 Ruksana Huda
Ruksana Huda
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 30 January 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1110499
This article is part of the Research Topic Global Excellence in Inflammatory diseases: North America 2021 View all 8 articles
Myasthenia gravis (MG) is a neuromuscular autoimmune disorder characterized by chronic but intermittent fatigue of the eye- and general body muscles. Muscle weakness is caused primarily by the binding of an autoantibody to the acetylcholine receptors, resulting in blockage of normal neuromuscular signal transmission. Studies revealed substantial contributions of different proinflammatory or inflammatory mediators in the pathogenesis of MG. Despite these findings, compared to therapeutic approaches that target autoantibody and complements, only a few therapeutics against key inflammatory molecules have been designed or tested in MG clinical trials. Recent research focuses largely on identifying unknown molecular pathways and novel targets involved in inflammation associated with MG. A well-designed combination or adjunct treatment utilizing one or more selective and validated promising biomarkers of inflammation as a component of targeted therapy may yield better treatment outcomes. This review briefly discusses some preclinical and clinical findings of inflammation associated with MG and current therapy approaches and suggest the potential of targeting important inflammatory marker(s) along with current monoclonal antibody or antibody fragment based targeted therapies directed to a variety of cell surface receptors.
Myasthenia gravis (MG) is an autoimmune disorder characterized by muscle weakness and fatigue associated with autoantibodies binding to acetylcholine receptors (AChRs) distributed at the neuromuscular endplates (1). Autoantibody (IgG1 and IgG3 isotypes) binding reduces the availability of functional acetylcholine receptors (AChRs) to Acetylcholine through receptor blockade and internalization. Receptor depletion is further intensified through myocyte destructions by activated complement and membrane attack complexes (2, 3). About 40% of AChR autoantibody-seronegative patients showed the presence of either muscle-specific tyrosine kinase (MuSK) (predominantly IgG4 isotype) or LDL receptor-related protein (LRP)4 (complement-activating IgG1 isotype) -specific autoantibodies that were known to disperse postsynaptic AChR clusters (4, 5) to cause AChR deficiencies.
Inflammation has long been regarded as an important contributing factor for both generalized and ocular MG pathogenesis, occurring in about 80% and 50% of MG patients, respectively (6, 7). The mediators of inflammation, predominantly cytokines and chemokines are released mostly by inflammatory monocytes and macrophages in the affected postsynaptic neuromuscular junction (NMJ) or thymic tissue (8–10), which then move also into the peripheral circulation. Subsequently, AChR-specific T-helper cells infiltrate the NMJ or thymic microenvironment and interact with B cells to induce its autoimmune activation (11). In addition to the thymus, germinal centers (consisting mainly of B lymphocytes, T follicular helper cells (Tfh), and plasma cells) are also prominently formed in the secondary lymphoid organs, such as the spleen in experimental mouse model of MG (EAMG), to produce MG-specific autoantibody (12, 13). These cytokines directly or indirectly take part in priming and activating dendritic cells, AChR-specific T-helper cells, and other cells that orchestrate to promote maturation, proliferation, and differentiation of AChR-specific B cells into plasma cells for autoimmune development (14). Depending on the specificity of agonists and Toll-like receptors (TLRs) 3, 7, or 9 involved, several cytokines (e.g., Tumor necrosis factor/TNF) function in an autocrine or paracrine manner, controlled by the specific intracellular signaling pathways and predominantly by nuclear factor kappa B (NFκB), signal transducer and activators of transcription (STAT) and CAAT Enhancer Binding Protein (C/EBP) family of transcription factors (15). For example, IL-6 and IFN-γ are the STAT activator, whereas TNFα is an NFκB activator.
Increasing evidence shows that dysregulated inflammatory response is closely associated not only with MG but also with many autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), multiple sclerosis (MS), etc. (14). Recent RNA-seq analysis of peripheral blood mononuclear cells (PBMCs) from AChR-MG patients (16) and our nCounter transcriptome analysis of genes modulated by epigenetic inhibition of histone deacetylases in experimental mouse model of MG revealed occurrence of significant dysregulation of inflammatory molecules in association with altered autoantibody levels (17). Further, correlative evidence from such preclinical and clinical studies demonstrates that autoantibodies can also promote or suppress inflammation based on their immunoglobulin (Ig) isotypes and Fc domain interactions with other cells (18). The mechanisms include Fc receptor (FcR) binding of immune cells and their activations, complement-fixing, and immune complex deposition resulting in functional receptor depletion.
Traditional treatments such as plasma exchange, Thymectomy, and immunosuppressive drugs do not cure MG. Current targeted therapy using monoclonal antibodies and antibody fragments against various popular biomarkers demonstrated a variable degree of effectiveness (7). However, these monotherapies neither reduced expected levels of autoantibody nor showed significant clinical improvement or disease remission. Additionally, a small fraction of patients is refractory, exhibiting a relatively low response to targeted therapy (19). Therefore, in addition to the monoclonal antibody (mAb)-based therapies against conventional immune cell-associated biomarkers, there is also a need for adjunct therapies directed to new, precise, and reliable targets for inflammation that can ensure fulfilling the current gap in targeted therapy. Thus, finding and utilizing validated promising biomarkers for combination treatment might be critical for a highly effective treatment for MG.
