 Qijun Ruan
Qijun Ruan Peng Guan
Peng Guan Weijuan Qi1
Weijuan Qi1 Dandan Ma
Dandan Ma Jia Ni
Jia Ni- 1Department of Periodontics, Stomatological Hospital, School of Stomatology, Southern Medical University, Guangzhou, China
- 2Department of Endodontics, Stomatological Hospital, School of Stomatology, Southern Medical University, Guangzhou, China
Atherosclerosis (AS) is a chronic inflammatory disease, involving a pathological process of endothelial dysfunction, lipid deposition, plaque rupture, and arterial occlusion, and is one of the leading causes of death in the world population. The progression of AS is closely associated with several inflammatory diseases, among which periodontitis has been shown to increase the risk of AS. Porphyromonas gingivalis (P. gingivalis), presenting in large numbers in subgingival plaque biofilms, is the “dominant flora” in periodontitis, and its multiple virulence factors are important in stimulating host immunity. Therefore, it is significant to elucidate the potential mechanism and association between P. gingivalis and AS to prevent and treat AS. By summarizing the existing studies, we found that P. gingivalis promotes the progression of AS through multiple immune pathways. P. gingivalis can escape host immune clearance and, in various forms, circulate with blood and lymph and colonize arterial vessel walls, directly inducing local inflammation in blood vessels. It also induces the production of systemic inflammatory mediators and autoimmune antibodies, disrupts the serum lipid profile, and thus promotes the progression of AS. In this paper, we summarize the recent evidence (including clinical studies and animal studies) on the correlation between P. gingivalis and AS, and describe the specific immune mechanisms by which P. gingivalis promotes AS progression from three aspects (immune escape, blood circulation, and lymphatic circulation), providing new insights into the prevention and treatment of AS by suppressing periodontal pathogenic bacteria.
1 Introduction
P. gingivalis, a Gram-negative anaerobic bacterium, is the “dominant flora” in periodontitis (1, 2). P. gingivalis can stimulate the host immune response through virulence factors, including its structural components (fimbriae, LPS, etc.) and secretory components (gingipains and OMVs) (3). P. gingivalis fimbriae can enhance the inflammatory response and evade host immune clearance (4). P. gingivalis LPS is an important virulence factor with lipid A as its immune core, triggering a signaling pathway closely related to lipid A variants (5). P. gingivalis gingipains are powerful protein hydrolysers that assist P. gingivalis to evade host immunity (6). P. gingivalis OMVs are outer membrane vesicles containing multiple virulence factors that can circulate widely throughout the body and access areas of tissue not accessible to whole bacteria, thereby triggering an inflammatory response (7). P. gingivalis and its virulence factors can promote the development of a variety of systemic diseases such as cardiovascular disease, diabetes, Alzheimer’s disease, etc (8).
AS is a chronic inflammatory pathological change occurring in the walls of medium to large arteries, characterized by immune cell infiltration and lipid deposition, and poses a serious threat to human life and health. Recent single-cell sequencing studies have revealed abundant heterogeneity of immune cells in the AS plaque environment. For example, three macrophage subpopulations were identified in human and mouse AS plaques, including inflammatory, resident-like (TREM2hi), and TREM2hi macrophages. Among them, TREM2hi macrophages are a foam-like, anti-inflammatory type of macrophages and exhibit an osteoclast-like gene expression profile that may be associated with plaque calcification (9, 10). Similar studies point to the presence of a cluster in mouse AS plaques that matches the core marker profile of B1 cells (CD43highB220negCD11bhigh) but shows enriched TNF signaling and cell adhesion pathways that may be key cell types for promoting AS (11). Previously unknown clusters of naive T cells and ApoB+ T cells have also been found in plaques and are closely associated with vascular inflammation, but the exact mechanism is not yet clear (12). Another study found that vascular smooth muscle cells (VSMCs) phenotype-switching mechanisms play an important role in AS. During AS development, VSMCs can give rise to a novel intermediate cell with multidirectional differentiation potential, which can either differentiate into macrophage-like or fibrocartilage-like cells or revert to VSMCs (13). VSMCs and endothelial cells (ECs) in the core of human arterial AS lesions were found to drive cellular transdifferentiation through multiple genes, whereas VSMCs and ECs in the adjacent zone were involved in immune cell recruitment through C3 and MHC II molecules, respectively (14). More in-depth studies using single-cell sequencing pointed out that DHRS9 in macrophages is a key factor in AS formation (15), and CXCL3, GK, FPR1, and LST1 in monocytes are closely associated with plaque instability (16). The application of single-cell sequencing technology has deepened the understanding of cellular heterogeneity in AS lesions, and provided an important theoretical basis for further investigation of AS pathogenesis and the development of targeted therapeutic drugs. In addition, single-cell sequencing studies have the advantage of investigating the mechanisms of interaction between two or more cells in tissues, such as between immune cells and tissue cells, yet no relevant studies have been seen.
Existing researches show that P. gingivalis promotes AS progression through immune responses (17, 18). On the one hand, P. gingivalis evades host innate and adaptive immunity and, in various forms, circulates with blood and lymph and colonizes the arterial vessel wall, directly inducing local inflammation and lesions in blood vessels. On the other hand, after evading host immunity, P. gingivalis persistently stimulates the host immune response, induces systemic inflammatory mediators and autoimmune antibody production, disorders of lipid levels, and promotes AS progression. In this paper, we review the recent evidence on P. gingivalis promoting AS and the related immune response mechanisms to elucidate the potential mechanism of action and association between P. gingivalis and AS, and provide theoretical guidance for further in-depth studies.
2 Evidence of correlation between P. gingivalis and AS
2.1 Clinical studies
The American Heart Association (AHA) supports an independent association between periodontal disease (PD) and atherosclerotic vascular disease, but not a causal relationship between the two (19). Subsequent studies in the last decade also lack evidence to date for a causal relationship between PD and AS. Current clinical studies have mainly elaborated on the correlation between PD and AS by detecting the colonization of periodontal pathogens in AS plaques and serum levels of AS-associated inflammatory markers.
Clinical studies have shown that abundant P. gingivalis colonization can be detected in AS plaques of different artery types. A Meat analysis based on 1791 patients showed that P. gingivalis specifically localized to coronary AS plaques but not to other organs (20). A study of 58 patients with moderate or severe periodontitis with AS using a 16sRNA assay found P. gingivalis detection rates of 26.7% in carotid arteries and 39.3% in coronary arteries (21). Also using the detection method of 16sRNA assay, some studies have pointed out that the detection rate of periodontal pathogens including P. gingivalis in carotid AS lesions is only 21% (22). Another study using metagenomics techniques noted that P. gingivalis colonization was detected in the coronary or femoral arteries of 42 patients with AS who participated in the study and that P. gingivalis accounted for nearly 80% of all colonized bacterial species, but the study was notably deficient in that it did not assess the periodontal status of the participants (23). Some scholars believe that the difference in the detection rate of P. gingivalis in AS plaques may be due to demographic, geographical, and ethnic differences, as well as different sampling methods and laboratory testing methods of clinical samples, but there are no related statistical and methodological differences (24). The colonization and invasion of P. gingivalis into the arterial wall allows P. gingivalis to acquire a “privileged niche”. This “privileged niche” not only helps P. gingivalis to obtain proteins and iron substrates from the host but also separates P. gingivalis from humoral and cellular immunity, thus inducing endothelial dysfunction and promoting AS progression (25) (Table 1).
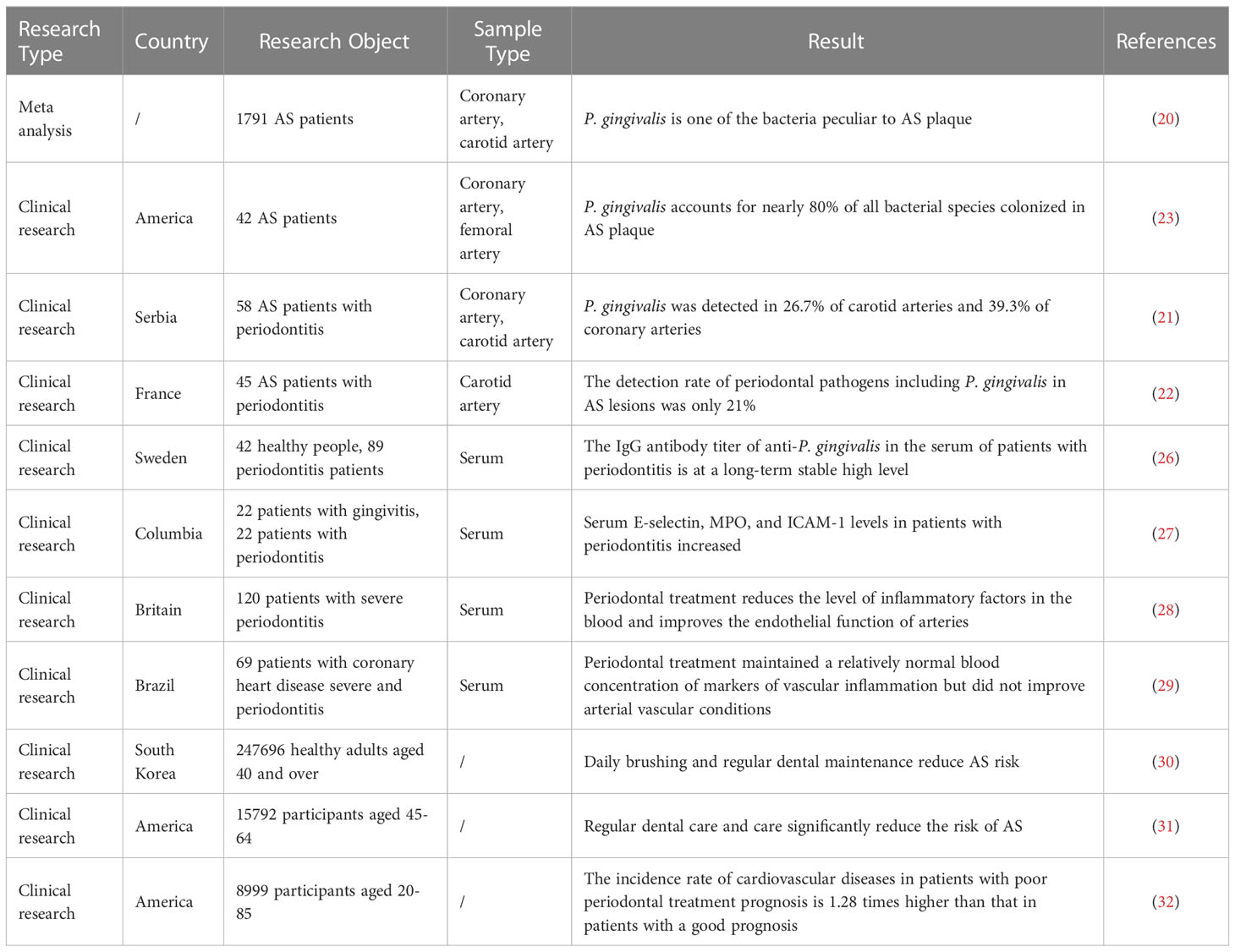
Table 1 Clinical Research Evidence of P. gingivalis and AS.
In patients with periodontitis, the presence of P. gingivalis colonization in the arterial vessel wall, as well as significantly higher serum anti-P. gingivalis antibody titers (26, 33) and inflammatory factor levels (27, 34) may lead to a concomitant increase in AS risk (35, 36). In contrast, routine oral maintenance and periodontal treatment significantly reduced the levels of AS-related inflammatory factors in the serum of the patient and significantly improved the endothelial function of the patient’s arterial vessels, leading to a consequent reduction in the risk of AS.
Several recent large population studies have evaluated the effect of routine oral maintenance and periodontal treatment on the progression of AS, and have reached similar conclusions. A Korean follow-up study based on 247,696 healthy adults aged 40 years and older showed that brushing more than once a day and regular professional dental cleanings reduced the risk of cardiovascular events, including AS, by 9% and 14%, respectively (30). A U.S. study of atherosclerosis risk in communities (ARIC) based on 15,792 participants aged 45-64 years over 15 years showed that regular dental care and attention significantly reduced the risk of AS (31). Another U.S. study based on 8,999 participants aged 20-85 years with 16.8 years of follow-up noted that the incidence of cardiovascular disease, including AS, was 1.28 times higher in those with a poor prognosis for periodontal treatment than in those with a good prognosis for periodontal treatment (32). A study of 120 patients with severe periodontitis showed lower levels of inflammatory factors in the blood after 24 hours of periodontal treatment compared to usual oral care, and a significant improvement in endothelial function of the arteries at six months (28). Plasma levels of AS-related risk molecules, including inflammatory factors (CRP, IL-6, TNF-α), thrombotic molecules (fibrinogen), and metabolic markers (triglycerides, TC, HDL-C, HbA1c, A-FABP), were significantly reduced within six months after periodontal treatment means to eliminate periodontal inflammation (37–40). However, it has also been shown that periodontal treatment, while maintaining relatively normal blood concentrations of vascular inflammatory markers, did not improve arterial vascular status in a short-term follow-up within three months after periodontal treatment and did not indicate an AS risk-reducing effect (29) (Table 1).
Based on the above clinical basis, we can speculate that P. gingivalis may promote AS by colonizing the arterial wall and causing abnormal serum inflammatory factor levels; however, there is still insufficient evidence from clinical studies on P. gingivalis and AS. Comparative studies on the colonization of P. gingivalis in the arterial canal wall before and after periodontal treatment are lacking. Current epidemiological data focus on medium-sized arteries such as coronary, carotid, and femoral arteries, however, the lack of studies on large arteries such as the aorta may be significantly associated with the availability of experimental samples. In addition, PD and AS each have multiple complex and critical risk factors that are pervasive and powerful contributors and difficult to fully exclude, including age, smoking, and diabetes mellitus (41). Therefore, future studies should increase the consideration of multiple confounding factors, including the criteria for admission to PD, standardized treatment regimens for PD, recurrence of PD and evolution of AS, and other important risk factors.
2.2 Animal studies
Animal studies have shown that ApoE-/- mice with oral P. gingivalis infection have elevated serum levels of cellular inflammatory factors (42) and significantly increased vascular reactivity (43), and P. gingivalis was detected to colonize the aorta, damaging the arterial endothelium (44) and increasing AS plaque (45). In addition, numerous studies have demonstrated that serum inflammatory mediators and lipoprotein levels were significantly abnormal in oral P. gingivalis-infected ApoE-/- mice, such as NLRP3, IL-6, IL-1β, TNF-α, intercellular adhesion molecule-1 (ICAM-1), vascular adhesion molecule 1 (VCAM-1), monocyte chemotactic protein(MCP), P-selectin, E-selectin, low-density lipoprotein (LDL), HDL, etc (46, 47) (Table 2).
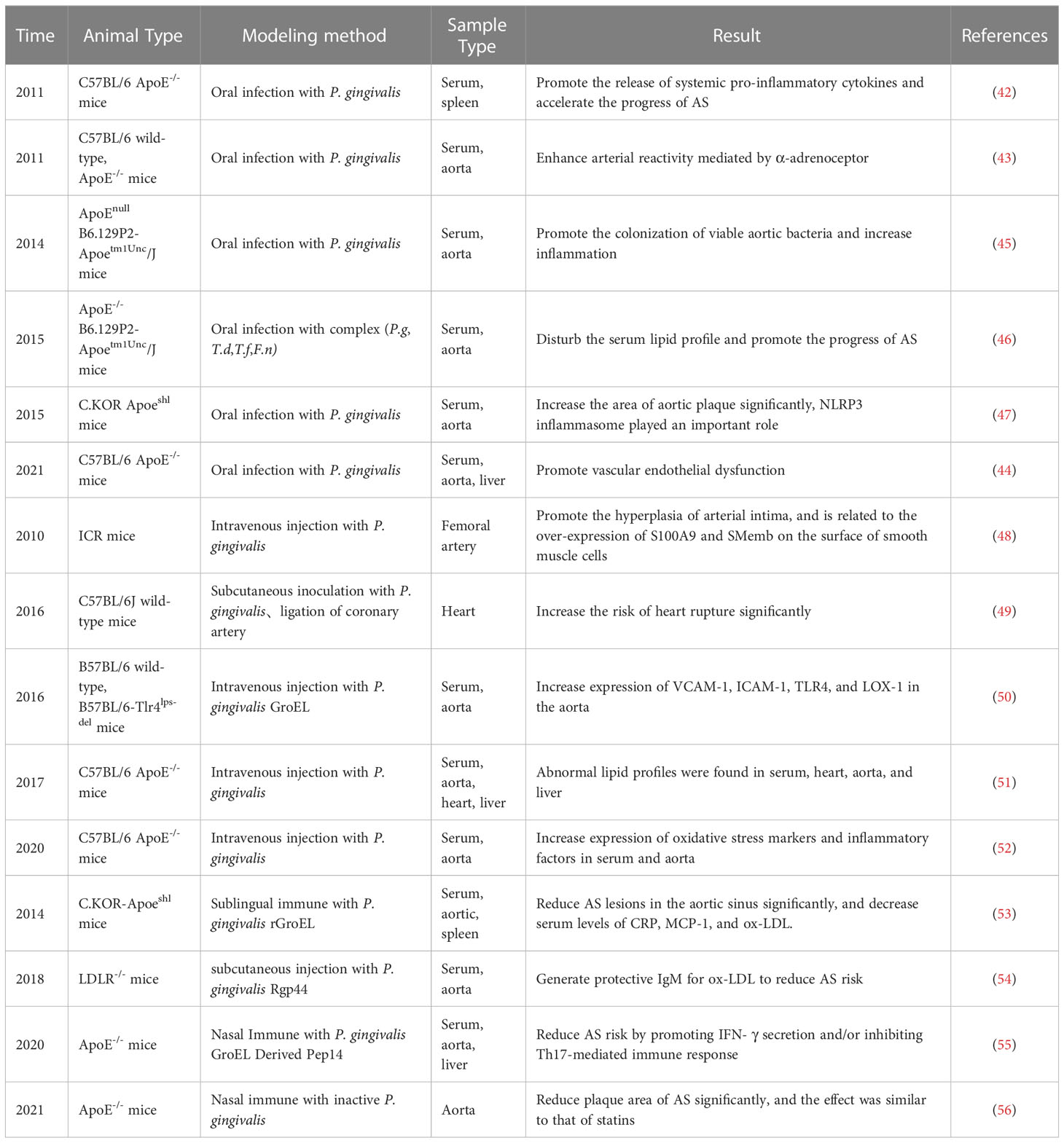
Table 2 Animal Research Evidence of P. gingivalis and AS.
