 Boyoung Shin
Boyoung Shin Ellen V. Rothenberg
Ellen V. Rothenberg
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 January 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1108368
This article is part of the Research Topic Transcriptional and Epigenetic Control of T and Innate Lymphoid Cell Development and Function View all 7 articles
T cells develop from multipotent progenitors by a gradual process dependent on intrathymic Notch signaling and coupled with extensive proliferation. The stages leading them to T-cell lineage commitment are well characterized by single-cell and bulk RNA analyses of sorted populations and by direct measurements of precursor-product relationships. This process depends not only on Notch signaling but also on multiple transcription factors, some associated with stemness and multipotency, some with alternative lineages, and others associated with T-cell fate. These factors interact in opposing or semi-independent T cell gene regulatory network (GRN) subcircuits that are increasingly well defined. A newly comprehensive picture of this network has emerged. Importantly, because key factors in the GRN can bind to markedly different genomic sites at one stage than they do at other stages, the genes they significantly regulate are also stage-specific. Global transcriptome analyses of perturbations have revealed an underlying modular structure to the T-cell commitment GRN, separating decisions to lose “stem-ness” from decisions to block alternative fates. Finally, the updated network sheds light on the intimate relationship between the T-cell program, which depends on the thymus, and the innate lymphoid cell (ILC) program, which does not.
Conventional T cells provide lifelong protection against infection and cancer by recognizing their cognate antigens and mediating effector functions. To ensure that the host can exert various immune responses in a context-specific manner, T cells have extensive diversity in their sub-lineages. The unique properties of T cells stem from the intersections of functional similarities with different types of immune cells (1, 2). Both T and B cells utilize antigen receptors whose diversities are achieved by DNA recombination and selection mechanisms. Distinct from B cells though, T cells can differentiate to various subsets showing functional parallelism with distinct types of helper-like innate lymphoid cells (ILCs) and natural killer (NK) cells, which lack recombined antigen receptors but produce effector cytokines and/or perform cytolytic functions in response to environmental signals. In addition, T cells possess proliferative potential and generate self-renewing long-lived populations, which are features shared with the multipotent hematopoietic progenitor cells and stem cells, respectively.
A recent global comparison of a wide range of hematopoietic cell types found that lineage-specific transcription factor motif “signatures” distinguished the active chromatin patterns for nearly every major analyzed hematopoietic lineage, suggesting the impacts of distinct lineage-determining transcription factors, but that T-lineage cells notably failed to show any cell type-specific “signature” (3). How, then, is the T-cell identity established and robustly maintained, despite functions broadly overlapping with those of other immune cells? We propose that this distinctive positioning as T cells can be supported by combinatorial actions of transcription factors, instead of relying on a lineage-specific “master regulator”. T cells utilize many transcription factors that are commonly employed by other types of hematopoietic cells for their own respective lineage specification and function. However, by precisely regulating combinatorial actions of these transcription factors over time under the influence of Notch signaling, and possibly also using epigenetic chromatin changes to inhibit reversibility, intrathymic hematopoietic precursors launch and lock down the specific T-lineage program. In this analytical review, we bring together recent evidence that assembles a newly clear picture of how this comes about.
Developmental progression to generate a specialized cell type requires continuous, ordered changes in gene expression (4, 5). As a regulatory program, development must thus be distinct both from the stable epigenetic mechanisms that maintain a mature cell type’s identity (e.g., superenhancers) and from random transcriptional “noise”, both of which have been much studied in cell lines (6, 7). While environmental signals are often essential to trigger and support development, the unidirectional regulatory cascade that emerges depends on the transcription factors that are present in the cell receiving the signals and their impacts on other transcription factors and cellular chromatin states. The genes encoding transcription factors and signaling components collectively determine the regulatory state of the cell, and then the genes they control (downstream genes) encoding effector molecules determine the functional identity of a cell.
All these genes, whether they encode signaling receptors, transcription factors, or effector proteins, are controlled by cis-regulatory modules encoded in the genomic DNA, such as enhancers, silencers, and insulators, largely via interactions with trans-acting sequence-specific transcription factors (8, 9). Specifically activated long noncoding RNAs (lncRNAs) can contribute to chromatin states as well, although their actions are still only characterized in a few cases (10–13). Therefore, the important components driving development, the genes and their regulatory modules, transfer information in a directional manner. For example, transcription factor “X” binds to the regulatory elements of target genes Y and Z, which in turn induces expression of other regulatory factors, “Y” and “Z”. Importantly, the activities of these cis-regulatory elements are never driven by single transcription factors alone, but rather by combinations of factors, even though one or another factor can be rate-limiting in a particular experimental situation (14–20). As a result, gene regulation cascades are not explained by a linear pathway but by a hierarchical network (21, 22). These features of developmental regulatory networks can be effectively captured by using topological network models, in which the functional interactions are represented as inputs and outputs. While there are some caveats about the interpretation of these networks for hematopoietic development (23, 24), topological models are indispensable for compiling evidence to explain cell state differences in terms of gene regulation mechanisms.
In this review, we will focus on the gene regulatory programs utilized in the early stages of thymic T-cell development, in which multipotent progenitor cells undergo definitive T-lineage commitment and establish T-cell identity. Some gene regulatory network (GRN) circuits have been shown to promote cell type stability, as in the pluripotency state of ES cells, while others have been shown to drive ultra-rapid, deterministic cascades of change, as in the early Drosophila embryo (25, 26). As shown below, early T cell development falls between these models. It includes both regulatory subcircuits that resist change and regulatory connections that enforce change; the stochastic balance between these network subcircuits is likely to underlie the distinctively asynchronous kinetics of T cell program entry (27, 28). We review the major regulatory genes that are involved in different sub-programs to establish T-cell fate, resolve coherent but distinct program modules that need to be deployed, and propose an updated gene regulatory network (GRN) model.
There are significant challenges and caveats of experimental strategies that are utilized to understand developmental GRNs. As transcription factors need to bind to specific sequences in the DNA in order to work, genome-wide transcription factor occupancy data should in principle help to map where the “direct” interactions of the putative regulatory factor occur. In the past fifteen years it has become relatively easy to map transcription factor binding across the genome by ChIP-seq (or CUT&RUN, or CUT&Tag) (29–31). Potential transcription factor inputs to active regulatory elements can also be mapped in the DNA even without direct evidence of transcription factor binding, based on the enrichment of their motifs in accessible chromatin in a given cell type (32–34). Mapping open chromatin by DNase-seq or ATAC-seq and using the cell type-specific enrichment of motifs predicted to be bound by given transcription factors (35–37) in the open chromatin has become a powerful way to predict which transcription factors may be important in that cell type (38–44). Even without any evidence that a particular target gene actually responds to the presence of the transcription factor itself, such predictive analysis can be a valuable step toward perceiving network relationships at a global level (39, 45). Adding measured evidence for actual transcription factor occupancy at sites around key genes, when possible, enables specific network predictions to be made (46, 47).
However, actually confronting binding-based or motif-based predictions with empirical tests of individual target gene responses to transcription factor activity perturbations has given a more complex picture. Binding should not be overinterpreted. Transcription factor-DNA interaction sites in a cell type are often much more numerous (order of 104 sites) than the number of genes that change expression in response to the loss or gain of a given transcription factor in that cell type (order of 102 genes) (48–51). This indicates that a given transcription factor’s occupancy is often dispensable for regulation of most of the genes that it binds. Even when the transcription factor may be required for the existence of the cell type, its binding to the promoter of a particular gene in that cell type can be functionally irrelevant for that gene. It is thus challenging to predict which binding sites are actually functional, or what mode of actions they mediate (activation vs. inhibition), solely based on the transcription factor occupancy pattern. In addition, it is not simple to assign a binding site to potential target genes, especially if the binding site is surrounded with multiple genes or when the binding site is distant from a promoter. Developmentally important enhancers of key genes in the T-cell gene regulatory network can be hundreds of kilobases (kb) away from the promoters (e.g. Bcl11b, Ets1, Gata3, Id2; also Myc) (52–59). Disrupting individual regulatory elements by deletion or a mutation of a specific motif sequences can reveal the enhancer-target gene link, but functional redundancy of regulatory elements can lead to underestimation (60–62).
For identification of the gene network linkages described here, therefore, we have required evidence from functional tests that acutely perturbed the levels of transcription factor proteins in a specific developmental context. This has been achieved by germline/conditional deletion using a Cre-loxP or CRISPR-Cas9 system, knocking down using RNA interference (RNAi), repression using CRISPR interference (CRISPRi), or acute overexpression/ectopic activation of the target gene, each of which was then followed by in vivo and/or in vitro phenotype scoring at the cellular, transcriptomic, and epigenomic levels. Furthermore, we have given greater weight to results from experiments where the role of a transcription factor was tested in a precise, relevant T-cell developmental context and time window. This was important because recent results of stage-specific perturbation tests have shown that the same transcription factor’s function may change or disappear entirely in a different context, or even at a different stage within the same lineage (49, 51, 63–68). We have sought to apply consistent statistical criteria to these expression differences and to emphasize relationships that have been independently confirmed.
Precise controlling of perturbation timing can be experimentally challenging, especially in the context of a developmental process, as the cells are constantly progressing forward to the next stage. Inadequately defined perturbation time-windows could lead cells to developmental deviation toward irrelevant fates prior to reaching the stages of interest, or allow cells to activate compensatory mechanisms. Wide perturbation time windows could also span states in which the same transcription factor plays different roles, which could complicate data interpretation. Also, although stage-specific perturbation approaches can capture the functional consequences of the perturbation effectively, it is difficult to dissect whether the phenotype is resulting from direct effect, indirect effect via gene network, or indirect effect via differential population survival. As noted above, simply detecting transcription factor binding in the vicinity of a possible target gene is not enough to prove a direct functional relationship.
These limitations and challenges are not completely avoided within the data sources that we have analyzed to construct the update of the early T-development GRN. However, support for specific sites through which a transcription factor could exert direct control of a target gene can be gleaned from the recent increase in available genome-wide transcription factor binding data together with measurements of local chromatin states such as chromatin accessibility, 3D chromatin structure, and changes in histone marks, when these data are coupled with analyses of transcription factor perturbation effects.
The current gene network model we present differs from previous versions (27, 69–74) in several ways. In particular, initial models for early T-cell developmental GRNs were based primarily on candidate gene measurements, due to technical considerations. Targeted assay systems such as qPCR were used to examine perturbation effects on sets of only 100-150 genes out of 10,000 expressed transcripts, focused mainly on high sensitivity monitoring of transcription factor coding genes. It is now routine to use RNA-seq to measure the entire transcriptome quantitatively in an unbiased manner with low numbers of input cells, both at the bulk population level and at the single-cell level. This reveals whole batteries of genes coregulated by a given transcription factor perturbation in the specific context, which help to identify the changes in developmental status that have been induced. Genome-wide transcriptome data processing pipelines also standardize accepted statistical criteria. Where available, single-cell transcriptome analyses are also useful to separate perturbation effects on cells within a lineage from perturbation effects on population balances between the lineage of interest and contaminants.
Another change has been the advent of better technology for acute loss of function as well as acute gain of function of transcription factor genes within a well-defined developmental time window. Whereas Cre excision required a separate mouse strain to be developed for each gene to be targeted, using Cas9-transgenic mice (75) as cell sources for in vitro T-cell differentiation has made it possible to use guide RNAs (gRNAs) targeting one or several genes anywhere in the genome to be introduced, to disrupt genes efficiently at any stage desired in the same genetic background. Previous analyses of transcription factor-target linkages in early T cell development have often depended on gain of function or ectopic expression experiments because these techniques change transcription factor levels acutely in a specific cell type with even faster kinetics. However, multiple recent examples have shown that transcription factor impacts on a network can differ markedly depending on the level of expression of the transcription factor protein (GATA3, PU.1, Runx), and levels may be hard to control in gain of function. While loss-of-function experiments can pose other problems (asynchronous, sometimes slow loss of targeted protein; potential viability losses in the affected cells; potential masking by redundancy), they measure effects of factors at their normal levels of expression. Thus, gain of function data have needed to be re-evaluated in light of corresponding loss of function data, and the manipulated levels of factor protein have needed to be compared to physiological levels.
To construct the network models shown below, we have compiled data from several sources where developmentally well-defined gain or loss of function perturbations were carried out, with all significantly responding genes from each perturbation study tabulated in Table S1. The studies used are described in Box 1. Table S1 also compiles evidence of local binding by the transcription factor of interest around each functional target gene, wherever this evidence was available. Note that different perturbation experiments used as sources focused on different developmental time windows and were more or less sensitive to loss or gain of a given factor’s activity, depending on the normal expression baseline at that timepoint. Where there was variation between different controls or inadequate expression of a gene within the time window tested, even repeatedly observed effects may have missed statistical significance cutoffs. Therefore, while we have generally depended on more than one corroborating piece of evidence for each connection shown, we have not required that all RNA-seq sources should score the same genes as “significantly” affected. Taken together, however, these results now provide a clearer view of the architecture of the T-cell specification gene regulatory network, showing its modular construction, and the coordination of changes in activities of its component subcircuits from stage to stage.
Box 1: Sources of data compiled in network models
Table S1 presents the main data and sources used to establish specific connections in the GRN models that follow. These sources constitute RNA-seq and microarray results from studies of germline deletion of genes encoding the high mobility group factor TCF1 (encoded by Tcf7) (28, 76, 77), the basic helix-loop-helix E proteins E2A (Tcf3) and HEB (Tcf12) (78, 79), and the later-activated zinc finger factor associated with T-cell lineage commitment, Bcl11b (28, 80). In addition, we used studies of acute, stage-specific deletions of the ETS family subgroup member PU.1 (encoded by Spi1, previously called Sfpi1 in mice) (28, 48); of Lmo2 (81); of GATA3 (28, 82, 83); of Bcl11a (28); of Erg (28); of Notch1 and Notch2 together (67); and of Runx1 and Runx3 together (51, 84). Although data for cells in the same developmental stages with complete disruption of Ikaros (Ikzf1) were not available, differentially expressed genes that responded to Ikaros (Ikzf1) zinc finger 4 deletion were also added (85). Finally, we included data from studies of acute gain of function of factors at stages after they would normally have been shut down, including PU.1 (Spi1) (48), and the transcription factor adaptor Lmo2 (86–88). In addition, supporting results came from studies introducing into pro-T cells acute antagonists of key transcription factors, including the natural E protein antagonist ID2 (89) or an artificially constructed dominant repressor form of PU.1 (90). Additional supporting results came from earlier perturbation studies knocking out E protein genes Tcf3 (E2A) and Tcf12 (HEB) or Bcl11b (71, 79) and studies utilizing progenitor or pro-T cell lines and acute T-cell malignancies to interrogate roles of the early-acting transcription factors Lmo2 and Hhex (81, 88, 91). For data on normal developmental expression dynamics of these genes in pro-T cells, RNA-seq and single-cell RNA-seq datasets were used (92–94), corroborated by highly curated microarray data (95). Data used were all from experiments in the mouse system, but the underlying gene expression patterns involved are largely conserved in human data (94, 96–103)(rev. in (103)). In addition to the data incorporated into Table S1, we consulted data from other studies for TCF1, GATA3, and Bcl11b target gene regulation as well (16, 63, 83, 90, 104, 105). Finally, note that positive and negative regulatory connections shown in the models indicate a measurable effect in the indicated developmental window, but usually not a pure Boolean function.
Thymic T-development begins as hematopoietic progenitor cells possessing lymphoid potentials migrate into the thymus and interact with the cortical epithelial cells providing Notch ligands, growth factors, and cytokine signaling (106–109). This drives the developmental program shown in Figure 1A. At early stages, intrathymic precursor cells undergo extensive cell proliferation and upregulate some of the T-lineage associated genes. However, they still preserve multipotentiality and can differentiate into non-T lineage cells, especially if Notch signaling is withdrawn. This uncommitted stage is referred to as “Phase 1”, which includes double-negative 1 (DN1, or Early T-cell precursor, ETP) and DN2a stages. In Phase 1, both ETP and DN2a cells express high levels of cKit and CD44, but CD25 surface expression distinguishes ETP (CD25-) from DN2a (CD25+)(Figure 1A). Recent single-cell transcriptomics studies reveal heterogeneity in Phase 1 population both in human and mouse. The pro-T cells comprising Phase 1 actually include multiple subsets of ETPs and populations transitioning to DN2a cells that display distinct gene regulatory programs, with patterns mostly conserved between mouse and human based on single-cell and bulk analyses (94–98, 110). Of interest, the intermediate-stage ETP populations, more than the most primitive ETPs, transiently express a set of non-T lineage-associated genes (e.g. Mpo, encoding myeloperoxidase) even though these cells are on the T-cell pathway (94). This suggests that these genes are induced as a part of a normal developmental progression program in ETPs, and multilineage priming occurs before T-lineage commitment.
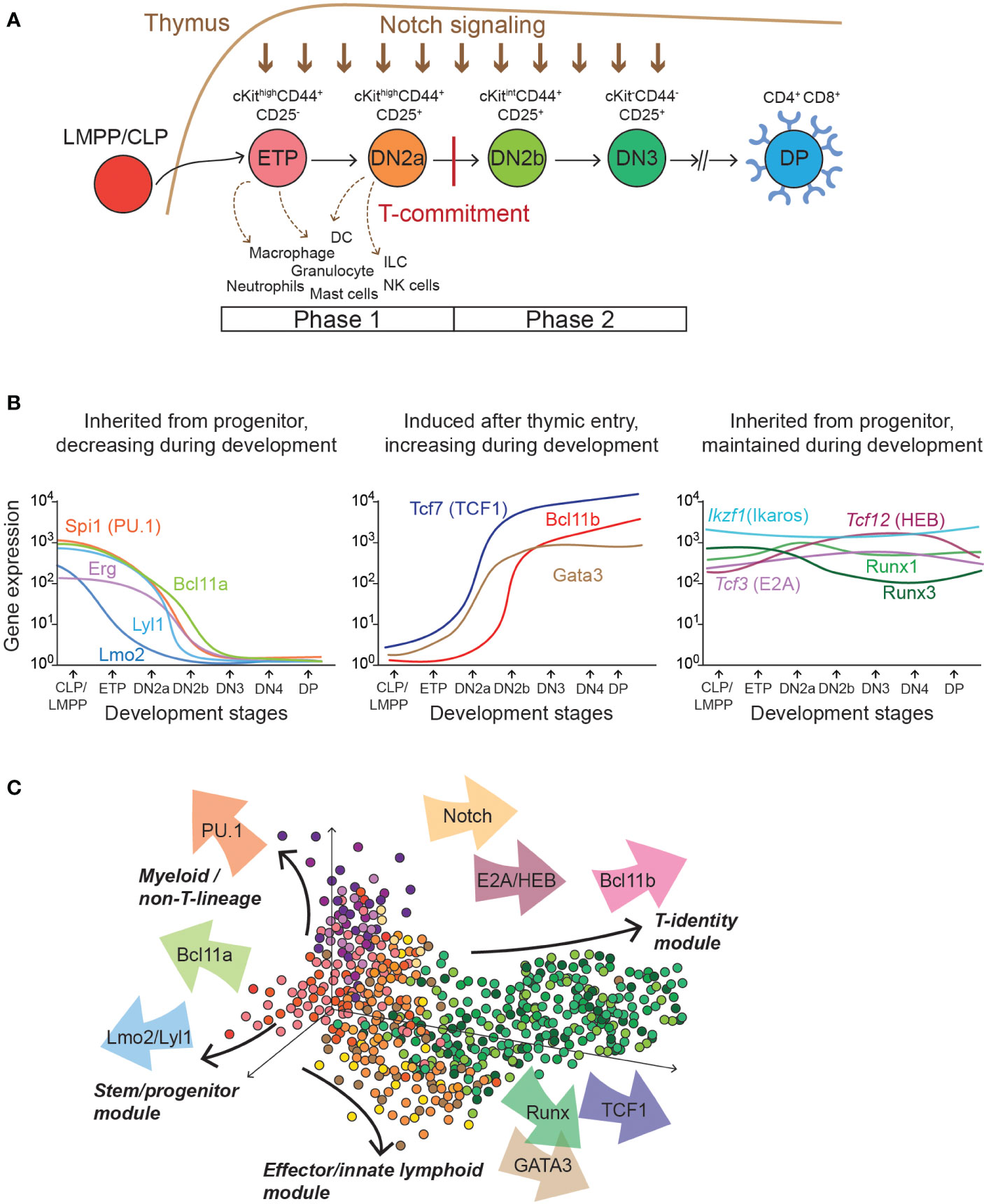
Figure 1 T cell development stages and transcription factor expression kinetics. (A) Diagram depicts different stages of early thymic T cell development that T-progenitor cells sequentially go through. Informative surface proteins that are utilized to define each stage are indicated (cKit, CD44, CD25 for ETP to DN3; CD4 and CD8 for DP). Developmental plasticity to generate alternative, non-T-lineage cells is shown with dotted arrows. Note that these alternative lineage potentials are silenced after T-lineage commitment. Lineage commitment to a T cell fate distinguishes Phase 1 (before T-lineage commitment) and Phase 2 (after T-lineage commitment). CLP, Common lymphoid progenitor, LMPP, lympho-myeloid primed progenitor. (B) Graphs show mRNA expression kinetics of important transcription factors involved in early T cell gene regulation programs. Left: Transcription factors inherited from the bone-marrow progenitor cells whose expressions gradually decline during T-development (Spi1, Bcl11a, Erg, Lyl1, and Lmo2). Middle: transcription factors upregulated in pro-T cells by thymic microenvironment (Tcf7, Gata3, and Bcl11b). Right: transcription factors expressed from the bone-marrow progenitor cells and stably and maintained during T cell development (Ikzf1, Tcf3, Tcf12, Runx1, and Runx3). Gene expression data was plotted by utilizing publicly available mRNA expression datasets for immune cells with curve smoothing (https://www.immgen.org) (93). (C) Diagram illustrates transcription factors (arrows) providing distinct forces to different gene expression program modules in individual cells.