Chronic inflammation in the thymus due to persistent microbial infection has been reported to occur in MG patients (20). Disruption of tolerance and generation of autoreactive cells in the thymus are seemingly the first step for AChR-MG pathogenesis. The changes are thought to be prompted by innate immune activation (e.g., TLRs) triggered in a host following a viral infection that leads to the production of an array of inflammatory molecules. Further supporting the reports, infections with certain microbes and viruses (including recent SARS-CoV-2) were found to initiate or exacerbate an autoimmune condition, likely through induction of interferon (IFN)-mediated immune response (21–24). Also, the antigenicity of exogenous molecules (e.g., a microbial protein) with significant homology with a host protein or host-derived endogenous protein associated with cell stress or cell damage is believed to act as an autoantigen to trigger such onset of the autoimmune response (25). A recent study of single-cell RNA sequencing analysis detected viral transcripts for Epstein-Barr virus and herpesvirus 6A in the thymoma of MG patients; however, the correlation with the virus was non-significant (26). Thus, the exact mechanism underlying the steps of immune activation in the thymus, contributed by infection and inflammation, is incompletely understood.
Inflammatory mediators, consisting mainly of interleukins, interferons, and chemokines from leukocytes, play a major role in immune modulation. Studies have shown the presence of infiltrated macrophages and monocytes at the neuromuscular junction in MG (27, 28). Local release of cytokines from these cells further recruits polymorpho neutrophils (PMN) and other immune cells that permeate into the specific site and start the inflammatory cascades for immune activation. This is supported by the findings of neuromuscular-related antigen expressions (including AChRs), the presence of germinal centers (GCs), and increased frequency of Tfh cells in the thymus, altered microRNAs, and IFN signaling in subpopulations of thymic epithelial cells in MG patients with thymoma (29, 30). Importantly, in addition to B cells for the autoantibody-producing prime role, subsets of T-lymphocytes (T helper cells) also play a crucial role in launching and maintaining chronic inflammation and enhancing the magnitude of inflammatory signals to influence class switching, somatic hypermutation, and differentiation of those B cells to generate autoantibody-producing plasma cells, leading to subsequent development and progression of MG disease (31).
Recent RNA-seq analysis of peripheral blood mononuclear cells (PBMCs) from 19 AChR-MG patients with a disease onset under the age of 50 years (AChR-EAMG) reported transcriptional dysregulation of inflammatory biomarkers (32). The inflammatory proteins important in this study were IL-4, NFκB2, IFN, and RelB, among others. Dysregulation of the molecules was also correlated with the altered AChR-specific antibody levels and microRNAs. Additional studies may dissect the mechanisms underlying the findings.
Our recent nCounter transcriptome results are also in line with literature showing a key role of an inflammatory signature in the pathogenesis of a mouse model of MG (17). We demonstrated an epigenetic influence in regulating expression levels of a key inflammatory cytokine, IL-6, from AChR-stimulated PBMCs in mouse model of MG. We further identified altered expression of many previously unknown genes associated with IL-6 suppression, which was further correlated with autoantibody reduction, by inhibiting an epigenetic molecule, histone deacetylase-2. Future directions in this field should include a comprehensive analysis of individual genes through gene deletion, and bioinformatics analysis of causal relationships of pathogenic and non-pathogenic gene sets affected by transcript level changes of the specific gene.
Cyto/chemokines are the small peptides produced virtually by all cells but primarily by myeloid cells in response to stimuli (Figure 1). They are critical modulators of inflammation and act as potent activators or inhibitors of immune responses (Table 1).
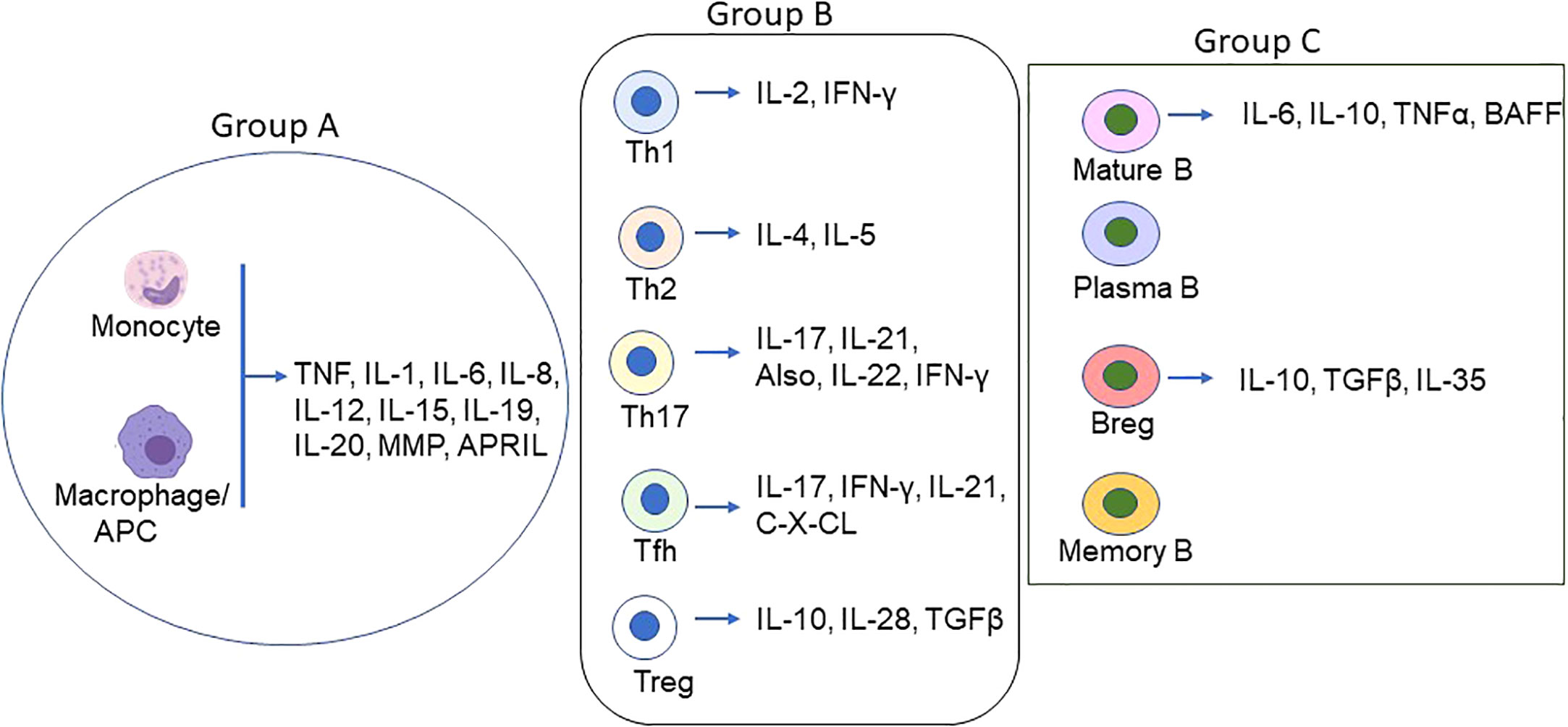
Figure 1 Schematic of Proinflammatory/inflammatory cytokines produced by main immune cell types in MG. Various cell types secret proinflammatory, inflammatory and inflammatory cytokines in addition to the groups (group A = monocytes/macrophages/DCs; group B = T lymphocytes, and group C = B lymphocytes) depicted above. The coordinated function of the cyto/chemokines prime activates DCs and AChR-specific Th/B cells and the ultimate differentiation of antigen-specific B cells to autoantibody-producing plasma cells and memory plasma B cells.
