
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Immunol. , 06 March 2023
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1100967
 Sarah E. Withers1,2
Sarah E. Withers1,2 Charlie F. Rowlands3,4Victor S. Tapia1,2Frances Hedley1,2
Charlie F. Rowlands3,4Victor S. Tapia1,2Frances Hedley1,2 Ioana-Emilia Mosneag1,2
Ioana-Emilia Mosneag1,2 Siobhan Crilly1,2
Siobhan Crilly1,2 Gillian I. Rice3,4
Gillian I. Rice3,4 Andrew P. Badrock5
Andrew P. Badrock5 Andrew Hayes6
Andrew Hayes6 Stuart M. Allan1,2,7
Stuart M. Allan1,2,7 Tracy A. Briggs3,4,7
Tracy A. Briggs3,4,7 Paul R. Kasher1,2,7*
Paul R. Kasher1,2,7*Aicardi-Goutières syndrome (AGS1-9) is a genetically determined encephalopathy that falls under the type I interferonopathy disease class, characterized by excessive type I interferon (IFN-I) activity, coupled with upregulation of IFN-stimulated genes (ISGs), which can be explained by the vital role these proteins play in self-non-self-discrimination. To date, few mouse models fully replicate the vast clinical phenotypes observed in AGS patients. Therefore, we investigated the use of zebrafish as an alternative species for generating a clinically relevant model of AGS. Using CRISPR-cas9 technology, we generated a stable mutant zebrafish line recapitulating AGS5, which arises from recessive mutations in SAMHD1. The resulting homozygous mutant zebrafish larvae possess a number of neurological phenotypes, exemplified by variable, but increased expression of several ISGs in the head region, a significant increase in brain cell death, microcephaly and locomotion deficits. A link between IFN-I signaling and cholesterol biosynthesis has been highlighted by others, but not previously implicated in the type I interferonopathies. Through assessment of neurovascular integrity and qPCR analysis we identified a significant dysregulation of cholesterol biosynthesis in the zebrafish model. Furthermore, dysregulation of cholesterol biosynthesis gene expression was also observed through RNA sequencing analysis of AGS patient whole blood. From this novel finding, we hypothesize that cholesterol dysregulation may play a role in AGS disease pathophysiology. Further experimentation will lend critical insight into the molecular pathophysiology of AGS and the potential links involving aberrant type I IFN signaling and cholesterol dysregulation.
Aicardi-Goutières syndrome (AGS) is a rare, type I interferonopathy that primarily presents as a severe childhood-onset neurological disease. To date, nine genetic subtypes have been identified that can be caused by mutations in TREX1 (AGS1), the three non-allelic components of RNASEH2 (RNASEH2A-C; AGS2-4), SAMHD1 (AGS5), ADAR1 (AGS6), IFIH1 (AGS7), LSM11 (AGS8) or RNU7-1 (AGS9) (1–6). Mutations in these genes lead to abnormal processing, sensing or metabolism of self-nucleic acids and subsequent activation of type I interferon (IFN-I) signaling and enhanced expression of interferon stimulated genes (ISGs). AGS patients exhibit a spectrum of neurological (and non-neurological) phenotypes, which can vary in severity, and most frequently include brain atrophy, intracranial calcification, microcephaly, white matter lesions, motor dysfunction and intellectual disability (7–9). Given the significant auto-inflammatory component of the disease, these phenotypes are thought to manifest due to a neurotoxic consequence of excessive IFN-I signaling, but to date this hypothesis remains unproven. Furthermore, AGS symptoms can present in a heterogeneous fashion, with additional neurological phenotypes observed in specific genetic subtypes, including ADAR1-related bilateral striatal necrosis (10, 11), ADAR1, IFIH1 and RNASEH2B-related spastic paraplegia (12) and SAMHD1-related cerebrovasculopathy and stroke (13–16).
Given AGS is a rare disease, the availability of patient-derived material for experimental investigation is limited. As such, animal models of AGS are utilized to help study disease mechanisms and for pre-clinical drug discovery. However, to date, few animal models of AGS successfully recapitulate both the inflammatory and neurological phenotypes associated with the disease (17). Previously, we have shown that morpholino-mediated knockdown of samhd1 in zebrafish embryos recapitulates aspects of the IFN-I and stroke phenotypes observed in AGS5 (18). In the present study, we expand on this work and characterize a novel CRISPR-Cas9 induced stable samhd1 mutant zebrafish line. The aim of this work was to further demonstrate the translational utility of zebrafish disease modelling for studying AGS and to make novel mechanistic observations associated with the disease.
Adult zebrafish husbandry was approved by The University of Manchester Animal Welfare and Ethical Review Board, and all experiments were performed in accordance with U.K Home Office regulations (PPL: P132EB6D7). The zebrafish used in this study were raised and maintained at The University of Manchester Biological Services Unit, under standard conditions, with adults housed in mixed sex tanks with a recirculating water supply maintained at 28°C under a 14/10 hour light/dark cycle, as previously described (19). Fertilized eggs were collected following natural spawning, and incubated at 28°C in fresh E3 medium. The embryos were staged according to standard guidelines (20). After termination of the experiment, all embryos were killed prior to protected status [5 days post fertilization (dpf)] using a lethal dose of Tricaine Methanesulfonate (MS222) and freezing at -20°C.
A guide RNA (gRNA) with no predicted off-target sequences was designed for exon 4 of samhd1 using CHOPCHOP (21). gRNA incorporating this target sequence was generated from a polymerase chain reaction (PCR) amplification (Forward primer: 5’- TAATACGACTCACTATAGGCGTCACATTAAGCAGCTCGGGTTTTAGAGC-3’; Reverse primer: 5’-AAAAGCACCGACTCGGTGCCACTTTTTCAAG-3’) including the remaining sequence of S. pyogenes chimeric single gRNA through in vitro transcription using a HiScribe T7 Quick kit (New England Biolabs). Precipitation of the gRNA and synthesis of Cas9 RNA was performed as previously described (22). The resulting RNAs were mixed with 0.05% phenol red, 120 mM KCl and 20mM HEPES, ph7.0, and ~1 nl of the mix was injected into the yolk of wild type (WT) AB fertilized eggs at the one cell stage to produce F0 crispants. To identify founders, F0 adults were individually crossed with wild type (WT) AB animals and the resulting embryos were assessed for indels using Hyp188I restriction analysis. This process identified a founder animal harboring a 23bp deletion in exon 4 (samhd1Δ23) that was predicted to lead to a frameshift. Following confirmation of germline transmission, the F0 samhd1Δ23 was crossed again with WT AB fish to produce an F1 generation. Following F1 heterozygous mutant incrosses, we ascertained that homozygotes (samhd1Δ23/Δ23) were viable and capable of breeding. Subsequent generations were genotyped following PCR and Sanger sequencing of fin clip genomic DNA, using primers that flanked the 23bp deletion (forward primer 5’-GTGTTTAATGACCCCATCCA-3’; reverse primer: 5’-CCTATGGAGTGCTCAAATCG-3’).
