 Zhangsheng Yang1
Zhangsheng Yang1 Tomas S. Cancio
Tomas S. Cancio Yansong Li
Yansong Li- 1Combat Casualty Care Research Team (CRT) 3, United States (US) Army Institute of Surgical Research, Joint Base San Antonio (JBSA)-Fort Sam Houston, TX, United States
- 2Division of Trauma Research, University of Texas Health Science Center at San Antonio, San Antonio, TX, United States
- 3United States (US) Army Burn Center, United States (US) Army Institute of Surgical Research, Joint Base San Antonio (JBSA)-Fort Sam Houston, TX, United States
- 4The Geneva Foundation, Immunological Damage Control Resuscitation Program, Tacoma, WA, United States
The hallmark of acute respiratory distress syndrome (ARDS) pathobiology is unchecked inflammation-driven diffuse alveolar damage and alveolar-capillary barrier dysfunction. Currently, therapeutic interventions for ARDS remain largely limited to pulmonary-supportive strategies, and there is an unmet demand for pharmacologic therapies targeting the underlying pathology of ARDS in patients suffering from the illness. The complement cascade (ComC) plays an integral role in the regulation of both innate and adaptive immune responses. ComC activation can prime an overzealous cytokine storm and tissue/organ damage. The ARDS and acute lung injury (ALI) have an established relationship with early maladaptive ComC activation. In this review, we have collected evidence from the current studies linking ALI/ARDS with ComC dysregulation, focusing on elucidating the new emerging roles of the extracellular (canonical) and intracellular (non-canonical or complosome), ComC (complementome) in ALI/ARDS pathobiology, and highlighting complementome as a vital nexus of the pathobiological connectome for ALI/ARDS via its crosstalking with other systems of the immunome, DAMPome, PAMPome, coagulome, metabolome, and microbiome. We have also discussed the diagnostic/therapeutic potential and future direction of ALI/ARDS care with the ultimate goal of better defining mechanistic subtypes (endotypes and theratypes) through new methodologies in order to facilitate a more precise and effective complement-targeted therapy for treating these comorbidities. This information leads to support for a therapeutic anti-inflammatory strategy by targeting the ComC, where the arsenal of clinical-stage complement-specific drugs is available, especially for patients with ALI/ARDS due to COVID-19.
Highlights
• ARDS is a life-threatening syndrome without specific pharmacotherapy for ARDS thus far.
• The role of profoundly destructive influence of unchecked ComC-driven air-blood-barrier failure, SIRS, immunothrombosis, meta-inflammation, PICS, and dysbiosis has been extensively studied in preclinical and clinical ARDS/ALI.
• ComC activation products (C3a, C4d, C5a, C5b-9, Bb) and their corresponding receptors provide useful diagnostic tools and promising therapeutic targets.
• With extreme heterogeneity in ARDS, the future of ARDS management strives towards identification of endotypes and theratypes through new methodologies and delivery of precision therapeutic interventions.
• Prospective studies are needed to confirm evidence of endotype-/theratype-driven therapeutic response prior to incorporation into clinical practice.
Open questions
• Is the composition of the complosome universal or cell specific?
• Which do the exact cellular subcompartments contain complement components, receptors and regulators for activation fragments?
• How does the complosome orchestrate the intracellular organelles (mitochondria, lysosomes, endoplasmic reticulum, etc.) and cytoskeleton?
• How does the complosome dance with the intracellular DAMPs and PAMPs?
• How does the complementome act in concert with other mutiomes to contribute to ARDS pathobiology?
1 Introduction
2022 marks the 55th year anniversary of the first description of acute respiratory distress syndrome (ARDS). Since then, substantial progress has been made in identifying the risk factors for and the pathogenic contributors to ARDS with an emphasis on the mechanisms of injury to the pulmonary endothelium and the epithelium. Despite considerable progress of pathophysiological mechanisms in the last five decades, no effective pharmacological therapeutics are available for ARDS patients yet. Current treatments still largely rely on supportive approaches of lung protective ventilation and a conservative fluid resuscitation (1, 2). Mortality remains high at 40%, and for patients who survive, recovery continues for months or even years. Recent studies have shown that uncontrolled complement cascade (ComC) activation may contribute to the pathological processes of ARDS (3–6). Therefore, modulation of the ComC may hold great promise for the treatment of acute lung injury (ALI) and ARDS. This review provides a broad overview of the ComC role in ALI and ARDS, discusses current understanding of the comprehensive molecular mechanistic insights of complement activation-mediated-ALI/ARDS, and highlights the newest developments in ComC pharmacological interventions for ARDS to help understand the rationale behind the current novel development of treatment strategies targeting ComC in ALI/ARDS patients.
2 ARDS
ARDS is a form of non-cardiogenic pulmonary edema, due to alveolar injury secondary to an inflammatory process. ARDS is a life-threatening condition of critically-ill patients characterized by acute onset of the illness, bilateral lung infiltrates, and respiratory distress due to severe arterial hypoxemia (7). This disorder is associated with pulmonary endothelial injury and alveolar epithelial damage. It carries a high mortality rate (8) without any effective pharmacological interventions.
2.1 Epidemiology
It has been reported that approximately 200,000 cases occur in the United States annually (8), with poor clinical outcomes with a pooled mortality rate of approximately 40% (8). Twenty-five percent of ARDS cases are classified as mild and 75% as moderate or severe. However, a third of the mild cases can progress to moderate or severe disease (9).
2.2 Pathophysiology and etiology
ARDS represents a stereotypic response to various etiologies. As shown in Table 1, its pathological changes have been described as three overlapping phases: exudative (acute), proliferative (sub-acute) and fibrotic (chronic) phases (11, 12). The hallmark of the acute phase is alveolar-capillary barrier dysfunction driven by immune-cell-mediated destruction of alveolar epithelium and vascular endothelium (11, 13), and is characterized by bilateral infiltration (14) (Figures 1A, B), interstitial and alveolar edema, capillary congestion, intra-alveolar hemorrhage, inflammatory cell infiltration, diffuse alveolar damage (DAD), intra-alveolar and intravascular fibrin deposition, hyaline membrane formation, and microthrombi (14, 15) (Figures 1C-G). These alterations lead to hypoxia, hyperinflammation, metabolic acidosis, pulmonary hypertension, and increased mortality. Traditionally, ARDS is recognized to be a neutrophil-driven disease. However, accumulating evidence has demonstrated the involvement of innate immune cells such as macrophages and platelets, and the adaptive immune system in the pathogenesis of ARDS. The proliferative phase occurs early (within the initial three days) and attempts to reduce the damage caused during the acute phase and restore pulmonary function with the regeneration of alveolar type II cells, differentiation of alveolar type II cells to alveolar type I cells, and the proliferation of fibroblasts and myofibroblasts. The fibrotic phase develops due to a failure of removal of alveolar collagen, which results in an increased dead-space fraction, high minute ventilation requirement, pulmonary hypertension, and further reduction of lung compliance. A hallmark of the damage seen in ARDS is that it is not uniform.