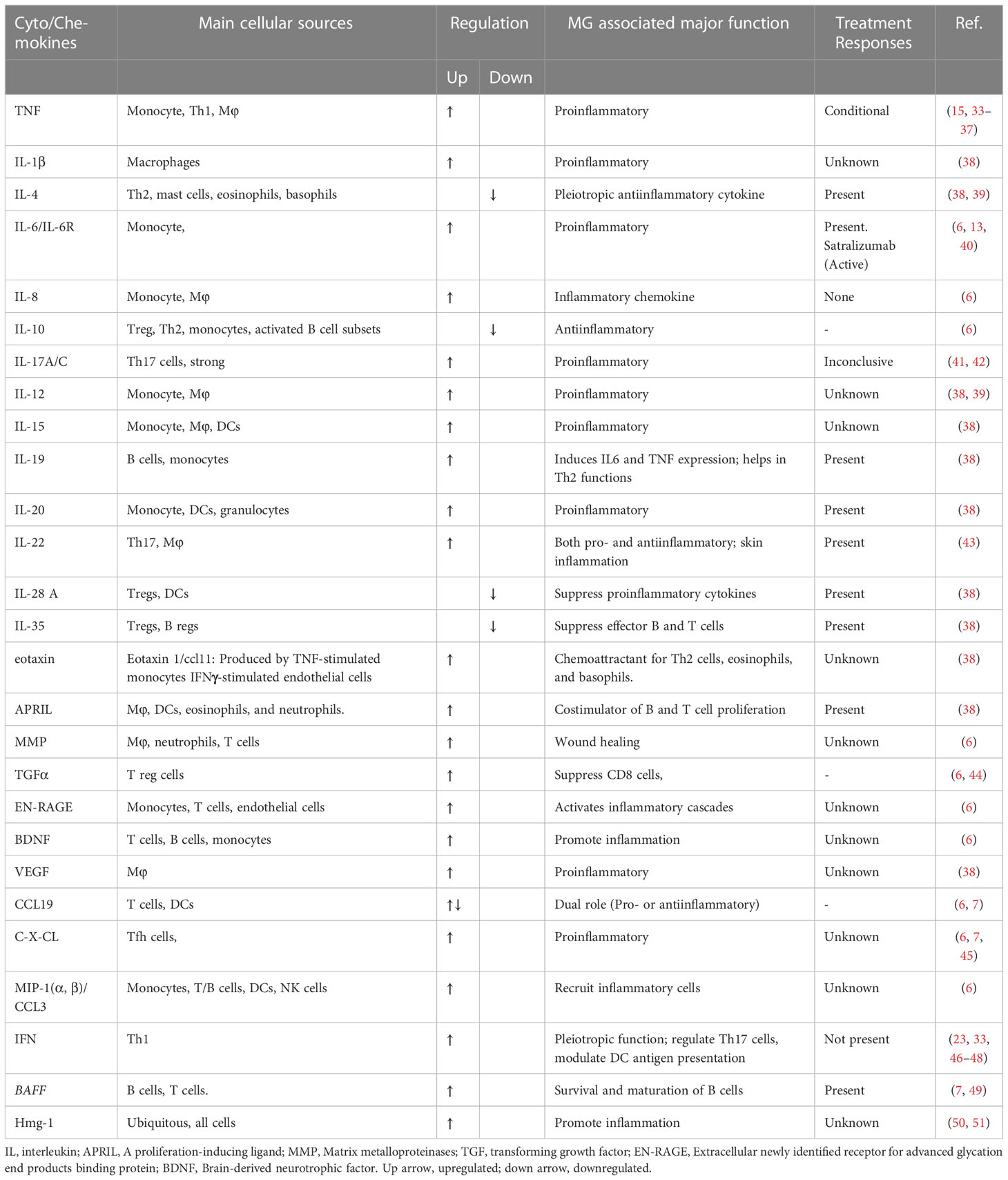
Table 1 Cyto/Chemokines: potent contributors of MG pathogenesis.