To generate the amino acid sequence, the zebrafish WT samhd1 transcript was obtained from Ensembl (23) and inputted into ExPASy for translation (24). For the samhd1Δ23/Δ23 sequence, the 23 bp deletion determined by Sanger sequencing was removed from the WT transcript before translation. For this study, all experiments were performed on an F4/F5 generation and comparisons were made between age matched WT and homozygous mutants.
Total RNA was extracted from pooled groups of whole larvae (n=20) or dissected larval heads (n=30) at 5 days post-fertilization (dpf) using a standard TRIzol (Invitrogen) method. Complementary DNA (cDNA) was synthesized from 800ng RNA as previously described (18). Quantitative PCR (qPCR) was performed on a StepOne Plus Real Time PCR machine (Applied Biosystems). To assess samhd1 expression in whole larvae, cDNA was combined with power SYBR green mastermix (Applied Biosystems) and primers (Eurofins Genomics) that flanked the 23bp deletion (Forward: 5’-AGAACATCATCTGCCGCCGG-3’; Reverse: 5’-CCAGTTCCTTCGCCCAGTCC-3’). Primers targeting the housekeeping gene hprt1 were used to normalize expression (Forward: 5’-GGACTTCATCCTCAAGAG-3’; Reverse: 5’-GTTCTAGCAGCGTCTTCATCG-3’). To assess expression of ISGs (isg15 (18), isg12 (Dr03140917_g1) and stat1b (Dr03151121_m1)) and the cholesterol biosynthesis gene hmgcrb (Dr03128326_m1) in dissected larval heads, a Taqman (Life Technologies) protocol was performed, as previously described (18).
Anaesthetized WT and samhd1Δ23/Δ23 embryos were fixed overnight in 4% paraformaldehyde (PFA; Alfa Aesar) at 2 dpf before washing in 1% PBS-Tritonx. To image the samples, 80% glycerol was added to the embryos which were placed into a nunc glass base dish 12 mm. The samples were individually imaged in a ventral orientation using a Leica M165FC light stereo microscope with DFC7000T camera, and processed using LAS-X v3.3.3.16958 software (Leica). To assess for microcephaly, the distance between the eyes was measured alongside full length of the embryo (μm). The head/body ratio was determined from these two values, and normalized to the average WT head/body ratio, to produce a microcephaly index (ratio). Measurements were obtained using Image J (version 1.52a). n=8 embryos per group, for each biological replicate, repeated 3 independent times.
At 2 dpf, PFA fixed WT and samhd1Δ23/Δ23 embryos were stained using the Click- iT Plus TUNEL assay kit (Invitrogen) following the manufacturer guidelines. For imaging, samples were mounted in a lateral position in 80% glycerol on a nunc glass base dish 12 mm (Thermo Fisher) and imaged on a Leica M205 FA Stereo fluorescence microscope using a 5x/0.50 PlanAPO LWD objective, captured using a DFC 365FX camera and processed using LAS AF v3.1.0.8587 software (Leica). Images were analyzed using the manual cell counting software on Image J (version 1.52a). Analyses were performed on n= 6-8 embryos from 3 independent replicates.
Swimming was measured from 3 – 5 dpf in WT and samhd1Δ23/Δ23 larvae using DanioVision camera chamber and ethovision XT software (Noldus, version 11), as previously described (25). Cumulative duration of movement was measured in 3 independent replicates, with n=22-24 larvae per replicate.
To assess for brain hemorrhages, WT and samhd1Δ23/Δ23 embryos (n=20 per group) were treated with atorvastatin (ATV; Merck) at 32 hours post-fertilization (hpf), as described previously (25). To visualize bleeds, ATV treated embryos were stained from 54 hpf using an o-dianisidine (Sigma) protocol (26). The percentage of embryos with hemorrhages within each treatment/genotype group was determined from 3 independent replicates.
Whole blood from AGS patients with confirmed mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1 or IFIH1 was taken to allow for whole genome RNA sequencing, as described previously (27, 28). In addition, samples from non-AGS patient age-matched controls were obtained. Briefly, AGS patients were identified through either direct clinical contact with the appropriate physicians, or through a referral process. The use of patient materials has been approved from the Leeds (East) research ethics committee (reference number: 07/Q1206/7) and South Centre – Hampshire A research ethics committee (reference number 17/SC/0026), whilst consent was also obtained from parents of the affected patients.
The Paxgene (PreAnalytix) whole blood RNA extraction method for RNA sequencing has been described previously, and the AGS patient RNA sequencing data-sets have been published elsewhere (27–29). RNA concentration was assessed with a spectrophotometer (FLUOstar Omega, Labtech), and 1 μg of mRNA from each sample was diluted into 20 μl nuclease-free water. Subsequently, the quality and integrity of the RNA samples were assessed using a 2200 TapeStation (Agilent Technologies). A poly-A enrichment library was then generated using the TruSeq® Stranded mRNA assay (Illumina), according to the manufacturer’s protocol, and 76 bp (+ indices) paired-end sequencing carried out on an Illumina HiSeq 4000. Raw sequencing output was demultiplexed (allowing one mismatch) and the Binary Base Call (BCL) sequence file format was converted to the text-based sequencing data file format (FASTQ) using Illumina’s bcl2fastq software, version 2.17.1.14. Low quality bases and adaptor sequences were trimmed using Trimmomatic, and reads aligned to the Genome Reference Consortium Human Build 37 (GRCh37) genome using the two-pass mode of the Spliced Transcripts Alignment to a Reference (STAR) aligner (v2.5.3a), as well as to the transcriptome according to the GENCODE v19 human gene annotation (downloaded from https://www.gencodegenes.org/human/release_19.html). For each patient sample, the RNA-Seq by Expectation Maximization (RSEM) software package (v1.3.0) was used to calculate gene expression values, in transcripts per million (TPM). Subsequently, fold change was determined from TPM values, and then Log transformed (Log2).
All data were analyzed using GraphPad Prism 8.1.2 (GraphPad software Inc). All zebrafish experiments, except from ATV treatment were analyzed using either an unpaired t test, or Mann Whitney test, depending on normality, established by the Shapiro Wilks normality test. ATV treatment was analyzed using a two-way ANOVA with sidak’s multiple comparisons test. AGS patient RNA seq data was analyzed using a one-way ANOVA with Dunnetts multiple comparisons test, comparing each AGS gene with the control group.
Following on from the samhd1 MO model, which successfully recapitulated aspects of the AGS5 phenotype, a stable mutant zebrafish line was subsequently generated to facilitate long term studies (18). To generate a stable samhd1 loss of function model, a gRNA was designed using the CHOPCHOP software, targeting exon 4 of the samhd1 gene (Figure 1A). A 23 bp deletion was confirmed by Sanger sequencing and PCR in the F1 generation (Figures 1B, C). To determine whether this mutation resulted in loss of transcript, RNA was harvested from WT and samhd1Δ23/Δ23 larvae at 5 dpf for qPCR analysis of the samhd1 gene, where the mutants exhibited almost a complete loss in samhd1 expression compared to WT larvae (P<0.05) (Figure 1D). Translation analysis of genomic sequences highlighted that the samhd1Δ23 mutation introduces a premature stop codon prior to the HD domain which is predicted to produce a truncated amino acid sequence (Supplementary Figure 1).