In addition to oral infection with P. gingivalis, some studies have also infected mice by subcutaneous inoculation or intravenous P. gingivalis injection. Animal studies have shown that subcutaneous inoculation with P. gingivalis accelerates AS and leads to a significant increase in mortality from cardiac rupture (49). Intravenous P. gingivalis injection revealed significant intimal hyperplasia and VSMCs proliferation in the aorta of mice (48). After intravenous P. gingivalis injection, ApoE-/- mice showed increased expression of oxidative stress markers and inflammatory factors in serum and aorta (52), and abnormal lipid profiles were found in serum, heart, aorta, and liver (51). Intravenous injection of recombinant P. gingivalis heat shock protein GroEL into C57BL/6 mice on a high-fat diet resulted in stronger expression of VCAM-1, ICAM-1, TLR4, and lectin-like ox-LDL receptor (LOX-1) (50) (Table 2).
Some recent studies have also conducted experiments on the relationship between P. gingivalis-related vaccines and AS. Heated ultrasound P. gingivalis-prepared vaccine significantly reduced AS plaque area in ApoE-/- mice on the background of high-fat diet and oral P. gingivalis infection employing nasal immunization, with effects comparable to those of statins (56). Similarly, sublingual immunization with P. gingivalis GroEL (53) or nasal immunization with its derivative peptide 14 (Pep14) (55) achieved inhibition of AS plaque formation. In addition, subcutaneous immunization of LDLR-/- mice with the A hemagglutinin domain (Rgp44) of P. gingivalis promotes the production of protective IgM against ox-LDL and reduces the risk of AS (54). There are few studies on the relationship between P. gingivalis-related vaccines and AS, and the mechanism of action is not fully understood and needs to be further investigated (Table 2).
Oral infection with P. gingivalis can lead to dysregulated intestinal flora (57), and dysregulated intestinal flora can also promote AS progression through metabolism-dependent pathways (58). Among them, Trimethylamine-N-oxide (TAMO) is one of the most important metabolites associated with dysregulated intestinal flora (59, 60). TMAO promotes foam cell production by upregulating macrophage CD36 and SR-A1 expression which impairs macrophage cholesterol reversal transport function (61). TMAO promotes the release of pro-inflammatory mediators through the activation of mitogen-activated protein kinase, extracellular signal-associated kinase, and NF-κB cascade pathways, which in turn induces an inflammatory response in ECs and VSMCs (62–64). TMAO exacerbates AS progression by promoting the release of intracellular Ca2+, leading to platelet aggregation and thrombosis (65).
However, there are still limitations in the establishment of current animal models. Intravenous injection of planktonic state P. gingivalis in an infectious manner not only hardly mimics the characteristics of inflammation triggered by normal plaque biofilm but also may lead to a sharp increase in the level of P. gingivalis in the circulatory system and trigger a strong stress response. Thus, the type and degree of the inflammatory response induced by intravenous P. gingivalis may not match the chronic state of infection in a real situation. Certainly, the intravenous approach may apply to the study model of acute systemic bacterial infections.
It is noteworthy that both clinical and animal studies have used 16srRNA, metagenomics sequencing to detect P. gingivalis g gene fragments in the arterial wall to determine colonization. But these techniques are impossible to assess the activity status and reproduction of P. gingivalis after colonization or to determine whether the gene fragments are from intact bacteria or P. gingivalis OMVs. These are limitations of the current assays. The activity of P. gingivalis colonized in the arterial wall or the different virulence factors released by dead bacteria may directly influence the degree of local inflammatory response in the vessel wall. In vitro studies have pointed out that live P. gingivalis induces monocyte adhesion to the endothelium and promotes the vascular inflammatory response by promoting the expression of adhesion molecules and pro-inflammatory factors in ECs, whereas heat-killed P. gingivalis does not trigger these effects (66, 67). Therefore, the conclusion that P. gingivalis colonizes the arterial vessel wall cannot yet be directly correlated with vascular inflammation using these techniques. Further culture and characterization of strains of AS tissue are needed to explore P. gingivalis activity after colonization. This is a central part of the follow-up study.
3 Pathogenicity of P. gingivalis and pathogenesis of AS
3.1 Pathogenicity of P. gingivalis
P. gingivalis is the “dominant flora” in periodontitis, which can reshape the symbiotic colonization of periodontal tissues and induce dysbiosis of periodontal microbial homeostasis (2). During disease development, P. gingivalis interacts with the host immune system through its unique virulence factors, resulting in a unique and complex pathogenic mechanism, such as induction of inflammatory response, activation of the complement system, promotion of apoptosis, and other biological processes (1). In addition, the virulence factors of P. gingivalis can interact with various host receptors to reshape the survival environment or escape host immune killing, allowing them to persist in host tissues (3).
3.1.1 P. gingivalis gingipains
P. gingivalis gingipains have potent proteolytic activity and play a key role in disrupting the host immune response. P. gingivalis gingipains include arginine-specific gingipains (RgpA and RgpB), and lysine-specific gingipains (Kgp). P. gingivalis gingipains cause host immune dysregulation and inflammatory responses to occur by activating matrix metalloproteinases, inactivating immunosuppressants, degrading immunomodulatory factors, and cleaving immune cell receptors (6). P. gingivalis gingipains degrade the junctional adhesion molecule (JAM1) of gingival epithelial cells, disrupting epithelial barrier function and increasing the ability of bacteria and their products such as LPS and peptidoglycan (PGN) to locally invade and penetrate the peripheral blood (68). P. gingivalis gingipains selectively degrade the macrophage surface innate immune receptor CD14, resulting in the hyporesponsiveness to bacterial challenge (69). P. gingivalis gingipains can degrade neutrophil-derived α-defensins and β-defensins, disrupting the host’s innate immune function and facilitating bacterial escape (70). In addition, P. gingivalis gingipains have a significant disruptive effect on the complement system. On the one hand, P. gingivalis gingipains inhibited the bactericidal effect of the complement system by degrading C3, C4, and C5 to inhibit complement activation and the formation of membrane attack complexes (71). On the other hand, P. gingivalis gingipains release the allergenic toxin C5a by cleaving complement C5, causing stronger inflammation (72). C5a induced by P. gingivalis gingipains can also subtly evade immune clearance via the C5aR-TLR2 crosstalk pathway (73).
3.1.2 P. gingivalis LPS
P. gingivalis LPS consists of lipid A, core oligosaccharide, and O-specific polysaccharide (6). P. gingivalis LPS can trigger innate immune responses by activating TLRs (74). The virulence properties of P. gingivalis LPS are determined by lipid A properties, thus different properties of lipid A can lead to different innate immune responses and the production of inflammatory factors. P. gingivalis LPS1690 with penta-acylated lipid A mainly activates the NF-κB signaling pathway, while P. gingivalis LPS1435/1449 with tetra-acylated lipid A mainly induces p38/MAPK and ERK1/2 signaling pathways (74). P. gingivalis LPS induces M1-type polarization in macrophages and promotes the expression of several pro-inflammatory factors, such as TNF-α, IL-1β, and IL-6 (75). P. gingivalis LPS promotes the progression of periodontal inflammation by inducing pyroptosis in gingival fibroblasts (76). P. gingivalis LPS triggers mitochondria-mediated apoptosis by regulating XBP1 expression (77). P. gingivalis LPS promotes platelet proliferation and thrombosis by activating platelet Cdc42 (78).
3.1.3 P. gingivalis fimbriae
The fimbriae of P. gingivalis are divided into long fimbriae (FimA) and short fimbriae (Mfa1), both of which have enhanced inflammatory responses and evasion of host immune clearance, although each has its mechanism of action with the host. FimA acts through the characteristic P. gingivalis peptidilarginine deiminase (PPAD)-dependent activation of TLR2, induction of NF-ĸB and MAPK signaling pathways, and promotion of pro-inflammatory factor production (79). FimA interacts with complement receptor 3 (CR3) in macrophages, leading to ERK1/2 phosphorylation and inhibition of IL-12 production to promote the survival of P. gingivalis (80). FimA induces cAMP-dependent protein kinase A (PKA) activation via instigating macrophage CXCR4/TLR2 co-association, which in turn inhibits TLR2-mediated antimicrobial responses (81). The binding of FimA to CXCR4 induces CR3 activation via phosphatidylinositol-3 kinase (PI3K) and inhibits the antibacterial response in macrophages (82). Similar to FimA, Mfa1 induces the production of pro-inflammatory factors through the activation of TLRs (83, 84). Mfa1 inhibits the autophagy of DCs through the DC-SIGN-TLR2 crosstalk pathway, evading intracellular killing and leading to long-term survival within DCs (85).
3.2 Pathogenesis of AS
AS is a chronic cardiovascular disease that threatens human health and is characterized by lipid deposition in parts of the artery, accompanied by VSMCs and fibrous matrix proliferation, which gradually forms an AS plaque. AS is often considered a chronic inflammatory disease because inflammation plays an important role in all stages of AS development (86). The AS-associated inflammation is mainly mediated by pro-inflammatory factors, inflammatory signaling pathways, bioactive lipids, and adhesion molecules (87).
The main triggers of AS are endothelial damage, abnormal lipid metabolism, and hemodynamic impairment. In the early stages of AS, these pathological factors activate ECs (88). When ECs are activated, they express a variety of pro-inflammatory factors and adhesion molecules, including MCP-1, IL-8, ICAM-1, VCAM-1, E-selectin, and P-selectin, which attract lymphocytes and monocytes that bind to ECs and infiltrate the arterial wall, promoting the progression of the inflammatory response (89). Among them, pro-inflammatory monocytes expressing high levels of Ly6C or Gr-1 preferentially accumulate at damaged endothelial sites (90). Immune cells residing in the vessel wall participate in the inflammatory response process in the vessel wall together with the attracted immune cells. Large amounts of LDL are modified to ox-LDL and accumulate in the vessel wall, while macrophages in the vessel wall take up ox-LDL and convert it to foam cells, leading to the formation of AS plaques (91). Other types of immune cells, such as dendritic cells (DCs), T cells, B cells, and neutrophils are also involved in the progression of the inflammatory response within the plaque (92). In the advanced stages of AS, large numbers of macrophages and pro-inflammatory factors infiltrate the vessel wall, secrete matrix metalloproteinases (MMPs), and degrade collagen fibers in the extracellular matrix, leading to plaque rupture, hemorrhage, and thrombosis (89).
3.3 Pathogen-associated molecular patterns and damage-associated molecular patterns play a “bridging” role between P. gingivalis and AS
PAMPs are conserved pathogenic molecular structures shared by pathogenic microorganisms, while DAMPs are substances released into the intercellular space or blood circulation upon stimulation of tissues or cells (93). These substances bridge the gap between periodontitis and AS, allowing us to further understand the relationship between oral and systemic diseases (94).
P. gingivalis contains multiple PAMPs, including LPS and PGN, which initiate the inflammatory response of innate immunity by relying on the recognition of host cell pattern-recognition receptors (PRRs), such as NLRs and TLRs. P. gingivalis LPS induces the release of pro-inflammatory factors by activating NF-κB and MAPK signaling pathways in macrophages (95). P. gingivalis LPS promotes high expression of angiotensin II (Ang II) and IL-6 in ECs and accelerates ECs dysfunction (67). P. gingivalis LPS promotes monocyte chemotaxis and adhesion by increasing the expression of chemotactic and adhesion molecules in ECs through Akt and NF-κB signaling pathways (96). PGN promotes ICAM-1 production by monocytes through the activation of TLR2 and NF-κB pathways and induces monocyte migration and adhesion to the vascular endothelium (97). PGN promotes the upregulation of VCAM-1 through the NOD1-RIP2-NF-κB axis, inducing myeloid cells to recruit to the endothelium and leading to endothelial dysfunction (98). PGN can also mediate the over-expression of adhesion molecules in ECs through innate peptidoglycan recognition protein 1 (99). PGN induces the production of pro-inflammatory cytokines through TLR2 and CD14 and increases the susceptibility of AS plaques (100).
Meanwhile, periodontal pathogens further promote the progression of AS by activating inflammatory caspases that induce cell death and release various DAMPs, such as HSP60, cardiolipin, alarmins (S100 protein), and high mobility group box 1 (HMGB1) (101). HSP60 promotes immune cell migration and adhesion to the endothelium by stimulating the expression of E-selectin, VCAM-1, and ICAM-1 in ECs, and also induces endothelial inflammatory responses by activating TLRs (mainly TLR4) in innate immune cells (macrophages, DCs) (102). HSP60 induces DCs maturation and activates Th1 and Th17 cells in an MHC-II-dependent manner, promoting the release of pro-inflammatory mediators (103). HSP60 induces the activation of specific CD4+CD25+CD45RO+ T cells, which bind to ECs expressing HSP60 and adhesion molecules (VCAM-1 and E-selectin), forming susceptible sites of AS lesions (104). HSP60 induces the proliferation of VSMCs via TLR2 and TLR4 (105). The released cardiolipin may be oxidized by P. gingivalis to become oxidized cardiolipin (106, 107). Oxidized cardiolipin increases the expression levels of ICAM-1 and VCAM-1 in ECs and induces migration and adhesion of immune cells. At the same time, oxidized cardiolipin could also activate 5-lipoxygenase and induce leukotriene B4 production by increasing intracellular calcium concentration in macrophages and neutrophils, promoting inflammatory responses and exacerbating AS progression (108). P. gingivalis could promote VSMCs from contractile phenotype to synthetic phenotype by upregulating the expression of S100A9 in VSMCs (109). The circular RNA PPP1CC of P. gingivalis could promote VSMCs pyroptosis through the HMGB1/TLR9/AIM2 axis, which in turn increases AS plaque vulnerability (110).
4 Immune mechanism of P. gingivalis to promote AS progression
Immunity is an important line of defense of the organism against pathogenic invasion. P. gingivalis expresses a variety of virulence factors that stimulate host immune responses and play an important role in promoting AS progression. On the one hand, P. gingivalis evades host innate and adaptive immunity, internalizes in host tissues, and, in various forms, circulates with blood and lymph and colonizes arterial vessel walls, causing local inflammation and lesions; on the other hand, P. gingivalis persistently stimulates host immune responses in the process of host immune evasion and systemic dissemination, inducing systemic inflammatory mediators and disruption of lipid levels, and promoting AS progression. Based on the possible key role of P. gingivalis-associated PAMPs and DAMPs in promoting AS progression, the following literature will be reviewed from three aspects involving immune response: immune escape, blood circulation, and lymphatic circulation.
4.1 Immune escape
Immune escape is an important pathway for P. gingivalis to invade and survive in the host for a long time. A recent paper has reviewed P. gingivalis evasion of host immune killing through various pathways but did not elucidate the possible potential association and mechanism of P. gingivalis with AS during immune evasion (111). In this section, we will review the potential associations and mechanisms of P. gingivalis with AS during immune evasion in terms of dysregulation of the complement system and disruption of immune cell function.
4.1.1 P. gingivalis interferes with the function of the complement system
The complement system is a major part of the innate immune system and is activated by the hydrolytic cascade reaction of serine proteases. In inflamed vessels, the complement system can be activated by conjugates of CRP and modified LDL (112), inducing apoptosis of ECs, promoting the proliferation of VSMCs, inducing the release of procoagulant and adhesive factors, recruiting immune cells, and accelerating AS progression (113).
Studies suggest that the virulence factors of P. gingivalis may promote AS progression by interfering with the function of the complement system and, in turn, by promoting AS progression. P. gingivalis gingipains cleave complement C5 into biologically active C5a and C5b fragments (114, 115) and induce M1-type polarization of macrophages via the C5a pathway to promote inflammatory factor secretion (116). C5a is present in AS plaques and acts as a pro-AS molecule (117–119). In the early plaque formation stage, C5a activates mast cells in the arterial wall, promotes secretion of fibrinogen activator inhibitor (PAI-1), inhibits fibrinolysis and extracellular protein hydrolysis, and accelerates thrombus and AS plaque formation (120, 121). In the advanced stage of AS, C5a induces apoptosis of ECs and VSMCs, and expression of MMP1 and MMP9 in plaques, leading to VSMCs-dependent collagen loss, fibrous cap thinning, and plaque rupture (118, 122, 123). In addition, C5a accumulation enhanced NLRP3 inflammatory vesicle activation in AS plaques and decreased plaque stability (124). In contrast, a significant reduction in AS plaque area was observed after treatment of ApoE-/- mice with C5aR antagonists (125, 126). Notably, P. gingivalis cleavage of C5 produced large amounts of C5a, which activated C5aR, triggered cross-talk signal between TLR2 and C5aR (127), suppressed macrophage immune function, increased P. gingivalis survival, and led to ubiquitinated degradation of myeloid differentiation factor (Myd88) (127–129), which is also a positive regulator of foam cell formation in AS (130). Therefore, it is speculated that the crosstalk between TLR2-C5aR may be closely related to the promotion of foam cell formation by P. gingivalis. However, it has not been reported yet and needs further in-depth study.
The above studies suggest that P. gingivalis gingipains may promote AS plaque formation and rupture by interfering with complement C5a function (Figure 1). However, the current studies on the promotion of AS development by P. gingivalis through interference with the complement system are very limited, mainly focusing on the effects of P. gingivalis gingipains with C5a. However, whether similar effects and mechanisms exist for other virulence factors of P. gingivalis, whether other components of the complement system are involved in AS progression, and whether P. gingivalis-mediated TLR2-C5aR crosstalk is associated with foam cell formation is not fully understood and need to be further investigated in depth.