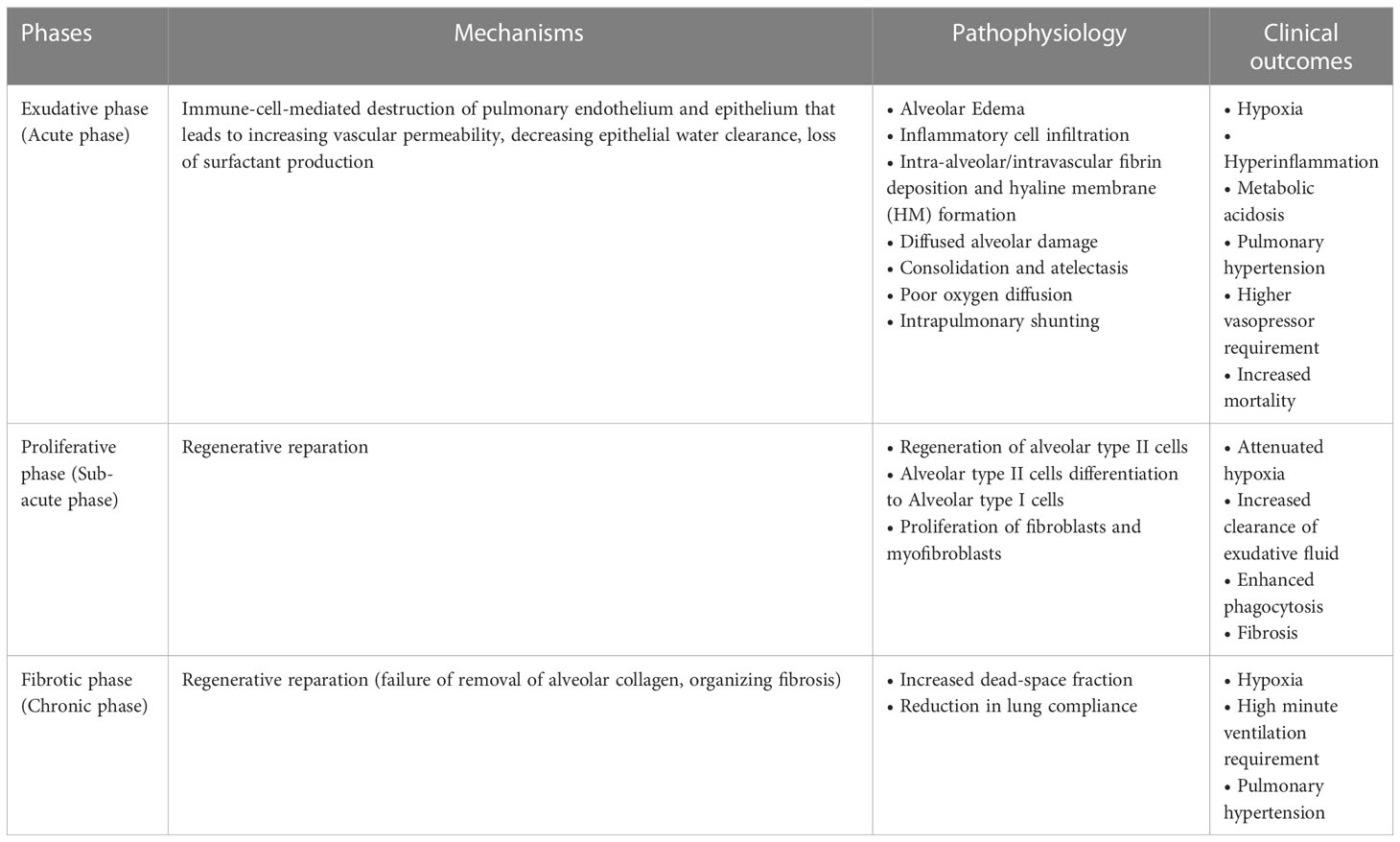
Table 1 Pathophysiology of ARDS (10).
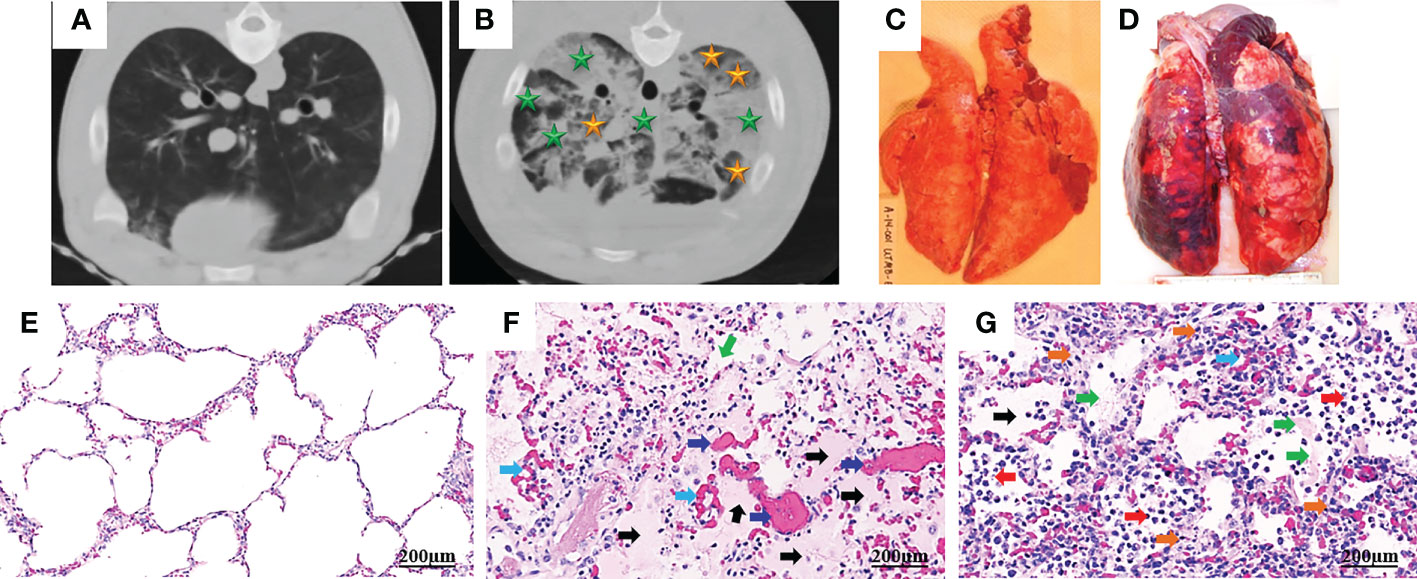
Figure 1 Computed tomography (CT) and histopathology of ARDS following smoke inhalation and burn in pigs. Anesthetized Yorkshire female pigs (35-50kg) were injured by a combination of wood bark smoke inhalation (the cooled smoke delivered with a dose of 20-30 liters at a tidal volume of 30 ml/kg) and a 40% total body surface area deep burn, and observed under a standard ARDSNet mechanical ventilation for 48 hours. The lung tissues were harvested at the end of study for macro-/micro-pathological evaluation and CT scan was conducted at the end of study. Chest CT from a non-injured lung (A) and an ARDS lung showing bilateral infiltration (B, golden star, ground glass opacities; green star, consolidation areas). Macropathology from a non-injured lung (C) and an ARDS lung (D). Lung from a non-injured pig (E) and an ARDS pig (F, G), stained with hematoxylin and eosin. The histological alterations of ARDS are characterized by alveolar (black arrow) and interstitial (golden arrow) edema, hyaline membrane formation (green arrows), inflammatory cell infiltration (red arrow), thrombosis (dark blue arrow), and vascular congestion (light blue arrow) (14, 15).