Upon sustained exposure to Notch-ligand and other thymic microenvironmental signals, progenitor cells intrinsically commit to a T cell fate, and the developmental plasticity to non-T-lineages is terminally blocked. After T-lineage commitment, pro-T cells establish a T-cell identity gene expression program and start to rearrange some forms of T cell receptor (TCR) genes. For conventional T cells, successful gene rearrangement for expression of TCRβ chain is assured by quality control at the β-selection checkpoint during DN3 stage. Other T cell precursors rearrange and express genes encoding TCRγ and TCRδ instead, to become γδ T cells. These stages from commitment to β-selection are collectively referred to as “Phase 2”, which is comprised of DN2b (cKitint CD25+ cells) and DN3a (cKitlow CD25+ CD28- CD27low) stages (Figure 1A) (111–113). Further development depends on the cells’ TCR interactions. Following β-selection, most developing T cells accumulate as CD4+CD8+ (“DP”) cells, while they complete their TCRα gene rearrangements and express for the first time the TCRαβ that they will use forever, if they are allowed to live. However, they undergo an ultra-stringent selection process to reject all cells with inadequate or highly autoreactive TCR specificity. The rare surviving cells can finally mature, undergoing divergent programs of positive selection into CD4 or CD8 single-positive cells before they emerge from the thymus, associated with “helper” or “cytotoxic” function respectively. Notably however, most core T-identity program genes that pro-T cells activate in Phase 2 are irreversibly maintained throughout all later stages of T cell development and immunological responses.
As the thymus-seeding precursor cells migrate from the bone marrow and first enter the thymus, they express transcription factors inherited from their progenitor cells in the bone marrow. Expression of many such legacy transcription factors is maintained across multiple cell divisions. While many progenitor-associated factors are turned off eventually, some legacy transcription factors maintain their expression throughout thymic T cell development, but these often adopt new roles during stage transitions by occupying different sets of genomic regions from those in hematopoietic progenitor cells. The co-existence of “inherited transcription factors” together with “newly induced regulators” in the thymic precursor cells generates numerous possible combinatorial inputs to different target genes which dynamically change their expression during developmental progression in Phase 1 and Phase 2 (Figure 1B). This review focuses on key driving regulatory factors and responders composing the Phase 1- and Phase 2-GRNs, in which many components show dynamic changes throughout developmental progression.
The newly proposed GRN that we present here highlights the observation that in each stage of early T-lineage development, subsets of target genes for a given transcription factor are often regulated in parallel ways, collectively composing a specific module that often has coherent biological function (28, 114). While the definition of a module is not precise, we use this framework to describe fairly discrete components of the developmental process which are regulated distinctly even if the cell is expressing other sets of genes at the same time. One module (defined by expression of a group of genes) may remain active consistently throughout a series of stages while other modules are sharply changing activity, or a module can be affected coherently by a perturbation that does not affect genes in other modules within the same cells. As shown below, this gives the overall process of early T cell development an “assembled” quality. Key transcription factors often appear to play roles in activity of an entire module, even while the individual target genes they act upon also have other regulatory inputs. However, it is noteworthy that a transcription factor does not define the module on its own. The same transcription factor may even work in more than one module, as shown below. Instead, different transcription factor ensembles can be seen to define distinct modules, working together to drive expression of shared target genes. They establish positive and negative feedback loops within a module, for stabilization, or between different modules, which results in dynamic gene network behavior. This modular structure is significant because the timing of transitions that individual cells make along the pathway is very asynchronous, with cells showing an ability to linger in any of several states for several cell cycles before progressing (27). The slow pace of differentiation implies that regulation of the modules that predominate in any particular stage is fairly stable; however, developmental progression depends on eventually breaking this stability. In developmental gene networks, a regulatory state is often stabilized when different transcription factors with concordant effects on the same module also support each other’s expression (115), and some examples of this are shown below. In contrast, inter-module inhibition or repression between regulators can make expression of different modules unstable and eventually incompatible.
Mutually exclusive expression of different regulators generates sharp cell-fate boundaries in binary cell fate decisions and in embryos, for example (115, 116). However, one notable feature of the T cell developmental network linkages is that the repression which is detected is often incomplete. Many repressive interactions in this system cause a dampening of expression levels but not a silencing of the target genes. This “soft” repression function enables factors with mutually antagonistic activities to coexist in the cells for days and multiple cell divisions, and it enables activities of opposing modules to overlap within an individual cell at the same time. Therefore, distinct sets of transcription factors can pull and push activities of their target modules in different directions in the same cell (Figure 1C), and the resultant sum of the vectorized “forces” instructs cell fate (28). In the following, we review both the regulatory circuits that maintain coherent module activity, and also the sources of antagonists that finally keep them from persisting.
The architecture of the modular subprograms within the T-cell GRN has become much clearer through recent work. The distinct subprograms that comprise the early T-cell GRNs can be categorized as 1) the T-identity module 2), the stem or progenitor module, 3) conditional access to alternative fates as a side effect of the stem/progenitor module, and 4) a cell survival and proliferation module. While the T-identity module needs to be successfully installed to provide constitutive expression of the core T-cell genes, the second module must be silenced in order to convert multipotent precursors to T-lineage committed cells. Finally, the cell survival and proliferation program in Phase 1 and Phase 2 ensures the production of sufficient number of immature T cells to accommodate later positive and negative selection. Because the T-cell identity module introduces the initiators and overall structure of the entire process, it is discussed first.
Commitment to the T-cell fate involves two major events: 1) terminal blockade of alternative lineages, concomitant with 2) acquisition of T-identity gene expression. The gene network instructing the constitutive expression of the T-lineage defining genes will be referred to as the T-identity module. Successful establishment of the T-identity module is pivotal because the core T-identity established in early thymic developmental phases is robustly maintained even after these progenitor cells become mature T cells, long after they leave the Notch ligand-rich thymic environment. The “T-cell markers” include CD3 clusters and TCR signaling mediators, as well as transcription factors necessary for the induction and maintenance of these marker genes. The T-identity module requires activities of Notch signaling, TCF1, GATA3, Runx transcription factors, and Ikaros starting from Phase 1, then receives critical positive inputs from the E protein complex and Bcl11b as pro-T cells progress through Phase 2. Expression patterns of the key factors in different immune cell populations are shown in Figure 2A (data from www.immgen.org (93)).
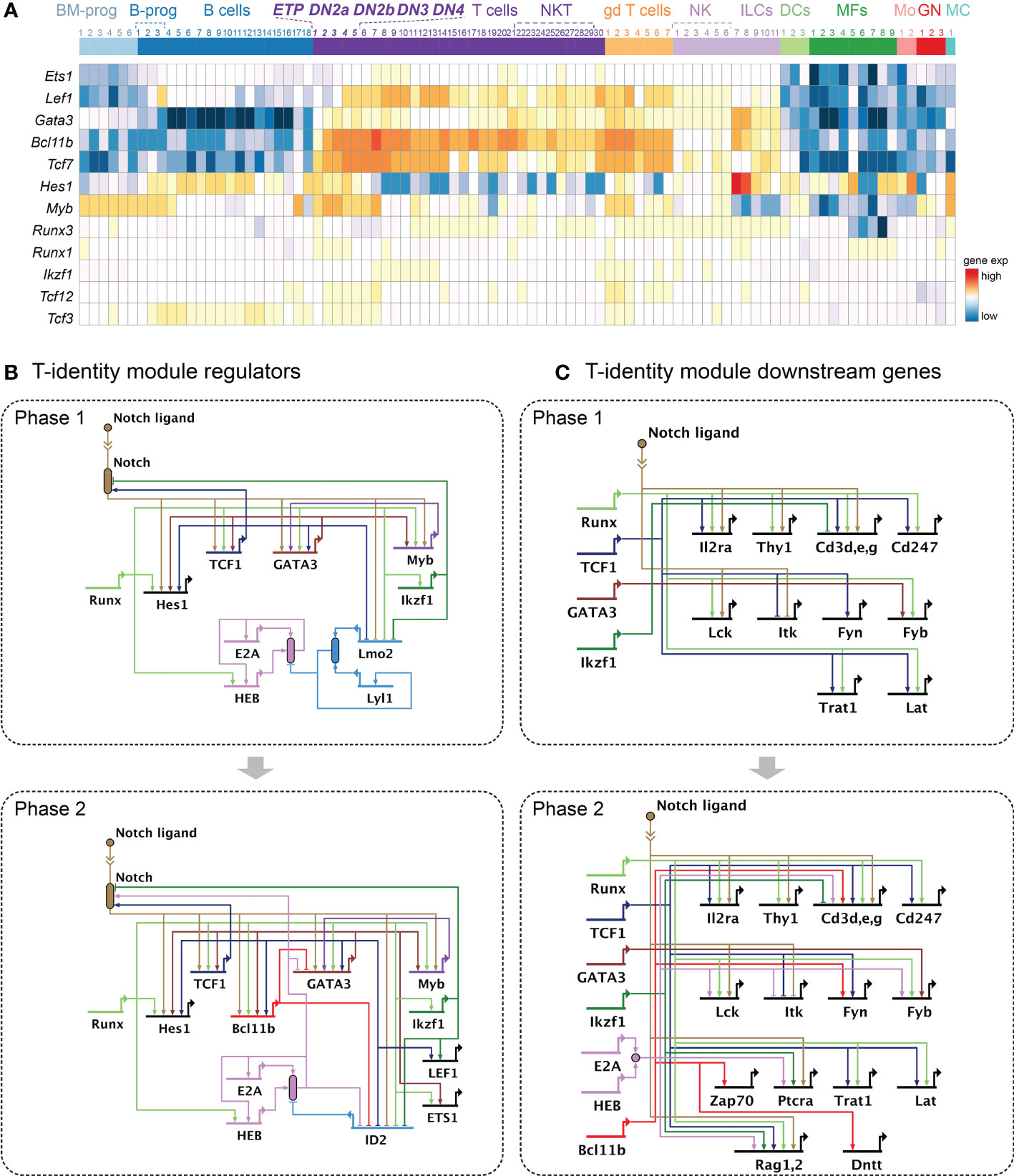
Figure 2 Gene regulatory networks driving the T-identity module. (A) Heatmap shows expression patterns of key transcription factors supporting the T-identity module across different immune cell types, using the ImmGen MyGeneSet tool (https://www.immgen.org) (93). Bone marrow progenitor (BM progen), B cells precursors (B progen), B cells, T cells, γδ T cells ("gd T cells"), natural killer cells (NK), Innate lymphoid cells (ILC)s, Dendritic cells (DC), Macrophage (MF), Monocyte (Mo), Granulocyte (GN), Mast cell (MC). See Table S2 for the number keys. (B, C) BioTapestry models (71, 117) of gene regulatory network relationships described in the text. Evidence for all connections shown is in Table S1. Structure of BioTapestry models: Genes are shown with regulatory regions (horizontal lines) distinct from their encoded outputs (bent arrow at “promoter”). Connections with arrows represent positive regulation. Connections ending in blocking lines represent negative regulation. When gene products combine to produce a functional unit, or when activity is not a simple function of transcriptional output, a “bubble” symbol is placed to represent the emergent function from their collective activities. Both negative and positive regulation can impinge on such activity bubbles, e.g. ID factors inhibiting E protein activity without necessarily inhibiting the expression of the E protein coding genes themselves. Double chevron symbol indicates ligand-receptor interactions, here used to represent Notch ligand—Notch interaction as a source of regulatory input to other genes. Arrows consisting of dotted lines show decreasing or weak activities. These conventions are used also in Figures 4B, C, 5B, C. (B) Current model for the T-identity module regulator transcription factors in Phase 1 (top) and Phase 2 (bottom). (C) Current model for the T-identity module downstream molecules in Phase 1 (top) and Phase 2 (bottom). Expression of Il2ra (CD25) indicates developmental progression from ETP to DN2 and DN3 stages in mice. Surface markers: Thy1, Cd3 clusters genes (Cd3d, Cd3e, Cd3g), Cd247; TCR signaling molecules: Lck, Itk, Fyn, Fyn, Trat1, Lat, Zap70, Ptcra; TCR rearrangement molecules: Rag1, Rag2, Dntt.
Notch signaling is an absolute requirement for initiation and establishment of the T-identity module. In the absence of Notch signaling, B cells develop in the thymus instead of T cells, whereas constitutive expression of intracellular domain of the Notch (ICN) in the bone-marrow progenitor cells induce extrathymic T cell development (118–120). In addition, many if not all thymic seeding progenitors need to be primed by some level of Notch signaling in the bone marrow. Whereas Jagged-class Notch ligands expressed in the bone marrow do not signal as strongly as the Delta-class Notch ligands in the thymic microenvironment (121–123), this prior experience seems to be important for the cells to acquire competence to initiate the T cell program (124) A thymic seeding progenitor population that had received Notch signaling before thymic entry also exists in humans, supporting a physiological contribution of Notch signaling to T-lineage specification from the prethymic stages in both humans and mice (97). The stromal cells in the thymic cortex express the strongest ligands for signaling through Notch1, mainly Delta-like 4 (Dll4), as well as Dll1 and to a lesser extent Jagged 2 (Jag2). These engage with Notch receptors (Notch 1-Notch3) on the thymic precursor cells with varied affinities (122, 125). This ligand-receptor interaction induces proteolytic cleavages in the Notch receptor, releasing the ICN to the cytoplasm. Then, the Notch ICN interacts with DNA-binding transcription factor RBPJκ and functions as co-activator, recruiting other transcriptional coactivators and chromatin modifying enzymes (122, 126).
Although Notch1 is most strongly expressed throughout, acute deletion of different Notch family genes in pro-T cells shows that Notch1 and Notch2 cooperate to induce the T-identity module in Phase 1 cells by activating genes encoding essential transcription factors (e.g., Tcf7, Myb, Gata3, Hes1), the core-T cell marker genes (Cd3g, Cd3e, Thy1) and the useful DN2-DN3 stage marker Il2ra (in mouse; not in human at this stage). Later, in Phase 2, Notch signaling induces genes necessary for TCR rearrangement and signaling (Rag1, Rag2, Lck, Ptcra) (67)(Figures 2B, C). Notch actions in the T-cell identity module are not hit-and-run; the signals must be sustained through lineage commitment and then to sustain most cells’ viability into the beginning of β-selection [rev. by (127)]. Notably, however, the target genes regulated by Notch signals either positively or negatively change markedly between Phase 1, early Phase 2, and the end of Phase 2 (67). The shifting but essential roles of Notch signaling in pro-T cells provide an example of context-dependent shifts in regulator deployment which are seen for other factors as well (15, 49, 51).
Notch-activated targets in Phase 1 cells include genes encoding TCF1 (Tcf7) and GATA3, which are pivotal for instituting the T-identity module (Figures 2A, B top). The activation of Tcf7 by Notch signaling appears to be direct, although its maintenance becomes Notch-independent in Phase 2 (67, 76). The functional importance of TCF1 and GATA3 in T-lineage specification is demonstrated by transgenic animal models that lack Tcf7 or Gata3 expression. Tcf7- or Gata3-deficiency in pro-T cells abrogates T-cell development from the earliest Phase 1 stage (76, 77, 82, 104, 128–130). Tcf7 deletion in bone-marrow derived progenitors using Vav1-Cre caused developmental arrest at ETP stage and allowed abnormal transcriptome clusters to accumulate among thymocytes in steady state, based on single-cell RNA-seq (scRNA-seq) (63). In accord with these results, another recent single-cell transcriptome study using dual guide-RNAs (gRNAs) to disrupt Tcf7 or Gata3 specifically in Phase 1 cells showed that the precursor cells lacking TCF1 completely failed to enter the normal T-cell developmental trajectory, while those lacking GATA3 failed to progress properly (28). While Tcf7 and Gata3 both depend on inputs from Notch signaling and Runx family transcription factors to be turned on, they also create a possible stabilization circuit for early T-cell specification by positively regulating each other as well (28, 77, 82)(Figure 2B).
Despite these positive feedbacks, acute Tcf7 deletion results in different impacts on the developing pro-T cell population than acute Gata3 deletion in the same Phase 1 developmental time window, based on scRNA-seq data. TCF1 and GATA3 regulate distinct target genes, indicating that these factors do not perform redundant functions. Also, studies using gain-of-function approaches suggest that TCF1 overexpression is completely different from the effect of high dosage GATA3 in pro-T cells. A high level of TCF1 in the mouse bone-marrow progenitor cells can upregulate essential genes in the T-identity module, such as Gata3 and Bcl11b, even without Notch signaling, causing cells apparently to bypass the ETP stage (77). In contrast, elevated GATA3 levels block T cell development, promoting deviation to an alternative, mast-cell lineage in the absence of Notch signaling, and killing pro-T cells if Notch signaling is sustained (79, 82, 83, 131). This difference in part involves the different impacts these factors have on genes within proliferation and survival modules used in Phase 1 cells, including effects on Kit, Il7r and Spi1 (see below).
In pro-T cells, TCF1 and GATA3 instruct T-cell development by upregulating many T-program genes (Notch1, Notch3, Hes1, Gata3, Bcl11b, Lef1, Ets2, Il2ra, all Cd3 genes, Cd247, Tcrb, Lat, Fyn, Rag1, Rag2, and Trat1 by TCF1; Myb, Ets1, Tcf7, and Bcl11b by GATA3)(Figure 2C). Importantly, both TCF1 and GATA3 are required together for the initial induction of Bcl11b, which will be important in Phase 2, suggesting that multiple transcription factor inputs are non-redundantly required (16).
As TCF1 and GATA3 are necessary to initiate the T-lineage specification program, the positive regulatory factors inducing these transcription factors are also critical. TCF1 and GATA3 positively regulate each other and Runx transcription factors also provide supportive inputs, as described below. The critical regulatory elements of the Tcf7 gene in T-lineage cells, 30-40kb upstream of its promoter regions (132), are also occupied by RBPJκ, Runx factors, GATA3, and TCF1, consistent with direct positive regulation by these factors (Figure 3 left, in "DN2b/DN3" samples; note that both Tcf7 and Gata3 in this figure are transcribed from right to left). Similarly, an enhancer region 280 kb downstream of the Gata3 gene, which is known to be necessary for Gata3 expression in the T-lineage (54), is occupied by RBPJκ, Runx factors, TCF1, Bcl11b, and GATA3 by Phase 2 (Figure 3 right, “[i.e., "DN2b/DN3"] samples).