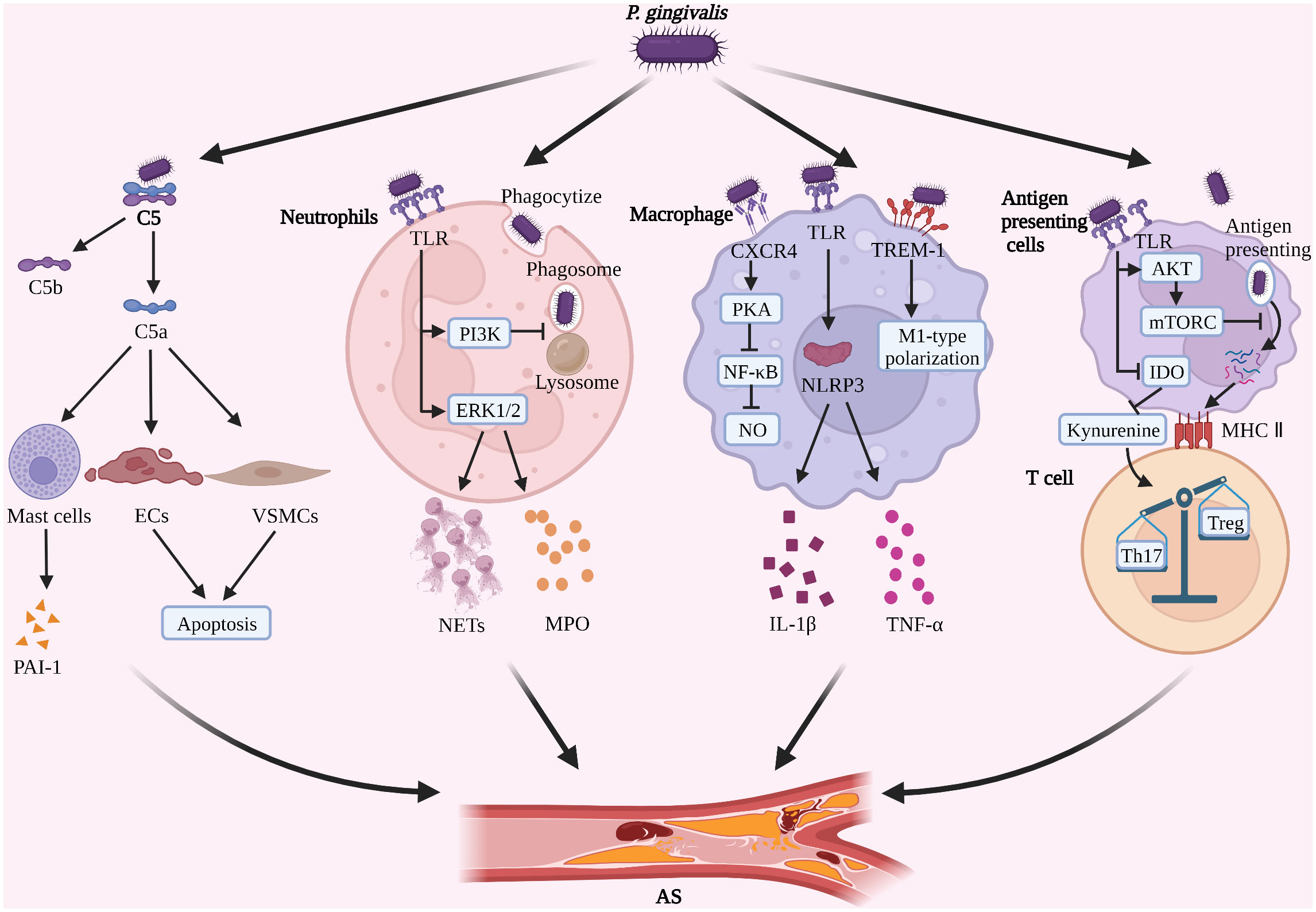
Figure 1 P. gingivalis induces immune escape and promotes AS progression (1). P. gingivalis degrades complement C5, promotes PAI-1 secretion by mast cells, and induces apoptosis of ECs and VSMCs (2). P. gingivalis inhibits phagosome-lysosome fusion in neutrophils, induces the formation of NETs, and increases secretion of MPO (3). P. gingivalis inhibits NO formation in macrophages and promotes M1-type polarization and activation of NLRP3 (4). P. gingivalis inhibits the antigen presentation process and induces Th17/Treg imbalance. (Created with BioRender.com).
4.1.2 P. gingivalis inhibits the antimicrobial function of immune cells
P. gingivalis inhibits the phagocytosis, surveillance, and clearance functions of immune cells through various mechanisms, evades host immune killing, survives in the host for a long time, repeatedly stimulates the body’s immune system, leads to a persistent low-level inflammatory state in the host, and promotes the progression of AS.
4.1.2.1 Neutrophils
Neutrophils are a class of innate immune cells that are the first to reach the site of P. gingivalis infection and can constitute an important barrier against P. gingivalis by producing proteases, antimicrobial peptides, and extracellular traps (NETs) (131, 132), as well as being important regulators of vascular inflammation (133).
It was found that P. gingivalis can evade immune killing by neutrophils. P. gingivalis activates the non-MyD88-dependent TLR2-PI3K signaling pathway in neutrophils, which both reduces the phagocytosis of P. gingivalis by neutrophils and blocks intracellular phagosome-lysosome fusion, thereby increasing the intracellular survival of P. gingivalis (129). It was also noted that P. gingivalis stimulated neutrophils to form NETs in a gingipains-dependent manner, but the antimicrobial components (histone protease, LL-37, etc.) in the formed NETs were hydrolyzed by gingipains, resulting in the lack of antimicrobial activity of NETs and the inability to achieve P. gingivalis clearance (132). In addition, the OMVs secreted by P. gingivalis can coat the neutrophils without being internalized, while the gingipains carried can degrade LL-37 and myeloperoxidase (MPO), which have secreted antimicrobial particle activity, and thus achieve the effect of avoiding neutrophil killing from a distance (134). The above pathways provide an opportunity for P. gingivalis to evade neutrophil immune killing, however, the efficacy of the various pathways has not been conclusively established.
However, it should not be overlooked that P. gingivalis still has a continuous stimulatory effect on neutrophils after evading immune killing, causing neutrophils to develop immune tolerance and inhibiting phagocytosis of P. gingivalis, while possibly promoting the progression of AS. P. gingivalis LPS-tolerant neutrophils have reduced phagocytosis of P. gingivalis, but significantly increased NETs formation and increased extracellular MPO levels, which may be related to neutrophils’ immune reconstitution (135). However, as already mentioned, P. gingivalis gingipains can degrade MPO (134). The “antagonism” between the pathogenicity of evaded phagocytosed P. gingivalis and the antimicrobial capacity of tolerant neutrophils determines the final fate of P. gingivalis. Furthermore, it has been shown that excess NETs promote plaque instability by directly inducing the death of ECs (136, 137), and that plasma MPO levels are positively correlated with the risk of AS (138). It is suggested that P. gingivalis may accelerate the progression of AS by promoting the production of NETs and MPO by tolerogenic neutrophils (Figure 1). P. gingivalis gingipain R and some host-derived proteases inhibit the nonphlogistical clearance of apoptotic cells by macrophages through hydrolytic modification of apoptotic neutrophil surface protein ligands, leading to local accumulation of apoptotic neutrophils and secondary necrosis (139). The process of nonphlogistical clearance of apoptotic cells by macrophages is called “efferocytosis” and defective efferocytosis exacerbates AS progression (140).
4.1.2.2 Macrophages
Macrophages are a class of intrinsic immune cells with strong phagocytic capacity and antigen-presenting function, which can exert antimicrobial effects by phagocytosing P. gingivalis and P. gingivalis-infected cells, secreting pro-inflammatory cytokines (141), and are also major players in the formation of foam cells and mediating AS plaque stability (142).
It was found that P. gingivalis can undergo repeated cycling behavior in and out of cells in macrophages and successfully avoid macrophage killing (143), which may be related to the following mechanisms. First, the high heme concentration in the inflammatory environment can convert P. gingivalis surface lipid A to a tetraacylated form without TLR4 agonistic activity, limiting macrophage activation (144–146). Second, various specific virulence factors of P. gingivalis play an important role in interfering with macrophage immune responses. P. gingivalis gingipains degrade caspase-1, IL-1β, and CD14, inhibit the activation of a TLR2/4 signaling pathway in macrophages (69, 147), and suppress the bactericidal effect of inflammasomes (148). P. gingivalis FimA (long or major fimbriae) bind to CXCR4, both through PI3K signaling activates CR3 on macrophages contributing to P. gingivalis immune evasion (82, 149), and also induces cAMP-dependent protein kinase A (PKA) signaling, inhibits TLR2/1-mediated NF-κB activation and NO production, suppresses macrophage activation, and improves P. gingivalis survival and virulence (81). P. gingivalis sialidase inhibits macrophage IL-12 expression (150), thereby suppressing NK cell activation and the Th1 to Th2 phenotype switch, which in turn reduces the clearance of P. gingivalis (151). In addition, besides inhibiting the endocytic digestion of macrophages, P. gingivalis can also inhibit the autophagy of macrophages to a certain extent, leading to the immune escape of P. gingivalis. It has been shown that P. gingivalis can induce autophagy in macrophages and promote the clearance of P. gingivalis by macrophages (152). However, it has also been shown that different P. gingivalis LPS variants (LPS1690, LPS1435/1449) exert different promoting or inhibiting effects on autophagy. Among them, the dominance of different P. gingivalis LPS variants depend on factors such as temperature, growth cycle, and heme chloride level (5, 153). LPS1690 induces macrophages to produce giant LC3-positive vesicles and melanoregulin puncta (cargo sorting protein), promoting lysosome maturation and autophagic response, while the presence of LPS1435/1449 significantly inhibited the above effects (154). Therefore, it is speculated that the different occupancy of LPS1690 and LPS1435/1449 in P. gingivalis may regulate the autophagy of macrophages and thus affect the survival of P. gingivalis. However, this has not been reported and needs to be further investigated in depth.
P. gingivalis, which survives in macrophages, induces inflammatory vesicle activation and M1-type polarization in macrophages (155). Animal studies have shown that oral inoculation of P. gingivalis can activate NLRP3 inflammasomes in macrophages in AS plaques in a gingipains-independent manner, promote the release of IL-1β and TNF-α, and accelerate AS progression (47, 148). In vitro studies have revealed that P. gingivalis LPS may induce M1-type polarization of macrophages via pattern recognition receptor triggering receptors expressed on myeloid cells-1(TREM-1) and its downstream signaling pathways to promote the secretion of multiple inflammatory factors and accelerate AS progression (75, 156, 157) (Figure 1). However, recent studies have also indicated that P. gingivalis LPS-induced tolerant macrophages represent an intermediate state between M1/M2 polarization and function as M2-like cells to limit the inflammatory response (158), suggesting that P. gingivalis LPS tolerant macrophages may be closely related to tissue repair. This may be related to the different macrophage sources, stimulation factors, and temporal phases, and needs to be analyzed in further studies. In addition, the foam-like, anti-inflammatory TREM2hi macrophage subpopulation identified by recent single-cell sequencing studies may be associated with plaque calcification (9). However, no studies related to P. gingivalis and TREM2hi macrophage subsets have been reported. As a key cell type in host immune response and AS plaque formation, macrophages may have important intersections with multiple immune and metabolic mechanisms, but there is no uniform conclusion yet, and further in-depth studies are needed.
4.1.2.3 T cells
T cells are the main performers of adaptive immunity in the body and are also important regulators of AS plaque formation, development, and late stability (159). Notably, the hypoxic and ischemic environment of AS plaques may also have an impact on T cell metabolic status and function, which in turn affects AS progression (160, 161).
It is suggested that P. gingivalis may evade T cell immune killing by inhibiting antigen presentation function and thus T cell activation, proliferation, and antimicrobial function. In terms of inhibiting antigen presentation, P. gingivalis promotes IL-10 secretion by macrophages, downregulates the expression of MHC-II molecules (162), and promotes PD-L1 and PD-1 binding on the surface of macrophages and CD4+ T cells, inhibiting antigen presentation and T cell activation (162–164). In addition, P. gingivalis interferes with DCs autophagy and apoptotic processes by activating the Akt/mTOR axis, thereby promoting survival in DCs and inhibiting P. gingivalis processing and presentation by DCs (165, 166). In addition to inhibiting antigen presentation function, P. gingivalis can also directly inhibit T cell activation and proliferation. P. gingivalis and its Rgp-gingipains inhibit T cell proliferation by suppressing protein kinase C (PKC) and p38 phosphorylation and inhibiting transcription factor activator protein-1 (AP-1), thereby downregulating IL-2 gene expression and accumulation (167). In addition, P. gingivalis may inhibit the development and proliferation of Treg cells by decreasing the secretion of TGF-β1, which may be mainly related to the type II FimA of P. gingivalis (34). However, this study did not compare the effects of other types of FimA at the same time, and the mechanism has not been clarified, which needs further in-depth study. Based on the above study, we found that P. gingivalis suppressed host adaptive immunity from the process of inhibiting antigen presentation, T cell proliferation, and activation, providing a possibility for the long-term survival of P. gingivalis in the host.
AS is also regulated by T-lymphocyte subsets. Among them, Th17 cells have pro-atherogenic effects and Treg cells have anti-atherogenic effects (168), thus Th17/Treg balance is closely related to AS progression. A clinical study with 1251 patients suggested that P. gingivalis may regulate tryptophan and kynurenine metabolism by inhibiting indoleamine 2,3 dioxygenase (IDO) activity in antigen-presenting cells, thereby promoting Th17 and inhibiting Treg proliferation, resulting in Th17/Treg imbalance and accelerating AS progression (169), but this mechanism needs to be further confirmed by in vitro experiments (Figure 1). Animal studies suggest that oral infection with P. gingivalis may promote Th17/Treg imbalance by increasing IL-6 expression in DCs, resulting in increasing plaque size and decreasing stability in AS (170). However, the role of P. gingivalis with other T cell subsets (e.g. γδ T cells, naive T cells, ApoB+ T cells) as well as Th cell subsets (e.g. Th9 and Th22) is unknown and needs to be further investigated in depth (12, 168).
In summary, P. gingivalis evades host immune cell killing through various pathways, survives in the host for a long time, induces a prolonged state of low inflammation (171), and provides an opportunity to promote the progression of AS. Although the molecular mechanism regarding the selective inhibition of immune elimination by P. gingivalis without suppressing the inflammatory response remains unclear, this phenomenon suggests a potentially significant threat following P. gingivalis evasion of immune killing. Therefore, targeting the inhibition or enhancement of one of the key links may provide a new strategy to enhance the body’s immunity and combat AS.
4.2 Blood circulation
P. gingivalis may enter the blood circulation locally from the oral cavity and then undergo systemic dissemination. Notably, P. gingivalis circulating with the blood may colonize the arterial endothelium, induce endothelial damage, recruit and activate platelets, promote foam cell formation and vessel wall calcification, and eventually lead to plaque rupture (172, 173) (Figure 2).
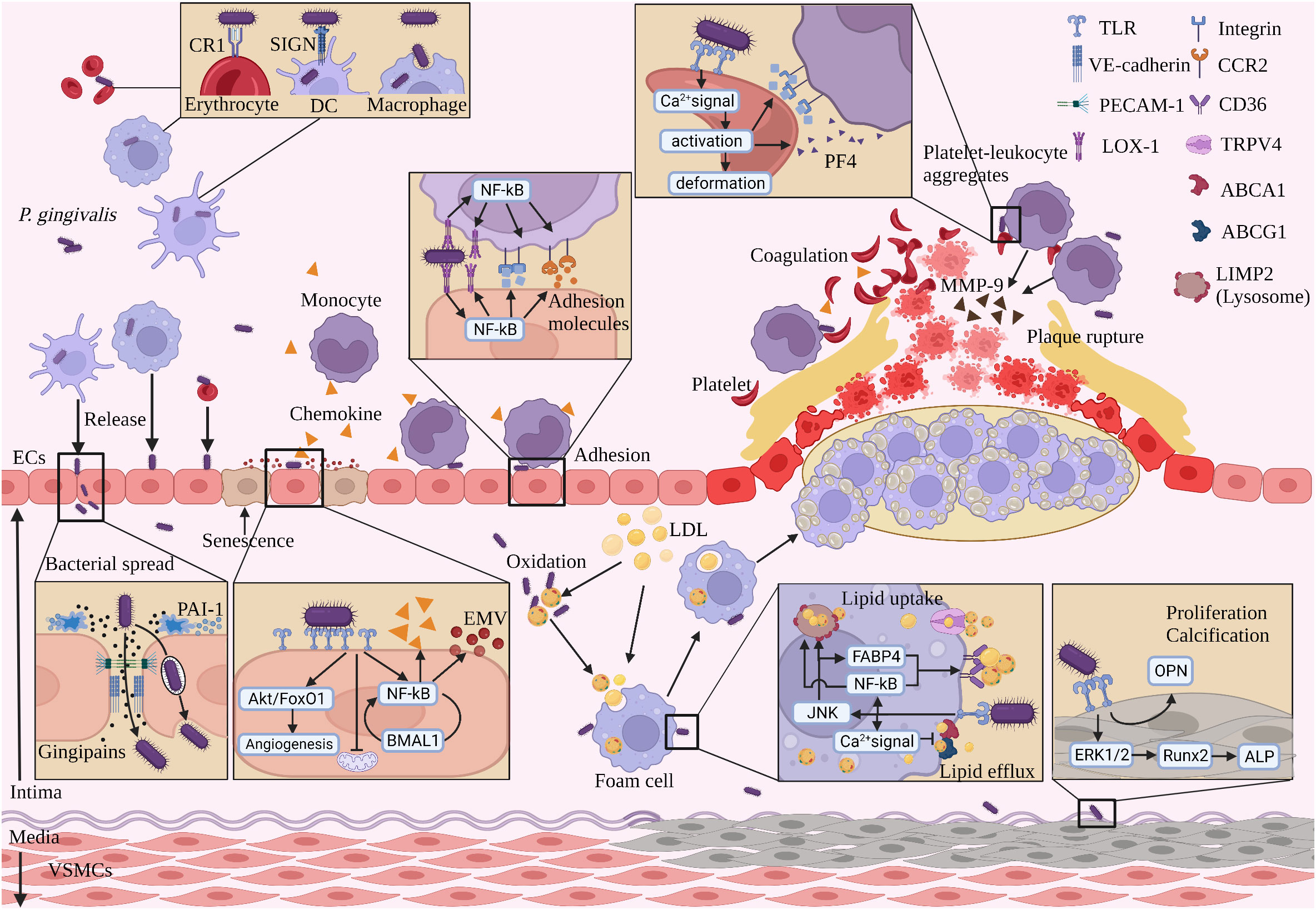
Figure 2 The process of P. gingivalis promoting AS with blood circulation. (1) P. gingivalis spreads to the vascular endothelium mainly in four forms with blood circulation. (2) P. gingivalis spreads by secreting gingipains breaking intercellular junction protein, being captured and released by ECs. (3) P. gingivalis stimulates ECs to produce chemokines and EMVpg, which recruit monocytes and induce senescence in adjacent ECs, respectively. (4) P. gingivalis mediates monocyte adhesion to the vascular endothelium. (5) P. gingivalis promotes lipid uptake and inhibits lipid efflux from subendothelial macrophages. (6) P. gingivalis activates platelets, promotes coagulation, and induces adhesion with leukocytes to form platelet-leukocyte aggregates. (7) P. gingivalis induces proliferation and calcification of VSMCs. (8) P. gingivalis induces monocytes to secrete MMP-9, promoting AS plaque rupture. (Created with BioRender.com).