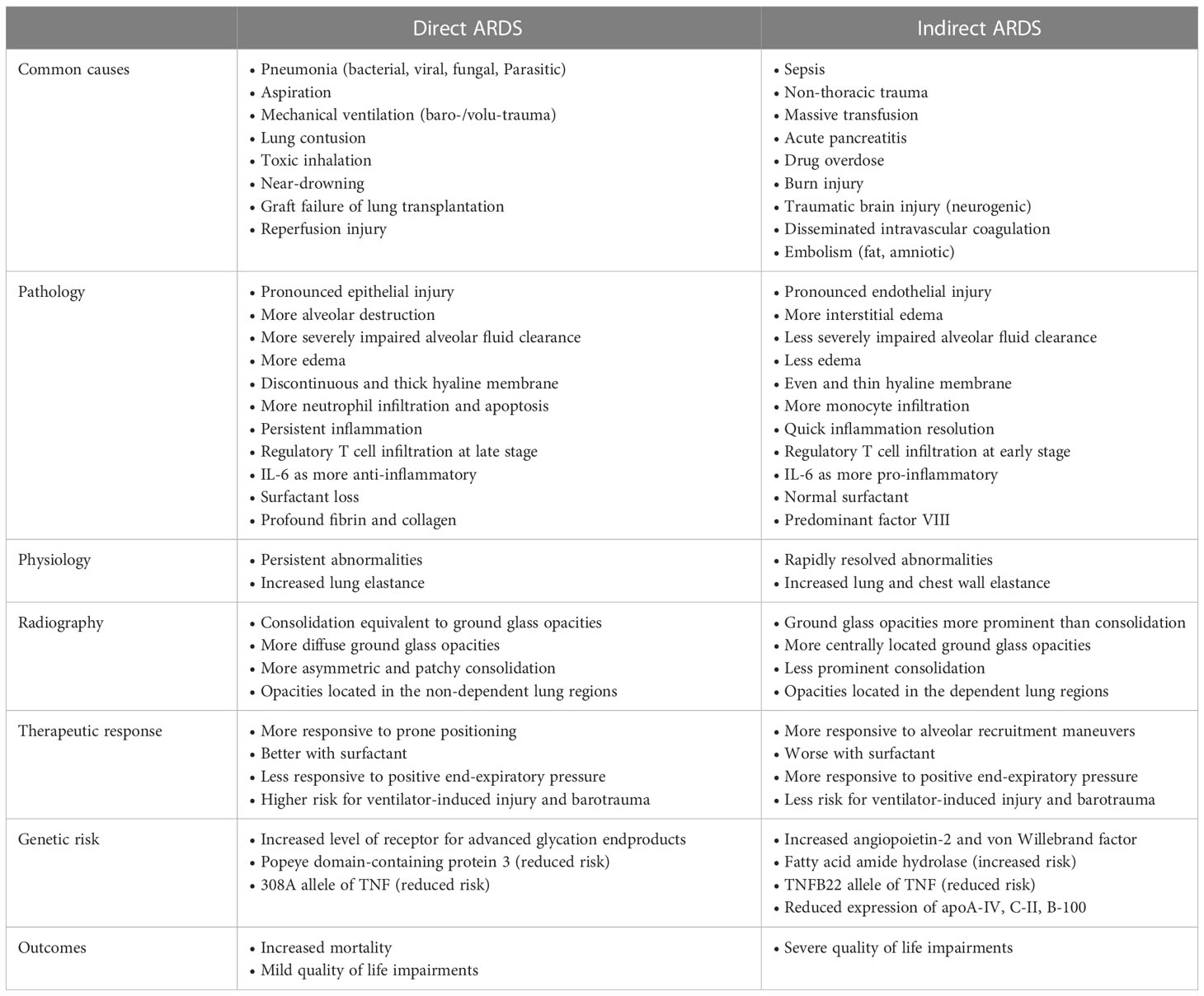
ARDS has many risk factors with heterogeneous etiologies. Generally, ARDS is classified into two major types: direct lung injury affecting the pulmonary epithelium and indirect lung injury disrupting the pulmonary vascular endothelium. Greater understanding of the differences between direct and indirect lung injury may help development of new therapies targeted at specific subgroups of patients with ARDS. The common causes of ARDS are summarized in Table 2. There are certain common pathophysiological differences between direct and indirect lung injury (10) (Table 2).
2.3 Diagnosis
No single diagnostic test confirms or refutes a diagnosis of ARDS. As shown in Table 3, the current diagnosis of ARDS is based on the Berlin Consensus Criteria in 2012 according to the timing, hypoxemia, positive end-expiratory pressure (PEEP), chest radiograph, and origin of edema. Safe, inexpensive, and point-of-care tools such as ultrasound imaging and pulse oximetry, can be implemented as diagnostic modalities for monitoring pulmonary infiltration and measuring SpO2/FiO2 ratio, respectively. In 2016, investigators proposed alternative criteria (SpO2/FiO2 ratio and lung ultrasound imaging) for ARDS diagnosis-the so-called Kigali modification of the Berlin Criteria. There is no biomarker for ARDS recommended in clinical practice. With the evolution of clinical care and increasing recognition of the global burden of ARDS, and the emerging new methodologies, the advent of these new methods (multiomics, big data, systems biology, and multiplexed point-of-care testing) combined with insights into biomarker strategies will likely yield robust information that will address the dynamic and complex interaction between risk factors, phenotypes, endotypes, and expression modulators and facilitate the development of precision medicine for ARDS patients through the endotype-/theratype-driven approach (Table 3).
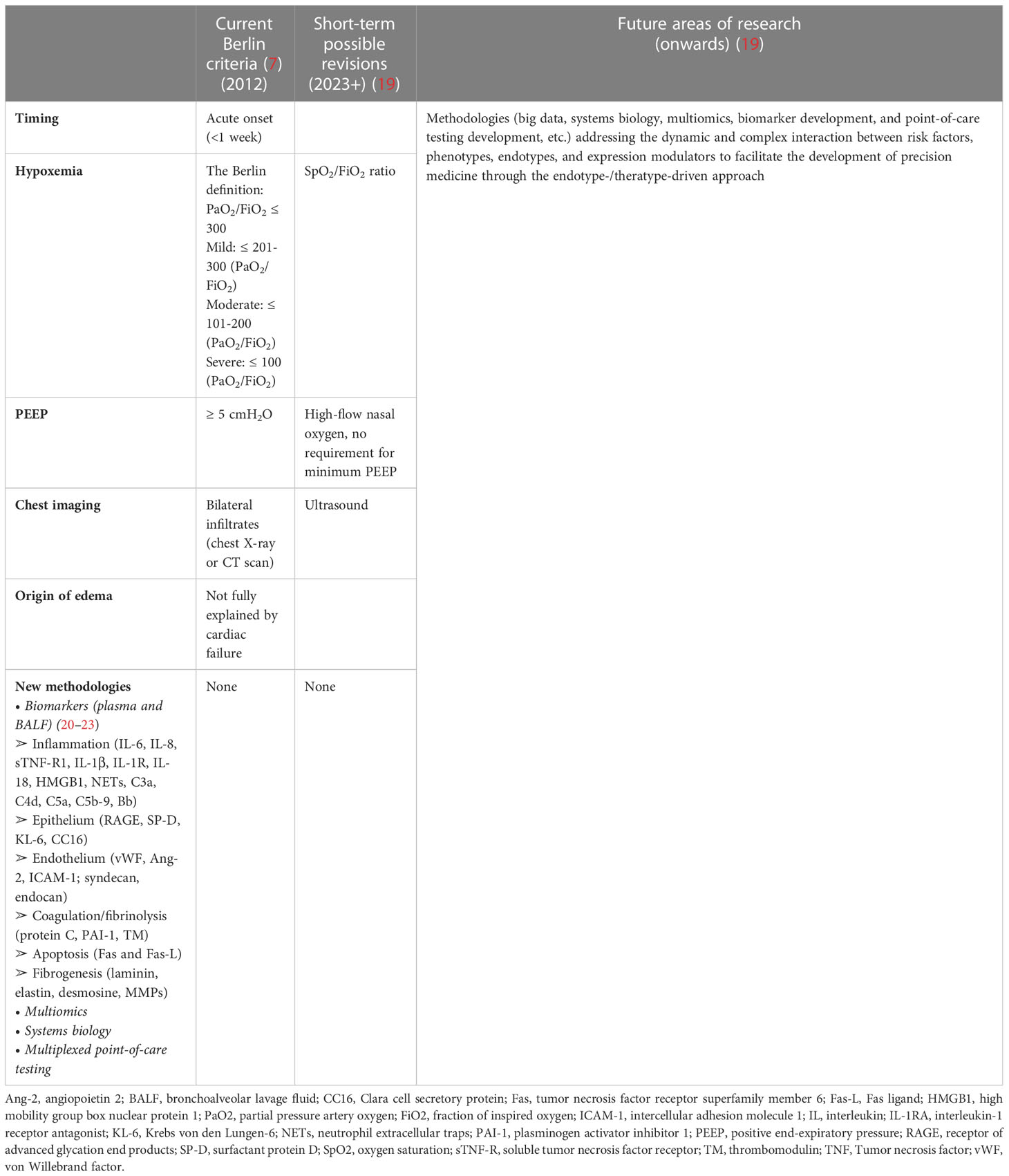
Table 3 Current ARDS criteria and future direction.
2.4 Treatment and management
ARDS requires urgent intubation and ventilation, careful assessment and treatment of the underlying causes and continual reassessment for potential complications. The major treatments are divided into supportive managements and specific managements. Supportive therapies include venous thromboembolism (VTE) prophylaxis, nutrition, mobilization, and sedation. Specific managements comprise lung protective mechanic ventilation, fluid-conservative management, neuromuscular blockade, prone positioning, and extracorporeal membrane oxygenation (ECMO) (24). Despite these standards of care, ARDS is still associated with poor clinical outcomes with a mortality rate of approximately 40%. The lack of effective pharmacotherapies presents a continuing challenge in the field.