Cytokine producing role of nearly all immune cells is known; however, FcR-expressing monocytes/macrophages are the primary sources of the proinflammatory/inflammatory cytokines and chemokines, including mainly TNF, IL-1, IL-6, IL-8, and IL-12 causing histopathological injuries in target tissues, autoantigen expressions, and autoantibody production in MG and other autoimmune diseases (38, 52). IL-6 is a pleiotropic cytokine, released mainly from monocytes and macrophages but also from other cells, including B cells, muscle, and epithelial cells. High levels of IL-6 are associated with the pathogenesis of many diseases. Mice with an acquired or genetic deficiency of IL-6 are resistant to MG, and anti-IL6 antibodies reduced autoantibody levels and disease symptoms in a rat model of EAMG (13, 53). Following binding with its receptor, IL6 receptor (IL6-R), CD26, or soluble IL6-R, the complex binds to CD136, which dimerizes and activates intracellular kinases. The role of Granulocyte-macrophage colony-stimulating factor (GM-CSF), produced by macrophages, endothelium, and other cells, as pro- or anti-inflammatory cytokine is controversial.
Another less-studied intracellular DNA binding protein with a proinflammatory cytokine-like role in MG pathogenesis is the High mobility group box 1 (HMGB1) (50). Extracellular HMGB1 secreted passively from monocytes and other cell types bind to TLR4 of monocytes and macrophages and induce ehnanced production of TNF-α, IL-1, IL-6, IL-8, macrophage inflammatory protein (MIP), C-reactive protein (CRP) and others to amplify inflammatory responses.
Although the role of neutrophils has not been studied well for MG pathogenesis, they play a significant damaging role in many autoimmune diseases, such as anti-glomerular basement membrane nephritis and lupus nephritis (54).
T lymphocytes or T cells play a role in the autoantibody synthesis of B cells through the secretion of various cytokines following stimulation with AChR in MG. Of T cells, T-helper cells 2 (Th2) secret IL-4 and IL-5 that helps in T and B cell growth. Th1 cells produce IL-2 and IFN-γ. T helper 17 (Th17) cells secret mostly IL-17, IL-21, and IL-22 but also IL-23 and IFN-γ (55), and their frequency increases with MG disease severity. Evidence demonstrating stimulus-mediated polarization of Th cells from one subtype to another, such as Th1 to Th17 cells and vice versa (56), might play a crucial role in the pathogenesis of MG.
T follicular helper (Tfh) cells are a subset of CD4 cells (CD4+CXCR5+PD1+) that also secrete IL-17 and IFN-γ. An increase in Tfh cells in correlation with increased numbers of plasma cells and AChR-specific autoantibody titer has been reported from generalized MG patients (57). Tfh cell frequencies were found to decrease after treatment. The-secreted cytokine, IL-21, has been reported to be elevated in MG patients with higher QMG scores and anti-AChR antibody (12). T regulatory cells (Tregs) are indispensable for maintaining peripheral tolerance through suppressing CD4 cells. Reduced expressions of Treg markers (e.g., FoxP3 and IL-10) cause Treg dysfunction and are reported to be associated with increased levels of proinflammatory cytokines, IL-6, IL-17 in MG patients, and IFN-γ (46, 58).
A crucial pathogenic role of B cells in autoantibody-mediated autoimmune disease is known. In addition to producing anti-AChR autoantibody, B cells play an important role in the presentation of autoantigen, secretion of proinflammatory cytokines, interaction with T cells through expressing costimulatory molecules, and formation of ectopic germinal centers in autoimmune diseases. Mature B cells are mainly located in the secondary lymphoid organs but also in peripheral circulation and tissues (tissue-resident B cells). Various cell surface markers, e.g., CD269 and CD138/Syndecan-1 on plasma cells, CD19, CD20, CD21, CD268, and CD79b on mature B cells, and CD27 on memory B cells are present or upregulated on B cell surface during disease progression and are useful targets for current B cell depletion therapies for MG (7). In contrast to T reg cells, B regulatory cells (Bregs) cells lack specific surface markers and are recognized by the secretion of IL-10. Reportedly, CD19+IL-10+ (more precisely, CD19+CD5+CD1d+ and CD19+CD24+CD38+ subsets) Breg cell numbers are significantly lower in peripheral blood and thymus of MG patients compared to healthy controls, and that correlates with disease severity (59–61).
Dendritic cells are differentiated from monocytes and are the professional antigen-presenting cells (APCs) that promote B- and T-cell activations and differentiation. Percentages of plasmacytoid DCs (pDCs) are known to increase significantly over myeloid DCs (another DC subset) in naïve MG patients (62) compared to healthy controls. Activated DCs migrate to secondary lymphoid organs and secret IL-12, IL-6, TNF- and many chemokines, and CD303+ pDCs produce high levels of IFN-α.
Proteins belonging to the CC and CXC family of chemokine ligands and receptors, secreted by peripheral blood mononuclear cells, lymph node cells, macrophages, and thymic GCs also contribute to inflammation in MG (7, 45). Based on in silico analyses, potential therapeutic chemokine targets for MG could be CXCR2, CXCR3, CXCL1, CXCl3, CCL, CCL19, and CCL20 (7).
Fc receptors (FCRs) of some hematopoietic cells that bind to the Fc domain of many Immunoglobulin (Ig) subclasses play an important role in the pathogenesis of MG and other autoimmune diseases. Classical FcRs are activating Fcγ(gamma)Rs (CD64, CD32a, CD16a/b) and inhibitory FcγRs (CD32b), and nonclassical FcR includes only FcRn. Autoantibody bound to an autoantigen, i.e., immune complex (IC), can lead to inflammation through the aggregation of FCRs (45). Targeting FcR is a novel therapeutic approach for some autoimmune diseases. In a mouse model of MG, recombinant Fc multimers (IgG Fc analogue) have been shown to effectively block complement activation by a monoclonal antibody against FcγR. Recombinant soluble FcγRs have not been tested in MG. A clinical trial is underway evaluating a humanized anti-FcγRIIA antibody (VIB 9600) that was recently shown to be effective in inhibiting IC-induced production of type 1 IFN by plasmacytoid dendritic cells and TNF and IL-6 by monocytes (18). The antibody has not been tested for MG. Several monoclonal antibodies against an IgG recycling receptor, neonatal Fc Receptor (FcRn) (63), are currently in MG clinical trial, and some show promise. FcRn blockade has been shown to decrease circulating IgG levels in mice and humans (64) as opposed to the finding of a group showing that despite reducing inflammation, FcRn blockade did not alter the levels of circulating autoantibody in the rheumatoid arthritis (RA) model. A decrease in inflammatory disease burden, as determined in this study by clinical scoring of histopathologic arthritic inflammation and animal mobility, affirms that FcRn is also directly associated with IgG IC-associated inflammation (65). Molecular mechanisms underlying this correlation or association, however, require more research.