4.2.1 Pathways of P. gingivalis into the blood circulation
The pathways of P. gingivalis into the blood circulation include three main parts, which are oral physical injury, disruption of interepithelial cell junctions by virulence factors, and immune escape.
Oral mucosal damage caused by physical stimuli such as daily activities (brushing, flossing) and oral treatments (periodontal therapy, tooth extraction) can lead to bacteremia (174). Inflammatory periodontal tissue swelling and bleeding states may also lead to the infiltration of oral bacteria into the bloodstream (175). P. gingivalis’ various virulence factors play an important role in disrupting gingival epithelial junctional structures. P. gingivalis gingipains degrade E-cadherin, JAM1, and ocludin, which maintain gingival epithelial integrity and barrier function (68, 176, 177). P. gingivalis LPS reduces E-cadherin expression in the gingival epithelium by inducing the production of TNF-α and ROS production reduces E-cadherin expression in the gingival epithelium (178). P. gingivalis and its LPS may lead to a dramatic decrease in the expression of endogenous grainyhead-like 2 (GRHL2), an epithelial-specific transcription factor that regulates the expression of connexins (179). The P. gingivalis that enters the bloodstream also needs to evade the killing effect of various host immune cells and immune responses before it can successfully survive and multiply in the host, triggering a persistent host inflammatory response.
Systemic dissemination can occur after P. gingivalis has successfully entered the bloodstream and evaded host immune killing. There are four ways for P. gingivalis to disseminate with the blood circulation: first, it is planktonic; second, it binds to the CR1 immune adhesion receptor on the erythrocyte membrane (180); third, it binds to the SIGN receptor on DCs and enters the DCs and survives intracellularly (181); fourth, it survives in macrophages and can enter and exit the cells repeatedly (143). P. gingivalis in the oral cavity can follow the above four ways to reach the AS lesion site (Figure 2).
4.2.2 P. gingivalis induces vascular endothelial dysfunction
P. gingivalis-induced dysfunction of vascular ECs is the first step in promoting AS (182, 183). Activation and dysfunction of ECs occur in AS-prone areas when stimulated by abnormal lipids, pro-inflammatory mediators, promoting secretion of pro-adhesive cytokines (184), which in turn leads to AS. Endothelial dysfunction includes changes in tissue function of contraction, spreading, barrier (185) and phenotypic changes in ECs (184). P. gingivalis and its virulence factors induce endothelial dysfunction in three main ways (Figure 2).
4.2.2.1 P. gingivalis directly disrupts the connecting proteins and barrier function between ECs
P. gingivalis gingipains directly damage the vascular endothelium by degrading the endothelial adhesion molecules PECAM-1 and VE-cadherin (186, 187). Because PECAM-1 and VE-cadherin are essential for maintaining endothelial integrity and continuity, degradation of these proteins would lead to loss of endothelial integrity, increase permeability, and increase risk of direct contact of irritant molecules with deeper tissues of the vessel wall, triggering vascular inflammation (188, 189). P. gingivalis gingipains also hydrolyze Plasminogen Activator Inhibitor-1(PAI-1) produced by ECs, which in turn delays vascular endothelial wound healing (190). In the AS study, PAI-1 promoted the expression of Vitronectin (VN) in VSMCs by binding to LDL receptor-related protein 1(LRP1) (191), which in turn induced ECs migration and regulated vascular remodeling and healing (192, 193). Then, whether P. gingivalis inhibits vascular endothelial self-healing through hydrolysis of PAI-1 remains to be investigated.
4.2.2.2 P. gingivalis affects the biological function of ECs
P. gingivalis not only enters and survives in ECs mediated by ICAM-1 but also releases from ECs and infects neighboring cells (194, 195). Repeated stimulation with P. gingivalis increases the expression of pro-inflammatory molecules (IL-6, MCP-1, GM-CSF) and vasoconstrictor molecules (Ang II) in ECs, increases monocyte adhesion to the endothelium and inhibits endothelial diastolic function (67). Similarly, a systemic Th1-type immune response occurred in mice immunized with P. gingivalis antigen, increasing the sensitivity of ECs to AngII and exacerbating the inhibition of endothelial diastolic function (196). However, the immunization method using intraperitoneal injection of P. gingivalis in this study failed to realistically mimic the natural infection state, and the mechanism of action of the Th1-type immune response and vascular endothelial sensitivity has not been fully clarified. P. gingivalis reduces NO production and inhibits endothelial diastolic function by inhibiting activation of the GSK-3β/BH4/eNOS/Nrf2 pathway in ECs (197). In addition, P. gingivalis-induced ERS occurring in ECs could both promote apoptosis and also induce an autophagic response to inhibit apoptosis (198, 199), and this paradoxical phenomenon may be related to the potential mechanism of P. gingivalis-promoting AS, but the further in-depth study is still needed.
P. gingivalis regulates the cytosolic molecules and intracellular pathways of ECs. In terms of cytosolic molecules, P. gingivalis GroEL upregulates TLR-4 expression on the cytosolic membrane of ECs, leading to hypersensitivity of ECs to P. gingivalis GroEL (50), promoting the expression of adhesion molecules (ICAM-1, VCAM-1), inducing monocyte adhesion and infiltration, and promoting AS progression (200). However, it has also been shown that TLR4 plays a protective role in AS progression (201), but the actual role of TLR4 still needs to be elucidated in the highly variable and dynamic inflammatory environment induced by P. gingivalis. Deeper studies revealed that P. gingivalis-induced inflammatory responses almost disappeared when targeted to inhibit the adapter MAL or TRAM of the Toll/IL-1 receptor (TIR) domain on ECs (202). In terms of intracellular pathways, P. gingivalis inhibits ECs proliferation, and promotes endothelial-mesenchymal transitions and apoptosis in a TLR-NF-κB axis dependent manner, compromising endothelial integrity and leading to the loss of ECs’ ability to repair themselves (203). Endothelial-mesenchymal transition means that activated ECs can be transformed into ectopic cell types, such as ECs into fibroblasts and calcified cells, promoting AS progression (204, 205). P. gingivalis LPS induces the secretion of multiple pro-inflammatory factors by macrophages in the vessel wall. These pro-inflammatory factors promote endothelial-mesenchymal transition in ECs by activating the p38-Erk1/2-p65 signaling pathway in ECs (206). In addition, P. gingivalis induces persistent oxidative stress and inflammatory responses in ECs through the NF-kB-BMAL1-NF-kB signaling loop with positive feedback (207). P. gingivalis LPS promotes the angiogenic function of endothelial progenitor cells through the Akt/FoxO1 signaling pathway (208). The enhanced angiogenic function of endothelial progenitor cells promotes AS plaque expansion and progression, as well as increases the incidence of subsequent complications such as bleeding, rupture, and thrombosis (209). Deeper studies have shown that P. gingivalis promotes mitochondrial mtDNA damage, increased mtROS production, and leads to endothelial dysfunction by inducing phosphorylation and translocation of mitochondrial dynamin-related protein (Drp1) in ECs (210).
The mortality and type of death of P. gingivalis-induced ECs were influenced by the lipid load and inflammatory status of ECs. mortality was higher in the P. gingivalis-induced ox-LDL pretreatment group than in the TNF-α pretreatment group, and apoptosis occurred mainly in the ox-LDL pretreatment group, whereas necrosis occurred mainly in the TNF-α pretreatment group. It suggests a synergistic relationship between P. gingivalis infection and AS risk factors (dyslipidemia, systemic inflammation), which together promote endothelial injury and accelerate AS progression (211). In addition, P. gingivalis stimulated ECs to produce microvesicular (EMVPg) shedding, while EMVPg may induce the conversion of adjacent endothelium to a senescent phenotype through JNK/AKT and STAT signaling pathways, promoting endothelial injury (212). It indicates that EMVPg has significant autocrine pro-inflammatory properties. However, current studies are very limited, and it is expected to be a new indicator of vascular inflammation.
4.2.2.3 P. gingivalis induces migration and adhesion of immune cells to the endothelium
P. gingivalis LPS promotes vascular inflammation and promotes AS progression through the expression of the chemokine RANTES in ECs (213, 214), which can induce leukocyte infiltration to sites of inflammation and is positively correlated with plaque instability (215, 216). P. gingivalis promotes the expression of lectin-like oxidized low-density lipoprotein on the cytosol of ECs and monocytes by promoting receptor-1 (LOX-1) expression, which in turn regulates ligand expression of MCP-1, ICAM-1 and E-selectin on ECs and receptor expression of CCR2 and integrin αMβ2 on monocytes, inducing monocyte migration and adhesion to ECs (217). Similarly, P. gingivalis and its outer membrane vesicles promote the expression of chemotactic proteins (CXCL1, CXCL2, and CXCL8) and adhesion molecules (e.g. E-selectin) in ECs (218). In addition, P. gingivalis promotes the secretion of macrophage migration inhibitory factor (MIF) by ECs, while MIF binds to the CD74/CXCR4 receptor complex on ECs, increases ICAM-1 expression, and promotes monocyte-ECs adhesion (219, 220). Immune cells migrate and adhere to the endothelium and then secrete multiple inflammatory factors, inducing endothelial dysfunction and promoting AS progression.
4.2.3 P. gingivalis induces pro-coagulant effects
When vascular ECs are activated or endothelial dysfunction is present, platelets adhere to the vessel wall and are activated, releasing large amounts of chemokines that mediate the recruitment of circulating leukocytes, platelets, and coagulation factors to the vascular endothelium, promoting local inflammation and coagulation and accelerating the progression of AS (221, 222).
P. gingivalis activates and aggregates platelets by increasing the intraplatelet Ca2+ concentration, accelerating blood clotting and thrombosis. P. gingivalis gingipains also have a hydrolytic effect on chemokines (RANTES, MIF) released after platelet activation, inhibiting the recruitment of immune cells, preventing the clearance of P. gingivalis by immune cells, and producing persistent stimulation (223). Platelets activated by P. gingivalis express P-selectin on their surface, which binds to the receptor P-selectin glycoprotein-1 on leukocytes, forming platelet-leukocyte aggregates that not only converge a variety of leukocytes (neutrophils, monocytes) to the site of inflammation and clear bacteria but also induce a coagulation-inflammatory series of responses (224). Further studies have indicated that P. gingivalis induces increased platelet-neutrophil aggregate formation, enhances platelet-neutrophil interactions, and induces the release of NETs, which in turn promotes late thrombosis in AS (225, 226). However, no studies on the effects of P. gingivalis on other types of leukocyte-platelet interactions have been seen.
In addition, P. gingivalis virulence factors promote coagulation. P. gingivalis LPS activates platelet GTPase Cdc42 and accelerates actin assembly, which in turn induces platelet shape change and proliferation and promotes coagulation (78). P. gingivalis LPS promotes platelet secretion of platelet factor 4 (PF4), which is known to recruit immune cells and promote AS (221, 227). P. gingivalis gingipains promote P-selectin expression in platelets, which in turn promotes the adhesion of leukocytes and platelets to the endothelium, thereby promoting AS progression (228) (Figure 2).
4.2.4 P. gingivalis induces foam cell formation
Macrophages are capable of uptake and clearance of modified lipoproteins. P. gingivalis increases intracellular lipid accumulation by interfering with lipid metabolic processes in macrophages and promotes the conversion of macrophages into “foam cells” (229, 230). The formation and accumulation of subendothelial foam cells is a key process in the formation of AS (142). The mechanism of P. gingivalis-induced foam cell formation is divided into three main parts (Figure 2):
4.2.4.1 P. gingivalis induces lipid modification or peroxidation
P. gingivalis gingipains induce lipid modification and peroxidation of LDL/VLDL through hydrolysis of ApoE and ApoB-100 and enhancement of oxidative stress pathways (231, 232), while LDL receptors in the liver do not recognize modified LDL/VLDL, resulting in circulating LDL/VLDL is not cleared and continues to accumulate, promoting elevated lipids (233). Similarly, Pep19, a peptide derived from P. gingivalis GroEL, can also induce LDL peroxidation (234). P. gingivalis can also induce oxidation of HDL, where the oxidized HDL not only loses its protective function against AS but also promotes the release of TNF-α and MMP-9 from monocytes, triggering a pro-inflammatory response (235). An increase in circulating modified or peroxidized lipids is an important prerequisite for triggering foam cells, and macrophage cells initiate repair mechanisms to increase the uptake of lipids (142).
4.2.4.2 P. gingivalis promotes lipid uptake by macrophages
Macrophage uptake of lipids is positively correlated with the number and function of scavenger receptors (CD36/SR-B2) (236). P. gingivalis promotes lipid uptake through the trans-activation of the CD36 promoter via the ERK/NF-κB pathway, which in turn upregulates CD36 expression in macrophages (237). In addition, P. gingivalis released large amounts of IL-1β in the form of activated CD36/SR-B2-TLR2, which in turn promoted lipid uptake by macrophages (238). Interestingly, the study also showed that IL-1β produced by P. gingivalis-activated CD36/SR-B2-TLR2 promoted cell pyroptosis, while ox-LDL inhibited IL-1β production and prevented cell pyroptosis in a CD36/SR-B2-dependent manner. Thus, macrophages in the vessel wall were stimulated by multiple stimuli of P. gingivalis LPS, ox-LDL, and IL-1β to increase lipid uptake and promote foam cell formation, but at the same time foam cells in the vessel wall were allowed to persist because pyroptosis was inhibited (238). This study provides a comprehensive analysis of the complex mechanisms underlying foam cell formation and survival in the vessel wall, suggesting that CD36/SR-B2 plays a diverse role in P. gingivalis-mediated AS and may be one of the important targets for regulating AS.
It was also found that the cytosolic mechanosensitive channel TRPV4 plays a key role in mediating P. gingivalis-promoted lipid uptake by macrophages. Under the stimulation of P. gingivalis LPS, the activity of the mechanosensitive channel TRPV4 in macrophages was significantly increased, the Ca2+ internal channel was activated, and the uptake of ox-LDL was increased. And this process was independent of the CD36 expression level (239). It is suggested that TRPV4 is another important uptake pathway independent of the scavenger receptor.
In addition, P. gingivalis can also affect the expression and function of fatty acid binding proteins in macrophages. It was shown that P. gingivalis upregulates the expression of fatty acid binding protein 4 (FABP4) through activation of the JNK pathway and forms a positive feedback loop that promotes lipid uptake and increases intracellular lipid accumulation by macrophages (240, 241).
4.2.4.3 P. gingivalis inhibits lipid efflux from macrophages
P. gingivalis LPS promotes lipid accumulation in macrophages by activating the JNK-c-Jun/AP-1 pathway to up-regulate CD36 (lipid uptake) expression, while down-regulating ATP-binding cassette transporterA1 (lipid efflux) expression by enhancing calpain activity (230). Secondly, P. gingivalis may inhibit cholesterol efflux by activating NF-κB and JNK signaling pathways, which in turn upregulates lysosomal integral membrane protein 2 (LIMP2) expression levels in macrophages (242). In addition, P. gingivalis inhibited the activity of cholesterol efflux-related enzymes (ABCG1 and CYP46A1) and promoted lipid accumulation in macrophages by enhancing Ca2+ signaling and promoting ROS production (243). Current studies on the inhibition of lipid efflux from macrophages by P. gingivalis are relatively limited. It mainly involves the functions of membrane transport proteins and receptors, and related enzymes, and its effects are attributed to the triggering and persistence of inflammatory signaling pathways.
4.2.5 P. gingivalis induces vascular calcification
Vascular calcification is the pathological deposition of hydroxyapatite minerals in the vascular system in the vessel wall, which in turn promotes AS progression. VSMCs are the key cell type involved in vascular calcification and exhibit phenotypic conversion. Various immune cells infiltrate the lesion in the early stages of AS formation, producing pro-inflammatory factors and regulatory molecules that induce the migration of contractile phenotype VSMCs, originally located in the interstitial layer, to the intimal layer and convert to a synthetic phenotype that contributes to the deposition of hydroxyapatite minerals in the vessel wall (244). VSMCs of the synthetic phenotype in the intimal layer are the first cells to appear at the site of impending AS lesions (245).
P. gingivalis promotes calcium deposition in VSMCs and promotes the transdifferentiation of VSMCs to osteoblast-like cells (246). It is hypothesized that various inflammatory cytokines (TNF-α, IL-β) induced by P. gingivalis promote vascular calcification by upregulating the expression of osteogenic-related genes (ALP, RUNX2) in VSMCs (247, 248), but further validation is needed. In addition, different virulence factors of P. gingivalis also play an important role in inducing vascular calcification. P. gingivalis LPS stimulates VSMCs proliferation and calcification, leading to vascular calcification (249). P. gingivalis gingipains induce VSMCs proliferation and conversion to synthetic type, increase bone bridge protein (OPN) expression, and promote vascular calcification (250). P. gingivalis OMVs promote vascular calcification by activating the ERK1/2- Runx2 signaling pathway to promote calcification in VSMCs (251).
In recent years, some studies using complex cell models to simulate the complex environment in vivo have also emerged. P. gingivalis LPS promoted calcification in VSMCs co-cultured with human periodontal ligament cells. This study indicates that the calcification effect is not only derived from the stimulation of P. gingivalis LPS, but also the secretion of various pro-inflammatory factors by P. gingivalis LPS-stimulated human periodontal ligament cells (252). This study somewhat mimics the in vivo environment in which periodontitis and cardiovascular disease coexist. There is also one that simulates the relationship between P. gingivalis and the calcification of VSMCs under hyperglycemic conditions. Under hyperglycemic conditions, P. gingivalis enhanced smad1/5/8-runx2 signaling by activating TLR-4 and ERK1/2-p38 signaling and promoting bone morphogenetic protein 4 (BMP4) autocrine regulation, which in turn induced calcification in VSMCs (253) (Figure 2).
4.2.6 P. gingivalis promotes plaque rupture
When AS is advanced, immune cells recruited by P. gingivalis infiltrate the vessel wall and secrete MMPs, such as MMP-1 and MMP-9, which degrade the collagen fibers of the plaque fibrous cap, leading to plaque rupture and bleeding (254).
P. gingivalis Mfa-1(short or minor fimbriae) induces differentiation of blood monocytes into DCs and promotes the expression of MMP-9 which in turn increases the risk of plaque rupture (255). Similarly, P. gingivalis LPS promoted MMP-9 expression and activity in monocytes (256). In addition, P. gingivalis gingipains hydrolyze the complement component C5, leading to local accumulation of C5a accumulation (114, 257). C5a is the only biologically active fragment following the action of P. gingivalis gingipains that induces increased expression of MMP1 and MMP9 in macrophages in plaques (114, 118), leading to degradation of the extracellular matrix and rupture of plaques (123). Studies on the promotion of AS plaque rupture by P. gingivalis are scarce and remain to be investigated (Figure 2).