3 Complement system
The complement system is an integral component of the innate immune system and a major initiator of inflammatory response. It has key roles in the recognition and elimination of invading pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by enhancing inflammation, opsonization, phagocytosis, the formation of neutrophil extracellular traps (NETs), and cellular lysis via bridging innate and adaptive immune immunity. However, exaggerated complement activation can contribute to tissue damage and organ dysfunction.
3.1 Key complement components and complement regulators
The complement system has key roles in both the innate and adaptive immune responses. It is composed of nearly 60 complement proteins that interact in a proteolytic cascade. As shown in Table 4, complement proteins can be divided into two broad categories: complement components that are involved in activation and complement regulators that inhibit complement activation. Traditionally, three different initiation pathways are known to activate the ComC. Complement can also be activated by extrinsic proteases including coagulation cascade (CoaC) proteases and neutrophil-/macrophage-derived proteases. The complement pathways are tightly regulated by complement inhibitors presented in the soluble form (blood) and the solid form (membrane bound).
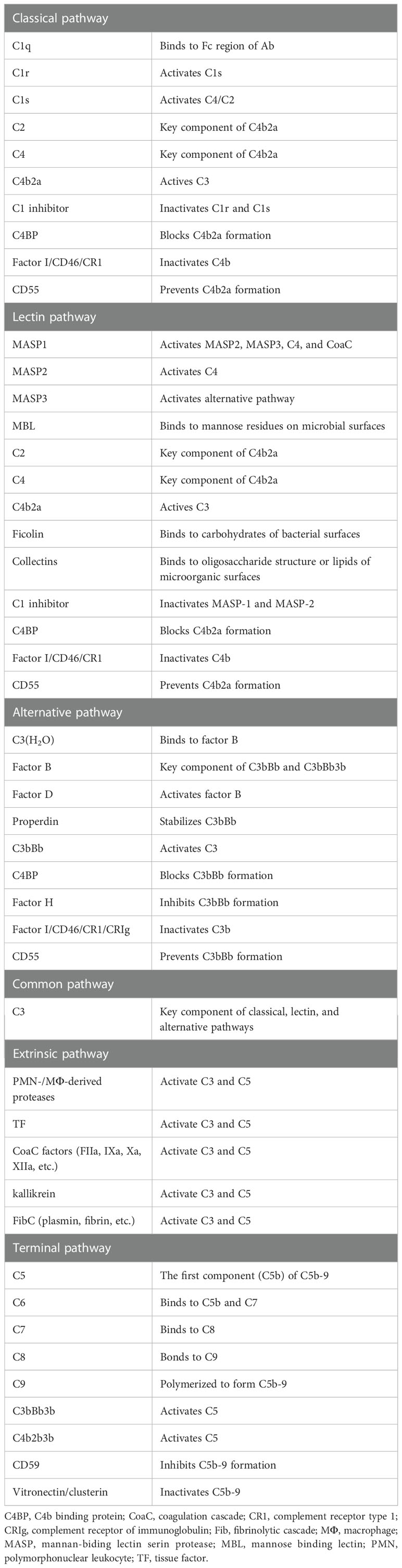
Table 4 Key complement components and their functions.
3.2 Complement activation pathways
The activated complement provides a critical link between the innate and adaptive immune responses. As shown in Table 4, the cascade has 4 initiating arms: the classical, lectin, alternative and extrinsic (driven by activated coagulation/fibrinolytic/kinin proteolytic enzymes) pathways. Each pathway is activated in a distinct mechanism. The classical pathway (CP) is initiated by binding pattern recognition proteins (PRPs) to immune complexes (ICs), C-reactive protein (CRP), serum amyloid P (SAP), DAMPs, and/or PAMPs, whereas the lectin pathway (LP) is homologous to the classical pathway and triggered by binding PRPs to exposed aberrant carbohydrates on foreign, damaged or necrotic cells. Unlike the CP and LP, the alternative pathway (AP) amplifies the initial response and maintains a low level of activity by a mechanism known as “tickover” (25). However, the AP is always primed to respond quickly and vigorously to pathogens or injury, and accounts for as much as 80% of C5a and C5b-9 production.
In addition, the extrinsic pathway that activates ComC has been described by Huber-Lang et al. (26). Traditionally, the plasma cascades, including the ComC, coagulation cascade (CoaC), fibrinolytic cascade (FibC), and kinin cascade (KinC), are described as separate cascades. Nevertheless, these systems belong to complex inflammatory networks (27) and exhibit similar characteristics regarding specialized functions of their activators and inhibitors. Expanding evidence indicates multiple interactions among these cascades under physiological and pathological conditions. It has been documented that active components of the CoaC (factor IIa, IXa, Xa, XIa, XIIa) (26, 28), FibC (plasmin) (29–31), and kinC (kallikrein) (32) can cleave and/or activate proteins (C3, C5) of the ComC, leading to the generation of ComC activation products (C3a, C3b, C5a, and C5b-9).