Of various FcRn mAbs, Efgartigimod (ARGX-113; Argenx, Breda, the Netherlands) or VYVGART™ was recently approved by U.S. Food and Drug Administration (FDA) for the treatment of AChR seropositive generalized myasthenia gravis (gMG) in adult patients.
Most of the current targeted therapy for MG showed good success in clinical trials. Monotherapies targeting various inflammatory mediators (such as IL-6 or IL6-receptors) are currently active in clinical trials. Tocilizumab (TCZ; RoActemra® or Actemra®; Roche, Basel, Switzerland), also known as atlizumab, is a recombinant humanized mAb against the membrane bound or soluble IL6 receptor. Tocilizumab treatment has shown clinical efficacy in MG patients refractory to rituximab and other diseases, including rheumatoid arthritis (RA), juvenile idiopathic arthritis, Castleman’s disease, and Crohn’s disease (33, 40). However, some solo anti-inflammatory therapeutic treatments did not produce striking outcomes. For example, etanercept (a TNF-α antagonist decoy receptor) was beneficial for patients with low plasma levels of IL6 and interferon (IFN)γ (34), but it exacerbated MG in a patient with RA (35) and also reactivated tuberculosis (36, 37). Other anti-IL6 antibodies (e.g., sarilumab, sirukumab, and siltuximab were tested in treating RA or other diseases but not in MG (66). Humanized anti-IFNα mAbs (Rontalizumab, developed by Genentech (San Francisco, CA, USA) and anifrolumab, developed by Medimmune (Gaithersburg, MD, USA) produced contradictory results in MG or mice model of MG (47, 48), and therefore no clinical trial in MG was conducted.
Investigations are also ongoing to assess novel treatments for MG. Strategies to design a targeted therapy paired with novel molecules targeting inflammation can be beneficial.
Under the influence of local cytokines, CD4+ T cells, through expressions of the lineage-specific transcription factor, can differentiate into Th1, Th2, Th17, or Tregs (e.g., T-bet or TBX21 for Th1, GATA-3 for Th2, Fos-like antigen 2 or Fosl2 for Th17, and FoxP3 for Tregs) (44). Such phenotypic and functional plasticity of Th cells have not been studied in MG, and thorough future investigations in this line remain to be seen. Strategies to manipulate properties of immune cell plasticity through designing appropriate biologic, inhibitor, or microRNA, and thus increasing or equilibrating the number of Tregs and reducing Th1/Th17 type inflammatory cytokines (67) could be a potential therapeutic strategy for MG. One recent study, however, failed to find any observed clinical benefit of using an IL-17-specific monoclonal antibody (Secukinumab) in patients with MuSK-MG (41). Treatment nonresponse to the IL-17-directed therapy could be due to prior exposure to immunosuppressive therapies in the patients. Further, it has recently been shown that not all Th17 cells producing IL-17 are pathogenic or induce autoimmune responses (68). More research in this line is necessary to dissect the mechanism of action of IL-17 antagonists. Expansion of IL-10-producing B-regulatory (B-reg) cells through expressing inhibitory coreceptors such as CD22 (69–71) and suppressing B cell activation is another mechanism to investigate and exploit for developing future B cell-directed therapy.
In conclusion, both the basic- and clinical research data show evidence of a clear association of inflammation with MG. Utilizing new technologies and highly efficient tools for research and data collection, it is possible to conduct a more comprehensive mechanistic study about the role of inflammation in autoimmune development or modulation of the autoimmune response in preclinical models of MG and MG. The results will provide deeper insights to help design more precise and effective therapies. Future therapy approaches for MG may also assess the effectiveness of combination therapy targeting both components, autoantibody and inflammation, responsible for MG pathogenesis.
The author reviewed the literature, contributed solely to the writing, and approved the manuscript for publication.
RH’s research has been supported partly by grants from the Association Francaise contre les Myopathies (AFM), France; the Myasthenia Gravis Foundation of America (MGFA), and Conquer Myasthenia Gravis, USA.
The author thanks Dr. Ashley E. Smith for editing the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MG, myasthenia gravis; AChR, acetylcholine receptor; MuSK, muscle specific tyrosine kinase; LRP, LDL receptor-related protein; NMJ, neuromuscular junction; Tfh, T follicular helper cells; TLR, toll-like receptor; TNF, tumor necrosis factor; NFκB, nuclear factor kappa B; STAT, signal transducers and activators of transcription; C/EBP, CAAT enhancer binding protein; IFN, interferon; RA, rheumatoid arthritis, SLE, systemic lupus erythematosus, MS, multiple sclerosis, PBMC, peripheral blood mononuclear cells, FcR, Fc receptor; mAb, monoclonal antibody; GC, germinal center; IL, interleukin; NFKB2, NFκB subunit 2; Th, T-helper cells, CD, cluster of differentiation; IL6-R, IL-6 receptor; IC, immune complex; Treg, T regulatory cells; Breg, B regulatory cells; DC, Dendritic cells.
1. Howard JF Jr. Myasthenia gravis: The role of complement at the neuromuscular junction. Ann N Y Acad Sci (2018) 1412(1):113–28. doi: 10.1111/nyas.13522
2. Hughes BW, Moro De Casillas ML, Kaminski HJ. Pathophysiology of myasthenia gravis. Semin Neurol (2004) 24(1):21–30. doi: 10.1055/s-2004-829585
3. Huda R, Tüzün E, Christadoss P. Targeting complement system to treat myasthenia gravis. Rev Neurosci (2014) 25(4):575–83. doi: 10.1515/revneuro-2014-0021
4. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol (2011) 69(2):418–22. doi: 10.1002/ana.22312
5. Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: Update on disease types, models, and mechanisms. F1000Res (2016) 5:F1000. doi: 10.12688/f1000research.8206.1
6. Molin CJ, Westerberg E, Punga AR. Profile of upregulated inflammatory proteins in sera of myasthenia gravis patients. Sci Rep (2017) 7:39716. doi: 10.1038/srep39716
7. Huda R. New approaches to targeting b cells for myasthenia gravis therapy. Front Immunol (2020) 11:240. doi: 10.3389/fimmu.2020.00240
8. Tüzün E, Huda R, Christadoss P. Complement and cytokine based therapeutic strategies in myasthenia gravis. J Autoimmun (2011) 37(2):136–43. doi: 10.1016/j.jaut.2011.05.006
9. Hohlfeld R, Wekerle H. Reflections on the "intrathymic pathogenesis" of myasthenia gravis. J Neuroimmunol (2008) 201-202:21–7. doi: 10.1016/j.jneuroim.2008.05.020
10. Schönbeck S, Padberg F, Hohlfeld R, Wekerle H. Transplantation of thymic autoimmune microenvironment to severe combined immunodeficiency mice. a new model of myasthenia gravis. J Clin Invest (1992) 90(1):245–50. doi: 10.1172/JCI115843
11. Fichtner ML, Jiang R, Bourke A, Nowak RJ, O'Connor KC. Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol (2020) 11:776. doi: 10.3389/fimmu.2020.00776
12. Zhang X, Liu S, Chang T, Xu J, Zhang C, Tian F, et al. Intrathymic Tfh/B cells interaction leads to ectopic GCs formation and anti-AChR antibody production: Central role in triggering MG occurrence. Mol Neurobiol (2016) 53(1):120–31. doi: 10.1007/s12035-014-8985-1
13. Deng C, Goluszko E, Tüzün E, Yang H, Christadoss P. Resistance to experimental autoimmune myasthenia gravis in IL-6-deficient mice is associated with reduced germinal center formation and C3 production. J Immunol (2002) 169(2):1077–83. doi: 10.4049/jimmunol.169.2.1077
14. Duan L, Rao X, Sigdel KR. Regulation of inflammation in autoimmune disease. J Immunol Res (2019) 2019:7403796. doi: 10.1155/2019/7403796
15. Caldwell AB, Cheng Z, Vargas JD, Birnbaum HA, Hoffmann A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes Dev (2014) 28(19):2120–33. doi: 10.1101/gad.244749.114
16. Jin W, Yang Q, Peng Y, Yan C, Li Y, Luo Z, et al. Single-cell RNA-seq reveals transcriptional heterogeneity and immune subtypes associated with disease activity in human myasthenia gravis. Cell Discovery (2021) 7(1):85. doi: 10.1038/s41421-021-00314-w
17. Bahauddin A, Ivannikov M, Wang Z, Jamaluddin M, Curtis K, Ibtehaj N, et al. Histone deacetylase isoforms differentially modulate inflammatory and autoantibody responses in a mouse model of myasthenia gravis. Front Neurol (2022) 12:804113. doi: 10.3389/fneur.2021.804113
18. Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding fc receptor involvement in inflammatory diseases: From mechanisms to new therapeutic tools. Front Immunol (2019) 10:811. doi: 10.3389/fimmu.2019.00811
19. Feng X, Song Z, Wu M, Liu Y, Luo S, Zhao C, et al. Efficacy and safety of immunotherapies in refractory myasthenia gravis: A systematic review and meta-analysis. Front Neurol (2021) 12:725700. doi: 10.3389/fneur.2021.725700
20. Leopardi V, Chang YM, Pham A, Luo J, Garden OA. A systematic review of the potential implication of infectious agents in myasthenia gravis. Front Neurol (2021) 12:618021. doi: 10.3389/fneur.2021.618021
21. Delly F, Syed MJ, Lisak RP, Zutshi D. Myasthenic crisis in COVID-19. J Neurol Sci (2020) 414:116888. doi: 10.1016/j.jns.2020.116888
22. Sacchi MC, Tamiazzo S, Stobbione P, Agatea L, De Gaspari P, Stecca A, et al. SARS-CoV-2 infection as a trigger of autoimmune response. Clin Transl Sci (2021) 14(3):898–907. doi: 10.1111/cts.12953
23. Psarras A, Emery P, Vital EM. Type I interferon-mediated autoimmune diseases: Pathogenesis, diagnosis and targeted therapy. Rheumatol (Oxford) (2017) 56(10):1662–75. doi: 10.1093/rheumatology/kew431
24. Ibtehaj N, Huda R. High-dose BAFF receptor specific mAb-siRNA conjugate generates fas-expressing b cells in lymph nodes and high-affinity serum autoantibody in a myasthenia mouse model. Clin Immunol (2017) 176:122–30. doi: 10.1016/j.clim.2017.01.005
25. Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol (2012) 42(1):102–11. doi: 10.1007/s12016-011-8294-7
26. Yasumizu Y, Ohkura N, Murata H, Kinoshita M, Funaki S, Nojima S, et al. Myasthenia gravis-specific aberrant neuromuscular gene expression by medullary thymic epithelial cells in thymoma. Nat Commun (2022) 13(1):4230. doi: 10.1038/s41467-022-31951-8