4.3 Lymphatic circulation
The initial lymphatic vessels, the beginning segment of the lymphatic tract, lacking continuous basement membrane and perivascular wall cells, may serve as an ideal pathway for pathogen transmission through the lymphatic circulation (258–260). P. gingivalis may enter the submandibular and submental lymph nodes through the initial lymphatic vessels, and they may subsequently drain down the superficial/deep jugular lymph nodes to the jugular venous trunk for further metastasis to more distant organs, such as AS plaques, liver, etc (261). P. gingivalis that circulates with lymph may accelerate AS progression by inhibiting T cell and B cell activation and cholesterol reversal of transport function and promoting Th17/Treg imbalance in the spleen.
4.3.1 P. gingivalis inhibits the immune function of the lymphatic system
DCs, as the cells with the highest antigen-presenting capacity, are important for the activation of T and B cells in the lymphatic system. P. gingivalis gingipains hydrolyze the chemokine CCL21 in periodontal tissue and inhibit the entry of DCs into the initial lymphatic vessels (262, 263), which in turn reduces the activation of T and B cells in the lymphatic system by DCs (264). At the same time, P. gingivalis enters and survives in DCs by binding to SIGN receptors on DCs through its fimbriae, which helps P. gingivalis evade immune killing by the lymphatic system and promotes AS progression (255). P. gingivalis that survives in DCs does not affect CCR7 expression on the cytosol, but significantly upregulates CXCR4 expression (265). CCR7 mediates the homing of DCs to secondary lymphoid organs, whereas CXCR4 mediates the migration of DCs to sites of active vascular remodeling, such as AS (266). Thus, P. gingivalis may “hijack” DCs and enrich intracellular P. gingivalis-containing DCs in inflammatory vascular sites.
E. coli LPS can impair the recruitment of immune cells (DCs, macrophages) associated with lymphatic vessels and reduce the contractility of lymphatic vessels, affecting the function of the lymphatic pump (267). However, whether P. gingivalis LPS has similar functions has not been reported and needs to be urgently investigated.
4.3.2 P. gingivalis attenuates the cholesterol reversal function of lymphatic vessels
Lymphatic vessels can efficiently reverse cholesterol transport from multiple tissues (268), including the arterial wall, a process that is positively dependent on HDL uptake and transport by scavenger receptor class B type I (SR-BI) on lymphatic ECs (269). The density of capillary lymphatic vessels present in AS lesions increase with plaque progression (270), and proliferating capillary lymphatic vessels may inhibit AS progression by reversing cholesterol transport (271). P. gingivalis may generate large amounts of ROS by interfering with the function of the mitochondrial respiratory chain, leading to endoplasmic reticulum stress (ERS). Subsequently, it triggers an unfolded protein response (UPR), which promotes CHOP gene expression and suppresses SR-BI expression (272–274). This in turn inhibits the process of cholesterol reversal in the arterial wall and accelerates the progression of AS. However, there is a lack of direct studies on the inhibition of SR-BI expression on lymphoid ECs by P. gingivalis.
SR-BI is present in the liver, macrophages, and ECs. In addition to its function of uptake and transport of HDL, SR-BI also has an induced anti-inflammatory effect (275). For example, SR-BI binding to HDL inhibits the NF-κB pathway, decreases the inflammatory response of macrophages to LPS, and significantly reduces the secretion of various pro-inflammatory factors (276). However, the role of SR-BI in P. gingivalis promoting AS and its mechanism need further study.
4.3.3 P. gingivalis induces Th17/Treg imbalance in the spleen
The spleen is a peripheral lymphoid organ dominated by T cells and B cells, and is the center of cellular and humoral immunity in the body. (277). Among them, Th17 promotes AS progression, while Treg inhibits AS progression. Therefore, Th17/Treg balance can control inflammation and may play an important role in plaque stability (278).
The DNA genome of P. gingivalis can be found in the spleen of Apoeshl mice orally infected with P. gingivalis (47), and P. gingivalis promotes Th17 proliferation and differentiation by upregulating the expression of IL-6 in DCs in the spleen, leading to Th17/Treg imbalance, increasing plaque area and decreasing plaque stability (170). Intravenous administration of P. gingivalis to ApoE-/- mice showed a significant increase in Th17 and Th17-related molecules in the spleen and heart, as well as a significant increase in AS plaque area (279). In addition, P. gingivalis LPS did not induce the expression of NK cell CD69 in the spleen (280), which in turn promoted Th17 differentiation and inhibited Treg differentiation, leading to Th17/Treg imbalance (281), but the mechanism needs to be further investigated.
5 Conclusion and outlook
In recent years, the relationship between P. gingivalis and AS has received increasing attention. P. gingivalis can occur immune escape, promote local inflammation and plaque formation in blood vessels, inhibit immune defense and cholesterol transport function of the lymphatic system, and promote the progression of AS. This paper summarizes the recent research results related to the promotion of AS by P. gingivalis through immune response and provides new insights to further reveal the potential mechanisms and associations between P. gingivalis and AS.
In addition, this paper shows the different cell subpopulations associated with AS discovered by single-cell sequencing technology in recent years, which updates the understanding of the nature and function of cell subpopulations in AS. It provides the conditions to precisely grasp the cellular level of AS pathogenesis. However, unfortunately, single-cell sequencing studies on P. gingivalis and AS have not been reported yet. It is believed that shortly, the use of single-cell sequencing technology will certainly provide important theoretical support for the prevention and treatment of P. gingivalis to accelerate the progression of AS.
Author contributions
QR and PG wrote the manuscript. WQ, JL, MX, LX and SZ made the figures and edited the manuscript. DM, and JN administrated and supervised the whole research project. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No. 81970930), Chinese Medicine Research Projects in Guangdong Province (No.20222144), Hygiene and Health Appropriate Technology Promotion Project in Guangdong Province (No. 202106241649075233), The Research Incubation Program of Southern Medical University Dental Hospital (No. PY2021015), Postdoctoral Research Fund of Southern Medical University Dental Hospital (No. PY2018031).
Acknowledgments
Figures were created with BioRender software (https://biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Reyes L. Porphyromonas gingivalis. Trends Microbiol (2021) 29(4):376–7. doi: 10.1016/j.tim.2021.01.010
2. Olsen I, Lambris JD, Hajishengallis G. Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol (2017) 9(1):1340085. doi: 10.1080/20002297.2017.1340085
3. Chopra A, Bhat SG, Sivaraman K. Porphyromonas gingivalis adopts intricate and unique molecular mechanisms to survive and persist within the host: a critical update. J Oral Microbiol (2020) 12(1):1801090. doi: 10.1080/20002297.2020.1801090
4. Jia L, Han N, Du J, Guo L, Luo Z, Liu Y. Pathogenesis of important virulence factors of porphyromonas gingivalis via toll-like receptors. Front Cell Infect Microbiol (2019) 9:262. doi: 10.3389/fcimb.2019.00262
5. Olsen I, Singhrao SK. Importance of heterogeneity in porhyromonas gingivalis lipopolysaccharide lipid a in tissue specific inflammatory signalling. J Oral Microbiol (2018) 10(1):1440128. doi: 10.1080/20002297.2018.1440128
6. Lunar Silva I, Cascales E. Molecular strategies underlying porphyromonas gingivalis virulence. J Mol Biol (2021) 433(7):166836. doi: 10.1016/j.jmb.2021.166836
7. Xie H. Biogenesis and function of porphyromonas gingivalis outer membrane vesicles. Future Microbiol (2015) 10(9):1517–27. doi: 10.2217/fmb.15.63
8. Carter CJ, France J, Crean S, Singhrao SK. The porphyromonas gingivalis/Host interactome shows enrichment in GWASdb genes related to alzheimer's disease, diabetes and cardiovascular diseases. Front Aging Neurosci (2017) 9:408. doi: 10.3389/fnagi.2017.00408
9. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res (2018) 122(12):1661–74. doi: 10.1161/CIRCRESAHA.117.312509
10. Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol (2020) 250(5):705–14. doi: 10.1002/path.5392
11. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res (2018) 122(12):1675–88. doi: 10.1161/CIRCRESAHA.117.312513
12. Winkels H, Wolf D. Heterogeneity of T cells in atherosclerosis defined by single-cell RNA-sequencing and cytometry by time of flight. Arterioscler Thromb Vasc Biol (2021) 41(2):549–63. doi: 10.1161/ATVBAHA.120.312137
13. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation (2020) 142(21):2060–75. doi: 10.1161/CIRCULATIONAHA.120.048378
14. Alsaigh T, Evans D, Frankel D, Torkamani A. Decoding the transcriptome of calcified atherosclerotic plaque at single-cell resolution. Commun Biol (2022) 5(1):8–9. doi: 10.1038/s42003-022-04056-7
15. Xu J, Zhou H, Cheng Y, Xiang G. Identifying potential signatures for atherosclerosis in the context of predictive, preventive, and personalized medicine using integrative bioinformatics approaches and machine-learning strategies. EPMA J (2022) 13(3):433–49. doi: 10.1007/s13167-022-00289-y
16. Qin W, Gan F, Liang R, Li J, Lai X, Dai Y, et al. Identification of monocyte-associated genes related to the instability of atherosclerosis plaque. Oxid Med Cell Longev (2022) 2022:3972272. doi: 10.1155/2022/3972272
17. Czerniuk MR, Surma S, Romanczyk M, Nowak JM, Wojtowicz A, Filipiak KJ. Unexpected relationships: Periodontal diseases: Atherosclerosis-plaque destabilization? from the teeth to a coronary event. Biol (Basel) (2022) 11(2):4–7. doi: 10.3390/biology11020272
18. Zhang J, Xie M, Huang X, Chen G, Yin Y, Lu X, et al. The effects of porphyromonas gingivalis on atherosclerosis-related cells. Front Immunol (2021) 12:766560. doi: 10.3389/fimmu.2021.766560
19. Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, et al. Periodontal disease and atherosclerotic vascular disease: Does the evidence support an independent association?: a scientific statement from the American heart association. Circulation (2012) 125(20):2520–44. doi: 10.1161/CIR.0b013e31825719f3
20. Chhibber-Goel J, Singhal V, Bhowmik D, Vivek R, Parakh N, Bhargava B, et al. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes (2016) 2:7. doi: 10.1038/s41522-016-0009-7
21. Pavlic V, Peric D, Kalezic IS, Madi M, Bhat SG, Brkic Z, et al. Identification of periopathogens in atheromatous plaques obtained from carotid and coronary arteries. BioMed Res Int (2021) 2021:9986375. doi: 10.1155/2021/9986375
22. Brun A, Nuzzo A, Prouvost B, Diallo D, Hamdan S, Meseguer E, et al. Oral microbiota and atherothrombotic carotid plaque vulnerability in periodontitis patients. A cross-sectional study. J Periodontal Res (2021) 56(2):339–50. doi: 10.1111/jre.12826
23. Mougeot JC, Stevens CB, Paster BJ, Brennan MT, Lockhart PB, Mougeot FK. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J Oral Microbiol (2017) 9(1):1281562. doi: 10.1080/20002297.2017.1281562
24. Kregielczak A, Dorocka-Bobkowska B, Slomski R, Oszkinis G, Krasinski Z. Periodontal status and the incidence of selected bacterial pathogens in periodontal pockets and vascular walls in patients with atherosclerosis and abdominal aortic aneurysms. PloS One (2022) 17(8):e0270177. doi: 10.1371/journal.pone.0270177
25. Kozarov E. Bacterial invasion of vascular cell types: Vascular infectology and atherogenesis. Future Cardiol (2012) 8(1):123–38. doi: 10.2217/fca.11.75
26. Papapanou PN, Neiderud AM, Disick E, Lalla E, Miller GC, Dahlen G. Longitudinal stability of serum immunoglobulin G responses to periodontal bacteria. J Clin Periodontol (2004) 31(11):985–90. doi: 10.1111/j.1600-051X.2004.00599.x
27. Ramirez JH, Parra B, Gutierrez S, Arce RM, Jaramillo A, Ariza Y, et al. Biomarkers of cardiovascular disease are increased in untreated chronic periodontitis: a case control study. Aust Dent J (2014) 59(1):29–36. doi: 10.1111/adj.12139
28. Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N Engl J Med (2007) 356(9):911–20. doi: 10.1056/NEJMoa063186
29. Saffi MAL, Rabelo-Silva ER, Polanczyk CA, Furtado MV, Montenegro MM, Ribeiro IWJ, et al. Periodontal therapy and endothelial function in coronary artery disease: A randomized controlled trial. Oral Dis (2018) 24(7):1349–57. doi: 10.1111/odi.12909
30. Park SY, Kim SH, Kang SH, Yoon CH, Lee HJ, Yun PY, et al. Improved oral hygiene care attenuates the cardiovascular risk of oral health disease: A population-based study from Korea. Eur Heart J (2019) 40(14):1138–45. doi: 10.1093/eurheartj/ehy836
31. Sen S, Giamberardino LD, Moss K, Morelli T, Rosamond WD, Gottesman RF, et al. Periodontal disease, regular dental care use, and incident ischemic stroke. Stroke (2018) 49(2):355–62. doi: 10.1161/STROKEAHA.117.018990
32. Holmlund A, Lampa E, Lind L. Poor response to periodontal treatment may predict future cardiovascular disease. J Dent Res (2017) 96(7):768–73. doi: 10.1177/0022034517701901
33. Lakio L, Antinheimo J, Paju S, Buhlin K, Pussinen PJ, Alfthan G. Tracking of plasma antibodies against aggregatibacter actinomycetemcomitans and porphyromonas gingivalis during 15 years. J Oral Microbiol (2009) 1:6–7. doi: 10.3402/jom.v1i0.1979
34. Yang J, Wu J, Liu Y, Huang J, Lu Z, Xie L, et al. Porphyromonas gingivalis infection reduces regulatory T cells in infected atherosclerosis patients. PLoS One (2014) 9(1):e86599. doi: 10.1371/journal.pone.0086599
35. Joshi C, Bapat R, Anderson W, Dawson D, Cherukara G, Hijazi K. Serum antibody response against periodontal bacteria and coronary heart disease: Systematic review and meta-analysis. J Clin Periodontol (2021) 48(12):1570–86. doi: 10.1111/jcpe.13550
36. Liljestrand JM, Paju S, Pietiainen M, Buhlin K, Persson GR, Nieminen MS, et al. Immunologic burden links periodontitis to acute coronary syndrome. Atherosclerosis (2018) 268:177–84. doi: 10.1016/j.atherosclerosis.2017.12.007
37. Teeuw WJ, Slot DE, Susanto H, Gerdes VE, Abbas F, D'Aiuto F, et al. Treatment of periodontitis improves the atherosclerotic profile: A systematic review and meta-analysis. J Clin Periodontol (2014) 41(1):70–9. doi: 10.1111/jcpe.12171
38. Li X, Tse HF, Yiu KH, Zhang C, Jin LJ. Periodontal therapy decreases serum levels of adipocyte fatty acid-binding protein in systemically healthy subjects: A pilot clinical trial. J Periodontal Res (2013) 48(3):308–14. doi: 10.1111/jre.12009
39. Fu YW, Li XX, Xu HZ, Gong YQ, Yang Y. Effects of periodontal therapy on serum lipid profile and proinflammatory cytokines in patients with hyperlipidemia: A randomized controlled trial. Clin Oral Investig (2016) 20(6):1263–9. doi: 10.1007/s00784-015-1621-2
40. D'Aiuto F, Gkranias N, Bhowruth D, Khan T, Orlandi M, Suvan J, et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: A 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol (2018) 6(12):954–65. doi: 10.1016/S2213-8587(18)30038-X
41. Aarabi G, Eberhard J, Reissmann DR, Heydecke G, Seedorf U. Interaction between periodontal disease and atherosclerotic vascular disease–fact or fiction? Atherosclerosis (2015) 241(2):555–60. doi: 10.1016/j.atherosclerosis.2015.04.819
42. Miyauchi S, Maekawa T, Aoki Y, Miyazawa H, Tabeta K, Nakajima T, et al. Oral infection with porphyromonas gingivalis and systemic cytokine profile in C57BL/6. KOR-ApoE shl mice J Periodontal Res (2012) 47(3):402–8. doi: 10.1111/j.1600-0765.2011.01441.x
43. Pereira RB, Vasquez EC, Stefanon I, Meyrelles SS. Oral p. gingivalis infection alters the vascular reactivity in healthy and spontaneously atherosclerotic mice. Lipids Health Dis (2011) 10:80. doi: 10.1186/1476-511X-10-80
44. Parvaneh M, Witting PK, Ku J, Moradi T, Eroglu E, Freedman B, et al. Periodontitis induces endothelial dysfunction in mice. Sci Rep (2021) 11(1):14993. doi: 10.1038/s41598-021-94418-8
45. Velsko IM, Chukkapalli SS, Rivera MF, Lee JY, Chen H, Zheng D, et al. Active invasion of oral and aortic tissues by porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS One (2014) 9(5):e97811. doi: 10.1371/journal.pone.0097811
46. Chukkapalli SS, Velsko IM, Rivera-Kweh MF, Zheng D, Lucas AR, Kesavalu L. Polymicrobial oral infection with four periodontal bacteria orchestrates a distinct inflammatory response and atherosclerosis in ApoE null mice. PLoS One (2015) 10(11):e0143291. doi: 10.1371/journal.pone.0143291
47. Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Activation of the NLRP3 inflammasome in porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis (2015) 73(4):1–7. doi: 10.1093/femspd/ftv011
48. Hokamura K, Inaba H, Nakano K, Nomura R, Yoshioka H, Taniguchi K, et al. Molecular analysis of aortic intimal hyperplasia caused by porphyromonas gingivalis infection in mice with endothelial damage. J Periodontal Res (2010) 45(3):337–44. doi: 10.1111/j.1600-0765.2009.01242.x
49. Shiheido Y, Maejima Y, Suzuki JI, Aoyama N, Kaneko M, Watanabe R, et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J Mol Cell Cardiol (2016) 99:123–37. doi: 10.1016/j.yjmcc.2016.03.017
50. Huang CY, Shih CM, Tsao NW, Lin YW, Shih CC, Chiang KH, et al. The GroEL protein of porphyromonas gingivalis regulates atherogenic phenomena in endothelial cells mediated by upregulating toll-like receptor 4 expression. Am J Transl Res (2016) 8(2):384–404.
