
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 February 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1114350
This article is part of the Research Topic Community Series in Epigenetics of the Immune Component of Inflammation, Volume II View all 10 articles
 Yu Shan1,2,3†
Yu Shan1,2,3† Jianan Zhao1,2,3†
Jianan Zhao1,2,3† Yixin Zheng1,2,3
Yixin Zheng1,2,3 Shicheng Guo4,5*
Shicheng Guo4,5* Steven J. Schrodi4,5*
Steven J. Schrodi4,5* Dongyi He1,2,3,6*
Dongyi He1,2,3,6*Rheumatoid arthritis (RA) is a highly disabling chronic autoimmune disease. Multiple factors contribute to the complex pathological process of RA, in which an abnormal autoimmune response, high survival of inflammatory cells, and excessive release of inflammatory factors lead to a severe chronic inflammatory response. Clinical management of RA remains limited; therefore, exploring and discovering new mechanisms of action could enhance clinical benefits for patients with RA. Important bidirectional communication occurs between the brain and immune system in inflammatory diseases such as RA, and circulating immune complexes can cause neuroinflammatory responses in the brain. The gamma-aminobutyric acid (GABA)ergic system is a part of the nervous system that primarily comprises GABA, GABA-related receptors, and GABA transporter (GAT) systems. GABA is an inhibitory neurotransmitter that binds to GABA receptors in the presence of GATs to exert a variety of pathophysiological regulatory effects, with its predominant role being neural signaling. Nonetheless, the GABAergic system may also have immunomodulatory effects. GABA/GABA-A receptors may inhibit the progression of inflammation in RA and GATs may promote inflammation. GABA-B receptors may also act as susceptibility genes for RA, regulating the inflammatory response of RA via immune cells. Furthermore, the GABAergic system may modulate the abnormal pain response in RA patients. We also summarized the latest clinical applications of the GABAergic system and provided an outlook on its clinical application in RA. However, direct studies on the GABAergic system and RA are still lacking; therefore, we hope to provide potential therapeutic options and a theoretical basis for RA treatment by summarizing any potential associations.
Rheumatoid arthritis (RA) is an autoimmune-mediated chronic inflammatory joint disease characterized by pain and swelling in the joints of the hands and feet (primarily in the toe), proximal interphalangeal joints, and wrists. Unlike the “hard” swelling of osteoarthritis, this swelling is usually “soft”, and is attributed to synovitis and fluid accumulation (1, 2). The global incidence of RA is approximately 0.24% (3), with a positive correlation with age. Furthermore, RA is a chronic, long-term joint disorder that may persist for decades and even for life (4, 5); however, the pathogenesis of RA remains unclear. Nonetheless, it is understood that in RA, immune tolerance to autologous proteins (such as collagen, wave proteins, and fibrinogen) is disrupted for various reasons, resulting in the formation of autoantibodies against autoantigens, such as anti-citrullinated peptide antibodies, anti-immunoglobulin G antibodies, and autoantigens that cross-react with bacterial or viral antigens (6, 7). It occurs when the immune system mistakenly attacks the tissues in the joints, leading to inflammation, swelling, and destruction of the joint structure (8).
Neovascularization is another hallmark of RA synovitis, in which multiple synovial infiltrates of the joint occur and endothelial cells are activated; furthermore, an expansion of synovial fibroblasts and macrophage-like cells leads to the proliferation of the supra-synovial layer and invasion of the periarticular bone at the cartilage junction, ultimately leading to bone erosion and cartilage degeneration (9). RA is incurable and patients must be treated with disease-modifying anti-rheumatic drugs (DMARDs) to relieve clinical symptoms, improve somatic function, and inhibit the progression of joint damage (10). Commonly used DMARDs include methotrexate, leflunomide, and sulfonamides. Early treatment with combined methotrexate and glucocorticoids followed by other DMARDs with the inhibitors of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), or Janus kinase can improve prognosis (11). Treatment aims to reduce disease activity by at least 50% within three months and achieve remission or reduced disease activity within six months, and can prevent RA-related disability with sequential drug therapy if necessary (12, 13). Therefore, understanding and identifying novel targets is important for the clinical management and treatment of RA. There is some evidence to suggest that the GABAergic system may be involved in the development and progression of RA (14). Some studies have found that gamma-aminobutyric acid (GABA) levels are lower in people with RA compared to healthy individuals (15). Additionally, GABA has anti-inflammatory and pain-relieving effects, and some researchers have suggested that abnormal GABA signaling may contribute to the immune system dysregulation that occurs in RA (16).
The GABAergic system is a network of neurons in the brain and central nervous system that produce and use the neurotransmitter GABA (17). GABA is an inhibitory neurotransmitter that helps to regulate the activity of neurons and maintain balance in the nervous system (18). It acts by binding to specific receptors on neurons, causing them to reduce their activity. Some studies have explored the potential role of GABA signaling in RA-related inflammation and pain. For example, some research has suggested that GABA signaling may be involved in the downregulation of T-cell autoimmunity and antigen-presenting cells (APC) activity, which play a role in the development of RA-related inflammation (19). Other studies have found that GABA agonists, which are medications that enhance GABA signaling, may be effective in reducing inflammation and pain in animal models (20). It is important to note that the relationship between the GABAergic system and RA is complex and not fully understood. More research is needed to fully understand the function of the GABAergic system in RA and its potential as a target for treatment. However, the findings of some studies suggest that medications that enhance GABA signaling may be a promising approach for managing RA-related inflammation and pain.
The GABAergic system comprises the following components: GABA, glutamate decarboxylase (GAD), GABA-A receptors, GABA-B receptors, and GABA transporters (GATs). GAD is predominantly composed of two isoforms, GAD65 and GAD67. The genes of these isoforms are distributed differently and have multiple functions, and GABA acts by binding to their receptors (17, 21). The balance of the GABAergic system is essential for many physiological aspects, including blood pressure, baroreceptor function, human growth hormone release, weight regulation and feeding, respiratory function, brain function, kidney function, vision, and pancreatic function (22). The most prominent feature of RA is synovial inflammation, which is closely associated with abnormal expression of immune cells and inflammatory factors. Pro-inflammatory cytokines, including tumor necrosis factor (TNF) and IL-6, induce RANKL, prostaglandins, and matrix metalloproteinases, which mediate the pain and swelling of the disease; furthermore, RANKL, TNF, and IL-6 stimulate bone and cartilage (23). Recent studies have shown that immune system cells can produce GABA and express GABA-A ion channels, GATs, and GABA-B receptors (24). The synovium also contains a GABAergic system; specifically, synovial macrophage-like A cells possess a GABA-producing system alongside GAD and GABA-B receptor subunits. Furthermore, the GABAergic system appears to play a functional role in the synovium (14). GABAergic components have been reported to negatively regulate the immune response by affecting the production of pro-inflammatory cytokines and activation of signaling pathways (16, 25). Overall, GABAergic components may offer a new therapeutic approach for inflammatory and autoimmune diseases, including RA. However, the GABAergic system in the synovium has yet to be studied. In this article, we explore the role of GABAergic components in the regulation of inflammation and pain in RA by summarizing the major components of the GABAergic system and their physiological functions, alongside the potential links that exist with RA.
GABA receptors are widely expressed in the central nervous system (CNS) and occupy approximately 20% of the synapses in the cerebral cortex, hippocampus, thalamus, and cerebellum. These are closely linked to the control of cognitive functions such as memory, language, and attention (17); they are also located in the retina, thalamus, hippocampus, pituitary, and gastrointestinal tract, and are associated with visual processing, sleep-wake rhythm regulation, pain perception, memory, learning, hormone regulation, and neuroendocrine gastrointestinal secretion (26). GATs are present in the presynaptic membrane. GAT-1 and GAT-3 are abundantly expressed in the CNS, whereas GAT-2 and betaine/gamma-aminobutyric acid transporter (BGT-1) are expressed in tissues such as the liver, kidney, and intestine (26–28).
GABA is the main inhibitory neurotransmitter in the CNS and plays a key role in controlling excitability, plasticity, and network synchronization in the CNS. GABA is present in the CNS alongside many other organs, such as the pancreas, pituitary gland, testes, gastrointestinal tract, ovaries, placenta, uterus, and adrenal medulla (29, 30). GABA synthesis requires GAD, which consists of two isomers (GAD65 and GAD67), encoded by a single gene on chromosomes 10 and 2, respectively, and is the rate-limiting enzyme that catalyzes the conversion of glutamate to the inhibitory neurotransmitter GABA (31). GABA is primarily synthesized via two pathways: direct synthesis from glutamic acid catalyzed by GAD65 or GAD67, and synthesis from glutamic acid produced by trichloroacetic acid catalyzed by GAD67. GABA acts through its receptors, primarily GABA-A and GABA-B (32). The GABA-A receptor is an ion-affinity receptor that binds to GABA and opens its complete chloride channel. In contrast, GABA-B receptors are G protein-coupled metabotropic receptors that have a negative effect on presynaptic voltage-activated calcium channels and a positive effect on postsynaptic inwardly rectifying potassium channels (33). GABA signaling in the synaptic cleft can be reuptaken by high-affinity GATs into the presynaptic membrane, thus avoiding GABA overexpression (34).
GABA receptors are the major inhibitory neurotransmitters of the vertebrate CNS and are expressed by many neurons in the CNS, as well as other cell types in the periphery, with two main types: GABA-A and GABA-B receptors (35). GABA-A receptors (ionotropic receptors) are ligand-gated ion channels that bind to GABA and open the Cl- channel. GABA-A receptors include many subunit types: α (one-six), β (one-three), γ (one-three), δ, ϵ, θ, π, and ρ (one-three). The most common GABA-A receptor subunit types in the brain are α1, β2, and γ2 (36). GABA-A receptors are recruited to increase acetylcholine release and facilitate transmission; however, GABA-B receptors are activated at high GABA concentrations, thereby decreasing acetylcholine release and transmission (36, 37).
GABA-B receptors (metabotropic receptors) are G-protein coupled receptors that have a negative effect on presynaptic voltage-activated Ca2+ channels but a positive effect on postsynaptic inwardly corrected K+ channels (38). GABA-B receptors consist of B1 and B2 subunits, and it is generally believed that B1a/B2 heterodimers are localized to presynaptic neurons, while B1b/B2 heterodimers are localized to postsynaptic neurons. GABA-B receptors bind to G protein subsets, which consequently regulate specific ion channels, such as calcium channels, and trigger cyclic adenosine monophosphate cascade responses (39). GABA-B receptors modulate their inhibitory effects by activating the inwardly regulated K+ channels, inactivating voltage-stalled Ca2+ channels, and inhibiting adenylyl cyclase. Postsynaptic receptors induce slow inhibitory postsynaptic currents that hyperpolarize the membrane and shunt excitatory currents by gating a particular type of K+ channel (39, 40).
The GAT family, which includes GAT-1, GAT-2, GAT-3, and BGT-1, are important regulators of intracellular and extracellular GABA concentrations; specifically, these transporters mediate the secondary active transport of ion-coupled GABA across the plasma membrane (41). GATs inhibit GABA signaling by translocating GABA to cells and reducing extracellular GABA concentrations, and are potential drug targets in various diseases associated with dysregulated GABA delivery. GAT-1 and GAT-3 are predominantly expressed in the CNS where they regulate GABA activity (42), whereas GAT-2 is mainly located in peripheral organs, especially in the liver and kidney, and is thought to be a GABA and taurine transporter in the liver (43). GAT-1 is a highly conserved molecule encoded by solute carrier family 6 member 1, which transports GABA via Na+ and Cl-. As the major GAT in the brain, in addition to being involved in a wide range of brain functions, GAT-1 has been implicated in the pathophysiology of several neuropsychiatric disorders, including anxiety disorders, depression, epilepsy, Alzheimer’s disease, and schizophrenia (44).
The GABAergic system may act as a bridge between the nervous system and the immune system. As an inhibitory neurotransmitter in the CNS, the main role of GABA is to reduce the excitability of neurons throughout the nervous system (18). Activation of GABA and other ligands by the GABA receptor induces a conformational change in the receptor that increases axonal K+ conductance, thereby accelerating action potential repolarization and leading to transient secondary inhibition of calcium by decreasing the degree of calcium channel activation (45, 46). GABAergic system plays an important role in the function of immune cells mainly being influenced by GABA signaling. Recent studies have highlighted the immune function of GABA, suggesting cross-talk between the nervous and immune systems (47, 48). T cells express many neurotransmitter receptors, and their expression is regulated by T cell receptor activation, cytokines, and neurotransmitters themselves (49). The GABAergic system can negatively regulate immune responses, especially T cell–mediated immune responses, by affecting the production of pro-inflammatory cytokines and the activation of signaling pathways, such as mitogen-activated protein kinase and nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) pathways (50). These results may indicate that the GABAergic system offers a new therapeutic approach for inflammatory and autoimmune diseases and requires further evaluation, especially regarding RA treatment.
Central and peripheral GABA-A receptors play a major role in inflammation; specifically, GABA inhibition reverses the pathological pain state in mice, and GABA-A reduces the production of pro-inflammatory mediators, thereby ameliorating the symptoms of inflammation (51). GABA/GABA-A receptors may induce RA inflammation by affecting T cell and macrophage populations. Mechanistically, GABA receptors firstly interact with KCNN4 to induce Ca2+ entry, resulting in activation of nuclear factor κB signal, and finally promotes macrophage invasion by inducing CXCL5 and CCL20 expression (52). This functional GABAergic system acts as a regulator of T cell activation (16). GABA is not only an inhibitory neurotransmitter but also an immunomodulator. GABA binding to GABA-A receptors directly affects the function of antigen-presenting cells and inhibits the production of inflammatory cytokines by T cells (53). Binding of GABA to GABA-A receptors also inhibits the proliferation of antigen-specific T cells and suppresses the production of IL-6, IL-12, inducible nitric oxide synthase, IL-1β, and TNF-α (52, 54). Furthermore, activation of GABA-A receptors inhibits the release of TNF-α and IL-6 from alveolar macrophages induced by lipopolysaccharide (55).
P38/MAPK may be a key player in the link between the GABA/GABA-A axis and inflammation in RA. P38 is a tyrosine phosphoprotein kinase isolated and purified from mammalian cells stimulated by endotoxin; this enzyme is the most important member of the mitogen-activated protein kinase (MAPK) family in terms of inflammatory response mediation (56). Key enzymes of the P38/MAPK pathway include mitogen-activated protein kinase kinase (MKK)-3, MKK6, and transforming growth factor-β activating kinase (TAK) (57). TAK activates MKK4, which in turn activates P38/MAPK that phosphorylates and activates many protein kinases and transcription factors (58). P38/MAPK activates signal transducer-activator of transcription 4 (STAT4) and NF-κB and promotes the release of inflammatory factors such as TNF-α, IL-1β, and IL-6 (59). Inflammatory stimuli, such as TNF-α, platelet-activating factor, and interleukins, induce P38 activation in endogenous immune cells such as monocytes, endothelial cells, and neutrophils (60, 61). When synovial disruption begins, somatic afferent pain signals received by the spinal cord could lead to stress-induced kinase release; these pain and cytokine signals activate P38/MAPK, which induces the upregulation and release of pro-inflammatory cytokines such as IL-1, IL-6, and matrix metallopeptidase 3 (MMP3) into the periphery (62, 63). A relationship exists between the CNS and peripheral immune response; specifically, afferent pain signals help the CNS propagate the inflammatory response, which may influence the development of peripheral arthritis (64). GABA is a major inhibitory neurotransmitter in the CNS that downregulates P38 activity to reduce the production of proinflammatory cytokines. For example, GABA prevents IL-6 release by inhibiting P38/MAPK in glioma cells, potentially affecting the inflammatory response in RA (65, 66).
The GABAergic system has been suggested to be involved in the pain response in RA. Pain is one of the most important manifestations of RA, and is thought to be caused by joint inflammation (67). Inflammatory pain is predominantly due to local joint synovial inflammation caused by pro-inflammatory cytokines. This inflammation leads to activation of afferent nociceptive fibers and transmission of “pain” signals to the dorsal horn of the spinal cord, via the spinothalamic tract, to the thalamus (68, 69). However, clinical studies have shown that even when inflammation is controlled, patients with RA may continue to experience non-inflammatory pain (70). The main factors causing non-inflammatory pain are yet to be determined. Nonetheless, they are understood to be related to structural changes in the patient’s joint environment, continued disease progression, ectopic secretions, and peripheral nerve damage and dysfunction due to increased excitability of damaged afferent injury receptors, which manifest as neuropathic pain (71, 72). GABA is an inhibitory neurotransmitter; therefore, selective loss of GABAergic interneurons after peripheral nerve injury is thought to be the underlying cause of inhibitory signal loss in RA. Furthermore, reduced inhibitory neurotransmission is a key feature of chronic pain states (73). There are two possible causes for this decrease in inhibitory neurotransmission: a decrease in GABA and its synthase caused by apoptosis of GABAergic neurons in the spinal cord (74, 75) and depletion of GABA in the synaptic gap (76). GABA-A and -B receptors and GAT proteins appear to be involved in the pathophysiology of the chronic pain associated with RA (73). Currently, several studies have investigated the positive modulators of these two receptors and used them for various types of pain; furthermore, a successful testing phase in animal models has recently been achieved (77). GABA-A and glycine receptors are key elements of the spine that control injury perception and pain. Impaired function of these receptors may contribute to the development of chronic pain; therefore, restoring their normal function through aggressive modulators has become an effective treatment for chronic pain syndromes (78). In the context of RA, the pain response in patients with RA may be modulated through the GABAergic system. Therefore, some small-molecule inhibitors or activators currently targeting the GABAergic system may be potentially beneficial in RA.
Neutrophil is one of the key inflammatory effector cells of RA.GABA-B receptors are expressed in neutrophils and play an important role as chemotactic receptors in the inflammatory response (79). GABA-B receptors are also metabotropic receptors. Unlike ionotropic GABA-A receptors, which utilize rapid synaptic transmission, GABA-B receptors are heterodimers consisting of subunits encoded by GABBR1 and GABBR2 (80). The coding region of GABA type B receptor subunit (GABBR)-1 is located on chromosome 6, 6p21.3, and the major histocompatibility complex (MHC) located in this region is associated with multiple sclerosis, Alzheimer’s disease, schizophrenia, narcolepsy, epilepsy, and RA (81). The specific MHC, class II, DR beta 1 alleles are strongly associated with susceptibility to RA; this susceptibility is likely due to their role in presenting arthritogenic polypeptides. However, the linkage disequilibrium of the GABBR1 gene polymorphism with these alleles is not as expected, as the distance between the two motifs is three Mb (82). Thus, the observed genetic association with this region suggests that GABBR1 plays an independent role in genetic susceptibility to RA (83).
In yeast glycan-induced arthritis, GABA-B receptors are involved in neutrophil migration to the knee, similar to GABA; this migration may be associated with P38/MAPK activation in the spinal cord (84). Spinal cord inhibitory signaling may downregulate P38/MAPK and reduce pro-inflammatory cytokine production. Furthermore, any changes that affect this negative regulation, such as SNP alleles or haplotypes in GABBR1, may allow P38/MAPK to continue and worsen RA pathology in an uncontrolled manner (82). Although GABBR1 polymorphisms have not been experimentally characterized in RA patients, computational analysis suggests that GABBR1 encoding multiple isoforms, mutations, and several genes that potentially affect selectively spliced protein structures may be associated with RA progression (85).
Members of the GAT family, including GAT-1, GAT-2, GAT-3, and BGT-1, may contribute to inflammation. Multiple inflammatory factors, such as IL-6, IL-1β, and TNF-α are present in RA (86). The expression of GAT-1 and GAT-3 is closely associated with inflammatory factors, such as IL-6, IL-1β, and TNF-α, and may show a positive correlation with inflammation (87). IL-1β and TNF-α were found to upregulate GAT-1 and GAT-3 expression through the MAPK pathway, which subsequently increased IL-6 levels and further upregulated GAT-1 and GAT-3 expression; alternatively, inhibition of IL-1β and TNF-α receptors attenuated this GAT-1 and GAT-3 expression (88). GATs may be associated with subpopulations of lymphoid T cells and have been determined to regulate cytokine production and T cell proliferation. The gene transcripts of two cotransporters, GAT-1 and GAT-2, have been detected in immune cells and identified in human peripheral blood lymphocytes (89). GABA inhibits Th1 cell–mediated DTH responses in vivo and participates in T cell immunity via the GAT and GABA receptors. For example, GAT-1 is expressed only on antigen-activated T cells and downregulates the proliferation and expression of CD4+ T cells (90). These findings suggest that GAT-1 is a key regulator of the T cell–mediated immune response. Additional evidence suggests that T cells expressing GAT-2 and GAT-2 deficiency promote T helper 17 cell (Th17) responses through the activation of GABA-mammalian target of rapamycin signaling; specifically, in a mouse model of infection, GAT-2 deficiency was observed to enhance the differentiation of naive T cells into Th1 cells (91).
GAT-2 is primarily pro-inflammatory in RA. Furthermore, Interferon (IFN)-γ is an important effector of RA (92) that induces GAT-2 expression in macrophages (93). A systematic evaluation and meta-analysis of common trace metals in RA found that serum copper levels were elevated in RA and higher in patients with active RA. These levels were positively correlated with erythrocyte sedimentation rate and morning stiffness, and were negatively correlated with hemoglobin levels and considered to be adjunctive markers for disease assessment (94, 95). Therefore, GAT-2 may promote inflammatory responses by being associated with abnormally high copper levels in RA. GAT-2 deficiency attenuates macrophage-mediated inflammatory responses in vivo, including lipopolysaccharide-induced sepsis, infection-induced pneumonia, and high-fat diet-induced obesity (25, 96). GAT-2 deficiency also decreases IL-1β production in pro-inflammatory macrophages. This mechanism may involve enhancement of the betaine/S-adenosylmethionine/hypoxanthine metabolic pathway by increasing DNA methylation in its promoter region to inhibit the expression of the transcription factor zinc finger protein 354C (ZNF354C). This zinc finger protein suppresses inflammasome formation and inhibits M1 macrophage polarization by targeting the expression of oxidative phosphorus-related genes (93). According to previous reports, activation of the GABAergic system in macrophages enhances autophagy activation, phagosome maturation, and antimicrobial response to Mycobacterium infection (97). Macrophage autophagy in the context of RA is an important mechanism that may co-mediate the abnormal immune response of macrophages with the GABAergic system (98).
Relevant clinical trials of the major components of the GABAergic system mainly involve GABA, GABA receptor agonists and antagonists, and GABA agonists and antagonists (Table 1). Although there have been no direct clinical studies in the context of RA, ongoing clinical trials are currently providing information regarding the development of clinical small-molecule GABA drugs for RA. First, GABA and GABA-A receptor agonists can inhibit the immune response of immune cells to stimuli, which may be important for remission of inflammation (99). For example, honokiol (a GABA-A receptor modulator) relieves inflammatory arthritis and allergic asthma by affecting cytokine expression of cytokines (100). Additionally, a series of immunological abnormalities have been reported in T1DM patients, involving production of autoantibodies, glutamic acid decarboxylase (GAD-65), tyrosine phosphatase-associated islet antigen 2, zinc transporter protein 8, and insulin; furthermore, there is an altered ability of regulatory T cells (Treg cells) to inhibit the action of effector T cells, which play a key role in the immune destruction process (101). GABA also acts on GABA-A receptors in pancreatic alpha cells, thereby inhibiting glucagon secretion, suppressing inflammation, and increasing the number of regulatory T cells. In RA, Treg cells are important for the proliferation of inflammatory cells (Th17) and suppression of inflammatory factor secretion. Therefore, GABA has potential therapeutic implications for RA by increasing the number of regulatory T cells. Furthermore, the application of activators or inhibitors of the GABAergic system may have potential therapeutic effects on the pain response in RA. For example, studies based on animal models of inflammatory or neuropathic pain have found that selective positive modulators of α2 and α3 subtypes of GABA-A receptors may reverse the loss of postsynaptic GABA-A receptor-mediated spinal cord inhibition, leading to analgesia (102). GABA-B receptors are highly expressed in the structure of the pain pathway, suggesting their involvement in different levels of pain signaling; thus, they have long been considered valuable targets for the treatment of chronic pain (103). Activation of GABA-B receptors leads to hyperpolarization and reduced firing frequency, increased mediation of inhibitory neurotransmission, and production of analgesia in the brain and spinal cord (104). The GABA-B receptor agonist, baclofen, is widely used in clinical practice for the treatment of chronic pain. In the presence of reduced endogenous GABAergic tone, the GABA-B receptor agonist baclofen exerts an anti-injury sensory effect and releases GABA from the cortical precursors of GABAergic interneurons transplanted into the medial ganglion bulge, ultimately reversing neuropathic abnormal pain (105). GAT proteins are highly dynamic in different cell types and are responsible for the recycling and reuptake of GABA, thereby terminating inhibitory signaling (106). Studies have reported that inhibition of GAT reuptake enhances GABAergic neurotransmission and has an inhibitory effect on the release of aspartate and glutamate in the dorsal spinal cord, thereby inhibiting the release of pro-nociceptive neurotransmitters to achieve analgesia (107).
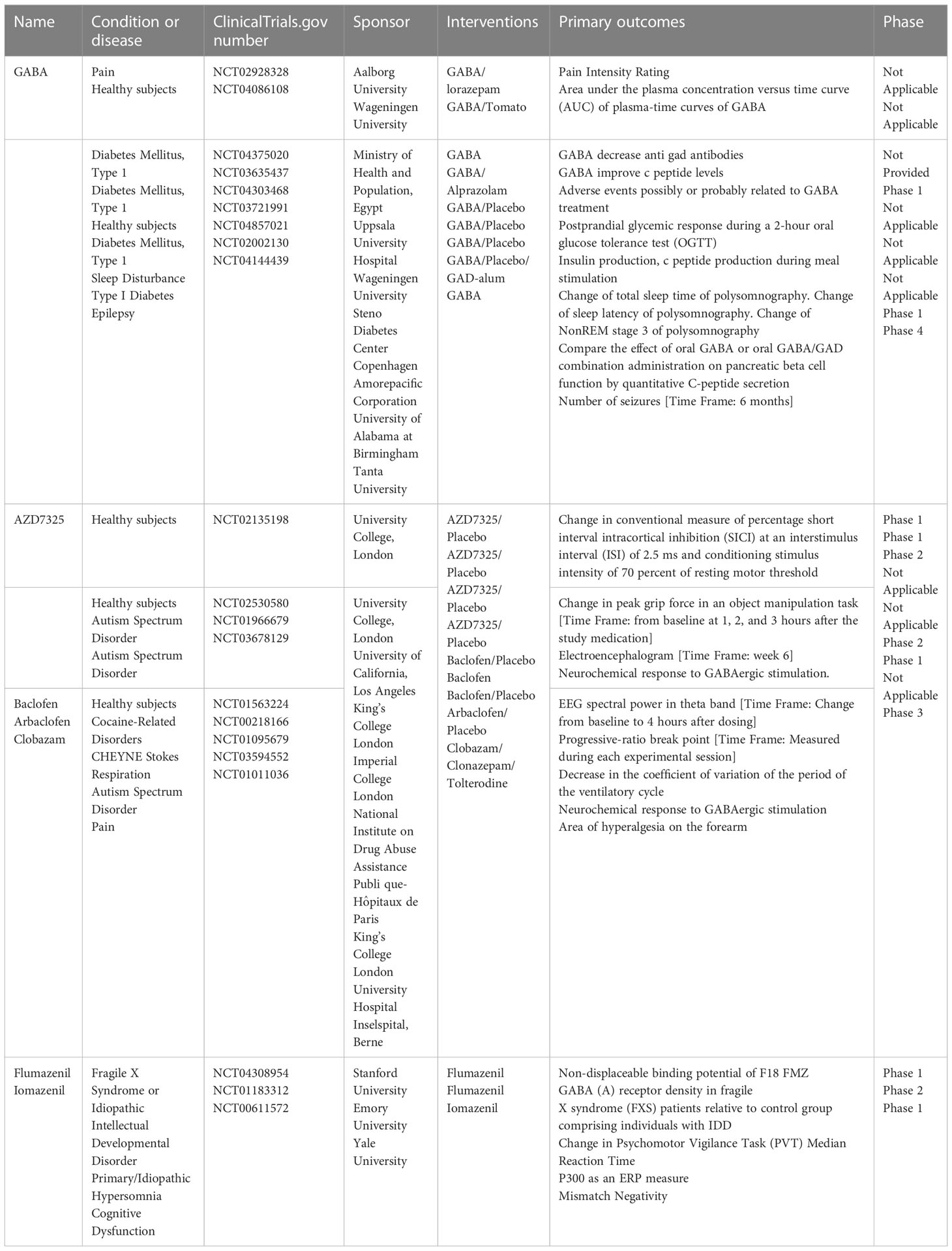
Table 1 Clinical trials of GABAergic system components.
This article focuses on the physiological role of the GABAergic system, its components, and the possible mechanisms of its influence in RA pathology (Figure 1). RA is an inflammatory disease that manifests in peripheral joints and connects the nervous system to the immune system via spinal P38/MAPK. GABA is not only a neurotransmitter, but also an immunomodulator that downregulates P38/MAPK activity to reduce the production of pro-inflammatory cytokines in RA joints, inhibit pro-inflammatory T cell value, and increase the number of regulatory T cells. Through MAPK phosphorylation, the GABA-A receptor inhibits the production of inflammatory cytokines via T cells and suppresses the proliferation of effector T cells, thereby inhibiting the inflammatory progression of RA. In contrast, the GABA-B receptor may be relevant to the pathogenesis of RA due to its unique coding region. GAT expression is positively correlated with inflammatory factors, with an increase in GATs promoting high expression of inflammatory factors and vice versa. GAT-2 deficiency enhances the differentiation of naive T cells into Th1 cells. The GABAergic system also plays an important role in analgesia, as GABA is an inhibitory neurotransmitter that negatively regulates pain signaling; similarly, its derivative, gabapentin, is also used in the treatment of pain. Both GABA receptors and its modulators are potential targets for pain treatment; consequently, studies surrounding the use of GAT inhibitors for pain treatment are ongoing. Therefore, we can see that the GABAergic system plays an important role in the pathogenesis and disease progression of RA, and in addition, GABA has excellent prospects for application in the control of RA-related pain. Nonetheless, there is still no relevant research about the direct link between GABA and the pathogenesis of RA. Further exploration regarding other aspects of the research, such as the nervous and immune systems, could improve our understanding of the overproduction of cytokines, underlying genetic information, and related signaling pathways in RA or neurodegeneration, which will ultimately provide a multidisciplinary understanding of RA and neurological diseases.
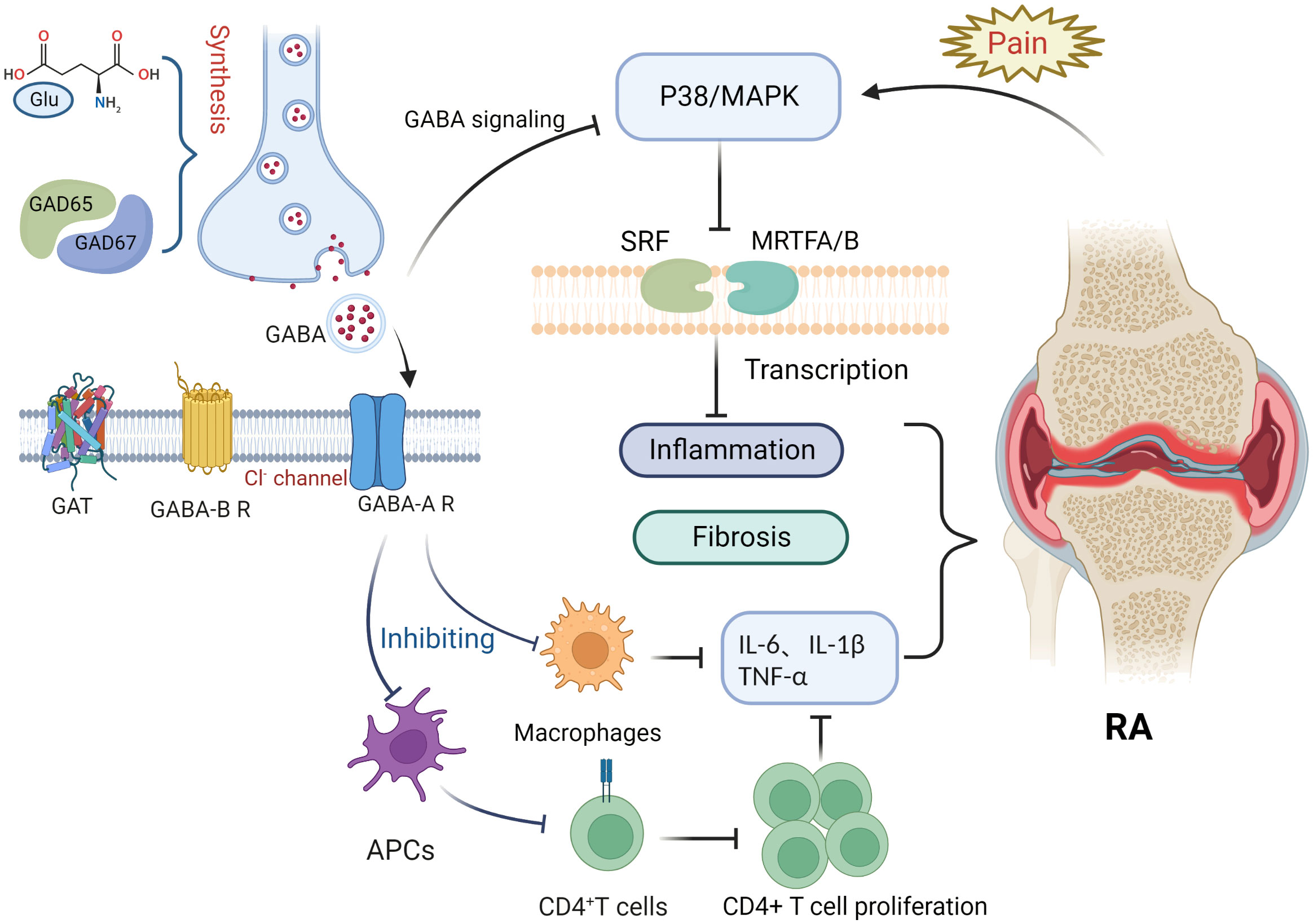
Figure 1 Potential role of the gamma-aminobutyric acid (GABA)ergic system in rheumatoid arthritis. Glutamate interacts with glutamate decarboxylase (GAD65 and GAD67) to produce gamma-aminobutyric acid (GABA). Binding of GABA to GABA-A receptors inhibits macrophage activation and decreases the release of inflammatory factors such as IL-6, IL-1β, and TNF-α. Antigen presentation by antigen-presenting cells, however, is impaired, inhibiting CD4+ T cell proliferation and differentiation and reducing the expression of inflammatory factors such as IL-6, IL-1β, and TNF-α. Pain signaling activates the P38/MAPK pathway, whereas GABA binding to GABA-A receptors inhibits P38/MAPK. The P38/MAPK signaling pathway contributes to inflammation and is involved in the activation of myocardin-related transcription factor A (MRTFA), myocardin-related transcription factor B (MRTFB), and serum response factor (SRF) that played key roles in fibroblast activation.
YS and JZ are responsible for the collection, collation, and writing of the original manuscript. YZ is responsible for the collection. SG, SS, and DH are responsible for the concept development, revision, and manuscript review. All authors contributed to the article and approved the submitted version.
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), National Key Research and Development Project (2018YFC1705200 and 2018YFC1705203), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance, and Shanghai He Dongyi Famous Chinese Medicine Studio Construction Project (SHGZS-202220).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
GABA, gamma-aminobutyric acid; RA, rheumatoid arthritis; GAT, GABA transporter; DMARDs, disease-modifying anti-rheumatic drugs; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; GAD, glutamate decarboxylase; BGT-1, betaine/gamma-aminobutyric acid transporter; CNS, central nervous system; NF-κB, nuclear factor kappa-light-chain enhancer of activated B cells; MAPK, mitogen-activated protein kinase; TAK, transforming growth factor-β activating kinase; STAT4, signal transducer-activator of transcription 4; MMP3, matrix metallopeptidase 3; MHC, major histocompatibility complex; GABBR-1, gamma-aminobutyric acid type B receptor subunit 1; mTOR, mammalian target of rapamycin; Th17, T helper 17; IFN, interferon; ZNF354C, zinc finger protein 354C; KCNN4, potassium calcium-activated channel subfamily N member 4; CXCL5, C-X-C motif chemokine ligand 5; CCL20, C-C motif chemokine ligand 20; APC, antigen-presenting cells.
1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (London England). (2016) 388(10055):2023–38. doi: 10.1016/s0140-6736(16)30173-8
2. Radu AF, Bungau SG. Management of rheumatoid arthritis: An overview. Cells. (2021) 10(11):2857. doi: 10.3390/cells10112857
3. Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of rheumatoid arthritis 1990-2017: a systematic analysis of the global burden of disease study 2017. Ann Rheum Dis (2019) 78(11):1463–71. doi: 10.1136/annrheumdis-2019-215920
4. van der Woude D, van der Helm-van Mil AHM. Update on the epidemiology, risk factors, and disease outcomes of rheumatoid arthritis. Best Pract Res Clin Rheumatol (2018) 32(2):174–87. doi: 10.1016/j.berh.2018.10.005
5. Xu L, Chang C, Jiang P, Wei K, Zhang R, Jin Y, et al. Metabolomics in rheumatoid arthritis: Advances and review. Front Immunol (2022) 13:961708. doi: 10.3389/fimmu.2022.961708
6. Zhao J, Wei K, Jiang P, Chang C, Xu L, Xu L, et al. G-Protein-Coupled receptors in rheumatoid arthritis: Recent insights into mechanisms and functional roles. Front Immunol (2022) 13:907733. doi: 10.3389/fimmu.2022.907733
7. Zhao J, Guo S, Schrodi SJ, He D. Cuproptosis and cuproptosis-related genes in rheumatoid arthritis: Implication, prospects, and perspectives. Front Immunol (2022) 13:930278. doi: 10.3389/fimmu.2022.930278
8. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol (2021) 22(1):10–8. doi: 10.1038/s41590-020-00816-x
9. Smith MH, Berman JR. What is rheumatoid arthritis? Jama. (2022) 327(12):1194. doi: 10.1001/jama.2022.0786
10. Wei K, Jiang P, Zhao J, Jin Y, Zhang R, Chang C, et al. Biomarkers to predict DMARDs efficacy and adverse effect in rheumatoid arthritis. Front Immunol (2022) 13:865267. doi: 10.3389/fimmu.2022.865267
11. Zhao J, Wei K, Chang C, Xu L, Jiang P, Guo S, et al. DNA Methylation of T lymphocytes as a therapeutic target: Implications for rheumatoid arthritis etiology. Front Immunol (2022) 13:863703. doi: 10.3389/fimmu.2022.863703
12. Cush JJ. Rheumatoid arthritis: Early diagnosis and treatment. Med Clinics North America. (2021) 105(2):355–65. doi: 10.1016/j.mcna.2020.10.006
13. Heckert SL, Bergstra SA, Matthijssen XME, Goekoop-Ruiterman YPM, Fodili F, Ten Wolde S, et al. Joint inflammation tends to recur in the same joints during the rheumatoid arthritis disease course. Ann Rheum Dis (2022) 81(2):169–74. doi: 10.1136/annrheumdis-2021-220882
14. Tamura S, Watanabe M, Kanbara K, Yanagawa T, Watanabe K, Otsuki Y, et al. Expression and distribution of GABAergic system in rat knee joint synovial membrane. Histol histopathology. (2009) 24(8):1009–19. doi: 10.14670/hh-24.1009
15. Liu B, Guo H, Li L, Geng Q, Zhao N, Tan Y, et al. Serum metabolomics analysis of deficiency pattern and excess pattern in patients with rheumatoid arthritis. Chin Med (2022) 17(1):71. doi: 10.1186/s13020-022-00632-5
16. Bhandage AK, Barragan A. GABAergic signaling by cells of the immune system: More the rule than the exception. Cell Mol Life Sci CMLS. (2021) 78(15):5667–79. doi: 10.1007/s00018-021-03881-z
17. Zhang W, Xiong BR, Zhang LQ, Huang X, Yuan X, Tian YK, et al. The role of the GABAergic system in diseases of the central nervous system. Neuroscience. (2021) 470:88–99. doi: 10.1016/j.neuroscience.2021.06.037
18. Ngo DH, Vo TS. An updated review on pharmaceutical properties of gamma-aminobutyric acid. Molecules (Basel Switzerland). (2019) 24(15):2678. doi: 10.3390/molecules24152678
19. Tian J, Yong J, Dang H, Kaufman DL. Oral GABA treatment downregulates inflammatory responses in a mouse model of rheumatoid arthritis. Autoimmunity. (2011) 44(6):465–70. doi: 10.3109/08916934.2011.571223
20. Shao FB, Fang JF, Wang SS, Qiu MT, Xi DN, Jin XM, et al. Anxiolytic effect of GABAergic neurons in the anterior cingulate cortex in a rat model of chronic inflammatory pain. Mol brain. (2021) 14(1):139. doi: 10.1186/s13041-021-00849-9
21. Li J, Chen L, Guo F, Han X. The effects of GABAergic system under cerebral ischemia: Spotlight on cognitive function. Neural plasticity. (2020) 2020:8856722. doi: 10.1155/2020/8856722
22. Jahangir M, Zhou JS, Lang B, Wang XP. GABAergic system dysfunction and challenges in schizophrenia research. Front Cell Dev Biol (2021) 9:663854. doi: 10.3389/fcell.2021.663854
23. Neumann E, Frommer K, Diller M, Müller-Ladner U. [Rheumatoid arthritis]. Z fur Rheumatologie. (2018) 77(9):769–75. doi: 10.1007/s00393-018-0500-z
24. Zhang B, Vogelzang A, Miyajima M, Sugiura Y, Wu Y, Chamoto K, et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature. (2021) 599(7885):471–6. doi: 10.1038/s41586-021-04082-1
25. Xia Y, He F, Wu X, Tan B, Chen S, Liao Y, et al. GABA transporter sustains IL-1b production in macrophages. Sci advances. (2021) 7(15):eabe9274. doi: 10.1126/sciadv.abe9274
26. Wisden W, Yu X, Franks NP. GABA receptors and the pharmacology of sleep. Handb Exp Pharmacol (2019) 253:279–304. doi: 10.1007/164_2017_56
27. Boddum K, Jensen TP, Magloire V, Kristiansen U, Rusakov DA, Pavlov I, et al. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat Commun (2016) 7:13572. doi: 10.1038/ncomms13572
28. Brohan J, Goudra BG. The role of GABA receptor agonists in anesthesia and sedation. CNS Drugs (2017) 31(10):845–56. doi: 10.1007/s40263-017-0463-7
29. Miller MW. GABA as a neurotransmitter in gastropod molluscs. Biol bulletin. (2019) 236(2):144–56. doi: 10.1086/701377
30. Sarasa SB, Mahendran R, Muthusamy G, Thankappan B, Selta DRF, Angayarkanni J. A brief review on the non-protein amino acid, gamma-amino butyric acid (GABA): Its production and role in microbes. Curr Microbiol (2020) 77(4):534–44. doi: 10.1007/s00284-019-01839-w
31. Rocco BR, Sweet RA, Lewis DA, Fish KN. GABA-synthesizing enzymes in calbindin and calretinin neurons in monkey prefrontal cortex. Cereb Cortex (New York NY 1991). (2016) 26(5):2191–204. doi: 10.1093/cercor/bhv051
32. Roth FC, Draguhn A. GABA metabolism and transport: Effects on synaptic efficacy. Neural plasticity. (2012) 2012:805830. doi: 10.1155/2012/805830
33. Xu B, Sai N, Gilliham M. The emerging role of GABA as a transport regulator and physiological signal. Plant Physiol (2021) 187(4):2005–16. doi: 10.1093/plphys/kiab347
34. Zaręba P, Gryzło B, Mazur G, Malawska B. Development, recent achievements and current directions of research into GABA uptake inhibitors. Curr medicinal Chem (2021) 28(4):750–76. doi: 10.2174/0929867325666191010120236
35. Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol (2015) 88(1):203–17. doi: 10.1124/mol.115.097998
36. Crocetti L, Guerrini G. GABA(A) receptor subtype modulators in medicinal chemistry: An updated patent review (2014-present). Expert Opin Ther patents. (2020) 30(6):409–32. doi: 10.1080/13543776.2020.1746764
37. Scott S, Aricescu AR. A structural perspective on GABA(A) receptor pharmacology. Curr Opin Struct Biol (2019) 54:189–97. doi: 10.1016/j.sbi.2019.03.023
38. Evenseth LSM, Gabrielsen M, Sylte I. The GABA(B) receptor-structure, ligand binding and drug development. Molecules (Basel Switzerland). (2020) 25(13):3093. doi: 10.3390/molecules25133093
39. Shaye H, Stauch B, Gati C, Cherezov V. Molecular mechanisms of metabotropic GABA(B) receptor function. Sci advances. (2021) 7(22):eabg3362. doi: 10.1126/sciadv.abg3362
40. Nieto A, Bailey T, Kaczanowska K, McDonald P. GABA(B) receptor chemistry and pharmacology: Agonists, antagonists, and allosteric modulators. Curr topics Behav neurosciences. (2022) 52:81–118. doi: 10.1007/7854_2021_232
41. Schijns O, Karaca Ü, Andrade P, de Nijs L, Küsters B, Peeters A, et al. Hippocampal GABA transporter distribution in patients with temporal lobe epilepsy and hippocampal sclerosis. J Chem neuroanatomy. (2015) 68:39–44. doi: 10.1016/j.jchemneu.2015.07.004
42. Ghirardini E, Wadle SL, Augustin V, Becker J, Brill S, Hammerich J, et al. Expression of functional inhibitory neurotransmitter transporters GlyT1, GAT-1, and GAT-3 by astrocytes of inferior colliculus and hippocampus. Mol brain. (2018) 11(1):4. doi: 10.1186/s13041-018-0346-y
43. Fu J, Zhang Q, Wu Z, Hong C, Zhu C. Transcriptomic analysis reveals a sex-dimorphic influence of GAT-2 on murine liver function. Front Nutr (2021) 8:751388. doi: 10.3389/fnut.2021.751388
44. Fattorini G, Melone M, Conti F. A reappraisal of GAT-1 localization in neocortex. Front Cell Neurosci (2020) 14:9. doi: 10.3389/fncel.2020.00009
45. Ashby JA, McGonigle IV, Price KL, Cohen N, Comitani F, Dougherty DA, et al. GABA binding to an insect GABA receptor: A molecular dynamics and mutagenesis study. Biophys J (2012) 103(10):2071–81. doi: 10.1016/j.bpj.2012.10.016
46. Leresche N, Lambert RC. GABA receptors and T-type Ca(2+) channels crosstalk in thalamic networks. Neuropharmacology. (2018) 136(Pt A):37–45. doi: 10.1016/j.neuropharm.2017.06.006
47. Auteri M, Zizzo MG, Serio R. GABA and GABA receptors in the gastrointestinal tract: From motility to inflammation. Pharmacol Res (2015) 93:11–21. doi: 10.1016/j.phrs.2014.12.001
48. Huang D, Wang Y, Thompson JW, Yin T, Alexander PB, Qin D, et al. Cancer-cell-derived GABA promotes β-catenin-mediated tumour growth and immunosuppression. Nat Cell Biol (2022) 24(2):230–41. doi: 10.1038/s41556-021-00820-9
49. Ganor Y, Levite M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J Neural Transm (Vienna Austria 1996). (2014) 121(8):983–1006. doi: 10.1007/s00702-014-1167-5
50. Wang YY, Sun SP, Zhu HS, Jiao XQ, Zhong K, Guo YJ, et al. GABA regulates the proliferation and apoptosis of MAC-T cells through the LPS-induced TLR4 signaling pathway. Res veterinary science. (2018) 118:395–402. doi: 10.1016/j.rvsc.2018.04.004
51. Luo Y, Kusay AS, Jiang T, Chebib M, Balle T. Delta-containing GABA(A) receptors in pain management: Promising targets for novel analgesics. Neuropharmacology. (2021) 195:108675. doi: 10.1016/j.neuropharm.2021.108675
52. Jiang SH, Zhu LL, Zhang M, Li RK, Yang Q, Yan JY, et al. GABRP regulates chemokine signalling, macrophage recruitment and tumour progression in pancreatic cancer through tuning KCNN4-mediated Ca(2+) signalling in a GABA-independent manner. Gut. (2019) 68(11):1994–2006. doi: 10.1136/gutjnl-2018-317479
53. Sparrow EL, James S, Hussain K, Beers SA, Cragg MS, Bogdanov YD. Activation of GABA(A) receptors inhibits T cell proliferation. PloS One (2021) 16(5):e0251632. doi: 10.1371/journal.pone.0251632
54. Passarelli MK, Pirkl A, Moellers R, Grinfeld D, Kollmer F, Havelund R, et al. The 3D OrbiSIMS-label-free metabolic imaging with subcellular lateral resolution and high mass-resolving power. Nat Methods (2017) 14(12):1175–83. doi: 10.1038/nmeth.4504
55. Crowley T, Cryan JF, Downer EJ, O'Leary OF. Inhibiting neuroinflammation: The role and therapeutic potential of GABA in neuro-immune interactions. Brain behavior immunity. (2016) 54:260–77. doi: 10.1016/j.bbi.2016.02.001
56. Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 pathway: From biology to cancer therapy. Int J Mol Sci (2020) 21(6):1913. doi: 10.3390/ijms21061913
57. Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of p38 MAPK in synaptic function and dysfunction. Int J Mol Sci (2020) 21(16):5624. doi: 10.3390/ijms21165624
58. Davies C, Tournier C. Exploring the function of the JNK (c-jun n-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans (2012) 40(1):85–9. doi: 10.1042/bst20110641
59. Denny WA. Inhibitors and activators of the p38 mitogen-activated MAP kinase (MAPK) family as drugs to treat cancer and inflammation. Curr Cancer Drug targets. (2022) 22(3):209–20. doi: 10.2174/1568009622666220215142837
60. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J (2011) 30(8):1536–48. doi: 10.1038/emboj.2011.69
61. Johnson MD, Reeder JE, O'Connell M. p38MAPK activation and DUSP10 expression in meningiomas. J Clin Neurosci Off J Neurosurgical Soc Australasia. (2016) 30:110–4. doi: 10.1016/j.jocn.2015.12.031
62. Li Z, Dai A, Yang M, Chen S, Deng Z, Li L. p38MAPK signaling pathway in osteoarthritis: Pathological and therapeutic aspects. J Inflammation Res (2022) 15:723–34. doi: 10.2147/jir.S348491
63. Awasthi A, Raju MB, Rahman MA. Current insights of inhibitors of p38 mitogen-activated protein kinase in inflammation. Medicinal Chem (Shariqah (United Arab Emirates)). (2021) 17(6):555–75. doi: 10.2174/1573406416666200227122849
64. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Sci (New York NY). (2016) 353(6301):766–71. doi: 10.1126/science.aag2638
65. Wang Y, Mack JA, Maytin EV. CD44 inhibits α-SMA gene expression via a novel G-actin/MRTF-mediated pathway that intersects with TGFβR/p38MAPK signaling in murine skin fibroblasts. J Biol Chem (2019) 294(34):12779–94. doi: 10.1074/jbc.RA119.007834
66. Yang Y, Lian YT, Huang SY, Yang Y, Cheng LX, Liu K. GABA and topiramate inhibit the formation of human macrophage-derived foam cells by modulating cholesterol-metabolism-associated molecules. Cell Physiol Biochem Int J Exp Cell physiology biochemistry Pharmacol (2014) 33(4):1117–29. doi: 10.1159/000358681
67. Zhang A, Lee YC. Mechanisms for joint pain in rheumatoid arthritis (RA): from cytokines to central sensitization. Curr osteoporosis Rep (2018) 16(5):603–10. doi: 10.1007/s11914-018-0473-5
68. Salaffi F, Giacobazzi G, Di Carlo M. Chronic pain in inflammatory arthritis: Mechanisms, metrology, and emerging targets-a focus on the JAK-STAT pathway. Pain Res management. (2018) 2018:8564215. doi: 10.1155/2018/8564215
69. Yam MF, Loh YC, Tan CS, Khadijah Adam S, Abdul Manan N, Basir R. General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int J Mol Sci (2018) 19(8):2164. doi: 10.3390/ijms19082164
70. Ishida M, Kuroiwa Y, Yoshida E, Sato M, Krupa D, Henry N, et al. Residual symptoms and disease burden among patients with rheumatoid arthritis in remission or low disease activity: A systematic literature review. Modern Rheumatol (2018) 28(5):789–99. doi: 10.1080/14397595.2017.1416940
71. Walsh DA, McWilliams DF. Mechanisms, impact and management of pain in rheumatoid arthritis. Nat Rev Rheumatol (2014) 10(10):581–92. doi: 10.1038/nrrheum.2014.64
72. McWilliams DF, Walsh DA. Pain mechanisms in rheumatoid arthritis. Clin Exp Rheumatol (2017) 35 Suppl 107(5):94–101.
73. Benson C, Mifflin K, Kerr B, Jesudasan SJ, Dursun S, Baker G. Biogenic amines and the amino acids GABA and glutamate: Relationships with pain and depression. Modern Trends pharmacopsychiatry. (2015) 30:67–79. doi: 10.1159/000435933
74. Fu Q, Shi D, Zhou Y, Zheng H, Xiang H, Tian X, et al. MHC-I promotes apoptosis of GABAergic interneurons in the spinal dorsal horn and contributes to cancer induced bone pain. Exp neurology. (2016) 286:12–20. doi: 10.1016/j.expneurol.2016.09.002
75. Lorenzo LE, Magnussen C, Bailey AL, St Louis M, De Koninck Y, Ribeiro-da-Silva A. Spatial and temporal pattern of changes in the number of GAD65-immunoreactive inhibitory terminals in the rat superficial dorsal horn following peripheral nerve injury. Mol pain. (2014) 10:57. doi: 10.1186/1744-8069-10-57
76. Allen NJ, Káradóttir R, Attwell D. Reversal or reduction of glutamate and GABA transport in CNS pathology and therapy. Pflugers Archiv Eur J Physiol (2004) 449(2):132–42. doi: 10.1007/s00424-004-1318-x
77. Kannampalli P, Sengupta JN. Role of principal ionotropic and metabotropic receptors in visceral pain. J Neurogastroenterol motility. (2015) 21(2):147–58. doi: 10.5056/jnm15026
78. Munro G, Hansen RR, Mirza NR. GABA(A) receptor modulation: Potential to deliver novel pain medicines? Eur J Pharmacol (2013) 716(1-3):17–23. doi: 10.1016/j.ejphar.2013.01.070
79. Kniazeff J. The different aspects of the GABA(B) receptor allosteric modulation. Adv Pharmacol (San Diego Calif). (2020) 88:83–113. doi: 10.1016/bs.apha.2020.02.003
80. Frangaj A, Fan QR. Structural biology of GABA(B) receptor. Neuropharmacology. (2018) 136(Pt A):68–79. doi: 10.1016/j.neuropharm.2017.10.011
81. Enoch MA, Hodgkinson CA, Shen PH, Gorodetsky E, Marietta CA, Roy A, et al. GABBR1 and SLC6A1, two genes involved in modulation of GABA synaptic transmission, influence risk for alcoholism: Results from three ethnically diverse populations. Alcoholism Clin Exp Res (2016) 40(1):93–101. doi: 10.1111/acer.12929
82. Burfoot RK, Jensen CJ, Field J, Stankovich J, Varney MD, Johnson LJ, et al. SNP mapping and candidate gene sequencing in the class I region of the HLA complex: Searching for multiple sclerosis susceptibility genes in tasmanians. Tissue Antigens (2008) 71(1):42–50. doi: 10.1111/j.1399-0039.2007.00962.x
83. Zhao X, Qin S, Shi Y, Zhang A, Zhang J, Bian L, et al. Systematic study of association of four GABAergic genes: Glutamic acid decarboxylase 1 gene, glutamic acid decarboxylase 2 gene, GABA(B) receptor 1 gene and GABA(A) receptor subunit beta2 gene, with schizophrenia using a universal DNA microarray. Schizophr Res (2007) 93(1-3):374–84. doi: 10.1016/j.schres.2007.02.023
84. Wu J, Xu Y, Pu S, Jiang W, Du D. p38/MAPK inhibitor modulates the expression of dorsal horn GABA(B) receptors in the spinal nerve ligation model of neuropathic pain. Neuroimmunomodulation. (2011) 18(3):150–5. doi: 10.1159/000323141
85. Li WC, Bai L, Xu Y, Chen H, Ma R, Hou WB, et al. Identification of differentially expressed genes in synovial tissue of rheumatoid arthritis and osteoarthritis in patients. J Cell Biochem (2019) 120(3):4533–44. doi: 10.1002/jcb.27741
86. Zhao J, Guo S, Schrodi SJ, He D. Molecular and cellular heterogeneity in rheumatoid arthritis: Mechanisms and clinical implications. Front Immunol (2021) 12:790122. doi: 10.3389/fimmu.2021.790122
87. Hernandez-Rabaza V, Cabrera-Pastor A, Taoro-Gonzalez L, Gonzalez-Usano A, Agusti A, Balzano T, et al. Neuroinflammation increases GABAergic tone and impairs cognitive and motor function in hyperammonemia by increasing GAT-3 membrane expression. reversal by sulforaphane by promoting M2 polarization of microglia. J neuroinflammation. (2016) 13(1):83. doi: 10.1186/s12974-016-0549-z
88. Gao D, Yu H, Li B, Chen L, Li X, Gu W. Cisplatin toxicology: The role of pro-inflammatory cytokines and GABA transporters in cochlear spiral ganglion. Curr Pharm design. (2019) 25(45):4820–6. doi: 10.2174/1381612825666191106143743
89. Kinjo A, Sassa M, Koito T, Suzuki M, Inoue K. Functional characterization of the GABA transporter GAT-1 from the deep-sea mussel bathymodiolus septemdierum. Comp Biochem Physiol Part A Mol Integr Physiol (2019) 227:1–7. doi: 10.1016/j.cbpa.2018.08.016
90. Serrats J, Sawchenko PE. CNS activational responses to staphylococcal enterotoxin b: T-lymphocyte-dependent immune challenge effects on stress-related circuitry. J Comp neurology. (2006) 495(2):236–54. doi: 10.1002/cne.20872
91. Ding X, Chang Y, Wang S, Yan D, Yao J, Zhu G. Transcriptomic analysis of the effect of GAT-2 deficiency on differentiation of mice naïve T cells into Th1 cells in vitro. Front Immunol (2021) 12:667136. doi: 10.3389/fimmu.2021.667136
92. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol (2018) 14(4):214–28. doi: 10.1038/nrrheum.2018.31
93. Paul AM, Branton WG, Walsh JG, Polyak MJ, Lu JQ, Baker GB, et al. GABA transport and neuroinflammation are coupled in multiple sclerosis: Regulation of the GABA transporter-2 by ganaxolone. Neuroscience. (2014) 273:24–38. doi: 10.1016/j.neuroscience.2014.04.037
94. Ma Y, Zhang X, Fan D, Xia Q, Wang M, Pan F. Common trace metals in rheumatoid arthritis: A systematic review and meta-analysis. J Trace elements Med Biol Organ Soc Minerals Trace Elements (GMS). (2019) 56:81–9. doi: 10.1016/j.jtemb.2019.07.007
95. Chakraborty M, Chutia H, Changkakati R. Serum copper as a marker of disease activity in rheumatoid arthritis. J Clin Diagn Res JCDR. (2015) 9(12):Bc09–11. doi: 10.7860/jcdr/2015/14851.7001
96. Nakanishi T, Fukuyama Y, Fujita M, Shirasaka Y, Tamai I. Carnitine precursor γ-butyrobetaine is a novel substrate of the na(+)- and cl(-)-dependent GABA transporter Gat2. Drug Metab pharmacokinetics. (2011) 26(6):632–6. doi: 10.2133/dmpk.dmpk-11-nt-053
97. Wu C, Qin X, Du H, Li N, Ren W, Peng Y. The immunological function of GABAergic system. Front bioscience (Landmark edition). (2017) 22(7):1162–72. doi: 10.2741/4539
98. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
99. Jin Z, Mendu SK, Birnir B. GABA is an effective immunomodulatory molecule. Amino Acids (2013) 45(1):87–94. doi: 10.1007/s00726-011-1193-7
100. Wang XD, Wang YL, Gao WF. Honokiol possesses potential anti-inflammatory effects on rheumatoid arthritis and GM-CSF can be a target for its treatment. Int J Clin Exp pathology. (2015) 8(7):7929–36.
101. Ilonen J, Lempainen J, Veijola R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat Rev Endocrinology. (2019) 15(11):635–50. doi: 10.1038/s41574-019-0254-y
102. Mirza NR, Munro G. The role of GABA(A) receptor subtypes as analgesic targets. Drug News perspectives. (2010) 23(6):351–60. doi: 10.1358/dnp.2010.23.6.1489909
103. Zhou YQ, Chen SP, Liu DQ, Manyande A, Zhang W, Yang SB, et al. The role of spinal GABAB receptors in cancer-induced bone pain in rats. J pain. (2017) 18(8):933–46. doi: 10.1016/j.jpain.2017.02.438
104. Malcangio M. GABA(B) receptors and pain. Neuropharmacology. (2018) 136(Pt A):102–5. doi: 10.1016/j.neuropharm.2017.05.012
105. Bráz JM, Wang X, Guan Z, Rubenstein JL, Basbaum AI. Transplant-mediated enhancement of spinal cord GABAergic inhibition reverses paclitaxel-induced mechanical and heat hypersensitivity. Pain. (2015) 156(6):1084–91. doi: 10.1097/j.pain.0000000000000152
106. Gryzło B, Zaręba P, Malawska K, Mazur G, Rapacz A, Ła Tka K, et al. Novel functionalized amino acids as inhibitors of GABA transporters with analgesic activity. ACS Chem Neurosci (2021) 12(16):3073–100. doi: 10.1021/acschemneuro.1c00351
Keywords: rheumatoid arthritis, gamma-aminobutyric acid(GABA)ergic, GABA relatedreceptors, GABA transporter (GAT) systems, inflammation
Citation: Shan Y, Zhao J, Zheng Y, Guo S, Schrodi SJ and He D (2023) Understanding the function of the GABAergic system and its potential role in rheumatoid arthritis. Front. Immunol. 14:1114350. doi: 10.3389/fimmu.2023.1114350
Received: 02 December 2022; Accepted: 26 January 2023;
Published: 07 February 2023.
Edited by:
Haitao Wang, National Cancer Institute (NIH), United StatesReviewed by:
Zhiyong Liu, The Rockefeller University, United StatesCopyright © 2023 Shan, Zhao, Zheng, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Steven J. Schrodi, U2Nocm9kaUB3aXNjLmVkdQ==; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.