Appropriate activation of the ComC play an important role in host hemostasis, defense against infection, immune complexes and damaged cell clearance, priming adaptive immunity, regeneration, and metabolic reprogramming. However, excessive ComC activation could trigger systemic inflammatory response syndrome, cellular damage, and alveolar vascular/epithelial hyperpermeability leading to ARDS and multiple-organ failure (Figures 2, 3).
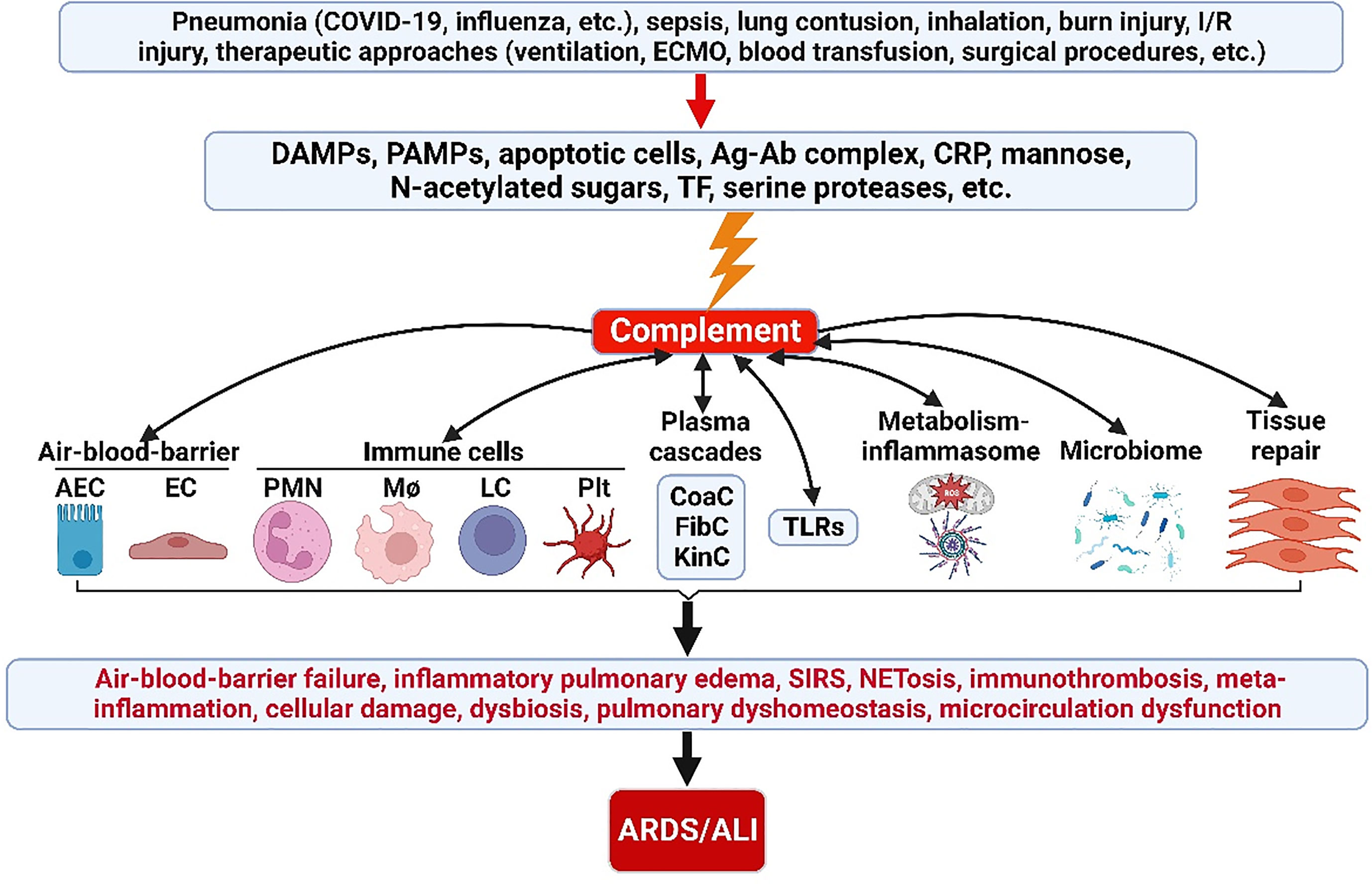
Figure 2 ComC in the pathogenesis of ARDS/ALI. A variety of acute insults trigger complement activation via DAMPs, PAMPs, apoptotic cells, Ag-Ab complex, CRP, mannose, N-acetylated sugars, TF and serine proteases. The activated ComC resides upstream of immunity and acts as a vital nexus of the pathobiological connectome for ARDS/ALI. AEC, alveolar epithelial cell; Ag-Ab, antigen-antibody; ALI, acute lung injury; ARDS, acute respiratory distress syndrome; CoaC, coagulation cascade; ComC, complement cascade; CRP, C-reactive protein; DAMPs, damage-associated molecular patterns; EC, endothelial cell; ECMO, extracorporeal membrane oxygenation; FibC, fibrinolytic cascade; I/R, ischemia/reperfusion; KinC, kinin cascade; LC, lymphocyte; NETosis, neutrophil extracellular trap formation; PAMPs, pathogen-associated molecular patterns; PICS, persistent inflammation/immunosuppression and catabolism syndrome; plt, platelet; PMN polymorphonuclear neutrophil; SIRS, systemic inflammatory response syndrome; TF, tissue factor; TLRs, toll-like receptors; MΦ, macrophage.
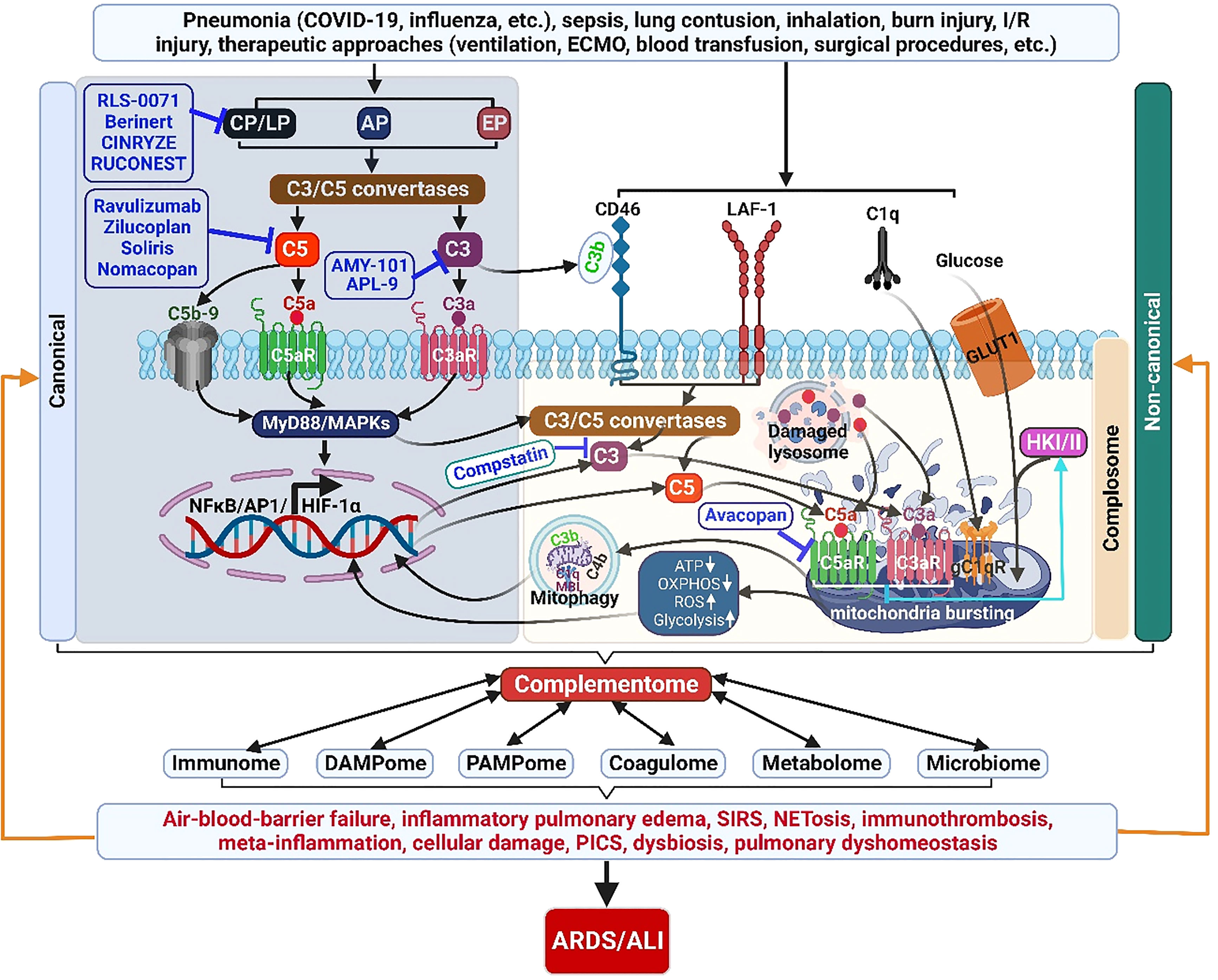
Figure 3 The complementome role and therapeutic targets in ALI/ARDS. Acute insults can induce exaggerated ComC activation locally and systemically. Complement proteins are produced and released intrahepatically providing the systemic extracellular ComC. Complement proteins are also synthesized extrahepatically (locally) by other cells and the intracellular local ComC plays a pivotal role in cell homeostasis and metabolisms. The activated complementome acts as a pivotal nexus to activate other systems of the immunome, coagulatome, metabolome, and microbiome via that results in air-blood barrier failure, inflammatory pulmonary edema, SIRS, immunothrombosis, meta-inflammation, PICS, and dysbiosis, ultimately leading to the development of ARDS/ALI. ALI, acute lung injury; AP, alternative pathway; AP1, activator protein 1; ARDS, acute respiratory distress syndrome; C3aR, C3a receptor; C5aR, C5a receptor; CP, classical pathway; ECMO, extracorporeal membrane oxygenation; EP, extrinsic pathway; HIF-1α, hypoxia-induced factor 1-alpha; GLUT1, glucose transporter 1; HKI/II, hexokinase I/II; I/R, ischemia/reperfusion, LAF-1, leukocyte adhesion molecule 1; LP, lectin pathway; NETosis, neutrophil extracellular trap formation; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; OXPHOS, oxidative phosphorylation; PICS, persistent inflammation/immunosuppression and catabolism syndrome; ROS, reactive oxidation species; SIRS, systemic inflammatory response syndrome.