51. Xuan Y, Shi Q, Liu GJ, Luan QX, Cai Y. Porphyromonas gingivalis Infection Accelerates Atherosclerosis Mediated by Oxidative Stress and Inflammatory Responses in ApoE-/- Mice. Clin Lab (2017) 63(10):1627–37. doi: 10.7754/Clin.Lab.2017.170410
52. Xuan Y, Cai Y, Wang XX, Shi Q, Qiu LX, Luan QX. [Effect of porphyromonas gingivalis infection on atherosclerosis in apolipoprotein-e knockout mice]. Beijing Da Xue Xue Bao Yi Xue Ban (2020) 52(4):743–9. doi: 10.19723/j.issn.1671-167X.2020.04.028
53. Hagiwara M, Kurita-Ochiai T, Kobayashi R, Hashizume-Takizawa T, Yamazaki K, Yamamoto M. Sublingual vaccine with GroEL attenuates atherosclerosis. J Dent Res (2014) 93(4):382–7. doi: 10.1177/0022034514523784
54. Kyrklund M, Kummu O, Kankaanpaa J, Akhi R, Nissinen A, Turunen SP, et al. Immunization with gingipain a hemagglutinin domain of porphyromonas gingivalis induces IgM antibodies binding to malondialdehyde-acetaldehyde modified low-density lipoprotein. PLoS One (2018) 13(1):e0191216. doi: 10.1371/journal.pone.0191216
55. Joo JY, Cha GS, Kim HJ, Lee JY, Choi J. Atheroprotective nasal immunization with a heat shock protein 60 peptide from porphyromonas gingivalis. J Periodontal Implant Sci (2020) 50(3):159–70. doi: 10.5051/jpis.2020.50.3.159
56. Ha HS, Kim TY, Han SJ, Sung HJ, Seo KY, Ha JW. Anti-atherosclerotic vaccination against porphyromonas gingivalis as a potential comparator of statin in mice. PeerJ (2021) 9:e11293. doi: 10.7717/peerj.11293
57. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral administration of porphyromonas gingivalis alters the gut microbiome and serum metabolome. mSphere (2018) 3(5):1–9. doi: 10.1128/mSphere.00460-18
58. Qian B, Zhang K, Li Y, Sun K. Update on gut microbiota in cardiovascular diseases. Front Cell Infect Microbiol (2022) 12:1059349. doi: 10.3389/fcimb.2022.1059349
59. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med (2013) 368(17):1575–84. doi: 10.1056/NEJMoa1109400
60. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell (2015) 163(7):1585–95. doi: 10.1016/j.cell.2015.11.055
61. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature (2011) 472(7341):57–63. doi: 10.1038/nature09922
62. Liu M, Han Q, Yang J. Trimethylamine-n-oxide (TMAO) increased aquaporin-2 expression in spontaneously hypertensive rats. Clin Exp Hypertens (2019) 41(4):312–22. doi: 10.1080/10641963.2018.1481420
63. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine n-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc (2016) 5(2):1. doi: 10.1161/JAHA.115.002767
64. Ke Y, Li D, Zhao M, Liu C, Liu J, Zeng A, et al. Gut flora-dependent metabolite trimethylamine-n-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med (2018) 116:88–100. doi: 10.1016/j.freeradbiomed.2018.01.007
65. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell (2016) 165(1):111–24. doi: 10.1016/j.cell.2016.02.011
66. Roth GA, Moser B, Roth-Walter F, Giacona MB, Harja E, Papapanou PN, et al. Infection with a periodontal pathogen increases mononuclear cell adhesion to human aortic endothelial cells. Atherosclerosis (2007) 190(2):271–81. doi: 10.1016/j.atherosclerosis.2006.03.018
67. Viafara-Garcia SM, Morantes SJ, Chacon-Quintero Y, Castillo DM, Lafaurie GI, Buitrago DM. Repeated porphyromonas gingivalis W83 exposure leads to release pro-inflammatory cytokynes and angiotensin II in coronary artery endothelial cells. Sci Rep (2019) 9(1):19379. doi: 10.1038/s41598-019-54259-y
68. Takeuchi H, Sasaki N, Yamaga S, Kuboniwa M, Matsusaki M, Amano A. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1. PLoS Pathog (2019) 15(11):e1008124. doi: 10.1371/journal.ppat.1008124
69. Wilensky A, Tzach-Nahman R, Potempa J, Shapira L, Nussbaum G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J Innate Immun (2015) 7(2):127–35. doi: 10.1159/000365970
70. Carlisle MD, Srikantha RN, Brogden KA. Degradation of human alpha- and beta-defensins by culture supernatants of porphyromonas gingivalis strain 381. J Innate Immun (2009) 1(2):118–22. doi: 10.1159/000181015
71. Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol (2000) 2000:24:153–92. doi: 10.1034/j.1600-0757.2000.2240108.x
72. Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. The C5a receptor impairs IL-12-dependent clearance of porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol (2011) 186(2):869–77. doi: 10.4049/jimmunol.1003252
73. Hajishengallis G, Lambris JD. Complement and dysbiosis in periodontal disease. Immunobiology (2012) 217(11):1111–6. doi: 10.1016/j.imbio.2012.07.007
74. Xu W, Zhou W, Wang H, Liang S. Roles of porphyromonas gingivalis and its virulence factors in periodontitis. Adv Protein Chem Struct Biol (2020) 120:45–84. doi: 10.1016/bs.apcsb.2019.12.001
75. Veloso P, Fernández A, Astorga J, González-Quintanilla D, Castro A, Escobar A, et al. Lipopolysaccharide from porphyromonas gingivalis, but not from porphyromonas endodontalis, induces macrophage M1 profile. Int J Mol Sci (2022) 23(17):1. doi: 10.3390/ijms231710011
76. Li YY, Cai Q, Li BS, Qiao SW, Jiang JY, Wang D, et al. The effect of porphyromonas gingivalis lipopolysaccharide on the pyroptosis of gingival fibroblasts. Inflammation (2021) 44(3):846–58. doi: 10.1007/s10753-020-01379-7
77. Lv YT, Zeng JJ, Lu JY, Zhang XY, Xu PP, Su Y. Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) influences adipocytes injuries through triggering XBP1 and activating mitochondria-mediated apoptosis. Adipocyte (2021) 10(1):28–37. doi: 10.1080/21623945.2020.1856527
78. Senini V, Amara U, Paul M, Kim H. Porphyromonas gingivalis lipopolysaccharide activates platelet Cdc42 and promotes platelet spreading and thrombosis. J Periodontol (2019) 90(11):1336–45. doi: 10.1002/JPER.18-0596
79. Wielento A, Bereta GP, Łagosz-Ćwik KB, Eick S, Lamont RJ, Grabiec AM, et al. TLR2 activation by porphyromonas gingivalis requires both PPAD activity and fimbriae. Front Immunol (2022) 13:823685. doi: 10.3389/fimmu.2022.823685
80. Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of porphyromonas gingivalis and negates its virulence in vivo. J Immunol (2007) 179(4):2359–67. doi: 10.4049/jimmunol.179.4.2359
81. Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A (2008) 105(36):13532–7. doi: 10.1073/pnas.0803852105
82. Hajishengallis G, McIntosh ML, Nishiyama SI, Yoshimura F. Mechanism and implications of CXCR4-mediated integrin activation by porphyromonas gingivalis. Mol Oral Microbiol (2013) 28(4):239–49. doi: 10.1111/omi.12021
83. Takahashi Y, Cueno ME, Kamio N, Iinuma T, Hasegawa Y, Imai K. Porphyromonas gingivalis Mfa1 fimbria putatively binds to TLR2 and induces both IL-6 and IL-8 production in human bronchial epithelial cells. Biochem Biophys Res Commun (2022) 589:35–40. doi: 10.1016/j.bbrc.2021.12.003
84. Takayanagi Y, Kikuchi T, Hasegawa Y, Naiki Y, Goto H, Okada K, et al. Porphyromonas gingivalis Mfa1 induces chemokine and cell adhesion molecules in mouse gingival fibroblasts via toll-like receptors. J Clin Med (2020) 9(12):1. doi: 10.3390/jcm9124004
85. El-Awady AR, Miles B, Scisci E, Kurago ZB, Palani CD, Arce RM, et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog (2015) 10(2):e1004647. doi: 10.1371/journal.ppat.1004647
86. Libby P. Inflammation in atherosclerosis. Nature (2002) 420(6917):868–74. doi: 10.1038/nature01323
87. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules (2018) 8(3):1. doi: 10.3390/biom8030080
88. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol (2015) 209(1):13–22. doi: 10.1083/jcb.201412052
89. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers (2019) 5(1):56. doi: 10.1038/s41572-019-0106-z
90. Swirski FK, Nahrendorf M, Libby P. Mechanisms of myeloid cell modulation of atherosclerosis. Microbiol Spectr (2016) 4(4):3. doi: 10.1128/microbiolspec.MCHD-0026-2015
91. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond) (2018) 132(12):1243–52. doi: 10.1042/CS20180306
92. Libby P. Inflammation in atherosclerosis-no longer a theory. Clin Chem (2021) 67(1):131–42. doi: 10.1093/clinchem/hvaa275
93. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol (2020) 15:493–518. doi: 10.1146/annurev-pathmechdis-012419-032847
94. Zhu X, Huang H, Zhao L. PAMPs and DAMPs as the bridge between periodontitis and atherosclerosis: The potential therapeutic targets. Front Cell Dev Biol (2022) 10:856118. doi: 10.3389/fcell.2022.856118
95. Nativel B, Couret D, Giraud P, Meilhac O, d'Hellencourt CL, Viranaïcken W, et al. Porphyromonas gingivalis lipopolysaccharides act exclusively through TLR4 with a resilience between mouse and human. Sci Rep (2017) 7(1):15789. doi: 10.1038/s41598-017-16190-y
96. Wang X, Liu Y, Zhang S, Ouyang X, Wang Y, Jiang Y, et al. Crosstalk between akt and NF-κB pathway mediates inhibitory effect of gas6 on monocytes-endothelial cells interactions stimulated by p. gingivalis-LPS J Cell Mol Med (2020) 24(14):7979–90. doi: 10.1111/jcmm.15430
97. Xie Y, Li Y, Cai X, Wang X, Li J. Interleukin-37 suppresses ICAM-1 expression in parallel with NF-κB down-regulation following TLR2 activation of human coronary artery endothelial cells. Int Immunopharmacol (2016) 38:26–30. doi: 10.1016/j.intimp.2016.05.003
98. González-Ramos S, Paz-García M, Rius C, Del Monte-Monge A, Rodríguez C, Fernández-García V, et al. Endothelial NOD1 directs myeloid cell recruitment in atherosclerosis through VCAM-1. FASEB J (2019) 33(3):3912–21. doi: 10.1096/fj.201801231RR
99. Jin Y, Huang H, Shu X, Liu Z, Lu L, Dai Y, et al. Peptidoglycan recognition protein 1 attenuates atherosclerosis by suppressing endothelial cell adhesion. J Cardiovasc Pharmacol (2021) 78(4):615–21. doi: 10.1097/FJC.0000000000001100
100. Nijhuis MM, van der Graaf Y, Melief MJ, Schoneveld AH, de Kleijn DP, Laman JD, et al. IgM antibody level against proinflammatory bacterial peptidoglycan is inversely correlated with extent of atherosclerotic disease. Atherosclerosis (2004) 173(2):245–51. doi: 10.1016/j.atherosclerosis.2003.12.005
101. Jun HK, Jung YJ, Choi BK. Treponema denticola, porphyromonas gingivalis, and tannerella forsythia induce cell death and release of endogenous danger signals. Arch Oral Biol (2017) 73:72–8. doi: 10.1016/j.archoralbio.2016.09.010
102. Krishnan-Sivadoss I, Mijares-Rojas IA, Villarreal-Leal RA, Torre-Amione G, Knowlton AA, Guerrero-Beltrán CE. Heat shock protein 60 and cardiovascular diseases: An intricate love-hate story. Med Res Rev (2021) 41(1):29–71. doi: 10.1002/med.21723
103. Rahman M, Steuer J, Gillgren P, Végvári Á, Liu A, Frostegård J. Malondialdehyde conjugated with albumin induces pro-inflammatory activation of T cells isolated from human atherosclerotic plaques both directly and Via dendritic cell-mediated mechanism. JACC Basic Transl Sci (2019) 4(4):480–94. doi: 10.1016/j.jacbts.2019.03.009
104. Almanzar G, Öllinger R, Leuenberger J, Onestingel E, Rantner B, Zehm S, et al. Autoreactive HSP60 epitope-specific T-cells in early human atherosclerotic lesions. J Autoimmun (2012) 39(4):441–50. doi: 10.1016/j.jaut.2012.07.006
105. de Graaf R, Kloppenburg G, Kitslaar PJ, Bruggeman CA, Stassen F. Human heat shock protein 60 stimulates vascular smooth muscle cell proliferation through toll-like receptors 2 and 4. Microbes Infect (2006) 8(7):1859–65. doi: 10.1016/j.micinf.2006.02.024
106. Schenkein HA, Papapanou PN, Genco R, Sanz M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontol 2000 (2020) 83(1):90–106. doi: 10.1111/prd.12304
107. Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal (2010) 12(8):1009–59. doi: 10.1089/ars.2009.2597
108. Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, et al. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by annexin A5. Atherosclerosis (2014) 235(2):592–8. doi: 10.1016/j.atherosclerosis.2014.05.913
109. Inaba H, Hokamura K, Nakano K, Nomura R, Katayama K, Nakajima A, et al. Upregulation of S100 calcium-binding protein A9 is required for induction of smooth muscle cell proliferation by a periodontal pathogen. FEBS Lett (2009) 583(1):128–34. doi: 10.1016/j.febslet.2008.11.036
110. Liu J, Wang Y, Liao Y, Zhou Y, Zhu J. Circular RNA PPP1CC promotes porphyromonas gingivalis-lipopolysaccharide-induced pyroptosis of vascular smooth muscle cells by activating the HMGB1/TLR9/AIM2 pathway. J Int Med Res (2021) 49(3):300060521996564. doi: 10.1177/0300060521996564
111. Zheng S, Yu S, Fan X, Zhang Y, Sun Y, Lin L, et al. Porphyromonas gingivalis survival skills: Immune evasion. J Periodontal Res (2021) 56(6):1007–18. doi: 10.1111/jre.12915
112. Torzewski M, Enzymatically modified LDL. Atherosclerosis and beyond: paving the way to acceptance. Front Biosci (Landmark Ed) (2018) 23(7):1257–71. doi: 10.2741/4642
113. Vlaicu SI, Tatomir A, Rus V, Mekala AP, Mircea PA, Niculescu F, et al. The role of complement activation in atherogenesis: The first 40 years. Immunol Res (2016) 64(1):1–13. doi: 10.1007/s12026-015-8669-6
114. Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from porphyromonas (Bacteroides) gingivalis. J Biol Chem (1992) 267(26):18902–7. doi: 10.1016/S0021-9258(19)37046-2
115. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
116. Hou Y, Yu H, Liu X, Li G, Pan J, Zheng C, et al. Gingipain of porphyromonas gingivalis manipulates M1 macrophage polarization through C5a pathway. In Vitro Cell Dev Biol - Animal (2017) 53(7):593–603. doi: 10.1007/s11626-017-0164-z
117. An G, Ren G, An F, Zhang C. Role of C5a-C5aR axis in the development of atherosclerosis. Sci China Life Sci (2014) 57(8):790–4. doi: 10.1007/s11427-014-4711-5
118. Speidl WS, Kastl SP, Hutter R, Katsaros KM, Kaun C, Bauriedel G, et al. The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB J (2011) 25(1):35–44. doi: 10.1096/fj.10-156083
119. Speidl WS, Exner M, Amighi J, Kastl SP, Zorn G, Maurer G, et al. Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur Heart J (2005) 26(21):2294–9. doi: 10.1093/eurheartj/ehi339
120. Wezel A, Quax PH, Kuiper J, Bot I. The role of mast cells in atherosclerosis. Hamostaseologie (2015) 35(2):113–20. doi: 10.5482/HAMO-14-08-0034
121. Wojta J, Huber K, Valent P. New aspects in thrombotic research: complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol Haemost Thromb (2003) 33(5-6):438–41. doi: 10.1159/000083842
122. Patel S, Thelander EM, Hernandez M, Montenegro J, Hassing H, Burton C, et al. ApoE(-/-) mice develop atherosclerosis in the absence of complement component C5. Biochem Biophys Res Commun (2001) 286(1):164–70. doi: 10.1006/bbrc.2001.5276
123. Wezel A, de Vries MR, Lagraauw HM, Foks AC, Kuiper J, Quax PH, et al. Complement factor C5a induces atherosclerotic plaque disruptions. J Cell Mol Med (2014) 18(10):2020–30. doi: 10.1111/jcmm.12357
124. Niyonzima N, Bakke SS, Gregersen I, Holm S, Sandanger O, Orrem HL, et al. Cholesterol crystals use complement to increase NLRP3 signaling pathways in coronary and carotid atherosclerosis. EBioMedicine (2020) 60:102985. doi: 10.1016/j.ebiom.2020.102985
125. An G, Li B, Liu X, Zhang M, Gao F, Zhao Y, et al. Overexpression of complement component C5a accelerates the development of atherosclerosis in ApoE-knockout mice. Oncotarget (2016) 7(35):56060–70. doi: 10.18632/oncotarget.11180
126. Manthey HD, Thomas AC, Shiels IA, Zernecke A, Woodruff TM, Rolfe B, et al. Complement C5a inhibition reduces atherosclerosis in ApoE-/- mice. FASEB J (2011) 25(7):2447–55. doi: 10.1096/fj.10-174284
127. Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal (2010) 3(109):ra11. doi: 10.1126/scisignal.2000697
128. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe (2014) 15(6):768–78. doi: 10.1016/j.chom.2014.05.012
129. Makkawi H, Hoch S, Burns E, Hosur K, Hajishengallis G, Kirschning CJ, et al. Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front Cell Infect Microbiol (2017) 7:359. doi: 10.3389/fcimb.2017.00359
130. Shaik-Dasthagirisaheb YB, Huang N, Baer MT, Gibson FC 3rd. Role of MyD88-dependent and MyD88-independent signaling in porphyromonas gingivalis-elicited macrophage foam cell formation. Mol Oral Microbiol (2013) 28(1):28–39. doi: 10.1111/omi.12003
