
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 08 March 2023
Sec. Viral Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1112505
This article is part of the Research TopicNew Therapeutic Approaches for SARS-CoV-2/COVID-19View all 17 articles
 Latifa Zekri1,2,3,4*
Latifa Zekri1,2,3,4* Natalia Ruetalo5
Natalia Ruetalo5 Mary Christie6Carolin Walker1,2Timo Manz1,2
Mary Christie6Carolin Walker1,2Timo Manz1,2 Hans-Georg Rammensee1,2,4
Hans-Georg Rammensee1,2,4 Helmut R. Salih2,3,4
Helmut R. Salih2,3,4 Michael Schindler5†Gundram Jung1,2,4†
Michael Schindler5†Gundram Jung1,2,4†Despite the successful development of vaccines and neutralizing antibodies to limit the spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerging variants prolong the pandemic and emphasize the persistent need to develop effective antiviral treatment regimens. Recombinant antibodies directed to the original SARS-CoV-2 have been successfully used to treat established viral disease. However, emerging viral variants escape the recognition by those antibodies. Here we report the engineering of an optimized ACE2 fusion protein, designated ACE2-M, which comprises a human IgG1 Fc domain with abrogated Fc-receptor binding linked to a catalytically-inactive ACE2 extracellular domain that displays increased apparent affinity to the B.1 spike protein. The affinity and neutralization capacity of ACE2-M is unaffected or even enhanced by mutations present in the spike protein of viral variants. In contrast, a recombinant neutralizing reference antibody, as well as antibodies present in the sera of vaccinated individuals, lose activity against such variants. With its potential to resist viral immune escape ACE2-M appears to be particularly valuable in the context of pandemic preparedness towards newly emerging coronaviruses.
In the past two years, the Coronavirus disease 2019 (COVID-19) pandemic has claimed several millions of lives worldwide and has caused enormous -and unprecedented- social and economic damage (1, 2). Fortunately -and unprecedented as well- efficient vaccines have been developed and administered to millions of individuals in less than two years, and currently it appears that vaccination has become the cornerstone for the control of the pandemic worldwide (1, 3).
In the face of this truly remarkable success, the development of reagents for the treatment of established viral infections remains challenging. A growing understanding and appropriate treatment of the hyper-inflammatory and -coagulatory states occurring in the course of moderate and severe disease resulted in a significant reduction in mortality rates in treated patients. In addition, reagents with direct antiviral activity have been developed. Such reagents can be divided into two classes, small molecules with antiviral activity and neutralizing antiviral antibodies. For the latter, the tools of modern recombinant antibody technology, i. e., phage display, and single-cell cloning, have been used to generate optimized monoclonal antibodies with potent neutralizing capacity, directed to the receptor-binding domain (RBD) of the viral spike protein (S-protein) that binds to the ACE2 receptor on target cells (4–8). Several of these reagents have received approval for use during the early stages of infection. As of today, however, their activity in more advanced stages has been limited. Indeed, antibody-dependent enhancement (ADE), e.g., by non-neutralizing antibodies binding to viral particles, was reported to promote their Fc-mediated uptake by cells carrying Fc-receptors (FcRs), such as alveolar macrophages (9, 10).
However, a major limitation for the therapeutic activity of antibodies are recent mutations in SARS-CoV-2 variants that not only confer enhanced affinity to ACE2 and thus increased infectivity but also prevent the binding of antibodies raised against the B.1 S-protein (11–13).
A recombinant antibody approved for treatment of limited disease, REGN 10933 (14) exemplifies this strikingly. It strongly binds to the RBD of the B.1 S-protein but fails to bind to the S-protein encoded by known variants of concern (VOCs), such as the Beta and Omicron variants. The latter escapes effective neutralization by five of seven mAbs approved for treatment of COVID-19 (15–17). At the same time, the S-protein of the Omicron variant gained affinity towards the ACE2 protein (18, 19), resulting in increased infectivity.
In principle, the conceptual weakness of neutralizing antibodies directed to the RBD domain of the S-protein discussed above might be overcome by recombinant Fc-based fusion proteins comprising the “natural” binding partner of the RBD domain, the ACE2 protein. In contrast to RBD binding antibodies, the neutralizing capacity of such proteins would not be impaired but rather strengthened by affinity gaining mutations in the RBD. Moreover, since the RBD ACE2 interaction is mediated by a dimeric form of ACE2, an Fc based format may promote ACE2 dimerization (20). Despite this conceptual advantage, the construction of such fusion proteins faces challenges as well: first, the affinity of recombinant ACE2 to viral S-proteins is lower than that of most antibodies. Second, the enzymatic activity of physiologically expressed ACE2 is critical for the proper function of the renin-angiotensin-aldosterone system (RAAS). This system is of vital importance, among others, for blood pressure regulation, and high doses of enzymatically active protein might induce uncontrollable side effects. Although it has been suggested that ACE2 may function as a “rescue protein” in the course of the SARS-CoV-2 infection (21), we share the view expressed in a paper Khodarahmi et al. (22), that recommend the use of enzymatically inactive ACE2 if blockade of the S-protein is intended.
Based on the considerations outlined above, we have constructed and characterized an ACE2-Fc fusion protein designated ACE2-M, that carries mutations to:
i. abrogate FcR binding and complement activation by the Fc domain
ii. deplete enzymatic activity of the ACE2 protein
iii. enhance the apparent affinity of the ACE2 S-protein interaction
Here we evaluate the capability of ACE2-M to bind and to neutralize various virus variants. This activity was benchmarked against the therapeutic antibody REGN 10933 and the serum of vaccinated individuals.
The human ACE2 extracellular domain (aa 18-740; Gene ID: 59272) and the variable domain sequences of the REGN 10933 antibody (14) were codon-optimized for expression by Chinese hamster cells using the GeneArt GeneOptimizer tool (Thermo Fisher Scientific, Regensburg, Germany). VH, VL, and ACE2 sequences (ACE2 wild-type (ACE2) or the indicated mutants, ACE2-RR, ACE2-K, ACE2-M) were synthesized de novo at GeneArt (Thermo Fisher Scientific, Regensburg, Germany). ACE-2 coding sequences were fused at their C-terminus to a human Igγ1 Hinge- Fc domain via a flexible (GGGGS)3 linker. Modifications in the CH2 domain consisting of the amino acid substitutions and deletions E233P; L234V; L235A; ΔG236; D265G; A327Q; A330S (EU index), which abrogate FcR binding and complement fixation, were introduced as described (23). REGN 10933 variable sequences were inserted into a human Igγ1 backbone comprising CH1‐CH2‐CH3‐ or CΚ‐constant domain sequences as described (23). All constructs were transiently transfected and produced using the ExpiCHO™ Expression System (Thermo Fisher Scientific, Regensburg, Germany) according to the manufacturer’s instructions and were then purified by HiTrap™ MabSelect™ SuRe columns (Cytiva, Freiburg, Germany), before being subjected to preparative and analytical size exclusion chromatography (SEC) using HiLoad™ 16/600 Superdex 200 pg and Superdex™ 200 Increase 10/300 GL columns (Cytiva Freiburg, Germany), respectively. Endotoxin levels of samples, as determined by a limulus amebocyte lysate assay (Endosafe®Charles River, Charleston, SC), were < 0.5 EU/ml. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as previously described (23).
