tumorigenesis in mice by modulating AMPK activation and associated biological responses. Mol carcinogenesis (2015) 54(4):291–300. doi: 10.1002/mc.22099
50. Kim HJ, Kim SK, Kim BS, Lee SH, Park YS, Park BK, et al. Apoptotic effect of quercetin on HT-29 colon cancer cells via the AMPK signaling pathway. J Agric Food Chem (2010) 58(15):8643–50. doi: 10.1021/jf101510z
51. Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med (2014) 6(4):519–38. doi: 10.1002/emmm.201302734
52. Cheng J, Huang T, Li Y, Guo Y, Zhu Y, Wang Q, et al. AMP-activated protein kinase suppresses the in vitro and in vivo proliferation of hepatocellular carcinoma. PloS One (2014) 9(4):e93256. doi: 10.1371/journal.pone.0093256
53. Zou YF, Xie CW, Yang SX, Xiong JP. AMPK activators suppress breast cancer cell growth by inhibiting DVL3-facilitated wnt/β-catenin signaling pathway activity. Mol Med Rep (2017) 15(2):899–907. doi: 10.3892/mmr.2016.6094
54. Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS, Lim JS. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol (2012) 83(3):385–94. doi: 10.1016/j.bcp.2011.11.008
55. Kang S, Kim JE, Song NR, Jung SK, Lee MH, Park JS, et al. The ginsenoside 20-O-β-D-glucopyranosyl-20 (S)-protopanaxadiol induces autophagy and apoptosis in human melanoma via AMPK/JNK phosphorylation. PloS One (2014) 9(8):e104305. doi: 10.1371/journal.pone.0104305
56. Vara-Ciruelos D, Dandapani M, Russell FM, Grzes KM, Atrih A, Foretz M, et al. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep (2019) 27(3):690–698.e4. doi: 10.1016/j.celrep.2019.03.067
57. Pan Y, Zhang F, Zhao Y, Shao D, Zheng X, Chen Y, et al. Berberine enhances chemosensitivity and induces apoptosis through dose-orchestrated AMPK signaling in breast cancer. J Cancer (2017) 8(9):1679. doi: 10.7150/jca.19106
58. Dong H, Huang J, Zheng K, Tan D, Chang Q, Gong G, et al. Metformin enhances the chemosensitivity of hepatocarcinoma cells to cisplatin through AMPK pathway. Oncol Lett (2017) 14(6):7807–12. doi: 10.3892/ol.2017.7198
59. Bi Y, Li H, Yi D, Sun Y, Bai Y, Zhong S, et al. Cordycepin augments the chemosensitivity of human glioma cells to temozolomide by activating AMPK and inhibiting the AKT signaling pathway. Mol pharmaceutics (2018) 15(11):4912–25. doi: 10.1021/acs.molpharmaceut.8b00551
60. Gao Y, Chen DL, Zhou M, Zheng ZS, He MF, Huang S, et al. Cordycepin enhances the chemosensitivity of esophageal cancer cells to cisplatin by inducing the activation of AMPK and suppressing the AKT signaling pathway. Cell Death Dis (2020) 11(10):1–12. doi: 10.1038/s41419-020-03079-4
61. Luo F, Zhao J, Liu S, Xue Y, Tang D, Yang J, et al. Ursolic acid augments the chemosensitivity of drug-resistant breast cancer cells to doxorubicin by AMPK-mediated mitochondrial dysfunction. Biochem Pharmacol (2022) 205:115278. doi: 10.1016/j.bcp.2022.115278
62. Zhu P, Wang L, Xu P, Tan Q, Wang Y, Feng G, et al. GANT61 elevates chemosensitivity to cisplatin through regulating the hedgehog, AMPK and cAMP pathways in ovarian cancer. Future Medicinal Chem (2022) 14(7):479–500. doi: 10.4155/fmc-2021-0310
63. Ji C, Yang B, Yang YL, He SH, Miao DS, He L, et al. Exogenous cell-permeable C6 ceramide sensitizes multiple cancer cell lines to doxorubicin-induced apoptosis by promoting AMPK activation and mTORC1 inhibition. Oncogene (2010) 29(50):6557–68. doi: 10.1038/onc.2010.379
64. Park JB, Lee JS, Lee MS, Cha EY, Kim S, Sul JY. Corosolic acid reduces 5−FU chemoresistance in human gastric cancer cells by activating AMPK. Mol Med Rep (2018) 18(3):2880–8. doi: 10.3892/mmr.2018.9244