131. Herrero-Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol (2022) 19(2):177–91. doi: 10.1038/s41423-021-00832-3
132. Bryzek D, Ciaston I, Dobosz E, Gasiorek A, Makarska A, Sarna M, et al. Triggering NETosis via protease-activated receptor (PAR)-2 signaling as a mechanism of hijacking neutrophils function for pathogen benefits. PLoS Pathog (2019) 15(5):e1007773. doi: 10.1371/journal.ppat.1007773
133. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol (2020) 17(6):327–40. doi: 10.1038/s41569-019-0326-7
134. du Teil Espina M, Fu Y, van der Horst D, Hirschfeld C, Lopez-Alvarez M, Mulder LM, et al. Coating and corruption of human neutrophils by bacterial outer membrane vesicles. Microbiol Spectr (2022) 10(5):e0075322. doi: 10.1128/spectrum.00753-22
135. Gu JY, Liu YJ, Zhu XQ, Qiu JY, Sun Y. Effects of endotoxin tolerance induced by porphyromonas gingivalis lipopolysaccharide on inflammatory responses in neutrophils. Inflammation (2020) 43(5):1692–706. doi: 10.1007/s10753-020-01243-8
136. Moschonas IC, Tselepis AD. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis (2019) 288:9–16. doi: 10.1016/j.atherosclerosis.2019.06.919
137. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7(2):e32366. doi: 10.1371/journal.pone.0032366
138. Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA (2001) 286(17):2136–42. doi: 10.1001/jama.286.17.2136
139. Guzik K, Bzowska M, Smagur J, Krupa O, Sieprawska M, Travis J, et al. A new insight into phagocytosis of apoptotic cells: proteolytic enzymes divert the recognition and clearance of polymorphonuclear leukocytes by macrophages. Cell Death Differ (2007) 14(1):171–82. doi: 10.1038/sj.cdd.4401927
140. Doran AC, Yurdagul A Jr., Tabas I. Efferocytosis in health and disease. Nat Rev Immunol (2020) 20(4):254–67. doi: 10.1038/s41577-019-0240-6
141. Papadopoulos G, Shaik-Dasthagirisaheb YB, Huang N, Viglianti GA, Henderson AJ, Kantarci A, et al. Immunologic environment influences macrophage response to porphyromonas gingivalis. Mol Oral Microbiol (2017) 32(3):250–61. doi: 10.1111/omi.12168
142. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl) (2017) 95(11):1153–65. doi: 10.1007/s00109-017-1575-8
143. Werheim ER, Senior KG, Shaffer CA, Cuadra GA. Oral pathogen porphyromonas gingivalis can escape phagocytosis of mammalian macrophages. Microorganisms (2020) 8(9):1. doi: 10.3390/microorganisms8091432
144. Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, et al. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog (2014) 10(7):e1004215. doi: 10.1371/journal.ppat.1004215
145. Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, Billharz RJ, et al. Porphyromonas gingivalis lipopolysaccharide lipid a heterogeneity: differential activities of tetra- and penta-acylated lipid a structures on e-selectin expression and TLR4 recognition. Cell Microbiol (2006) 8(5):857–68. doi: 10.1111/j.1462-5822.2005.00672.x
146. Herath TD, Darveau RP, Seneviratne CJ, Wang CY, Wang Y, Jin L. Tetra- and penta-acylated lipid a structures of porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-kappaB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS One (2013) 8(3):e58496. doi: 10.1371/journal.pone.0058496
147. Castro SA, Collighan R, Lambert PA, Dias IH, Chauhan P, Bland CE, et al. Porphyromonas gingivalis gingipains cause defective macrophage migration towards apoptotic cells and inhibit phagocytosis of primary apoptotic neutrophils. Cell Death Dis (2017) 8(3):e2644. doi: 10.1038/cddis.2016.481
148. Okano T, Ashida H, Suzuki S, Shoji M, Nakayama K, Suzuki T. Porphyromonas gingivalis triggers NLRP3-mediated inflammasome activation in macrophages in a bacterial gingipains-independent manner. Eur J Immunol (2018) 48(12):1965–74. doi: 10.1002/eji.201847658
149. Hajishengallis G, Wang M, Bagby GJ, Nelson S. Importance of TLR2 in early innate immune response to acute pulmonary infection with porphyromonas gingivalis in mice. J Immunol (2008) 181(6):4141–9. doi: 10.4049/jimmunol.181.6.4141
150. Yang X, Pan Y, Xu X, Tong T, Yu S, Zhao Y, et al. Sialidase deficiency in porphyromonas gingivalis increases IL-12 secretion in stimulated macrophages through regulation of CR3, IncRNA GAS5 and miR-21. Front Cell Infect Microbiol (2018) 8:100. doi: 10.3389/fcimb.2018.00100
151. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3(2):133–46. doi: 10.1038/nri1001
152. Park MH, Jeong SY, Na HS, Chung J. Porphyromonas gingivalis induces autophagy in THP-1-derived macrophages. Mol Oral Microbiol (2017) 32(1):48–59. doi: 10.1111/omi.12153
153. Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, et al. Temperature-dependent modulation of porphyromonas gingivalis lipid a structure and interaction with the innate host defenses. Infect Immun (2011) 79(3):1187–93. doi: 10.1128/IAI.00900-10
154. Blasi I, Korostoff J, Dhingra A, Reyes-Reveles J, Shenker BJ, Shahabuddin N, et al. Variants of porphyromonas gingivalis lipopolysaccharide alter lipidation of autophagic protein, microtubule-associated protein 1 light chain 3, LC3. Mol Oral Microbiol (2016) 31(6):486–500. doi: 10.1111/omi.12141
155. Lin J, Huang D, Xu H, Zhan F, Tan X. Macrophages: A communication network linking porphyromonas gingivalis infection and associated systemic diseases. Front Immunol (2022) 13:952040. doi: 10.3389/fimmu.2022.952040
156. Liu L, Yang Y, Zhou JY, Wu YF, Zhao L. [Porphyromonas gingivalis lipopolysaccharide regulates macrophage polarization via triggering receptors expressed on myeloid cells-1]. Zhonghua Kou Qiang Yi Xue Za Zhi (2021) 56(2):175–81. doi: 10.3760/cma.j.cn112144-20200210-00042
157. Yang Y, Chen SS, Xu CM, Wu YF, Zhao L. [Expression of triggering receptors expressed by myeloid cells-1 in macrophages stimulated by porphyromonas gingivalis-lipopolysaccharide]. Hua Xi Kou Qiang Yi Xue Za Zhi (2018) 36(5):475–81. doi: 10.7518/hxkq.2018.05.003
158. Gu JY, Fu ZB, Jia-Lu C, Liu YJ, Cao XZ, Sun Y. Endotoxin tolerance induced by porphyromonas gingivalis lipopolysaccharide alters macrophage polarization. Microb Pathog (2022) 164:105448. doi: 10.1016/j.micpath.2022.105448
159. Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity (2017) 47(4):621–34. doi: 10.1016/j.immuni.2017.09.008
160. Xu Y, Chaudhury A, Zhang M, Savoldo B, Metelitsa LS, Rodgers J, et al. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest. (2016) 126(7):2678–88. doi: 10.1172/JCI85834
161. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
162. Gaddis DE, Maynard CL, Weaver CT, Michalek SM, Katz J. Role of TLR2-dependent IL-10 production in the inhibition of the initial IFN-gamma T cell response to porphyromonas gingivalis. J Leukoc Biol (2013) 93(1):21–31. doi: 10.1189/jlb.0512220
163. Mittal SK, Cho KJ, Ishido S, Roche PA. Interleukin 10 (IL-10)-mediated immunosuppression: March-i induction regulates antigen presentation by macrophages but not dendritic cells. J Biol Chem (2015) 290(45):27158–67. doi: 10.1074/jbc.M115.682708
164. Kuol N, Stojanovska L, Nurgali K, Apostolopoulos V. PD-1/PD-L1 in disease. Immunotherapy (2018) 10(2):149–60. doi: 10.2217/imt-2017-0120
165. Meghil MM, Tawfik OK, Elashiry M, Rajendran M, Arce RM, Fulton DJ, et al. Disruption of immune homeostasis in human dendritic cells via regulation of autophagy and apoptosis by porphyromonas gingivalis. Front Immunol (2019) 10:2286. doi: 10.3389/fimmu.2019.02286
166. Meghil MM, Ghaly M, Cutler CW. A tale of two fimbriae: How invasion of dendritic cells by porphyromonas gingivalis disrupts DC maturation and depolarizes the T-Cell-Mediated immune response. Pathogens (2022) 11(3):5–6. doi: 10.3390/pathogens11030328
167. Khalaf H, Bengtsson T. Altered T-cell responses by the periodontal pathogen porphyromonas gingivalis. PLoS One (2012) 7(9):e45192. doi: 10.1371/journal.pone.0045192
168. Saigusa R, Winkels H, Ley K. T Cell subsets and functions in atherosclerosis. Nat Rev Cardiol (2020) 17(7):387–401. doi: 10.1038/s41569-020-0352-5
169. Yang J, Hao T, Liu Y, Huang J, Wu W, Wu J, et al. Th17/Treg balance and indoleamine 2,3 dioxygenase activity in periodontitis-associated atherosclerotic patients. J Int Med Res (2022) 50(2):3000605221080877. doi: 10.1177/03000605221080877
170. Yang J, Wu J, Zhang R, Yao M, Liu Y, Miao L, et al. Porphyromonas gingivalis oral infection promote T helper 17/Treg imbalance in the development of atherosclerosis. J Dent Sci (2017) 12(1):60–9. doi: 10.1016/j.jds.2016.10.003
171. Kramer CD, Genco CA. Microbiota, immune subversion, and chronic inflammation. Front Immunol (2017) 8:255. doi: 10.3389/fimmu.2017.00255
172. Bale BF, Doneen AL, Vigerust DJ. High-risk periodontal pathogens contribute to the pathogenesis of atherosclerosis. Postgrad Med J (2017) 93(1098):215–20. doi: 10.1136/postgradmedj-2016-134279
173. McNicol A, Israels SJ. Mechanisms of oral bacteria-induced platelet activation. Can J Physiol Pharmacol (2010) 88(5):510–24. doi: 10.1139/Y10-029
174. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol (2006) 33(6):401–7. doi: 10.1111/j.1600-051X.2006.00924.x
175. Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000 (2014) 64(1):57–80. doi: 10.1111/prd.12002
176. Katz J, Sambandam V, Wu JH, Michalek SM, Balkovetz DF. Characterization of porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect Immun (2000) 68(3):1441–9. doi: 10.1128/IAI.68.3.1441-1449.2000
177. Katz J, Yang QB, Zhang P, Potempa J, Travis J, Michalek SM, et al. Hydrolysis of epithelial junctional proteins by porphyromonas gingivalis gingipains. Infect Immun (2002) 70(5):2512–8. doi: 10.1128/IAI.70.5.2512-2518.2002
178. Abe-Yutori M, Chikazawa T, Shibasaki K, Murakami S. Decreased expression of e-cadherin by porphyromonas gingivalis-lipopolysaccharide attenuates epithelial barrier function. J Periodontal Res (2017) 52(1):42–50. doi: 10.1111/jre.12367
179. Chen W, Alshaikh A, Kim S, Kim J, Chun C, Mehrazarin S, et al. Porphyromonas gingivalis impairs oral epithelial barrier through targeting GRHL2. J Dent Res (2019) 98(10):1150–8. doi: 10.1177/0022034519865184
180. Belstrøm D, Holmstrup P, Damgaard C, Borch TS, Skjødt MO, Bendtzen K, et al. The atherogenic bacterium porphyromonas gingivalis evades circulating phagocytes by adhering to erythrocytes. Infect Immun (2011) 79(4):1559–65. doi: 10.1128/IAI.01036-10
181. Meghil MM, Cutler CW. Oral microbes and mucosal dendritic cells, "Spark and flame" of local and distant inflammatory diseases. Int J Mol Sci (2020) 21(5):2–7. doi: 10.3390/ijms21051643
182. Pirro M, Bianconi V, Paciullo F, Mannarino MR, Bagaglia F, Sahebkar A. Lipoprotein(a) and inflammation: A dangerous duet leading to endothelial loss of integrity. Pharmacol Res (2017) 119:178–87. doi: 10.1016/j.phrs.2017.02.001
183. Deshpande RG, Khan MB, Genco CA. Invasion of aortic and heart endothelial cells by porphyromonas gingivalis. Infect Immun (1998) 66(11):5337–43. doi: 10.1128/IAI.66.11.5337-5343.1998
184. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res (2016) 118(4):620–36. doi: 10.1161/CIRCRESAHA.115.306301
185. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: From mechanism to pharmacotherapies. Pharmacol Rev (2021) 73(3):924–67. doi: 10.1124/pharmrev.120.000096
186. Farrugia C, Stafford GP, Potempa J, Wilkinson RN, Chen Y, Murdoch C, et al. Mechanisms of vascular damage by systemic dissemination of the oral pathogen porphyromonas gingivalis. FEBS J (2021) 288(5):1479–95. doi: 10.1111/febs.15486
187. Farrugia C, Stafford GP, Murdoch C. Porphyromonas gingivalis outer membrane vesicles increase vascular permeability. J Dent Res (2020) 99(13):1494–501. doi: 10.1177/0022034520943187
188. Morris G, Puri BK, Olive L, Carvalho A, Berk M, Walder K, et al. Endothelial dysfunction in neuroprogressive disorders-causes and suggested treatments. BMC Med (2020) 18(1):305. doi: 10.1186/s12916-020-01749-w
189. Mussbacher M, Schossleitner K, Kral-Pointner JB, Salzmann M, Schrammel A, Schmid JA. More than just a monolayer: the multifaceted role of endothelial cells in the pathophysiology of atherosclerosis. Curr Atheroscler Rep (2022) 24(6):483–92. doi: 10.1007/s11883-022-01023-9
190. Song LT, Tada H, Nishioka T, Nemoto E, Imamura T, Potempa J, et al. Porphyromonas gingivalis gingipains-mediated degradation of plasminogen activator inhibitor-1 leads to delayed wound healing responses in human endothelial cells. J Innate Immun (2022) 14(4):306–19. doi: 10.1159/000519737
191. Luo M, Ji Y, Luo Y, Li R, Fay WP, Wu J. Plasminogen activator inhibitor-1 regulates the vascular expression of vitronectin. J Thromb Haemost (2017) 15(12):2451–60. doi: 10.1111/jth.13869
192. Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost (2005) 3(8):1879–83. doi: 10.1111/j.1538-7836.2005.01420.x
193. Ismail AA, Shaker BT, Bajou K. The plasminogen-activator plasmin system in physiological and pathophysiological angiogenesis. Int J Mol Sci (2021) 23(1):8–9. doi: 10.3390/ijms23010337
194. Reyes L, Getachew H, Dunn WA, Progulske-Fox A. Porphyromonas gingivalis W83 traffics via ICAM1 in microvascular endothelial cells and alters capillary organization in vivo. J Oral Microbiol (2020) 12(1):1742528. doi: 10.1080/20002297.2020.1742528
195. Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell-to-cell transmission of porphyromonas gingivalis. BMC Microbiol (2008) 8:26. doi: 10.1186/1471-2180-8-26
196. Czesnikiewicz-Guzik M, Nosalski R, Mikolajczyk TP, Vidler F, Dohnal T, Dembowska E, et al. Th1-type immune responses to porphyromonas gingivalis antigens exacerbate angiotensin II-dependent hypertension and vascular dysfunction. Br J Pharmacol (2019) 176(12):1922–31. doi: 10.1111/bph.14536
197. Sampath C, Okoro EU, Gipson MJ, Chukkapalli SS, Farmer-Dixon CM, Gangula PR. Porphyromonas gingivalis infection alters Nrf2-phase II enzymes and nitric oxide in primary human aortic endothelial cells. J Periodontol (2021) 92(7):54–65. doi: 10.1002/JPER.20-0444
198. Hirasawa M, Kurita-Ochiai T. Porphyromonas gingivalis induces apoptosis and autophagy via ER stress in human umbilical vein endothelial cells. Mediators Inflamm (2018) 2018:1967506. doi: 10.1155/2018/1967506
199. Durand E, Scoazec A, Lafont A, Boddaert J, Al Hajzen A, Addad F, et al. In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation: A clue to the understanding of the mechanisms of thrombotic plaque erosion. Circulation (2004) 109(21):2503–6. doi: 10.1161/01.CIR.0000130172.62481.90
200. Pan S, Lei L, Chen S, Li H, Yan F. Rosiglitazone impedes porphyromonas gingivalis-accelerated atherosclerosis by downregulating the TLR/NF-kappaB signaling pathway in atherosclerotic mice. Int Immunopharmacol (2014) 23(2):701–8. doi: 10.1016/j.intimp.2014.10.026
201. Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal AG, et al. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J Immunol (2012) 189(7):3681–8. doi: 10.4049/jimmunol.1201541
202. Bugueno IM, Benkirane-Jessel N, Huck O. Implication of Toll/IL-1 receptor domain containing adapters in porphyromonas gingivalis-induced inflammation. Innate Immun (2021) 27(4):324–42. doi: 10.1177/17534259211013087
203. Xie M, Tang Q, Yu S, Sun J, Mei F, Zhao J, et al. Porphyromonas gingivalis disrupts vascular endothelial homeostasis in a TLR-NF-κB axis dependent manner. Int J Oral Sci (2020) 12(1):28. doi: 10.1038/s41368-020-00096-z
204. Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125(12):4514–28. doi: 10.1172/JCI82719
205. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol (2018) 9:294. doi: 10.3389/fimmu.2018.00294
206. Suh JS, Kim S, Bostrom KI, Wang CY, Kim RH, Park NH. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial-mesenchymal transition in mice. Int J Oral Sci (2019) 11(3):21. doi: 10.1038/s41368-019-0054-1
207. Xie M, Tang Q, Nie J, Zhang C, Zhou X, Yu S, et al. BMAL1-downregulation aggravates porphyromonas gingivalis-induced atherosclerosis by encouraging oxidative stress. Circ Res (2020) 126(6):e15–29. doi: 10.1161/CIRCRESAHA.119.315502
208. Deng H, Gong Y, Chen Y, Zhang G, Chen H, Cheng T, et al. Porphyromonas gingivalis lipopolysaccharide affects the angiogenic function of endothelial progenitor cells via Akt/FoxO1 signaling. J Periodontal Res (2022) 57(4):859–68. doi: 10.1111/jre.13024