27. Maselli RA, Richman DP, Wollmann RL. Inflammation at the neuromuscular junction in myasthenia gravis. Neurology (1991) 41(9):1497–504. doi: 10.1212/wnl.41.9.1497
28. Lefeuvre CM, Payet CA, Fayet OM, Maillard S, Truffault F, Bondet V, et al. Risk factors associated with myasthenia gravis in thymoma patients: The potential role of thymic germinal centers. J Autoimmun (2020) 106:102337. doi: 10.1016/j.jaut.2019.102337
29. Song Y, Zhou L, Miao F, Chen G, Zhu Y, Gao X, et al. Increased frequency of thymic T follicular helper cells in myasthenia gravis patients with thymoma. J Thorac Dis (2016) 8(3):314–22. doi: 10.21037/jtd.2016.03.03
30. Sengupta M, Wang BD, Lee NH, Marx A, Kusner LL, Kaminski HJ. MicroRNA and mRNA expression associated with ectopic germinal centers in thymus of myasthenia gravis. PloS One (2018) 13(10):e0205464. doi: 10.1371/journal.pone.0205464
31. Çebi M, Durmus H, Aysal F, Özkan B, Gül GE, Çakar A, et al. CD4+ T cells of myasthenia gravis patients are characterized by increased IL-21, IL-4, and IL-17A productions and higher presence of PD-1 and ICOS. Front Immunol (2020) 11:809. doi: 10.3389/fimmu.2020.00809
32. Barzago C, Lum J, Cavalcante P, Srinivasan KG, Faggiani E, Camera G, et al. A novel infection- and inflammation-associated molecular signature in peripheral blood of myasthenia gravis patients. Immunobiology (2016) 221(11):1227–36. doi: 10.1016/j.imbio.2016.06.012
33. Kremer JM, Blanco R, Halland AM, Brzosko M, Burgos-Vargas R, Mela CM, et al. Clinical efficacy and safety maintained up to 5 years in patients with rheumatoid arthritis treated with tocilizumab in a randomised trial. Clin Exp Rheumatol (2016) 34(4):625–33.
34. Tüzün E, Meriggioli MN, Rowin J, Yang H, Christadoss P. Myasthenia gravis patients with low plasma IL-6 and IFN-gamma benefit from etanercept treatment. J Autoimmun (2005) 24(3):261–8. doi: 10.1016/j.jaut.2005.01.013
35. Fee DB, Kasarskis EJ. Myasthenia gravis associated with etanercept therapy. Muscle Nerve (2009) 39:866–70. doi: 10.1002/mus.21280
36. Miossec P. Réactivation de la tuberculose au cours des traitements par inhibiteurs du TNF [Reactivation of tuberculosis during treatment with inhibitors of TNF]. Rev Prat (2018) 68(5):537–40.
37. Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, et al. Tumor necrosis factor neutralization results in disseminated disease in acute and latent mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheumatol (2010) 62(2):340–50. doi: 10.1002/art.27271
38. Uzawa A, Kuwabara S, Suzuki S, Imai T, Murai H, Ozawa Y, et al. Roles of cytokines and T cells in the pathogenesis of myasthenia gravis. Clin Exp Immunol (2021) 203(3):366–74. doi: 10.1111/cei.13546
39. Huan X, Zhao R, Song J, Zhong H, Su M, Yan C, et al. Increased serum IL-2, IL-4, IL-5 and IL-12p70 levels in AChR subtype generalized myasthenia gravis. BMC Immunol (2022) 23(1):26. doi: 10.1186/s12865-022-00501-8
40. Jonsson DI, Pirskanen R, Piehl F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscul Disord (2017) 27(6):565–8. doi: 10.1016/j.nmd.2017.03.007
41. Papi C, Granata G, Galluzzo M, Iorio R. Myasthenia gravis associated with muscle-specific kinase antibodies in a patient treated with interleukin-17 inhibitor. Neurol Sci (2022). doi: 10.1007/s10072-022-06498-7
42. Roche JC, Capablo JL, Larrad L, Gervas-Arruga J, Ara JR, Sánchez A, et al. Increased serum interleukin-17 levels in patients with myasthenia gravis. Muscle Nerve (2011) 44(2):278–80. doi: 10.1002/mus.22070
43. Zheng S, Dou C, Xin N, Wang J, Wang J, Li P, et al. Expression of interleukin-22 in myasthenia gravis. Scand J Immunol (2013) 78(1):98–107. doi: 10.1111/sji.12057
44. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: Regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178(11):6725–9. doi: 10.4049/jimmunol.178.11.6725
45. Feferman T, Aricha R, Mizrachi K, Geron E, Alon R, Souroujon MC, et al. Novel CXCL13 transgenic mouse: inflammation drives pathogenic effect of CXCL13 in experimental myasthenia gravis. J Neuroimmunol (2009) 209:87–95. doi: 10.1016/j.jneuroim.2009.01.021
46. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
47. Congeni JP, Kirkpatrick RB. Pegylated interferon induced myasthenia crisis–a case report. J Clin Neuromuscul Dis (2013) 14:123–5. doi: 10.1097/CND.0b013e318285257f
48. Baik SJ, Kim TH, Kim HI, Rhie JY. Myasthenia crisis induced by pegylated-interferon in patient with chronic hepatitis c: A case report. Med (Baltimore) (2016) 95:e3782. doi: 10.1097/MD.0000000000003782
49. Ragheb S, Lisak RP. B-cell-activating factor and autoimmune myasthenia gravis. Autoimmune Dis (2011) 2011:939520. doi: 10.4061/2011/939520