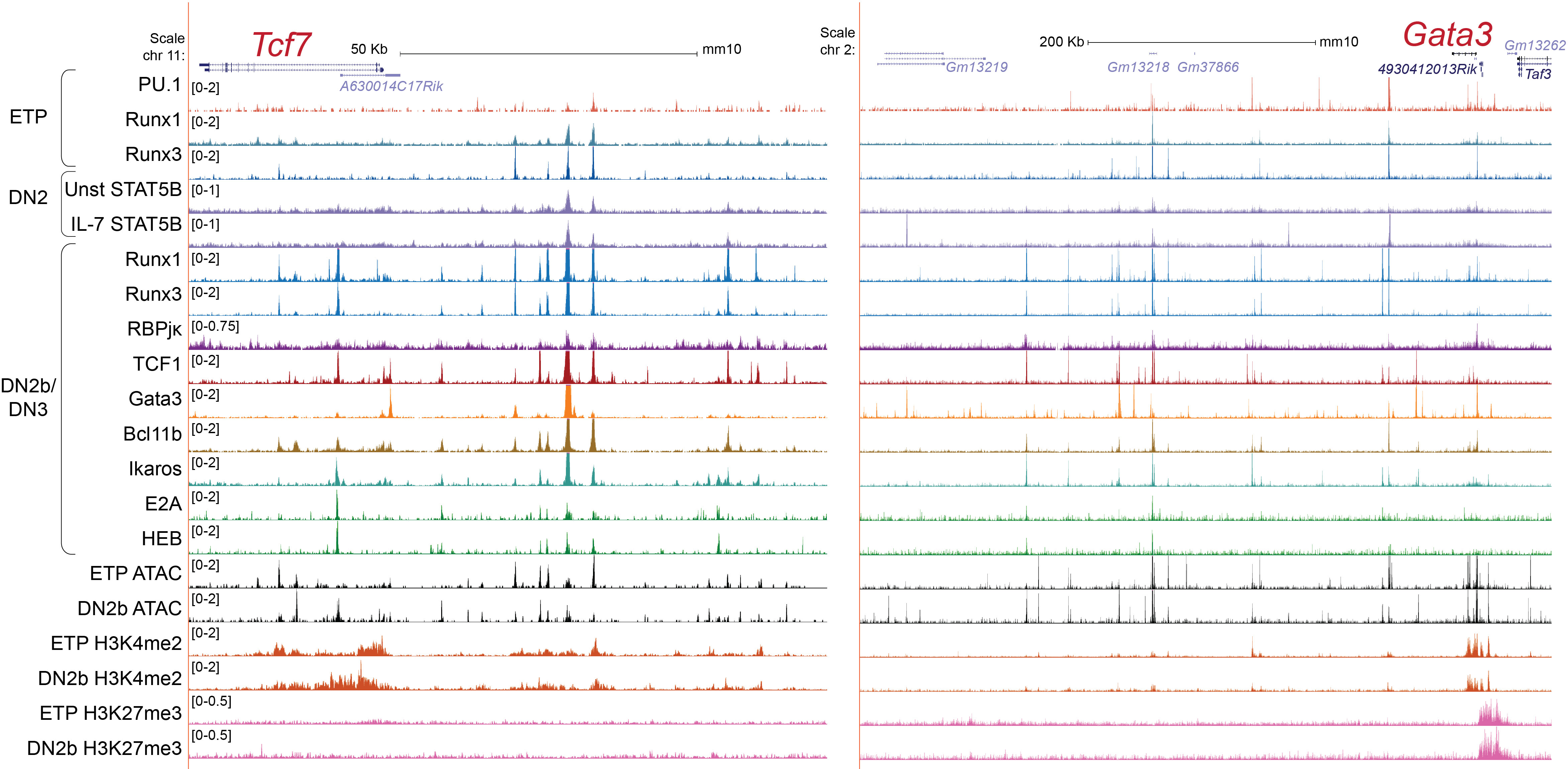
Figure 3 Transcription factors and chromatin state markers in Tcf7 and Gata3 enhancer regions. UCSC genome browser tracks show transcription factor occupancy, histone markers, and chromatin accessibility profiles near the Tcf7 locus (left) and Gata3 locus (right) in the indicated stage (67, 84, 93, 101, 133, 134). H3K4me2 represents active enhancers and H3K27me3 marks repressive chromatin regions. Unst STAT5B: unstimulated STAT5B, IL-7 STAT5B: IL-7 stimulated STAT5B.
The strong force that TCF1 exerts to drive the T-cell program involves reprogramming of chromatin accessibility and long-range looping. TCF1 overexpression in fibroblasts opens the chromatin regions near the T lineage-associated genes, which are naturally demarcated by repressive histone marks in fibroblasts (135). Recent studies demonstrate a potent role of TCF1 in chromatin architecture remodeling in pro-T cells, later DP thymocytes, and mature T cells (136, 137). In Phase 2, TCF1 occupies key sites in evolutionary conserved topologically associating domains (TAD), i.e. regions of chromatin containing clusters of regulatory elements that interact within the TAD but are usually insulated from other TADs. When TCF1 binds to the inter-TAD sites along with CTCF (a transcription factor that can anchor chromatin architecture), this co-occupancy weakens insulation of the TAD boundaries and enables intermingling of TADs, potentially allowing new enhancer-promoter interactions. In addition, TCF1 establishes long-range looping around the T cell genes and marks the surrounding regions with H3K27ac (136).
Runx transcription factors are broadly expressed in all hematopoietic lineage cells, but they exert context-specific functions by switching their interaction sites across the genome. The expression of Runx1 and Runx3 is established prior to the thymic entry, and these two paralogs are co-expressed within individual Phase 1 and Phase 2 pro-T cells alike (51, 93, 94). The sum of Runx1 and Runx3 activities, measured by total binding by ChIP-seq, is maintained stably throughout the early T-lineage developmental process, suggesting that overall Runx availability between Phase 1 and Phase 2 does not dynamically change (51). However, the DNA binding profiles of Runx factors from hematopoietic stem cells (HSCs), ETP, DN3, DP, naïve CD4 T cells, and regulatory T cells (Tregs) demonstrate that a large fraction of the Runx binding sites displays highly cell type-specific occupancy (84). The sites occupied by Runx factors in ETPs are almost as different from those in DN3 cells as the sites occupied in completely different hematopoietic lineages, explaining the fact that Runx factors positively or negatively regulate substantially different sets of target genes in Phase 1 pro-T cells than in Phase 2 pro-T cells (51). Thus, the gene network role of Runx factors within the T-identity module changes with increased developmental progression, like the role of Notch signaling.
Due to functional redundancy between Runx1 and Runx3 in the pro-T cell stages, their roles in early T-developmental stages were previously under-detected when only one of the paralogs was disturbed. However, upon disruption of CBFβ, the common co-factor of all Runx factors, almost all stages of pro-T cells disappear, demonstrating the importance of Runx factors in early stages of thymic T-development, and NK cell development as well (138). Accordingly, acute deletion of Runx1 and Runx3 together in Phase 1 or Phase 2 pro-T cells results in a severe developmental block and dysregulation of developmentally important genes. The Runx target genes defined by stage-specific loss-of-function and/or gain-of-function reveal the striking positive influence of Runx1 and Runx3 on regulating the core T-cell identity genes, which include Tcf7, Gata3, Hes1, Bcl11b, Myb, Ikzf1, Tcf12, Lef1, and Ets1 (transcription factors); Cd3g, Cd3d, Cd3e, Cd247, and Thy1 (T cell surface markers); Lck, Lat, Fyb, Trat1, and Rag2 (TCR signaling and rearrangement molecules) (51)(Figures 2B, C).
Of interest, cell-type-preferential Runx binding sites show distinct, stage-specific enrichment profiles for motifs of other factors, including PU.1, E2A, ETS, or TCF/HMG factors, suggesting that Runx factors may be cooperating with distinct partners to be recruited to these different binding sites in a cell-type specific manner (84). Indeed, Runx factors are identified as “popular” functional collaborators of other factors in Phase 1 and Phase 2, as they work with PU.1 in Phase 1, and at least with GATA3 and Bcl11b in Phase 2 (48, 71, 133). As Notch/RBPJκ binding sites are frequently co-enriched with Runx motifs (67), and there is substantial overlap between Notch and Runx-regulated target genes (Figure 2), it is likely that Runx interacts functionally at some sites with Notch/RBPJκ as well. Runx interacting co-factors can indeed alter Runx binding site choices. PU.1 binding accompanies Runx binding at a major fraction of its Phase 1 sites. If added to Phase 2 cells, where Runx binds different sites, PU.1 recruits Runx1 to its binding loci even though these sites possess lower quality Runx motif sequences than the starting Phase 2 sites (133). This PU.1-mediated Runx redistribution actually depletes Runx occupancy from the higher quality Runx binding sites, but this can be rescued by increasing the Runx availability levels (84). Notably, introducing additional Runx1 to Phase 1 pro-T cells results in precocious Runx DNA binding to post-commitment specific sites, which hastens activation of some Phase 2 targets of Runx factors and accompanies substantial acceleration in developmental progression. These results show that multiple co-factors may compete for the limited amount of Runx factors, and that this competition has a strong impact on the Runx DNA binding site choices as well as T-development speed.
A zinc finger protein, Ikaros is encoded by Ikzf1 and highly expressed in lymphoid progenitor cells. Thymic progenitor cells maintain a stable level of Ikzf1 expression throughout all stages, and Ikaros is necessary for normal T-development as well as B and innate lymphoid cell development (139–142). An impaired form of T cell development survives a complete Ikzf1 null mutation (143), but this is likely due to redundancy with its relative, Ikzf2, which is also expressed in early T-cell precursors (144, 145). Disruption of zinc finger 4 of Ikzf1 affects DNA binding, resulting in different developmental defects, which range from DN2/DN3 cellularity loss, abnormal β-selection, and delayed progression, to T-cell lymphoma (85, 142). Notably, Ikaros target genes are highly context dependent, and distinct sets of genes were uniquely regulated in Phase 1 vs. Phase 2, and beyond. The positive targets affected by the zinc finger 4 deletion at least include Lef1, Spib, Ptcra, Cd8a, Cd8b1, Rag1 and Rag2, while its negative targets include Cd4, all relevant both to Phase 2 (Figures 2B, C lower panels) and to the orderly transition of the cells succeeding in β-selection to later stages. Other evidence has also indicated that Ikaros can act as a quantitative damper on Notch signaling in this transition (146, 147). Some Notch-induced genes antagonized by Ikaros acquire H3K27me3 histone marks in DN3 stage by Ikaros-mediated Polycomb repressive complex 2 recruitments to these regions (134). However, the importance of Ikzf1 for prethymic lymphoid precursor development (148) makes it important to revisit the targets and gene regulation mechanisms of Ikaros by stage-specific perturbation specifically in the earlier T-lineage context as well.
E proteins are class I basic helix-loop-helix (bHLH) family members and among them, E2A (encoded by Tcf3), E2-2 (encoded by Tcf4), and HEB (encoded by Tcf12) contribute to early thymic T cell development (78, 149–153). As E proteins form homodimers or heterodimers with other bHLH or HLH family member proteins, their activity is regulated by the expression of the other heterodimerization partners as well as of the E protein coding genes themselves. For instance, ID-family transcription factors harboring an HLH domain but not a basic region specifically antagonize the DNA-binding activity of E proteins (154). Also, under pre-leukemic conditions, Lmo1, Lmo2, Lyl1, and SCL are suggested to sequester E protein activity in thymic progenitor cells (155–157). In pro-T cells, E2A/HEB activity sharply increases between DN2a and DN3, as measured by target gene effects (Figures 2B, C, compare upper vs lower panels), even though E2A itself is relatively unchanging across Phase 1 and Phase 2, and HEB level only increases by about fourfold (78, 93). Since there is no detectable decrease in ID protein expression across this interval, it is possible that before this transition, stem/progenitor-associated partners like Lyl1 or SCL were previously sequestering these E proteins into complexes with alternative activities (see below).
The E2A/HEB heterodimer is essential for activating many genes involved in the T-identity module, especially as the cells move into Phase 2 (Notch1 Notch3, Tcf3, Tcf12, Cd3d, Cd3e, Fyb, Ptcra, Lck, Rag1, Rag2). E2A and HEB directly bind to enhancer regions near Notch1, Notch3, Rag1, and Rag2, and these regions require E proteins to increase chromatin accessibility during the Phase 1 to Phase 2 transition (78). A recent study shows that E2A activates expression of the Rag1 and Rag2 recombinase genes through distinct enhancer regions in pro-T cells, transforming the spatial organization of chromatin around the Rag1-Rag2 locus (158). E protein activity subsequently enforces proliferation arrest and TCR gene rearrangement quality control at the β-selection checkpoint (159).
The onset of Bcl11b expression in thymic pro-T cells precisely marks commitment to the T cell fate (16). This is a relatively late event which is tightly linked to the Phase 1-Phase 2 transition (Figures 2B, C, lower). Bcl11b-deficient animals reveal that many genes crucial for T cell identity are Bcl11b-dependent (Cd3g, Cd3d, Cd3e, Lat, Ptcra, Zap70, Notch3, Hes1, Dntt) (80). Consistent with these results, Bcl11b deficient pro-T cells fail to assemble TCRβ and cannot progress beyond Phase 2 due to a defect in establishing the T-cell program (71, 80, 160). Many genes likely to be direct activation targets of Bcl11b (possessing Bcl11b binding near their putative regulatory domain) are also sensitive to the deletion of Runx1, indicating that this partner factor may also participate in Bcl11b-mediated positive gene regulation (80).
Bcl11b not only requires multiple positive inputs for its expression (TCF1, GATA3, Notch, and Runx) but also needs to undergo a major chromatin change (161), involving a compartment flip of at least 1 megabase (162) and removal of Polycomb repression marks (163), in order to be activated. This makes its advent later than might be predicted from gene network considerations alone. Like germline Bcl11b knockouts (164–166), disruption of the Bcl11b locus at pre-commitment stage (using Vav1-Cre or retroviral Cre) or immediately post-commitment (using proximal Lck-Cre) results in impaired developmental progression from DN2 to DN3 stage (80, 167).
The impact of Bcl11b has a notable overlap with the effects of E proteins. However, their overlapping target genes require inputs from both Bcl11b and E proteins, as E proteins and Bcl11b do not directly regulate each other nor bind frequently to linked sites. Bcl11b does protect E protein activity indirectly by repressing Id2, but this effect may only account for a select minority of the overlapping gene regulation effects (80). Thus, the T-identity module is initiated from Phase 1 by activities of Notch, TCF1, GATA3, Ikaros, and Runx factors, and further specified and strengthened by E protein factors and Bcl11b in Phase 2.
Entry into the T-cell program also requires additional factors which still require better characterization to determine the genes they regulate in early pro-T cells specifically. These other critical regulators include the bifunctional transcription factor Myb (168–170), the zinc finger repressor Gfi1 (171, 172), and ETS (E twenty-six) family transcription factors, which bind a motif that is among the most highly enriched in the active chromatin regions in all early pro-T cell subsets (51). Although single knockouts of ETS factors have not given strong phenotypes in pro-T cells, a large number of ETS family members with near-identical DNA binding specificities are expressed in overlapping patterns in these cells (173, 174), indicating that their function is probably reinforced by redundancy. Future work is needed to place these factors in the T-identity gene network module, and to elucidate how the T-identity module is robustly maintained later throughout all subsets of conventional T cells.
The T-cell identity program is not established on a blank slate, but rather intrudes on a previously established progenitor-associated gene regulation program when the cells enter the strong Notch signaling environment of the thymus. Many transcription factors handed down from hematopoietic stem cells to pro-T cells contribute to the progenitor program in Phase 1, then they are repressed after T-lineage commitment. A failure of silencing the stem/progenitor module regulators in the later T-developmental stages is often linked to various types of leukemia as well as failure of developmental progression (175). This group of transcription factors includes PU.1, Lmo2, Lyl1, Hhex, Bcl11a, Erg, Mef2c, Meis1, Hoxa9, and possibly also Etv6, Mycn, and Hopx (Figure 4A), and these factors regulate both common and unique sets of genes (28, 48, 90–92, 175–183).
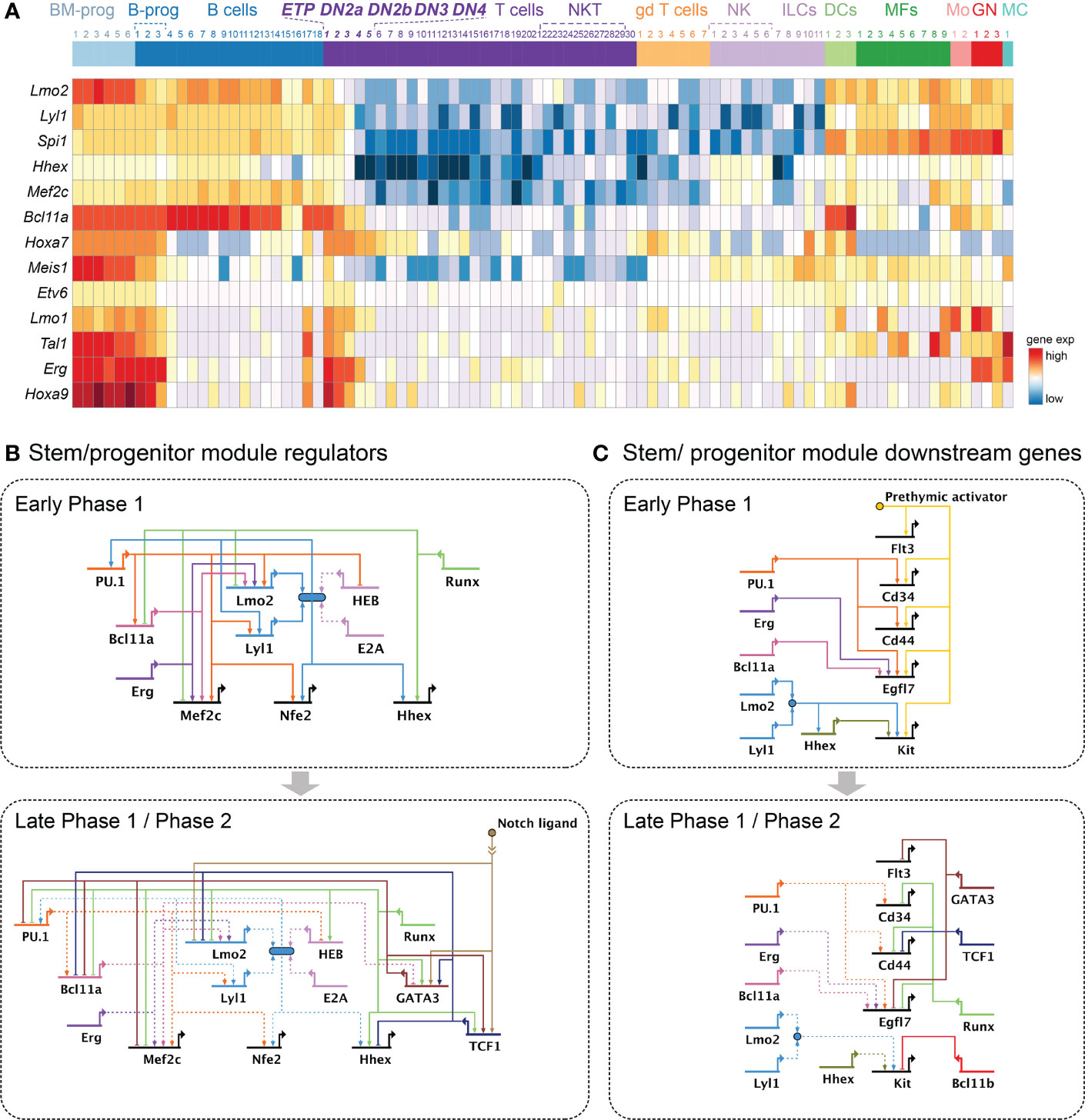
Figure 4 Gene regulatory networks controlling the stem or progenitor module. (A) Heatmap illustrates expression patterns of important transcription factors involved in the stem or progenitor module. Heatmap includes the same immune cell populations as Figure 2A. (B, C). Gene regulatory network models as in Figure 2. (B) Current model for key transcription factors shaping the stem or progenitor module in Phase 1 (top) and Phase 2 (bottom). (C) Current model for the stem or progenitor module downstream molecules in Phase 1 (top) and Phase 2 (bottom). Growth factor receptors: Flt3, Kit; Growth factor molecule: Egfl7; ETP marker genes: Cd34, Cd44.
The best-studied legacy regulator involved in the progenitor module is an ETS family transcription factor, PU.1, encoded by Spi1. PU.1 also acts in a pioneer factor-like way to set much of the chromatin accessibility landscape in Phase 1 cells (48). This mode of action is similar to its mechanism of function in myeloid cells (184), although deployed to somewhat different sites. The expression of PU.1 is highest in ETP, then gradually declines as pro-T cells transition to Phase 2. PU.1 provides strong and broad positive inputs to a large number of other progenitor-associated transcription factor coding genes (Lmo2, Lyl1, Bcl11a, and Mef2c)(Figure 4B) and genes encoding progenitor cell-related surface molecules (Cd34, Cd44, and Egfl7)(Figure 4C). The DNA binding profile of PU.1 in hematopoietic stem and progenitor cells (HSPC) vs. thymic precursor cells (ETP, DN2a, and DN2b cells) shows that at least 50% of the PU.1 DNA binding sites in HSPCs remain occupied by PU.1 in ETPs, which may provide continuity with PU.1’s function supporting the stem/progenitor program in pro-T cells (84).
Many of the genes functionally regulated by PU.1 in Phase 1 pro-T cells encode molecules used in myeloid cells as well, including receptors for myeloid-cell promoting cytokines (185). In hematopoiesis, the importance of PU.1 for myeloid lineage and dendritic cell fates is well-known, where it establishes chromatin accessibility and induces lineage specifying gene expression through collaborations with C/EBPα and IRF4/8 (15, 186–192). However, strong Notch signaling in the thymus restricts the full actions of PU.1, ensuring that pro-T cells do not actively follow the myeloid developmental path (89, 193, 194). In part, Notch signaling acts to silence expression of the partner PU.1 uses for several myeloid programs, C/EBPα, via the Notch-activated repressor Hes1 (105). Thus, in the context of strong Notch signaling, even moderately elevated levels of PU.1 can be tolerated within early T-cell precursors (89).
PU.1 maintains the stem and progenitor state both by direct positive regulation of targets in the same module, and by inhibition, direct or indirect, of the activity of factors promoting progression to T-cell commitment. The single-cell perturbation analysis study previously cited found that while PU.1 did not affect TCF1 or GATA3, it reduced the expression of later-acting progression-associated regulators, including HEB (Tcf12) (28) (Figure 4B), with strong impacts on their predicted target genes as identified by the SCENIC algorithm (39). Thus, without diverting T lineage development, PU.1 could act as a brake on the speed of differentiation along the T-cell pathway.