4 ComC as a pivotal integrator for ARDS pathobiological connectome
4.1 Complement in lung homeostasis
The ComC is part of the innate sensor and effector systems that are considered the host’s ´first line of defense´ with a number of biological functions. An intact ComC plays an integrative role in host defense and homeostasis by interacting with microorganisms, removing damaged cells, crosstalking with toll-like receptors (TLRs), bridging with innate and adaptive immune cells, interplaying with the CoaC, promoting tissue repair, and regulating inflammatory resolution. The pulmonary airway is exposed to inhaled pathogens and allergens on a daily basis: Airway epithelial cells and macrophages play a central role in this defense against pathogens and allergens via various host mediators including the ComC. Although the liver is the primary site for the synthesis of circulating complement proteins (except C1q, C7, and factor D), various studies in both animals and humans have demonstrated that alveolar epithelial cells, airway epithelial cells and alveolar macrophages synthesize a number of complement components. In particular, alveolar type II epithelial cells produce complement proteins C2, C3, C4, C5, C6, C7, C8, C9, factor B, factor H, factor I, and C1s inhibitor (33, 34), whereas human bronchiolar epithelial cells [C3, Decay-accelerating factor (DAF), membrane cofactor protein (MCP), and CD59] and lung fibroblasts (C1s, C3, C4, C5, C6, C8, and C9) can synthesize complement proteins (34, 35). Alveolar macrophages have the potential to synthesize the complement proteins of the functional alternative and terminal pathways (36), whereas the alveolar macrophages under a disease condition produced more complement components than their healthy counterparts (37). Furthermore, alveolar macrophage-derived serine proteinases cleaved local synthesized C5 into C5a, which initiates pulmonary inflammation (38). This local production not only significantly contributes to the systemic pool of complement, but also forms fully functioning complement pathways in the pulmonary niche thereby influencing local complement processes. The pulmonary synthesis of complement proteins in the microenvironment plays an important role in host defense against infection, clearance of ICs and damaged cells, and regulation of metabolism. Researchers reported that C1q and mannan-binding lectin (MBL) bound to and enhanced apoptotic cell uptake by alveolar macrophages in vivo (39). Chang et al. revealed that MBL was present in the lung of healthy wild type mice and MBL deficiency increased susceptibility to influenza A virus infection (40). However, excessive local ComC activation increases pulmonary vascular permeability, initiates thromboinflammation, and causes pneumonic cell damage. Our previous studies have demonstrated that excessive pulmonary complement synthesis and ComC activation contribute to pulmonary inflammation and ALI in a swine model of traumatic hemorrhage (41–44). Furthermore, one would expect prolonged activation by C3a and C5a under such pathological conditions because of the absence of carboxypeptidase N (a major anaphylatoxin inactivator) in the pulmonary interstitial space (45).
4.2 Evidence and mechanism of ComC dysregulation in ALI/ARDS
Accumulating evidence is showing that overwhelming or deregulated complement activation may fuel a cytokine storm, endotheliopathy and neutrophil extracellular traps (NETs)-driven thromboinflammation leading to collateral tissue damage. In patients, complement activation has been detected in plasma (46–49) and bronchoalveolar lavage (BAL) fluid (47, 50, 51), as demonstrated by C3a, C3c, C5a and C5b-9 increase. Besides C5a, C3a and C3adesArg (a variation of C3a that plays a central role in the metabolism of adipose tissue) were also elevated in the plasma and BAL fluid obtained from patients with ARDS compared to that obtained from patients that did not develop ARDS (6, 47, 52). However, it seems that C5adesArg was not significantly changed during ARDS (6). In addition, Zilow and colleagues reported a more sensitive assay by monitoring the C3a/C3 ratio in the plasma and BAL fluids to predict the risk to ARDS at an early stage (53, 54). Furthermore, in a particular subtype of ARDS patients, such as trauma related patients, complement activation was also detected via increased levels of C3a and C4a in plasma (4, 55). Fosse et al. suggested that different types of ARDS may have different patterns of complement activation (5). In trauma associated patients, complement activation occurred immediately after the injury and correlated strongly with severity scores of lung injury, whereas other types of ARDS (such as in sepsis patients), complement activation occurred at a relatively later stage (5). A recent study demonstrated widespread complement activation characterized by excessive C3a generation, C5b-9 formation, and C3d, C4d, MASP-2 deposition in lung tissues as well as prominent increase in blood C5a in proportion to the severity and high expression of C5aR in blood and pulmonary myeloid cells in patients with severe COVID-19 (56–59). These insights suggest that blockade of the ComC could be used to prevent the neutrophil/monocyte infiltration, endotheliopathy, and thromboinflammation associated with ARDS.
However, the mechanism of ComC activation in ARDS/ALI is largely unknown. Recently, compelling evidence reveals that DAMPs and PAMPs are activators of the ComC in other diseases and conditions. Ratajczak et al. demonstrated that high mobility group box protein 1 (HMGB1) and S100 trigger the complement lectin pathway activation via binding to the MBL during mobilization of hematopoietic stem/progenitor cells (60). Similarly, HMGB1 release activated the classical pathway by binding to C1q in a mouse model of brain ischemia/reperfusion injury (61). Furthermore, pattern-recognition molecules such as MBL, ficolins, and collectins binding to invading pathogens or damaged cells initiate ComC activation via the lectin pathway (62–65). Therefore, we postulate that the release of DAMPs and/or PAMPs after ARDS/ALI may activate the ComC. Another important factor in regards to ComC activation in the setting of ARDS/ALI is respiratory acidosis-induced ComC activation. Low extracellular pH favors the activation of the ComC and CoaC (66–73). Respiratory acidosis and metabolic acidosis (ischemic conditions) significantly lower the local and/or systemic pH and therefore might lead to complement activation.
To study the pathogenesis of ARDS, animal models are an appropriate approach. No single model can mimic all the pathophysiological changes that are observed in patients (74), but complement activation associated with ARDS development have been explored in several experimental animal models, including related to conditions of sepsis (75), IgG immune complex (76), blunt chest trauma (77, 78), transfusion-related acute lung injury (TRALI) (79) and influenza H5N1 viral infection induced-ALI/ARDS (80). Besides the correlation of complement activation with ARDS development, several other studies involving complement-mediated ARDS pathogenesis have been conducted in animals. Till et al. demonstrated that intravenously injected cobra venom factor (CVF) induces lung injury in rats (81). While direct administration of C5a has been shown to induce alveolar inflammations in guinea pigs (82), rats (76) and rabbits (83). Furthermore, using genetic manipulation of ComC, studies have showed reduced lung damage via complement-mediation: SARS-COVID-infected C3-/- mice had reduced pulmonary infiltration of neutrophils and monocytes, reduced systemic inflammation, and less respiratory dysfunction (3); C5-/- mice exhibited a reduction of lung injuries in an ICs-induced ALI model (84); C5aR-/- and C5L2-/- mice had attenuated lung injuries in three models of acute lung injury induced by LPS, IgG immune complexes, C5a, and bacterial polyphosphates (85, 86); and significantly increased survival, increased release of interferon- γ, and decreased production of IL-10 accompanied by improved pathogen clearance were observed in mild-moderate septic models of C5aR1-deficient mice (87). Additionally, pharmacological inhibition of complement levels and complement activation in preclinical and clinical ALI/ARDS demonstrated beneficial effects: C3 inhibition with AMY-101 in ex vivo whole blood infection models reduced IL-6 release (88); anti-C5 antibody (eculizumb) treatment attenuated ALI in a non-human primate septic model (89); C3 inhibitor (AMY-101) was safe and associated with a favorable clinical outcome in a patient with COVID-19 pneumonia with systemic hyperinflammation (90); anti-C5a antibody (IFX-1) treatment led to increased lung oxygenation and decreased systemic inflammation in COVID-19 ARDS patients (58, 59), attenuated ALI in patients with influenza H7N9 viral infection (91); and administration of IFX-1 in patients with severe sepsis and septic shock selectively neutralized C5a without detectable safety issues (92). E culizumab treatment in severe COVID-19 patients with pneumonia/ARDS led to a significant drop in inflammatory markers and duration of disease (93). Taken together, these studies demonstrated that complement activation is associated with ARDS development in both patients and in animal models.
4.3 Complement function in the pathogenesis of ARDS
Excessive complement activation in ARDS can be detected very early in both plasma and pulmonary tissue (49, 94–97). However, the mechanism of initiation of complement activation in ARDS is unclear. Several possible mechanisms have been proposed: (1) classical pathway activation via C1q binding to the antigen-antibody complex, CRP, SAP and apoptotic cells (98); (2) lectin pathway activation through MBL interaction with PAMPs (for example, the spike protein of SARS-CoV-2, viral RNA, LPS, and lipoteichoic acid) (99); (3) alternative pathway activation by means of DAMPs-TLRs (88).