50. Maimaitiming B, Uzawa A, Ozawa Y, Yasuda M, Kojima Y, Kanai T, et al. High mobility group box 1 is involved in the pathogenesis of passive transfer myasthenia gravis model. Neuroreport (2021) 32(9):803–7. doi: 10.1097/WNR.0000000000001665
51. Uzawa A, Kawaguchi N, Kanai T, Himuro K, Kuwabara S. Serum high mobility group box 1 is upregulated in myasthenia gravis. J Neurol Neurosurg Psychiatry (2015) 86(6):695–7. doi: 10.1136/jnnp-2014-309232
52. Poëa-Guyon S, Christadoss P, Le Panse R, Guyon T, De Baets M, Wakkach A, et al. Effects of cytokines on acetylcholine receptor expression: implications for myasthenia gravis. J Immunol (2005) 174(10):5941–9. doi: 10.4049/jimmunol.174.10.5941
53. Aricha R, Mizrachi K, Fuchs S, Souroujon MC. Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis. J Autoimmun (2011) 36:135–41. doi: 10.1016/j.jaut.2010.12.001
54. Fu X, Liu H, Huang G, Dai SS. The emerging role of neutrophils in autoimmune-associated disorders: effector, predictor, and therapeutic targets. MedComm (2020) 2(3):402–13. doi: 10.1002/mco2.69
55. Yi JS, Russo MA, Raja S, Massey JM, Juel VC, Shin J, et al. Inhibition of the transcription factor ROR-γ reduces pathogenic Th17 cells in acetylcholine receptor antibody positive myasthenia gravis. Exp Neurol (2020) 325:113146. doi: 10.1016/j.expneurol.2019.113146
56. Caza T, Landas S. Functional and phenotypic plasticity of CD4(+) T cell subsets. BioMed Res Int (2015) 2015:521957. doi: 10.1155/2015/521957
57. Zhang CJ, Gong Y, Zhu W, Qi Y, Yang CS, Fu Y, et al. Augmentation of circulating follicular helper T cells and their impact on autoreactive b cells in myasthenia gravis. J Immunol (2016) 197(7):2610–7. doi: 10.4049/jimmunol.1500725
58. Saito R, Onodera H, Tago H, Suzuki Y, Shimizu M, Matsumura Y, et al. Altered expression of chemokine receptor CXCR5 on T cells of myasthenia gravis patients. J Neuroimmunol (2005) 170(1-2):172–8. doi: 10.1016/j.jneuroim.2005.09.001
59. Karim MR, Zhang HY, Yuan J, Sun Q, Wang YF. Regulatory b cells in seropositive myasthenia gravis versus healthy controls. Front Neurol (2017) 8:43. doi: 10.3389/fneur.2017.00043
60. Lin Y, Chang T, Lin J, Sun C, Wei C, Zhao J, et al. Regulatory b cells are decreased and functionally impaired in myasthenia gravis patients. Front Neurol (2022) 13:808322. doi: 10.3389/fneur.2022.808322
61. Guptill JT, Yi JS, Sanders DB, Guidon AC, Juel VC, Massey JM, et al. Characterization of b cells in muscle-specific kinase antibody myasthenia gravis. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e77. doi: 10.1212/NXI.0000000000000077
62. Chen P, Li Y, Huang H, Li Y, Huang X, Chen Z, et al. Imbalance of the two main circulating dendritic cell subsets in patients with myasthenia gravis. Clin Immunol (2019) 205:130–7. doi: 10.1016/j.clim.2018.10.012
63. Chen B, Vousden KA, Naiman B, Turman S, Sun H, Wang S, et al. Humanised effector-null FcγRIIA antibody inhibits immune complex-mediated proinflammatory responses. Ann Rheum Dis (2019) 78(2):228–37. doi: 10.1136/annrheumdis-2018-213523
64. Kuo TT, Aveson VG. Neonatal fc receptor and IgG-based therapeutics. MAbs (2011) 3(5):422–30. doi: 10.4161/mabs.3.5.16983
65. Blumberg LJ, Humphries JE, Jones SD, Pearce LB, Holgate R, Hearn A, et al. Blocking FcRn in humans reduces circulating IgG levels and inhibits IgG immune complex-mediated immune responses. Sci Adv (2019) 5(12):eaax9586. doi: 10.1126/sciadv.aax9586
66. Raimondo MG, Biggioggero M, Crotti C, Becciolini A, Favalli EG. Profile of sarilumab and its potential in the treatment of rheumatoid arthritis. Drug Des Devel Ther (2017) 11:1593–603. doi: 10.2147/DDDT.S100302
67. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol (2016) 16(3):149–63. doi: 10.1038/nri.2015.18
68. Wu X, Tian J, Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front Immunol (2018) 9:1112. doi: 10.3389/fimmu.2018.01112
69. Li XL, Liu Y, Cao LL, Li H, Yue LT, Wang S, et al. Atorvastatin-modified dendritic cells in vitro ameliorate experimental autoimmune myasthenia gravis by up-regulated treg cells and shifted Th1/Th17 to Th2 cytokines. Mol Cell Neurosci (2013) 56:85–95. doi: 10.1016/j.mcn.2013.03.005
70. Lu J, Li J, Zhu TQ, Zhang L, Wang Y, Tian FF, et al. Modulation of b cell regulatory molecules CD22 and CD72 in myasthenia gravis and multiple sclerosis. Inflammation (2013) 36(3):521–8. doi: 10.1007/s10753-012-9573-z
Keywords: myasthenia gravis, inflammation, autoimmune diseases, AChR, autoantibody, IL-6
Citation: Huda R (2023) Inflammation and autoimmune myasthenia gravis. Front. Immunol. 14:1110499. doi: 10.3389/fimmu.2023.1110499
Received: 28 November 2022; Accepted: 13 January 2023;
Published: 30 January 2023.
Edited by:
Linda L. Kusner, George Washington University, United StatesReviewed by:
Raffaele Iorio, Agostino Gemelli University Polyclinic (IRCCS), ItalyCopyright © 2023 Huda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruksana Huda, cmh1ZGFAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.