Other critical regulators of the stem or progenitor module are Lmo2 and the bHLH factor Lyl1. These form a complex with E protein (e.g. E2A or HEB), a GATA family factor, and an Ldb1 “bridge”, in which the Lyl1/E protein heterodimer interacts with DNA and the association of Lmo2 with Lyl1 stabilizes the complex (155, 156, 195, 196). In hematopoietic stem and progenitor cells, the complex often involves the related bHLH factor SCL instead of Lyl1 (197, 198). Lyl1 (or SCL) heterodimerization with an E protein like E2A or HEB confers an altered DNA binding specificity, such that Lyl1/E2A or SCL/E2A dimers bind different genomic sites than E protein/E protein dimers (156). Of note, the Lyl1/E2A complex in prethymic precursors appears to be important to support lymphoid programs and T cell potential, and to generate true lymphomyeloid primed progenitors (LMPPs) expressing genes shared with ETPs, such as Flt3, Dntt, Bcl11a, and even Notch1 itself (199). During early T cell development, Lyl1 is important to make ETP to DN2a progression possible and to turn on Gfi1 (200), which encodes a zinc finger repressor protein that is crucial for survival of early lymphoid progenitors in the presence of Notch signals (172). Recent studies also show a supportive role of Lmo2 in maintaining the T-lineage competence of immortalized progenitor-like cells (81, 195). Thus, although silenced in mature T cells, Lmo2 and Lyl1 contribute to the T-lineage developmental competence of hematopoietic progenitors.
However, overexpression of Lmo2 or Lyl1 in thymocytes does not drive the T cell program. Instead, it causes T-cell lymphoma/leukemia, and when Lmo2 is involved, it causes increased expression of genes associated with progenitor fates (86–88, 201–203)(Figures 4B, C, top). Under these conditions, the Lmo2/Lyl1 protein complex directly activates Hhex, which upregulates Kit expression (encoding the growth factor receptor cKit) and promotes self-renewal (91) at the expense of T-lineage differentiation. The Lyl1/Lmo2 complex can also upregulate Spi1 (encoding PU.1) itself (88). Such gain of function phenotypes suggest that Lyl1 and Lmo2 could be regulating the same genes in normal Phase 1 pro-T cells, although the definitive tests still need to be done. Of note, Lmo2 expression normally declines in ETPs much earlier than expression of Lyl1. However, PU.1 provides positive input to both, in a positive feedback circuit (Figure 4B, top): acute disruption of Spi1 in Phase 1 pro-T cells downregulates both Lmo2 and Lyl1 expression, while ectopic expression of Spi1 in early Phase 2 increases Lmo2 expression (48, 89). This suggests that PU.1 at high levels can re-activate Lmo2 for a limited time even after T-lineage commitment.
Acute CRISPR/Cas9-mediated target gene disruption in Phase 1 pro-T cells has demonstrated previously unknown roles of Bcl11a and Erg (28), both factors that have been difficult to study in T-lineage cells with conventional knockouts because of their essential roles in hematopoietic progenitor cell survival. Bcl11a is a zinc finger factor that has expression closely associated with lymphoid potential in progenitors, in B lineage cells, dendritic cells (especially pDC), and Phase 1 pro-T cells, then parallels PU.1 (Spi1) in its downregulation in T lineage cells. Thus, its expression pattern in pro-T cells is the opposite of that of its relative Bcl11b. Germline Bcl11a integrity is known to be essential for lymphoid development, but has mainly been implicated in survival for lymphoid cells (92, 180). Erg, an ETS factor similar to Fli1, is expressed specifically in stem and progenitor cells, granulocytes, mast cells, and pro-T cells through Phase 1, shutting off later than PU.1 and Bcl11a in Phase 2 [data for both from (93)]. At the single-cell transcriptome level, Bcl11a and Erg both contribute to the stem and progenitor module (28, 92, 94).
The recent data from single-cell transcriptome analysis shows that acute loss of Bcl11a in ETPs accelerates T lineage developmental progression along a largely physiological path. The loss of Bcl11a causes downregulation of progenitor-associated genes (Mef2c, Lmo2, Hoxa7, and Egfl7) together with upregulation of the T-lineage promoting regulatory gene Gata3 (28). While Bcl11a knockout effects parallel those of Spi1 (PU.1) knockouts, they are not identical, indicating distinct if overlapping sets of targets. In contrast to Bcl11a, the proto-oncogene Erg has not been implicated in any positive role specific to lymphocyte development before, as it is a sharply dose-sensitive regulator that is indispensable for survival of hematopoietic stem cells and for the embryo (181, 204). With acute Cas9-mediated deletion and in the presence of anti-apoptotic Bcl2, however, a striking effect of Erg knockout can be seen specifically in Phase 1 pro-T cells. First, expression of other stem/progenitor signature genes (Mef2c, Lmo1, Lmo2, Egfl7, and Meis1) is markedly decreased in the Erg-knockout cells. Second, the loss of Erg most prominently shifts the entire developmental trajectory of the Phase 1 pro-T cells to a novel path of development, one in which normally-unused genes Klf4, Id1, and others increase expression aberrantly (28). Thus, in contrast to Bcl11a and PU.1, Erg does not only sustain the stem/progenitor program but also blocks a particular cryptic, alternative pathway that was not known to be available before in T-cell developmental conditions.
The stem/progenitor module regulators in Phase 1 thymocytes very likely are preserved from the particular minority of multipotent progenitors that achieve success in colonizing the thymus, and so it is likely that some of these factors contribute positively to this very early event. As noted above, progenitor factors PU.1, Bcl11a, and Lyl1 are crucial for the generation of bone marrow cells competent to go to the thymus, and the Lyl1 partner Lmo2 is similarly essential to preserve T cell developmental potential in immortalized progenitor cells. However, it is not clear exactly which of their direct gene regulatory targets are the decisive ones for priming the ability to enter the thymus, thus separating T cell precursors from most ILC precursors. More inputs are also needed to explain the expression patterns of Flt3, Cd34, Cd44, Egfl7, and possibly Kit. Thus, a crucial early potentiating mechanism for T-cell development remains undefined.
Other factors expressed during the Phase 1 period could also contribute important regulatory inputs to targets in the stem/progenitor module, including Mef2c, Meis1, Hoxa9, Etv6, Hhex, and Mycn. These are all implicated in T-ALL (reviewed in (205)), similarly to Lyl1 and Lmo2 (86–88, 91), but are not yet adequately explored in the normal pro-T cell context. Hoxa9 is implicated in generating lymphoid-competent multipotent precursors (206) and has been reported to synergize with Runx1 to drive induced pluripotent cells towards a T cell pathway (207). Hhex is also required for generation of lymphoid-competent cells and has been suggested to play roles in sustaining a T-lineage competent state (208, 209), although it delays T-lineage developmental progression by stimulating extra self-renewal (91). Mef2c, also robustly expressed in early ETPs (94), may actively oppose the T cell program. Like PU.1 at high levels, Mef2c is reported to block T cell development by antagonizing Notch signaling (182), although it remains to be seen whether this is also true of Mef2c at natural levels in nonmalignant cells. One problem with determining the roles of these factors in normal pro-T cells has been technical: as the roles of these factors are confined to early stages, it has been difficult to disrupt or neutralize them within the T-lineage differentiation context faster than they are downregulated naturally. Thus, while the stem/progenitor module presented here already includes mutual activation and feed-forward network circuit motifs (PU.1 and Lmo2/Lyl1; PU.1 and Bcl11a on common targets) that could produce metastable persistence, it is likely that other module participants could further reinforce this system property.
The repressive forces antagonizing the stem or progenitor module also begin in Phase 1, and they fully suppress this program in Phase 2. This antagonism is mediated by a distinct set of T identity-associated regulators, primarily Runx transcription factors (Runx1 and Runx3), TCF1, and GATA3 (Figure 4, lower panels). Although PU.1 utilizes Runx transcription factors as its predominant binding partners, Runx factor activities most commonly oppose the actions of PU.1 on individual shared functional target genes (48, 51, 133). Runx transcription factors are also potent at counteracting the stem/progenitor module by repressing the majority of key transcription factor coding genes supporting this program (Mef2c, Lmo2, Lyl1, Bcl11a, Tal1, and Meis1) in a highly concentration-sensitive way (84). As the cells pass from Phase 1 to Phase 2, Runx1 begins to collaborate with GATA3 to repress Spi1 (PU.1) (51, 210) and turns off the downstream target genes of PU.1 as well, Cd34 and Cd44 (51).
TCF1 and GATA3 are not only indispensable for the T-identity program but also a source of discrete inhibitory inputs to the stem or progenitor module. Intrathymic Notch signaling activates TCF1 and GATA3 early in the ETP stage (76, 77, 82, 128, 132, 135, 211, 212), and they are co-expressed with genes associated with the progenitor module in individual ETPs and DN2a cells in mouse (94). Acute deletion of Tcf7 causes pro-T cells to stay behind at a progenitor-like state with sustained high expression of the genes associated with the stem/progenitor module (Cd34, Mef2c, Bcl11a, Lmo2, and Hhex), instead of progressing toward the T-lineage commitment stage. The contribution of GATA3 to this module is partially mediated by its repression of Bcl11a within the Phase 1 cells (28)(Figure 4B, lower). In addition, it plays a seemingly direct role in silencing Spi1 during T-lineage commitment. A recent study shows that in mice, GATA3 and Runx1 together interact with a specific regulatory element in intron 2 of Spi1 (PU.1) to cause and maintain PU.1 repression in Phase 2 (210). Although a different mechanism is apparently used in humans (213), forced expression of GATA3 in pro-T cells silences Spi1 prematurely (and blocks access to myeloid developmental fates) in mouse and human cells alike (83, 102, 104). Moreover, GATA3 inhibits an additional set of progenitor genes (Hopx, Hoxa9, and Flt3), all associated with self-renewal (28), which are not strongly repressed by Runx factors (Figures 4B, C, lower).
Together, the activity of the stem and progenitor module in early period of Phase 1 is maintained by the integrative efforts from PU.1, Lmo2 and Lyl1 complex, Bcl11a, and Erg, and potentially also Hhex, Etv6, Meis1, and Hoxa9. However, Notch signaling and Runx factors start to offset this activity from early ETP stage. As Notch and Runx factors activate TCF1 and GATA3, these factors provide additive force against the progenitor module. As a result, the major positive regulators in this module are downregulated with Lmo2 first, followed by Hhex, Mef2c, Bcl11a, and then Spi1, Lyl1, and last Erg. This process pushes the Phase 1 precursor cells finally out of the progenitor state as they develop.
Developmental potential for the B cell and myeloid cell fates depends on factors exogenous to the T cell program, like EBF1 and Pax5 for the B cell program, C/EBP factors for the neutrophil and macrophage programs, and IRF family factors for macrophage and dendritic cell programs. The B-cell option becomes unavailable relatively quickly for T-cell cohorts that have access to it at all (214), potentially associated with the early loss of Mef2c (182), and the myeloid-related options are less prominent by the time pro-T cells reach the DN2a stage. Instead, the NK and ILC lineage potentials become more preferred alternative lineage choices in DN2a cells, especially in the absence of Notch signaling (16, 94). The gene network components supporting the NK and ILC fates will be referred to as “the effector and innate lymphoid module” here. Notably, the effector/innate lymphoid module is often fueled by the same transcription factors necessary for the T-identity module, such as Runx factors, TCF1, and GATA3, whose expression is maintained in all T-lineage cells. The distinctive transcription factors separating the effector and innate lymphoid module from the T lineage include ID family proteins (mostly Id2), PLZF (encoded by Zbtb16), NFIL3, RORα, and Fos/Jun family members (Fos, Fosb, Jun) (215–225). Although some of these can be activated by acute stimuli, these genes are generally not highly expressed in ETPs, with a few exceptions (Figure 5A).
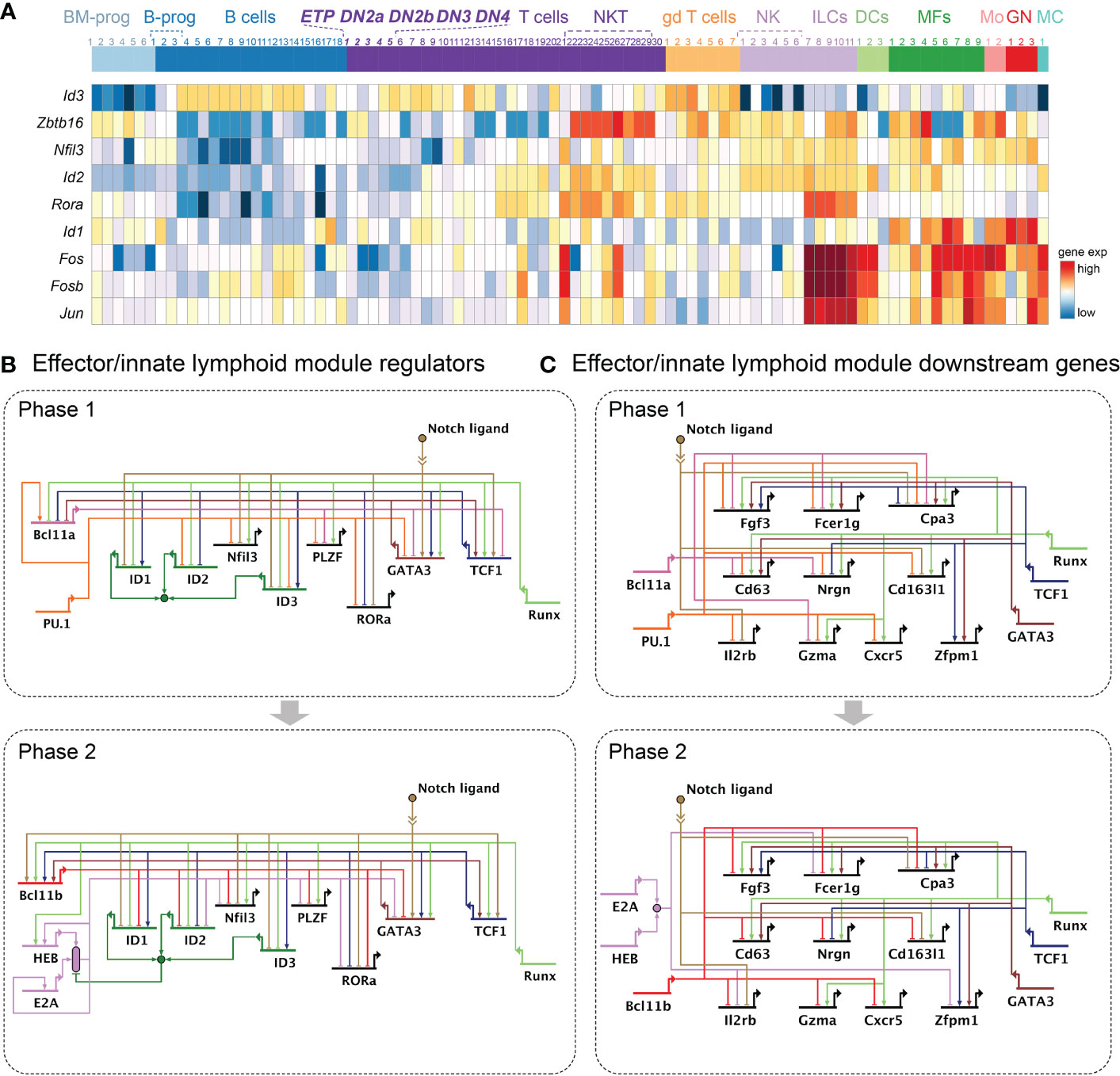
Figure 5 Gene regulatory networks regulating the effector or innate lymphoid module. (A) Heatmap represents expression patterns of transcription factors supporting the effector or innate lymphoid module. Heatmap shows the same immune cell populations displayed in Figure 2A. (B, C). Gene regulatory network models as in Figure 2. (B) Current model for transcription factors regulating the effector or innate lymphoid module in Phase 1 (top) and Phase 2 (bottom). (C) Current model for the effector or innate lymphoid module downstream molecules in Phase 1 (top) and Phase 2 (bottom). Growth factor, immunoglobulin, scavenger, or chemokine receptors (Fgf3, Fcer1g, Cd163l1, Il2rb, Cxcr5), enzymes (Cpa, Gzma), and signal transduction associated molecules (Cd63, Nrgn) involved in immune cell effector function and innate lymphoid cells are shown. Not all genes discussed in the text are shown.
Runx1 and Runx3 generally antagonize the stem/progenitor module in ETPs and generally favor T-lineage progression, but they also globally activate another distinctive program, including a suite of regulatory genes associated with effector and innate lymphoid cells plus downstream genes associated with innate lymphoid cells, γδ T cells, and other effectors. In fact, Runx factors, especially Runx3, are indispensable for the normal development and functions of ILCs (218, 226–228). A modest increase of Runx1 levels in Phase 1 pro-T cells quickly upregulates the innate lymphoid cell-associated regulators Zbtb16 and Nfil3. In addition, effector genes associated with innate-lymphoid cells (Fcer1g, Nrgn), γδ T cells (Cd163l1), and mast cells (Cpa3, Cd63), and the rarely-expressed Fgf3, are upregulated by Runx1 overexpression and inhibited by Runx1/Runx3 double knockout (84). Although this gene set does not mimic markers of a known mature cell type, its highly coherent regulation, described in this section, suggests a specific alternative developmental program. The association with NK or ILC programming, based on Zbtb16 and Nfil3, is supported by the developmental behavior of Runx-overexpressing cells (84). Consistently, if Notch signaling is withdrawn, Runx1 overexpression in ETPs causes preferential diversion to the NK-associated lineage at the expense of the myeloid lineage choice. However, Runx factors do inhibit all ID proteins (Id1, Id2, Id3) as long as Notch signaling is present, keeping the full innate program conditionally on hold. Hence, the activity of Runx proteins in Phase 1 guides pro-T cells out of the progenitor state and blocks the myeloid program, while driving cells towards lymphoid fates by upregulating both T- and ILC-associated genes (84).
Although GATA3 and TCF1 are essential for generating functional T cells, recent studies have shown their roles in ILC development as well (132, 229–233). GATA3 is necessary for development of all helper-like ILC subsets and their functions (232–234), and ILCs may express much higher levels of Gata3 than pro-T cells (Figure 2A). The early ILC progenitor (EILP) population is also dependent on TCF1 upregulation, in which TCF1 provides positive inputs to key ILC genes while blocking DC development from EILP (229, 235). During early thymic T-development, GATA3 and TCF1 positively function in the effector and innate lymphoid module by activating the gene encoding the GATA cofactor Zfpm1 (by TCF1 and GATA3)(Figure 5B, top), and also activating non-T effector molecules Cpa3, Cd63, and Fcer1g (by GATA3), and Fgf3 (by TCF1 and GATA3)(Figure 5C, top; Table S1). Of interest, TCF1-regulated genes in EILPs often overlap with TCF1 targets in pro-T cells, suggesting that it generally promotes a T or innate lymphoid program. Many of the non-T targets are inhibited by Bcl11b in pro-T cells in Phase 2 (Table S1, also see below). The physiological relevance of this pathway is shown by the fact that normal ETP-DN2a pro-T cells within the mouse thymus transiently express all of these genes (in a “wave”) before commitment to the T-cell pathway (94). This suggests that TCF1 may provide some extent of common inputs to the effector and ILC program in both EILPs and Phase 1 pro-T cells.
If these genes truly provide access to ILC potential, then they should be silenced to ensure lineage specification to the T cell fate. First, Notch signaling widely represses the genes associated with the innate lymphoid/effector module (Id1, Id3, Nfil3, Rora, Il2rb, and Cpa3, Cd163l) (Figures 5B,C, lower panels). In addition, this module gets repressive inputs from two distinct, seemingly opposite groups of transcription factors. The first group, consisting of PU.1 and Bcl11a, inhibits the effector and common lymphoid program in Phase 1, and then hands over this job to the second group of transcription factors in Phase 2, which are E protein complexes and Bcl11b.
PU.1 shows strikingly powerful opposition to the effector and innate lymphoid module by inhibiting ID proteins, Jun family members, as well as other ILC-associated transcription factors (Zbtb16, Id2, Id3, Nfil3, Jun, Jund, Rora, and Pou2af1). In addition, multiple downstream molecules in this module (Fcer1g, Fgf3, Gzma, Gzmb, Il2rb, Cd63, Nrgn, Cxcr5, Cd163l1, and Cpa3) are negatively regulated by PU.1 (28, 48). Similar to PU.1, but to a weaker extent, Bcl11a also functions to inhibit expression of the lymphoid/effector module (Zbtb16, Gzma, Gzmb, Cd63, Zfp105, Fgf3, Cpa3, Fcer1g, and Nrgn). Notably, Bcl11a and GATA3 inhibit each other (28). This suggests that PU.1 and Bcl11a are not generally permissive to all of the non-T lineage potentials in Phase 1, but in fact provide a critical counterbalance to keep pro-T cells from deviating to the ILC-lineage when excessive activity of Runx factors, GATA3, and TCF1 pushes the cell state towards the effector like-lymphoid lineage.