Upon activation, complement products induce a profound inflammatory response, of which the main contributors are anaphylatoxins C3a and C5a. In physiological conditions, C3a and C5a are rapidly degraded by enzymatic cleavage of the C-terminal arginine residue by plasma carboxypeptidase N and R to generate cleavage products C3adesArg and C5adesArg (100). These cleavage products are involved in ARDS pathogenesis as described by Huber-Lang et al. (6), although the anaphylactic activity of C5adesArg and C3adesArg seems much lower. In the setting of ARDS, C3a interacts with its receptors C3aR, while C5a engages with its two receptors, the C5aR1 (101) and C5L2 (102) to sense the DAMPs/PAMPs signals. These receptors are highly expressed on neutrophils, macrophages, and lymphocytes. All these inflammatory cells are significantly activated in the lungs and are effector cells which perform their biological roles via multiple mechanisms. Furthermore, excessive C5b-9 formation can directly damage pulmonary cells, including alveolar epithelial cells and endothelial cells during ARDS as described below.
4.3.1 Complement-mediated alveolar-capillary barrier dysfunction
Profound pulmonary edema and alveolar-capillary injury are major phenomena of the pathophysiological alterations associated with ARDS/ALI. Alveolar-capillary barrier dysfunction in ARDS/ALI highlights the pathophysiological role of the alveolar-capillary barrier in gas exchange, pulmonary fluid balance, immune cell trafficking, inflammatory mediators, noninflammatory signaling, mechanical injury, and lung repair (103). Alveolar epithelial cells (AEC) and pulmonary endothelial cells synthesize most of the ComC proteins (98, 104), so they not only significantly contribute to the systemic pool of complement but also form fully functioning complement pathways in the pulmonary niche, thereby contributing to alveolar-capillary barrier dysfunction if the ComC in the pulmonary system is excessively activated. As a result, increased bronchoalveolar lavage (BAL) fluid levels of C3a were observed in ARDS patients (105). C5a and C5b-9 potentiated alveolar-capillary barrier dysfunction in experimental rodent ARDS models (16, 106), whereas inhibition of C5a-C5aR interaction reversed alveolar epithelial barrier dysfunction and inflammation in a preclinical animal ALI model (107). Moreover, the COVID-19 spike protein can directly activate the lectin pathway by binding to MASP-2 (88). There was substantial deposition of activated complement products (C5b-9, C4d, MASP-2) and their co-localization with COVID-19 spike glycoproteins in the microvasculature of interalveolar septa, and pulmonary microvascular injury was associated with elevated plasma C5a and endothelial C5b-9 deposition in COVID-19 patients with ARDS (57), suggesting a key role of complement-mediated catastrophic microvascular injury syndrome in the pathogenesis of severe COVID-19. Furthermore, C5b-9 formation can cause pulmonary endotheliopathy by activating two independent molecular pathways: the inflammatory molecular pathway and the microthrombotic molecular pathway. This occurs as C5b-9 triggers inflammatory cytokine release and promotes either exocytosis of large von Willebrand factor multimers in the case of the inflammatory nuclear pathway or promotes platelet activation in the case of the microthrombotic pathway (108). More importantly, treatment with a lectin-pathway inhibitor (narsoplimab) in 6 COVID-19 patients with ARDS resulted in a rapid and sustained reduction of circulating endothelial cell counts and serum cytokines and an increase in survival (109). It appears that complement activation participates in regulating alveolar-capillary functions. The mechanism by which the ComC induced pulmonary endotheliopathy remains unclear. Given the function of the ComC on the endothelium, neutrophils, monocytes/macrophages, and coagulation, downstream contributors such as the shedding of complement regulatory proteins from pulmonary endothelium (110), C5b-9-mediated endotheliopathy, NETs-induced platelet-leukocyte aggregates, the cytokine storm, tissue factor (TF)-triggered extrinsic CoaC activation could all have played a role (88, 111).
4.3.2 Complement and inflammation
4.3.2.1 Complement mediates neutrophil activation in ARDS
Complement-mediated neutrophil activation has been proposed as an important pathogenic mechanism causing acute microvascular lung injury in ARDS, an element of the disease which has been extensively studied (11, 112, 113). Complement activation generates anaphylatoxins C3a and C5a. Engagement of C3a and C5a with their receptors on neutrophils leads to neutrophil activation. Activated neutrophils are capable of ingesting microorganisms or particles (phagocytosis), and releasing an assortment of proteinases through degranulation and generation of neutrophil extracellular traps (NETs) composed of chromatin and serine proteases (114). Severe pulmonary neutrophil activation and infiltration lead to lung injuries (95, 115). This pathway explains the major mechanism for alveolar epithelial and capillary endothelial injuries, as evidenced by several studies: 1) Neutrophils were the dominant inflammatory cell type recruited into the lungs during ARDS development, particularly in the acute phase (13, 116, 117); 2) C5a administration to animals led to induction of rapid influx of neutrophils into the lung, and degree of neutrophil activation was associated with disease severity during ARDS (81, 83); 3) The neutrophil swarming in the lungs was largely reduced by blocking C5 interaction with C5aR1 or C5L2; and 4) The induction of inflammatory neutrophils in lungs was reduced greatly and the pathologies were alleviated synchronously in C5aR or C5L2 knockout mice (78, 118, 119).
Neutrophils are granulocytes that contain various biological mediators in their granules. Upon activation, several inflammatory mediators are released into the surrounding milieus, including proteases, reactive oxygen species (ROS), pro-inflammatory cytokines, and pro-coagulant molecules. During ARDS, these molecules contribute to increased vascular permeability and a sustained loss of normal epithelial and endothelial barrier functions (120–122). In addition, recent studies have reported that complement activation-mediated lung injury are via NETing neutrophils and necroinflammation (49, 123) and the effectors of extracellular histones (115, 124, 125) in ARDS patients and preclinical animal ALI. It has been proposed that C5a activates neutrophils to form NETs, and that NETs release extracellular histones to exert biological effects. A key observation that links C5a with extracellular histones is that, in C5aR-/- and C5L2-/- mice, lung injuries were attenuated, and level of extracellular histones was reduced by more than 90%. This observation indicates that the appearance of extracellular histones was C5a receptor-dependent (85). In the same experimental settings, depletion of neutrophils in the wild type mice resulted in a 70% reduction in histone levels in BAL fluid (85) suggesting that C5a induced extracellular histone release was largely through neutrophils. Moreover, blockade of C5aR or C3 disrupted NET formation and reduced TF expression in neutrophils (49). In addition, it was reported that circulating histones can rise to toxic levels and serve as mediators of other distant organ damages, although the lung was the organ most susceptible to damage (126). Altogether, current finding indicates that C5a- and C3a-triggered NETosis and extracellular histones release in neutrophils contribute to ARDS development.
In addition, C5a is also reported to cause activation of inflammasomes during ARDS through neutrophils (127). Activated inflammasomes trigger the assembling of the active form of caspase-1. Once active, caspase-1 cleaves cytokine precursors (pro–IL-1β and pro–IL-18) to their mature biologically active and secreted forms (IL-1β and IL-18). Both proinflammatory cytokines IL-1β and IL-18 have been linked to ARDS development (128–130), which indicates that inflammasome activation is a critical factor that contributes to ARDS pathogenesis. Taken together, the aforementioned studies highlight that neutrophil activation by complement pathway is a key mechanism of lung injuries.
