 Sheng Wan1
Sheng Wan1 Wen Xu
Wen Xu Cuiping Guan
Cuiping Guan Xiuzu Song
Xiuzu Song
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 02 May 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1111547
This article is part of the Research TopicMethods in Autoimmune and Autoinflammatory Disorders: 2022View all 11 articles
Cytotoxic T lymphocyte has been a concern for the etiopathogenesis of alopecia areata (AA), some recent evidence suggests that the regulatory T (Treg) cell deficiency is also a contributing factor. In the lesional scalp of AA, Treg cells residing in the follicles are impaired, leading to dysregulated local immunity and hair follicle (HF) regeneration disorders. New strategies are emerging to modulate Treg cells’ number and function for autoimmune diseases. There is much interest to boost Treg cells in AA patients to suppress the abnormal autoimmunity of HF and stimulate hair regeneration. With few satisfactory therapeutic regimens available for AA, Treg cell-based therapies could be the way forward. Specifically, CAR-Treg cells and novel formulations of low-dose IL-2 are the alternatives.
▪ Treg cells act as an indispensable component to maintain self-tolerance and immune homeostasis.
▪ Defective Treg cells are a typical feature of almost all autoimmune diseases, including AA.
▪ Skin-resident Treg cells are the HF-IP guardians. They localize to the HFSC niche and promote hair regeneration, which is the basis for Treg cell-based therapies for AA.
▪ Antigen-specific Treg cells could be generated for trials of AA-targeted therapy, and potential autoantigens need to be identified urgently.
▪ Low-dose IL-2 in combination with other treatments is expected to enhance the efficacy of therapy for AA.
It is well acknowledged that alopecia areata (AA), which causes non-scarring hair loss, is a T cell-mediated autoimmune phenomenon affecting approximately 2% of the population (1, 2). Patients with severe AA have poor quality of life and suffer from high levels of anxiety and depression (3). Certain issues cause hair follicle immune privilege (HF-IP) to fall apart, which is thought to be the driver of AA (4). However, the exact pathogenesis of AA remains to be fully characterized. The use of nonspecific immunosuppressants were the mainstay of treatment for the majority of AA patients in the past. Although novel therapies have progressed rapidly in recent years (5), they are still unsatisfactory and new treatment options are essential. New evidence suggests that several other cells have been implicated besides the “central players”, CD8+ T cells, such as T-helper (TH) 17 cells, natural killer (NK) cells, mast cells, plasmacytoid dendritic cells (pDCs), and regulatory T (Treg) cells (6–9). Treg cell is one of the regulatory lymphocyte subsets, maintaining self-tolerance and immune homeostasis of the body. Interestingly, defects in Treg cell function are strongly linked to various autoimmune disorders (10, 11) and AA is no exception (12). Therefore, by enhancing the function of Treg cells, restoration of immune tolerance in patients with autoimmune illnesses has now been achieved (13, 14), but for AA, there are few relevant studies.
The current review focuses on Treg cells and their close association with AA, and specific roles in HF have been discussed with an emphasis on the therapeutic potential of Treg cells. We aim to broaden the horizon of AA research and offer some suggestions for therapeutic use.
Self-tolerance, the state of unresponsiveness to self-tissues (antigens), is pivotal in the field of immunology. The breakdown of this state results in tissue inflammation and autoimmunity. Central tolerance (recessive) and peripheral tolerance (dominant) are the two mechanism categories (15). It is believed that regulatory T cells, in particular, induce and maintain peripheral tolerance.
Early in the 1960s, neonatal mice with thymectomy were found to have wasting syndrome, which closely resembles graft-versus-host reactions (16). Further research suggested the existence of “suppressor T cells” in lymphocytes (17–19), but the immunological field remained skeptical of them (20). Until 1995, a subset of CD4+ T cells that constitutively express interleukin-2 (IL-2) receptor α-chain (CD25) were first described by Sakaguchi et al. (21); these CD4+CD25+ T cells potently repressed the function of conventional T (Tconv) cells. The concept of regulatory T (Treg) cells was thus formally put forward. However, as CD25 is upregulated in all activated T cells, it is thought that CD25+ Treg cells are simply Tconv in an active state. Finally, the forkhead box P3 (Foxp3) discovery laid the foundation for Treg cell biology (22, 23). This transcription factor is essential for the development, phenotype maintenance, and activity of Treg cells (24–28). Since that, CD4/CD25/Foxp3 have become reliable phenotypic markers to identify Treg cells (in a narrow sense) and this field has been facilitated largely.
In fact, CD4+CD25+FOXP3+ Treg cells are just one of the classic population of Treg cells. To date, several subsets of Treg cells have been described. However, whether any definitive phenotypic marker exists remains controversial (29, 30). The terms of these subsets are not uniform and oftentimes confusing. A recommendation has been proposed to simplify the nomenclature but limited progress has been made (31). Some researchers classify them according to their developmental origin: thymus-derived Treg (tTreg) cells and peripherally derived Treg (pTreg) cells (32, 33). Others divide them into natural Treg (nTreg) cells and induced Treg (iTreg) cells (34, 35), and still, others argue that there are three subgroups of Treg cells, namely, nTreg cells, pTreg cells, and iTreg cells (36, 37). Undoubtedly, both nTreg cells and pTreg cells refer to CD4+CD25+FOXP3+ Treg cells (29, 33), which are naturally occurring Treg cells derived from the thymus as a separate lineage. They are induced by the thymus’s TCR signal and costimulatory molecules (36, 38). These CD4+CD25+Foxp3+ Treg cells constitute 5%–10% of CD4+ T cells (34, 39). pTreg cells develop from naïve CD4+ T cells in the periphery after contact with antigens and in the presence of specific factors such as transforming growth factor-beta (TGF-β) and IL-2 (29, 40). Some of them circulate through the blood and peripheral lymphoid organs. While others exist in non-lymphoid tissues, these Treg cells are what we commonly call “tissue Treg cell” or “tissue-resident Treg cell” (this notion was advanced in 2009) (41). Moreover, these cells fit the criteria for effector memory cells (CD45RO+); thus, they are also named memory Treg cells (42). The iTreg cells could be induced by TGF-β in vitro (36, 37). Many researchers conflate the latter two and regard them as “iTreg cells”; hence, it is not easy to understand whether they are referring to the Treg cells generated in vitro or in vivo.
Regarding the function of these subsets, it was not initially thought that iTreg cells had sufficient suppressive activity compared to nTreg cells (35, 43) due to the loss of Foxp3 expression in iTreg cells, resulting from failing to fully demethylate the Treg cell-specific demethylated region (TSDR). Then, by exploring the plasticity and stability of iTreg cells (34, 40), recent studies have established the position of iTreg cells in immunological tolerance. More details of the types of Treg cells have been summarized (30, 44); no further description will be given here, and they are also not strictly differentiated in this review for the convenience of follow-up discussion.
Treg cell-mediated suppression serves as a vital mechanism for negative regulation of immune-mediated inflammation. There are concise descriptions of the three main categories of mechanisms involved.
1. Cell–cell contact. Treg cells constitutively express cytotoxic T lymphocyte-associated protein 4 (CTLA-4). CTLA-4 competes with CD28, a T-cell costimulatory molecule, for CD80/CD86 on antigen-presenting cells (APCs) and downregulates expression of the latter, resulting in the inhibition of Tconv cells (45, 46). Furthermore, CTLA-4 upregulates indoleamine 2,3-dioxygenase (IDO), resulting in cell cycle arrest and increased sensitivity to apoptosis in effector T (Teff) cells, and dysfunctional APCs (47, 48). Like CTLA-4, lymphocyte activation gene 3 (LAG-3) is highly expressed on the surface of Treg cells, which inhibits the function of dendritic cells (DCs) (49, 50). Moreover, Treg cells could directly induce apoptosis of target cells via cell contact, attributing to their release of cytotoxic factors such as granzymes (51, 52).
2. Secretion of inhibitory cytokines. Treg cells secrete some inhibitory cytokines such as IL-10, TGF-β, and IL-35, which suppress both Teff cells and APCs. Major histocompatibility complex (MHC) class II molecules of APCs are downregulated in the presence of IL-10 (53). TGF-β may inhibit T cells and (or) maintain Foxp3 expression in Treg cells (54), and IL-35 could reduce T-cell proliferation (55).
3. Metabolic disruption of Teff cells. Treg cells scarcely produce IL-2 but consume IL-2 from the surroundings via their high-affinity IL-2 receptor, resulting in cytokine deprivation-induced apoptosis of Teff cells (56, 57). Besides that, Treg cells express CD39 and CD73, which generate adenosine and cyclic adenosine monophosphate (cAMP); the former could increase intracellular cAMP of Teff by adenosine receptor 2A, disrupting their metabolism (58, 59).
Presumptive T cell-mediated autoimmune illness of the skin with destruction to the hair follicle (HF) is known as alopecia areata (AA). However, the pathobiology of this chronic, relapsing hair-loss disorder is not entirely known. IFN-γ, IL-15, and CD8+NKG2D+ T cells have long been identified as the core contributors to pathological processes, but numerous studies have shown that they are not the only drivers and several other cell populations may be the new “player”. Treg cells, regulating the immune response and maintaining peripheral tolerance. Thus, an important new frontier is the role of regulatory lymphocytes in maintaining the HF immune privilege (HF-IP). Meanwhile, the discovery of tissue-resident Treg cells will give us a thorough grasp of Treg cells’ roles in HF. Although the mechanism of Treg cell weakening in AA has not been illuminated, it may be an exciting subject for future studies. To avoid redundancy, we will analyze the contributions of Treg cells in AA pathogenesis.
Many previous studies have revealed defects in the frequencies and functions of Treg cells in virtually all the common systemic autoimmune disorders (11, 60, 61), but little is known about AA. A genome-wide association study in AA identified several genes controlling the activation and proliferation of Treg cells (62). With the growing appreciation of Treg cells, related articles have been published. Circulating Treg cells from AA patients have been found to have impaired inhibitory activity (63). In other words, Teff cells are relatively dominant. Some researchers proposed that the imbalance between TH17 cells and Treg cells is crucial in the pathogenesis (64–66). However, some of the results in the literature are controversial. Two excellent reviews (67, 68) gave an overview of Treg cells in autoimmune skin diseases including AA, vitiligo, psoriasis, and systemic sclerosis. Interestingly, almost all of them were linked to abnormal Treg cell function. Thus, there is a reasonable prospect that it may be a “universal law” in the pathogenesis of the autoimmune disease. This raises a question: why does the imbalance occur? Next, we seek to offer a perspective of Treg cells in lesional HF.
Gilhar et al. (69) theorized that the IFN-γ “storm” and CD8+ T cells might break the delicate balance of immune cells, and that there will be tremendous and complex cytokines in the environment after the HF under immune attack (Figures 1A, B). TGF-β and IL-6 together stimulate the development of pathogenic TH17 cells from naive T cells, and IL-6 can inhibit the production of Foxp3+ Treg cells generated by TGF-β (70). Moreover, due to the plasticity of Treg cells, IL-6 and IL-1β could also reverse their suppressive function or result in their conversion to TH17 cells (29, 71). This phenomenon has been demonstrated in some other disorders (72–74). Considering that some reports on AA found that the percentage of circulating Treg cells was normal, Treg cells’ intrinsic defects may be another main reason, such as defects in suppression, survival, or stability. As we mentioned earlier, CD39 is a key component in Treg cell suppressive machinery. However, there was significantly reduced CD39 and HLA-DR expression on circulating Treg cells and HF Treg cells in AA patients, and Hamed et al. (12) speculated that impaired Treg cell function is mainly due to a defect in cell–cell contact and CD39-mediated suppression. The opposite is true in most cancers (75). CD39+ Treg cells are strong suppressors of TH17 cells and NK cells in antitumor immunity (76, 77). Moreover, Conteduca et al.’s study suggested that Foxp3 and inducible costimulatory ligand (ICOSLG) polymorphisms may predispose to AA by decreasing its mRNA expression (78), which is due to the destruction of the stability of Treg cells. In conclusion, there could be many reasons for defective Treg cell suppression.
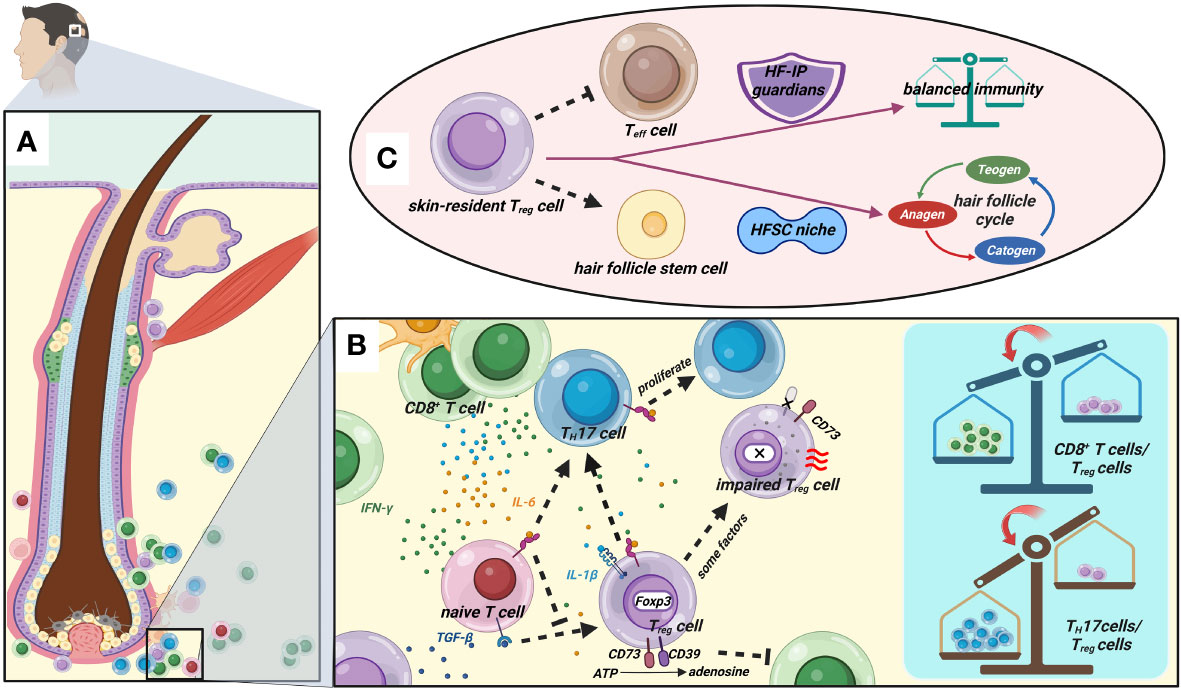
Figure 1 Great importance of Treg cells in AA. (A) The anagen HF of AA patients. (B) There are tremendous amounts cytokines in the HF after immune attack. IL-6 inhibits the generation of Foxp3+ Treg cells induced by TGF-β and induces the differentiation of pathogenic TH17 cells. IL-6 and IL-1β could reverse Treg cells’ suppressive function or result in their conversion to TH17 cells. Some issues lead to intrinsic defects of Treg cells such as loss of expression of CD39 and Foxp3. Eventually, impairment of Treg cells contributes to the imbalance of immunocyte populations including CD8+ T cells/Treg cells and TH17 cells/Treg cells. (C) Skin-resident Treg cells have dual roles in maintaining immune homeostasis as HF-IP guardians and localizing in the HFSC niche to drive the cyclic growth of HF. (Figure1 created with BioRender.com).
IP was proposed in the 1940s as a relative and dynamic special status of self-tolerance, which occurs in tissues and organs including the eye, testis, heart valves (79–81), and, of course, HF (82, 83). There are at least two distinct areas of relative IP in HF: anagen hair bulb and the bulge region (4). Mechanisms of HF IP (these two IP areas are slightly different) are thought to downregulate the expression of MHC I and β2-microglobulin (84), and the secretion of immunosuppressive cytokines and neuropeptides (“IP guardians”) such as TGF-β1/2, IL-10, alpha-melanocyte-stimulating hormone (α-MSH), calcitonin gene-related peptide (CGRP), and vasoactive intestinal peptide (VIP) (85–87). Anagen hair follicles’ immune privilege maintaining mechanism collapses in patients with AA, but the cause is a matter of debate (88). Recently, there has been a suspicion that Treg cells may also be a type of key HF-IP guardian. Perifollicular Treg cells create a local immunoinhibitory environment to maintain HF-IP. This peripheral tolerance is critical for HF because some HF-associated antigens are not expressed in the thymus (89).
One of the most important advances in this topic concerns tissue-resident Treg cells, a special population of Treg cells. Tissue-resident Treg cells populate specific peripheral tissues in the body, performing non-immunological functions and being devoted to maintain tissue homeostasis and wound repair (29, 90–93). Skin is the largest organ and is home to a large proportion of the body’s tissue-resident Treg cells, and skin-resident Treg cells are beginning to be understood (42, 94, 95). They predominantly reside in the dermis (96) and promote wound healing and tolerance to the skin microbiota (97–99). Surprisingly, skin-resident Treg cells preferentially localized to hair follicles and were most abundant in the scalp and face (with high hair follicle density), whereas Tconv cells displayed a more diverse distribution (42).
Furthermore, Ali et al. found that follicular Treg cells of mice reside within 0–5 μm of bulge hair follicle stem cells (HFSCs) (9). We are aware that HFSCs are responsible for the cyclic proliferation of hair follicles, so do skin-resident Treg cells control hair growth via HFSC? Inevitably, the authors further demonstrated that skin-resident Treg cells with Jagged 1 expression promote HFSC proliferation and differentiation to drive HF cycling through the Notch signaling pathway. A new study reported that glucocorticoids induce glucocorticoid receptor (GR) in skin-resident Treg cells to produce TGF-β3, which activates HFSCs and promotes hair regeneration (100). Given that HF is a sophisticated mini-organ of the skin and offers a “niche” for stem cells (including HFSC) (101), some investigators suggested that skin-resident Treg cells might occupy a specialized HFSC niche (9, 102). Almost all Treg cells (greater than 95%) in human skin have an effector memory phenotype (42); that is to say, a substantial part of Treg cells in HF promote both immunosuppression and HF regeneration (Figure 1C). Actually, the dual role of skin-resident Treg cells has been proposed by Maryanovich et al. (103). Taken together, given the HF damage is reversible in AA, we believe that protecting and restoring the functioning skin-resident Treg cells might be the cornerstone of AA management.
To date, the majority of traditional treatments for AA are of limited efficacy (especially unsustainable remission) with a high risk of adverse effects (5, 104, 105). As an established therapy with marked curative effect, corticosteroids (including topical, intralesional, and systemic therapy) have been widely used to treat AA (106). Nevertheless, they could increase the risk of folliculitis and skin atrophy (105, 107). Methotrexate and cyclosporine may cause more severe systemic adverse effects. Allergic reactions in the form of severe dermatitis, lymphadenopathy, and urticaria are often associated with contact immunotherapy (105). Minoxidil as a monotherapy might be insufficient to achieve obvious hair regrowth (108). As for innovative strategies, Janus kinase (JAK) inhibitors appear to be most successful (109–111), while further large-scale studies are required to confirm its safety and durability (112, 113). Therefore, seeking new ways is highly desirable for patients suffering from AA. Notably, it has been widely accepted that AA is an autoimmune disease (114–117), although this concept remains a hypothesis (autoimmune target antigen has yet to be defined) and even a few people challenge it (69). AA is also related to various other autoimmune disorders (118). In fact, Treg cell-based therapies have emerged as a new avenue in various human autoimmune disorders, which aim to restore balanced immunity. As Treg cells are important regulators of HFSC function, Treg cell-based therapies could be a type of treatment that stimulates HF regeneration. Here, we will review some existing related studies and discuss the prospects and challenges of Treg cell-based AA therapies.
The impact of AA is frequently underestimated and perhaps dismissed as only a “cosmetic problem” (116, 119). Coupled with the high price of cell products, little attention has, to our knowledge, been paid to cellular therapy for AA, but that could be the way to go. Previously, adoptive transfer of polyclonal Treg cells has been investigated extensively; these trials fully demonstrate its feasibility and safety (120–122). However, polyclonal Treg cells have been limited by the difficulty in producing quantities sufficient for clinical use and systemic immunosuppression (such as inadvertent suppression of immune responses to infection or malignancies) (68, 123), while antigen-specific Treg cells could overcome these difficulties. They migrate to lesions and respond to their cognate antigen to provide more effective protection from autoimmune activity (124–127). Chimeric antigen receptor (CAR) and T-cell receptor (TCR) could redirect Treg cells. Specifically, unlike TCR-Treg cells, CAR-Treg cells can bypass HLA restriction (128, 129) and target more flexibly (antigens recognized by CARs also include non-protein and soluble targets) (130, 131). Expectedly, CAR technology might be available to AA.
Except for graft versus host disease (GvHD, with a very clear target), selecting a suitable antigen is critical when constructing a CAR for most autoimmune diseases, although this process is time-consuming and sometimes difficult (129, 132). Owing to the great efforts of researchers, a variety of CARs have been developed and show great potential for treating various diseases in preclinical studies. Elinav et al. (133) were one of the first to develop a CAR to cure mouse colitis. MacDonald and colleagues (134) transduced human Treg cells with a CAR that targets the HLA-A2 and found that A2-CAR-expressing Treg cells ameliorated the progression of GvHD in a mouse model. CAR-Treg cells have also been reported to be engineered with specificity for myelin oligodendrocyte glycoprotein to prevent experimental autoimmune encephalomyelitis (EAE, a model relating to multiple sclerosis in humans) (135). Excitingly, CAR-Treg cells have been tested in studies on autoimmune skin disorders. A recent study described the curative effect of CAR-Treg cells targeting ganglioside D3 to provide antigen-specific immune tolerance for vitiligo (136). Furthermore, the UK and US authorized the first CAR-Treg cell clinical trial (STEADFAST, NCT04817774) for kidney transplant patients (137, 138).
Regrettably, owing to the hurdle that the exact HF autoantigen(s) is still disputed, there is hardly any research on the therapeutic use of CAR-Treg cells for AA. Given the frequent clinical observations that AA seems to target gray hairs rather than pigmented hairs and nonpigmented hair regrowth, melanocyte antigen epitopes have long been suspected as potential targets (139–143), such as tyrosinase (TYR), tyrosinase-related protein-1 (TRP-1), TRP-2, glucoprotein100 (gp100, premelanosome protein analog), Melan-A (also known as melanoma antigen recognized by T cells 1 Leu27 analog, MART-1), and melanocortin-1-receptor (MC1R) (140, 144). Why hair loss happens rather than just graying could not be proven. That is to say, the destruction of melanocytes is only part of the story of immunoreactive HF. Our recently published review (145) argued that melanocytes might only be the initiating factor of autoimmune attack. Wang et al. (146) deduced that activated CTLs secrete multiple inflammatory cytokines harmful to keratinocytes even without direct cell–cell contact. IFN-γ upregulates MHC I and MHC II molecules in HF, potentially triggering antigen presentation in other cell populations. Many researchers have discovered keratinocyte-derived autoantigens, such as trichohyalin (expressed in the growing HF’s inner root sheath) and keratin (expressed in the anagen HF pre-cortical zone) (146–148). Although the authors speculated that these proteins were the major autoantigens in human AA, definitive evidence is lacking. We think that the above elusive melanocyte- and keratinocyte-targeting antigens should be used to exploit CAR-Treg cells and initially investigate their effects on AA in animal models because of their great significance: some autoantigens of AA could be confirmed on the one hand, and the effectiveness of CAR-Treg cells could be examined on the other hand (Figure 2A).
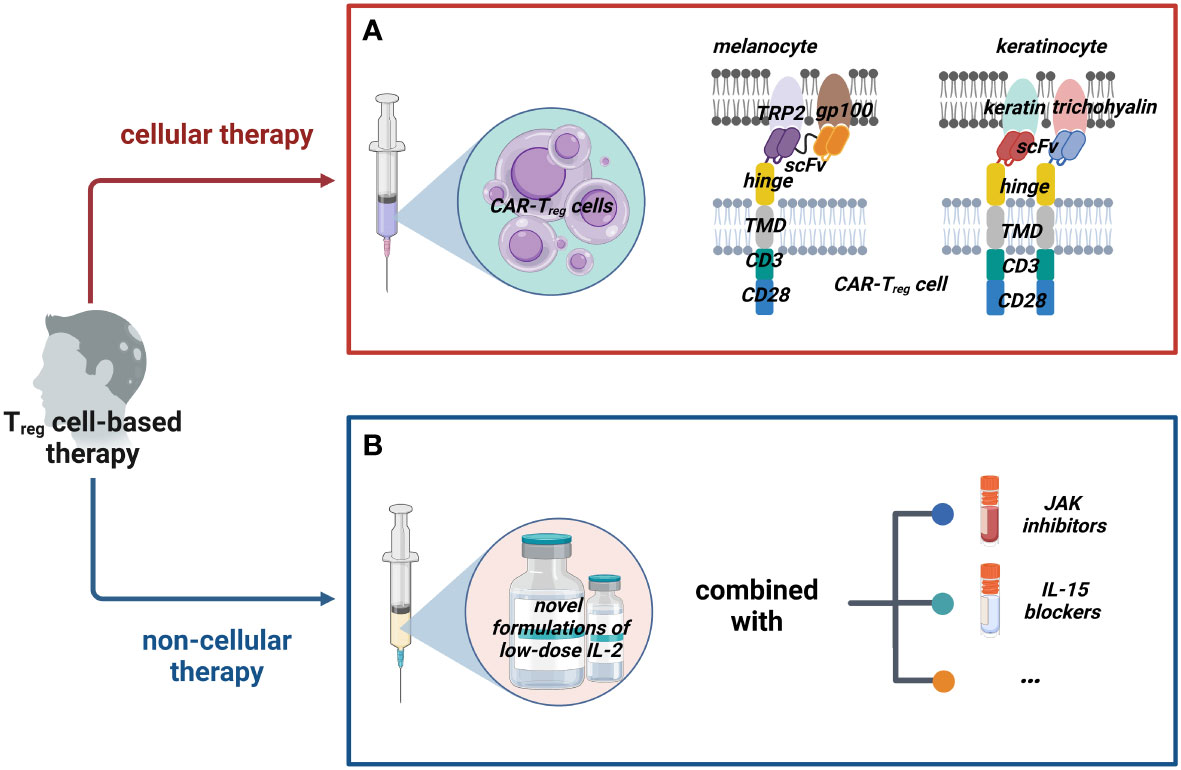
Figure 2 Possible Treg cell-based therapies for AA. We classified possible Treg cell-based therapy into two groups: cellular therapy and non-cellular therapy. (A) Cellular therapies such as adoptive transfer of CAR-Treg cells that could target dual (or multiple) potential antigens on melanocytes and keratinocytes with appropriate costimulatory domains should be tried. (B) Non-cellular therapies mainly work by inducing Treg cells; novel formulations of IL-2 are constantly being developed and they could be combined with other drugs such as JAK inhibitors and IL-15 blockers to enhance the efficacy. (Figure2 created with BioRender.com).
Specifically, for CARs, the following aspects need to be considered. In general, increasing CAR’s specificity could enhance Treg cells’ effect. As a result, most people focused only on mono-specific CAR-Treg cells, which sometimes fall short of our expectations. For example, some scientists generated a CAR against human insulin and found that the Treg cells were suppressive and long-lived, but did not prevent type 1 diabetes (T1D) in mice (149). Mohseni et al. provided a new perspective in their review (138); the methods of implementing universal recognition of CAR-Treg cells were summarized: mixing CAR-Treg cells against different antigens, constructing a Treg cell with several CARs or one multi-specific CAR against different antigens, and building UniCAR-Treg cells. Perhaps, these novel CAR-Treg cells targeting multiple antigens in HF will make a difference, which awaits confirmation. It should be pointed out that choosing the appropriate costimulatory domain is also crucial. The costimulatory domains of CAR-Treg cells currently used are the same as those used in conventional CAR-T cells, but they could result in different effector functions (150). A typical example is that 4-1BB may reduce the suppressive function of CAR-Treg cells (151, 152). This means that CD28 instead of 4-1BB should be considered the costimulatory domain of CAR-Treg cells for AA. The relevant mechanism needs to be better understood; perhaps the presence of one or several costimulatory domains contributes to the optimal function in AA.
All in all, the identification of the specific autoantigen(s) is a prerequisite for targeted therapy. Only our comprehensive knowledge about the target antigens in AA could open the door to antigen-specific Treg cell therapy for it.
As an alternative strategy for Treg cell transfer, some agents could also increase Treg cell numbers and support Treg cell function. Compared to transferring living cells, this approach will bring two obvious advantages: improved cost-effectiveness and lower risk for adverse events. In a deeper sense, for AA, these therapeutics may induce naïve T cells to develop into Foxp3+ Treg cells to re-establish immune homeostasis and activate skin-resident Treg cells to promote HF regeneration in the lesional scalp.
Nowadays, there are lots of existing therapeutic agents that target Treg cells, such as IL-2, TGF-β, rapamycin, or CTLA-4Ig. Foxp3 expression and histone acetylation are induced by TGF-signaling (153–155). The PI3K–AKT–mTOR axis activation is a crucial negative regulator of Treg cells (156, 157), and rapamycin could promote the Treg cell expansion (158–161). IL-2 is the most widely used during the last couple of years. Treg cell activation and proliferation highly depend on IL-2 production, mainly by Tconv cells, a key mechanism to prevent T cells’ overexpansion (162–164). Tconv cells can be induced to express Foxp3 in the presence of IL-2 (43, 165), whereas anti-IL-2 antibodies could impair Treg cells (166). In vivo, IL-2 is crucial for Treg cell survival, proliferation, and stability (167–169), suggesting that IL-2 is an important agent for regulating Treg cells.
Encouragingly, some promising data have been achieved with IL-2 to treat AA. Subcutaneous injection of low-dose recombinant IL-2 allowed the recruitment of Treg cells inside the lesional scalp skin in four of five patients with severe AA refractory to prior systemic therapies, as shown by Castela et al. (170), but the remaining patient did not show improvement. In addition to AA, low-dose IL-2 has positively affected other autoimmune diseases, including systemic lupus erythematosus, T1D, hepatitis C virus-induced vasculitis, and immune thrombocytopenia (171–175). Theoretically speaking, Treg cells can efficiently compete for a limited amount of IL-2 with effector cells because the former expresses a higher-affinity IL-2 receptor, and is sensitive to low levels of IL-2 (56, 176, 177). However, the dose of IL-2 should be treated with caution, because its slight increase could activate conventional memory T cells and NK cells, namely, off-target complications (172, 178). Patients must be administered IL-2 regularly and at short intervals because of its short half-life (179). Consequently, the IL-2 therapy protocol in AA should be optimized in Castela et al.’s study. To address the drawbacks of utilizing pure IL-2, many investigators have improved IL-2 therapy. For example, controlled release formulations (IL-2/TGF-β1/rapamycin) (180), dual-acting cytokine fusion protein (IL2-EHD2-sc-mTNFR2) (181), long-lived IL-2 mutein [IgG-(IL-2N88D)2] (182), and IL-2/UFKA-20 complex (183) have been developed successively. These novel formulations achieved superior Treg-expanding properties and selectivity.
Recently, some negative results attracted our attention. In 2020, 43 adult patients with severe AA completed a multicentric randomized placebo-controlled trial with a 52-week follow-up period (184). Unfortunately, despite significantly increasing peripheral Treg cells due to IL-2 therapy, these patients failed to achieve noticeable hair regrowth. The authors hypothesized that the limitation of increasing Treg cells only to the naïve subset (without skin-homing capabilities) is partly responsible for poor efficacy. Not coincidentally, another study reports a similar phenomenon: the Treg cells of mice injected with the IL-2 cytokine antibody complex (consisting of human IL-2, anti-hIL-2 antibody, and mouse IL-2 Fc) were 8–10 times higher than those of the control group. However, this administration cannot reverse AA on the established mouse model (185). As the authors note, the treatment of IL-2 confronted the problem of whether combination therapy is needed. Indeed, co-medication has shown promise in other autoimmune diseases (186). The IL-2 therapy combined with the application of JAK inhibitors (187, 188), blocking IL-15 transpresentation (189), and more would be worth investigating in future studies (Figure 2B). Additionally, Ferreira et al. (131) suggested that IL-2 therapy could be combined with cellular therapy of Treg cells. Three clinical trials are currently testing whether this hypothesis could increase efficacy in T1D (NCT02772679), steroid-refractory chronic GvHD (NCT01937468), and amyotrophic lateral sclerosis (NCT03241784), which could also provide us some insight.
Autoimmune disorders go together with the impairment of Treg cells. Suppressing abnormal autoimmunity by boosting Treg cells in patients is a rational approach. An increasing number of clinical trials on them are being conducted to evaluate safety (including possible side effects) and efficacy. Although the pathogenesis of AA remains incompletely understood, Treg cells have been considered to be involved. It should be pointed out that many questions need to be answered: “what is the exact autoantigen of AA?”, “do autoantigen responses play a primary role in AA pathobiology?”, “are the defects of Treg cell the cause or effect of HF-IP collapse?”, and so on. However, this does not deny the strong link between Treg cells and AA. Treg cells play a major role in establishing and maintaining local self-tolerance as HF-IP guardians. Moreover, recent studies on tissue-resident Treg cells revealed their unique biological functions. In particular, Treg cells located in the HFSC niche could promote hair follicle regeneration, allowing us to better understand the greater potential of Treg cell-based therapies in AA than other autoimmune skin diseases. Owing to its unique pathophysiology, AA is difficult to manage medically. Despite the promising drug JAK inhibitors, long-term efficacy is still limited. Thus, there is no harm in conducting a Treg cell-based therapy investigation; this work could guide clinical practice. We believe that Treg cell-based therapy is the next logical step for AA management, which ultimately may improve patient outcomes.
SW, CG and XS participated in manuscript writing. CG, XS, WX and BX collected the literature and provided general idea. CG contributed to manuscript editing. All authors contributed to the article and approved the submitted version.
This work was supported by research funding from the National Natural Science Foundation of China (No. 81872517), the Zhejiang Provincial Natural Science Foundation of China (No. LY21H110001), the Hangzhou Medical Key Discipline Construction Project [No (2021).21-3], the Science and Technology Special Project of Biomedicine and Health Industry in Hangzhou (No. 2021WJCY156), and the Zhejiang Medical and Health Research Project (2023KY189).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AA, alopecia areata; APC, antigen-presenting cell; cAMP, cyclic adenosine monophosphate; CAR, chimeric antigen receptor; CGRP, calcitonin gene-related peptide; CTLA-4, cytotoxic T lymphocyte-associated protein 4; DCs, dendritic cells; EAE, experimental autoimmune encephalomyelitis; Foxp3, forkhead box P3; GR, glucocorticoid receptor; GvHD, graft versus host disease; gp100, glucoprotein100; HF, hair follicle; HFSC, hair follicle stem cell; ICOSLG, inducible costimulatory ligand; IDO, indoleamine 2,3-dioxygenase; IL, interleukin; IP, immune privilege; JAK, Janus kinase; LAG-3, lymphocyte activation gene 3; MC1R, melanocortin-1-receptor; MHC, major histocompatibility complex; NK cell, natural killer cell; pDCs, plasmacytoid dendritic cells; T1D, type 1 diabetes; Tconv cell, conventional T cell; Teff cell, effector T cell; Treg cell, regulatory T cell; TH17 cell, T helper 17 cell; TCR, T-cell receptor; TGF-β, transforming growth factor-beta; TRP, tyrosinase-related protein; TSDR, demethylate the Treg cell-specific demethylated region; TYR, tyrosinase; VIP, vasoactive intestinal peptide; α-MSH, alpha-melanocyte-stimulating hormone.
1. Safavi KH, Muller SA, Suman VJ, Moshell AN, Melton LJ 3rd. Incidence of alopecia areata in Olmsted county, Minnesota, 1975 through 1989. Mayo Clin Proc (1995) 70:628–33. doi: 10.4065/70.7.628
2. Lee HH, Gwillim E, Patel KR, Hua T, Rastogi S, Ibler E, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol (2020) 82:675–82. doi: 10.1016/j.jaad.2019.08.032
3. Toussi A, Barton VR, Le ST, Agbai ON, Kiuru M. Psychosocial and psychiatric comorbidities and health-related quality of life in alopecia areata: a systematic review. J Am Acad Dermatol (2021) 85:162–75. doi: 10.1016/j.jaad.2020.06.047
4. Bertolini M, McElwee K, Gilhar A, Bulfone-Paus S, Paus R. Hair follicle immune privilege and its collapse in alopecia areata. Exp Dermatol (2020) 29:703–25. doi: 10.1111/exd.14155
5. Wang ECE, Dai Z, Christiano AM. Novel therapies for alopecia areata: the era of rational drug development. J Allergy Clin Immunol (2018) 141:499–504. doi: 10.1016/j.jaci.2017.10.028
6. Ghraieb A, Keren A, Ginzburg A, Ullmann Y, Schrum AG, Paus R, et al. iNKT cells ameliorate human autoimmunity: lessons from alopecia areata. J autoimmunity. (2018) 91:61–72. doi: 10.1016/j.jaut.2018.04.001
7. Tanemura A, Oiso N, Nakano M, Itoi S, Kawada A, Katayama I. Alopecia areata: infiltration of Th17 cells in the dermis, particularly around hair follicles. Dermatol (Basel Switzerland). (2013) 226:333–6. doi: 10.1159/000350933
8. Abou Rahal J, Kurban M, Kibbi AG, Abbas O. Plasmacytoid dendritic cells in alopecia areata: missing link? J Eur Acad Dermatol Venereology JEADV (2016) 30:119–23. doi: 10.1111/jdv.12932
9. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. (2017) 169:1119–29.e11. doi: 10.1016/j.cell.2017.05.002
10. Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev (2011) 10:744–55. doi: 10.1016/j.autrev.2011.05.004
11. Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and–yet–unanswered questions. Autoimmun Rev (2015) 14:105–16. doi: 10.1016/j.autrev.2014.10.012
12. Hamed FN, Åstrand A, Bertolini M, Rossi A, Maleki-Dizaji A, Messenger AG, et al. Alopecia areata patients show deficiency of FOXP3+CD39+ T regulatory cells and clonotypic restriction of treg TCRβ-chain, which highlights the immunopathological aspect of the disease. PloS One (2019) 14:e0210308. doi: 10.1371/journal.pone.0210308
13. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (Orlando Fla.). (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
14. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and b-cell responses. Blood. (2015) 125:1107–15. doi: 10.1182/blood-2014-04-566786
15. Xing Y, Hogquist KA. T-Cell tolerance: central and peripheral. Cold Spring Harbor Perspect Biol (2012) 4. doi: 10.1101/cshperspect.a006957
16. McIntire KR, Sell S, Miller JF. PATHOGENESIS OF THE POST-NEONATAL THYMECTOMY WASTING SYNDROME. Nature. (1964) 204:151–5. doi: 10.1038/204151a0
17. Fujimoto S, Greene M, Sehon AH. Immunosuppressor T cells in tumor bearing host. Immunol Commun (1975) 4:201–17. doi: 10.3109/08820137409055774
18. Berendt MJ, North RJ. T-Cell-mediated suppression of anti-tumor immunity. an explanation for progressive growth of an immunogenic tumor. J Exp Med (1980) 151:69–80. doi: 10.1084/jem.151.1.69
19. Bursuker I, North RJ. Generation and decay of the immune response to a progressive fibrosarcoma. II. failure to demonstrate postexcision immunity after the onset of T cell-mediated suppression of immunity. J Exp Med (1984) 159:1312–21. doi: 10.1084/jem.159.5.1312
20. Janeway CA Jr. Do suppressor T cells exist? a reply. Scandinavian J Immunol (1988) 27:621–3. doi: 10.1111/j.1365-3083.1988.tb02392.x
21. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
22. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi: 10.1038/83784
23. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
24. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
25. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi: 10.1038/ni909
26. Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol (2007) 8:359–68. doi: 10.1038/ni1445
27. Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol (2007) 8:457–62. doi: 10.1038/ni1455
28. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. (2007) 445:936–40. doi: 10.1038/nature05563
29. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
30. Dhamne C, Chung Y, Alousi AM, Cooper LJ, Tran DQ. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol (2013) 4:253. doi: 10.3389/fimmu.2013.00253
31. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi: 10.1038/ni.2554
32. Robertson SA, Green ES, Care AS, Moldenhauer LM, Prins JR, Hull ML, et al. Therapeutic potential of regulatory T cells in preeclampsia-opportunities and challenges. Front Immunol (2019) 10:478. doi: 10.3389/fimmu.2019.00478
33. Martin-Moreno PL, Tripathi S, Chandraker A. Regulatory T cells and kidney transplantation. Clin J Am Soc Nephrol CJASN. (2018) 13:1760–4. doi: 10.2215/cjn.01750218
34. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol (2013) 4:152. doi: 10.3389/fimmu.2013.00152
35. Edinger M. Regulatory T cells for the prevention of graft-versus-host disease: professionals defeat amateurs. Eur J Immunol (2009) 39:2966–8. doi: 10.1002/eji.200940030
36. Zhang Y, Guo J, Jia R. Treg: a promising immunotherapeutic target in oral diseases. Front Immunol (2021) 12:667862. doi: 10.3389/fimmu.2021.667862
37. Göschl L, Scheinecker C, Bonelli M. Treg cells in autoimmunity: from identification to treg-based therapies. Semin immunopathology. (2019) 41:301–14. doi: 10.1007/s00281-019-00741-8
38. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
39. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6:295–307. doi: 10.1038/nri1806
40. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol (2016) 37:803–11. doi: 10.1016/j.it.2016.08.012
41. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15:930–9. doi: 10.1038/nm.2002
42. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest (2014) 124:1027–36. doi: 10.1172/jci72932
43. Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, et al. Alloantigen-specific de novo-induced Foxp3+ treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol (2009) 39:3091–6. doi: 10.1002/eji.200939432
44. Beissert S, Schwarz A, Schwarz T. Regulatory T cells. J Invest Dermatol (2006) 126:15–24. doi: 10.1038/sj.jid.5700004
45. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Sci (New York N.Y.). (2008) 322:271–5. doi: 10.1126/science.1160062
46. Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J autoimmunity. (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
47. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi: 10.1038/nri1457
48. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol (2012) 3:51. doi: 10.3389/fimmu.2012.00051
49. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
50. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
51. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme b-dependent, perforin-independent mechanism. J Immunol (2005) 174:1783–6. doi: 10.4049/jimmunol.174.4.1783
52. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme b and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
53. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol (2004) 22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622
54. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol (2005) 6:338–44. doi: 10.1038/ni1180
55. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
56. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8:1353–62. doi: 10.1038/ni1536
57. Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O'Garra A, et al. CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol (2005) 17:279–88. doi: 10.1093/intimm/dxh207
58. Park A, Govindaraj C, Xiang SD, Halo J, Quinn M, Scalzo-Inguanti K, et al. Substantially modified ratios of effector to regulatory T cells during chemotherapy in ovarian cancer patients return to pre-treatment levels at completion: implications for immunotherapy. Cancers. (2012) 4:581–600. doi: 10.3390/cancers4020581
59. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology. (2013) 2:e26246. doi: 10.4161/onci.26246
60. Wang M, Liu C, Bond A, Yang J, Zhou X, Wang J, et al. Dysfunction of regulatory T cells in patients with ankylosing spondylitis is associated with a loss of Tim-3. Int immunopharmacology. (2018) 59:53–60. doi: 10.1016/j.intimp.2018.03.032
61. van Roon JA, Hartgring SA, van der Wurff-Jacobs KM, Bijlsma JW, Lafeber FP. Numbers of CD25+Foxp3+ T cells that lack the IL-7 receptor are increased intra-articularly and have impaired suppressive function in RA patients. Rheumatol (Oxford England). (2010) 49:2084–9. doi: 10.1093/rheumatology/keq237
62. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. (2010) 466:113–7. doi: 10.1038/nature09114
63. Shin BS, Furuhashi T, Nakamura M, Torii K, Morita A. Impaired inhibitory function of circulating CD4+CD25+ regulatory T cells in alopecia areata. J Dermatol science. (2013) 70:141–3. doi: 10.1016/j.jdermsci.2013.01.006
64. Han YM, Sheng YY, Xu F, Qi SS, Liu XJ, Hu RM, et al. Imbalance of T-helper 17 and regulatory T cells in patients with alopecia areata. J Dermatol (2015) 42:981–8. doi: 10.1111/1346-8138.12978
65. Loh SH, Moon HN, Lew BL, Sim WY. Role of T helper 17 cells and T regulatory cells in alopecia areata: comparison of lesion and serum cytokine between controls and patients. J Eur Acad Dermatol Venereology JEADV. (2018) 32:1028–33. doi: 10.1111/jdv.14775
66. Elela MA, Gawdat HI, Hegazy RA, Fawzy MM, Abdel Hay RM, Saadi D, et al. B cell activating factor and T-helper 17 cells: possible synergistic culprits in the pathogenesis of alopecia areata. Arch Dermatol Res (2016) 308:115–21. doi: 10.1007/s00403-016-1617-z
67. Ujiie H. Regulatory T cells in autoimmune skin diseases. Exp Dermatol (2019) 28:642–6. doi: 10.1111/exd.13535
68. Mukhatayev Z, Ostapchuk YO, Fang D, Le Poole IC. Engineered antigen-specific regulatory T cells for autoimmune skin conditions. Autoimmun Rev (2021) 20:102761. doi: 10.1016/j.autrev.2021.102761
69. Gilhar A, Laufer-Britva R, Keren A, Paus R. Frontiers in alopecia areata pathobiology research. J Allergy Clin Immunol (2019) 144:1478–89. doi: 10.1016/j.jaci.2019.08.035
70. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
71. Shevach EM. Foxp3(+) T regulatory cells: still many unanswered questions-a perspective after 20 Years of study. Front Immunol (2018) 9:1048. doi: 10.3389/fimmu.2018.01048
72. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol (2009) 183:3170–6. doi: 10.4049/jimmunol.0803721
73. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
74. Singh K, Gatzka M, Peters T, Borkner L, Hainzl A, Wang H, et al. Reduced CD18 levels drive regulatory T cell conversion into Th17 cells in the CD18hypo PL/J mouse model of psoriasis. J Immunol (2013) 190:2544–53. doi: 10.4049/jimmunol.1202399
75. Moesta AK, Li XY, Smyth MJ. Targeting CD39 in cancer. Nat Rev Immunol (2020) 20:739–55. doi: 10.1038/s41577-020-0376-4
76. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
77. Ahlmanner F, Sundström P, Akeus P, Eklöf J, Börjesson L, Gustavsson B, et al. CD39(+) regulatory T cells accumulate in colon adenocarcinomas and display markers of increased suppressive function. Oncotarget. (2018) 9:36993–7007. doi: 10.18632/oncotarget.26435
78. Conteduca G, Rossi A, Megiorni F, Parodi A, Ferrera F, Tardito S, et al. Single nucleotide polymorphisms in the promoter regions of Foxp3 and ICOSLG genes are associated with alopecia areata. Clin Exp Med (2014) 14:91–7. doi: 10.1007/s10238-012-0224-3
79. Hori J, Yamaguchi T, Keino H, Hamrah P, Maruyama K. Immune privilege in corneal transplantation. Prog retinal eye Res (2019) 72:100758. doi: 10.1016/j.preteyeres.2019.04.002
80. Chen Q, Deng T, Han D. Testicular immunoregulation and spermatogenesis. Semin Cell Dev Biol (2016) 59:157–65. doi: 10.1016/j.semcdb.2016.01.019
81. Hill MA, Kwon JH, Gerry B, Hardy WA, Walkowiak OA, Kavarana MN, et al. Immune privilege of heart valves. Front Immunol (2021) 12:731361. doi: 10.3389/fimmu.2021.731361
82. Christoph T, Müller-Röver S, Audring H, Tobin DJ, Hermes B, Cotsarelis G, et al. The human hair follicle immune system: cellular composition and immune privilege. Br J Dermatol (2000) 142:862–73. doi: 10.1046/j.1365-2133.2000.03464.x
83. Paus R, Nickoloff BJ, Ito T. A 'hairy' privilege. Trends Immunol (2005) 26:32–40. doi: 10.1016/j.it.2004.09.014
84. Meyer KC, Klatte JE, Dinh HV, Harries MJ, Reithmayer K, Meyer W, et al. Evidence that the bulge region is a site of relative immune privilege in human hair follicles. Br J Dermatol (2008) 159:1077–85. doi: 10.1111/j.1365-2133.2008.08818.x
85. Ito N, Ito T, Kromminga A, Bettermann A, Takigawa M, Kees F, et al. Human hair follicles display a functional equivalent of the hypothalamic-pituitary-adrenal axis and synthesize cortisol. FASEB J Off Publ Fed Am Societies Exp Biol (2005) 19:1332–4. doi: 10.1096/fj.04-1968fje
86. Böhm M, Bodó E, Funk W, Paus R. α-melanocyte-stimulating hormone: a protective peptide against chemotherapy-induced hair follicle damage? Br J Dermatol (2014) 170:956–60. doi: 10.1111/bjd.12759
87. Wahl SM, Wen J, Moutsopoulos N. TGF-beta: a mobile purveyor of immune privilege. Immunol Rev (2006) 213:213–27. doi: 10.1111/j.1600-065X.2006.00437.x
88. Rajabi F, Drake LA, Senna MM, Rezaei N. Alopecia areata: a review of disease pathogenesis. Br J Dermatol (2018) 179:1033–48. doi: 10.1111/bjd.16808
89. Jadeja SD, Tobin DJ. Autoantigen discovery in the hair loss disorder, alopecia areata: implication of post-translational modifications. Front Immunol (2022) 13:890027. doi: 10.3389/fimmu.2022.890027
90. Delacher M, Imbusch CD, Hotz-Wagenblatt A, Mallm JP, Bauer K, Simon M, et al. Precursors for nonlymphoid-tissue treg cells reside in secondary lymphoid organs and are programmed by the transcription factor BATF. Immunity. (2020) 52:295–312.e11. doi: 10.1016/j.immuni.2019.12.002
91. Muñoz-Rojas AR, Mathis D. Tissue regulatory T cells: regulatory chameleons. Nat Rev Immunol (2021) 21:597–611. doi: 10.1038/s41577-021-00519-w
92. Köhne M, Beyer M. ATAC-ing human tissue treg cells. Immunity. (2021) 54:605–7. doi: 10.1016/j.immuni.2021.03.014
93. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
94. Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. (2011) 480:538–42. doi: 10.1038/nature10664
95. Boothby IC, Cohen JN, Rosenblum MD. Regulatory T cells in skin injury: At the crossroads of tolerance and tissue repair. Sci Immunol (2020) 5. doi: 10.1126/sciimmunol.aaz9631
96. Chow Z, Mueller SN, Deane JA, Hickey MJ. Dermal regulatory T cells display distinct migratory behavior that is modulated during adaptive and innate inflammation. J Immunol (2013) 191:3049–56. doi: 10.4049/jimmunol.1203205
97. Mathur AN, Zirak B, Boothby IC, Tan M, Cohen JN, Mauro TM, et al. Treg-cell control of a CXCL5-IL-17 inflammatory axis promotes hair-Follicle-Stem-Cell differentiation during skin-barrier repair. Immunity. (2019) 50:655–67.e4. doi: 10.1016/j.immuni.2019.02.013
98. Kalekar LA, Cohen JN, Prevel N, Sandoval PM, Mathur AN, Moreau JM, et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aaw2910
99. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. (2015) 43:1011–21. doi: 10.1016/j.immuni.2015.10.016
100. Liu Z, Hu X, Liang Y, Yu J, Li H, Shokhirev MN, et al. Glucocorticoid signaling and regulatory T cells cooperate to maintain the hair-follicle stem-cell niche. Nat Immunol (2022) 23:1086–97. doi: 10.1038/s41590-022-01244-9
101. Hu XM, Li ZX, Zhang DY, Yang YC, Fu SA, Zhang ZQ, et al. A systematic summary of survival and death signalling during the life of hair follicle stem cells. Stem Cell Res Ther (2021) 12:453. doi: 10.1186/s13287-021-02527-y
102. Ali N, Rosenblum MD. Regulatory T cells in skin. Immunology. (2017) 152:372–81. doi: 10.1111/imm.12791
103. Maryanovich M, Frenette PS. T-Regulating hair follicle stem cells. Immunity. (2017) 46:979–81. doi: 10.1016/j.immuni.2017.06.011
104. Zhou C, Li X, Wang C, Zhang J. Alopecia areata: an update on etiopathogenesis, diagnosis, and management. Clin Rev Allergy Immunol (2021) 61:403–23. doi: 10.1007/s12016-021-08883-0
105. Strazzulla LC, Wang EHC, Avila L, Lo Sicco K, Brinster N, Christiano AM, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol (2018) 78:15–24. doi: 10.1016/j.jaad.2017.04.1142
106. Shreberk-Hassidim R, Ramot Y, Gilula Z, Zlotogorski A. A systematic review of pulse steroid therapy for alopecia areata. J Am Acad Dermatol (2016) 74:372–4. doi: 10.1016/j.jaad.2015.09.045
107. Yee BE, Tong Y, Goldenberg A, Hata T. Efficacy of different concentrations of intralesional triamcinolone acetonide for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol (2020) 82:1018–21. doi: 10.1016/j.jaad.2019.11.066
108. Freire PCB, Riera R, Martimbianco ALC, Petri V, Atallah AN. Minoxidil for patchy alopecia areata: systematic review and meta-analysis. J Eur Acad Dermatol Venereology JEADV. (2019) 33:1792–9. doi: 10.1111/jdv.15545
109. Ibrahim O, Bayart CB, Hogan S, Piliang M, Bergfeld WF. Treatment of alopecia areata with tofacitinib. JAMA Dermatol (2017) 153:600–2. doi: 10.1001/jamadermatol.2017.0001
110. Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol (2017) 76:29–32. doi: 10.1016/j.jaad.2016.09.006
111. Phan K, Sebaratnam DF. JAK inhibitors for alopecia areata: a systematic review and meta-analysis. J Eur Acad Dermatol Venereology JEADV. (2019) 33:850–6. doi: 10.1111/jdv.15489
112. Gilhar A, Keren A, Paus R. JAK inhibitors and alopecia areata. Lancet (London England). (2019) 393:318–9. doi: 10.1016/s0140-6736(18)32987-8
113. Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R, et al. Efficacy and safety of tofacitinib, an oral janus kinase inhibitor, in the treatment of psoriasis: a phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol (2012) 167:668–77. doi: 10.1111/j.1365-2133.2012.11168.x
114. Randall VA. Is alopecia areata an autoimmune disease? Lancet (London England) (2001) 358:1922–4. doi: 10.1016/s0140-6736(01)06943-4
115. Pratt CH, King LE Jr., Messenger AG, Christiano AM, Sundberg JP. Alopecia areata. nature reviews. Dis primers. (2017) 3:17011. doi: 10.1038/nrdp.2017.11
116. Gilhar A, Etzioni A, Paus R. Alopecia areata. New Engl J Med (2012) 366:1515–25. doi: 10.1056/NEJMra1103442
117. Gilhar A, Kalish RS. Alopecia areata: a tissue specific autoimmune disease of the hair follicle. Autoimmun Rev (2006) 5:64–9. doi: 10.1016/j.autrev.2005.07.001
118. Chu SY, Chen YJ, Tseng WC, Lin MW, Chen TJ, Hwang CY, et al. Comorbidity profiles among patients with alopecia areata: the importance of onset age, a nationwide population-based study. J Am Acad Dermatol (2011) 65:949–56. doi: 10.1016/j.jaad.2010.08.032
119. Strazzulla LC, Wang EHC, Avila L, Lo Sicco K, Brinster N, Christiano AM, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol (2018) 78:1–12. doi: 10.1016/j.jaad.2017.04.1141
120. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (Orlando Fla.). (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
121. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
122. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (London England). (2020) 395:1627–39. doi: 10.1016/s0140-6736(20)30167-7
123. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplantation. (2013) 19:1271–3. doi: 10.1016/j.bbmt.2013.06.004
124. Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest (2008) 118:3619–28. doi: 10.1172/jci33185
125. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Trans Med (2011) 3:83ra42. doi: 10.1126/scitranslmed.3002076
126. Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. (2007) 109:827–35. doi: 10.1182/blood-2006-05-025460
127. Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ treg. Eur J Immunol (2009) 39:1108–17. doi: 10.1002/eji.200839073
128. Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc Trans (2016) 44:342–8. doi: 10.1042/bst20150247
129. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) treg: a promising approach to inducing immunological tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
130. Riet T, Chmielewski M. Regulatory CAR-T cells in autoimmune diseases: progress and current challenges. Front Immunol (2022) 13:934343. doi: 10.3389/fimmu.2022.934343
131. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug discovery. (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
132. Harris DT, Kranz DM. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci (2016) 37:220–30. doi: 10.1016/j.tips.2015.11.004
133. Elinav E, Waks T, Eshhar Z. Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology. (2008) 134:2014–24. doi: 10.1053/j.gastro.2008.02.060
134. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126:1413–24. doi: 10.1172/jci82771
135. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J neuroinflammation. (2012) 9:112. doi: 10.1186/1742-2094-9-112
136. Mukhatayev Z, Dellacecca ER, Cosgrove C, Shivde R, Jaishankar D, Pontarolo-Maag K, et al. Antigen specificity enhances disease control by tregs in vitiligo. Front Immunol (2020) 11:581433. doi: 10.3389/fimmu.2020.581433
137. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-treg) cells: engineering and applications. Biomedicines. (2022) 10. doi: 10.3390/biomedicines10020287
138. Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, Lombardi G. The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Front Immunol (2020) 11:1608. doi: 10.3389/fimmu.2020.01608
139. Paus R, Slominski A, Czarnetzki BM. Is alopecia areata an autoimmune-response against melanogenesis-related proteins, exposed by abnormal MHC class I expression in the anagen hair bulb? Yale J Biol Med (1993) 66:541–54.
140. Gilhar A, Landau M, Assy B, Shalaginov R, Serafimovich S, Kalish RS. Melanocyte-associated T cell epitopes can function as autoantigens for transfer of alopecia areata to human scalp explants on prkdc(scid) mice. J Invest Dermatol (2001) 117:1357–62. doi: 10.1046/j.0022-202x.2001.01583.x
141. Bertolini M, Rossi A, Paus R. Cover image: are melanocyte-associated peptides the elusive autoantigens in alopecia areata? Br J Dermatol (2017) 176:1106. doi: 10.1111/bjd.15288
142. Trautman S, Thompson M, Roberts J, Thompson CT. Melanocytes: a possible autoimmune target in alopecia areata. J Am Acad Dermatol (2009) 61:529–30. doi: 10.1016/j.jaad.2009.01.017
143. Asz-Sigall D, Ortega-Springall MF, Smith-Pliego M, Rodríguez-Lobato E, Martinez-Velasco MA, Arenas R, et al. White hair in alopecia areata: clinical forms and proposed physiopathological mechanisms. J Am Acad Dermatol (2019). doi: 10.1016/j.jaad.2018.12.047
144. Kemp EH, Sandhu HK, Weetman AP, McDonagh AJ. Demonstration of autoantibodies against tyrosine hydroxylase in patients with alopecia areata. Br J Dermatol (2011) 165:1236–43. doi: 10.1111/j.1365-2133.2011.10597.x
145. Xie B, Sun J, Song X. Hair follicle melanocytes initiate autoimmunity in alopecia areata: a trigger point. Clin Rev Allergy Immunol (2022). doi: 10.1007/s12016-022-08954-w
146. Wang EHC, Yu M, Breitkopf T, Akhoundsadegh N, Wang X, Shi FT, et al. Identification of autoantigen epitopes in alopecia areata. J Invest Dermatol (2016) 136:1617–26. doi: 10.1016/j.jid.2016.04.004
147. Leung MC, Sutton CW, Fenton DA, Tobin DJ. Trichohyalin is a potential major autoantigen in human alopecia areata. J Proteome Res (2010) 9:5153–63. doi: 10.1021/pr100422u
148. Erb U, Freyschmidt-Paul P, Zöller M. Tolerance induction by hair-specific keratins in murine alopecia areata. J leukocyte Biol (2013) 94:845–57. doi: 10.1189/jlb.0413196
149. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J autoimmunity. (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
150. Gille I, Claas FHJ, Haasnoot GW, Heemskerk MHM, Heidt S. Chimeric antigen receptor (CAR) regulatory T-cells in solid organ transplantation. Front Immunol (2022) 13:874157. doi: 10.3389/fimmu.2022.874157
151. Dawson NAJ, Rosado-Sánchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Trans Med (2020) 12. doi: 10.1126/scitranslmed.aaz3866
152. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight (2019) 5. doi: 10.1172/jci.insight.126194
153. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol (2008) 9:194–202. doi: 10.1038/ni1549
154. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
155. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol (2004) 172:5149–53. doi: 10.4049/jimmunol.172.9.5149
156. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance t(reg) cell anabolic metabolism for suppression. Nat Immunol (2016) 17:1459–66. doi: 10.1038/ni.3577
157. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T Cell receptor signaling controls Foxp3 expression via PI3K, akt, and mTOR. Proc Natl Acad Sci United States America. (2008) 105:7797–802. doi: 10.1073/pnas.0800928105
158. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. (2010) 33:301–11. doi: 10.1016/j.immuni.2010.09.002
159. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
160. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
161. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. (2008) 111:453–62. doi: 10.1182/blood-2007-06-094482
162. Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol (Orlando Fla.). (2013) 146:248–61. doi: 10.1016/j.clim.2013.01.004
163. James CR, Buckle I, Muscate F, Otsuka M, Nakao M, Oon J, et al. Reduced interleukin-2 responsiveness impairs the ability of treg cells to compete for IL-2 in nonobese diabetic mice. Immunol Cell Biol (2016) 94:509–19. doi: 10.1038/icb.2016.7
164. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi: 10.1038/cr.2016.151
165. Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol (2007) 178:4022–6. doi: 10.4049/jimmunol.178.7.4022
166. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
167. D'Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol (2005) 6:1152–9. doi: 10.1038/ni1264
168. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6:1142–51. doi: 10.1038/ni1263
169. Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol (2011) 186:6329–37. doi: 10.4049/jimmunol.1100061
170. Castela E, Le Duff F, Butori C, Ticchioni M, Hofman P, Bahadoran P, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol (2014) 150:748–51. doi: 10.1001/jamadermatol.2014.504
171. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann rheumatic diseases. (2016) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
172. Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes. (2015) 64:2172–83. doi: 10.2337/db14-1322
173. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. New Engl J Med (2011) 365:2067–77. doi: 10.1056/NEJMoa1105143
174. Zhang J, Ruan Y, Xu X, Wang H, Tao Q, Lu J, et al. Therapeutic potential of low-dose IL-2 in immune thrombocytopenia: an analysis of 3 cases. cytometry. part b. Clin cytometry (2018) 94:428–33. doi: 10.1002/cyto.b.21601
175. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann rheumatic diseases. (2019) 78:209–17. doi: 10.1136/annrheumdis-2018-214229
176. Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci United States America. (2010) 107:3058–63. doi: 10.1073/pnas.0812851107
177. Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC. The structure of interleukin-2 complexed with its alpha receptor. Sci (New York N.Y.). (2005) 308:1477–80. doi: 10.1126/science.1109745
178. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments tregs yet transiently impairs β-cell function. Diabetes. (2012) 61:2340–8. doi: 10.2337/db12-0049
179. Donohue JH, Rosenberg SA. The fate of interleukin-2 after in vivo administration. J Immunol (1983) 130:2203–8.
180. Jhunjhunwala S, Balmert SC, Raimondi G, Dons E, Nichols EE, Thomson AW, et al. Controlled release formulations of IL-2, TGF-β1 and rapamycin for the induction of regulatory T cells. J Controlled release Off J Controlled Release Society. (2012) 159:78–84. doi: 10.1016/j.jconrel.2012.01.013
181. Padutsch T, Sendetski M, Huber C, Peters N, Pfizenmaier K, Bethea JR, et al. Superior treg-expanding properties of a novel dual-acting cytokine fusion protein. Front Pharmacol (2019) 10:1490. doi: 10.3389/fphar.2019.01490
182. Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J autoimmunity. (2018) 95:1–14. doi: 10.1016/j.jaut.2018.10.017
183. Karakus U, Sahin D, Mittl PRE, Mooij P, Koopman G, Boyman O. Receptor-gated IL-2 delivery by an anti-human IL-2 antibody activates regulatory T cells in three different species. Sci Trans Med (2020) 12. doi: 10.1126/scitranslmed.abb9283
184. Le Duff F, Bouaziz JD, Fontas E, Ticchioni M, Viguier M, Dereure O, et al. Low-dose IL-2 for treating moderate to severe alopecia areata: a 52-week multicenter prospective placebo-controlled study assessing its impact on T regulatory cell and NK cell populations. J Invest Dermatol (2021) 141:933–6.e6. doi: 10.1016/j.jid.2020.08.015
185. Lee E, Kim M, Lee YJ. Selective expansion of tregs using the IL-2 cytokine antibody complex does not reverse established alopecia areata in C3H/HeJ mice. Front Immunol (2022) 13:874778. doi: 10.3389/fimmu.2022.874778
186. Kolios AGA, Tsokos GC, Klatzmann D. Interleukin-2 and regulatory T cells in rheumatic diseases. Nat Rev Rheumatol (2021) 17:749–66. doi: 10.1038/s41584-021-00707-x
187. Hordinsky M, Kaplan DH. Low-dose interleukin 2 to reverse alopecia areata. JAMA Dermatol (2014) 150:696–7. doi: 10.1001/jamadermatol.2014.510
188. Ju W, Zhang M, Jiang JK, Thomas CJ, Oh U, Bryant BR, et al. CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood. (2011) 117:1938–46. doi: 10.1182/blood-2010-09-305425
Keywords: alopecia areata, regulatory T cell, autoimmune disease, immune homeostasis, hair follicle, Treg cell-based therapy, CAR-Treg cell, low-dose IL-2
Citation: Wan S, Xu W, Xie B, Guan C and Song X (2023) The potential of regulatory T cell-based therapies for alopecia areata. Front. Immunol. 14:1111547. doi: 10.3389/fimmu.2023.1111547
Received: 29 November 2022; Accepted: 07 April 2023;
Published: 02 May 2023.
Edited by:
Amra Adrovic, Koç University Hospital, TürkiyeReviewed by:
You Jeong Lee, Seoul National University, Republic of KoreaCopyright © 2023 Wan, Xu, Xie, Guan and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuzu Song, c29uZ3hpdXp1QHNpbmEuY29t; Cuiping Guan, aW1nY3BAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.