SARS-CoV-2 full-length trimeric spike proteins corresponding to the B.1 (B.1.126), Alpha (B.1.1.7), Beta (B1.351), Gamma (P.1), and Delta (B.1.617.2) were purchased from BioServ, (Sheffield, UK). The Trimeric S-protein corresponding to Omicron (B.1.1.529) or Omicron subvariant BA.5 and BQ1.1 were obtained from Sino Biological (Beijing, China).
Enzymatic activity of ACE2 fusion proteins was measured using the ACE2 Activity Assay Kit (Fluorometric) (BioVision, Milpitas, CA) according to the manufacturer’s instructions. The proteins were diluted in assay buffer to 22.7, 4.54, and 0.91 nM final concentration. Fluorescence was measured using a Wallac 1420 Victor 2 Multi-Label Counter (Perkin-Elmer, Waltham, MA).
The indicated S trimeric proteins were coated on 96-well plates at 1µg/ml, 4°C overnight. After washing, wells were blocked with PBS containing 3% BSA for 1 hour at room temperature. Next, a serial dilution of the indicated ACE2 fusion proteins, REGN 10933 or serum antibodies were pre-mixed with 150 nM of His-tagged ACE2 wild-type protein (BioLegend, San Diego, CA) and added to the plates. In case of the Omicron variants (Sino Biological), the His-tagged ACE2 protein was additionally biotinylated using the One-Step Biotinylation Kit (Miltenyi, Cologne, Germany) according to the manufacturer’s instructions. For visualization, a Penta-His HRP conjugate (1:1000) (Qiagen, Hilden, Germany) or mouse anti-Biotin HRP conjugate (1:1000) (Invitrogen, Waltham, MA) were used. Unbound HRP-conjugated antibodies were removed by washing, TMB substrate was added, and absorbance was measured at 450nm.
Sera were collected from 8 healthy donors (25-65 years of age). All donors received a first dose of Vaxzevria vaccine and then a second dose of Comirnaty or Spikevax. Sera were collected 40-45 days after the second dose and anti-SARS-CoV-2 antibody concentrations were measured using the Euroimmun Anti-SARS CoV-2 ELISA IgG kit (Euroimmun, Luebeck, Germany). Briefly, serum samples were diluted at 1:100 and 1:1000 and ELISA was performed following manufacturer’s instructions. A titration of a reference neutralizing antibody (REGN 10933) was used to calibrate the assay.
Trimeric S-proteins were analyzed for their binding to ACE2-RR or ACE2-M fusion proteins using an Octet HTX system (Sartorius, Goettingen, Germany). Assays were run with a sensor offset of 3 mm and an acquisition rate of 5 Hz on AHC biosensors in 16-channel mode. Microplates were loaded with 60 μL per well of assay buffer consisting of PBS with 0.05% Tween-20 and 0.1% BSA. Sensors were equilibrated in assay buffer for 10 min. Following a baseline step of 60s, the analyte S-proteins were loaded for 120 s. Association was measured for 150s and dissociation for 300s. Regeneration of the sensors was performed using 10 mM Glycine pH 1.5. Data evaluation was done using Octet Analysis HT Software. The reference subtraction was performed to consider the potential dissociation of analyte loaded onto the biosensor. Data traces were aligned to the baseline, followed by an inter-step correction for the dissociation step. Savitzky-Golay filtering was applied to the data and the curves were fitted globally using a 1:1 binding model (with Rmax unlinked by sensor).
All experiments with SARS-CoV-2 viruses were conducted in a Biosafety Level 3 laboratory at the University Hospital Tübingen. The SARS-CoV-2 strain icSARS-CoV-2-mNG (24) was obtained from the World Center for Emerging Viruses and Arboviruses (WRCEVA) of the UTMB (University of Texas Medical Branch, Galveston, TX, USA). SARS-CoV-2 B.1.126 (parental D614G), referred as B.1, and SARS-CoV-2 B.1.351 (Beta), were isolated from patient samples and variant identity was confirmed by next-generation sequencing of the entire viral genome as described before (25, 26). SARS-CoV-2 B.1.1.529 (Omicron) was isolated from a throat swab collected in December 2021 at the Institute for Medical Virology and Epidemiology of Viral Diseases, University Hospital Tübingen, from a PCR-positive patient. Fifty microliters of patient material were diluted in medium and used directly to inoculate 150,000 Caco-2 cells in a six-well plate. 48 hours post-infection (hpi), the supernatant was collected, centrifuged, and stored at -80°C. After two consecutive passages, an RNA sample from the supernatant was prepared, and NGS confirmed that the clinical isolate belongs to the lineage B.1.1.529. All virus stocks were generated in Caco-2 cells collecting supernatants 48-72 hpi. Multiplicity of infection determination (MOI) was conducted by titration using serial dilutions of both virus stocks. The number of infectious virus particles per ml was calculated as (MOI × cell number)/(infection volume), where MOI = -ln (1-infection rate).
Caco-2 (Human Colorectal adenocarcinoma, ATCC HTB-37) cells were cultured at 37°C with 5% CO2 in DMEM containing 10% FCS, 2 mM L-glutamine, 100 mg/ml penicillin-streptomycin and 1% NEAA. Neutralization assays using clinical isolates were performed as described in Wagner et al., 2021. Briefly, cells were co-incubated with the clinical isolate SARS-CoV-2 B.1.126, SARS-CoV-2 B.1.351 (Beta), or SARS-CoV-2 B.1.1.529 (Omicron), at MOI of 0.7-4.0, and serial dilutions of the ACE2 protein designs. 48 hpi, cells were fixed with 80% acetone, and immunofluorescence (IF) staining was performed using an anti-SARS-CoV-2 nucleocapsid antibody (GeneTex, Cat No. GTX135357) and goat anti-rabbit Alexa594-conjugated secondary antibody. Cells were counterstained with DAPI solution and images were taken with the Cytation3 (BioTek). Infection rates were calculated as the ratio of Alexa594-positive over DAPI-positive cells, which were automatically counted by the Gen5 software (BioTek). Inhibitory concentration 50 (IC50) was calculated as the half-maximal inhibitory dose using four-parameter nonlinear regression (GraphPad Prism).