209. Li Calzi S, Neu MB, Shaw LC, Kielczewski JL, Moldovan NI, Grant MB. EPCs and pathological angiogenesis: when good cells go bad. Microvasc Res (2010) 79(3):207–16. doi: 10.1016/j.mvr.2010.02.011
210. Xu T, Dong Q, Luo Y, Liu Y, Gao L, Pan Y, et al. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int J Oral Sci (2021) 13(1):28. doi: 10.1038/s41368-021-00134-4
211. Bugueno IM, Khelif Y, Seelam N, Morand DN, Tenenbaum H, Davideau JL, et al. Porphyromonas gingivalis differentially modulates cell death profile in ox-LDL and TNF-alpha pre-treated endothelial cells. PLoS One (2016) 11(4):e0154590. doi: 10.1371/journal.pone.0154590
212. Bugueno IM, Zobairi El-Ghazouani F, Batool F, El Itawi H, Anglès-Cano E, Benkirane-Jessel N, et al. Porphyromonas gingivalis triggers the shedding of inflammatory endothelial microvesicles that act as autocrine effectors of endothelial dysfunction. Sci Rep (2020) 10(1):1778. doi: 10.1038/s41598-020-58374-z
213. Qi X, Zhao L, Chen S, Meng S, Wu Y. [Effects of porphyromnonas gingivalis lipopolysaccharide on the expression of RANTES and fractalkine in human urnbilical vein endothelial cells]. Hua Xi Kou Qiang Yi Xue Za Zhi (2016) 34(2):194–9. doi: 10.7518/hxkq.2016.02.018
214. Braunersreuther V, Steffens S, Arnaud C, Pelli G, Burger F, Proudfoot A, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol (2008) 28(6):1090–6. doi: 10.1161/ATVBAHA.108.165423
215. Marino AP, Silva AA, Santos PV, Pinto LM, Gazinelli RT, Teixeira MM, et al. CC-chemokine receptors: a potential therapeutic target for trypanosoma cruzi-elicited myocarditis. Mem Inst Oswaldo Cruz. (2005) 100 Suppl 1:93–6. doi: 10.1590/s0074-02762005000900015
216. Lv YB, Jing J, Li JM, Zhong JP, Fang L, Yang B. Assessment of RANTES levels as the indicators of plaque vulnerability in rabbit models of atherosclerosis. Pathol Res Pract (2014) 210(12):1031–7. doi: 10.1016/j.prp.2014.03.012
217. Li Q, Liu J, Liu W, Chu Y, Zhong J, Xie Y, et al. LOX-1 regulates p. gingivalis-induced monocyte migration and adhesion to human umbilical vein endothelial cells. Front Cell Dev Biol (2020) 8:596. doi: 10.3389/fcell.2020.00596
218. Ho MH, Guo ZM, Chunga J, Goodwin JS, Xie H. Characterization of innate immune responses of human endothelial cells induced by porphyromonas gingivalis and their derived outer membrane vesicles. Front Cell Infect Microbiol (2016) 6:139. doi: 10.3389/fcimb.2016.00139
219. Wu Y, Xu W, Hou J, Liu Y, Li R, Liu J, et al. Porphyromonas gingivalis-induced MIF regulates intercellular adhesion molecule-1 expression in EA.hy926 cells and monocyte-endothelial cell adhesion through the receptors CD74 and CXCR4. Inflammation (2019) 42(3):874–83. doi: 10.1007/s10753-018-0942-0
220. Xu W, Pan Y, Xu Q, Wu Y, Pan J, Hou J, et al. Porphyromonas gingivalis ATCC 33277 promotes intercellular adhesion molecule-1 expression in endothelial cells and monocyte-endothelial cell adhesion through macrophage migration inhibitory factor. BMC Microbiol (2018) 18(1):16. doi: 10.1186/s12866-018-1156-1
221. Bakogiannis C, Sachse M, Stamatelopoulos K, Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine (2019) 122:154157. doi: 10.1016/j.cyto.2017.09.013
222. Chen WA, Fletcher HM, Gheorghe JD, Oyoyo U, Boskovic DS. Platelet plug formation in whole blood is enhanced in the presence of porphyromonas gingivalis. Mol Oral Microbiol (2020) 35(6):251–9. doi: 10.1111/omi.12314
223. Klarstrom Engstrom K, Khalaf H, Kalvegren H, Bengtsson T. The role of porphyromonas gingivalis gingipains in platelet activation and innate immune modulation. Mol Oral Microbiol (2015) 30(1):62–73. doi: 10.1111/omi.12067
224. Zhan Y, Lu R, Meng H, Wang X, Hou J. Platelet activation and platelet-leukocyte interaction in generalized aggressive periodontitis. J Leukoc Biol (2016) 100(5):1155–66. doi: 10.1189/jlb.4A1115-526RR
225. Chen WA, Fletcher HM, Payne KJ, Aka S, Thornburg MB, Gheorghe JD, et al. Platelet and neutrophil responses to porphyromonas gingivalis in human whole blood. Mol Oral Microbiol (2021) 36(3):202–13. doi: 10.1111/omi.12336
226. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
227. Brousseau-Nault M, Kizhakkedathu JN, Kim H. Chronic periodontitis is associated with platelet factor 4 (PF4) secretion: A pilot study. J Clin Periodontol (2017) 44(11):1101–11. doi: 10.1111/jcpe.12771
228. Jockel-Schneider Y, Kobsar A, Stellzig-Eisenhauer A, Vogel U, Stork S, Frantz S, et al. Wild-type isolates of porphyromonas gingivalis derived from periodontitis patients display major variability in platelet activation. J Clin Periodontol (2018) 45(6):693–700. doi: 10.1111/jcpe.12895
229. Pollreisz A, Huang Y, Roth GA, Cheng B, Kebschull M, Papapanou PN, et al. Enhanced monocyte migration and pro-inflammatory cytokine production by porphyromonas gingivalis infection. J Periodontal Res (2010) 45(2):239–45. doi: 10.1111/j.1600-0765.2009.01225.x
230. Li XY, Wang C, Xiang XR, Chen FC, Yang CM, Wu J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol Rep (2013) 30(3):1329–36. doi: 10.3892/or.2013.2600
231. Lonn J, Ljunggren S, Klarstrom-Engstrom K, Demirel I, Bengtsson T, Karlsson H. Lipoprotein modifications by gingipains of porphyromonas gingivalis. J Periodontal Res (2018) 53(3):403–13. doi: 10.1111/jre.12527
232. Miyakawa H, Honma K, Qi M, Kuramitsu HK. Interaction of porphyromonas gingivalis with low-density lipoproteins: implications for a role for periodontitis in atherosclerosis. J Periodontal Res (2004) 39(1):1–9. doi: 10.1111/j.1600-0765.2004.00697.x
233. Hashimoto M, Kadowaki T, Tsukuba T, Yamamoto K. Selective proteolysis of apolipoprotein b-100 by arg-gingipain mediates atherosclerosis progression accelerated by bacterial exposure. J Biochem (2006) 140(5):713–23. doi: 10.1093/jb/mvj202
234. Joo JY, Cha GS, Chung J, Lee JY, Kim SJ, Choi J. Peptide 19 of porphyromonas gingivalis heat shock protein is a potent inducer of low-density lipoprotein oxidation. J Periodontol (2017) 88(2):e58–64. doi: 10.1902/jop.2016.160402
235. Kim HJ, Cha GS, Kim HJ, Kwon EY, Lee JY, Choi J, et al. Porphyromonas gingivalis accelerates atherosclerosis through oxidation of high-density lipoprotein. J Periodontal Implant Sci (2018) 48(1):60–8. doi: 10.5051/jpis.2018.48.1.60
236. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med (2014) 46:e99. doi: 10.1038/emm.2014.38
237. Liang DY, Liu F, Chen JX, He XL, Zhou YL, Ge BX, et al. Porphyromonas gingivalis infected macrophages upregulate CD36 expression via ERK/NF-kappaB pathway. Cell Signal (2016) 28(9):1292–303. doi: 10.1016/j.cellsig.2016.05.017
238. Brown PM, Kennedy DJ, Morton RE, Febbraio M. CD36/SR-B2-TLR2 dependent pathways enhance porphyromonas gingivalis mediated atherosclerosis in the ldlr KO mouse model. PLoS One (2015) 10(5):e0125126. doi: 10.1371/journal.pone.0125126
239. Gupta N, Goswami R, Alharbi MO, Biswas D, Rahaman SO. TRPV4 is a regulator in p. gingivalis lipopolysaccharide-induced exacerbation of macrophage foam cell formation. Physiol Rep (2019) 7(7):e14069. doi: 10.14814/phy2.14069
240. Furuhashi M. Fatty acid-binding protein 4 in cardiovascular and metabolic diseases. J Atheroscler Thromb (2019) 26(3):216–32. doi: 10.5551/jat.48710
241. Furuhashi M, Fuseya T, Murata M, Hoshina K, Ishimura S, Mita T, et al. Local production of fatty acid-binding protein 4 in Epicardial/Perivascular fat and macrophages is linked to coronary atherosclerosis. Arterioscler Thromb Vasc Biol (2016) 36(5):825–34. doi: 10.1161/ATVBAHA.116.307225
242. Yang Y, He X, Xia S, Liu F, Luo L. Porphyromonas gingivalis facilitated the foam cell formation via lysosomal integral membrane protein 2 (LIMP2). J Periodontal Res (2021) 56(2):265–74. doi: 10.1111/jre.12812
243. Rho JH, Kim HJ, Joo JY, Lee JY, Lee JH, Park HR. Periodontal pathogens promote foam cell formation by blocking lipid efflux. J Dent Res (2021) 100(12):1367–77. doi: 10.1177/00220345211008811
244. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res (2018) 114(4):590–600. doi: 10.1093/cvr/cvy010
245. Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol (2008) 28(5):812–9. doi: 10.1161/ATVBAHA.107.159327
246. Park HJ, Kim Y, Kim MK, Park HR, Kim HJ, Bae SK, et al. Infection of porphyromonas gingivalis increases phosphate-induced calcification of vascular smooth muscle cells. Cells (2020) 9(12):4–7. doi: 10.3390/cells9122694
247. Park HJ, Kim Y, Kim MK, Hwang JJ, Kim HJ, Bae SK, et al. Inhibition of gastrin-releasing peptide attenuates phosphate-induced vascular calcification. Cells (2020) 9(3):5–9. doi: 10.3390/cells9030737
248. Lencel P, Hardouin P, Magne D. Do cytokines induce vascular calcification by the mere stimulation of TNAP activity? Med Hypotheses (2010) 75(6):517–21. doi: 10.1016/j.mehy.2010.07.011
249. Liu G, Deng J, Zhang Q, Song W, Chen S, Lou X, et al. Porphyromonas gingivalis lipopolysaccharide stimulation of vascular smooth muscle cells activates proliferation and calcification. J Periodontol (2016) 87(7):828–36. doi: 10.1902/jop.2016.150602
250. Cao C, Ji X, Luo X, Zhong L. Gingipains from porphyromonas gingivalis promote the transformation and proliferation of vascular smooth muscle cell phenotypes. Int J Clin Exp Med (2015) 8(10):18327–34.
251. Yang WW, Guo B, Jia WY, Jia Y. Porphyromonas gingivalis-derived outer membrane vesicles promote calcification of vascular smooth muscle cells through ERK1/2-RUNX2. FEBS Open Bio (2016) 6(12):1310–9. doi: 10.1002/2211-5463.12151
252. Li J, Deng J, Shang S, Liu G, Song W, Sun P, et al. Effect of porphyromonas gingivalis lipopolysaccharide on calcification of human umbilical artery smooth muscle cells co-cultured with human periodontal ligament cells. Exp Ther Med (2021) 21(6):655. doi: 10.3892/etm.2021.10988
253. Chen TC, Lin CT, Chien SJ, Chang SF, Chen CN. Regulation of calcification in human aortic smooth muscle cells infected with high-glucose-treated porphyromonas gingivalis. J Cell Physiol (2018) 233(6):4759–69. doi: 10.1002/jcp.26268
254. Babaniamansour P, Mohammadi M, Babaniamansour S, Aliniagerdroudbari E. The relation between atherosclerosis plaque composition and plaque rupture. J Med Signals Sens (2020) 10(4):267–73. doi: 10.4103/jmss.JMSS_48_19
255. Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, et al. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol (2012) 189(6):3178–87. doi: 10.4049/jimmunol.1201053
256. Chen TY, Kuo PJ, Lin CY, Hung TF, Chiu HC, Chiang CY, et al. Porphyromonas gingivalis lipopolysaccharide and gingival fibroblast augment MMP-9 expression of monocytic U937 cells through cyclophilin a. J Periodontol (2022) 93(3):449–57. doi: 10.1002/JPER.19-0740
257. Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from porphyromonas gingivalis on the human complement system. J Immunol (2007) 178(11):7242–50. doi: 10.4049/jimmunol.178.11.7242
258. Jackson DG. Leucocyte trafficking via the lymphatic vasculature- mechanisms and consequences. Front Immunol (2019) 10:471. doi: 10.3389/fimmu.2019.00471
259. Alitalo K. The lymphatic vasculature in disease. Nat Med (2011) 17(11):1371–80. doi: 10.1038/nm.2545
260. Voisin MB, Nourshargh S. Neutrophil trafficking to lymphoid tissues: physiological and pathological implications. J Pathol (2019) 247(5):662–71. doi: 10.1002/path.5227
261. Amodini Rajakaruna G, Umeda M, Uchida K, Furukawa A, Yuan B, Suzuki Y, et al. Possible translocation of periodontal pathogens into the lymph nodes draining the oral cavity. J Microbiol (2012) 50(5):827–36. doi: 10.1007/s12275-012-2030-8
262. Kuroshima S, Sawa Y, Yamaoka Y, Notani K, Yoshida S, Inoue N. Expression of cys-cys chemokine ligand 21 on human gingival lymphatic vessels. Tissue Cell (2004) 36(2):121–7. doi: 10.1016/j.tice.2003.10.004
263. Papadakou P, Bletsa A, Yassin MA, Karlsen TV, Wiig H, Berggreen E. Role of hyperplasia of gingival lymphatics in periodontal inflammation. J Dent Res (2017) 96(4):467–76. doi: 10.1177/0022034516681762
264. Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, et al. DC Mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med (2011) 208(10):2141–53. doi: 10.1084/jem.20102392
265. Miles B, Zakhary I, El-Awady A, Scisci E, Carrion J, O'Neill JC, et al. Secondary lymphoid organ homing phenotype of human myeloid dendritic cells disrupted by an intracellular oral pathogen. Infect Immun (2014) 82(1):101–11. doi: 10.1128/IAI.01157-13
266. Murad HAS, Rafeeq MM, Alqurashi TMA. Role and implications of the CXCL12/CXCR4/CXCR7 axis in atherosclerosis: still a debate. Ann Med (2021) 53(1):1598–612. doi: 10.1080/07853890.2021.1974084
267. Chakraborty S, Zawieja SD, Wang W, Lee Y, Wang YJ, von der Weid PY, et al. Lipopolysaccharide modulates neutrophil recruitment and macrophage polarization on lymphatic vessels and impairs lymphatic function in rat mesentery. Am J Physiol Heart Circ Physiol (2015) 309(12):H2042–57. doi: 10.1152/ajpheart.00467.2015
268. Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. (2013) 123(4):1571–9. doi: 10.1172/JCI63685
269. Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab (2013) 17(5):671–84. doi: 10.1016/j.cmet.2013.04.002
270. Rademakers T, van der Vorst EP, Daissormont IT, Otten JJ, Theodorou K, Theelen TL, et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci Rep (2017) 7:45263. doi: 10.1038/srep45263
271. Martel C, Randolph GJ. Atherosclerosis and transit of HDL through the lymphatic vasculature. Curr Atheroscler Rep (2013) 15(9):354. doi: 10.1007/s11883-013-0354-4
272. Wang X, Luan Q, Chen Q, Zhao L, Guo Y. Mitochondrial polymorphisms and dysfunction related to aggressive periodontitis: a pilot study. Oral Dis (2014) 20(5):490–8. doi: 10.1111/odi.12163
273. Choi CH, Spooner R, DeGuzman J, Koutouzis T, Ojcius DM, Yilmaz Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol (2013) 15(6):961–76. doi: 10.1111/cmi.12089
274. Sun Y, Zhang D, Liu X, Li X, Liu F, Yu Y, et al. Endoplasmic reticulum stress affects lipid metabolism in atherosclerosis Via CHOP activation and over-expression of miR-33. Cell Physiol Biochem (2018) 48(5):1995–2010. doi: 10.1159/000492522
275. Linton MF, Tao H, Linton EF, Yancey PG. SR-BI: A multifunctional receptor in cholesterol homeostasis and atherosclerosis. Trends Endocrinol Metab (2017) 28(6):461–72. doi: 10.1016/j.tem.2017.02.001
276. Song GJ, Kim SM, Park KH, Kim J, Choi I, Cho KH. SR-BI mediates high density lipoprotein (HDL)-induced anti-inflammatory effect in macrophages. Biochem Biophys Res Commun (2015) 457(1):112–8. doi: 10.1016/j.bbrc.2014.12.028
277. Lewis SM, Williams A, Eisenbarth SC. Structure and function of the immune system in the spleen. Sci Immunol (2019) 4(33):1. doi: 10.1126/sciimmunol.aau6085
278. He X, Liang B, Gu N. Th17/Treg imbalance and atherosclerosis. Dis Markers (2020) 2020:8821029. doi: 10.1155/2020/8821029
279. Cai Y, Kobayashi R, Hashizume-Takizawa T, Kurita-Ochiai T. Porphyromonas gingivalis infection enhances Th17 responses for development of atherosclerosis. Arch Oral Biol (2014) 59(11):1183–91. doi: 10.1016/j.archoralbio.2014.07.012
280. Wang Y, Zhang W, Xu L, Jin JO. Porphyromonas gingivalis lipopolysaccharide induced proliferation and activation of natural killer cells in vivo. Molecules (2016) 21(8):1–5. doi: 10.3390/molecules21081086
Keywords: porphyromonas gingivalis, atherosclerosis, immune escape, blood circulation, lymphatic circulation
Citation: Ruan Q, Guan P, Qi W, Li J, Xi M, Xiao L, Zhong S, Ma D and Ni J (2023) Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front. Immunol. 14:1103592. doi: 10.3389/fimmu.2023.1103592
Received: 22 November 2022; Accepted: 01 March 2023;
Published: 14 March 2023.
Edited by:
Aditi Chopra, Manipal College of Dental Sciences, Manipal, IndiaReviewed by:
Krzysztof Guzik, Jagiellonian University, PolandSasanka Chukkapalli, Texas A&M University College Station, United States
Copyright © 2023 Ruan, Guan, Qi, Li, Xi, Xiao, Zhong, Ma and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dandan Ma, bWRkQHNtdS5lZHUuY24=; Jia Ni, NDQxNTY5MDA4QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship