
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1111369
This article is part of the Research TopicAdenosine Pathways in Cancer Immunity and ImmunotherapyView all 14 articles
 Chifei Kang1,2†
Chifei Kang1,2† Luyu Liu3†Chengyu Wu4Lingyun Li1Xiao Jia1Wendi Xie1Siyu Chen1Xinying Wu1Huaxiao Zheng1
Luyu Liu3†Chengyu Wu4Lingyun Li1Xiao Jia1Wendi Xie1Siyu Chen1Xinying Wu1Huaxiao Zheng1 Jingxin Liu1*Rongsong Li2*Bin Zeng1*
Jingxin Liu1*Rongsong Li2*Bin Zeng1*Extracellular adenosine (eADO) signaling has emerged as an increasingly important regulator of immune responses, including tumor immunity. eADO is mainly produced from extracellular ATP (eATP) hydrolysis. eATP is rapidly accumulated in the extracellular space following cell death or cellular stress triggered by hypoxia, nutrient starvation, or inflammation. eATP plays a pro-inflammatory role by binding and activating the P2 purinergic receptors (P2X and P2Y), while eADO has been reported in many studies to mediate immunosuppression by activating the P1 purinergic receptors (A1, A2A, A2B, and A3) in diverse immune cells. Consequently, the hydrolysis of eATP to eADO alters the immunosurveillance in the tumor microenvironment (TME) not only by reducing eATP levels but also by enhancing adenosine receptor signaling. The effects of both P1 and P2 purinergic receptors are not restricted to immune cells. Here we review the most up-to-date understanding of the tumor adenosinergic system in all cell types, including immune cells, tumor cells, and stromal cells in TME. The potential novel directions of future adenosinergic therapies in immuno-oncology will be discussed.
Adenosine (ADO) is a metabolic intermediate involved in the ATP catabolism pathway and the synthesis of some important signaling molecules, such as cyclic adenosine monophosphate (cAMP) (1). Extracellular nucleotides, including purines and pyrimidines, have been unequivocally reported as signaling molecules involved in several systems such as blood pressure regulation, platelet activation, cardiovascular system remodeling, neurotransmission, anti-cell death, promotion of cell growth, and immunoregulation (2). Under physiological conditions, both ATP and ADO are usually at low levels in the extracellular space (3). Several cell conditions and stresses like cell membrane damage, ischemia, inflammation, and cancer could trigger the massive release of endogenous ATP in controlled manners such as regulated vesicular exocytosis and ion channel/transporter-mediated release but also in a direct cell-lytic way through cell destruction (Figure 1) (4–6). Thus, the accumulation of extracellular ATP (eATP) actually functions as a danger sign or nominated Danger-Associated Molecular Pattern (DAMP) to attract phagocytic cells to immigrate to the inflammatory sites and caution the whole immune system about the presence of pathogen-associated molecules and cell/tissue damage (7, 8). The activation of inflammation achieved by eATP is notably mediated through P2 purinergic receptors, including ligand-gated receptors (P2X) and metabotropic nucleotide-selective receptors (P2Y) (9, 10). Most family members of P2Y receptors promote oncogenic processes directly in tumor cells, while P2Y receptors in immune cells regulate these processes indirectly (11). Recent studies suggested that eATP activates P2X purinoceptor 7 (P2X7) expressed on macrophages, dendritic cells (DCs), granulocytes, T cells, and B cells to promote the formation of the NLRP3 inflammasome and the release of inflammatory cytokines such as IL-1β and IL-18 to enhance anti-tumor immunity (12–14). However, eATP is rapidly hydrolyzed to extracellular adenosine (eADO) in the tumor microenvironment (TME) since solid tumors normally have higher levels of ectonucleotidases than non-tumor tissues (15, 16).
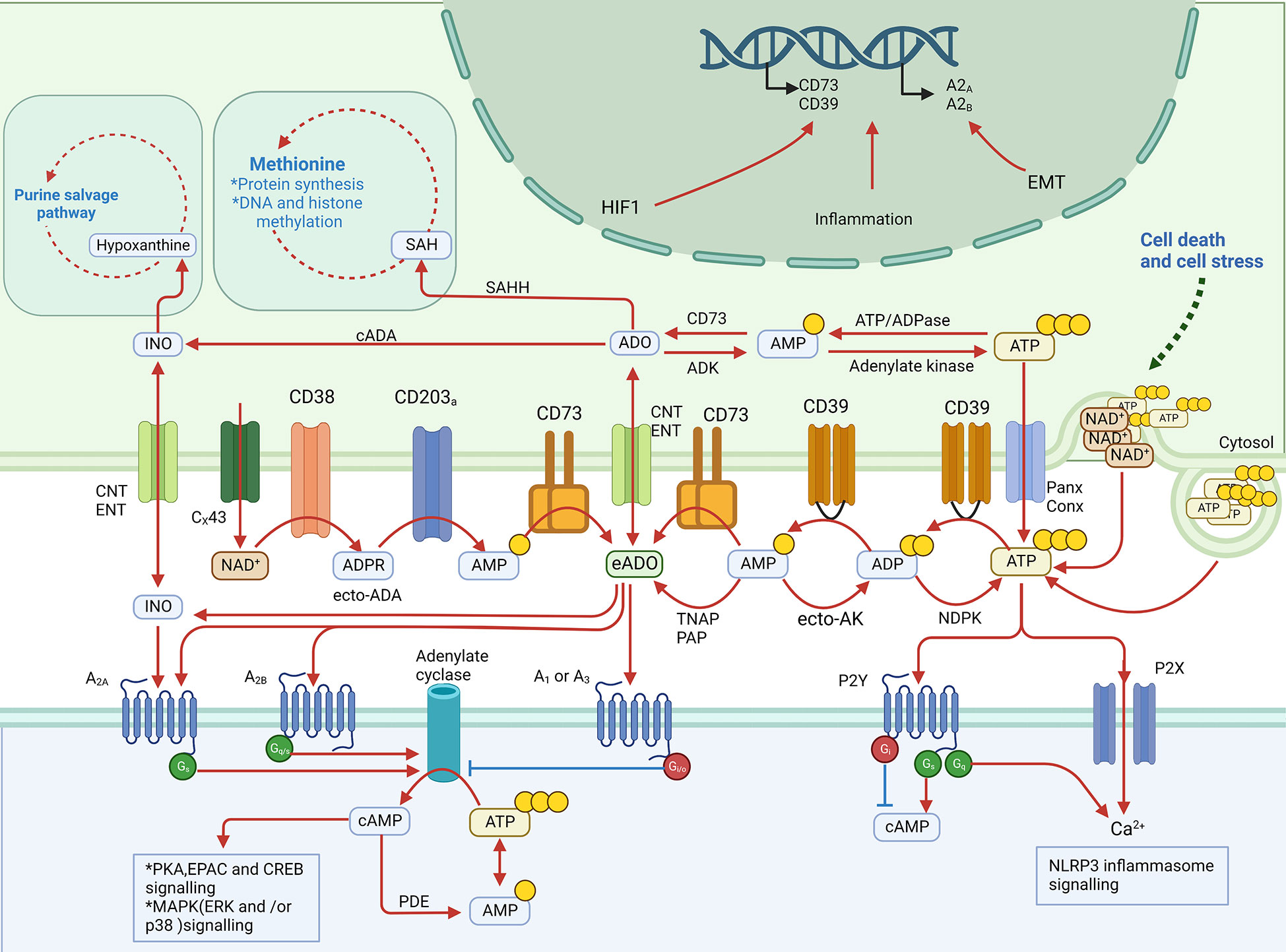
Figure 1 eADO metabolic pathways: production, degradation, and signaling.
eADO is primarily derived from the sequential hydrolysis of eATP mediated by several established ectonucleotidases (5). In a canonical route, eATP is hydrolyzed to extracellular ADP and AMP sequentially by CD39, which is known as ectonucleoside triphosphate diphosphohydrolase 1, and AMP is finally hydrolyzed to eADO by CD73, which is known as 5′-nucleotidase (17). However, the fate of eAMP is not limited to producing eADO; eAMP can also be phosphorylated sequentially to eATP by secreted or membrane-associated adenylate kinase (ecto-AK) and nucleoside diphosphate kinase (NDPK) (18).
The non-classical eADO production pathway is mediated by CD38, which is known as NAD+ ectohydrolase, and CD203a, which is known as ectonucleotide pyrophosphatase (19). Extracellular nicotinamide dinucleotide (NAD) released via gap junction protein connexin 43 (Cx43) regulation can be hydrolyzed to nicotinamide and ADP-ribose (ADPR) by CD38 (20, 21). Then CD203a consumes the ADPR to generate inorganic pyrophosphate and AMP, which are hydrolyzed by CD73 to eADO as mentioned above (19). In addition to CD73, prostatic acid phosphatase (22) and tissue-non-specific alkaline phosphatase (TNAP) were reported to hydrolyze eAMP to eADO (23, 24).
Analogous to eATP, in the extracellular space, the half-life of eADO is very short. The eADO molecule can be catalyzed directly into inosine by adenosine deaminase (ADA) and then into hypoxanthine by purine nucleoside phosphorylase (PNP) on the cell surface (25). eADO could also be transported into cells via concentrative nucleoside transporters (CNT1/2) or equilibrative nucleoside transporters (ENT1/2) (26). Inside cells, adenosine also has several metabolic pathways. The fundamental route is that intracellular ADO is phosphorylated by cytosolic adenylate kinase (ADK) to AMP, followed by conversion to ATP (27). Intracellular ADO could also be converted by cytosolic ADA (cADA) into inosine or by S-adenosyl-homocysteine hydrolase (SAHH) into S-adenosyl-homocysteine (SAH) involved in the methionine cycle (28). In conclusion, the eATP–CD39–CD73 pathway is the fundamental factor determining the concentration of eADO, but alternative ecto-enzymes also regulate metabolism, counteracting ATP-regenerating regulation.
Although the half-life of eADO is short, the concentration of eADO could remain high in TME. Cancer cell death due to rapid growth or chemotherapy contributes to ATP release and then eADO accumulation in the extracellular space (29). In addition to cancer cells, Treg cell deaths also provide ATP and CD39/CD73 to supply eADO production for immunosuppression in TME (30). Other than immune cells, cancer-associated fibroblasts (CAFs) in TME were reported to highly express CD73 induced by A2B receptor activation to sustain a high level of eADO concentration in colorectal cancer (31). Under physiological conditions, ADO plays a role in balancing the immune system’s activation and overreaction. However, in TME, all cell types are also regulated by adenosine signaling and involved in eADO production, which ultimately builds up the role of eADO as a tumor cell growth supporter.
eADO has its own specific receptors, which are P1 purinergic receptors. The P1 receptor family is composed of four G protein-coupled receptors: A1, A2A, A2B, and A3 (15, 32). These four receptors have different affinities for eADO. According to affinity, they can be roughly divided into two groups: A1, A2A, and A3 have affinities for eADO in the nanomolar range (100–310 nM), while A2B has a comparatively low affinity for eADO in the micromolar range (15 µM) (33). The common primary function of P1 receptor family members is to regulate adenylate cyclase activity, which means modulating the intracellular cAMP concentration (34). A1 and A3, which are Gi/o(Gi/Go)-coupled adenosine receptors, implement inhibition of adenylate cyclase to decrease the intracellular level of cAMP. In contrast, A2A and A2B, as Gq/s(Gq/Gs)-coupled adenosine receptors, increase the intracellular level of cAMP, which could potently dampen the immune response in some immune cells (35). A2A receptor is generally expressed on most immune cells—monocytes, macrophages, DCs, neutrophils, natural killer (NK) cells, T cells, and natural killer T (NKT) cells; meanwhile, A2B receptor is primarily highly expressed on macrophages and DCs (7).
In T cells (Figure 2), the pioneering work that provided evidence on the role of A2A-mediated immunosuppression in cancer can be traced to 20 years ago (36, 37). eADO binds to the A2A receptor to stimulate the accumulation of cAMP, leading to the activation of the cAMP-dependent protein kinase A (38) signaling pathway, which negatively regulates the activation of T-cell receptor (TCR)-dependent transmembrane signaling via providing an OFF signal to activated immune cells (36). In addition to the cAMP/PKA pathway, eADO receptors can also function through cAMP-independent pathways such as DAG/PKC, MAPK (ERK and/or p38), and PI3K/AKT/mTOR pathways (39). In T cells, the eADO-activated A2A receptor signaling-cAMP/PKA cascade triggers the direct inhibition of TCR activation via non-receptor tyrosine kinase (CSK). In addition, CSK inhibits CD28-mediated PI3K/AKT/mTORC pathways to decrease T cell protein synthesis, proliferation, and survival (40). PKA also phosphorylates the cAMP response element binding protein (CREB) to dampen the transcription activity of TCR downstream NF-κB (41, 42). In addition, PKA could activate SHP-2 and EPAC to impair T cell IL-2 receptor downstream signaling by inhibiting STAT5 and JAK, respectively, to suppress T-cell activation, survival, proliferation, and cytokine production (43–45). PKA inhibits KCa3.1 potassium channels, which causes extracellular Ca2+ cannot flux in through the calcium release-activated channels (CRAC) to suppress the upregulation of NFAT regulated genes which encode factors such as granzyme B (GzmB), IFNγ, TNF, IL-6, IL-17, IL-2, and IL-2R which are crucial to T-cell function and expansion (46). A2A receptor activation was also reported to upregulate the expression of T-cell suppressive receptors such as programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte antigen 4 (CTLA4), and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) so that T-cell immunosuppression is potentially enhanced (47, 48).
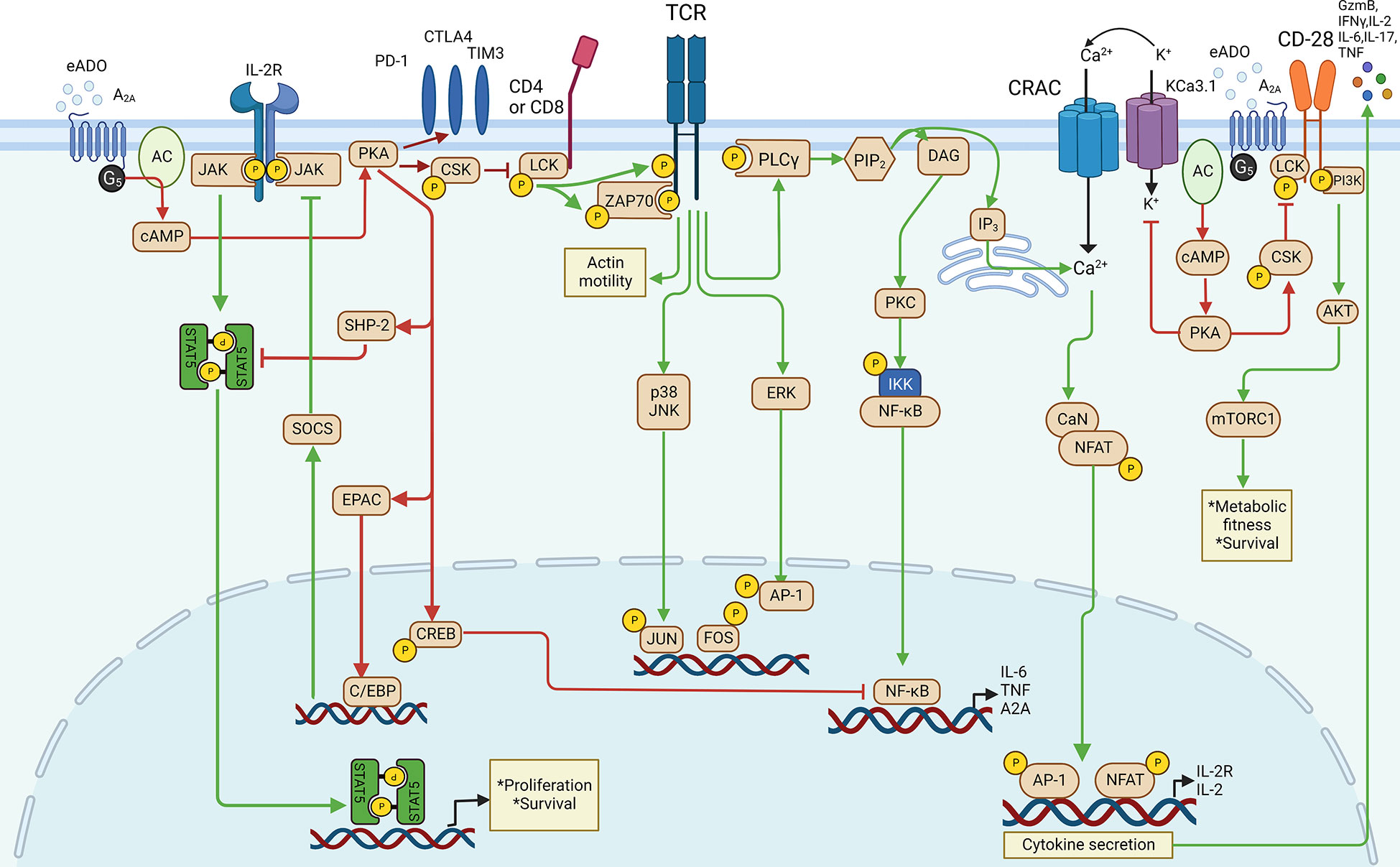
Figure 2 eADO/adenosine signaling in T cell.
In other immune cells (Figure 3), such as B cells, NF-κB, the downstream factor of the B cell receptor (BCR), and Toll-like receptor 4 (TLR4), are suppressed by PKA from activated A2A receptor signaling, hence disrupting B cell survival (49). PKA from A2A receptor signaling decreases production of IFNγ and perforin, which is the Fas ligand, to dampen the maturation and activity of NK cells (50, 51). A2A receptor activation reduces IFNγ production in NKT cells and inhibits NKT cell activation (52). In non-professional antigen-presenting cells (APCs), such as fibroblasts, A2B receptor-induced cAMP can suppress IFNγ-stimulated STAT1 activity and inhibit CIITA through upregulating TGFβ. The combined effects of this A2B receptor signaling lead to a decrease of MHC II transcription, which attenuates tumor immune response (53). In macrophages, the expression level of both A2A and A2B receptors is promoted by Toll-like receptor signaling (54, 55). Activation of both A2A and A2B receptor signaling favors the shift of macrophages towards a tolerogenic tumor-promoting “M2” phenotype polarization accompanied by increased production of immunosuppressive IL-10, IL-6, and VEGF as well as a decrease in pro-inflammatory IL-12 and THF (38, 56). Similarly, in dendritic cells (DCs), both A2A and A2B-mediated cAMP/PKA signaling enhance the production of IL-10, IL-6, VEGF, and TGFβ plus indoleamine 2,3-dioxygenase (IDO), cyclooxygenase 2 (COX2), and arginase 1/2 (ARG 1/2), which meanwhile dampen the expression of IL-12 and TNF (57). Based on most currently known data, in a sense, the A2A receptor elicits immunosuppression in both lymphocytes and myeloid cells. In contrast, the A2B receptor elicits immunosuppression mainly from myeloid cells.
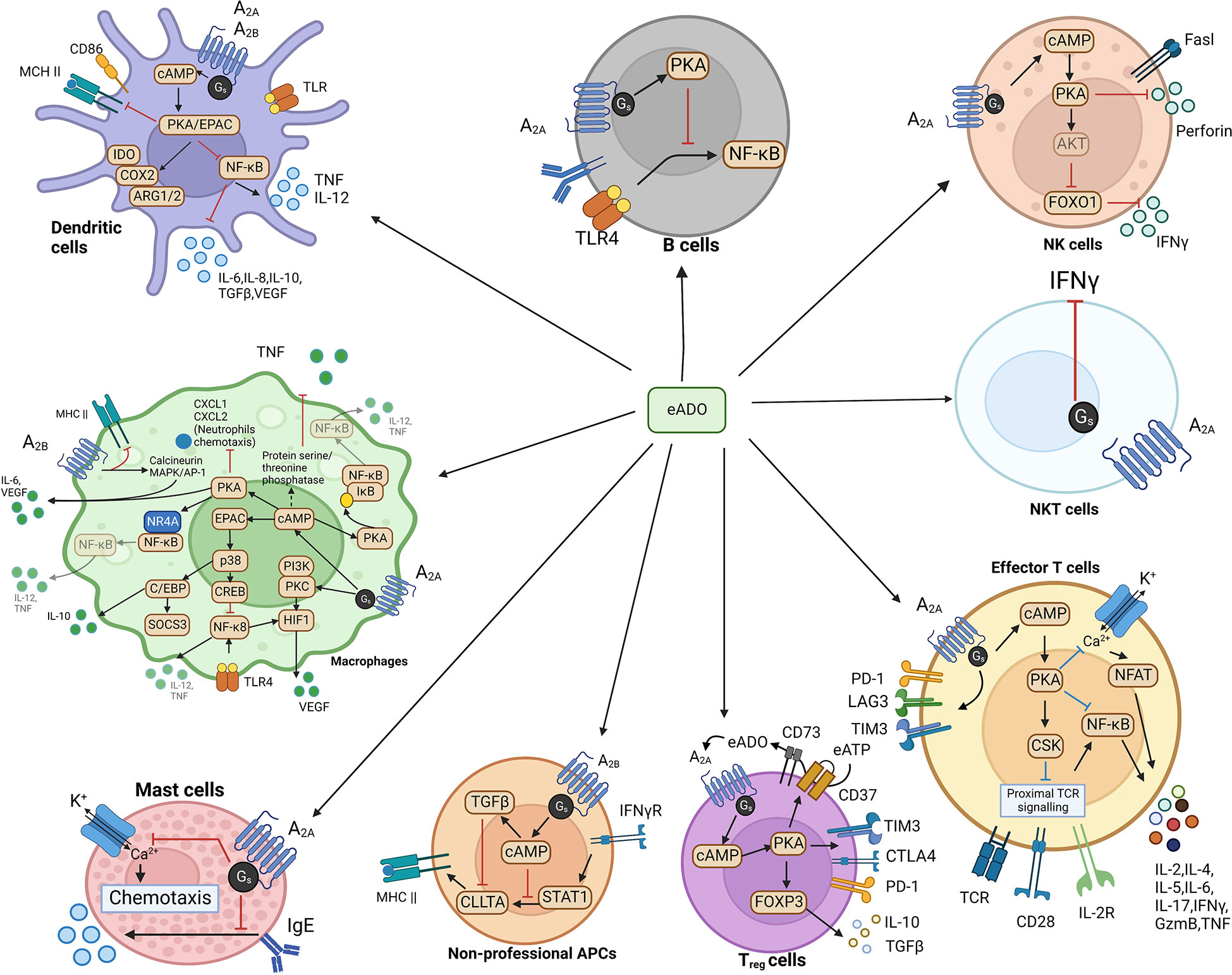
Figure 3 eADO/adenosine signaling in various immune cells.
In cancer cells: In the extracellular space of solid tumor TME, the concentration of eATP is considerably high due to both the passive release from tumor cell necrosis and the active secretion from tumor cells and other TME cells. Many factors, such as mechanical stress, starvation, hypoxia, and chronic inflammation, could induce this active secretion of ATP (12, 58). eATP binds to both P2X and P2Y, notably P2X7 expressed in immune cells in TME such as DCs, macrophages, B cells, and T cells (6, 12). The activation of P2X7 could promote calcium influx to enhance NLRP3 inflammasome formation, leading to antitumor immunity promotion (29, 59). In this regard, TME seems to provide a strategy to promote the inflammatory response, which could potentially contribute to antitumor activity. In fact, tumor cells express a higher level of ectonucleotidases such as CD39 and CD73 to execute the hydrolysis of eATP to eADO. In addition to the most reported transcription factor, hypoxia-inducible factor 1 (HIF1), several proteins such as TGFβ, TNF, IL-2, and IL-6 could enhance the expression of CD39 and CD73 (16, 60). As mentioned above, CD39 and CD73 are also generally expressed in immune cells; thus, ectonucleotidases from both tumor cells and immune cells together produce a large amount of eADO in the TME niche.
HIF1 as a transcription factor was found to increase the expression of CD39, CD73, A2A, and A2B as well as suppress the expression of both ENTs and adenylate kinase, leading to eADO accumulation in solid tumor TME, which is normally hypoxic (61–66). The upregulation of CD39, CD73, and A2B in various cancers was reported to positively correlate with poor prognosis in patients (60, 67). In particular, there have already been tremendous studies showing that high expression of CD39 and CD73 consistently correlated with poor prognosis in patients with those high incidence rates and malignant cancers such as ovarian, gastric, rectal, breast cancers (including TNBC), hepatocellular carcinoma (HCC), and non-small-cell lung cancers (NSCLCs) (68–73). This is the rationale that supports many current and ongoing clinical trials targeting CD39 and CD73.
More studies uncovered the molecular mechanisms involved in CD73 upregulation in cancer cells in addition to the regulation of HIF1 and TGFβ. Epithelial-to-Mesenchymal Transition (EMT) factors such as WNT/β-catenin pathway activators and TWIST were found to upregulate the expression of CD73 in human tumors (74). Mutations or upregulation of TP53, KRAS, BRAF, and EGFR also positively correlated with increased expression of CD73 in various human tumors (73, 75–77). In tumor cells, especially those with an EMT phenotype, CD73 and some factors like TGFβ form a positive feedback loop in that TGFβ signaling increases CD73 expression and CD73 produces more eADO stimulating A2A and A2B receptor pathways to favor TGFβ production and secretion; thus, CD73/eADO receptor signaling contributes to EMT promotion in cancer cells (78). Since the high concentration of extracellular NAD+ is present in the TME niche in some cancer types, probably due in part to the altered metabolism in cancer cells, the non-classical eADO production pathway mediated by CD38 also plays an influential role in eADO signaling in several solid tumors (22, 79).
The effects of eADO are not limited to immune cells to implement immunosuppression but also on cancer cells directly to regulate tumor proliferation, growth, anti-apoptosis, and metastasis. The PI3K/AKT/mTORC signaling pathway could be promoted upon eADO-mediated A2A receptor signaling to promote cell proliferation, tumor progression, and metastasis in melanoma, hepatocellular carcinoma, and gastric cancer (80–82). The A2B receptor was found to stimulate different downstream signaling compared to A2A in cancer cells. In TNBC cells, activation of the A2B receptor occurs notably via the ERK1/2-MAPK pathway. Knockdown of the A2B receptor in TNBC cells suppresses cancer cell proliferation and lung metastasis (67). A2B receptor signaling could activate FOS-related antigen 1 (FRA-1) and the small GTPase RAP1B to enhance TNBC cells’ lung metastasis in mouse models (83, 84). An intriguing finding is that A2B receptor signaling is constitutively activated in prostate cancer cells to promote cancer cell proliferation in vitro. However, activation is not dependent on the availability of the A2B receptor ligand, eADO. This study suggested potential adenosine-independent signaling under the A2B receptor in cancer cells (85). The EMT process has an unequivocal interaction with adenosine signaling. Enhancing EMT levels leads to increased CD73 expression and thus eADO receptor signaling, which in turn promotes the EMT process in ovarian cancer (68, 78). Cancer cells with an EMT phenotype usually exhibit cell stemness, which is suggested as a potential cancer stem cell. In breast cancer and glioblastoma, hypoxia-induced A2B receptor activation results in the maintenance of self-renewing tumor cells in the mouse model (86, 87). In a hepatocellular carcinoma study, CD73 was found to be upregulated, leading to A2A receptor activation, which results in cancer cells’ EMT and stemness promotion through increasing SOX9 expression and activity (88).
Not surprisingly, drugs designed to target the adenosine signaling pathway have been blooming vigorously for the last decade. Strategies for targeting adenosine signaling pathway could generally be classified into two groups: ① inhibition of adenosine production and prevention of ATP degradation simultaneously in TME via targeting CD73 and/or CD39; and ② interruption of adenosine signaling through blocking A2A and A2B receptors. According to ongoing pre-clinical research and clinical trials, drugs targeting the CD73 and A2A receptors are the mainstream adenosine pathway inhibitors. Most CD73 inhibitors are monoclonal antibodies for potential pharmacological application, whereas small-molecule inhibitors are currently the only available clinical drugs targeting A2A and A2B receptors since they are G protein-coupled receptors (GPCRs) with specific conformations notoriously difficult for antibody binding.
Targeting A2A and/or A2B in cancer: A2A antagonists were initially developed for neurological disorders such as Parkinson’s disease or adult attention deficit hyperactivity disorder (ADHD) (89, 90). Their evaluations in clinical trials suggested a great tolerability and safety profile. The available preliminary data of several A2A antagonists in clinical trials with cancer patients showed good tolerability and exhibited some effects. They are CPI-444 (Corvus), PBF-509 (Novartis/Pablobiofarma), EOS100850 (iTeos), MK-3814 (Merck), AZD4635 (AstraZeneca/Heptares), and a dual A2A and A2B antagonist AB928 (Arcus) (91–96). PBF-1129 (Pablobiofarma), a selective A2B antagonist, has also been developed and is being tested in a clinical trial involving NSCLC cancer patients. In two clinical trials, CPI-444 was administered alone and in combination with Atezolizumab (PD-L1 antibody, Genentech) in patients with renal and advanced metastatic castration-resistant prostate cancer (91). Most common adverse events are in grades 1–2, including fatigue, pruritus, nausea, diarrhea, rash, vomiting, and anemia as well as several in grades 3–4, such as decreased appetite, anemia, arthralgia, and peripheral edema. A better outcome (median progression-free survival of 5.8 months versus 4.1 months and overall survival of 90% versus 55% at 20 weeks) was observed with the A2A antagonist CPI-444 plus the anti-PD-L1 antibody atezolizumab compared to CPI-444 alone in patients with advanced-stage renal cell carcinoma (91). Similar results have been reported in patients with mCRPC: 57% of patients (eight of 14) experienced disease control, with five partial responses and two stable disease responses.
Targeting CD73 and/or CD39: There are several anti-CD73 monoclonal antibodies in phase I/II clinical trials currently, including MEDI9447 (MedImmune), BMS-986179 (BMS), NZV930 (Novartis), and CPI-006 (Corvus), as well as a small molecule inhibitor, AB680 (Arcus) (97–99). In these clinical trials, CD73 inhibitors were administered alone and in combination with PD-1/PD-L1 monoclonal antibodies. Most adverse events were mild, and most outcomes indicated a decreased primary tumor expansion rate, less metastasis formation, and an improved survival rate (99). In addition to CD73, monoclonal antibodies and small-molecule antagonists to CD39 and CD38 are also under development (22, 99).
Targeting drugs are listed in Table 1.
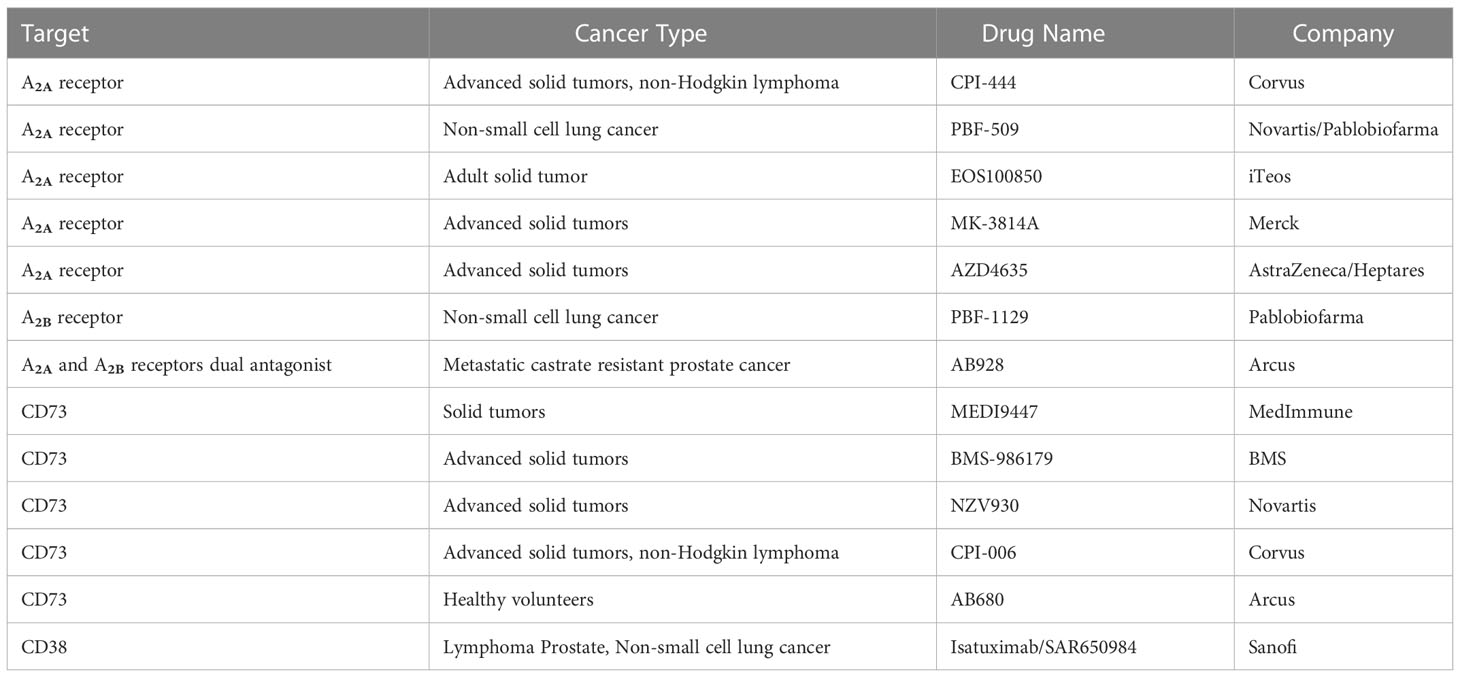
Table 1 Representative eADO pathway-targeting drugs which were involved in the most recent clinical trials.
The existing controversial effects of adenosine blockage in cancer: The prevalent view is that eADO production and eADOA/ARs signaling activation are associated with poor clinical outcomes. However, it is not substantial for every type of cancer. A group found that in endometrial carcinoma, CD73 played a critical role in tumor suppression (100), whereas another group reported that in endometrial carcinoma, the loss of CD73 is essential for tumor progression (101). Although several studies found a link between A2A expression or activation and poor outcomes in breast cancer, Vasiukov et al. revealed a positive correlation between A2A receptor gene expression and better survival data in basal-type breast cancer and TNBC patients (102). In addition, adenosine receptors (ARs) also exhibit both stimulatory and inhibitory effects in melanoma (80). A similar contradictory effect of adenosine receptors on hepatocellular carcinoma progression has also been reported (103). More mechanisms and pre-clinical studies are necessary to provide fundamental knowledge for adenosine targeting therapy.
Specificity issue in adenosine receptor blockage: As mentioned, adenosine receptors are members of the GPCR family. The conformational complexity of GPCR gives rise to the difficulty of developing antibodies to target the receptors. The currently available pharmacological inhibitors of ARs are small molecules that have the notorious disadvantage of engaging of multiple targets (poly-pharmacology). Several compounds, which were previously confirmed as binding interactors of A1, A2, and A3 receptors, were found to have intracellular binding targets (104, 105). In addition, the putative selective A2B receptor agonist BAY 60-6583 was reported to have other binding molecules to increase CAR-T cell activity independently of the A2B receptor (106).
There is still a large amount of work to be done to pursue better safety and efficacy in adenosine signaling targeting therapy.
Both eATP and eADO are important signal molecules in the physiological processes of cells and tissues. Tissue damage or various cell stresses such as hypoxia, starvation, and mechanical stress, which are common in the TME niche, could stimulate eATP accumulation and rapid hydrolysis to eADO. This would lead to dramatically increased eADO. This eATP–eADO metabolic pathway is involved in pathological shifts in several aspects: rapid eATP degradation dampens the inflammatory response; accumulation of eADO triggers immunosuppression; and it promotes tumor cell proliferation and EMT.
In adenosine signaling, pre-clinical studies suggested the CD39–CD73–A2A receptor pathway is an attractive and tractable therapeutic target for cancer treatment. Inhibitors targeting the CD73 and A2A receptors exhibited good tolerability and achieved some therapeutic effects in some clinical trials. However, several knowledge gaps are worthy of exploring to assist further pre-clinical and clinical trial design (1): What are the potential compensation pathways for the inhibition of eADO signaling? They are probably not limited to intracellular ADO release and ADO-independent adenosine receptor activation. (2) More combined therapies, such as immune checkpoint blockers and adenosine signaling inhibitors, have shown better efficacy. (3) What are reliable biomarkers to indicate which patient subgroups have a higher chance of benefiting from treatments targeting eADO signaling? In conclusion, the adenosinergic system offers new therapeutic strategies aimed at limiting immunosuppression and potentiating antitumor immune responses.
BZ, RL, and JL, carried out the concepts, design, definition of intellectual content, literature search. CK and LLiu contributed writing and editing the manuscript. LLi, XJ, and HZ helped in formatting the manuscript. CW helped perform the manuscript with constructive discussions. WX, SC, and XW carried out the figures in manuscript. All authors contributed to the article and approved the submitted version.
This research received financial support by the Key Special Projects of National Key Research and Development Plan (2021YFA1301302), The Belt and Road Initiative and China-Africa Science and Technology Cooperation Project of Department of Science and Technology of Jiangxi Province (20202BDH80007), the National Natural Science Foundation of China (32200606), and the Natural Science Foundation of Top Talent of SZTU (GDRC202118), the Natural Science Foundation of Top Talent of SZTU (2020102), Shenzhen Natural Science Foundation (JCYJ20190813141001745, 20220718100823002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Moser GH, Schrader J, Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol (1989) 256(4 Pt 1):C799–806. doi: 10.1152/ajpcell.1989.256.4.C799
2. Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res (2019) 47(D1):D766–73. doi: 10.1093/nar/gky955
3. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57(13):2602–5.
4. Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal (2012) 8(3):359–73. doi: 10.1007/s11302-012-9304-9
5. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta (2008) 1783(5):673–94. doi: 10.1016/j.bbamcr.2008.01.024
6. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
7. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol (2016) 16(3):177–92. doi: 10.1038/nri.2016.4
8. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med (2012) 367(24):2322–33. doi: 10.1056/NEJMra1205750
9. Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev (1998) 50(3):413–92.
10. Bours MJ, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112(2):358–404. doi: 10.1016/j.pharmthera.2005.04.013
11. Woods LT, Forti KM, Shanbhag VC, Camden JM, Weisman GA. P2Y receptors for extracellular nucleotides: Contributions to cancer progression and therapeutic implications. Biochem Pharmacol (2021) 187:114406. doi: 10.1016/j.bcp.2021.114406
12. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18(10):601–18. doi: 10.1038/s41568-018-0037-0
13. Borges da Silva H, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature (2018) 559(7713):264–8. doi: 10.1038/s41586-018-0282-0
14. De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, et al. The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene (2019) 38(19):3636–50. doi: 10.1038/s41388-019-0684-y
15. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov (2013) 12(4):265–86. doi: 10.1038/nrd3955
16. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
17. Yegutkin GG, Henttinen T, Samburski SS, Spychala J, Jalkanen S. The evidence for two opposite, ATP-generating and ATP-consuming, extracellular pathways on endothelial and lymphoid cells. Biochem J (2002) 367(Pt 1):121–8. doi: 10.1042/bj20020439
18. Yegutkin GG, Henttinen T, Jalkanen S. Extracellular ATP formation on vascular endothelial cells is mediated by ecto-nucleotide kinase activities via phosphotransfer reactions. FASEB J (2001) 15(1):251–60. doi: 10.1096/fj.00-0268com
19. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology (2013) 2(9):e26246. doi: 10.4161/onci.26246
20. Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J (2001) 15(1):10–2. doi: 10.1096/fj.00-0566fje
21. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev (2008) 88(3):841–86. doi: 10.1152/physrev.00035.2007
22. Chen L, Diao LX, Yang YB, Yi XH, Rodriguez L, Li YL, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discovery (2018) 8(9):1156–75. doi: 10.1158/2159-8290.CD-17-1033
23. Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron (2008) 60(1):111–22. doi: 10.1016/j.neuron.2008.08.024
24. Street SE, Kramer NJ, Walsh PL, Taylor-Blake B, Yadav MC, King IF, et al. Tissue-nonspecific alkaline phosphatase acts redundantly with PAP and NT5E to generate adenosine in the dorsal spinal cord. J Neurosci (2013) 33(27):11314–22. doi: 10.1523/JNEUROSCI.0133-13.2013
25. Yegutkin GG. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit Rev Biochem Mol Biol (2014) 49(6):473–97. doi: 10.3109/10409238.2014.953627
26. Young JD. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans (2016) 44(3):869–76. doi: 10.1042/BST20160038
27. Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev (2013) 65(3):906–43. doi: 10.1124/pr.112.006361
28. Zimmerman TP, Wolberg G, Duncan GS, Elion GB. Adenosine analogues as substrates and inhibitors of s-adenosylhomocysteine hydrolase in intact lymphocytes. Biochemistry (1980) 19(10):2252–9. doi: 10.1021/bi00551a040
29. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma YT, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334(6062):1573–7. doi: 10.1126/science.1208347
30. Maj T, Wang W, Crespo J, Zhang HJ, Wang WM, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
31. Yu M, Guo G, Huang L, Deng LB, Chang CS, Achyut BR, et al. CD73 on cancer-associated fibroblasts enhanced by the A(2B)-mediated feedforward circuit enforces an immune checkpoint. Nat Commun (2020) 11(1):515. doi: 10.1038/s41467-019-14060-x
32. Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol Sci (2016) 37(6):419–34. doi: 10.1016/j.tips.2016.02.006
33. Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim Biophys Acta (2011) 1808(5):1290–308. doi: 10.1016/j.bbamem.2010.12.017
34. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of adenosine receptors: The state of the art. Physiol Rev (2018) 98(3):1591–625. doi: 10.1152/physrev.00049.2017
35. Kunzli BM, Bernlochner MI, Rath S, Kaser S, Csizmadia E, Enjyoji K, et al. Impact of CD39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal (2011) 7(2):231–41. doi: 10.1007/s11302-011-9228-9
36. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414(6866):916–20. doi: 10.1038/414916a
37. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev LD, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA (2006) 103(35):13132–7. doi: 10.1073/pnas.0605251103
38. Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J (2012) 26(1):376–86. doi: 10.1096/fj.11-190934
39. Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochim Biophys Acta (2011) 1808(5):1400–12. doi: 10.1016/j.bbamem.2010.09.020
40. Bjorgo E, Tasken K. Novel mechanism of signaling by CD28. Immunol Lett (2010) 129(1):1–6. doi: 10.1016/j.imlet.2010.01.007
41. Lukashev D, Ohta A, Apasov S, Chen JF. Cutting edge: Physiologic attenuation of proinflammatory transcription by the gs protein-coupled A2A adenosine receptor in vivo. J Immunol (2004) 173(1):21–4. doi: 10.4049/jimmunol.173.1.21
42. Sitkovsky MV. T Regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol (2009) 30(3):102–8. doi: 10.1016/j.it.2008.12.002
43. Zhang H, et al. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3',5'-monophosphate and phosphatases. J Immunol (2004) 173(2):932–44. doi: 10.4049/jimmunol.173.2.932
44. Raskovalova T, Lokshin A, Huang XJ, Su YY, Mandic M, Zarour HM, et al. Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase a type I signaling. Cancer Res (2007) 67(12):5949–56. doi: 10.1158/0008-5472.CAN-06-4249
45. Csoka B, et al. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J (2008) 22(10):3491–9. doi: 10.1096/fj.08-107458
46. Chimote AA, Balajthy A, Arnold MJ, Newton HS, Hajdu P, Qualtieri J, et al. A defect in KCa3.1 channel activity limits the ability of CD8(+) T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci Signal (2018) 11(527). doi: 10.1126/scisignal.aaq1616
47. Leone RD, Sun IM, Oh MH, Sun IH, Wen JY, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother (2018) 67(8):1271–84. doi: 10.1007/s00262-018-2186-0
48. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi: 10.3389/fimmu.2012.00190
49. Minguet S, Huber M, Rosenkranz L, Schamel WWA, Reth M, Brummer T, et al. Adenosine and cAMP are potent inhibitors of the NF-kappa b pathway downstream of immunoreceptors. Eur J Immunol (2005) 35(1):31–41. doi: 10.1002/eji.200425524
50. Raskovalova T, Lokshin A, Huang XJ, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res (2006) 36(1-3):91–9. doi: 10.1385/IR:36:1:91
51. Raskovalova T, et al. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol (2005) 175(7):4383–91. doi: 10.4049/jimmunol.175.7.4383
52. Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J, et al. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med (2006) 203(12):2639–48. doi: 10.1084/jem.20061097
53. Fang M, Xia J, Wu XY, Kong H, Wang H, Xie WP, et al. Adenosine signaling inhibits CIITA-mediated MHC class II transactivation in lung fibroblast cells. Eur J Immunol (2013) 43(8):2162–73. doi: 10.1002/eji.201343461
54. Cohen HB, Ward A, Hamidzadeh K, Ravid K, Mossern DM. IFN-gamma prevents adenosine receptor (A2bR) upregulation to sustain the macrophage activation response. J Immunol (2015) 195(8):3828–37. doi: 10.4049/jimmunol.1501139
55. Ramanathan M, Luo W, Csóka B, Haskó G, Lukashev D, Sitkovsky MV, et al. Differential regulation of HIF-1alpha isoforms in murine macrophages by TLR4 and adenosine A(2A) receptor agonists. J Leukoc Biol (2009) 86(3):681–9. doi: 10.1189/jlb.0109021
56. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation (2013) 36(4):921–31. doi: 10.1007/s10753-013-9621-3
57. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang YH, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood (2008) 112(5):1822–31. doi: 10.1182/blood-2008-02-136325
58. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res (2010) 70(3):855–8. doi: 10.1158/0008-5472.CAN-09-3566
59. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38(4):729–41. doi: 10.1016/j.immuni.2013.03.003
60. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
61. Eltzschig HK, et al. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood (2009) 113(1):224–32. doi: 10.1182/blood-2008-06-165746
62. Synnestvedt K, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110(7):993–1002. doi: 10.1172/JCI0215337
63. Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP, et al. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J (2006) 20(13):2242–50. doi: 10.1096/fj.06-6419com
64. Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo XL, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci USA (2009) 106(26):10684–9. doi: 10.1073/pnas.0901326106
65. Morote-Garcia JC, Rosenberger P, Nivillac NMI, Coe MR, Eltzschig HK, et al. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology (2009) 136(2):607–18. doi: 10.1053/j.gastro.2008.10.037
66. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med (2005) 202(11):1493–505. doi: 10.1084/jem.20050177
67. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res (2016) 76(15):4372–82. doi: 10.1158/0008-5472.CAN-16-0544
68. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res (2015) 75(21):4494–503. doi: 10.1158/0008-5472.CAN-14-3569
69. Cai XY, Wang XF, Li J, Dong JN, Liu JQ, Li NP, et al. Overexpression of CD39 and high tumoral CD39(+)/CD8(+) ratio are associated with adverse prognosis in resectable gastric cancer. Int J Clin Exp Pathol (2015) 8(11):14757–64.
70. Zhang B, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. The expression and clinical significance of CD73 molecule in human rectal adenocarcinoma. Tumour Biol (2015) 36(7):5459–66. doi: 10.1007/s13277-015-3212-x
71. Loi S, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A (2013) 110(27):11091–6. doi: 10.1073/pnas.1222251110
72. Cai XY, Ni XC, Yi Y, He HW, Wang JX, Fu YP, et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Med (Baltimore) (2016) 95(40):e4989. doi: 10.1097/MD.0000000000004989
73. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget (2017) 8(5):8738–51. doi: 10.18632/oncotarget.14434
74. Spychala J, Kitajewski J. Wnt and beta-catenin signaling target the expression of ecto-5'-nucleotidase and increase extracellular adenosine generation. Exp Cell Res (2004) 296(2):99–108. doi: 10.1016/j.yexcr.2003.11.001
75. Lawrence RT, Hernandez D, Miller CP, Haas KM, Irie HY. The proteomic landscape of triple-negative breast cancer. Cell Rep (2015) 11(4):630–44. doi: 10.1016/j.celrep.2015.03.050
76. Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther (2011) 10(2):336–46. doi: 10.1158/1535-7163.MCT-10-0750
77. Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res (2017) 77(17):4697–709. doi: 10.1158/0008-5472.CAN-17-0395
78. Lupia M, Angiolini F, Bertalot G, Freddi S, Sachsenmeier KF, Chisci E, et al. CD73 regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Rep (2018) 10(4):1412–25. doi: 10.1016/j.stemcr.2018.02.009
79. Horenstein AL, et al. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol Med (2016) 22:694–704. doi: 10.2119/molmed.2016.00198
80. Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, et al. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol (2002) 119(4):923–33. doi: 10.1046/j.1523-1747.2002.00111.x
81. Ma XL, Shen MN, Hu B, Wang BL, Yang WJ, Lv LH, et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110beta and predicts poor prognosis. J Hematol Oncol (2019) 12(1):37. doi: 10.1186/s13045-019-0724-7
82. Shi L, Wu Z, Miao J, Du S, Ai S, Xu E, et al. Adenosine interaction with adenosine receptor A2a promotes gastric cancer metastasis by enhancing PI3K-AKT-mTOR signaling. Mol Biol Cell (2019) 30(19):2527–34. doi: 10.1091/mbc.E19-03-0136
83. Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, et al. Identification of a pharmacologically tractable fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci U S A (2013) 110(13):5139–44. doi: 10.1073/pnas.1222085110
84. Ntantie E, Gonyo P, Lorimer EL, Hauser AD, Schuld N, McAllister D, et al. An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering. Sci Signal (2013) 6(277):ra39. doi: 10.1126/scisignal.2003374
85. Vecchio EA, Tan CYR, Gregory KJ, Christopoulos A, White PJ, May LT, et al. Ligand-independent adenosine A2B receptor constitutive activity as a promoter of prostate cancer cell proliferation. J Pharmacol Exp Ther (2016) 357(1):36–44. doi: 10.1124/jpet.115.230003
86. Lan J, Lub H, Samantab D, Salmanb S, Gregg Lua YL. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci U S A (2018) 115(41):E9640–8. doi: 10.1073/pnas.1809695115
87. Liu TZ, Wang X, Bai YF, Liao HZ, Qiu SC, Yang YQ, et al. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int J Biochem Cell Biol (2014) 49:8–16. doi: 10.1016/j.biocel.2014.01.007
88. Ma XL, Hu B, Tang WG, Xie SH, Ren N, Guo L, et al. CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J Hematol Oncol (2020) 13(1):11. doi: 10.1186/s13045-020-0845-z
89. Pinna A. Adenosine A2A receptor antagonists in parkinson's disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs (2014) 28(5):455–74. doi: 10.1007/s40263-014-0161-7
90. Franco R, Navarro G. Adenosine A(2A) receptor antagonists in neurodegenerative diseases: Huge potential and huge challenges. Front Psychiatry (2018) 9:68. doi: 10.3389/fpsyt.2018.00068
91. Fong L, Hotson A, Powderly J, Sznol M, Heist RS, Choueiri TK. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discovery (2020) 10(1):40–53. doi: 10.1158/2159-8290.CD-19-0980
92. Chiappori AA, et al. Phase I study of taminadenant (PBF509/NIR178), an adenosine 2A receptor antagonist, with or without spartalizumab (PDR001), in patients with advanced non-small cell lung cancer. Clin Cancer Res (2022) 28(11):2313–20. doi: 10.1158/1078-0432.CCR-21-2742
93. Cortiula F, et al. Immunotherapy in unresectable stage III non-small-cell lung cancer: state of the art and novel therapeutic approaches. Ann Oncol (2022) 33(9):893–908. doi: 10.1016/j.annonc.2022.06.013
94. Cutler DL, Tendolkar A, Grachev ID. Safety, tolerability and pharmacokinetics after single and multiple doses of preladenant (SCH420814) administered in healthy subjects. J Clin Pharm Ther (2012) 37(5):578–87. doi: 10.1111/j.1365-2710.2012.01349.x
95. Lim EA, et al. Phase ia/b, open-label, multicenter study of AZD4635 (an adenosine A2A receptor antagonist) as monotherapy or combined with durvalumab, in patients with solid tumors. Clin Cancer Res (2022) 28(22):4871–84. doi: 10.1158/1078-0432.CCR-22-0612
96. Seitz L, Jin L, Leleti M, Ashok D, Jeffrey J, Rieger A, et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Invest New Drugs (2019) 37(4):711–21. doi: 10.1007/s10637-018-0706-6
97. Herbst RS, et al. COAST: An open-label, phase II, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage III non-Small-Cell lung cancer. J Clin Oncol (2022) 40(29):3383–93. doi: 10.1200/JCO.22.00227
98. Lee DH. Update of early phase clinical trials in cancer immunotherapy. BMB Rep (2021) 54(1):70–88. doi: 10.5483/BMBRep.2021.54.1.242
99. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
100. Kurnit KC, Draisey A, Kazen RC, Chung C, Phan LH, Harvey JB, et al. Loss of CD73 shifts transforming growth factor-beta1 (TGF-beta1) from tumor suppressor to promoter in endometrial cancer. Cancer Lett (2021) 505:75–86. doi: 10.1016/j.canlet.2021.01.030
101. Bowser JL, Blackburn MR, Shipley GL, Molina JG, Dunner K, Broaddus RR, et al. Loss of CD73-mediated actin polymerization promotes endometrial tumor progression. J Clin Invest (2016) 126(1):220–38. doi: 10.1172/JCI79380
102. Vasiukov G, Novitskaya T, Zijlstra A, Owens P, Ye F, Zhao Z, et al. Myeloid cell-derived TGFbeta signaling regulates ECM deposition in mammary carcinoma via adenosine-dependent mechanisms. Cancer Res (2020) 80(12):2628–38. doi: 10.1158/0008-5472.CAN-19-3954
103. Ni S, Wei Q, Yang L. ADORA1 promotes hepatocellular carcinoma progression via PI3K/AKT pathway. Onco Targets Ther (2020) 13:12409–19. doi: 10.2147/OTT.S272621
104. Man S, Lu Y, Yin L, Cheng X, Ma L. Potential and promising anticancer drugs from adenosine and its analogs. Drug Discovery Today (2021) 26(6):1490–500. doi: 10.1016/j.drudis.2021.02.020
105. Jensen K, Johnson LAA, Jacobson PA, Kachler S, Kirstein MN, Lamba J, et al. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol (2012) 385(5):519–25. doi: 10.1007/s00210-011-0719-6
Keywords: adenosine, machinery and mechanisms, cancer, therapy, EADO
Citation: Kang C, Liu L, Wu C, Li L, Jia X, Xie W, Chen S, Wu X, Zheng H, Liu J, Li R and Zeng B (2023) The adenosinergic machinery in cancer: In-tandem insights from basic mechanisms to therapy. Front. Immunol. 14:1111369. doi: 10.3389/fimmu.2023.1111369
Received: 29 November 2022; Accepted: 31 January 2023;
Published: 23 February 2023.
Edited by:
Junjiang Fu, Southwest Medical University, ChinaReviewed by:
Mauricio Díaz-Muñoz, National Autonomous University of Mexico, MexicoCopyright © 2023 Kang, Liu, Wu, Li, Jia, Xie, Chen, Wu, Zheng, Liu, Li and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingxin Liu, bGl1amluZ3hpbkBzenR1LmVkdS5jbg==; Rongsong Li, bGlyb25nc29uZ0BzenR1LmVkdS5jbg==; Bin Zeng, emVuZ2JpbkBzenR1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.