Figure 1 Generation of the samhd1Δ23/Δ23 line. (A) gRNA sequence (red) designed to target a site within exon 4 of the samhd1 gene to initiate a 23 bp deletion in injected embryos. (B) Sanger sequencing of WT and samhd1Δ23/Δ23 mutants from fish derived from F1 generation incross. Green highlighted area on WT sequencing represents the site of the 23 bp deletion, causing a frameshift on the mutant sequence. (C) Amplification of samhd1 transcripts from mutant and WT zebrafish embryos using exon 4 specific primers, yielded a single PCR product with clearly visible reduction in band size, as a result of 23 bp deletion. Length of samhd1 exon 4 is 161 bp in WT embryos, with the mutants producing a band of 138 bp. (D) qPCR analysis of samhd1 gene expression in 5 dpf WT and samhd1Δ23/Δ23 larvae. Data analyzed using Mann Whitney U test and presented as median ± IQR (*P=0.0286). n=20 larvae per group, per biological replicate, repeated 4 times.
One of the primary hematological hallmarks of AGS is the excessive upregulation of IFN-I and the subsequent ISG signature, which is often used as a clinical diagnostic tool (7, 27, 30). Therefore, in an attempt to phenocopy this observation, we measured ISG expression in larval heads (Figure 2A). Variable expression of isg12, isg15 and stat1b was observed in samhd1Δ23/Δ23 larvae compared to the WTs. However, although variable, the samhd1Δ23/Δ23 larvae appeared to display increased expression of each ISG, although this did not reach statistical significance. Expression analysis of other ISGs displayed similar variability (data not shown).
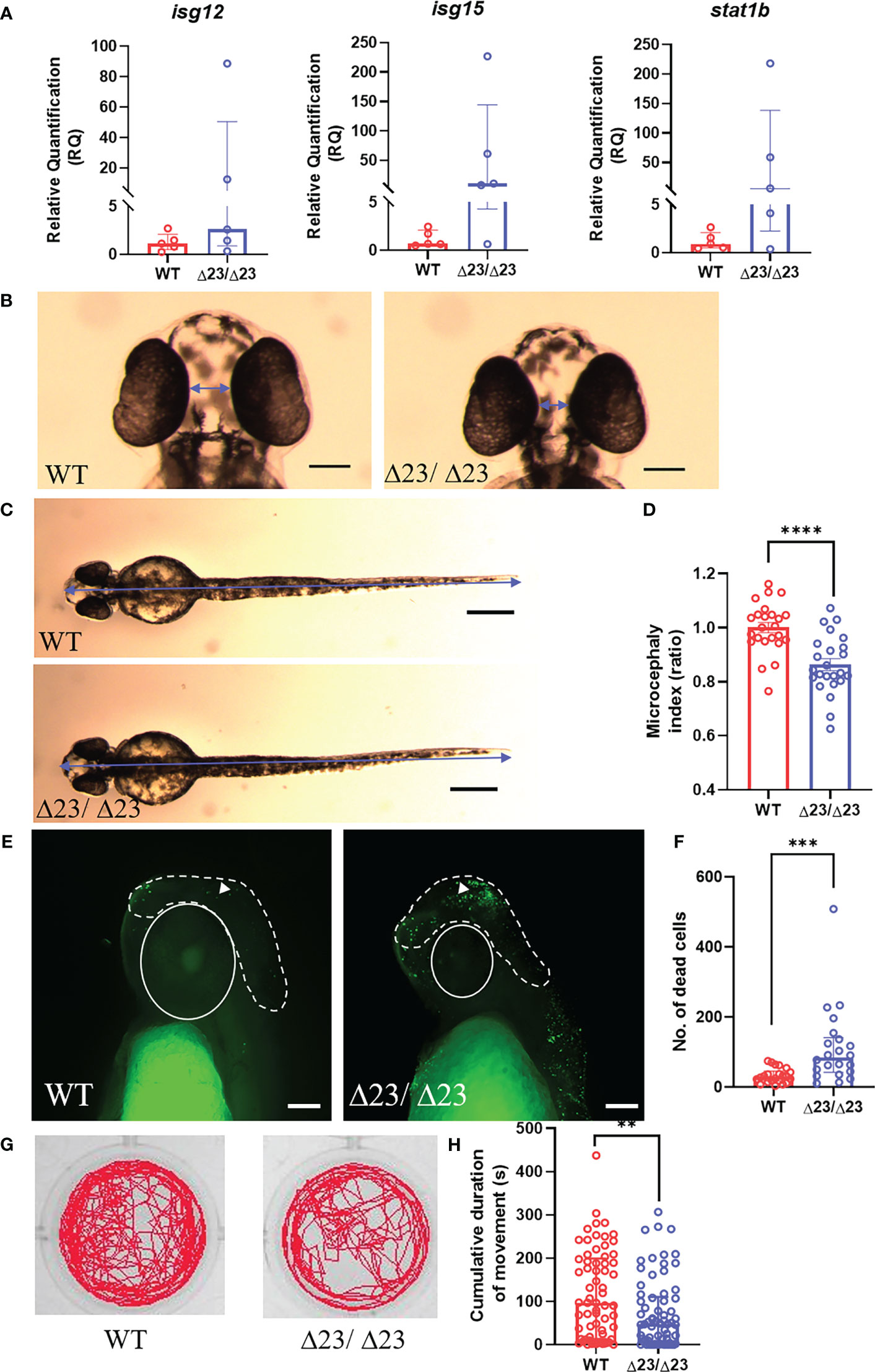
Figure 2 samhd1Δ23/Δ23 zebrafish exhibit neurological phenotypes. (A) Specific ISG expression (from taqman probes for isg12, isg15 and stat1b, normalized to the housekeeper gene hprt1) measured in 5 dpf WT and samhd1Δ23/Δ23 larval heads, n=30 per biological replicate, repeated 5 times. (B-D) Microcephaly phenotype identified by ventral measurements between the eyes (B) and full body length (C) (blue arrows) to generate a microcephaly index (ratio) determined by distance between the eyes and body length (D). Data analyzed using unpaired t test (****P<0.0001) presented as mean ± SEM. (E, F) Enhanced head cell death in samhd1Δ23/Δ23 embryos determined through TUNEL staining. White dotted line indicates area of interest for manual cell counting, excluding the eyes, mouth and ears. White triangle denotes TUNEL positive cell used for analysis. n=6-8 embryos per group, with 3 biological replicates. Scale bar= 100 μm. Quantification of number of dead cells (F). Data analyzed using Mann Whitney U test (***P<0.001) presented as median ± IQR. (G) Representative examples of locomotor tracks for 4 dpf samhd1Δ23/Δ23 and WT larvae. (H) Significant decrease in cumulative duration of movement at 4 dpf. n=22-24 larvae per group, with three biological replicates. Data analyzed using a Mann Whitney U test (**P<0.001).
