 Bing Wang
Bing Wang Can Chen
Can Chen Xia Liu1
Xia Liu1- 1Department of Rheumatology and Immunology, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China
- 2Department of Oncology, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease involving multiple organs. It is often called “immortal cancer” due to the difficulties in disease treatment. As the cornerstone of immune regulation, the programmed cell death protein 1 (PD-1) has been extensively studied in the context of chronic inflammation due to its ability of regulating immune response and immunosuppression. Recently, more and more studies on rheumatic immune related complications have also focused on PD-1 and proposed that the use of PD-1 agonist could inhibit the activation of lymphocytes and alleviate SLE disease activity. In this review, we summarized the role of PD-1 in SLE, implicating its potential application as a biomarker to predict SLE disease activity; we also proposed that the combination of PD-1 agonist and low-dose IL-2 may have better therapeutic efficacy, shining light on a new direction for developing specific treatment approaches.
1 Introduction
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease, which can affect multiple systems of the body and has high heterogeneity (1, 2). The pathogenesis of SLE involves abnormal activation of immune system and, lymphocytes, which then proliferate and attack various host tissues (3–5). Programmed death factor 1 and its ligand (PD-1/PD-L1) negative costimulatory molecules belong to the CD28/B7 family (6–8). PD-1 was initially found in the 2B4.11 clone of apoptotic mouse T cell hybridoma, and its structure was similar to that of CD28 molecule (1). Subsequent clinical trials on SLE showed that PD-1 is present on various immune cells and plays a role in inhibiting the activation of patients’ own lymphocytes (9, 10). The regulatory T (Treg) cells and effector T (Teff) cells regulate each other to maintain immune homeostasis (11). The loss of homeostatic balance or Treg function is associated with overactivated Teff, which responses against self-antigens and, causes autoimmune disease. As a famous co-signaling receptor, PD-1 plays an indispensable role in regulating the proliferation and activation of Teff and Treg cells, and thus it is involved in the development of autoimmune diseases (12). Recently, some studies showed that sPD-1(the soluble form of PD-1)played a role in autoimmune inflammation (13). Compared with other negative regulators with non-antigen specific characteristics, the absence of PD-1 can affect antigen specific autoimmune response (14). The expression levels of PD-1 and sPD-1 are significantly associated with the pathogenesis of SLE, and they can serve as independent biomarkers and prognostic factors of the disease progression (15–17). So far, the mechanisms of SLE pathogenesis are still unclear. This article reviewed the relationship between PD-1 and SLE, and proposed to use PD-1 as a predictive biomarker for SLE disease activity. We also discussed the potential of combining PD-1 agonist and low-dose interleukin-2 for the treatment of SLE and how this regimen might improve clinical outcomes. Finally, we explored the problem of off-target effect and the biosafety of targeted immune therapy.
2 Structure and biological function of PD-1
As a negative co-inhibitory receptor, PD-1(CD279) was originally identified in mice with different autoimmune phenotypes. It regulates the antigen reaction threshold of T cells and B cells in peripheral blood to balance immunity. PD-1 is an important member of the CD28/B7 family and the immunoglobulin superfamily, with 288 amino acid residues. It mainly exists in activated T lymphocytes, B lymphocytes, NK cells, monocytes, and dendritic cells. PD-1 is composed of an N-terminal IgV domain, a transmembrane domain, a cytoplasmic tail with two tyrosine-based signal motifs, and an 20-amino acids side chain. The two tyrosine-based signal sequences form a tyrosine-based immune receptor switch motif (ITSM) that ensures PD-1 performs its inhibitory function (6). PD-1 can suppress harmful immune reactions and maintain immune tolerance by blocking immune checkpoints.
PD-1 plays an inhibitory role in cellular immunity, humoral immunity and innate immunity. In cellular immunity, the activation of PD-1 pathway leads to the phosphorylation of its cytoplasmic domain tyrosine and the recruitment of homology 2-domain-containing protein tyrosine phosphatase (SHP)-2 at the tyrosine site. This cascade of SHP-2 recruitment then leads to the dephosphorylation of ZAP70 and inhibits the production of downstream interleukin (IL)-2, ultimately inducing T cell anergy. In humoral immunity, PD-1 pathway activation and B cell receptor signaling produce co-stimulation, which results in SHP-2 recruitment to these phosphorylated regions of PD-1 protein, leading to the inactivation of downstream molecules and inducing B cell anergy. In innate immunity, the activation of PD-1 pathway can inhibit the secretion of inflammatory factors and inhibit the function of NK cells (18).
There are two kinds of PD-1 ligands, PD-L1 and PD-L2. It is generally believed that the distribution of PD-L1 is much wider than that of PD-L2 (19). PD-L1 is also a type I transmembrane protein, which can interact with PD-1 through the extracellular domain, leading to the conformational change of PD-1 (20). When PD-1 binds to PD-L1, it can regulate the peripheral tolerance of immune cells and arrest T cells at G0/G1 phase.
In recent years, the research on PD-1 immune checkpoint has focused on its transcriptional and, post-transcriptional regulation. The high expression of PD-1 is a marker of the gradual loss of T cell function and T cell depletion (20). PD-1 releases sPD-L1 under the catalysis of matrix metalloproteinase. sPD-L1 can not only reflect the expression of PD-1, but also block the response of PD-1 to PD-L1 (21, 22). There is a significant overlap in genetic susceptibility between SLE and type 1 diabetes (T1D) (23), by using a nonobese diabetic (NOD)mice model, Mohammed Javeed I. Ansari et al. demonstrated that after PD-1–PD-L1 blockade, GAD-reactive T cells were in a highly active state and targeted host tissues, leading to increased destruction of self-organization (Figure 1). The sensitivity of Teff cells to PD-1-PD-L1 inhibitory pathway, provides a promising strategy of immunotherapy. Moreover, it is discovered that the parenchymal cells might down-regulate lymphocyte function at the inflammatory site (24). The membrane bound PD-1 protein can be hydrolyzed and released as the soluble PD-1 (sPD-1) (17), and recent studies have reported that the expression of sPD-1 is well correlated with the activity of autoimmune diseases (25).
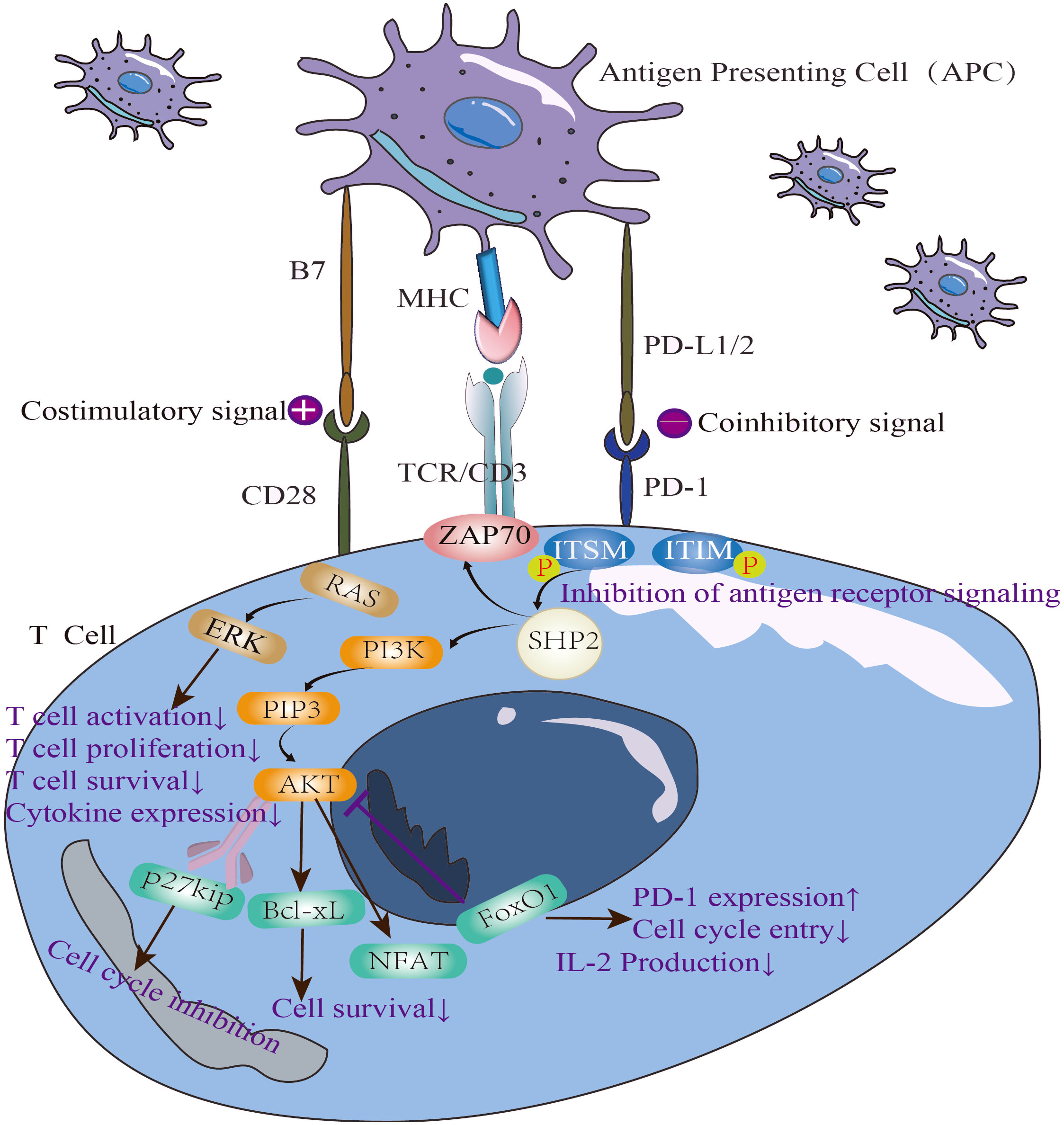
Figure 1 The immunosuppressive mechanism of PD-1/PD-L1 pathway and the negative correlation between PD-1 and IL-2.
3 Mechanism and implications of PD-1/PD-L1 axis
PD-1/PD-L1 both have soluble and membrane bound types. The soluble molecules are similar to cytokines and function through the circulatory system, while the membrane bound molecules participate in immune regulation by interacting with other membrane type molecules. The main molecular forms of PD-1/PD-L1 axis are mPD-1 and mPD-L1. The immunogenicity of antigen receptors often results in incomplete thymic negative selection, which can prevent autoimmune reactions that attack host tissues, and PD-1/PD-L1 axis plays a key role in this process.
Taku Okazaki et al. reported that when the co-inhibitory molecule PD-L1 is deficient, the threshold of antigen recognition reaction in T cells decreased, and the CD4+lymphocyte activity increased, which led to increased sensitivity of Teff to antigens (26). Shu Zhang et al. also found that the genetic polymorphism of PD-1/PD-L1 pathway was related to the genetic susceptibility to rheumatic diseases. Contrary to PD-1, the polymorphism of PD-L1 gene seems to have no clear relationship with SLE (27), but the lack of PD-L1 is related to increased disease activity (27). Moreover, it has been shown that the manipulation of PD-1/PD-L1 pathway can significantly alter disease severity in multiple animal models (18, 28).
PD-1/PD-L1 axis has been an important target of immunotherapy for many years, especially in the treatment of autoimmune diseases (29). PD-1 is overexpressed on the surface of activated T cells, which leads to the release of inflammatory factors, such as tumor necrosis factor-a (TNF-a) and interleukin. These inflammatory factors then result in PD-L1 overexpression in tissues, which in turn inhibits the activation of T cells (30, 31). An important sign of T cell activation and proliferation is aerobic glycolysis. Doumet Georges Helou et al. found that the lack of PD-1 led to significant up regulation of glycolytic genes (such as Hk1, Pkm and G6pdx), and increased glucose consumption, that is, blocking PD-1 can enhance glycolytic metabolism (32) (Figure 2).
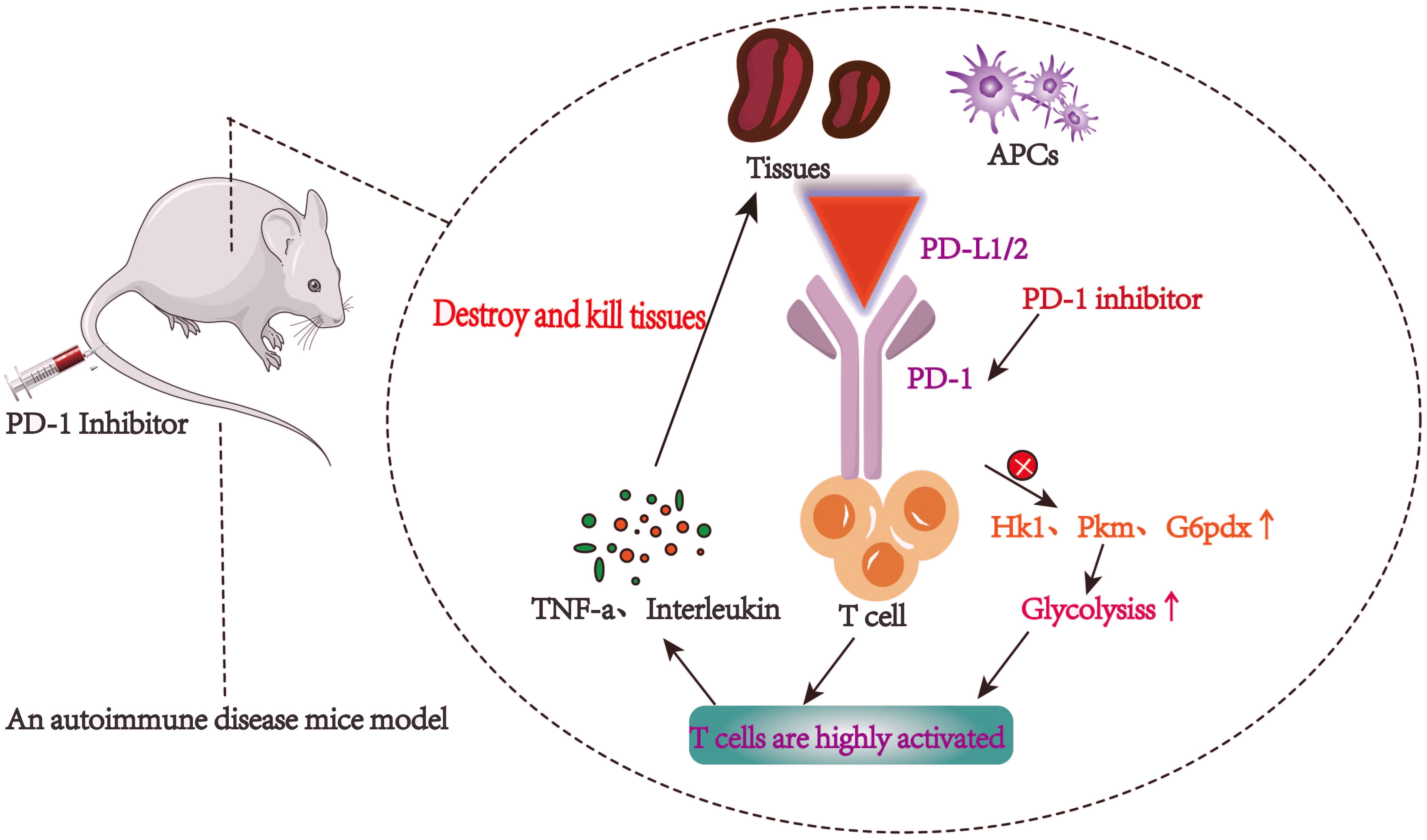
Figure 2 T cells are highly active and autoimmune inflammatory reaction is intensified after PD-1/PD-L1 pathway is blocked.
PD-1/PD-L1 axis is mainly involved in TH1 cell reaction, and they are significantly expressed on activated antigen presenting cells (APCs) (33–36). After induction by the major histocompatibility complex (MHC), PD-L1 can bind to CD28 and CTLA-4 (inhibitory receptors) on the surface of activated T cells and then inhibit T cell activation and proliferation (37, 38). As one of the most important immunosuppression checkpoints, PD-L1 and PD-1 inhibit the overall activation signal through ligand-ligand-cis interaction. Studies have shown that in patients with active SLE, the normal process of PD-L1 expression is impaired. Blocking PD-1 can affect the disease activity in lupus mouse model, but the lack of PD-L1 did not cause SLE, but rather aggravated the systemic autoimmunity of lupus susceptible mice (39). In addition, in the knock-in mouse model that lacked ligand cis-action, the autoimmunity of the mice was greatly attenuated (11).
Previous studies have found that the expression of PD-L1 may decrease during the onset of systemic lupus erythematosus(SLE) (40). The interaction between PD-1 and PD-L1 reduces the autoimmunity of surrounding normal tissues, and the disruption of this interaction is involved in the pathogenesis of autoimmune diseases (41). The controlled PD-L1 expression also maintains immune tolerance by inhibiting the activation of T cells to prevent damage to the host immune system. Daisuke Sugiura et al. proposed an effective strategy to trigger PD-1 function by targeting the cis-PD-L1-CD80 complex, which potentiates the PD-1/PD-L1 interaction to inhibit autoreactive T cells, thereby alleviating autoimmune diseases (42).
4 Regulation of PD-1 and sPD-1 expression in SLE
In recent years, genetics and metabolomics studies have found that smoking, ultraviolet light, diet, microbial flora disorders, interleukin polymorphisms and predisposition are influencing factors for the pathogenesis and severity of SLE (43, 44). Inflammatory cytokines such as IL-6 and TNF-a can activate NF- κB pathway and increase the miR-34a expression on the surface of peripheral blood mononuclear cells (PBMCs), which then inhibits the expression of Foxp3 (It is expressed in a subset of Treg cells with specific transcriptional functions that maintain immune homeostasis) and disrupts the immune homeostasis (45). Taku Okazaki et al. found that PD-1-deficient mice produced self-reactive antibodies, which were generated by the dysregulated gut microbiota and induced germinal center (GC) T cells with proinflammatory activities (26). Bing Wan et al. reported that in the synovial fluid of patients, there was a soluble splicing variant of PD-1(PD- 1Δex3) that can antagonize the co inhibition function of PD-1 (46). Consistently, the latest research showed that SLE was characterized by abnormal type I IFN signal transduction. In the lupus mouse model, type III IFN λ (IFN- λ) can induce active TLR7 on B cells and promote the development of tissue inflammation (47–49). The factors that induce soluble PD-1 include IF-γ, IFN-λ, TNF α, and IL17A. The serum level of sPD-1 in SLE patients with high disease activity is significantly higher compared to those with low disease activity (17). sPD-1 could block the PD-1/PD-L1 interaction, significantly enhance T cell immunity, and lead to adverse disease outcomes. An in vitro cohort study also found that in SLE patients, the expression level of serum sPD-1 was significantly increased, and there was a significant positive correlation between the expression level and SLEDAI score. Thus, high expression level of sPD-1 can predict disease activity and prognosis (13).
The main feature of unstimulated lupus B cells and T cells is the increased expression of PD-1. After the stimulation with IL-2 for 48 hours, the ability of B-cells in SLE patients to up-regulate PD-L1 expression is significantly reduced (50).In the serum of SLE patients, although the number of B cells is significantly expanding and the transcription level is increasing, the effector molecules connecting B cells and PD-1 are phosphorylated by tyrosine kinase and PD-1 cannot be effectively expressed. Therefore, the major identified defect of CD8+T cells in SLE patients is the suppressed expression of PD-1 (51–53).
The polymorphism of the regulatory intron in PD-1 gene is associated with an increased risk of SLE (18, 54). It has been shown that PD-1 cross-linking can inhibit the proliferation of T cells and the production of cytokines in the serum of normal people and lupus patients (Figure 1). Jin Lin et al. explored the potential relationship between the recently discovered T-cell subpopulation PD-1+CXCR5-CD4+T peripheral helper cells (Tph) and the pathogenesis of SLE through cohort study.They found that the number of Tph cells was positively correlated with SLE disease activity (16).
5 PD-1 expression in SLE and its correlation with clinical outcome
Targeting PD-1/PD-L1 is a promising treatment for chronic autoimmune diseases (Figure 3). Some tumor cases showed that, after the use of PD-1 pathway inhibitors in cancer therapy, the original SLE disease activity or lupus nephritis attack was induced, including new onset of SLE (55, 56).
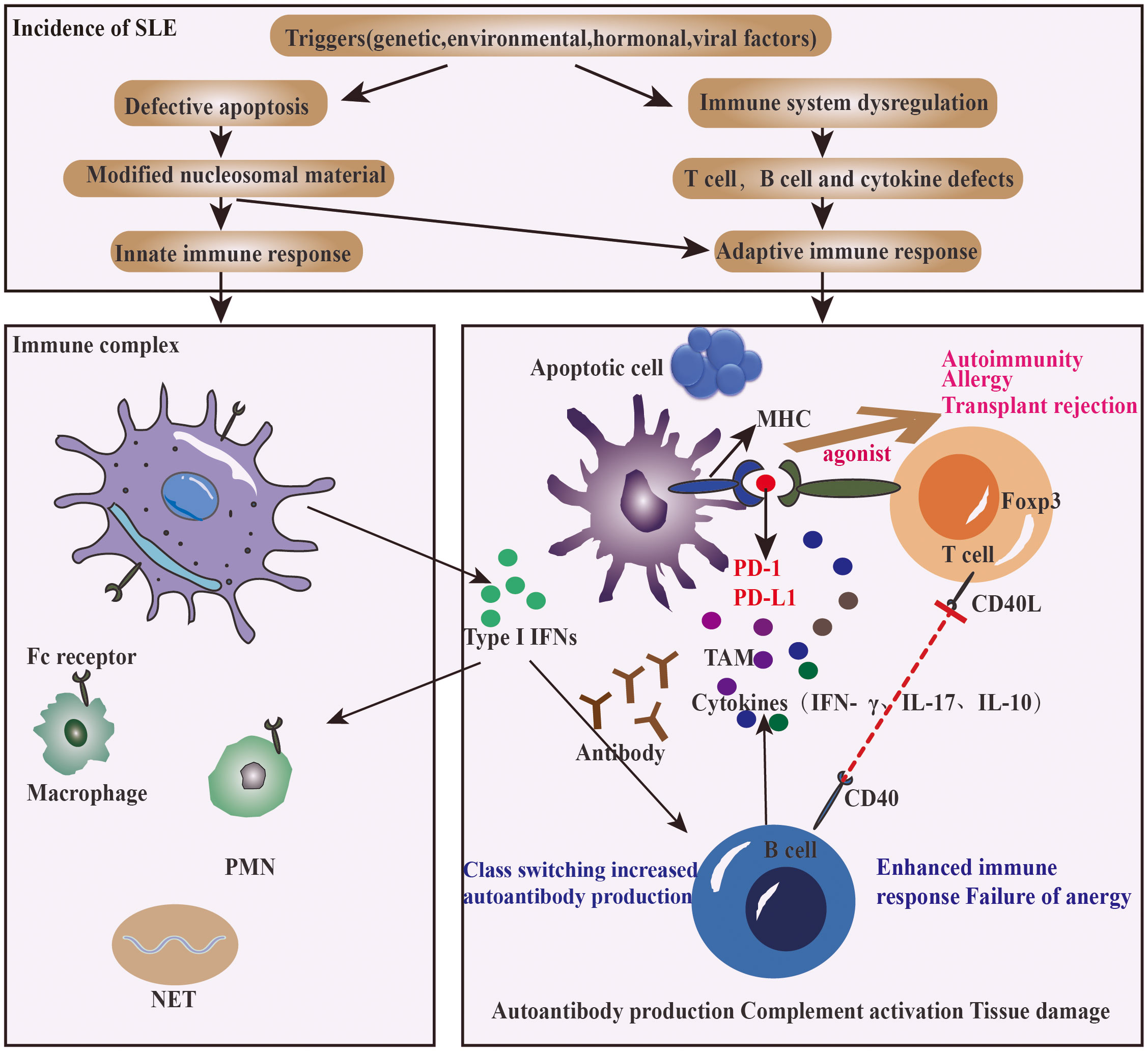
Figure 3 How PD-1 agonist improves the clinical outcomes of SLE.
It is known that in SLE, tumor associated macrophages (TAM) disorder occurs when the activation of PD-1 by its ligand is affected or blocked (57, 58). As one of the heterogeneous cell populations, TAM can consume amino acids to secrete immunosuppressants, which can promote cell migration, survival and exocytosis (59). TAM is also closely related to the pathogenesis of SLE. If the immune checkpoint is selectively blocked, the killing of tumor cells by TAM will be enhanced, but it will also lead to autoimmune diseases such as SLE and rheumatoid arthritis (51).
In SLE, the chronic inflammatory mediators will drive the activation of FOXP3 HELIOS Tregs derived from the thymus, followed by the compensatory restoration of immune balance (23). Ricardo C Ferreira et al. showed that stimulating PD-1 with specific agonists could effectively inhibit autoreactive T cells and selectively restore the homeostasis of Tregs cell population (23), confirming that PD-1 plays a central role in SLE diseases.
In addition, PD-1 knockout mice showed lupus like autoimmune symptoms, accompanied by an increase in immunoglobulin and a decrease in complement; the exogenous addition of serum from active lupus patients led to the proliferation of T cells in these mice. This lupus murine model suggests that PD-1 is a negative effector of T cell activation, and the biological function of PD-1 antibody is to block PD-1 pathway and increase the activity of lupus (60).
The common cause of death in SLE patients is renal failure. According to statistical analysis, 5-20% of SLE patients developed end-stage kidney disease within 10 years after initial diagnosis (61). Therefore, early and accurate diagnosis and treatment of lupus nephritis is important for patient prognosis (61). A large amount of abnormally activated complement and immunoglobulin are deposited in foot cells, mesangial cells, renal tubular epithelial cells, renal macrophages, and stromal cells of the kidney (62). When PD-1 agonist was used to activate PD-1/PD-L1 axis, the affinity between PD-1 and reactive T cells was improved, and Th17 was significantly inhibited in multiple organs, which led to reduced inflammatory cytokines secretion in serum, such as IFN-γ, IL-17 and IL-10. The deposition of immunoglobulin in kidney and the level of urinary protein were also reduced and the survival time of mice was prolonged (63).
6 PD-1 is a promising biomarker to predict disease activity
The SLE Disease Activity Index (SLEDAI) has been the most widely used indicator to evaluate the disease activity of SLE. The complement level, anti-double-stranded DNA antibody titer and other parameters are included in the scoring standard (64). However, this indicator is still very limited in detecting the changes of SLE disease activity (65). Diogo Jesus et al. applied multivariate linear regression analysis to develop and verify a better SLE disease activity score system(SLE-DAS), which can be used for continuous measurement of the changes in SLE disease activity (66). In clinical practice, there is still a lack of a gold standard for the evaluation of SLE disease activity. The combination of laboratory indicators, clinical characteristics, and doctor’s overall impression of patient’ status (PGA score) is the basic judgment of SLE disease activity (67, 68). Essentially, all variables that are confirmed to be related to the disease are considered (67–69).
At present, PD-1 expression level is a widely used and accepted biomarker to guide immunotherapy (70). Active inflammatory factors in SLE patients can activate NF-κB and induce the overexpression of IFNs, which in turn up-regulates the downstream transcription factors such as STAT1, STAT4, STAT6 (71–73). STAT family is a crucial part of many signaling pathways that regulate cell growth and pathogen resistance (74). STAT pathway involves IL-6 (gp130) receptor family, which participates in regulating the differentiation of B cells and the expression of PD-1/PD-L1 (75). Jingwen Shi et al. reported that PD-1 blocks the up regulation of Tfh cells by inhibiting the PI3K activity downstream of follicle-guidance receptor CXCR5, which can control the position and function of Tfh cells in a co-stimulation dependent manner (76). The circulating Tfh-like T cells were amplified in the serum of SLE patients and altered the aberrant germinal center activity in these patients (77), suggesting abnormal homeostasis of T cells and B cells. The expression of PD-1 in Tfh cells is also related to SLEDAI and anti-double-stranded DNA antibody positivity, and hence the PD-1 expression can be used to measure disease activity (78).
It has been found that there is almost no expression of PD-1 or PD-L1 in the peripheral serum mononuclear cells (PBMC) of patients who meet the criteria of active SLE. However, during the remission of lupus, the expression of PD-1 and PD-L1 return to normal, indicating that there is a potential inverse correlation between the expression of PD-1, PD-L1 and disease activity. Therefore, we speculate that PD-1/PD-L1 expression can be used as a promising biomarker to predict disease activity (40).
7 PD-1 agonist monotherapy
Identifying common pathogenic pathways and corresponding biomarkers that link abnormal cell activity to disease activity are essential for finding new therapeutic targets. At present, the standard treatment of SLE is mainly non-specific immunosuppressant, which often causes side effects such as infection and leukopenia. Developing biological agents targeting specific targets or co-inhibition pathways is an effective way to reduce the side effects (79).
As an crucial immune checkpoint for T cell activation, PD-1/PD-L1 pathway can be effectively activated by PD-1 agonists to treat autoimmune diseases (80). The simulation of the interaction between PD-L1 and PD-1 on the surface of target cells and T cells can be used to precisely realize PD-1/TCR co-clustering on the T cell (81, 82). Adam P Curnock et al. have developed tissue specific PD-1 agonists that can avoid systemic immunosuppression and only inhibit local T cells, which can be used to treat highly chronic inflammatory diseases (80).
Zhao P et al. established three groups of models (spontaneous T1D model disease delay study, cyclophosphamide (CP) accelerated T1D model disease delay study and α PD-1 accelerates T1D mode disease delay research) and showed that PD-1+ not only helped to control autoimmune diseases, but also maintained the normal adaptive immunity of the body (31, 83, 84).Therefore, we speculated that PD-1 agonists might be able to achieve specific inhibition of highly activated immune cells through PD-1 inhibition signals.
Adam P Curnock et al. developed a PD-1 agonist targeting T cell receptor, called PD-1 ImmTAAI molecule, which can specifically promote the binding between PD-1 and PD-L1 on target cells while avoiding systemic immunosuppression. So far, the PD-1 agonists developed for autoimmune diseases are in Phase II and III clinical trials (Table 1). These drugs can stimulate the physiological immunosuppressive pathway and restore immune regulation, and are expected to become a new treatment scheme for autoimmune and autoimmune inflammatory diseases (80).
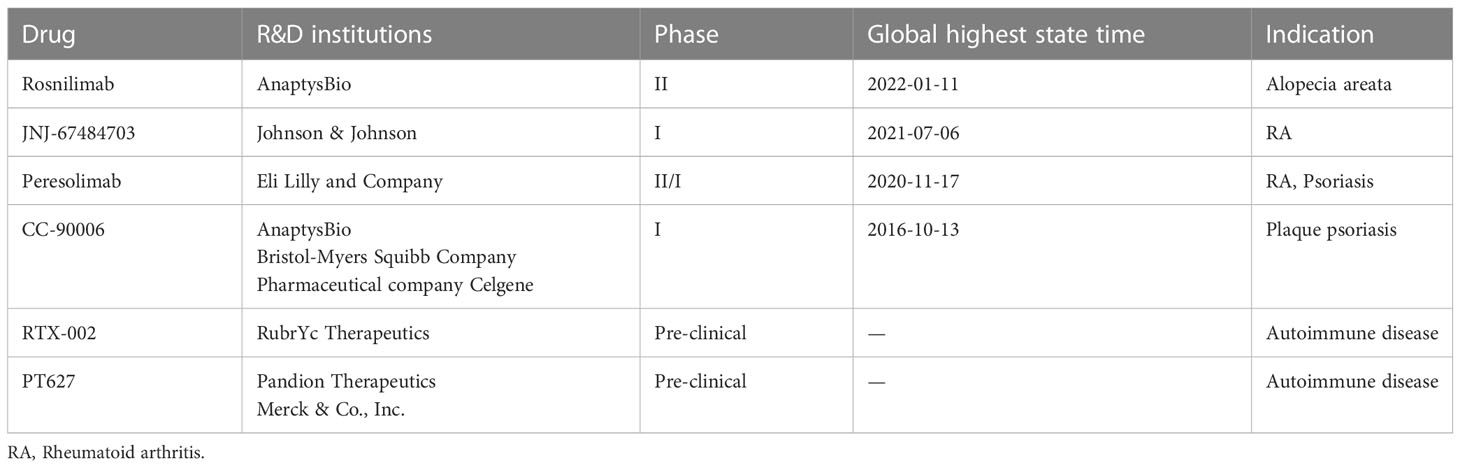
Table 1 PD-1 agonist antibody developed for autoimmune diseases registered in Insight Database.
8 The combination of PD-1 agonist and low-dose Interleukin-2
For autoimmune diseases, the use of a single immune checkpoint inhibitor (ICI) alone is not as effective as a combination of several drugs (23). Thereby, the combination of drugs may provide a better treatment efficacy for SLE patients (85). By using flow cytometry and qPCR, He Hao et al. showed that the proportion of PD-1 hi T follicular helper (Tfh) cells (P<0.01) increased in the serum of SLE patients, while IL-2, the cytokine that induced T-follicular helper cells to transform into T-follicular regulatory (Tfr)cells, was low. Therefore, administration of low-dose IL-2 supplementation may be able to relieve SLE by rebalancing Tfh and Tfr cells (86).
As an important inflammatory factor, interleukin-2 (IL-2) has been extensively studied in immune regulation. The latest research shows that different doses of IL-2 have different effects on immune regulation. High dose of IL-2 can promote the differentiation and expansion of effector and memory T cells, while low dose of IL-2 mainly act on regulatory T cells(Treg cells) (87). In the ex vivo coculture assays of Tfh and B cells, IL-2 stimulation can cause the downstream transcription factors STAT3 and STAT5 to selectively bind to FOXP3 and BCL6 gene loci and inhibit H3K27me3, which further suppressed the plasma mother cells (86). In addition, Ricardo C Ferreira et al. have shown that the FOXP3+HELIOS+Tregs in the serum of SLE patients have demethylated FOXP3, resulting in increased number of CD4+FOXP3+cells. This CD4+FOXP3+state can be maintained by IL-2 induction (23).
Jing He et al. first proposed to use low-dose IL-2 to selectively regulate T cell subsets to achieve the goals of balancing immunity and treating SLE (88). The safety and effectiveness of using low-dose IL-2 to treat SLE and restore immune balance have been confirmed (89). In vitro experiments showed that when IL-2 was used to stimulate B cells in SLE, it was found that although the proliferation of B cells was weakened, the ability of IL-2 to up-regulate PD-L1 was also decreased. As a result, when low-dose IL-2 is used to treat SLE, adding PD-1 agonist at the same time may provide better therapeutic effect (50). Moreover, there was evidence showing that PD-1 signaling pathway can inhibit the production of IL-2 (90). When PD-1 agonist was used to stimulate experimental mice, the expression of IL-2 was decreased, and a negative correlation between the expression of PD-1 and the production of IL-2 was identified (91). Therefore, it is also essential to supplement low dose of IL-2 when using PD-1 agonist alone. Studies have shown that PD-1 expression increases on Treg in response to low-dose IL-2 treatment, which is crucial for maintaining the stable regulation of Treg. The data suggests that the combination of low-dose IL-2 immunotherapy and PD-1 agonist can selectively correct the defects of Treg in vivo, and inhibit the proliferation of pathogenic auto-reactive PD-1 Teffs. Pre-clinical research is currently ongoing and the outcomes are highly anticipated (23).
In summary, the key to the treatment of SLE is regulating Tregs homeostasis. IL-2 can induce the expression of PD-1 on Tregs, and PD-1 can regulate the proliferation of IL-2–induced Treg (92). The impairment of PD-1 inhibitory function in SLE has been confirmed, and low dose IL-2 can restore its function (93). Therefore, the combination of PD-1 agonist and low-dose IL-2 is a promising treatment option for SLE.
9 Biosafety issues and future prospects
Although there is a strong reason to develop PD-1 agonist as a therapeutic treatment for SLE, the research progress in this field is limited due to the following biosafety issues and off-target concerns (94). When the disease is in different active states, PD-1 agonists have different effects on the disease (95). In addition, the effect of PD-1 is closely linked with the patient’s gene expression profile, disease activity indexes, pharmacokinetics, and the binding affinity between drugs and receptors.
PD-1 agonist has been shown to effectively inhibit autoimmunity and is used to treat SLE, but it can also bring off-target effect and side effects. PD-1 agonist can inhibit the immune reaction to viral infection (96, 97). When cellular immunity, humoral immunity and innate immunity are suppressed, the body’s ability to fight off virus is weakened. Second, PD-1 agonists may increase the risk of cancer. Activation of the PD-1 pathway can inactivate tumor-infiltrating lymphocytes, thus evading immune surveillance (98–102). Finally, the activation of PD-1 pathway leads to immunosuppression, thereby reducing the effectiveness of vaccination (101).
Although a single PD-1-targeted agents may have significant efficacy for SLE, the results from PD-1 knockout mice suggest that combination therapy is still required. At the current research stage, low-dose IL-2 combined with PD-1 agonist has been confirmed to be effective in treating SLE in pre-clinical trials, and in-depth research is required for the selection of drugs in clinical treatment. Moreover, this combination is also an important supplement to the precise treatment of autoimmune diseases, helping to maximize the therapeutic effect and minimize the side effects.
10 Conclusions
As a chronic inflammatory autoimmune disease, SLE often affects multiple organs of the body. Its disease evaluation and disease relief have always been the research focus of clinicians and scientists. For a long time, the treatment of autoimmune diseases such as SLE mainly targets the auto-reactive lymphocytes and/or the auto-antibodies secreted by them, but the non-selective inhibition of adaptive immunity often yield low efficacy. Different from other negative regulators, PD-1 exists on activated T cells and B cells and has antigenic specificity. Therefore, it is a promising immune target for the treatment of SLE. We found that the expression and activity of PD-1 and its pathway are tightly related to the incidence and disease activity of SLE. In addition, further studies also showed that the soluble form of PD-1, sPD-1, was negatively correlated with the disease activity of SLE. Therefore, we propose that PD-1 and sPD-1 may be potential biomarkers for predicting SLE disease activity. Based on the above reasoning, we also propose that the application of PD-1 agonist might be an effective approach to treat SLE. However, single drug therapy may not achieve satisfactory response rate. According to the literature review and the latest research progress, the combination of PD-1 agonist and low-dose IL-2 may have a broader therapeutic prospect and advantages.
Author contributions
BW wrote the first draft of the manuscript. CC contributed to conception, review and editing. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pauken KE, Torchia JA, Chaudhri A, Sharpe AH, Freeman GJ. Emerging concepts in PD-1 checkpoint biology. Semin Immunol (2021) 52:101480. doi: 10.1016/j.smim.2021.101480
2. Aringer M. Inflammatory markers in systemic lupus erythematosus. J Autoimmun (2020) 110:102374. doi: 10.1016/j.jaut.2019.102374
3. Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet (2019) 393(10188):2344–58. doi: 10.1016/S0140-6736(19)30546-X
4. Allen ME, Rus V, Szeto GL. Leveraging heterogeneity in systemic lupus erythematosus for new therapies. Trends Mol Med (2021) 27(2):152–71. doi: 10.1016/j.molmed.2020.09.009
5. Geraud A, Gougis P, Vozy A, Anquetil C, Allenbach Y, Romano E, et al. Clinical pharmacology and interplay of immune checkpoint agents: A yin-yang balance. Annu Rev Pharmacol Toxicol (2021) 61:85–112. doi: 10.1146/annurev-pharmtox-022820-093805
6. Ghosh C, Luong G, Sun Y. A snapshot of the PD-1/PD-L1 pathway. J Cancer (2021) 12(9):2735–46. doi: 10.7150/jca.57334
7. Karandikar NJ, Vanderlugt CL, Bluestone JA, Miller SD. Targeting the B7/CD28:CTLA-4 costimulatory system in CNS autoimmune disease. J Neuroimmunol (1998) 89(1-2):10–8. doi: 10.1016/S0165-5728(98)00058-7
8. Oosterwegel MA, Greenwald RJ, Mandelbrot DA, Lorsbach RB, Sharpe AH. CTLA-4 and T cell activation. Curr Opin Immunol (1999) 11(3):294–300. doi: 10.1016/S0952-7915(99)80047-8
9. Zhu Q, Li Y, Zhang L, Wang M, Chen Z, Shi J, et al. Patients with systemic lupus erythematosus show increased proportions of CD19CD20 b cells and secretion of related autoantibodies. Clin Rheumatol (2021) 40(1):151–65. doi: 10.1007/s10067-020-05220-2
10. Siwiec A, Majdan M. The role of the PD-1 protein in pathogenesis of autoimmune diseases, with particular consideration of rheumatoid arthritis and systemic lupus erythematosus. Postepy Hig Med Dosw (2015) 69:534–42. doi: 10.5604/17322693.1150784
11. Sugiura D, Maruhashi T, Okazaki I-M, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by -PD-L1/CD80 interactions is required for optimal T cell responses. Science (2019) 364(6440):558–66. doi: 10.1126/science.aav7062
12. Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and treg homeostasis in autoimmunity and tumor immunity. J Autoimmun (2018) 95:77–99. doi: 10.1016/j.jaut.2018.08.007
13. Du Y, Nie L, Xu L, Wu X, Zhang S, Xue J. Serum levels of soluble programmed death-1 (sPD-1) and soluble programmed death ligand 1(sPD-L1) in systemic lupus erythematosus: Association with activity and severity. Scand J Immunol (2020) 92(1):e12884. doi: 10.1111/sji.12884
14. Prokunina L, Castillejo-López C, Oberg F, Gunnarsson I, Berg L, Magnusson V, et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet (2002) 32(4):666–9. doi: 10.1038/ng1020
15. Hellbacher E, Sundström C, Molin D, Baecklund E, Hollander P. Expression of PD-1, PD-L1 and PD-L2 in lymphomas in patients with pre-existing rheumatic diseases-a possible association with high rheumatoid arthritis disease activity. Cancers (Basel) (2022) 14(6). doi: 10.3390/cancers14061509
16. Lin J, Yu Y, Ma J, Ren C, Chen W. PD-1+CXCR5-CD4+T cells are correlated with the severity of systemic lupus erythematosus. Rheumatol (Oxford) (2019) 58(12):2188–92. doi: 10.1093/rheumatology/kez228
17. Hirahara S, Katsumata Y, Kawasumi H, Kawaguchi Y, Harigai M. Serum levels of soluble programmed cell death protein 1 and soluble programmed cell death protein ligand 2 are increased in systemic lupus erythematosus and associated with the disease activity. Lupus (2020) 29(7):686–96. doi: 10.1177/0961203320916517
18. Zhang S, Wang L, Li M, Zhang F, Zeng X. The PD-1/PD-L pathway in rheumatic diseases. J Formos Med Assoc (2021) 120(1 Pt 1):48–59. doi: 10.1016/j.jfma.2020.04.004
19. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
20. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
21. Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med (2003) 197(9):1083–91. doi: 10.1084/jem.20021752
22. Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, et al. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine (2011) 56(2):231–8. doi: 10.1016/j.cyto.2011.06.004
23. Ferreira RC, Castro Dopico X, Oliveira JJ, Rainbow DB, Yang JH, Trzupek D, et al. Chronic immune activation in systemic lupus erythematosus and the autoimmune PTPN22 trp risk allele drive the expansion of FOXP3 regulatory T cells and PD-1 expression. Front Immunol (2019) 10:2606. doi: 10.3389/fimmu.2019.02606
24. Ansari MJI, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med (2003) 198(1):63–9. doi: 10.1084/jem.20022125
25. Greisen SR, Kragstrup TW, Thomsen JS, Hørslev-Pedersen K, Hetland ML, Stengaard-Pedersen K, et al. The programmed death-1 pathway counter-regulates inflammation-induced osteoclast activity in clinical and experimental settings. Front Immunol (2022) 13:773946. doi: 10.3389/fimmu.2022.773946
26. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat Immunol (2013) 14(12):1212–8. doi: 10.1038/ni.2762
27. Wang S-C, Lin C-H, Ou T-T, Wu C-C, Tsai W-C, Hu C-J, et al. Ligands for programmed cell death 1 gene in patients with systemic lupus erythematosus. J Rheumatol (2007) 34(4):721–5.
28. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
29. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
30. Wu Q, Jiang L, Li S-C, He Q-J, Yang B, Cao J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol Sin (2021) 42(1):1–9. doi: 10.1038/s41401-020-0366-x
31. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
32. Helou DG, Shafiei-Jahani P, Lo R, Howard E, Hurrell BP, Galle-Treger L, et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat Commun (2020) 11(1):3998. doi: 10.1038/s41467-020-17813-1
33. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
34. Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol (2002) 169(10):5538–45. doi: 10.4049/jimmunol.169.10.5538
35. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol (2003) 33(11):3117–26. doi: 10.1002/eji.200324270
36. Ding H, Wu X, Gao W. PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clin Immunol (2005) 115(2):184–91. doi: 10.1016/j.clim.2005.01.005
37. Bhatia S, Edidin M, Almo SC, Nathenson SG. Different cell surface oligomeric states of B7-1 and B7-2: Implications for signaling. Proc Natl Acad Sci USA (2005) 102(43):15569–74. doi: 10.1073/pnas.0507257102
38. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the b cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi: 10.1084/jem.174.3.561
39. Ou J-N, Wiedeman AE, Stevens AM. TNF-α and TGF-β counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep (2012) 2:295. doi: 10.1038/srep00295
40. Mozaffarian N, Wiedeman AE, Stevens AM. Active systemic lupus erythematosus is associated with failure of antigen-presenting cells to express programmed death ligand-1. Rheumatol (Oxford) (2008) 47(9):1335–41. doi: 10.1093/rheumatology/ken256
41. Lee J-M, Chen M-H, Chou K-Y, Chao Y, Chen M-H, Tsai C-Y. Novel immunoprofiling method for diagnosing SLE and evaluating therapeutic response. Lupus Sci Med (2022) 9(1). doi: 10.1136/lupus-2022-000693
42. Sugiura D, Okazaki I-M, Maeda TK, Maruhashi T, Shimizu K, Arakaki R, et al. PD-1 agonism by anti-CD80 inhibits T cell activation and alleviates autoimmunity. Nat Immunol (2022) 23(3):399–410. doi: 10.1038/s41590-021-01125-7
43. Liu E, Perl A. Pathogenesis and treatment of autoimmune rheumatic diseases. Curr Opin Rheumatol (2019) 31(3):307–15. doi: 10.1097/BOR.0000000000000594
44. Liu Z, Sun T, Song L, Xue X, Liu S, Kuang H, et al. Interleukin polymorphisms and predisposition of systemic lupus erythematosus: A meta-analysis. Int Arch Allergy Immunol (2021) 182(10):1008–16. doi: 10.1159/000507482
45. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
46. Wan B, Nie H, Liu A, Feng G, He D, Xu R, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol (2006) 177(12):8844–50. doi: 10.4049/jimmunol.177.12.8844
47. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, in moderate-to-Severe systemic lupus erythematosus. Arthritis Rheumatol (2017) 69(2):376–86. doi: 10.1002/art.39962
48. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50(4):907–23. doi: 10.1016/j.immuni.2019.03.025
49. Ye L, Schnepf D, Staeheli P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat Rev Immunol (2019) 19(10):614–25. doi: 10.1038/s41577-019-0182-z
50. Stefanski A-L, Wiedemann A, Reiter K, Hiepe F, Lino AC, Dörner T. Enhanced programmed death 1 and diminished programmed death ligand 1 up-regulation capacity of post-activated lupus b cells. Arthritis Rheumatol (2019) 71(9):1539–44. doi: 10.1002/art.40897
51. Shi H, Ye J, Teng J, Yin Y, Hu Q, Wu X, et al. Elevated serum autoantibodies against co-inhibitory PD-1 facilitate T cell proliferation and correlate with disease activity in new-onset systemic lupus erythematosus patients. Arthritis Res Ther (2017) 19(1):52. doi: 10.1186/s13075-017-1258-4
52. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11cT-bet b cells in SLE. Nat Commun (2018) 9(1):1758. doi: 10.1038/s41467-018-03750-7
53. Chen H, Peng Q, Yang H, Yin L, Shi J, Zhang Y, et al. Increased levels of soluble programmed death ligand 1 associate with malignancy in patients with dermatomyositis. J Rheumatol (2018) 45(6):835–40. doi: 10.3899/jrheum.170544
54. Bertsias GK, Nakou M, Choulaki C, Raptopoulou A, Papadimitraki E, Goulielmos G, et al. Genetic, immunologic, and immunohistochemical analysis of the programmed death 1/programmed death ligand 1 pathway in human systemic lupus erythematosus. Arthritis Rheumatol (2009) 60(1):207–18. doi: 10.1002/art.24227
55. Leonardi GC, Gainor JF, Altan M, Kravets S, Dahlberg SE, Gedmintas L, et al. Safety of programmed death-1 pathway inhibitors among patients with non-Small-Cell lung cancer and preexisting autoimmune disorders. J Clin Oncol (2018) 36(19):1905–12. doi: 10.1200/JCO.2017.77.0305
56. Hassel JC, Heinzerling L, Aberle J, Bähr O, Eigentler TK, Grimm M-O, et al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): Evaluation and management of adverse drug reactions. Cancer Treat Rev (2017) 57:36–49. doi: 10.1016/j.ctrv.2017.05.003
57. Orme JJ, Du Y, Vanarsa K, Mayeux J, Li L, Mutwally A, et al. Heightened cleavage of axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin Immunol (2016) 169:58–68. doi: 10.1016/j.clim.2016.05.011
58. Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva Medina T, et al. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol (2018) 19(6):571–82. doi: 10.1038/s41590-018-0107-1
59. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci (2018) 39(3):307–25. doi: 10.1016/j.tips.2017.11.007
60. Luo Q, Huang Z, Ye J, Deng Y, Fang L, Li X, et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res Ther (2016) 18:47. doi: 10.1186/s13075-016-0942-0
61. Anders H-J, Saxena R, Zhao M-H, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Primers (2020) 6(1):7. doi: 10.1038/s41572-019-0141-9
62. Jamaly S, Rakaee M, Abdi R, Tsokos GC, Fenton KA. Interplay of immune and kidney resident cells in the formation of tertiary lymphoid structures in lupus nephritis. Autoimmun Rev (2021) 20(12):102980. doi: 10.1016/j.autrev.2021.102980
63. Liao W, Zheng H, Wu S, Zhang Y, Wang W, Zhang Z, et al. The systemic activation of programmed death 1-PD-L1 axis protects systemic lupus erythematosus model from nephritis. Am J Nephrol (2017) 46(5):371–9. doi: 10.1159/000480641
64. Tripathy R, Panda AK, Das BK. Serum ferritin level correlates with SLEDAI scores and renal involvement in SLE. Lupus (2015) 24(1):82–9. doi: 10.1177/0961203314552290
65. Uribe AG, Vilá LM, McGwin G, Sanchez ML, Reveille JD, Alarcón GS. The systemic lupus activity measure-revised, the Mexican systemic lupus erythematosus disease activity index (SLEDAI), and a modified SLEDAI-2K are adequate instruments to measure disease activity in systemic lupus erythematosus. J Rheumatol (2004) 31(10):1934–40.