As pro-T cells progress to DN2 stage, PU.1 and Bcl11a expression declines, and the role of repressing the effector and innate lymphoid module devolves upon E protein complexes and Bcl11b. The E2A/HEB complex provides strong opposing force to the effector and innate lymphoid module by inhibiting expression of key ILC and innate-like lymphoid transcription factors (Zbtb16, Id2, Nfil3, Ikzf2, Sox5, Junb, and Rora) as well as by damping down expression of Gata3 (78, 79). In addition, E protein activity is important for Notch1 expression, which is also necessary for preventing ILC potential from being expressed (78, 236). These E protein effects would all be countered by ID2, explaining in part why ID2, despite its inability to bind DNA, is so important for NK and ILC development. Tcf3/Tcf12 (E2A and HEB) double-deficient ETPs display an abnormal chromatin accessibility signature, which mimics the ATAC-profile of ILC2-precursor cells in the bone marrow (78). In accordance with this, disruption of E protein function by deleting Tcf3 and/or Tcf12 or by overexpressing their antagonistic factor ID proteins results in abnormal ILC- and NK-like cell development in the thymus (78, 89, 236–238). These results reaffirm that the E2A/HEB complex is necessary to block developmental access to the innate lymphoid-like functionality.
However, later in T cell development, the balance of E proteins to ID proteins is repeatedly tipped whenever the TCR complex is stimulated (239–241). Thus, E protein activity levels alone cannot guarantee that the cells will stay within the T cell lineage long-term. Another indispensable transcription factor repressing the innate lymphoid/effector module is thus Bcl11b. The onset of Bcl11b expression in early thymic T-development closely coincides with T-lineage commitment, and Bcl11b plays pivotal roles afterward for establishing T-cell identity (16, 28, 80, 165, 166). The most abundant Bcl11b interaction co-factors include multiple repressor complexes, and, during T lineage commitment, Bcl11b broadly represses the NK and innate lymphoid cell programs (80, 166, 167). This is achieved by blocking expression of multiple regulators (Zbtb16, Id1, Id2, Nfil3, Rora, Zfp105, and Pou2af1) as well as downstream genes (Gzma, Cpa3, Il12rb, Fgf3, Cd163l1, Nrgn, Fcer1g, Cxcr5, Cd63, and Il2rb) in the effector and innate lymphoid modules (80).
A recent single-cell transcriptome analysis highlights that Bcl11b-deficient pro-T cells begin to swerve out of the conventional αβ T cell pathway with Id2 derepression and abnormal activation of AP-1 factor-associated genes (28). This leads to a cascade of regulatory changes, shown in part in Table S1 by separate comparisons between WT and Bcl11b-knockout cells in specific single-cell clusters: the slight initial differences detected as the paths first separate, within cluster 5; and the widening divergences mid-cascade in the comparisons between clusters 2 and 0. At the end of this cascade in the Bcl11b-knockout cells, further upregulation of Id2 culminates with a shutoff of Notch signaling and upregulation of Rora, as the cells leave the T cell pathway (28). Consistent with this dysregulated transcriptional program, Bcl11b deletion results in abnormal lineage-conversion to an NK-like population (called induced T-to-natural killer, ITNK cells) in the thymus, which express genes associated with conventional NK cells and show cytolytic activity to tumor cells and even to OP9 stromal cells (166, 167). Combinatorial deletion of Bcl11b and Id2 or Bcl11b and Zbtb16 shows that Bcl11b-mediated negative regulation on these transcription factors is necessary to block alternative lineage choices to NK- and ILC-like fates (80).
In summary, the effector and innate lymphoid module represents a battery of genes that are remarkably consistently regulated by the activities of factors that cut across their usual progenitor vs. T-identity roles. These genes are activated by three of the same inputs that drive the T-identity module, i.e., Runx factors and the Notch-induced factors GATA3 and TCF1, but they are restrained from expression normally by three distinct sets of antagonists: 1) the progenitor factors PU.1 and Bcl11a, 2) the climax T lineage commitment factors Bcl11b and E proteins, and 3) environmental Notch signaling itself. The Notch, GATA3, and TCF1 inputs form a classic “incoherent feed forward” motif (242) as the inducer and the factors it induces exert opposite effects on this module. The silencing of these genes by progenitor factors early switching over to Bcl11b and E proteins later clearly leaves a window of vulnerability, in which these genes may be activated transiently, and indeed this is when the genes in this module show a burst of activity in normal late-Phase 1 cells (precommitment DN2a cells). To the extent that these genes indeed indicate competence to switch to a different developmental branch than normal αβ T cells, this discontinuity in repression provides a window of opportunity.
The thymus generates about 50 million immature T cells per day, and then outputs 1-2 million mature T cells after selection per day in the young adult mouse, although only a few hematopoietic progenitor cells enter the thymus every day (243, 244). This yield is supported by remarkable cell expansion before various checkpoints and selections. Hence, the program supporting cell proliferation and survival is a critical component of Phase 1 and Phase 2 GRNs.
In Phase 1, growth factors Flt3-ligand and cKit-ligand are necessary to yield optimal number of output pro-T cells (110, 245–248). The expression of Flt3 receptor is limited to the least mature ETP population, whereas cKit is expressed in a range of cells from ETP to DN2a (cKithigh)/DN2b(cKitintermediate) stages until T-lineage committed cells eventually downregulate it in Phase 2 (94, 110, 249). Among two forms of cKit-ligand or stem cell factor (SCF), membrane bound and secreted forms, the membrane-bound form of SCF is necessary for sustaining the Phase 1 population size. The SCF in thymic endothelial cells is particularly important for the ETP population, whereas membrane-bound SCF in thymic epithelial cortex cells is more important for DN2 cells than ETPs (250). However, the exact molecular mediators linking Flt3 and cKit signaling to the proliferation/survival transcriptional programs remain unclear.
In late Phase 1 and Phase 2, IL-7 signaling plays an essential role in cell survival and proliferation. The exact time windows affected could be complex. Fate-mapping studies tracing the origin of thymic T cells suggest that all T cells are derived from precursors expressing IL-7 receptor (IL-7Rα) at some level; in steady state, mice deficient in IL-7 or IL-7Rα manifest severe defects in thymic cellularity due to cell cycle arrest and high apoptosis rates (251–255), with a relative emptying of the pro-T cell niches (256). Some thymus-seeding cells in humans as well as mice indeed come from IL-7Rα+ prethymic precursors (termed Common Lymphoid Precursors or IL-7R+ LMPPs) (97, 257–260), especially in fetal thymus (261, 262). However, the great majority of ETPs in postnatal mice do not appear to express this cytokine receptor at RNA level (93, 94). Instead, the expression of IL-7Rα in the thymic precursor cells sharply increases at the late stage of Phase 1 (ETP to Bcl11b- DN2a transition stage), peaks at around T-lineage commitment (Bcl11b+ DN2a and DN2b stage), and then gradually decreases as pro-T cells develop through DN3 and DN4 stages (263–265). During the DN2a/2b phases, Il7r expression overlaps with Kit expression. The dynamic kinetics of IL-7R expression coordinated with the developmental progression may provide the maximum efficiency for the cell expansion during the time that it is most needed.
In the case of IL-7R signaling, the main mediators are known. The Jak3-STAT5 pathway and Jak3-NFAT2 pathways are directly activated by IL-7 signaling, and both upregulate anti-apoptotic genes Bcl2 and Bcl2l1 (266, 267). Consequently, ectopic expression of Bcl2 or deletion of Bim (proapoptotic molecule) provides partial rescue for the severe cellularity loss in IL-7 signaling-deficient mice (266–268). STAT5 target genes specific to the pro-T cell compartment were defined in a recent study by using Cas9/CRISPR to knock out both Stat5a and Stat5b in pro-T cells expressing a Bcl2 transgene to minimize the survival defect in STAT5-knockout cells (269). In this system, both uncommitted and newly committed pro-T cells could be compared from control and double-knockout populations. Knocking out Stat5a and Stat5b simultaneously resulted in dysregulation of genes associated with cell cycle, cytokine signaling feedbacks, and the mTOR/metabolic pathway. Surprisingly, however, these knockouts showed minimal changes in key transcription factors specifying T-cell identity, either before or after commitment. This suggests that the cell survival/proliferation module in Phase 2 is not necessarily intimately linked to the cell-identity specifying modules, in contrast to results in peripheral T cells (270). Thus, cytokine responses via STAT5 do not appear to instruct pro-T cell state changes via lineage-specifying transcription factor changes.
Nevertheless, certain lineage specifying transcription factors also exert population effects in Phase 1 and/or Phase 2. The effects of Phase 1 and Phase 2 transcription factors on well-established growth control genes in these cells are summarized in Table 1, based on data in Table S1. Notch signaling, PU.1, Lmo2/Lyl1 complex, TCF1, and GATA3 are not only involved in the cell identity-specifying modules, but are also essential for regulating the size of the Phase 1 pool. Notch signaling induces proliferation of thymic progenitor cells both in humans and mice (271–273). Notch activation target genes include Myc, which is potent at driving cellular proliferation (67). Also, gene set enrichment analysis suggests that loss of Notch signaling enriches expression of genes associated with apoptotic programmed cell death pathways (67).
Table 1 Inputs to growth control genes from developmental network regulators.
In Phase 1, acute deletion of PU.1 can result in a ~5-10-fold reduction in Phase 1 cell number from the in vitro culture system, which cannot be fully restored by forced expression of anti-apoptotic molecule Bcl-xL (90). PU.1 activates genes associated with cell cycle progression (Ccnd1 and Cdk18) and supports expansion of both ETP and DN2 cells, suggesting that PU.1 is still needed for optimal cell proliferation even after some genes in T-identity modules are upregulated in DN2 stage (48, 90, 133). Another important early Phase 1 transcription factor involved in cellular proliferation is Lmo2. Lmo2 interacts with DNA replication complexes to regulate the DNA synthesis rate in hematopoietic progenitor cells, and it increases the pool of self-renewing population in Phase 1 in a gain-of-function setting (86, 274, 275). Also, Lmo2 upregulates Bcl2 expression in pro-T cells, which contributes to maintain the size of the Phase 1 cell pool (81), consistent with the effect of the Lmo2 partner Lyl1 on the ETP-DN2a pool size in vivo (200). Consistently, constitutive expression of Lmo2 in bone marrow progenitor cells results in higher frequencies of ETPs in cell cycle upon bone marrow chimera reconstitution, which leads to T-ALL due to the accumulated number of ETP-like cells in the thymus (275).
TCF1 is critically important for supporting survival and proliferation of T lineage-competent cells starting from Phase 1. In the absence of Tcf7, the overall thymic DN cellularity is severely impaired and thymic progenitor cells display reduced frequency of cells in cell cycle (276, 277). In addition, Tcf7-deficient ETPs downregulate genes associated with DNA damage response and show increased cell death; constitutive expression of survival factor Bcl-xL does not rescue the early developmental defect resulting from Tcf7-deficiency (76, 77). This effect is strong enough to raise the concern that even when supported with a Bcl2 transgene, many cells in the pro-T population that survive Tcf7 knockout could be irrelevant bystander cells if the true T-precursors were annihilated. Another key transcription factor positively contributing to the proliferation/survival module is GATA3 (82, 128, 278). Acute knockdown of GATA3 expression using shRNA causes severe loss of the ETP population, ascribed to a cell intrinsic defect in proliferative capacity. Of note, transgenic expression of anti-apoptotic factor Bcl2 can rescue ETP survival in these cases, but not sufficiently to restore normal T cell development (82).
In contrast, the recent study deleting Erg using CRISPR-Cas9 has highlighted a further unexpected role of Erg in regulating cell proliferation in pre-commitment stage (28). The loss of Erg in Phase 1 not only pushes the cells to exit faster from the ETP state, but also results in a large burst of progeny with upregulated Myc family activity (28), as assessed by the SCENIC algorithm (39). This suggests that Erg may function normally as a “brake” for the proliferation program in Phase 1.
At the later stage of Phase 2, pro-T cells need to slow down cellular proliferation to prepare for TCRβ assembly, which involves cleavage and rearrangement of DNA strands in the G1 cell-cycle phase. The E protein complex provides antagonizing forces against the cell survival and proliferation module near the end of Phase 2. E2A/HEB double knockout mice and E2A/LAT double knockout mice (transgenic mice unable to signal through TCRβ) display hypercellularity in the thymus, because of aberrant proliferation of DN3 cells which cannot maintain cell cycle arrest (159, 279). Hence, E protein-deficient mice are highly susceptible to T-cell lymphoma (149, 280).
In summary, cell cycle and survival controls greatly shape the output populations of the T-cell specification gene regulatory network, and the network regulators indeed influence cell cycle state directly. As shown in Table 1, the positive and negative regulatory inputs to these growth control genes extend across the dichotomy between progenitor-associated and T-specification factors, emphasizing the importance of modulating expression of this gene set correctly. However, the main environmental signals controlling proliferation via IL-7R and STAT5 apparently do not directly affect the network architecture itself.
In classic gene network analysis, it is assumed that any sites with binding motifs for specific factors can be bound any time that those factors are expressed. However, this is not necessarily true in post-embryonic mammalian developmental systems, where chromatin configurations can pose a constraint. Examples from reprogramming to pluripotency have clearly shown that a concerted, multi-factor, stepwise process is needed to alter chromatin states to make many transcription factor binding sites accessible (281–283). In the T cell specification gene regulatory network, at least one important gene reveals how strong the accessibility constraint can be, separate from the availability of trans-acting factors, to affect the response of a target gene to its regulators. The Bcl11b gene requires positive inputs from four known factors, TCF1, Runx1, GATA3, and Notch, but all four of these inputs are available already in ETP stage, whereas Bcl11b is not activated normally until late DN2a stage, which is days later in cells from young adult mice. When the two different alleles of Bcl11b were tagged with distinct fluorescent reporter proteins in the same cell nucleus, longitudinal live imaging and flow cytometry showed that the two alleles do not necessarily get activated at the same time. Instead, in individual cells from the same mouse, the two Bcl11b alleles were activated in a non-coordinated, stochastic manner in which activation of the second allele could be delayed by for 1-4 day(s) after the first (161). This suggests that even though all trans-acting transcription factors may be fully capable to turn on one allele, slow cis-acting mechanisms affecting the chromatin state, such as removal of repressive histone marks and accumulation of cis-acting non-long coding RNA (10, 163), also need to operate properly on each allele separately before the gene responds. Such mechanisms could greatly affect the dynamics of network state switching even without affecting the topology of the regulatory relationships.
The mechanisms involved affect chromatin at two size scales. The mammalian genome has two different types of long-range interaction regions at large scales, the “A” and the “B” compartments, which correspond roughly to active and silent chromatin respectively (284, 285). Within a compartment, chromatin folds into topologically associated domains (TADs), in which DNA interactions such as enhancer-promoter looping preferentially occur within the same TAD (286, 287). During T-lineage commitment, pro-T cells reorganize compartments as well as the TADs within them (162). The genomic regions manifesting compartment reprogramming represent only a small fraction, but when this “compartment flip” occurs, it is near developmentally important genes. For instance, the major enhancer regions about ~850kb downstream of Bcl11b show a dramatic 3D compartment flipping as the gene is activated: these regions lie in an inactive compartment (compartment B) in HSPC, ETP, and DN2a stages, but the compartment status sharply changes to active (compartment A) across the region as Bcl11b turns on (162). Consistently, this genomic region also physically migrates from the nuclear lamina at the progenitor stage to the nuclear interior as pro-T cells reach the DN3 stage (10). Similarly, particular genomic regions near Phase 1-specific genes Meis1, Cd34, Mef2c, and Bcl11a display active-to-inactive compartment flip (A-to-B), while enhancer regions of Ets1 and Gata3 gradually convert from the inactive to the active compartment (B-to-A) as these genes are accessed by new enhancers.
Although not directly tested, these concerted large scale chromatin 3D architecture changes may create barriers for some transcription factors. For instance, Runx transcription factors can choose different stage-specific DNA binding sites across the genome as noted before (51), and interestingly, local chromatin accessibilities do not serve as major barriers. However, at a larger scale, Runx factor binding is almost excluded from the inactive B compartment, and even when Runx levels are experimentally raised, Runx1 still does not occupy genomic regions within the inactive compartments (84). It remains to be elucidated by which mechanism pro-T cells reprogram the compartment-level genome organization, and how this shapes transcription factor activities and gene expression program.
At a smaller scale, for accurate gene regulation, pro-T cells undergo dynamic, concerted changes in intra-TAD interactions, chromatin accessibilities (determined by ATAC-seq or DNase-seq), and re-establishment of histone marks (92, 93, 162). These changes are particularly pronounced during T-lineage commitment, i.e. the DN2-to-DN3 transition, suggesting that epigenetic marking is coordinated to drive cell state conversion (52, 93, 162). Chromatin accessibility in Phase 1 is largely governed by PU.1, which can bind closed chromatin sites and open them, then keep its binding sites from closing (48). As PU.1 expression declines when pro-T cells progress from ETP to DN2b stages, many PU.1 binding sites lose chromatin accessibility; this supports a pivotal role of PU.1 in maintaining the Phase 1-preferential open chromatin regions (48). By contrast, many of the long-range loops near the T-identity genes appear to be dependent on other factors, which include TCF1, Bcl11b, and E2A (136, 158, 162). Thus, the epigenetic dynamics modulating gene network action are affected by both current trans-acting transcription factor activity and baseline cis-regulatory chromatin states.
To convert progenitor cells possessing multilineage developmental potential to T-lineage committed cells, each cell needs to silence the stem/progenitor module and block activation of the effector/innate lymphoid module, while launching the T-identity module. These modules of coherently co-regulated genes are valuable to recognize as distinct units of developmental programming, because in various situations an effect on a population survival module, for example, can be completely distinct from an effect on a differentiation module per se, even if both could alter the output of the process. Noting that correct development requires both activation of a differentiation module and repression of a progenitor state module, potentially through separate mechanisms, can be valuable to shed light on perturbed development as seen in leukemias and lymphomas. As discussed above, different combinations of transcription factor activities generate distinct forces on each gene regulatory module, which in turn determines the state of a given cell. While the network models shown here are most likely lacking some key components that have not yet been closely studied, their structure already helps to bring several principles into focus.
Transcription factor cooperation within a module can be stabilized by positive feedback loops. For instance, TCF1 with GATA3, and GATA3 with Myb provide cycles of activating connections to each other, which support the T-identity. Similarly, PU.1 and Lmo2 positively regulate each other, and this positive feedback loop needs to be disrupted by receiving inputs from other transcription factors to silence the stem/progenitor module, in order for T-lineage development to progress. The relative stability of relationships within a module can provide insight into why cells appear to undergo multiple cell cycles within each substage in Phase 1 before moving to the next (74, 94, 243).
Notably, mutually repressive connections are also seen in this system, and they often result in inter-module antagonism. For example, Bcl11a and GATA3 repress each other, which generates antagonism between the stem/progenitor module vs. the T-identity module. In addition, E2A/HEB complex and ID2 inhibit each other, which is a critical determinant of gene expression programs favoring the T-identity module vs. the effector/innate lymphoid module. However, direct mutual repression is far less prevalent in these T-cell specification network circuits than would be expected from classic GATA-PU.1 opposition models of cell fate decision (116). In more frequent cases, activity of a factor like PU.1 inhibits expression of a T-identity gene like Tcf12 (HEB), while GATA3 clearly represses Spi1 in a dose-dependent way, in one-way antagonistic relationships.
The opposition between different modules is also driven by indirect circuits regulating target genes. Although Notch signaling does not regulate expression of PU.1 itself, Notch signaling broadly inhibits PU.1-activated target genes; whereas PU.1 downregulates Notch-induced genes (67, 89). Interestingly, Runx-inhibited target genes also show striking overlap with the genes positively regulated by PU.1, which is surprising, considering that PU.1 physically interacts with Runx factors to co-bind to PU.1 target genes (51, 133). Thus, collaborative binding does not necessarily imply concordant functional impacts. At DN2b stage, morever, Runx1 and GATA3 cooperatively repress PU.1 itself. Therefore, Notch signaling and Runx factors together oppose PU.1 activities, which generates antagonisms between the T-identity vs. the stem/progenitor module.
Notch signaling and Runx factors show mosaic patterns of cooperation. Notch and Runx factors commonly activate the genes necessary for the T-identity module and co-occupy shared genomic regions in Phase 2. However, Notch signaling opposes Runx activities supporting the effector and innate lymphoid module. Similarly, Notch signaling works in an incoherent feed-forward circuit to repress the effector and innate lymphoid module genes that are being activated by the Notch-activated factors TCF1 and GATA3. Thus, rather than working in a linear pathway, transcription factors act combinatorially to distinguish different programs.