Severe neurological manifestations are another important clinical feature of AGS, with patients presenting with a range of CNS symptoms (7, 31). To characterize the initiation of neurological phenotypes in the zebrafish model, we first measured head size, as microcephaly is a common clinical manifestation of AGS (32–34). The distance between the eyes after ventral imaging of fixed embryos at 2 dpf was used to measure microcephaly (Figure 2B). Full body length was also measured to determine a head to body length ratio, also referred to as a microcephaly index, normalized to the mean of the WT group (Figure 2C). The microcephaly index was significantly reduced by 15% in samhd1Δ23/Δ23 embryos (P<0.0001), thus indicating that loss of samhd1 is associated with smaller head size in zebrafish (Figure 2D).
Previous studies have suggested that microcephaly can be attributed to an increase in neuronal cell death (35). In an attempt to assess this, TUNEL staining was used to identify apoptotic cells in the head region. Manual counting of apoptotic cells in the indicated region (Figure 2E) revealed a significant increase in brain cell death in the samhd1Δ23/Δ23 embryos (P<0.001) (Figure 2F).
Subsequently, we aimed to establish whether the microcephaly and increased brain cell death in samhd1Δ23/Δ23 embryos also contributed to functional defects. From a clinical perspective, a wide range of motor disabilities are observed across all human AGS subtypes (7). Swimming behavior can be readily assessed in zebrafish larvae as a readout for motor dysfunction, as observed in other zebrafish models of disease (25). We tracked swimming behavior in the larvae between 3 - 5 dpf. No difference in movement was observed at either 3 dpf or 5 dpf (data not shown), however, a ~50% reduction in cumulative duration of movement was observed in the larvae at 4dpf (P<0.01), implying a transient physical deficit exists in the samhd1Δ23/Δ23 model (Figures 2G, H).
We next tested for AGS5-related cerebrovasculopathy, as the samhd1 MO model developed spontaneous cerebral hemorrhages (18). At baseline conditions, only a small proportion (5%) of samhd1Δ23/Δ23 embryos developed cerebral hemorrhages, whilst no hemorrhages were observed in the WT embryos.
Pharmacological inhibition of the 3-hydroxy-3-methylglutaryl-CoA reductase (hmgcr) pathway, using statins, has previously been shown to induce brain specific hemorrhages in zebrafish (25, 26). The hemorrhages arise due to a reduction in de novo cholesterol biosynthesis, resulting in lowered cholesterol essential for neurovascular integrity (26). As only a small percentage of embryos spontaneously hemorrhaged in the samhd1Δ23/Δ23 model at baseline, we tested whether the mutants may be more susceptible to brain hemorrhage following exposure to low concentrations of ATV. Following water bath incubation at ~30 hpf, ATV-induced hemorrhages were consistently identified from ~52 hpf. The incidence of hemorrhages increased in a dose-dependent manner for both mutant and WT groups, however, there was a significant increase in cerebral hemorrhage frequencies in the samhd1Δ23/Δ23 embryos at the lower doses of 0.25 μM (P<0.05) and 0.5 μM (P<0.05) ATV: 17% versus 38% and 63% versus 87% respectively (Figures 3A, B). We postulated that increased susceptibility to ATV-induced cerebral hemorrhages may indicate a deficiency in hmgcr expression in the homozygotes. Therefore, we measured hmgcr transcript by qPCR in larval heads and identified a ~50% reduction in samhd1Δ23/Δ23 larvae compared to WT (Figure 3C). Together, these data suggest that the samhd1Δ23/Δ23 embryos possess a genetically-induced susceptibility to cerebral hemorrhages that is associated with dysregulated cholesterol biosynthesis.
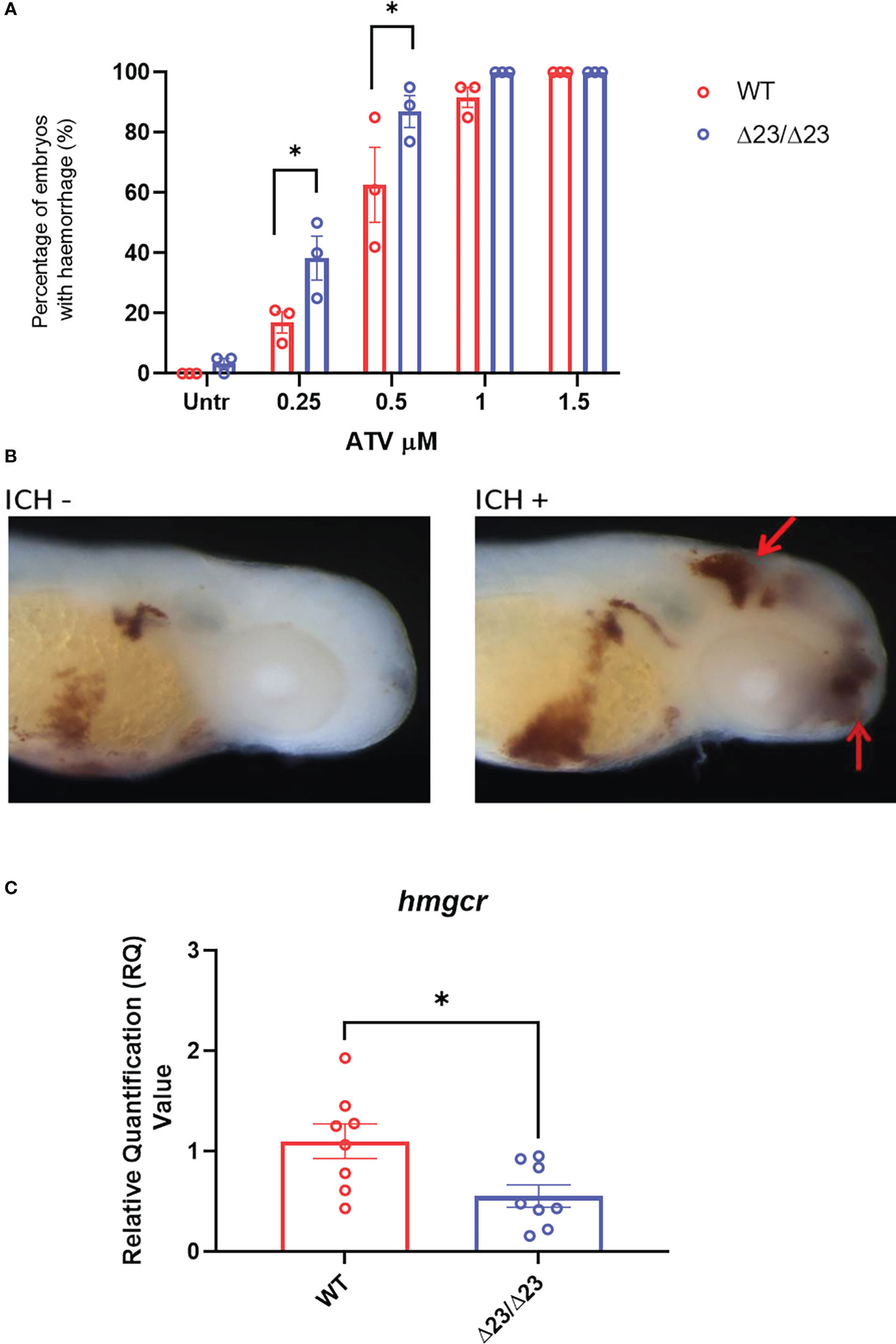
Figure 3 samhd1Δ23/Δ23 zebrafish exhibit cerebrovascular defects associated with cholesterol dysregulation. (A) WT and samhd1 Δ23/Δ23 embryos were treated with increasing concentrations (0.25-1.5 μM) of ATV added via water bath incubation and scored on the presence or absence of blood in the brain. N=20 embryos treated per group, with the percentage of animals per group which developed hemorrhages determined, repeated three times. Data analyzed using a two-way ANOVA with sidak’s multiple comparisons test (*P<0.05). (B) Representative images of embryos stained for o-dianisodine with and without brain hemorrhage, denoted by red arrow. (C) qPCR analysis of hmgcr expression in the WT and mutant larvae heads at 5 dpf. Data analyzed using unpaired t test (*P<0.05) and presented as mean ± SEM.
AGS5 patients frequently present with cerebrovascular disease (13–16). Furthermore, reductions in cholesterol have been associated with increased risk of hemorrhagic stroke in the general population (36–40). As such, we hypothesized that a reduction in cholesterol biosynthesis gene expression could be specifically associated with AGS5 and not observed across other AGS subtypes in which cerebrovasculopathy is not reported. To test this hypothesis and to validate the translational relevance of the samhd1Δ23/Δ23 zebrafish observation, we assessed the expression of several genes encoding enzymes involved in the multiple arms of the cholesterol biosynthesis pathway (Supplementary Figure 2) in existing RNA sequencing data obtained from AGS1-7 patient whole blood samples. This analysis revealed an overall increase in the expression of 9 of the 14 cholesterol biosynthesis genes measured (ACAT2, MVD, CYP51A, DHCR24, MSMO1, NSDHL, HSD17B, EBP, SC5D), compared to an age matched control group (Figure 4). This pattern was found across all AGS subtypes, excluding AGS1 patients, where no differences were observed. Although directionally inverse to the zebrafish model, these observations implicate defective cholesterol biosynthesis in the pathophysiology of AGS.
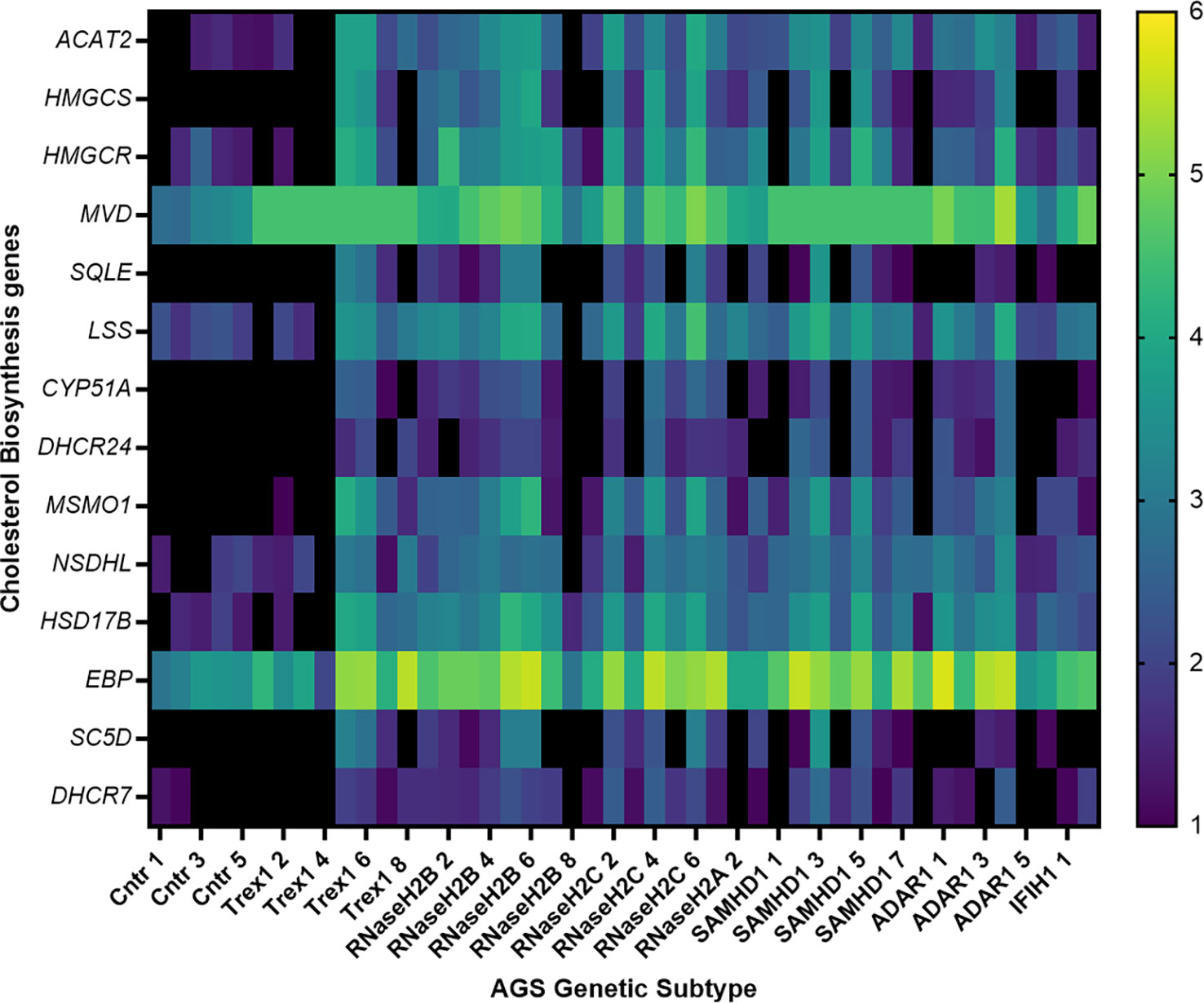
Figure 4 Transcriptional dysregulation of cholesterol biosynthesis genes in AGS patient blood. RNA from AGS1-7 patient whole blood was analyzed using the RSEM software to determine the TPM of each cholesterol biosynthesis gene for the AGS patients. Data was then normalized to an age-matched control group, and underwent a log2 transformation. The heat map scale on right hand side of figure indicates color changes representing 1-6 fold changes in expression. Sample sizes for each AGS subtype: Control n=5, TREX1 n=8, RNaseH2B n=8, RNaseH2C n=6, RNaseH2A n= 3, SAMHD1 n=8, ADAR1 n=6 and IFIH1 n=2. Data analyzed using a one-way ANOVA with Dunnetts multiple comparison test, comparing each AGS gene with the control group.
Here, we provide further evidence that zebrafish disease modelling represents a useful in vivo tool for studying AGS5, building upon the knowledge gained from the samhd1 MO zebrafish model previously described by our group (18). Furthermore, this work has provided new insight into AGS pathophysiology by implicating a potential role for defective cholesterol biosynthesis in the samhd1Δ23/Δ23 zebrafish and patient samples.
Loss of samhd1 produced a number of clinically relevant phenotypes in the zebrafish model, Firstly, the ISG response in larval heads was largely variable, and as such, we were unable to confirm a consistent ISG phenotype. However, high expression levels of ISGs were observed in a proportion of mutant larvae suggesting the discrepancy we observe may be attributed to distinct intra-larval variation. Indeed, heterogeneous ISG responses and symptom severity is also observed in the human condition (41). Intra-larval variation may be apparent due to the non-sterile aquatic environment the adult fish are housed in. Indeed, Rutherford and colleagues have previously suggested zebrafish are subjected to additional inflammagen exposure and other challenges from the water, compared to a sterile mouse environment (17). Upregulation of ISGs has been previously identified in a number of AGS animal models, resulting in multi-organ inflammation. However, the brain has remained largely unaffected, until relatively recently with the advent of newer murine models for Rnaseh2b and Adar1 mutations, and now the samhd1Δ23/Δ23 zebrafish model described here (42–45).
Due to the variation in ISG expression, we were unable to confirm that this upregulation is solely accountable for the presence of other observed neurological phenotypes, such as microcephaly, enhanced brain cell death and the locomotor deficits that were observed in the mutants. Future studies will assess the effects of IFN inhibition on these outcomes in this model. Interestingly, DNA damage, rather than IFN signaling has recently been hypothesized to be the cause of neural dysfunction in a neuro-progenitor conditional Rnaseh2b-/- mouse model (42, 46). Further study is required to determine whether DNA damage is the primary cause of these phenotypes in the zebrafish model.
The samhd1 MO model gave rise to a spontaneous brain hemorrhage phenotype, providing one of the first examples of a sub-type specific phenotype identified in a pre-clinical AGS model (18). For AGS5 this includes cerebral large artery disease, manifesting as stenosis, aneurysms, moyamoya, ischemic and hemorrhagic stroke (7, 13–15). As such, it has been hypothesized these cerebrovascular phenotypes reveal an as yet uncharacterized function of the SAMHD1 protein, relating to vascular homeostasis (14, 18). Moreover, the cerebrovascular defects found in AGS5 may be attributed to the spatial expression profile of SAMHD1. Whole tissue microarray from human donors identified constitutive expression of SAMHD1 within the vascular endothelium, including the CNS. However, expression profiles should also be established for the other AGS causative genes (47).
Whilst a small proportion of samhd1Δ23/Δ23 embryos did hemorrhage under baseline conditions, this was at a largely reduced frequency than observed in the previous samhd1 morphants (18). Therefore, we hypothesized that the stable mutants may possess a more subtle defect within the cerebrovasculature, which in isolation is insufficient to cause spontaneous hemorrhages, but may instead lead to an increased propensity to cerebral bleeds. To test this hypothesis, embryos were treated with a range of doses of the hemorrhage inducing drug ATV, where we observed a significant increase in hemorrhaging rates at the lowest doses in samhd1Δ23/Δ23 fish (25, 26). Given that ATV exerts its effects by targeting the cholesterol biosynthesis rate limiting enzyme, hmgcr, and inhibiting downstream cholesterol and lipid biosynthesis, this suggested the mutants may possess a baseline deficiency within the cholesterol biosynthesis pathway. Gene expression analysis of hmgcr highlighted a significant reduction in the samhd1 mutants compared to the WTs, thus providing novel evidence that a loss of samhd1 results in a genetic defect in cholesterol biosynthesis. Indeed, the relationship between increased cerebral hemorrhage rates and inhibition of cholesterol synthesis aligns with the clinical observation that reduced cholesterol levels are associated with increased risk of hemorrhagic stroke (36–40). Therefore, we hypothesized that this observation may account specifically for the cerebrovascular disease observed in AGS5 patients.
Patient whole blood RNA-seq analysis revealed the cholesterol biosynthesis gene dysregulation was not specific for SAMHD1-related AGS, indicating this observation cannot be directly attributed to the AGS5-related cerebrovasculopathy, as comparable effects were observed within AGS2-4 and 6-7 genetic subtypes. We propose that cholesterol biosynthesis dysregulation may be more broadly associated with aberrant IFN-I signaling, as highlighted by others (48). Notably, there were directional differences in dysregulated cholesterol gene expression identified between the samhd1Δ23/Δ23 larvae (downregulation) and patients (upregulation). We postulate these differences may reflect acute versus chronic exposure to aberrant IFN-I signaling, whereby a compensatory effect is initiated to counteract the inhibition of cholesterol synthesis over time. Such compensation is more likely feasible in patients over time, in comparison to the first five days of life in zebrafish. Moreover, we have only focused on cholesterol biosynthesis gene expression within the patients and zebrafish model, which does not fully portray the complex relationship between the antiviral response, initiated by IFN-I, and regulation of cholesterol.
As such, further research is required to understand this potential relationship between IFN-I signaling and cholesterol biosynthesis dysregulation specifically in AGS patients. We speculate that such a defect might contribute to certain pathological features of AGS, including white matter loss. The brain is cholesterol rich and a significant proportion is contained within myelin. Therefore, under disease conditions, defects in cholesterol may dysregulate myelin formation and lead to white matter degeneration. Indeed, in a mouse model of cerebrovascular disease, a high cholesterol diet was associated with white matter defects and cognitive decline (49).
Moreover, we appreciate that peripheral measurements of cholesterol biosynthesis gene expression do not directly represent the cholesterol lipid levels within the brain and CNS, and as such, future experimentation would involve taking plasma and CSF cholesterol measurements, whilst also investigating pathways other than cholesterol biosynthesis, such as cholesterol efflux. Whilst material is scarce, an interesting avenue for future studies would be to focus on assessing brain cholesterol levels in AGS post mortem tissue.
Irrespective of our focus on the periphery, previous literature has identified direct correlative relationships between plasma cholesterol levels and a number of different brain pathologies, including stroke (both ischemic and hemorrhagic), intracranial calcifications, and chronic neurodegenerative disorders (50, 51). Together, this demonstrates the importance of this RNA sequencing result in an attempt to understand aspects of the encephalopathic nature of AGS.
A limitation of the current study is that potential off-target effects initiated by the sgRNAs in zebrafish have not been fully assessed. Future studies could focus on using next generation sequencing to identify potential off-target candidates to further validate this model. Nevertheless, confirmation of cholesterol biosynthesis defects in both zebrafish and AGS patients provides confidence that this particular observation is likely bona fide.
To conclude, we have generated a stable mutant zebrafish model of AGS5, which exhibited several clinically relevant phenotypes, including neurological manifestations which have rarely been replicated in other pre-clinical models of AGS, thus reinforcing the usefulness of zebrafish as a pre-clinical species for type I interferonopathy research. The zebrafish model highlighted a potential novel association with AGS and dysregulation in cholesterol biosynthesis, which will spark further investigation into the role of cholesterol dysregulation in disease progression within AGS, whilst also increasing our understanding of the complexities surrounding IFN-I signaling and cholesterol homeostasis.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-9722?accession=E-MTAB-9722 and https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-7087?accession=E-MTAB-7087.
The studies involving human participants were reviewed and approved by Leeds (East) research ethics committee (reference number: 07/Q1206/7) South Centre-Hampshire A research ethics committee (reference number 17/SC/0026). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by The University of Manchester Animal Welfare and Ethical Review Board.
SW designed and performed experiments, analyzed the data, and wrote the manuscript. GR, VT, FH and I-EM designed and performed experiments. CR and AH performed data analysis. AB and SC provided technical expertise. SA and TB provided supervision and revised the manuscript. PK supervised the study and wrote the manuscript. All authors contributed to the article and approved the submitted version.
SW was funded by a MRC Doctoral Training Programme studentship (MR/N013751/1) and an English Welsh and Scottish railway (EWS) Exceptional contribution award. PK acknowledges funding from the MRC (MR/T03291X/1) and the Stroke Association (TSA LECT 2017/02). TB acknowledges funding from Medical Research Foundation (MRF-159-0002-ELP_BRIGG) and is supported by NIHR Manchester Biomedical Resource Centre.
We thank Prof. Yanick Crow for help with the initiation of this project and acknowledge funding from ERA-NET (MR/M501803/1). We also thank the aquatics staff at the Biological Services Unit at The University of Manchester for assistance with zebrafish care. Additionally, we thank the patients and their families for the donation of blood for this project. All authors are thankful to The Natalie Kate Moss Trust for their continued financial support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1100967/full#supplementary-material
1. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease Trex1 cause aicardi-goutieres syndrome at the Ags1 locus. Nat Genet (2006) 38(8):917–20. doi: 10.1038/ng1845
2. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in genes encoding ribonuclease H2 subunits cause aicardi-goutieres syndrome and mimic congenital viral brain infection. Nat Genet (2006) 38(8):910–6. doi: 10.1038/ng1842
3. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in aicardi-goutieres syndrome implicate Samhd1 as regulator of the innate immune response. Nat Genet (2009) 41(7):829–32. doi: 10.1038/ng.373
4. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in Adar1 cause aicardi-goutieres syndrome associated with a type I interferon signature. Nat Genet (2012) 44(11):1243–8. doi: 10.1038/ng.2414
5. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-Function mutations in Ifih1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet (2014) 46(5):503–9. doi: 10.1038/ng.2933
6. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. Cgas-mediated induction of type I interferon due to inborn errors of histone pre-mrna processing. Nat Genet (2020) 52(12):1364–72. doi: 10.1038/s41588-020-00737-3
7. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, adar, and Ifih1. Am J Med Genet A (2015) 167A(2):296–312. doi: 10.1002/ajmg.a.36887
8. Adang L, Gavazzi F, De Simone M, Fazzi E, Galli J, Koh J, et al. Developmental outcomes of aicardi goutieres syndrome. J Child Neurol (2020) 35(1):7–16. doi: 10.1177/0883073819870944
9. Vanderver A, Prust M, Kadom N, Demarest S, Crow YJ, Helman G, et al. Early-onset aicardi-goutieres syndrome: Magnetic resonance imaging (Mri) pattern recognition. J Child Neurol (2015) 30(10):1343–8. doi: 10.1177/0883073814562252
10. La Piana R, Uggetti C, Olivieri I, Tonduti D, Balottin U, Fazzi E, et al. Bilateral striatal necrosis in two subjects with aicardi-goutieres syndrome due to mutations in Adar1 (Ags6). Am J Med Genet A (2014) 164A(3):815–9. doi: 10.1002/ajmg.a.36360
11. Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in Adar1. J Med Genet (2014) 51(2):76–82. doi: 10.1136/jmedgenet-2013-102038
12. Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJ, et al. Mutations in Adar1, Ifih1, and Rnaseh2b presenting as spastic paraplegia. Neuropediatrics (2014) 45(6):386–93. doi: 10.1055/s-0034-1389161
13. Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, et al. Intracerebral Large artery disease in aicardi-goutieres syndrome implicates Samhd1 in vascular homeostasis. Dev Med Child Neurol (2010) 52(8):725–32. doi: 10.1111/j.1469-8749.2010.03727.x
14. Thiele H, du Moulin M, Barczyk K, George C, Schwindt W, Nurnberg G, et al. Cerebral arterial stenoses and stroke: Novel features of aicardi-goutieres syndrome caused by the Arg164x mutation in Samhd1 are associated with altered cytokine expression. Hum Mutat (2010) 31(11):E1836–50. doi: 10.1002/humu.21357
15. du Moulin M, Nurnberg P, Crow YJ, Rutsch F. Cerebral vasculopathy is a common feature in aicardi-goutieres syndrome associated with Samhd1 mutations. Proc Natl Acad Sci U.S.A. (2011) 108(26):E232. doi: 10.1073/pnas.1104699108
16. Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, et al. Homozygous mutation in Samhd1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci U.S.A. (2011) 108(13):5372–7. doi: 10.1073/pnas.1014265108
17. Rutherford HA, Kasher PR, Hamilton N. Dirty fish versus squeaky clean mice: Dissecting interspecies differences between animal models of interferonopathy. Front Immunol (2020) 11:623650. doi: 10.3389/fimmu.2020.623650
18. Kasher PR, Jenkinson EM, Briolat V, Gent D, Morrissey C, Zeef LA, et al. Characterization of Samhd1 morphant zebrafish recapitulates features of the human type I interferonopathy aicardi-goutieres syndrome. J Immunol (2015) 194(6):2819–25. doi: 10.4049/jimmunol.1403157
19. Westerfield M. The zebrafish book. a guide for the laboratory use of zebrafish (Danio rerio). 4th Ed. Eugene: Univ. of Oregon Press (2000).
20. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn (1995) 203(3):253–310. doi: 10.1002/aja.1002030302
21. Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. Chopchop: A Crispr/Cas9 and talen web tool for genome editing. Nucleic Acids Res (2014) 42(Web Server issue):W401–7. doi: 10.1093/nar/gku410
22. Badrock AP, Uggenti C, Wacheul L, Crilly S, Jenkinson EM, Rice GI, et al. Analysis of U8 snorna variants in zebrafish reveals how bi-allelic variants cause leukoencephalopathy with calcifications and cysts. Am J Hum Genet (2020) 106(5):694–706. doi: 10.1016/j.ajhg.2020.04.003
23. Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, et al. Ensembl 2022. Nucleic Acids Res (2022) 50(D1):D988–D95. doi: 10.1093/nar/gkab1049
24. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. Expasy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res (2003) 31(13):3784–8. doi: 10.1093/nar/gkg563
25. Crilly S, Njegic A, Laurie SE, Fotiou E, Hudson G, Barrington J, et al. Using zebrafish larval models to study brain injury, locomotor and neuroinflammatory outcomes following intracerebral haemorrhage. F1000Res (2018) 7:1617. doi: 10.12688/f1000research.16473.2
26. Eisa-Beygi S, Hatch G, Noble S, Ekker M, Moon TW. The 3-Hydroxy-3-Methylglutaryl-Coa reductase (Hmgcr) pathway regulates developmental cerebral-vascular stability Via prenylation-dependent signalling pathway. Dev Biol (2013) 373(2):258–66. doi: 10.1016/j.ydbio.2012.11.024
27. Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. Assessment of interferon-related biomarkers in aicardi-goutieres syndrome associated with mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, and adar: A case-control study. Lancet Neurol (2013) 12(12):1159–69. doi: 10.1016/S1474-4422(13)70258-8
28. Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, et al. Reverse-transcriptase inhibitors in the aicardi-goutieres syndrome. N Engl J Med (2018) 379(23):2275–7. doi: 10.1056/NEJMc1810983
29. Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in Stat2. Sci Immunol (2019) 4(42). doi: 10.1126/sciimmunol.aav7501
30. Garau J, Cavallera V, Valente M, Tonduti D, Sproviero D, Zucca S, et al. Molecular genetics and interferon signature in the Italian aicardi goutieres syndrome cohort: Report of 12 new cases and literature review. J Clin Med (2019) 8(5). doi: 10.3390/jcm8050750
31. Livingston JH, Crow YJ. Neurologic phenotypes associated with mutations in Trex1, Rnaseh2a, Rnaseh2b, Rnaseh2c, Samhd1, Adar1, and Ifih1: Aicardi-goutieres syndrome and beyond. Neuropediatrics (2016) 47(6):355–60. doi: 10.1055/s-0036-1592307
32. Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol (1984) 15(1):49–54. doi: 10.1002/ana.410150109
33. Lanzi G, Fazzi E, D'Arrigo S. Aicardi-goutieres syndrome: A description of 21 new cases and a comparison with the literature. Eur J Paediatr Neurol (2002) 6 Suppl A:A9–22. doi: 10.1053/ejpn.2002.0568. discussion A3-5, A77-86.
34. Orcesi S, La Piana R, Fazzi E. Aicardi-goutieres syndrome. Br Med Bull (2009) 89:183–201. doi: 10.1093/bmb/ldn049
35. Stankiewicz P, Khan TN, Szafranski P, Slattery L, Streff H, Vetrini F, et al. Haploinsufficiency of the chromatin remodeler bptf causes syndromic developmental and speech delay, postnatal microcephaly, and dysmorphic features. Am J Hum Genet (2017) 101(4):503–15. doi: 10.1016/j.ajhg.2017.08.014
36. Valappil AV, Chaudhary NV, Praveenkumar R, Gopalakrishnan B, Girija AS. Low cholesterol as a risk factor for primary intracerebral hemorrhage: A case-control study. Ann Indian Acad Neurol (2012) 15(1):19–22. doi: 10.4103/0972-2327.93270
37. Wang X, Dong Y, Qi X, Huang C, Hou L. Cholesterol levels and risk of hemorrhagic stroke: A systematic review and meta-analysis. Stroke (2013) 44(7):1833–9. doi: 10.1161/STROKEAHA.113.001326
38. Phuah CL, Raffeld MR, Ayres AM, Viswanathan A, Greenberg SM, Biffi A, et al. Subacute decline in serum lipids precedes the occurrence of primary intracerebral hemorrhage. Neurology (2016) 86(22):2034–41. doi: 10.1212/WNL.0000000000002716
39. Chen YW, Li CH, Yang CD, Liu CH, Chen CH, Sheu JJ, et al. Low cholesterol level associated with severity and outcome of spontaneous intracerebral hemorrhage: Results from Taiwan stroke registry. PloS One (2017) 12(4):e0171379. doi: 10.1371/journal.pone.0171379
40. Sun L, Clarke R, Bennett D, Guo Y, Walters RG, Hill M, et al. Causal associations of blood lipids with risk of ischemic stroke and intracerebral hemorrhage in Chinese adults. Nat Med (2019) 25(4):569–74. doi: 10.1038/s41591-019-0366-x
41. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med (2016) 213(12):2527–38. doi: 10.1084/jem.20161596
42. Aditi, Downing SM, Schreiner PA, Kwak YD, Li Y, Shaw TI, et al. Genome instability independent of type I interferon signaling drives neuropathology caused by impaired ribonucleotide excision repair. Neuron (2021) 109(24):3962–79 e6. doi: 10.1016/j.neuron.2021.09.040
43. Guo X, Wiley CA, Steinman RA, Sheng Y, Ji B, Wang J, et al. Aicardi-goutieres syndrome-associated mutation at Adar1 gene locus activates innate immune response in mouse brain. J Neuroinflamm (2021) 18(1):169. doi: 10.1186/s12974-021-02217-9
44. Inoue M, Nakahama T, Yamasaki R, Shibuya T, Kim JI, Todo H, et al. An aicardi-goutieres syndrome-causative point mutation in Adar1 gene invokes multiorgan inflammation and late-onset encephalopathy in mice. J Immunol (2021), 8(5). doi: 10.4049/jimmunol.2100526
45. de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, et al. Adar1 interaction with z-rna promotes editing of endogenous double-stranded rna and prevents Mda5-dependent immune activation. Cell Rep (2021) 36(6):109500. doi: 10.1016/j.celrep.2021.109500
46. Lavin MF, Yeo AJ. DNA Damage rather than type I ifn signaling is the primary mediator of neural dysfunction in aicardi-goutieres syndrome after Rnaseh2 disruption. Neuron (2021) 109(24):3897–900. doi: 10.1016/j.neuron.2021.11.019
47. Schmidt S, Schenkova K, Adam T, Erikson E, Lehmann-Koch J, Sertel S, et al. Samhd1's protein expression profile in humans. J Leukoc Biol (2015) 98(1):5–14. doi: 10.1189/jlb.4HI0714-338RR
48. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting cholesterol biosynthetic flux spontaneously engages type I ifn signaling. Cell (2015) 163(7):1716–29. doi: 10.1016/j.cell.2015.11.045
49. Tong XK, Trigiani LJ, Hamel E. High cholesterol triggers white matter alterations and cognitive deficits in a mouse model of cerebrovascular disease: Benefits of simvastatin. Cell Death Dis (2019) 10(2):89. doi: 10.1038/s41419-018-1199-0
50. Vance JE. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis Model Mech (2012) 5(6):746–55. doi: 10.1242/dmm.010124
Keywords: Aicardi Goutières syndrome, cholesterol, zebrafish disease models, type I interferonopathy, SAMHD1
Citation: Withers SE, Rowlands CF, Tapia VS, Hedley F, Mosneag I-E, Crilly S, Rice GI, Badrock AP, Hayes A, Allan SM, Briggs TA and Kasher PR (2023) Characterization of a mutant samhd1 zebrafish model implicates dysregulation of cholesterol biosynthesis in Aicardi-Goutières syndrome. Front. Immunol. 14:1100967. doi: 10.3389/fimmu.2023.1100967
Received: 17 November 2022; Accepted: 20 February 2023;
Published: 06 March 2023.
Edited by:
Alexandre Belot, Université Claude Bernard Lyon 1, FranceReviewed by:
Sébastien Viel, Hospices Civils de Lyon, FranceCopyright © 2023 Withers, Rowlands, Tapia, Hedley, Mosneag, Crilly, Rice, Badrock, Hayes, Allan, Briggs and Kasher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul R. Kasher, cGF1bC5rYXNoZXJAbWFuY2hlc3Rlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.