65. Seo Y, Kim J, Park SJ, Park JJ, Cheon JH, Kim WH, et al. Metformin suppresses cancer stem cells through AMPK activation and inhibition of protein prenylation of the mevalonate pathway in colorectal cancer. Cancers (2020) 12(9):2554. doi: 10.3390/cancers12092554
66. Zheng L, Yang W, Wu F, Wang C, Yu L, Tang L, et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular CarcinomaAnticancer effect of therapeutic Metformin/AMPK activation on HCC. Clin Cancer Res (2013) 19(19):5372–80. doi: 10.1158/1078-0432.CCR-13-0203
67. Tang G, Guo J, Zhu Y, Huang Z, Liu T, Cai J, et al. Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. Int J Oncol (2018) 52(6):1899–911. doi: 10.3892/ijo.2018.4343
68. Zhou X, Kuang Y, Liang S, Wang L. Metformin inhibits cell proliferation in SKM-1 cells via AMPK-mediated cell cycle arrest. J Pharmacol Sci (2019) 141(4):146–52. doi: 10.1016/j.jphs.2019.10.003
69. Duan W, Chen K, Jiang Z, Chen X, Sun L, Li J, et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett (2017) 385:225–33. doi: 10.1016/j.canlet.2016.10.019
70. Orecchioni S, Reggiani F, Talarico G, Mancuso P, Calleri A, Gregato G, et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer (2015) 136(6):E534–44. doi: 10.1002/ijc.29193
71. Huang Y, Zhou S, He C, Deng J, Tao T, Su Q, et al. Phenformin alone or combined with gefitinib inhibits bladder cancer via AMPK and EGFR pathways. Cancer Commun (2018) 38(1):1–14. doi: 10.1186/s40880-018-0319-7
72. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W, et al. ROS signaling under metabolic stress: cross-talk between AMPK and AKT pathway. Mol Cancer (2017) 16(1):1–12. doi: 10.1186/s12943-017-0648-1
73. Wang J, Xia S.A., Zhu Z. Synergistic effect of phenformin in non-small cell lung cancer (NSCLC) ionizing radiation treatment. Cell Biochem biophysics (2015) 71(2):513–8. doi: 10.1007/s12013-014-0283-z
74. Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci (2009) 106(31):12932–7. doi: 10.1073/pnas.0906606106
75. Theodoropoulou S, Kolovou PE, Morizane Y, Kayama M, Nicolaou F, Miller JW, et al. Retinoblastoma cells are inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. FASEB J (2010) 24(8):2620–30. doi: 10.1096/fj.09-152546
76. Awwad O, Coperchini F, Pignatti P, Denegri M, Massara S, Croce L, et al. The AMPK-activator AICAR in thyroid cancer: Effects on CXCL8 secretion and on CXCL8-induced neoplastic cell migration. J Endocrinol Invest (2018) 41(11):1275–82. doi: 10.1007/s40618-018-0862-8
77. Wu Y, Qi Y, Liu H, Wang X, Zhu H, Wang Z. AMPK activator AICAR promotes 5-FU-induced apoptosis in gastric cancer cells. Mol Cell Biochem (2016) 411(1):299–305. doi: 10.1007/s11010-015-2592-y
78. Sui X, Xu Y, Yang J, Fang Y, Lou H, Han W, et al. Use of metformin alone is not associated with survival outcomes of colorectal cancer cell but AMPK activator AICAR sensitizes anticancer effect of 5-fluorouracil through AMPK activation. PloS One (2014) 9(5):e97781. doi: 10.1371/journal.pone.0097781
79. Rae C, Mairs RJ. AMPK activation by AICAR sensitizes prostate cancer cells to radiotherapy. Oncotarget (2019) 10(7):749. doi: 10.18632/oncotarget.26598
80. Sauer H, Engel S, Milosevic N, Sharifpanah F, Wartenberg M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-jun n-terminal kinase. Int J Oncol (2012) 40(2):501–8. doi: 10.3892/ijo.2011.1230
81. Su CC, Hsieh KL, Liu PL, Yeh HC, Huang SP, Fang SH, et al. AICAR induces apoptosis and inhibits migration and invasion in prostate cancer cells through an AMPK/mTOR-dependent pathway. Int J Mol Sci (2019) 20(7):1647. doi: 10.3390/ijms20071647
82. Morishita M, Kawamoto T, Hara H, Onishi Y, Ueha T, Minoda M, et al. AICAR induces mitochondrial apoptosis in human osteosarcoma cells through an AMPK-dependent pathway. Int J Oncol (2017) 50(1):23–30. doi: 10.3892/ijo.2016.3775
83. Cheng XL, Zhou TY, Li B, Li MY, Li L, Li ZQ, et al. Methotrexate and 5-aminoimidazole-4-carboxamide riboside exert synergistic anticancer action against human breast cancer and hepatocellular carcinoma. Acta Pharmacologica Sin (2013) 34(7):951–9. doi: 10.1038/aps.2013.16
84. Fodor T, Szántó M, Abdul-Rahman O, Nagy L, Dér Á, Kiss B, et al. Combined treatment of MCF-7 cells with AICAR and methotrexate, arrests cell cycle and reverses warburg metabolism through AMP-activated protein kinase (AMPK) and FOXO1. PloS One (2016) 11(2):e0150232. doi: 10.1371/journal.pone.0150232
85. O’Brien AJ, Villani LA, Broadfield LA, Houde VP, Galic S, Blandino G, et al. Salicylate activates AMPK and synergizes with metformin to reduce the survival of prostate and lung cancer cells ex vivo through inhibition of de novo lipogenesis. Biochem J (2015) 469(2):177–87. doi: 10.1042/BJ20150122
86. Tsakiridis EE, Broadfield L, Marcinko K, Biziotis OD, Ali A, Mekhaeil B, et al. Combined metformin-salicylate treatment provides improved anti-tumor activity and enhanced radiotherapy response in prostate cancer; drug synergy at clinically relevant doses. Trans Oncol (2021) 14(11):101209. doi: 10.1016/j.tranon.2021.101209
87. Karalis TT, Chatzopoulos A, Kondyli A, Aletras AJ, Karamanos NK, Heldin P, et al. Salicylate suppresses the oncogenic hyaluronan network in metastatic breast cancer cells. Matrix Biol plus (2020) 6:100031. doi: 10.1016/j.mbplus.2020.100031
88. Shoda K, Tsuji S, Nakamura S, Egashira Y, Enomoto Y, Nakayama N, et al. Canagliflozin inhibits glioblastoma growth and proliferation by activating AMPK. Cell Mol Neurobiol (2022), 1–14. doi: 10.1007/s10571-022-01221-8
89. Kaji K, Nishimura N, Seki K, Sato S, Saikawa S, Nakanishi K, et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int J Cancer (2018) 142(8):1712–22. doi: 10.1002/ijc.31193
90. Schiliro C, Firestein BL. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells (2021) 10(5):1056. doi: 10.3390/cells10051056
91. Ohshima K, Morii E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites (2021) 11(1):28. doi: 10.3390/metabo11010028
92. Liberti MV, Locasale JW. The warburg effect: How does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
93. Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin Cancer Biol (2018) 53(2018):265–81. doi: 10.1016/j.semcancer.2018.10.002
94. Varghese E, Samuel SM, Líšková A, Samec M, Kubatka P, Büsselberg D. Targeting glucose metabolism to overcome resistance to anticancer chemotherapy in breast cancer. Cancers (2020) 12(8):2252. doi: 10.3390/cancers12082252
95. Rao E, Zhang Y, Zhu G, Hao J, Persson XM, Egilmez NK, et al. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget (2015) 6(10):7944. doi: 10.18632/oncotarget.3501
96. Angin Y, Beauloye C, Horman S, Bertrand L. Regulation of carbohydrate metabolism, lipid metabolism, and protein metabolism by AMPK. AMP-Activated Protein Kinase (2016) 107, 23–43. doi: 10.1007/978-3-319-43589-3_2
97. Hardie G. AMPK-a nutrient and energy sensor with roles in diabetes, cancer and viral infection. BMC Proc (2012) 6(Suppl 3):O10. doi: 10.1186/1753-6561-6-S3-O10
98. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol (2003) 2(4):1–16.
99. Muraleedharan R, Dasgupta B. AMPK in the brain: its roles in glucose and neural metabolism. FEBS J (2022) 289(8):2247–62. doi: 10.1111/febs.16151
100. Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J Pathol (2013) 230(4):350–5. doi: 10.1002/path.4218
101. Ros S, Schulze A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2, 6-bisphosphatases in cancer metabolism. Cancer Metab (2013) 1(1):1–10. doi: 10.1186/2049-3002-1-8
102. Kim T, Davis J, Zhang AJ, He X, Mathews ST. Curcumin activates AMPK and suppresses gluconeogenic gene expression in hepatoma cells. Biochem Biophys Res Commun (2009) 388(2):377–82. doi: 10.1016/j.bbrc.2009.08.018
103. Palma FR, Ratti BA, Paviani V, Coelho DR, Miguel R, Danes JM, et al. AMPK-deficiency forces metformin-challenged cancer cells to switch from carbohydrate metabolism to ketogenesis to support energy metabolism. Oncogene (2021) 40(36):5455–67. doi: 10.1038/s41388-021-01943-x
104. Wang C, Ma J, Zhang N, Yang Q, Jin Y, Wang Y. The acetyl-CoA carboxylase enzyme: a target for cancer therapy? Expert Rev Anticancer Ther (2015) 15(6):667–76. doi: 10.1586/14737140.2015.1038246
105. Liu S, Jing F, Yu C, Gao L, Qin Y, Zhao J. AICAR-induced activation of AMPK inhibits TSH/SREBP-2/HMGCR pathway in liver. PloS One (2015) 10(5):e0124951. doi: 10.1371/journal.pone.0124951
106. Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget (2015) 6(10):7365. doi: 10.18632/oncotarget.3629
107. Choi YK, Park K-G. Metabolic roles of AMPK and metformin in cancer cells. Molecules Cells (2013) 36(4):279–87. doi: 10.1007/s10059-013-0169-8
108. Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto SI, et al. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc Natl Acad Sci (2006) 103(19):7282–7. doi: 10.1073/pnas.0602205103
109. Melnik BC, Schmitz G. Metformin: An inhibitor of mTORC1 signaling. J Endocrinol Diabetes Obes (2014) 2(2):1029.
110. Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, et al. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and n-ras proto-oncogenes in mice. Hepatology (2015) 61(1):200–13. doi: 10.1002/hep.27396
111. Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, et al. Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene (2016) 35(36):4675–88. doi: 10.1038/onc.2015.515
112. Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR, et al. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differentiation (2012) 19(3):501–10. doi: 10.1038/cdd.2011.119
113. Jeon S-M, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature (2012) 485(7400):661–5. doi: 10.1038/nature11066
114. Gurd BJ. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl physiol nutrition Metab (2011) 36(5):589–97. doi: 10.1139/h11-070
115. Shen JJ, Zhan YC, Li HY, Wang Z. Ouabain impairs cancer metabolism and activates AMPK-src signaling pathway in human cancer cell lines. Acta Pharmacologica Sin (2020) 41(1):110–8. doi: 10.1038/s41401-019-0290-0
116. Yung MM, Siu MK, Ngan HY, Chan DW, Chan KK. Orchestrated action of AMPK activation and combined VEGF/PD-1 blockade with lipid metabolic tunning as multi-target therapeutics against ovarian cancers. Int J Mol Sci (2022) 23(12):6857. doi: 10.3390/ijms23126857
117. Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science (2010) 329(5996):1201–5. doi: 10.1126/science.1191241
118. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331(6016):456–61. doi: 10.1126/science.1196371
119. Hardie D, Pan D. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans (2002) 30(6):1064–70. doi: 10.1042/bst0301064
120. Yun CW, Jeon J, Go G, Lee JH, Lee SH. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int J Mol Sci (2020) 22(1):179. doi: 10.3390/ijms22010179
121. Chmurska A, Matczak K, Marczak A. Two faces of autophagy in the struggle against cancer. Int J Mol Sci (2021) 22(6):2981. doi: 10.3390/ijms22062981
122. Laderoute KR, Calaoagan JM, Chao WR, Dinh D, Denko N, Duellman S, et al. 5′-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J Biol Chem (2014) 289(33):22850–64. doi: 10.1074/jbc.M114.576371
123. Ríos M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, et al. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res (2013) 73(8):2628–38. doi: 10.1158/0008-5472.CAN-12-0861
124. Chaube B, Malvi P, Singh SV, Mohammad N, Viollet B, Bhat MK. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discovery (2015) 1(1):1–11. doi: 10.1038/cddiscovery.2015.63
125. Sadria M, Seo D, Layton AT. The mixed blessing of AMPK signaling in cancer treatments. BMC Cancer (2022) 22(1):1–16. doi: 10.1186/s12885-022-09211-1
126. Chang WH, Lai AG. An integrative pan-cancer investigation reveals common genetic and transcriptional alterations of AMPK pathway genes as important predictors of clinical outcomes across major cancer types. BMC Cancer (2020) 20(1):1–15. doi: 10.1186/s12885-020-07286-2
127. Wu Y, Yan B, Xu W, Guo L, Wang Z, Li G, et al. Compound c enhances the anticancer effect of aspirin in HER-2-positive breast cancer by regulating lipid metabolism in an AMPK-independent pathway. Int J Biol Sci (2020) 16(4):583. doi: 10.7150/ijbs.39936
128. Lemos C, Schulze VK, Baumgart SJ, Nevedomskaya E, Heinrich T, Lefranc J, et al. The potent AMPK inhibitor BAY-3827 shows strong efficacy in androgen-dependent prostate cancer models. Cell Oncol (2021) 44(3):581–94. doi: 10.1007/s13402-020-00584-8
129. Sas Z, Cendrowicz E, Weinhäuser I, Rygiel TP. Tumor microenvironment of hepatocellular carcinoma: Challenges and opportunities for new treatment options. Int J Mol Sci (2022) 23(7):3778. doi: 10.3390/ijms23073778
130. Wei R, Liu S, Zhang S, Min L, Zhu S. Cellular and extracellular components in tumor microenvironment and their application in early diagnosis of cancers. Analytical Cell Pathol (2020) 2020:1–13. doi: 10.1155/2020/6283796
131. Whiteside T. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008) 27(45):5904–12. doi: 10.1038/onc.2008.271
132. Mafi S, Mansoori B, Taeb S, Sadeghi H, Abbasi R, Cho WC, et al. mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front Immunol (2022) 12-2021, 5724. doi: 10.3389/fimmu.2021.774103
133. Cai DL, Jin L-P. Immune cell population in ovarian tumor microenvironment. J Cancer (2017) 8(15):2915. doi: 10.7150/jca.20314
134. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi: 10.1038/nrc3237
135. Dabi YT, Andualem H, Degechisa ST, Gizaw ST. Targeting metabolic reprogramming of T-cells for enhanced anti-tumor response. Biologics: Targets Ther (2022) 16:35. doi: 10.2147/BTT.S365490
136. Pokhrel RH, Acharya S, Ahn JH, Gu Y, Pandit M, Kim JO, et al. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol Cancer (2021) 20(1):1–15. doi: 10.1186/s12943-021-01420-9
137. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi: 10.1016/j.immuni.2014.12.030
138. Ma EH, Poffenberger MC, Wong AH, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
139. Yang K, Chi H. AMPK helps T cells survive nutrient starvation. Immunity (2015) 42(1):4–6. doi: 10.1016/j.immuni.2014.12.029
140. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPK α1: A glucose sensor that controls CD 8 T-cell memory. Eur J Immunol (2013) 43(4):889–96. doi: 10.1002/eji.201243008
141. Gao Y, Päivinen P, Tripathi S, Domènech-Moreno E, Wong IP, Vaahtomeri K, et al. Inactivation of AMPK leads to attenuation of antigen presentation and immune evasion in lung AdenocarcinomaInactivation of AMPK leads to immune evasion in lung cancer. Clin Cancer Res (2022) 28(1):227–37. doi: 10.1158/1078-0432.CCR-21-2049
142. Braverman E, Dobbs A, Monlish D, Brown RA, Byersdorfer CA. Increasing AMPK activity supports enhanced oxidative metabolism, proliferation, and in vitro recovery of human CD4+ T cells. Am Assoc Immnol (2020) 204(1_Supplement):240–2. doi: 10.4049/jimmunol.204.Supp.240.2
143. Lepez A, Pirnay T, Denanglaire S, Perez-Morga D, Vermeersch M, Leo O, et al. Long-term T cell fitness and proliferation is driven by AMPK-dependent regulation of reactive oxygen species. Sci Rep (2020) 10(1):1–14. doi: 10.1038/s41598-020-78715-2
144. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor MicroenvironmentsSpatial heterogeneity of TAMs in tumors. Cancer Res (2018) 78(19):5492–503. doi: 10.1158/0008-5472.CAN-18-1367
145. Stopforth RJ, Ward ES. The role of antigen presentation in tumor-associated macrophages. Crit Reviews™ Immunol (2020) 40(3):205–24. doi: 10.1615/CritRevImmunol.2020034910
146. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol (2008) 181(12):8633–41. doi: 10.4049/jimmunol.181.12.8633
147. Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget (2017) 8(13):20706. doi: 10.18632/oncotarget.14982
148. Fan K, Lin L, Ai Q, Wan J, Dai J, Liu G, et al. Lipopolysaccharide-induced dephosphorylation of AMPK-activated protein kinase potentiates inflammatory injury via repression of ULK1-dependent autophagy. Front Immunol (2018) 9:1464. doi: 10.3389/fimmu.2018.01464
149. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5′-monophosphate–activated protein kinase regulates IL-10–mediated anti-inflammatory signaling pathways in macrophages. J Immunol (2015) 194(2):584–94. doi: 10.4049/jimmunol.1401024
150. Mancini SJ, White AD, Bijland S, Rutherford C, Graham D, Richter EA, et al. Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Mol Cell Endocrinol (2017) 440:44–56. doi: 10.1016/j.mce.2016.11.010
151. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
152. Gao X, Sui H, Zhao S, Gao X, Su Y, Qu P. Immunotherapy targeting myeloid-derived suppressor cells (MDSCs) in tumor microenvironment. Front Immunol (2021) 11:585214. doi: 10.3389/fimmu.2020.585214
153. Trillo-Tinoco J, Sierra RA, Mohamed E, Cao Y, de Mingo-Pulido Á, Gilvary DL, et al. AMPK alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res (2019) 79(19):5034–47. doi: 10.1158/0008-5472.CAN-19-0880
154. Salminen A, Kauppinen A, Kaarniranta K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J Mol Med (2019) 97(8):1049–64. doi: 10.1007/s00109-019-01795-9
155. Miko E, Meggyes M, Doba K, Barakonyi A, Szereday L. Immune checkpoint molecules in reproductive immunology. Front Immunol (2019) 10:846. doi: 10.3389/fimmu.2019.00846
156. Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol Cell (2021) 81(11):2317–2331.e6. doi: 10.1016/j.molcel.2021.03.037
157. Fan Z, Wu C, Chen M, Jiang Y, Wu Y, Mao R, et al. The generation of PD-L1 and PD-L2 in cancer cells: From nuclear chromatin reorganization to extracellular presentation. Acta Pharm Sin B (2021) 12(3):1041–53. doi: 10.1016/j.apsb.2021.09.010
158. Afzal MZ, Mercado RR, Shirai K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J ImmunoTher Cancer (2018) 6(1):1–10. doi: 10.1186/s40425-018-0375-1
159. Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J immunother Cancer (2021) 9(5):1–7. doi: 10.1136/jitc-2020-002088
160. Lalau J-D. Lactic acidosis induced by metformin. Drug Saf (2010) 33(9):727–40. doi: 10.2165/11536790-000000000-00000
161. Kim I, He Y-Y. Targeting the AMP-activated protein kinase for cancer prevention and therapy. Front Oncol (2013) 3:175. doi: 10.3389/fonc.2013.00175
162. Hardie DG. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes (2013) 62(7):2164–72. doi: 10.2337/db13-0368
163. Nath LR, Swetha M, Vijayakurup V, Thangarasu AK, Haritha NH, Shabna A, et al. Blockade of uttroside b-induced autophagic pro-survival signals augments its chemotherapeutic efficacy against hepatocellular carcinoma. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.812598
164. HWANG, Kwak JT, Lin DW, Kim SK, Kim HM, Park YM, O J. Resveratrol induces apoptosis in chemoresistant cancer cells via modulation of AMPK signaling pathway. Ann New York Acad Sci (2007) 1095(1):441–8. doi: 10.1196/annals.1397.047
165. Rashid A, Liu C, Sanli T, Tsiani E, Singh G, Bristow RG, et al. Resveratrol enhances prostate cancer cell response to ionizing radiation. modulation of the AMPK, akt and mTOR pathways. Radiat Oncol (2011) 6(1):1–12. doi: 10.1186/1748-717X-6-144
166. Lin JN, Lin VC, Rau KM, Shieh PC, Kuo DH, Shieh JC, et al. Resveratrol modulates tumor cell proliferation and protein translation via SIRT1-dependent AMPK activation. J Agric Food Chem (2010) 58(3):1584–92. doi: 10.1021/jf9035782
167. Yu R, Zhang ZQ, Wang B, Jiang HX, Cheng L, Shen LM. Berberine-induced apoptotic and autophagic death of HepG2 cells requires AMPK activation. Cancer Cell Int (2014) 14(1):1–8. doi: 10.1186/1475-2867-14-49
168. Su Q, Peng M, Zhang Y, Xu W, Darko KO, Tao T, et al. Quercetin induces bladder cancer cells apoptosis by activation of AMPK signaling pathway. Am J Cancer Res (2016) 6(2):498. doi: 10.3390/cancers11121960
169. Li W, Hua B, Saud SM, Lin H, Hou W, Matter MS, et al. Berberine regulates AMP-activated protein kinase signaling pathways and inhibits colon tumorigenesis in mice. Mol carcinogenesis (2015) 54(10):1096–109. doi: 10.1002/mc.22179
170. Tong W, Wang Q, Sun D, Suo J. Curcumin suppresses colon cancer cell invasion via AMPK-induced inhibition of NF-κB, uPA activator and MMP9. Oncol Lett (2016) 12(5):4139–46. doi: 10.3892/ol.2016.5148
171. Tang Q, Wu J, Zheng F, Chen Y, Hann SS. Emodin increases expression of insulin-like growth factor binding protein 1 through activation of MEK/ERK/AMPKα and interaction of PPARγ and Sp1 in lung cancer. Cell Physiol Biochem (2017) 41(1):339–57. doi: 10.1159/000456281
172. Li W, Wang D, Li M, Li B. Emodin inhibits the proliferation of papillary thyroid carcinoma by activating AMPK. Exp Ther Med (2021) 22(4):1–9. doi: 10.3892/etm.2021.10509
173. Hwang JT, Ha J, Park IJ, Lee SK, Baik HW, Kim YM, et al. Apoptotic effect of EGCG in HT-29 colon cancer cells via AMPK signal pathway. Cancer Lett (2007) 247(1):115–21. doi: 10.1016/j.canlet.2006.03.030
174. Zhang H, Kim E, Yi J, Hai H, Kim H, Park S, et al. [6]-gingerol suppresses oral cancer cell growth by inducing the activation of AMPK and suppressing the AKT/mTOR signaling pathway. Vivo (2021) 35(6):3193–201. doi: 10.21873/invivo.12614
175. Zhang F, Thakur K, Hu F, Zhang JG, Wei ZJ. 10-gingerol, a phytochemical derivative from “tongling white ginger”, inhibits cervical cancer: insights into the molecular mechanism and inhibitory targets. J Agric Food Chem (2017) 65(10):2089–99. doi: 10.1021/acs.jafc.7b00095
176. Fan J, Yang X, Bi Z. 6-gingerol inhibits osteosarcoma cell proliferation through apoptosis and AMPK activation. Tumor Biol (2015) 36(2):1135–41. doi: 10.1007/s13277-014-2723-1
177. Lee SR, Kwon SW, Lee YH, Kaya P, Kim JM, Ahn C, et al. Dietary intake of genistein suppresses hepatocellular carcinoma through AMPK-mediated apoptosis and anti-inflammation. BMC Cancer (2019) 19(1):1–12. doi: 10.1186/s12885-018-5222-8
178. Jeon H, Huynh DT, Baek N, Nguyen TL, Heo KS. Ginsenoside-Rg2 affects cell growth via regulating ROS-mediated AMPK activation and cell cycle in MCF-7 cells. Phytomedicine (2021) 85:153549. doi: 10.1016/j.phymed.2021.153549
179. Han M, Gao H, Ju P, Gao MQ, Yuan YP, Chen XH, et al. Hispidulin inhibits hepatocellular carcinoma growth and metastasis through AMPK and ERK signaling mediated activation of PPARγ. Biomed Pharmacother (2018) 103:272–83. doi: 10.1016/j.biopha.2018.04.014
180. Woo SM, Seo SU, Kim SH, Nam JO, Kim S, Park JW, et al. Hispidulin enhances TRAIL-mediated apoptosis via CaMKKβ/AMPK/USP51 axis-mediated bim stabilization. Cancers (2019) 11(12):1960.
181. Yang JM, Hung CM, Fu CN, Lee JC, Huang CH, Yang MH, et al. Hispidulin sensitizes human ovarian cancer cells to TRAIL-induced apoptosis by AMPK activation leading to mcl-1 block in translation. J Agric Food Chem (2010) 58(18):10020–6. doi: 10.1021/jf102304g
182. Brookens SK, Cho SH, Basso PJ, Boothby MR. AMPKα1 in b cells dampens primary antibody responses yet promotes mitochondrial homeostasis and persistence of b cell memory. J Immunol (2020) 205(11):3011–22. doi: 10.4049/jimmunol.1901474
Keywords: AMPK signaling, cancer metabolism, tumor microenvironment, immune response, T cells, macrophages, immune checkpoints, phytochemicals
Citation: Keerthana CK, Rayginia TP, Shifana SC, Anto NP, Kalimuthu K, Isakov N and Anto RJ (2023) The role of AMPK in cancer metabolism and its impact on the immunomodulation of the tumor microenvironment. Front. Immunol. 14:1114582. doi: 10.3389/fimmu.2023.1114582
Received: 02 December 2022; Accepted: 03 February 2023;
Published: 15 February 2023.
Edited by:
Barbara Wegiel, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesReviewed by:
Leonard Maggi, Washington University in St. Louis, United StatesCopyright © 2023 Keerthana, Rayginia, Shifana, Anto, Kalimuthu, Isakov and Anto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruby John Anto, cmphbnRvQHJnY2IucmVzLmlu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.