The effector and innate lymphoid module is actively suppressed by different cohorts of transcription factors throughout Phase 1 and Phase 2. In Phase 1, PU.1, Bcl11a, and Notch signaling collectively inhibits this module, but then E2A/HEB complex and Bcl11b take over this task as Notch signaling continues in Phase 2. It is important to note that E2A/HEB activity is regulated by competitive complex formation. In Phase 1, Lmo2 and Lyl1 must partner with E protein, which may sequester the E2A to bind alternative genomic sites. In addition, ID proteins (mainly ID2) expressed at any stage are also capable of trapping bHLH factors into non-DNA binding complexes. Thus, liberating E2A/HEB heterodimer activity by inhibiting their competitors is critical for suppressing the effector and innate lymphoid module and supporting the T-identity module.
Together, the gene regulatory network modules and transcription factors shaping each module activities reviewed here highlight how transcription factors collaborate to initiate, stabilize, synergize, oppose, or silence different gene expression programs. These intra-module and inter-module dynamics suggest how T-cell fate specification is instructed by gene regulatory network architecture.
BS compiled the data from published sources, generated the network models, and wrote the paper. ER advised on sources, edited the network models and wrote the paper. Both authors planned the project before writing and edited the manuscript before submission. All authors contributed to the article and approved the submitted version.
BS gratefully acknowledges support from an Irvington Postdoctoral Research Fellowship from the Cancer Research Institute and a Baxter Postdoctoral Fellowship from Caltech; ER gratefully acknowledges support from the Edward B. Lewis Professorship in Biology. The recent cited work on gene regulatory networks from the Rothenberg lab was supported by grants to ER from the USPHS: R01AI135200, R01HD100039, and R01AI151704.
The authors would like to thank Drs. Mary Yui, Tom Sidwell, Berthold Göttgens, and the late Eric Davidson for valuable insights and critical dialogue about gene networks, and Dr. Warren Leonard (NHLBI) and his group for generously encouraging us to examine how STAT5 roles related to the specification gene network.
ER has been on the Scientific Advisory Board for Century Therapeutics and has advised Kite Pharma and A2 Biotherapeutics. However, ER also declares that the work in this paper is conceptually and scientifically unrelated to those advisory roles.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1108368/full#supplementary-material
Supplementary Table 1 | Comprehensive datasets for genes affected by transcription factor perturbations, with Accession Numbers of sources, and with transcription factor binding data where available.
Supplementary Table 2 | Heatmap keys describing different immune cell populations shown in in Figures 2, 4, 5.
1. Rothenberg EV. Programming for T-lymphocyte fates: Modularity and mechanisms. Genes Dev (2019) 33(17-18):1117–35. doi: 10.1101/gad.327163.119
2. Rothenberg EV. Logic and lineage impacts on functional transcription factor deployment for T-cell fate commitment. Biophys J (2021) 120(19):4162–81. doi: 10.1016/j.bpj.2021.04.002
3. Maslova A, Ramirez RN, Ma K, Schmutz H, Wang C, Fox C, et al. Deep learning of immune cell differentiation. Proc Natl Acad Sci U S A. (2020) 117(41):25655–66. doi: 10.1073/pnas.2011795117
4. Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, et al. A genomic regulatory network for development. Science. (2002) 295(5560):1669–78. doi: 10.1126/science.1069883
5. Davidson EH. Emerging properties of animal gene regulatory networks. Nature. (2010) 468(7326):911–20. doi: 10.1038/nature09645
6. Chalancon G, Ravarani CN, Balaji S, Martinez-Arias A, Aravind L, Jothi R, et al. Interplay between gene expression noise and regulatory network architecture. Trends Genet (2012) 28(5):221–32. doi: 10.1016/j.tig.2012.01.006
7. Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. (2010) 467(7312):167–73. doi: 10.1038/nature09326
8. Furlong EEM, Levine M. Developmental enhancers and chromosome topology. Science. (2018) 361(6409):1341–5. doi: 10.1126/science.aau0320
9. Spitz F, Furlong EEM. Transcription factors: From enhancer binding to developmental control. Nat Rev Genet (2012) 13(9):613–26. doi: 10.1038/nrg3207
10. Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, et al. Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell. (2017) 171(1):103–19 e18. doi: 10.1016/j.cell.2017.09.001
11. Dueva R, Akopyan K, Pederiva C, Trevisan D, Dhanjal S, Lindqvist A, et al. Neutralization of the positive charges on histone tails by RNA promotes an open chromatin structure. Cell Chem Biol (2019) 26(10):1436–49 e5. doi: 10.1016/j.chembiol.2019.08.002
12. Schertzer MD, Braceros KCA, Starmer J, Cherney RE, Lee DM, Salazar G, et al. lncRNA-induced spread of Polycomb controlled by genome architecture, RNA abundance, and CpG island DNA. Mol Cell (2019) 75(3):523–37.e10. doi: 10.1016/j.molcel.2019.05.028
13. Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. (2013) 341(6147):1237973. doi: 10.1126/science.1237973
14. Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: Genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell (2010) 7(4):532–44. doi: 10.1016/j.stem.2010.07.016
15. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell (2010) 38(4):576–89. doi: 10.1016/j.molcel.2010.05.004
16. Kueh HY, Yui MA, Ng KK, Pease SS, Zhang JA, Damle SS, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol (2016) 17(8):956–65. doi: 10.1038/ni.3514
17. Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EE. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. (2009) 462(7269):65–70. doi: 10.1038/nature08531
18. Buchler NE, Gerland U, Hwa T. On schemes of combinatorial transcription logic. Proc Natl Acad Sci U S A. (2003) 100(9):5136–41. doi: 10.1073/pnas.0930314100
19. Soler E, Andrieu-Soler C, de Boer E, Bryne JC, Thongjuea S, Stadhouders R, et al. The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev (2010) 24(3):277–89. doi: 10.1101/gad.551810
20. Hannah R, Joshi A, Wilson NK, Kinston S, Gottgens B. A compendium of genome-wide hematopoietic transcription factor maps supports the identification of gene regulatory control mechanisms. Exp Hematol (2011) 39(5):531–41. doi: 10.1016/j.exphem.2011.02.009
21. Erwin DH, Davidson EH. The evolution of hierarchical gene regulatory networks. Nat Rev Genet (2009) 10(2):141–8. doi: 10.1038/nrg2499
22. Yu H, Gerstein M. Genomic analysis of the hierarchical structure of regulatory networks. Proc Natl Acad Sci U S A. (2006) 103(40):14724–31. doi: 10.1073/pnas.0508637103
23. Rothenberg EV, Göttgens B. How haematopoiesis research became a fertile ground for regulatory network biology as pioneered by Eric Davidson. Curr Opin Hematol (2021) 28(1):1–10. doi: 10.1097/MOH.0000000000000628
24. Rothenberg EV. Causal gene regulatory network modeling and genomics: Second-generation challenges. J Comput biol: J Comput Mol Cell Biol (2019) 26(7):703–18. doi: 10.1089/cmb.2019.0098
25. Calderon D, Blecher-Gonen R, Huang X, Secchia S, Kentro J, Daza RM, et al. The continuum of Drosophila embryonic development at single-cell resolution. Science (2022) 377(6606):eabn5800. doi: 10.1126/science.abn5800
26. Levine M, Davidson EH. Gene regulatory networks for development. Proc Natl Acad Sci U S A. (2005) 102(14):4936–42. doi: 10.1073/pnas.0408031102
27. Olariu V, Yui MA, Krupinski P, Zhou W, Deichmann J, Andersson E, et al. Multi-scale dynamical modeling of T cell development from an early thymic progenitor state to lineage commitment. Cell Rep (2021) 34(2):108622. doi: 10.1016/j.celrep.2020.108622
28. Zhou W, Gao F, Romero-Wolf M, Jo S, Rothenberg EV. Single-cell deletion analyses show control of pro-T cell developmental speed and pathways by Tcf7, Spi1, Gata3, Bcl11a, Erg, and Bcl11b. Sci Immunol (2022) 7(71):eabm1920. doi: 10.1126/sciimmunol.abm1920
29. Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. (2017) 6:e21856. doi: 10.7554/eLife.21856
30. Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun (2019) 10(1):1930. doi: 10.1038/s41467-019-09982-5
31. Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. (2007) 316(5830):1497–502. doi: 10.1126/science.1141319
32. Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods (2006) 3(7):511–8. doi: 10.1038/nmeth890
33. Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, et al. DNase-chip: A high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods (2006) 3(7):503–9. doi: 10.1038/nmeth888
34. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods (2013) 10(12):1213–8. doi: 10.1038/nmeth.2688
35. Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. (2014) 158(6):1431–43. doi: 10.1016/j.cell.2014.08.009
36. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res (2022) 50(D1):D165–D73. doi: 10.1093/nar/gkab1113
37. Sahu B, Hartonen T, Pihlajamaa P, Wei B, Dave K, Zhu F, et al. Sequence determinants of human gene regulatory elements. Nat Genet (2022) 54(3):283–94. doi: 10.1038/s41588-021-01009-4
38. Tanigawa Y, Dyer ES, Bejerano G. WhichTF is functionally important in your open chromatin data? PLoS Comput Biol (2022) 18(8):e1010378. doi: 10.1371/journal.pcbi.1010378
39. Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods (2017) 14(11):1083–6. doi: 10.1038/nmeth.4463
40. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. (2012) 489(7414):75–82. doi: 10.1038/nature11232
41. Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol (2014) 10(7):e1003731. doi: 10.1371/journal.pcbi.1003731
42. Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. (2018) 174(5):1309–24.e18. doi: 10.1016/j.cell.2018.06.052
43. Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. (2012) 150(6):1274–86. doi: 10.1016/j.cell.2012.04.040
44. Liu C, Omilusik K, Toma C, Kurd NS, Chang JT, Goldrath AW, et al. Systems-level identification of key transcription factors in immune cell specification. PLoS Comput Biol (2022) 18(9):e1010116. doi: 10.1371/journal.pcbi.1010116
45. Kamimoto K, Hoffmann CM, Morris SA. CellOracle: Dissecting cell identity via network inference and in silico gene perturbation. bioRxiv. 2020.02.17.947416; doi: 10.1101/2020.02.17.947416
46. Goode DK, Obier N, Vijayabaskar MS, Lie A Ling M, Lilly AJ, Hannah R, et al. Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Dev Cell (2016) 36(5):572–87. doi: 10.1016/j.devcel.2016.01.024
47. Schutte J, Wang H, Antoniou S, Jarratt A, Wilson NK, Riepsaame J, et al. An experimentally validated network of nine haematopoietic transcription factors reveals mechanisms of cell state stability. eLife. (2016) 5:e11469. doi: 10.7554/eLife.11469.094
48. Ungerbäck J, Hosokawa H, Wang X, Strid T, Williams BA, Sigvardsson M, et al. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res (2018) 28(10):1508–19. doi: 10.1101/gr.231423.117
49. Revilla IDR, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, et al. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B-lymphopoiesis. EMBO J (2012) 31(14):3130–46. doi: 10.1038/emboj.2012.155
50. McManus S, Ebert A, Salvagiotto G, Medvedovic J, Sun Q, Tamir I, et al. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J (2011) 30(12):2388–404. doi: 10.1038/emboj.2011.140
51. Shin B, Hosokawa H, Romero-Wolf M, Zhou W, Masuhara K, Tobin VR, et al. Runx1 and Runx3 drive progenitor to T-lineage transcriptome conversion in mouse T cell commitment via dynamic genomic site switching. Proc Natl Acad Sci U S A. (2021) 118(4):e2019655118. doi: 10.1073/pnas.2019655118
52. Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F, et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood. (2013) 122(6):902–11. doi: 10.1182/blood-2012-08-447839
53. Chandra A, Yoon S, Michieletto MF, Goldman N, Ferrari EK, Fasolino M, et al. A multi-enhancer hub at the Ets1 locus controls T cell differentiation and allergic inflammation through 3D genome topology. bioRxiv. 2022.10.28.514213; doi: 10.1101/2022.10.28.514213
54. Hosoya-Ohmura S, Lin YH, Herrmann M, Kuroha T, Rao A, Moriguchi T, et al. An NK and T cell enhancer lies 280 kilobase pairs 3’ to the Gata3 structural gene. Mol Cell Biol (2011) 31(9):1894–904. doi: 10.1128/MCB.05065-11
55. Mowel WK, McCright SJ, Kotzin JJ, Collet MA, Uyar A, Chen X, et al. Group 1 innate lymphoid cell lineage identity is determined by a cis-regulatory element marked by a long non-coding RNA. Immunity. (2017) 47(3):435–49 e8. doi: 10.1016/j.immuni.2017.08.012
56. Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. (2016) 354(6313):769–73. doi: 10.1126/science.aag2445
57. Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A. (2014) 111(46):E4946–53. doi: 10.1073/pnas.1407079111
58. Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A Notch1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med (2014) 20(10):1130–7. doi: 10.1038/nm.3665
59. Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. (2018) 553(7689):515–20. doi: 10.1038/nature25193
60. Osterwalder M, Barozzi I, Tissieres V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. (2018) 554(7691):239–43. doi: 10.1038/nature25461
61. Cannavo E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, et al. Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol (2016) 26(1):38–51. doi: 10.1016/j.cub.2015.11.034
62. Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, et al. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. (2013) 155(7):1507–20. doi: 10.1016/j.cell.2013.11.039
63. Wang F, Qi Z, Yao Y, Yu G, Feng T, Zhao T, et al. Exploring the stage-specific roles of tcf-1 in T cell development and malignancy at single-cell resolution. Cell Mol Immunol (2021) 18(3):644–59. doi: 10.1038/s41423-020-00527-1
64. Wei G, Abraham BJ, Yagi R, Jothi R, Cui K, Sharma S, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. (2011) 35(2):299–311. doi: 10.1016/j.immuni.2011.08.007
65. Niebuhr B, Kriebitzsch N, Fischer M, Behrens K, Gunther T, Alawi M, et al. Runx1 is essential at two stages of early murine B-cell development. Blood. (2013) 122(3):413–23. doi: 10.1182/blood-2013-01-480244
66. Michaud J, Simpson KM, Escher R, Buchet-Poyau K, Beissbarth T, Carmichael C, et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics (2008) 9:363. doi: 10.1186/1471-2164-9-363
67. Romero-Wolf M, Shin B, Zhou W, Koizumi M, Rothenberg EV, Hosokawa H. Notch2 complements Notch1 to mediate inductive signaling that initiates early T cell development. J Cell Biol (2020) 219(10):e202005093. doi: 10.1083/jcb.202005093
68. Hosokawa H, Romero-Wolf M, Yang Q, Motomura Y, Levanon D, Groner Y, et al. Cell type-specific actions of Bcl11b in early T-lineage and group 2 innate lymphoid cells. J Exp Med (2020) 217(1):e20190972. doi: 10.1084/jem.20190972
69. Kueh HY, Rothenberg EV. Regulatory gene network circuits underlying T cell development from multipotent progenitors. Wiley Interdiscip Rev Syst Biol Med (2012) 4(1):79–102. doi: 10.1002/wsbm.162
70. Georgescu C, Longabaugh WJ, Scripture-Adams DD, David-Fung ES, Yui MA, Zarnegar MA, et al. A gene regulatory network armature for T lymphocyte specification. Proc Natl Acad Sci U S A. (2008) 105(51):20100–5. doi: 10.1073/pnas.0806501105
71. Longabaugh WJR, Zeng W, Zhang JA, Hosokawa H, Jansen CS, Li L, et al. Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc Natl Acad Sci U S A. (2017) 114(23):5800–7. doi: 10.1073/pnas.1610617114
72. Cacace E, Collombet S, Thieffry D. Logical modeling of cell fate specification-application to T cell commitment. Curr Top Dev Biol (2020) 139:205–38. doi: 10.1016/bs.ctdb.2020.02.008
73. Ye Y, Kang X, Bailey J, Li C, Hong T. An enriched network motif family regulates multistep cell fate transitions with restricted reversibility. PLoS Comput Biol (2019) 15(3):e1006855. doi: 10.1371/journal.pcbi.1006855
74. Manesso E, Kueh HY, Freedman G, Rothenberg EV, Peterson C. Irreversibility of T-cell specification: Insights from computational modelling of a minimal network architecture. PLoS One (2016) 11(8):e0161260. doi: 10.1371/journal.pone.0161260
75. Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. (2014) 159(2):440–55. doi: 10.1016/j.cell.2014.09.014
76. Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, et al. T-Cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc Natl Acad Sci U S A. (2011) 108(50):20060–5. doi: 10.1073/pnas.1110230108
77. Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. (2011) 476(7358):63–8. doi: 10.1038/nature10279
78. Miyazaki M, Miyazaki K, Chen K, Jin Y, Turner J, Moore AJ, et al. The E--Id protein axis specifies adaptive lymphoid cell identity and suppresses thymic innate lymphoid cell development. Immunity. (2017) 46(5):818–34.e4. doi: 10.1016/j.immuni.2017.04.022
79. Xu W, Carr T, Ramirez K, McGregor S, Sigvardsson M, Kee BL. E2A transcription factors limit expression of Gata3 to facilitate T lymphocyte lineage commitment. Blood. (2013) 121(9):1534–42. doi: 10.1182/blood-2012-08-449447
80. Hosokawa H, Romero-Wolf M, Yui MA, Ungerback J, Quiloan MLG, Matsumoto M, et al. Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat Immunol (2018) 19(12):1427–40. doi: 10.1038/s41590-018-0238-4
81. Hirano KI, Hosokawa H, Koizumi M, Endo Y, Yahata T, Ando K, et al. LMO2 is essential to maintain the ability of progenitors to differentiate into T-cell lineage in mice. Elife. (2021) 10:e68227. doi: 10.7554/eLife.68227
82. Scripture-Adams DD, Damle SS, Li L, Elihu KJ, Qin S, Arias AM, et al. GATA-3 dose-dependent checkpoints in early T cell commitment. J Immunol (2014) 193(7):3470–91. doi: 10.4049/jimmunol.1301663
83. Taghon T, Yui MA, Rothenberg EV. Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat Immunol (2007) 8(8):845–55. doi: 10.1038/ni1486
84. Shin B, Zhou W, Wang J, Gao F, Rothenberg EV. Runx factors launch T-cell and innate lymphoid programs via direct and gene network-based mechanisms. bioRxiv. 2022.11.18.517146; doi: 10.1101/2022.11.18.517146
85. Arenzana TL, Schjerven H, Smale ST. Regulation of gene expression dynamics during developmental transitions by the Ikaros transcription factor. Genes Dev (2015) 29(17):1801–16. doi: 10.1101/gad.266999.115
86. Li L, Mitra A, Cui K, Zhao B, Choi S, Lee JY, et al. Ldb1 is required for Lmo2 oncogene-induced thymocyte self-renewal and T-cell acute lymphoblastic leukemia. Blood. (2020) 135(25):2252–65. doi: 10.1182/blood.2019000794
87. McCormack MP, Shields BJ, Jackson JT, Nasa C, Shi W, Slater NJ, et al. Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood. (2013) 122(12):2093–103. doi: 10.1182/blood-2012-09-458570
88. Cleveland SM, Smith S, Tripathi R, Mathias EM, Goodings C, Elliott N, et al. Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells (2013) 31(5):882–94. doi: 10.1002/stem.1345
89. Del Real MM, Rothenberg EV. Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and Gata3. Development (2013) 140(6):1207–19. doi: 10.1242/dev.088559
90. Champhekar A, Damle SS, Freedman G, Carotta S, Nutt SL, Rothenberg EV. Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev (2015) 29(8):832–48. doi: 10.1101/gad.259879.115
91. Shields BJ, Alserihi R, Nasa C, Bogue C, Alexander WS, McCormack MP. Hhex regulates Kit to promote radioresistance of self-renewing thymocytes in Lmo2-transgenic mice. Leukemia. (2015) 29(4):927–38. doi: 10.1038/leu.2014.292
92. Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med (2012) 209(13):2467–83. doi: 10.1084/jem.20121846
93. Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, et al. The cis-regulatory atlas of the mouse immune system. Cell. (2019) 176(4):897–912 e20. doi: 10.1016/j.cell.2018.12.036
94. Zhou W, Yui MA, Williams BA, Yun J, Wold BJ, Cai L, et al. Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst (2019) 9(4):321–37 e9. doi: 10.1016/j.cels.2019.09.008
95. Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, et al. The transcriptional landscape of αβ T cell differentiation. Nat Immunol (2013) 14(6):619–32. doi: 10.1038/ni.2590
96. Le J, Park JE, Ha VL, Luong A, Branciamore S, Rodin AS, et al. Single-cell RNA-seq mapping of human thymopoiesis reveals lineage specification trajectories and a commitment spectrum in T cell development. Immunity. (2020) 52(6):1105–18.e9. doi: 10.1016/j.immuni.2020.05.010
97. Lavaert M, Liang KL, Vandamme N, Park JE, Roels J, Kowalczyk MS, et al. Integrated scRNA-seq identifies human postnatal thymus seeding progenitors and regulatory dynamics of differentiating immature thymocytes. Immunity. (2020) 52(6):1088–104.e6. doi: 10.1016/j.immuni.2020.03.019
98. Cordes M, Cante-Barrett K, van den Akker EB, Moretti FA, Kielbasa SM, Vloemans SA, et al. Single-cell immune profiling reveals thymus-seeding populations, T cell commitment, and multilineage development in the human thymus. Sci Immunol (2022) 7(77):eade0182. doi: 10.1126/sciimmunol.ade0182
99. Zeng Y, Liu C, Gong Y, Bai Z, Hou S, He J, et al. Single-cell RNA sequencing resolves spatiotemporal development of pre-thymic lymphoid progenitors and thymus organogenesis in human embryos. Immunity. (2019) 51(5):930–48.e6. doi: 10.1016/j.immuni.2019.09.008
100. Ha VL, Luong A, Li F, Casero D, Malvar J, Kim YM, et al. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia. (2017) 31(11):2503–14. doi: 10.1038/leu.2017.70
101. Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. (2012) 149(2):467–82. doi: 10.1016/j.cell.2012.01.056
102. Roels J, Kuchmiy A, De Decker M, Strubbe S, Lavaert M, Liang KL, et al. Distinct and temporary-restricted epigenetic mechanisms regulate human αβ and γδ T cell development. Nat Immunol (2020) 21(10):1280–92. doi: 10.1038/s41590-020-0747-9
103. Rothenberg EV. Single-cell insights into the hematopoietic generation of T-lymphocyte precursors in mouse and human. Exp Hematol (2021) 95:1–12. doi: 10.1016/j.exphem.2020.12.005
104. Garcia-Perez L, Famili F, Cordes M, Brugman M, van Eggermond M, Wu H, et al. Functional definition of a transcription factor hierarchy regulating T cell lineage commitment. Sci Adv (2020) 6(31):eaaw7313. doi: 10.1126/sciadv.aaw7313
105. De Obaldia ME, Bell JJ, Wang X, Harly C, Yashiro-Ohtani Y, DeLong JH, et al. T Cell development requires constraint of the myeloid regulator C/EBP-α by the Notch target and transcriptional repressor Hes1. Nat Immunol (2013) 14(12):1277–84. doi: 10.1038/ni.2760
106. Ezine S, Weissman IL, Rouse RV. Bone marrow cells give rise to distinct cell clones within the thymus. Nature. (1984) 309(5969):629–31. doi: 10.1038/309629a0
107. Kyewski BA. Seeding of thymic microenvironments defined by distinct thymocyte-stromal cell interactions is developmentally controlled. J Exp Med (1987) 166(2):520–38. doi: 10.1084/jem.166.2.520
108. Felli MP, Maroder M, Mitsiadis TA, Campese AF, Bellavia D, Vacca A, et al. Expression pattern of Notch1, 2 and 3 and Jagged1 and 2 in lymphoid and stromal thymus components: Distinct ligand-receptor interactions in intrathymic T cell development. Int Immunol (1999) 11(7):1017–25. doi: 10.1093/intimm/11.7.1017
109. Prockop SE, Palencia S, Ryan CM, Gordon K, Gray D, Petrie HT. Stromal cells provide the matrix for migration of early lymphoid progenitors through the thymic cortex. J Immunol (2002) 169(8):4354–61. doi: 10.4049/jimmunol.169.8.4354
110. Sambandam A, Maillard I, Zediak VP, Xu L, Gerstein RM, Aster JC, et al. Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol (2005) 6(7):663–70. doi: 10.1038/ni1216
111. Taghon T, Yui MA, Pant R, Diamond RA, Rothenberg EV. Developmental and molecular characterization of emerging β- and γδ-selected pre-T cells in the adult mouse thymus. Immunity. (2006) 24(1):53–64. doi: 10.1016/j.immuni.2005.11.012
112. Teague TK, Tan C, Marino JH, Davis BK, Taylor AA, Huey RW, et al. CD28 expression redefines thymocyte development during the pre-T to DP transition. Int Immunol (2010) 22(5):387–97. doi: 10.1093/intimm/dxq020
113. Yui MA, Feng N, Rothenberg EV. Fine-scale staging of T cell lineage commitment in adult mouse thymus. J Immunol (2010) 185(1):284–93. doi: 10.4049/jimmunol.1000679
114. Rothenberg EV. Dynamic control of the T-cell specification gene regulatory network. Curr Opin Syst Biol (2019) 18:62–76. doi: 10.1016/j.coisb.2019.10.012
115. Peter IS, Davidson EH. Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS letters. (2009) 583(24):3948–58. doi: 10.1016/j.febslet.2009.11.060
116. Huang S, Guo YP, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev Biol (2007) 305(2):695–713. doi: 10.1016/j.ydbio.2007.02.036
117. Paquette SM, Leinonen K, Longabaugh WJ. BioTapestry now provides a web application and improved drawing and layout tools. F1000Res. (2016) 5:39. doi: 10.12688/f1000research.7620.1
118. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. (1999) 10(5):547–58. doi: 10.1016/S1074-7613(00)80054-0
119. Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med (2001) 194(7):1003–12. doi: 10.1084/jem.194.7.1003
120. Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. (1999) 11(3):299–308. doi: 10.1016/S1074-7613(00)80105-3
121. Fröbel J, Landspersky T, Percin G, Schreck C, Rahmig S, Ori A, et al. The hematopoietic bone marrow niche ecosystem. Front Cell Dev Biol (2021) 9:705410. doi: 10.3389/fcell.2021.705410
122. Hozumi K. Distinctive properties of the interactions between Notch and Notch ligands. Dev Growth Differ (2020) 62(1):49–58. doi: 10.1111/dgd.12641
123. Van de Walle I, De Smet G, Gartner M, De Smedt M, Waegemans E, Vandekerckhove B, et al. Jagged2 acts as a delta-like Notch ligand during early hematopoietic cell fate decisions. Blood. (2011) 117(17):4449–59. doi: 10.1182/blood-2010-06-290049
124. Chen ELY, Thompson PK, Zúñiga-Pflücker JC. RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat Immunol (2019) 20(11):1456–68. doi: 10.1038/s41590-019-0518-7
125. Han J, Zuniga-Pflucker JC. A 2020 view of thymus stromal cells in T cell development. J Immunol (2021) 206(2):249–56. doi: 10.4049/jimmunol.2000889
126. Kopan R, Ilagan MX. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell. (2009) 137(2):216–33. doi: 10.1016/j.cell.2009.03.045
127. Thompson PK, Zúñiga-Pflücker JC. On becoming a T cell, a convergence of factors kick it up a Notch along the way. Semin Immunol (2011) 23(5):350–9. doi: 10.1016/j.smim.2011.08.007
128. Hosoya T, Kuroha T, Moriguchi T, Cummings D, Maillard I, Lim KC, et al. GATA-3 is required for early T lineage progenitor development. J Exp Med (2009) 206(13):2987–3000. doi: 10.1084/jem.20090934
129. Pai SY, Truitt ML, Ting CN, Leiden JM, Glimcher LH, Ho IC. Critical roles for transcription factor GATA-3 in thymocyte development. Immunity. (2003) 19(6):863–75. doi: 10.1016/S1074-7613(03)00328-5
130. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, et al. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. (1995) 374(6517):70–4. doi: 10.1038/374070a0
131. Anderson MK, Hernandez-Hoyos G, Dionne CJ, Arias A, Chen D, Rothenberg EV. Definition of regulatory network elements for T-cell development by perturbation analysis with PU.1 and GATA-3. Devel Biol (2002) 246(12):103–21. doi: 10.1006/dbio.2002.0674
132. Harly C, Kenney D, Wang Y, Ding Y, Zhao Y, Awasthi P, et al. A shared regulatory element controls the initiation of Tcf7 expression during early T cell and innate lymphoid cell developments. Front Immunol (2020) 11:470. doi: 10.3389/fimmu.2020.00470
133. Hosokawa H, Ungerbäck J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, et al. Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity (2018) 48(6):1119–34.e7. doi: 10.1016/j.immuni.2018.04.024
134. Oravecz A, Apostolov A, Polak K, Jost B, Le Gras S, Chan S, et al. Ikaros mediates gene silencing in T cells through Polycomb Repressive Complex 2. Nat Commun (2015) 6:8823. doi: 10.1038/ncomms9823
135. Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity. (2018) 48(2):243–57.e10. doi: 10.1016/j.immuni.2018.01.012
136. Wang W, Chandra A, Goldman N, Yoon S, Ferrari EK, Nguyen SC, et al. TCF-1 promotes chromatin interactions across topologically associating domains in T cell progenitors. Nat Immunol (2022) 23(7):1052–62. doi: 10.1038/s41590-022-01232-z
137. Shan Q, Zhu S, Chen X, Liu J, Yuan S, Li X, et al. Tcf1-CTCF cooperativity shapes genomic architecture to promote CD8(+) T cell homeostasis. Nat Immunol (2022) 23(8):1222–35. doi: 10.1038/s41590-022-01263-6
138. Guo Y, Maillard I, Chakraborti S, Rothenberg EV, Speck NA. Core binding factors are necessary for natural killer cell development and cooperate with Notch signaling during T-cell specification. Blood. (2008) 112(3):480–92. doi: 10.1182/blood-2007-10-120261
139. Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science. (1992) 258(5083):808–12. doi: 10.1126/science.1439790
140. Wang JH, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity. (1996) 5(6):537–49. doi: 10.1016/S1074-7613(00)80269-1
141. Winandy S, Wu L, Wang JH, Georgopoulos K. Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by Ikaros. J Exp Med (1999) 190(8):1039–48. doi: 10.1084/jem.190.8.1039
142. Schjerven H, McLaughlin J, Arenzana TL, Frietze S, Cheng D, Wadsworth SE, et al. Selective regulation of lymphopoiesis and leukemogenesis by individual zinc fingers of Ikaros. Nat Immunol (2013) 14(10):1073–83. doi: 10.1038/ni.2707
143. Georgopoulos K, Winandy S, Avitahl N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu Rev Immunol (1997) 15:155–76. doi: 10.1146/annurev.immunol.15.1.155
144. Kelley CM, Ikeda T, Koipally J, Avitahl N, Wu L, Georgopoulos K, et al. Helios, A novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr Biol (1998) 8(9):508–15. doi: 10.1016/S0960-9822(98)70202-7
145. Dovat S, Montecino-Rodriguez E, Schuman V, Teitell MA, Dorshkind K, Smale ST. Transgenic expression of Helios in B lineage cells alters B cell properties and promotes lymphomagenesis. J Immunol (2005) 175(6):3508–15. doi: 10.4049/jimmunol.175.6.3508
146. Geimer Le Lay AS, Oravecz A, Mastio J, Jung C, Marchal P, Ebel C, et al. The tumor suppressor Ikaros shapes the repertoire of Notch target genes in T cells. Sci Signal (2014) 7(317):ra28. doi: 10.1126/scisignal.2004545
147. Chari S, Winandy S. Ikaros regulates Notch target gene expression in developing thymocytes. J Immunol (2008) 181(9):6265–74. doi: 10.4049/jimmunol.181.9.6265
148. Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. (2009) 30(4):493–507. doi: 10.1016/j.immuni.2009.01.014
149. Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, et al. E2A deficiency leads to abnormalities in αβ T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol (1997) 17(8):4782–91. doi: 10.1128/MCB.17.8.4782
150. Barndt RJ, Dai M, Zhuang Y. Functions of E2A-HEB heterodimers in T-cell development revealed by a dominant negative mutation of HEB. Mol Cell Biol (2000) 20(18):6677–85. doi: 10.1128/MCB.20.18.6677-6685.2000
151. Engel I, Johns C, Bain G, Rivera RR, Murre C. Early thymocyte development is regulated by modulation of E2A protein activity. J Exp Med (2001) 194(6):733–45. doi: 10.1084/jem.194.6.733
152. Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J Exp Med (2006) 203(5):1329–42. doi: 10.1084/jem.20060268
153. Bouderlique T, Pena-Perez L, Kharazi S, Hils M, Li X, Krstic A, et al. The concerted action of E2-2 and HEB is critical for early lymphoid specification. Front Immunol (2019) 10:455. doi: 10.3389/fimmu.2019.00455
154. Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell. (1990) 61(1):49–59. doi: 10.1016/0092-8674(90)90214-Y
155. Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nat Immunol (2000) 1(2):138–44. doi: 10.1038/77819
156. Miyamoto A, Cui X, Naumovski L, Cleary ML. Helix-loop-helix proteins LYL1 and E2a form heterodimeric complexes with distinctive DNA-binding properties in hematolymphoid cells. Mol Cell Biol (1996) 16(5):2394–401. doi: 10.1128/MCB.16.5.2394
157. Tremblay M, Herblot S, Lecuyer E, Hoang T. Regulation of pTα gene expression by a dosage of E2A, HEB, and SCL. J Biol Chem (2003) 278(15):12680–7. doi: 10.1074/jbc.M209870200
158. Miyazaki K, Watanabe H, Yoshikawa G, Chen K, Hidaka R, Aitani Y, et al. The transcription factor E2A activates multiple enhancers that drive Rag expression in developing T and B cells. Sci Immunol (2020) 5(51):eabb1455. doi: 10.1126/sciimmunol.abb1455
159. Engel I, Murre C. E2A proteins enforce a proliferation checkpoint in developing thymocytes. EMBO J (2004) 23(1):202–11. doi: 10.1038/sj.emboj.7600017
160. Inoue J, Kanefuji T, Okazuka K, Watanabe H, Mishima Y, Kominami R. Expression of TCRαβ partly rescues developmental arrest and apoptosis of αβ T cells in Bcl11b-/- mice. J Immunol (2006) 176(10):5871–9. doi: 10.4049/jimmunol.176.10.5871
161. Ng KKH, Yui MA, Mehta A, Siu S, Irwin B, Pease S, et al. A stochastic epigenetic switch controls the dynamics of T-cell lineage commitment. eLife. (2018) 7:e37851. doi: 10.7554/eLife.37851
162. Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, et al. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity. (2018) 48(2):227–42 e8. doi: 10.1016/j.immuni.2018.01.013
163. Pease NA, Nguyen PHB, Woodworth MA, Ng KKH, Irwin B, Vaughan JC, et al. Tunable, division-independent control of gene activation timing by a Polycomb switch. Cell Rep (2021) 34(12):108888. doi: 10.1016/j.celrep.2021.108888
164. Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, et al. Bcl11b is required for differentiation and survival of αβ T lymphocytes. Nat Immunol (2003) 4(6):533–9. doi: 10.1038/ni927
165. Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, et al. An essential developmental checkpoint for production of the T cell lineage. Science. (2010) 329(5987):93–6. doi: 10.1126/science.1188995
166. Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. (2010) 329(5987):85–9. doi: 10.1126/science.1188063
167. Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. (2010) 329(5987):89–93. doi: 10.1126/science.1188989
168. Lieu YK, Kumar A, Pajerowski AG, Rogers TJ, Reddy EP. Requirement of c-Myb in T cell development and in mature T cell function. Proc Natl Acad Sci U S A. (2004) 101(41):14853–8. doi: 10.1073/pnas.0405338101
169. Bender TP, Kremer CS, Kraus M, Buch T, Rajewsky K. Critical functions for c-Myb at three checkpoints during thymocyte development. Nat Immunol (2004) 5(7):721–9. doi: 10.1038/ni1085
170. Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, et al. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res (2011) 39(11):4664–79. doi: 10.1093/nar/gkr024
171. Yücel R, Karsunky H, Klein-Hitpass L, Möröy T. The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med (2003) 197(7):831–44. doi: 10.1084/jem.20021417
172. Phelan JD, Saba I, Zeng H, Kosan C, Messer MS, Olsson HA, et al. Growth factor independent-1 maintains Notch1-dependent transcriptional programming of lymphoid precursors. PLoS Genet (2013) 9(9):e1003713. doi: 10.1371/journal.pgen.1003713
173. David-Fung ES, Butler R, Buzi G, Yui MA, Diamond RA, Anderson MK, et al. Transcription factor expression dynamics of early T-lymphocyte specification and commitment. Dev Biol (2009) 325(2):444–67. doi: 10.1016/j.ydbio.2008.10.021
174. Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development. (1999) 126(1):3131–48. doi: 10.1242/dev.126.14.3131
175. Thoms JA, Birger Y, Foster S, Knezevic K, Kirschenbaum Y, Chandrakanthan V, et al. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood. (2011) 117(26):7079–89. doi: 10.1182/blood-2010-12-317990
176. Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev (2004) 18(19):2336–41. doi: 10.1101/gad.1239604
177. Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med (2011) 208(13):2571–9. doi: 10.1084/jem.20112239
178. Cai M, Langer EM, Gill JG, Satpathy AT, Albring JC, Kc W, et al. Dual actions of Meis1 inhibit erythroid progenitor development and sustain general hematopoietic cell proliferation. Blood. (2012) 120(2):335–46. doi: 10.1182/blood-2012-01-403139
179. Anderson KL, Perkin H, Surh CD, Venturini S, Maki RA, Torbett BE. Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor-derived dendritic cells. J Immunol (2000) 164(4):1855–61. doi: 10.4049/jimmunol.164.4.1855
180. Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol (2003) 4(6):525–32. doi: 10.1038/ni925
181. Xie Y, Koch ML, Zhang X, Hamblen MJ, Godinho FJ, Fujiwara Y, et al. Reduced Erg dosage impairs survival of hematopoietic stem and progenitor cells. Stem Cells (2017) 35(7):1773–85. doi: 10.1002/stem.2627
182. Canté-Barrett K, Meijer MT, Cordo V, Hagelaar R, Yang W, Yu J, et al. MEF2C opposes Notch in lymphoid lineage decision and drives leukemia in the thymus. JCI Insight (2022) 7(13):e150363. doi: 10.1172/jci.insight.150363
183. Izon DJ, Rozenfeld S, Fong ST, Komuves L, Largman C, Lawrence HJ. Loss of function of the homeobox gene Hoxa-9 perturbs early T-cell development and induces apoptosis in primitive thymocytes. Blood. (1998) 92(2):383–93. doi: 10.1182/blood.V92.2.383
184. Minderjahn J, Schmidt A, Fuchs A, Schill R, Raithel J, Babina M, et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat Commun (2020) 11(1):402. doi: 10.1038/s41467-019-13960-2
185. Rothenberg EV, Hosokawa H, Ungerback J. Mechanisms of action of hematopoietic transcription factor PU.1 in initiation of T-cell development. Front Immunol (2019) 10:228. doi: 10.3389/fimmu.2019.00228
186. McIvor Z, Hein S, Fiegler H, Schroeder T, Stocking C, Just U, et al. Transient expression of PU.1 commits multipotent progenitors to a myeloid fate whereas continued expression favors macrophage over granulocyte differentiation. Exp Hematol (2003) 31(1):39–47. doi: 10.1016/s0301-472x(02)01017-2
187. DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science (2000) 288(5470):1439–41. doi: 10.1126/science.288.5470.1439
188. Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science (2013) 341(6146):670–3. doi: 10.1126/science.1240831
189. Yeamans C, Wang D, Paz-Priel I, Torbett BE, Tenen DG, Friedman AD. C/EBPα binds and activates the PU.1 distal enhancer to induce monocyte lineage commitment. Blood (2007) 110(9):3136–42. doi: 10.1182/blood-2007-03-080291
190. Pundhir S, Bratt Lauridsen FK, Schuster MB, Jakobsen JS, Ge Y, Schoof EM, et al. Enhancer and transcription factor dynamics during myeloid differentiation reveal an early differentiation block in Cebpa null progenitors. Cell Rep (2018) 23(9):2744–57. doi: 10.1016/j.celrep.2018.05.012
191. Kim S, Bagadia P, Anderson DA 3rd, Liu TT, Huang X, Theisen DJ, et al. High amount of transcription factor IRF8 engages AP1-IRF composite elements in enhancers to direct type 1 conventional dendritic cell identity. Immunity. (2020) 53(4):759–74.e9. doi: 10.1016/j.immuni.2020.07.018
192. Carotta S, Dakic A, D’Amico A, Pang SH, Greig KT, Nutt SL, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity (2010) 32(5):628–41. doi: 10.1016/j.immuni.2010.05.005