For binding experiments, 3×106 Caco-2 cells were seeded in a T75 flask the day before infection, in a medium containing 5% FCS. Cells were infected with SARS-CoV-2-mNG, and 48 hpi cells were detached from the flask using Accutase, fixed with 2% PFA for 10 minutes at 37°C, and resuspended in FACS buffer (PBS, 1%FCS). 1×106 cells in 100 µL in FACS buffer were distributed in a U-shape 96-well plate. The plate was centrifuged at 600 g for 5 min and the buffer was removed by a fast decant. Cells were incubated for 1h at 4°C using 50 µl of 3-fold serial dilutions of ACE2 protein or REGN 10933, tested from 40 µg/ml following 12 dilution points. Cells were washed with 150 µl of FACS buffer/well, centrifuged, and the supernatant decanted. The washing step was repeated using 200 µl of FACS buffer/well. Subsequently, cells were incubated with 50 µl of a 1:200 dilution of the Secondary AB- R-Phycoerythrin (PE) conjugated affinity pure F(ab’)2 Fragment Goat-anti Human IgG-Fc gamma fragment (Jackson-Immuno) for 30 minutes at 4°C. The two washing steps were repeated, and the cells were resuspended using 100 µl of FACS buffer/well. Controls included: mock-infected cells incubated with the highest and lower protein concentrations; infected cells non-incubated as well as infected cells stained only with the secondary antibody. Alternatively, Caco-2 cells were infected with SARS-CoV-2 parental or Omicron variants and the same protocol described above was followed. After incubation with PE-secondary antibody, cells were permeabilized with 80% Acetone for 5 minutes at room temperature, washed as described before and immunofluorescence (IF) staining was performed using an anti-SARS-CoV-2 nucleocapsid antibody (GeneTex, Cat No. GTX135357) (1:1000, 1 h) and goat anti-rabbit Alexa594-conjugated secondary antibody (1:2000, 1 h). After the final washing steps, the cells were analyzed using a MACSQuant VYB (Miltenyi). FACS analysis was performed with MACS Quantify Software (Miltenyi) and Flowlogic (Miltenyi–Inivai).
ACE2 fusion proteins were constructed by fusing the ACE2 extracellular domain to a human IgG1 Fc fragment that was originally developed to prevent binding to FcRs by T cell activating bispecific antibodies. This modification consists of 6 mutations and one deletion in the CH2 domain of the human IgG1 Fc domain that completely abrogates binding to FcRs and ablates complement activation (23).
To avoid the introduction of undesired peptidase activity, two catalytically-inactive forms of ACE2 were generated. The first mutant, ACE2-RR, contains two substitutions in the catalytic pocket, H374R, and H378R, that prevent zinc binding within the active site of the protein (27). The second mutant, ACE2-K, has a single mutation at position 273 (R273K), which is critical for enzymatic activity (28). Fc-fusion proteins constructed with either of the two variants show the size expected for a dimeric molecule as demonstrated by SDS-PAGE and size exclusion chromatography (Supplementary Figures 1A–C). In contrast to ACE2-RR, ACE2-K retained some residual enzymatic activity and showed a slight loss in binding affinity to recombinant trimeric parental S-protein as measured by enzyme-linked immunosorbent assay (ELISA) (Supplementary Figures 1D, E). Thus, the ACE2-RR version of the fusion protein was selected for further modification.
To enhance binding to the S-protein, we introduced three additional mutations to the ACE2-RR-Fc fusion protein that are located at the S-protein binding surface of ACE2: T27Y, L79Y, and N330Y (Figures 1A, B). These mutations are similar to an ACE2 variant described by Chan and collaborators (29). ACE2-M, was produced as a stable dimer and had no detectable enzymatic activity (Figures 1C–E).
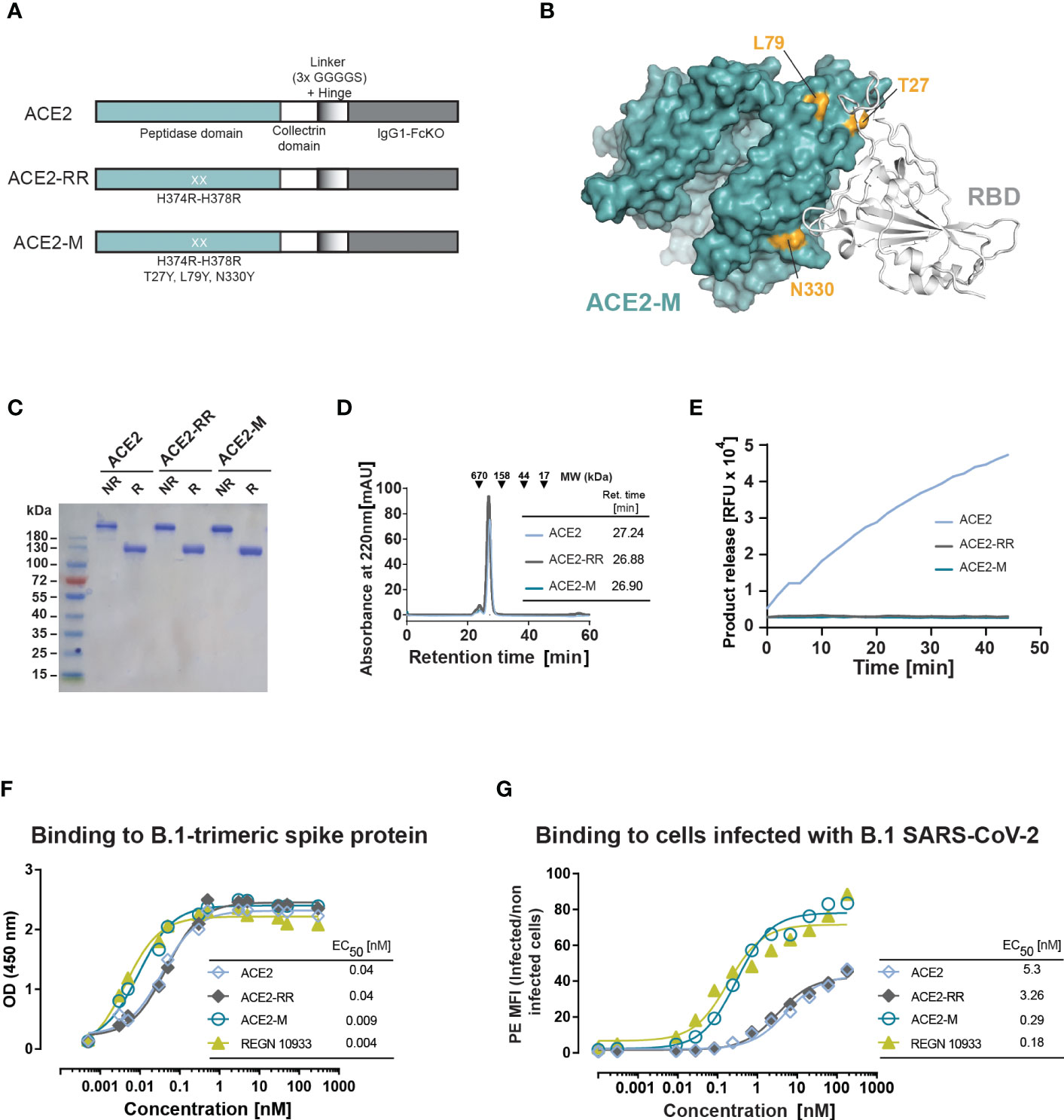
Figure 1 Design and biochemical characterization of ACE2-M. (A) Schematic representation of the ACE2 molecules used in this study. The ACE2 extracellular domain, comprising peptidase and collectrin domains, was fused to an IgG1-Hinge-Fc-Ko domain via a flexible linker (GGGGS)3 (ACE2 version). The H374R-H378R mutations were introduced to abolish ACE2 enzymatic activity (ACE2-RR version). Additional mutations were introduced to increase binding to the S-protein as indicated in (A, B). (B) Structure of ACE2 in complex with S-RBD (using PDB 6M0J) with residues substituted in ACE2-M highlighted in gold. (C) Coomassie-stained gel of the three generated fusion proteins. NR, non-reduced; R, reduced. (D) Superposed analytic chromatography profiles of the ACE2 proteins run on a Superdex S200 Increase 10/300GL column. The table indicates the corresponding retention times. (E) Enzymatic activity of the ACE2 fusion proteins (4.5 nM) as measured by cleavage of a fluorescent peptide substrate. (F) Binding of the fusion proteins and REGN 10933 to B.1 trimeric S-protein determined by ELISA. Results represent the standard deviation (SD) of n=3. EC50 as calculated by the GraphPad software. (G) Binding of the fusion proteins and REGN 10933 to Caco-2 cells infected with the recombinant infectious clone SARS-CoV-2 that expresses the mNeonGreen as a reporter gene. Binding was assessed by flow cytometry.
We next measured the binding affinity of recombinant trimeric S-protein and ACE2-M by ELISA, which was benchmarked against an Fc-fusion protein comprising the wild type ACE2 protein, as well as against the reference antibody REGN10933. In this assay, ACE2-M showed a 4-fold higher binding affinity than wild-type ACE2 fusion protein (Figure 1F). Interestingly, the apparent binding affinity measured by flow cytometry of Caco-2 cells infected with a recombinant parental virus resulted in a greater than 18-fold affinity enhancement (Figure 1G). In both assays, the S-protein apparent binding affinity of ACE2-M and REGN 10933 was similar. The differences between the affinities of antibodies and fusion proteins to S-proteins coated to an ELISA plate compared with those embedded in the membrane of a virus-infected cell are noteworthy. Obviously, binding to infected cells resembles the “physiological state” more closely, and may predict different neutralization capacities of the various fusion proteins more reliably as demonstrated below.
To test whether ACE2-M retains binding to the globally spreading SARS-CoV-2 VOCs Alpha (B.1.1.7), Beta (B1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529), structural analysis of ACE2-M and the respective S-RBD variants was performed (Supplementary Figure 2). This analysis revealed that the affinity-enhancing mutations introduced into the ACE2-M protein appear to be spared by the viral escape mechanism, probably because they form part of the RBD-binding interface. Those observations were further confirmed by measuring the binding kinetics of ACE2-M to all variants by biolayer interferometry technology (BLI). The results depicted in Supplementary Figure 3 demonstrate a superior apparent binding affinity of ACE2-M to spike variants. Again, the differences in affinities were less pronounced compared to those observed by flow cytometry of infected cells (Figure 1G).
Next, we tested the ability of ACE2-RR, ACE2-M, and REGN 10933 to inhibit binding of ACE2 to various S-proteins by competition ELISA. To this end, the fusion proteins and REGN 10933 were mixed with a saturating amount of His-tagged-ACE2 protein before binding to immobilized S-proteins was determined. In line with the BLI results, we observed an improved inhibition of binding by ACE2-M to all S-proteins (Figure 2A). In contrast, REGN 10933 failed to inhibit ACE2 binding to Beta, Gamma, and Omicron S-proteins. These results are consistent with previously published data, suggesting that the reduced binding of REGN 10933 to certain S variants is likely due to mutations in amino acids K417 and E484 of the S-protein (30, 31), found in the Beta, Gamma, and Omicron variants.
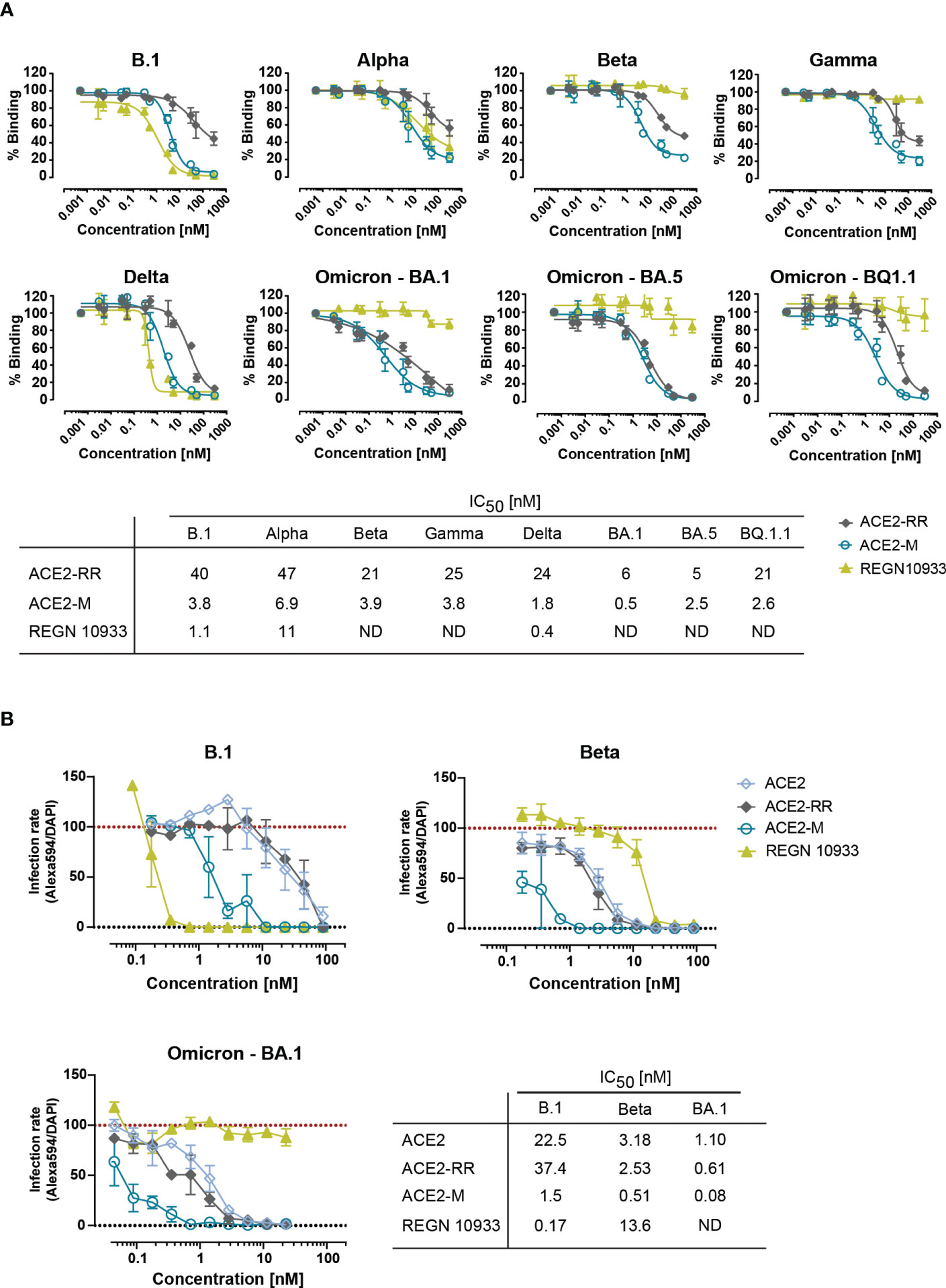
Figure 2 Competitive binding of fusion proteins and REGN 10933 to ACE2 and their capacity to neutralize parental and SARS-CoV-2 VOCs. (A) Competitive ELISA was performed by immobilizing the indicated full length S-proteins. The results depicted show the binding of 150 nM of His-tagged ACE2 wild-type protein, that was competed with the indicated concentrations of the ACE2 fusion molecules or REGN 10933 antibody. The error bars represent the SD of two independent experiments with technical replicates. The IC50 values summarized in the table were calculated using the GraphPad software. ND: Not detected. (B) Neutralization capacity was determined as described in material and methods. Briefly, Caco-2 cells were infected with the indicated clinical isolates of SARS-CoV-2. Infection rates, calculated as the number of infected cells (Alexa594+) over the total number of cells (DAPI+), were normalized to virus-only infection control. Mean and SEM values are calculated from three independent experiments with technical duplicates. Neutralization assay corresponding IC50 values were calculated using GraphPad software. ND, Not detectable.
Likewise, binding of ACE2 fusion proteins and REGN 10933 antibody to Caco-2 cells infected with authentic SARS-CoV-2 natural isolates corresponding to the parental strain (D614G) and Omicron (B.1.1.529) showed similar results (Supplementary Figure 4). REGN 10933 bound with high affinity to Caco-2 cells infected with the parental virus, in line with the results obtained with the recombinant virus in Figure 1G. However, REGN 10933 failed to bind to Caco-2 cells infected with the Omicron virus. In contrast, ACE2-M retains high-affinity binding to Caco-2 cells infected with either isolate. Unexpectedly, we observed a 4-fold lower MFI for binding to cells infected with Omicron vs. parental virus. This observation could be explained by the reduced replication capacity recently described for Omicron variant (32–34). The reduced replication seems to be due to the inefficient use of the cellular protease TMPRSS2, which promotes cell entry through plasma membrane fusion (33). We speculate that reduced replication in Caco-2 cells may result in a decreased expression of S-proteins on the cell surface.
To investigate the efficacy of ACE2-M in preventing viral infection, we performed a neutralization assay in Caco-2 cells using SARS-CoV-2 natural isolates from the parental B.1 as well as Beta, and Omicron strains. ACE2-M neutralized all SARS-CoV-2 infected cells at picomolar concentrations (Figure 2B and Supplementary Figure 5). ACE2-M activity was greater against Omicron than against the Beta variant or the parental B.1. In contrast, the activity of REGN 10933 against the Beta and Omicron variants was reduced and undetectable, respectively. Altogether, our results demonstrate that binding of ACE2-M to S-variants (vs. parental) is preserved or enhanced (in case of Omicron), while it is weakened or lost in the case of REGN 10933. Thus, these results confirm our “founding hypothesis” for the construction of ACE2 fusion proteins, namely that viral variants will mutate “away” from recognition by antibodies but “towards” recognition by ACE2 fusion proteins and hence to neutralization by such proteins.
To evaluate the binding efficiency of antibodies generated by active immunization to parental, Beta, Delta, and Omicron trimeric S-proteins, sera from 8 healthy fully vaccinated donors were used and evaluated in a competitive ELISA similar to that described above in Figure 2A. Similar to REGN 10933, antibodies present in the post-vaccination sera showed a reduction in binding to Beta, Delta, and, to a greater extent, to Omicron S-proteins (Figure 3A). These results suggest that antibodies generated after active immunization against the parental S-protein are less effective against new variants in accordance with recent reports (35–40).
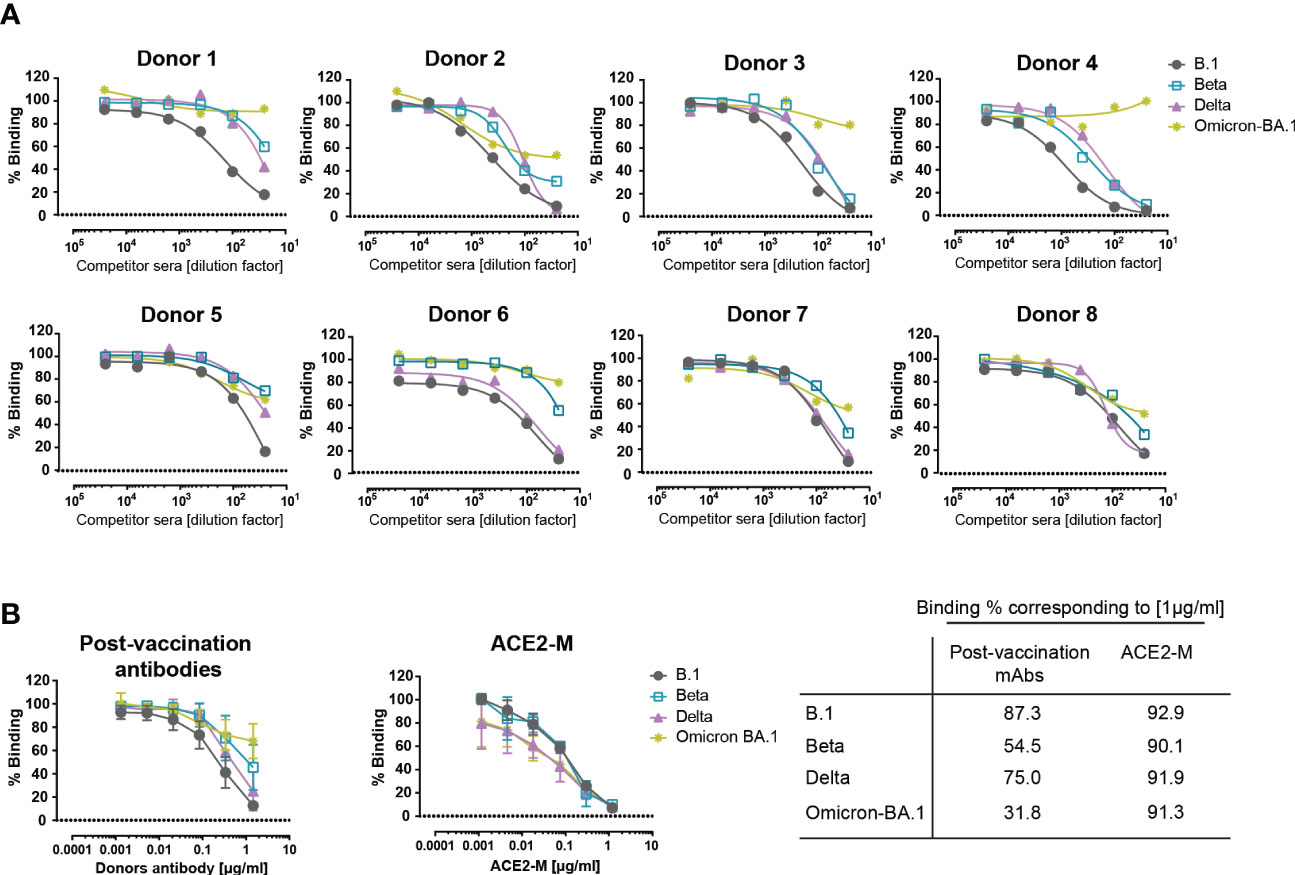
Figure 3 Competitive binding of antibodies in the sera of vaccinated donors and ACE2-M to various spike proteins. (A) Similar competition ELISA assay to the one described in Figure 1A was performed with sera from 8 healthy vaccinated individuals. Each serum sample was diluted in a range of 1/25 to 1/25600 and tested against His-tagged ACE2 for binding to SARS-CoV-2 B.1, Beta, Delta, and Omicron – BA.1 S-protein variants. (B) SARS-CoV-2 antibodies were quantified in donor sera using the Euroimmun ELISA. Equal amounts of antibody or ACE2-M were diluted in SARS-CoV-2 negative serum and tested in a competition ELISA for their ability to compete with wild-type ACE2-His for S-protein binding. The table summarizes the percentage binding achieved by a protein concentration of 1µg/ml.
Next, we defined the required amount of neutralizing antibodies and ACE2-M protein, to achieve a complete inhibition of binding to S-proteins. To this end, SARS-CoV-2 antibody concentrations present in the sera of vaccinated donors were quantified, adjusted, and compared in a competitive ELISA to the ACE2-M protein. Our results, depicted in Figure 3B, show that ACE2-M, at a concentration of 1 µg/ml, achieved almost complete binding inhibition of ACE2 wild type to all S-variants, however, only a partial binding inhibition could be obtained with antibodies in the various sera. Of note, a serum concentration of 1 µg/ml is easily reached during treatment of antiviral disease with suitable monoclonal antibodies (41, 42).
We have demonstrated that ACE2 fusion proteins are not subject to immune escape exerted by variants of the S-protein in line with recent publications (43–46). In fact, variants with increased affinity such as Omicron variants (BA.1, BA.5 and BQ1.1), are neutralized more effectively than the parental B.1. Recently, alternative receptors have been reported to interact with the SARS-CoV-2 virus (47–51). In cells with low ACE2 expression, it is thought that SARS-CoV-2 can enter the cells via several alternative receptors, but the entry mechanism remains to be defined. Although there is no direct evidence that SARS-CoV-2 escapes ACE2 treatment, the fact that the highly transmissible variant Omicron BA.1 has evolved to be less dependent on TMPRSS2, raises the possibility that alternative SARS-CoV-2 mutations may also contribute to viral evolution and may cause ACE2 immune escape.
To our knowledge, ACE2-M is the first engineered ACE2 fusion molecule combining modifications for Fc-attenuation, enzymatic depletion, and ACE2 affinity enhancement. Although animal studies are required to conclusively evaluate the importance of enzymatic depletion and Fc-attenuation, this molecule provides an important additional option for treatment of COVID-19 and other coronaviruses that use the ACE2 protein as entry receptor.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by ethics committee of the Faculty of Medicine of the Eberhard Karls Universitaet Tuebingen and of the University Hospital Tuebingen. The patients/participants provided their written informed consent to participate in this study.
GJ and LZ designed the study. LZ, NR, MS and GJ designed and interpreted the results. LZ produced and characterized ACE2 fusion proteins. MC designed the affinity maturated mutant and performed structural analysis. LZ and CW performed the in vitro binding and competition assays. NR performed the binding analysis on virus-infected cells and neutralization assays. TM and LZ conducted BLI analysis. HS and H-GR contributed to interpretation of the experimental results. LZ wrote the manuscript. GJ, MS, MC and NR critically revised the manuscript. All authors read and accepted the submitted manuscript.
This project was funded by the German Cancer Consortium (DKTK).
The authors thank Beate Pömmerl, Andrea Dobler and Ilona Hagelstein for expert technical assistance.
GJ, LZ, HS, MS, NR and MC are listed as inventors on the patent application “Modified ACE2 proteins with improved activity against SARS-CoV-2”, EP22198499, applicant German Cancer Research Center, Heidelberg and University of Tuebingen, Germany.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1112505/full#supplementary-material
1. Anderson RM, Vegvari C, Hollingsworth TD, Pi L, Maddren R, Ng CW, et al. The SARS-CoV-2 pandemic: Remaining uncertainties in our understanding of the epidemiology and transmission dynamics of the virus, and challenges to be overcome. Interface Focus (2021) 11(6):20210008. doi: 10.1098/rsfs.2021.0008
2. Cascella M, Rajnik M, Aleem A, Dulebohn SC, Di Napoli R. Features, evaluation, and treatment of coronavirus (COVID-19). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2022).
3. Kumar S, Caliskan DM, Janowski J, Faist A, Conrad BCG, Lange J, et al. Beyond vaccines: Clinical status of prospective COVID-19 therapeutics. Front Immunol (2021) 12:752227. doi: 10.3389/fimmu.2021.752227
4. Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science (2020) 369(6506):1014–8. doi: 10.1126/science.abd0831
5. Bertoglio F, Fuhner V, Ruschig M, Heine PA, Abassi L, Klunemann T, et al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep (2021) 36(4):109433. doi: 10.1016/j.celrep.2021.109433
6. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with covid-19. N Engl J Med (2021) 384(3):229–37. doi: 10.1056/NEJMoa2029849
7. Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: A randomized clinical trial. JAMA (2021) 325(7):632–44. doi: 10.1001/jama.2021.0202
8. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with covid-19. N Engl J Med (2021) 384(3):238–51. doi: 10.1056/NEJMoa2035002
9. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight (2019) 4(4):e123158. doi: 10.1172/jci.insight.123158
10. Nouailles G, Wyler E, Pennitz P, Postmus D, Vladimirova D, Kazmierski J, et al. Temporal omics analysis in Syrian hamsters unravel cellular effector responses to moderate COVID-19. Nat Commun (2021) 12(1):4869. doi: 10.1038/s41467-021-25030-7
11. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol (2021) 19(7):409–24. doi: 10.1038/s41579-021-00573-0
12. Hu J, Peng P, Cao X, Wu K, Chen J, Wang K, et al. Increased immune escape of the new SARS-CoV-2 variant of concern omicron. Cell Mol Immunol (2022) 19(2):293–5. doi: 10.1038/s41423-021-00836-z
13. Ao D, Lan T, He X, Liu J, Chen L, Baptista-Hon DT, et al. SARS-CoV-2 omicron variant: Immune escape and vaccine development. MedComm (2022) 3(1):e126. doi: 10.1002/mco2.126
14. Hansen J, Baum A, Pascal KE, Russo V, Giordano S, Wloga E, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science (2020) 369(6506):1010–4. doi: 10.1126/science.abd0827
15. Cameroni E, Bowen JE, Rosen LE, Saliba C, Zepeda SK, Culap K, et al. Broadly neutralizing antibodies overcome SARS-CoV-2 omicron antigenic shift. Nature (2022) 602(7898):664–70. doi: 10.1038/s41586-021-04386-2
16. McCallum M, Czudnochowski N, Rosen LE, Zepeda SK, Bowen JE, Walls AC, et al. Structural basis of SARS-CoV-2 omicron immune evasion and receptor engagement. Science (2022) 375(6583):864–8. doi: 10.1126/science.abn8652
17. VanBlargan LA, Errico JM, Halfmann PJ, Zost SJ, Crowe JE Jr., Purcell LA, et al. An infectious SARS-CoV-2 B.1.1.529 omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med (2022) 28(3):490–5. doi: 10.1038/s41591-021-01678-y
18. Han Y, Wang Z, Wei Z, Schapiro I, Li J. Binding affinity and mechanisms of SARS-CoV-2 variants. Comput Struct Biotechnol J (2021) 19:4184–91. doi: 10.1016/j.csbj.2021.07.026
19. Han P, Su C, Zhang Y, Bai C, Zheng A, Qiao C, et al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat Commun (2021) 12(1):6103. doi: 10.1038/s41467-021-26401-w
20. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
21. Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med (2020) 8(11):1154–8. doi: 10.1016/S2213-2600(20)30418-5
22. Khodarahmi R, Sayad B, Sobhani M. The ACE2 as a “rescue protein” or “suspect enzyme” in COVID-19: possible application of the “engineered inactive hrsACE2” as a safer therapeutic agent in the treatment of SARS-CoV-2 infection. J Iran Chem Soc (2020) 18(3):495–502. doi: 10.1007/s13738-020-02049-z
23. Zekri L, Vogt F, Osburg L, Muller S, Kauer J, Manz T, et al. An IgG-based bispecific antibody for improved dual targeting in PSMA-positive cancer. EMBO Mol Med (2021) 13(2):e11902. doi: 10.15252/emmm.201911902
24. Xie X, Muruato A, Lokugamage KG, Narayanan K, Zhang X, Zou J, et al. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe (2020) 27(5):841–8.e3. doi: 10.1016/j.chom.2020.04.004
25. Ruetalo N, Businger R, Althaus K, Fink S, Ruoff F, Pogoda M, et al. Antibody response against SARS-CoV-2 and seasonal coronaviruses in nonhospitalized COVID-19 patients. mSphere (2021) 6(1):e01145–20. doi: 10.1128/mSphere.01145-20
26. Becker M, Strengert M, Junker D, Kaiser PD, Kerrinnes T, Traenkle B, et al. Exploring beyond clinical routine SARS-CoV-2 serology using MultiCoV-ab to evaluate endemic coronavirus cross-reactivity. Nat Commun (2021) 12(1):1152. doi: 10.1038/s41467-021-20973-3
27. Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem (2004) 279(17):17996–8007. doi: 10.1074/jbc.M311191200
28. Guy JL, Jackson RM, Jensen HA, Hooper NM, Turner AJ. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J (2005) 272(14):3512–20. doi: 10.1111/j.1742-4658.2005.04756.x
29. Chan KK, Dorosky D, Sharma P, Abbasi SA, Dye JM, Kranz DM, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science (2020) 369(6508):1261–5. doi: 10.1126/science.abc0870
30. Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science (2021) 371(6531):850–4. doi: 10.1126/science.abf9302
31. Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature (2021) 593(7857):130–5. doi: 10.1038/s41586-021-03398-2
32. Shuai H, Chan JF, Hu B, Chai Y, Yuen TT, Yin F, et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 omicron. Nature (2022) 603(7902):693–9. doi: 10.1038/s41586-022-04442-5
33. Meng B, Abdullahi A, Ferreira I, Goonawardane N, Saito A, Kimura I, et al. Altered TMPRSS2 usage by SARS-CoV-2 omicron impacts infectivity and fusogenicity. Nature (2022) 603(7902):706–14. doi: 10.1038/s41586-022-04474-x
34. Bojkova D, Widera M, Ciesek S, Wass MN, Michaelis M, Cinatl J Jr. Reduced interferon antagonism but similar drug sensitivity in omicron variant compared to delta variant of SARS-CoV-2 isolates. Cell Res (2022) 32(3):319–21. doi: 10.1038/s41422-022-00619-9
35. Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, et al. Omicron extensively but incompletely escapes pfizer BNT162b2 neutralization. Nature (2022) 602(7898):654–6. doi: 10.1038/s41586-021-04387-1
36. Zhang L, Li Q, Liang Z, Li T, Liu S, Cui Q, et al. The significant immune escape of pseudotyped SARS-CoV-2 variant omicron. Emerg Microbes Infect (2022) 11(1):1–5. doi: 10.1080/22221751.2021.2017757
37. Dejnirattisai W, Shaw RH, Supasa P, Liu C, Stuart AS, Pollard AJ, et al. Reduced neutralisation of SARS-CoV-2 omicron B.1.1.529 variant by post-immunisation serum. Lancet (2022) 399(10321):234–6. doi: 10.1016/S0140-6736(21)02844-0
38. Edara VV, Manning KE, Ellis M, Lai L, Moore KM, Foster SL, et al. mRNA-1273 and BNT162b2 mRNA vaccines have reduced neutralizing activity against the SARS-CoV-2 omicron variant. bioRxiv (2021). doi: 10.1101/2021.12.20.473557
39. Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature (2022) 602(7898):657–63. doi: 10.1038/s41586-021-04385-3
40. VanBlargan L, Errico J, Halfmann P, Zost S, Crowe J, Purcell L, et al. An infectious SARS-CoV-2 B.1.1.529 omicron virus escapes neutralization by therapeutic monoclonal antibodies. Res Sq (2021). doi: 10.21203/rs.3.rs-1175516/v1
41. Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, et al. Ebola Virus can be effectively neutralized by antibody produced in natural human infection. J Virol (1999) 73(7):6024–30. doi: 10.1128/JVI.73.7.6024-6030.1999
42. Reuter SE, Evans AM, Ward MB. Reducing palivizumab dose requirements through rational dose regimen design. CPT Pharmacometrics Syst Pharmacol (2019) 8(1):26–33. doi: 10.1002/psp4.12364
43. Tsai TI, Khalili JS, Gilchrist M, Waight AB, Cohen D, Zhuo S, et al. ACE2-fc fusion protein overcomes viral escape by potently neutralizing SARS-CoV-2 variants of concern. Antiviral Res (2022) 199:105271. doi: 10.1016/j.antiviral.2022.105271
44. Chen Y, Sun L, Ullah I, Beaudoin-Bussieres G, Anand SP, Hederman AP, et al. Engineered ACE2-fc counters murine lethal SARS-CoV-2 infection through direct neutralization and fc-effector activities. Sci Adv (2022) 8(28):eabn4188. doi: 10.1126/sciadv.abn4188
45. Leach A, Ilca FT, Akbar Z, Ferrari M, Bentley EM, Mattiuzzo G, et al. A tetrameric ACE2 protein broadly neutralizes SARS-CoV-2 spike variants of concern with elevated potency. Antiviral Res (2021) 194:105147. doi: 10.1016/j.antiviral.2021.105147
46. Tang H, Gao L, Wu Z, Meng F, Zhao X, Shao Y, et al. Multiple SARS-CoV-2 variants exhibit variable target cell infectivity and ability to evade antibody neutralization. Front Immunol (2022) 13:836232. doi: 10.3389/fimmu.2022.836232
47. Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther (2020) 5(1):283. doi: 10.1038/s41392-020-00426-x
48. Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T, et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res (2021) 31(2):126–40. doi: 10.1038/s41422-020-00460-y
49. Gu Y, Cao J, Zhang X, Gao H, Wang Y, Wang J, et al. Receptome profiling identifies KREMEN1 and ASGR1 as alternative functional receptors of SARS-CoV-2. Cell Res (2022) 32(1):24–37. doi: 10.1038/s41422-021-00595-6
50. Hoffmann D, Mereiter S, Jin Oh Y, Monteil V, Elder E, Zhu R, et al. Identification of lectin receptors for conserved SARS-CoV-2 glycosylation sites. EMBO J (2021) 40(19):e108375. doi: 10.15252/embj.2021108375
Keywords: SARS-CoV-2 therapy, ACE-2, neutralizing antibodies, immune escape, fusion protein
Citation: Zekri L, Ruetalo N, Christie M, Walker C, Manz T, Rammensee H-G, Salih HR, Schindler M and Jung G (2023) Novel ACE2 fusion protein with adapting activity against SARS-CoV-2 variants in vitro. Front. Immunol. 14:1112505. doi: 10.3389/fimmu.2023.1112505
Received: 30 November 2022; Accepted: 22 February 2023;
Published: 08 March 2023.
Edited by:
Alfonso J. Rodriguez-Morales, Fundacion Universitaria Autónoma de las Américas, ColombiaReviewed by:
Wentao Li, Huazhong Agricultural University, ChinaCopyright © 2023 Zekri, Ruetalo, Christie, Walker, Manz, Rammensee, Salih, Schindler and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Latifa Zekri, bGF0aWZhLnpla3JpQGlmaXoudW5pLXR1ZWJpbmdlbi5kZQ==
†These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.