66. Jesus D, Matos A, Henriques C, Zen M, Larosa M, Iaccarino L, et al. Derivation and validation of the SLE disease activity score (SLE-DAS): A new SLE continuous measure with high sensitivity for changes in disease activity. Ann Rheum Dis (2019) 78(3):365–71. doi: 10.1136/annrheumdis-2018-214502
67. Inês L, Duarte C, Silva RS, Teixeira AS, Fonseca FP, da Silva JAP. Identification of clinical predictors of flare in systemic lupus erythematosus patients: A 24-month prospective cohort study. Rheumatol (Oxford) (2014) 53(1):85–9. doi: 10.1093/rheumatology/ket322
68. Gatto M, Saccon F, Zen M, Bettio S, Iaccarino L, Punzi L, et al. Success and failure of biological treatment in systemic lupus erythematosus: A critical analysis. J Autoimmun (2016) 74:94–105. doi: 10.1016/j.jaut.2016.06.014
69. Zen M, Iaccarino L, Gatto M, Saccon F, Larosa M, Ghirardello A, et al. Lupus low disease activity state is associated with a decrease in damage progression in Caucasian patients with SLE, but overlaps with remission. Ann Rheum Dis (2018) 77(1):104–10. doi: 10.1136/annrheumdis-2017-211613
70. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol (2021) 18(6):345–62. doi: 10.1038/s41571-021-00473-5
71. Curran CS, Gupta S, Sanz I, Sharon E. PD-1 immunobiology in systemic lupus erythematosus. J Autoimmun (2019) 97:1–9. doi: 10.1016/j.jaut.2018.10.025
72. Bally APR, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R, et al. NF-κB regulates PD-1 expression in macrophages. J Immunol (2015) 194(9):4545–54. doi: 10.4049/jimmunol.1402550
73. Rodríguez-García M, Porichis F, de Jong OG, Levi K, Diefenbach TJ, Lifson JD, et al. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J Leukoc Biol (2011) 89(4):507–15. doi: 10.1189/jlb.0610327
74. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
75. Bally APR, Austin JW, Boss JM. Genetic and epigenetic regulation of PD-1 expression. J Immunol (2016) 196(6):2431–7. doi: 10.4049/jimmunol.1502643
76. Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 controls follicular T helper cell positioning and function. Immunity (2018) 49(2):264–74. doi: 10.1016/j.immuni.2018.06.012
77. Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, et al. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154-CD40 interactions. J Clin Invest (2003) 112(10):1506–20. doi: 10.1172/JCI200319301
78. Choi J-Y, Ho J-e, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol (2015) 67(4):988–99. doi: 10.1002/art.39020
79. Ripoll È, Merino A, Herrero-Fresneda I, Aran JM, Goma M, Bolaños N, et al. CD40 gene silencing reduces the progression of experimental lupus nephritis modulating local milieu and systemic mechanisms. PloS One (2013) 8(6):e65068. doi: 10.1371/journal.pone.0065068
80. Curnock AP, Bossi G, Kumaran J, Bawden LJ, Figueiredo R, Tawar R, et al. Cell-targeted PD-1 agonists that mimic PD-L1 are potent T cell inhibitors. JCI Insight (2021) 6(20). doi: 10.1172/jci.insight.152468
81. Zhao Y, Lee CK, Lin C-H, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-heterodimer triggers the Co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity (2019) 51(6):1059–73. doi: 10.1016/j.immuni.2019.11.003
82. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
83. Torkildsen Ø, Myhr KM, Bø L. Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol (2016) 23 Suppl 1(Suppl 1):18–27. doi: 10.1111/ene.12883
84. McNamara C, Sugrue G, Murray B, MacMahon PJ. Current and emerging therapies in multiple sclerosis: Implications for the radiologist, part 2-surveillance for treatment complications and disease progression. AJNR Am J Neuroradiol (2017) 38(9):1672–80. doi: 10.3174/ajnr.A5148
85. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol (2017) 18(3):e143–e52. doi: 10.1016/S1470-2045(17)30074-8
86. Hao H, Nakayamada S, Yamagata K, Ohkubo N, Iwata S, Inoue Y, et al. Conversion of T follicular helper cells to T follicular regulatory cells by interleukin-2 through transcriptional regulation in systemic lupus erythematosus. Arthritis Rheumatol (2021) 73(1):132–42. doi: 10.1002/art.41457
87. Ma X, Nakayamada S, Kubo S, Sakata K, Yamagata K, Miyazaki Y, et al. Expansion of T follicular helper-T helper 1 like cells through epigenetic regulation by signal transducer and activator of transcription factors. Ann Rheum Dis (2018) 77(9):1354–61. doi: 10.1136/annrheumdis-2017-212652
88. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi: 10.1038/nm.4148
89. Lv C, Zhang H, Li Z, Zhang H, Zhang Y, Li R, et al. Low-dose IL-2 therapy compensates for metabolic shifts and reverses anxiety-like behavior in PD-1 deficiency-induced autoimmunity. Cell Mol Immunol (2021) 18(5):1336–8. doi: 10.1038/s41423-020-00562-y
90. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z, et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J Cell Biol (2020) 219(6). doi: 10.1083/jcb.201905085
91. Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S, et al. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol (2009) 182(11):6682–9. doi: 10.4049/jimmunol.0900080
92. Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood (2017) 129(15):2186–97. doi: 10.1182/blood-2016-09-741629
93. Kurata I, Mikami N, Ohyama A, Osada A, Kondo Y, Tsuboi H, et al. Impaired function of PD-1 follicular regulatory T cells in systemic lupus erythematosus. Clin Exp Immunol (2021) 206(1):28–35. doi: 10.1111/cei.13643
94. Ben Nasr M, Tezza S, D'Addio F, Mameli C, Usuelli V, Maestroni A, et al. PD-L1 genetic overexpression or pharmacological restoration in hematopoietic stem and progenitor cells reverses autoimmune diabetes. Sci Transl Med (2017) 9(416). doi: 10.1126/scitranslmed.aam7543
95. Stathopoulou C, Gangaplara A, Mallett G, Flomerfelt FA, Liniany LP, Knight D, et al. PD-1 inhibitory receptor downregulates asparaginyl endopeptidase and maintains Foxp3 transcription factor stability in induced regulatory T cells. Immunity (2018) 49(2):247–63. doi: 10.1016/j.immuni.2018.05.006
96. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol (2007) 19(7):813–24. doi: 10.1093/intimm/dxm057
97. Grakoui A, John Wherry E, Hanson HL, Walker C, Ahmed R. Turning on the off switch: regulation of anti-viral T cell responses in the liver by the PD-1/PD-L1 pathway. J Hepatol (2006) 45(4):468–72. doi: 10.1016/j.jhep.2006.07.009
98. Bryan CM, Rocklin GJ, Bick MJ, Ford A, Majri-Morrison S, Kroll AV, et al. Computational design of a synthetic PD-1 agonist. Proc Natl Acad Sci USA (2021) 118(29). doi: 10.1073/pnas.2102164118
99. Guo Q, Chen C, Wu Z, Zhang W, Wang L, Yu J, et al. Engineered PD-1/TIGIT dual-activating cell-membrane nanoparticles with dexamethasone act synergistically to shape the effector T cell/Treg balance and alleviate systemic lupus erythematosus. Biomaterials (2022) 285:121517. doi: 10.1016/j.biomaterials.2022.121517
100. Karamitopoulou E, Andreou A, Pahud de Mortanges A, Tinguely M, Gloor B, Perren A. PD-1/PD-L1-Associated immunoarchitectural patterns stratify pancreatic cancer patients into Prognostic/Predictive subgroups. Cancer Immunol Res (2021) 9(12):1439–50. doi: 10.1158/2326-6066.CIR-21-0144
101. Riley JL. PD-1 signaling in primary T cells. Immunol Rev (2009) 229(1):114–25. doi: 10.1111/j.1600-065X.2009.00767.x
Keywords: PD-1 agonist, systemic lupus erythematosus, targeted therapy, pathogenesis, biomarker
Citation: Wang B, Chen C, Liu X, Zhou S, Xu T and Wu M (2023) The effect of combining PD-1 agonist and low-dose Interleukin-2 on treating systemic lupus erythematosus. Front. Immunol. 14:1111005. doi: 10.3389/fimmu.2023.1111005
Received: 29 November 2022; Accepted: 27 February 2023;
Published: 08 March 2023.
Edited by:
Dong-Qing Ye, Anhui Medical University, ChinaReviewed by:
Ganesan Ramamoorthi, Moffitt Cancer Center, United StatesHaijing Wu, Second Xiangya Hospital, Central South University, China
Copyright © 2023 Wang, Chen, Liu, Zhou, Xu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Xu, ZGFpc3lfdGluZ3RpbmdAMTYzLmNvbQ==; Min Wu, d3V1bWluQDE2My5jb20=