193. Franco CB, Scripture-Adams DD, Proekt I, Taghon T, Weiss AH, Yui MA, et al. Notch/Delta signaling constrains reengineering of pro-T cells by PU. 1. Proc Natl Acad Sci U.S.A. (2006) 103(32):11993–8. doi: 10.1073/pnas.0601188103
194. Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBPα and PU.1 transcription factors. Immunity (2006) 25(5):731–44. doi: 10.1016/j.immuni.2006.09.011
195. Koizumi M, Kama Y, Hirano KI, Endo Y, Tanaka T, Hozumi K, et al. Transcription factor Zbtb1 interacts with bridging factor Lmo2 and maintains the T-lineage differentiation capacity of lymphoid progenitor cells. J Biol Chem (2022) 298(11):102506. doi: 10.1016/j.jbc.2022.102506
196. Goodings C, Tripathi R, Cleveland SM, Elliott N, Guo Y, Shyr Y, et al. Enforced expression of E47 has differential effects on Lmo2-induced T-cell leukemias. Leuk Res (2015) 39(1):100–9. doi: 10.1016/j.leukres.2014.11.016
197. Hoang T, Lambert JA, Martin R. SCL/TAL1 in hematopoiesis and cellular reprogramming. Curr Top Dev Biol (2016) 118:163–204. doi: 10.1016/bs.ctdb.2016.01.004
198. El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall-Ponsele E, Platonova O, et al. Structural basis for LMO2-driven recruitment of the SCL:E47bHLH heterodimer to hematopoietic-specific transcriptional targets. Cell Rep (2013) 4(1):135–47. doi: 10.1016/j.celrep.2013.06.008
199. de Pooter RF, Dias S, Chowdhury M, Bartom ET, Okoreeh MK, Sigvardsson M, et al. Cutting edge: Lymphomyeloid-primed progenitor cell fates are controlled by the transcription factor Tal1. J Immunol (2019) 202(10):2837–42. doi: 10.4049/jimmunol.1801220
200. Zohren F, Souroullas GP, Luo M, Gerdemann U, Imperato MR, Wilson NK, et al. The transcription factor Lyl-1 regulates lymphoid specification and the maintenance of early T lineage progenitors. Nat Immunol (2012) 13(8):761–9. doi: 10.1038/ni.2365
201. Zhong Y, Jiang L, Hiai H, Toyokuni S, Yamada Y. Overexpression of a transcription factor LYL1 induces T- and B-cell lymphoma in mice. Oncogene. (2007) 26(48):6937–47. doi: 10.1038/sj.onc.1210494
202. Smith S, Tripathi R, Goodings C, Cleveland S, Mathias E, Hardaway JA, et al. LIM domain only-2 (LMO2) induces T-cell leukemia by two distinct pathways. PLoS One (2014) 9(1):e85883. doi: 10.1371/journal.pone.0085883
203. McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science. (2010) 327(5967):879–83. doi: 10.1126/science.1182378
204. Loughran SJ, Kruse EA, Hacking DF, de Graaf CA, Hyland CD, Willson TA, et al. The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol (2008) 9(7):810–9. doi: 10.1038/ni.1617
205. Yui MA, Rothenberg EV. Developmental gene networks: A triathlon on the course to T cell identity. Nat Rev Immunol (2014) 14(8):529–45. doi: 10.1038/nri3702
206. Gwin KA, Shapiro MB, Dolence JJ, Huang ZL, Medina KL. Hoxa9 and Flt3 signaling synergistically regulate an early checkpoint in lymphopoiesis. J Immunol (2013) 191(2):745–54. doi: 10.4049/jimmunol.1203294
207. Guo R, Hu F, Weng Q, Lv C, Wu H, Liu L, et al. Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res (2020) 30(1):21–33. doi: 10.1038/s41422-019-0251-7
208. Goodings C, Smith E, Mathias E, Elliott N, Cleveland SM, Tripathi RM, et al. Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells (2015) 33(8):2628–41. doi: 10.1002/stem.2049
209. Jackson JT, Nasa C, Shi W, Huntington ND, Bogue CW, Alexander WS, et al. A crucial role for the homeodomain transcription factor Hhex in lymphopoiesis. Blood. (2015) 125(5):803–14. doi: 10.1182/blood-2014-06-579813
210. Hosokawa H, Koizumi M, Masuhara K, Romero-Wolf M, Tanaka T, Nakayama T, et al. Stage-specific action of Runx1 and GATA3 controls silencing of PU.1 expression in mouse pro-T cells. J Exp Med (2021) 218(8):e20202648. doi: 10.1084/jem.20202648
211. Hattori N, Kawamoto H, Fujimoto S, Kuno K, Katsura Y. Involvement of transcription factors TCF-1 and GATA-3 in the initiation of the earliest step of T cell development in the thymus. J Exp Med (1996) 184(3):1137–47. doi: 10.1084/jem.184.3.1137
212. Hozumi K, Negishi N, Tsuchiya I, Abe N, Hirano K, Suzuki D, et al. Notch signaling is necessary for GATA3 function in the initiation of T cell development. Eur J Immunol (2008) 38(4):977–85. doi: 10.1002/eji.200737688
213. Ebralidze AK, Guibal FC, Steidl U, Zhang P, Lee S, Bartholdy B, et al. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev (2008) 22(15):2085–92. doi: 10.1101/gad.1654808
214. Heinzel K, Benz C, Martins VC, Haidl ID, Bleul CC. Bone marrow-derived hemopoietic precursors commit to the T cell lineage only after arrival in the thymic microenvironment. J Immunol (2007) 178(2):858–68. doi: 10.4049/jimmunol.178.2.858
215. Malhotra N, Narayan K, Cho OH, Sylvia KE, Yin C, Melichar H, et al. A network of high-mobility group box transcription factors programs innate interleukin-17 production. Immunity. (2013) 38(4):681–93. doi: 10.1016/j.immuni.2013.01.010
216. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med (2014) 211(9):1733–40. doi: 10.1084/jem.20140145
217. Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, et al. NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep (2015) 10(12):2043–54. doi: 10.1016/j.celrep.2015.02.057
218. Mao AP, Ishizuka IE, Kasal DN, Mandal M, Bendelac A. A shared Runx1-bound Zbtb16 enhancer directs innate and innate-like lymphoid lineage development. Nat Commun (2017) 8(1):863. doi: 10.1038/s41467-017-00882-0
219. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. (2014) 157(2):340–56. doi: 10.1016/j.cell.2014.03.030
220. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. (2014) 508(7496):397–401. doi: 10.1038/nature13047
221. Ferreira ACF, Szeto ACH, Heycock MWD, Clark PA, Walker JA, Crisp A, et al. RORα is a critical checkpoint for T cell and ILC2 commitment in the embryonic thymus. Nat Immunol (2021) 22(2):166–78. doi: 10.1038/s41590-020-00833-w
222. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity. (2012) 37(3):463–74. doi: 10.1016/j.immuni.2012.06.012
223. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol (2018) 19(10):1093–9. doi: 10.1038/s41590-018-0201-4
224. Zook EC, Li ZY, Xu Y, de Pooter RF, Verykokakis M, Beaulieu A, et al. Transcription factor ID2 prevents E proteins from enforcing a naive T lymphocyte gene program during NK cell development. Sci Immunol (2018) 3(22):eaao2139. doi: 10.1126/sciimmunol.aao2139
225. Li ZY, Morman RE, Hegermiller E, Sun M, Bartom ET, Maienschein-Cline M, et al. The transcriptional repressor ID2 supports natural killer cell maturation by controlling TCF1 amplitude. J Exp Med (2021) 218(6):e20202032. doi: 10.1084/jem.20202032
226. Ebihara T, Song C, Ryu SH, Plougastel-Douglas B, Yang L, Levanon D, et al. Runx3 specifies lineage commitment of innate lymphoid cells. Nat Immunol (2015) 16(11):1124–33. doi: 10.1038/ni.3272
227. Miyamoto C, Kojo S, Yamashita M, Moro K, Lacaud G, Shiroguchi K, et al. Runx/Cbfβ complexes protect group 2 innate lymphoid cells from exhausted-like hyporesponsiveness during allergic airway inflammation. Nat Commun (2019) 10(1):447. doi: 10.1038/s41467-019-08365-0
228. Harly C, Cam M, Kaye J, Bhandoola A. Development and differentiation of early innate lymphoid progenitors. J Exp Med (2018) 215(1):249–62. doi: 10.1084/jem.20170832
229. Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol (2015) 16(10):1044–50. doi: 10.1038/ni.3248
230. Kasal DN, Bendelac A. Multi-transcription factor reporter mice delineate early precursors to the ILC and LTi lineages. J Exp Med (2021) 218(2):e20200487. doi: 10.1084/jem.20200487
231. Seillet C, Mielke LA, Amann-Zalcenstein DB, Su S, Gao J, Almeida FF, et al. Deciphering the innate lymphoid cell transcriptional program. Cell Rep (2016) 17(2):436–47. doi: 10.1016/j.celrep.2016.09.025
232. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. (2012) 37(4):634–48. doi: 10.1016/j.immuni.2012.06.020
233. Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity. (2014) 40(3):378–88. doi: 10.1016/j.immuni.2014.01.012
234. Serafini N, Klein Wolterink RG, Satoh-Takayama N, Xu W, Vosshenrich CA, Hendriks RW, et al. Gata3 drives development of RORγt+ group 3 innate lymphoid cells. J Exp Med (2014) 211(2):199–208. doi: 10.1084/jem.20131038
235. Harly C, Kenney D, Ren G, Lai B, Raabe T, Yang Q, et al. The transcription factor TCF-1 enforces commitment to the innate lymphoid cell lineage. Nat Immunol (2019) 20(9):1150–60. doi: 10.1038/s41590-019-0445-7
236. Qian L, Bajana S, Georgescu C, Peng V, Wang HC, Adrianto I, et al. Suppression of ILC2 differentiation from committed T cell precursors by E protein transcription factors. J Exp Med (2019) 216(4):884–99. doi: 10.1084/jem.20182100
237. Braunstein M, Anderson MK. HEB-deficient T-cell precursors lose T-cell potential and adopt an alternative pathway of differentiation. Mol Cell Biol (2011) 31(5):971–82. doi: 10.1128/MCB.01034-10
238. Wang HC, Qian L, Zhao Y, Mengarelli J, Adrianto I, Montgomery CG, et al. Downregulation of E protein activity augments an ILC2 differentiation program in the thymus. J Immunol (2017) 198(8):3149–56. doi: 10.4049/jimmunol.1602009
239. Jones ME, Zhuang Y. Stage-specific functions of E-proteins at the β-selection and T-cell receptor checkpoints during thymocyte development. Immunol Res (2011) 49(1-3):202–15. doi: 10.1007/s12026-010-8182-x
240. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12(11):749–61. doi: 10.1038/nri3307
241. Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol (2011) 12(12):1221–9. doi: 10.1038/ni.2158
242. Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet (2007) 8(6):450–61. doi: 10.1038/nrg2102
243. Manesso E, Chickarmane V, Kueh HY, Rothenberg EV, Peterson C. Computational modelling of T-cell formation kinetics: Output regulated by initial proliferation-linked deferral of developmental competence. J R Soc Interface. (2013) 10(78):20120774. doi: 10.1098/rsif.2012.0774
244. Krueger A, Ziętara N, Łyszkiewicz M. T Cell development by the numbers. Trends Immunol (2017) 38(2):128–39. doi: 10.1016/j.it.2016.10.007
245. Kenins L, Gill JW, Boyd RL, Hollander GA, Wodnar-Filipowicz A. Intrathymic expression of Flt3 ligand enhances thymic recovery after irradiation. J Exp Med (2008) 205(3):523–31. doi: 10.1084/jem.20072065
246. Rodewald HR, Waskow C, Haller C. Essential requirement for c-Kit and common gamma chain in thymocyte development cannot be overruled by enforced expression of Bcl-2. J Exp Med (2001) 193(12):1431–7. doi: 10.1084/jem.193.12.1431
247. Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-Kit in the maintenance of lymphopoiesis. Immunity. (2002) 17(3):277–88. doi: 10.1016/S1074-7613(02)00386-2
248. Massa S, Balciunaite G, Ceredig R, Rolink AG. Critical role for c-Kit (CD117) in T cell lineage commitment and early thymocyte development in vitro. Eur J Immunol (2006) 36(3):526–32. doi: 10.1002/eji.200535760
249. Ramond C, Berthault C, Burlen-Defranoux O, de Sousa AP, Guy-Grand D, Vieira P, et al. Two waves of distinct hematopoietic progenitor cells colonize the fetal thymus. Nat Immunol (2014) 15(1):27–35. doi: 10.1038/ni.2782
250. Buono M, Facchini R, Matsuoka S, Thongjuea S, Waithe D, Luis TC, et al. A dynamic niche provides Kit ligand in a stage-specific manner to the earliest thymocyte progenitors. Nat Cell Biol (2016) 18(2):157–67. doi: 10.1038/ncb3299
251. von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med (1995) 181(4):1519–26. doi: 10.1084/jem.181.4.1519
252. Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med (1994) 180(5):1955–60. doi: 10.1084/jem.180.5.1955
253. Schlenner SM, Madan V, Busch K, Tietz A, Laufle C, Costa C, et al. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity. (2010) 32(3):426–36. doi: 10.1016/j.immuni.2010.03.005
254. Hare KJ, Jenkinson EJ, Anderson G. An essential role for the IL-7 receptor during intrathymic expansion of the positively selected neonatal T cell repertoire. J Immunol (2000) 165(5):2410–4. doi: 10.4049/jimmunol.165.5.2410
255. Tani-ichi S, Shimba A, Wagatsuma K, Miyachi H, Kitano S, Imai K, et al. Interleukin-7 receptor controls development and maturation of late stages of thymocyte subpopulations. Proc Natl Acad Sci U S A. (2013) 110(2):612–7. doi: 10.1073/pnas.1219242110
256. Prockop SE, Petrie HT. Regulation of thymus size by competition for stromal niches among early T cell progenitors. J Immunol. (2004) 173(3):1604–11. doi: 10.4049/jimmunol.173.3.1604
257. Alhaj Hussen K, Vu Manh TP, Guimiot F, Nelson E, Chabaane E, Delord M, et al. Molecular and functional characterization of lymphoid progenitor subsets reveals a bipartite architecture of human lymphopoiesis. Immunity. (2017) 47(4):680–96.e8. doi: 10.1016/j.immuni.2017.09.009
258. Serwold T, Ehrlich LI, Weissman IL. Reductive isolation from bone marrow and blood implicates common lymphoid progenitors as the major source of thymopoiesis. Blood. (2009) 113(4):807–15. doi: 10.1182/blood-2008-08-173682
259. Saran N, Lyszkiewicz M, Pommerencke J, Witzlau K, Vakilzadeh R, Ballmaier M, et al. Multiple extrathymic precursors contribute to T-cell development with different kinetics. Blood. (2010) 115(6):1137–44. doi: 10.1182/blood-2009-07-230821
260. Paiva RA, Ramos CV, Leiria G, Martins VC. IL-7 receptor drives early T lineage progenitor expansion. J Immunol (2022) 209(10):1942–9. doi: 10.4049/jimmunol.2101046
261. Masuda K, Kubagawa H, Ikawa T, Chen CC, Kakugawa K, Hattori M, et al. Prethymic T-cell development defined by the expression of paired immunoglobulin-like receptors. EMBO J (2005) 24(23):4052–60. doi: 10.1038/sj.emboj.7600878
262. Berthault C, Ramond C, Burlen-Defranoux O, Soubigou G, Chea S, Golub R, et al. Asynchronous lineage priming determines commitment to T cell and B cell lineages in fetal liver. Nat Immunol (2017) 18(10):1139–49. doi: 10.1038/ni.3820
263. Wiede F, Dudakov JA, Lu KH, Dodd GT, Butt T, Godfrey DI, et al. PTPN2 regulates T cell lineage commitment and αβ versus γδ specification. J Exp Med (2017) 214(9):2733–58. doi: 10.1084/jem.20161903
264. Yu Q, Erman B, Park JH, Feigenbaum L, Singer A. IL-7 receptor signals inhibit expression of transcription factors TCF-1, LEF-1, and RORγt: Impact on thymocyte development. J Exp Med (2004) 200(6):797–803. doi: 10.1084/jem.20032183
265. Sudo T, Nishikawa S, Ohno N, Akiyama N, Tamakoshi M, Yoshida H, et al. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc Natl Acad Sci U S A. (1993) 90(19):9125–9. doi: 10.1073/pnas.90.19.9125
266. Patra AK, Avots A, Zahedi RP, Schuler T, Sickmann A, Bommhardt U, et al. An alternative NFAT-activation pathway mediated by IL-7 is critical for early thymocyte development. Nat Immunol (2013) 14(2):127–35. doi: 10.1038/ni.2507
267. Akashi K, Kondo M, von Freeden-Jeffry U, Murray R, Weissman IL. Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell. (1997) 89(7):1033–41. doi: 10.1016/S0092-8674(00)80291-3
268. Pellegrini M, Bouillet P, Robati M, Belz GT, Davey GM, Strasser A. Loss of bim increases T cell production and function in interleukin 7 receptor-deficient mice. J Exp Med (2004) 200(9):1189–95. doi: 10.1084/jem.20041328
269. Spolski R, Li P, Chandra V, Shin B, Liu C, Oh J, et al. Distinct super-enhancer elements differentially control Il2ra gene expression in a cell-type specific fashion. bioRxiv. 2022.11.18.516445; doi: 10.1101/2022.11.18.516445
270. Henriksson J, Chen X, Gomes T, Ullah U, Meyer KB, Miragaia R, et al. Genome-wide CRISPR screens in T helper cells reveal pervasive crosstalk between activation and differentiation. Cell. (2019) 176(4):882–96.e18. doi: 10.1016/j.cell.2018.11.044
271. Huang EY, Gallegos AM, Richards SM, Lehar SM, Bevan MJ. Surface expression of Notch1 on thymocytes: Correlation with the double-negative to double-positive transition. J Immunol (2003) 171(5):2296–304. doi: 10.4049/jimmunol.171.5.2296
272. Magri M, Yatim A, Benne C, Balbo M, Henry A, Serraf A, et al. Notch ligands potentiate IL-7-driven proliferation and survival of human thymocyte precursors. Eur J Immunol (2009) 39(5):1231–40. doi: 10.1002/eji.200838765
273. Garcia-Peydro M, de Yebenes VG, Toribio ML. Notch1 and IL-7 receptor interplay maintains proliferation of human thymic progenitors while suppressing non-T cell fates. J Immunol (2006) 177(6):3711–20. doi: 10.4049/jimmunol.177.6.3711
274. Gerby B, Tremblay CS, Tremblay M, Rojas-Sutterlin S, Herblot S, Hebert J, et al. SCL, LMO1 and Notch1 reprogram thymocytes into self-renewing cells. PLoS Genet (2014) 10(12):e1004768. doi: 10.1371/journal.pgen.1004768
275. Sincennes MC, Humbert M, Grondin B, Lisi V, Veiga DF, Haman A, et al. The LMO2 oncogene regulates DNA replication in hematopoietic cells. Proc Natl Acad Sci U S A. (2016) 113(5):1393–8. doi: 10.1073/pnas.1515071113
276. Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, Clevers HC. Critical involvement of tcf-1 in expansion of thymocytes. J Immunol (1998) 161(8):3984–91. doi: 10.4049/jimmunol.161.8.3984
277. Goux D, Coudert JD, Maurice D, Scarpellino L, Jeannet G, Piccolo S, et al. Cooperating pre-T-cell receptor and TCF-1-dependent signals ensure thymocyte survival. Blood. (2005) 106(5):1726–33. doi: 10.1182/blood-2005-01-0337
278. Hendriks RW, Nawijn MC, Engel JD, van Doorninck H, Grosveld F, Karis A. Expression of the transcription factor GATA-3 is required for the development of the earliest T cell progenitors and correlates with stages of cellular proliferation in the thymus. Eur J Immunol (1999) 29(6):1912–8. doi: 10.1002/(SICI)1521-4141(199906)29:06<1912::AID-IMMU1912>3.0.CO;2-D
279. Wojciechowski J, Lai A, Kondo M, Zhuang Y. E2A and HEB are required to block thymocyte proliferation prior to pre-TCR expression. J Immunol (2007) 178(9):5717–26. doi: 10.4049/jimmunol.178.9.5717
280. Parriott G, Kee BL. E protein transcription factors as suppressors of T lymphocyte acute lymphoblastic leukemia. Front Immunol (2022) 13:885144. doi: 10.3389/fimmu.2022.885144
281. Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. (2015) 161(3):555–68. doi: 10.1016/j.cell.2015.03.017
282. Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. (2012) 151(5):994–1004. doi: 10.1016/j.cell.2012.09.045
283. Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell. (2017) 168(3):442–59. doi: 10.1016/j.cell.2016.12.016
284. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. (2009) 326(5950):289–93. doi: 10.1126/science.1181369
285. Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. (2014) 159(7):1665–80. doi: 10.1016/j.cell.2014.11.021
286. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. (2012) 485(7398):376–80. doi: 10.1038/nature11082
Keywords: gene regulatory network, early T cell development, transcription factors, gene expression program, cell fate decision, thymus, epigenetic control, multipotency
Citation: Shin B and Rothenberg EV (2023) Multi-modular structure of the gene regulatory network for specification and commitment of murine T cells. Front. Immunol. 14:1108368. doi: 10.3389/fimmu.2023.1108368
Received: 26 November 2022; Accepted: 11 January 2023;
Published: 31 January 2023.
Edited by:
Mihalis Verykokakis, Alexander Fleming Biomedical Sciences Research Center, GreeceReviewed by:
Barbara L. Kee, The University of Chicago, United StatesCopyright © 2023 Shin and Rothenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boyoung Shin, Ym95b3VuZ0BjYWx0ZWNoLmVkdQ==; Ellen V. Rothenberg, ZXZyb3RoQGl0cy5jYWx0ZWNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.