4.3.2.2 Complement activates alveolar macrophages
Besides neutrophils, the complement system mediates activation of alveolar macrophages, inducing lung injury. Mounting evidence suggests that macrophages are another major inflammatory cell type activated in the lungs during ARDS, especially in the very early phase. Macrophage activation syndrome is considered to perform a leading role in cytokine storm syndrome during ARDS (131–133). Three classes of macrophages are identified in lung tissue: bronchial macrophages, alveolar macrophages, and interstitial macrophages (134). Of those, alveolar macrophages seem to be paramount, as the alveoli act as the port of entry to the body and are constantly exposed to foreign particles and infectious agents. Alveolar macrophages are the first line of defense against these agents. Findings have shown that alveolar macrophages express high levels of pattern recognition receptors (PRPs) during steady state (135). In addition, the function of alveolar macrophages is highly plastic. Upon stimulation, these macrophages can differentiate into classically activated macrophages (CAMs, also known as M1 macrophages) and alternatively activated macrophages (AAMs, also known as M2 macrophages). M1 macrophages produce high levels of pro-inflammatory cytokines (high levels of IL-12 and low levels of IL-10) in response to paracrine signaling from the Th1 cytokine INF-γ and in response to autocrine signaling by IFN-β, both of which may dominate during the acute phase of ARDS. The M2 macrophages release anti-inflammatory cytokines (high levels of IL-10, TGF-β and low levels of IL-12) in response to signals from Th2 cytokines such as IL-4 and IL-13. Macrophages with the M2 phenotype dominated the resolution phase of ARDS (131).
In the acute phase of ARDS, the resident alveolar macrophages are activated. Meanwhile, more macrophages are recruited into the alveolar compartment and function as pro-inflammatory cells, exacerbating inflammatory response. Kazmierowski et al. reported that during the first four hours of ARDS, alveolar macrophages are the principal respiratory cells (90%) initially recovered from BAL fluid. This indicates that macrophages are first responder inflammatory cells at the onset of lung injury (50). As proposed, anaphylatoxins (C3a, C5a) from complement activation products interact with their receptors (C3aR, C5aR1, C5L2) on macrophages and lead to macrophage activation (95, 98). Activated macrophages are highly phagocytic, and also release effector molecules such as cytokines and chemokines (136, 137), which can rapidly recruit neutrophils into the injury site (138). Activated complement products (C3a, C5a, and C5b-9) also function as “alarmins,” initiating secretion of both IL-1 and IL-18 at NLRP3-, ASC-, and caspase-1-dependent manner in macrophages via two signal pathways: (1) the priming signal, triggered by the signal axis of C3a/C5a-C3aR/C5aR-MAPKs-NF-kB/AP-1, leading to the transcriptional upregulation of canonical and non-canonical NLRP3 inflammasome components (139–141); and (2) the activation signal, which is elucidated through the cascades of C3a-C3aR-ERK1/2-ATP-P2X7, C5a-C5aR-mitochondria- ROS, and C5b-9-ATP influx/K+ efflux (140–144). Genetic deletion of C6 in mice resulted in a deficiency of NLRP3 inflammasomes in macrophages and neutrophils that resulted in less lung damage and improved survival after sepsis (145).
Rosseau et al. showed BAL fluid from ARDS patients with a predominant monocyte-like macrophages. These macrophages undergo phenotype switching during the disease progress in sepsis-, pancreatitis-, or severe pneumonia-induced ARDS (146). In a murine model of intestinal ischemia-induced ALI, Hu et al. observed that the binding of C5a produced during ALI to C5aR was increased on alveolar macrophages, leading to apoptosis of alveolar macrophages and development of ALI (147). C5a also induced ALI through STAT3-mediated macrophage activation after intrapulmonary deposition of IgG immune complexes in a rat model (148). Moreover, inhibition of C5a markedly diminished macrophage infiltration into lungs and reduced lung injury (91). This suggests that interruption of C5a/C5aR interaction reduces the infiltration of macrophages, and in turn ameliorated the disease in ARDS. Taken together, these data demonstrated that C5a/C5aR activates macrophages that play a pivotal role in lung tissue damage during ARDS. Besides direct interaction of C5a and C5aR on macrophages, the C5a-C5aR axis also indirectly stimulated alveolar macrophages to release TNFα and neutrophil accumulation in the lungs via Fc gamma receptors (FcγR)-mediated signal pathway (149).
On the other hand, macrophages also participate in anti-inflammatory and tissue-reparative phenotypes during the course of inflammation. Steinberg et al. characterized the alveolar inflammation in ARDS patients with multiple types of causes in survivors and non-survivors. They found that alveolar macrophages are increased in survivors of ARDS in BAL fluid. This indicates that macrophages provide a protective function during ARDS, and normal resolution of alveolar inflammation is associated with a favorable outcome (150). In addition, emerging evidence showed that macrophages play a role in anti-inflammation by producing anti-inflammatory cytokines (IL-10) via transcription factors (STAT3 and AP-1) (148, 151, 152). However, it is unclear whether the complement - for example, via the C5a-C5L2 or C5a-C5aR1 axis - participates in regulating the later phase of inflammation resolution.
4.3.2.3 Complement mediates lymphocyte activation
The complement system is part of the innate sensor and effector system. Accumulated evidence indicates that the complement system functions as a bridge between innate and adaptive immunity (153). Lymphocytes are considered to play major roles in adaptive immunity by inducing immune responses to protect the host. Likewise, lymphocytes highly affect disease progress and outcome during ARDS. In a mouse model, rapid influx of lymphocytes into the lung was observed during the early phase after LPS-induced lung injury. In an LPS induced lung inflammation model, Harris et al. reported an elevation of lymphocyte number over days 3-6 post LPS instillation (154). Morris et al. in an endotoxin induced lung injury model reported that the percentage of lymphocytes in the BAL fluid increased from 1.8% on day 1 post-endotoxin to more than 11% on days 3 and 5 (155). Nakajima et al. also reported a significant increase in CD4+ T cells in an LPS-induced ALI model (156). In addition, the indirect role of cytokines produced by lymphocytes contributed to ARDS pathogenesis. Cytokine IL-17A was the most characterized. Evidence from multiple studies supports the view that pro-inflammatory cytokine IL-17A contributed to ARDS pathogenesis (157–159). IL-17A enhances neutrophil recruitment, and also promotes production of inflammatory cytokines. CD4+ T cells, γδ T cells (160), and pulmonary innate lymphoid cells (ILC3s) (161) were the major source of IL-17A. Studies revealed that regulatory T cells (Tregs) also contribute to ARDS outcome. The Treg/CD4+ T cells ratio (162) and Th17/Treg ratios were also considered predicative parameters for ARDS patients (163). However, this point remains controversial because recent studies in COVID-19 ARDS patients demonstrated CD4+ lymphopenia (164, 165).
Although the ComC is considered to bridge the innate and adaptive immune systems, it remains largely unknown how the complement system regulates lymphocyte activation during ARDS. T cells and antigen-presenting cells (APCs) produce complement proteins and yield local complement-activated products, C3a and C5a. These anaphylatoxins can interact with C3aR and C5aR expressed on the T cells and APCs including dendritic cells and macrophages. These interactions trigger a series of events including recruitment of T cells, enhancement of T cell proliferation, prevention of T cell apoptosis (163, 166), release of cytokines (IL-12, IL-23), and upregulation of co-stimulatory molecules CD80 and CD86 (167). Genetic and pharmacological manipulation of complement activation attenuated T cell-mediated immunity and delayed allograft rejection in mice (166, 168). These findings suggest that C3a and C5a provide a vital bridge between innate and adaptive immunity, extending the roles of C3a and C5a in inflammation. Besides effector T cells, complement may also modulate other types of lymphocytes, Tregs and γδ T cells. Studies demonstrate that Treg subsets contributed to the resolutions of LPS-induced ALI, as reported by D’Alessio and colleagues (169). Significant accumulation of γδ T cells in lungs was observed during CLP-induced ALI (167). Complement C4 induced Treg differentiation via dendritic cells was observed in systemic lupus patients (170). However, there is a lack of experimental support to determine whether complement is involved in regulating these lymphocytes.
4.3.2.4 Complement-mediated platelet activation
In addition to clot formation and homeostasis, platelets also play an essential role as immunological multitaskers. Platelets contain complement factors and bear complement receptors, and can activate the ComC and vice versa. The ComC and platelets cooperate against pathogens and are involved in inflammatory diseases (171). Platelets are a key factor in the pathophysiology of ALI via the DAMPs/PAMPs-TLRs axis and platelet-neutrophil interaction at the site of injured endothelium (103). Increasing evidence demonstrates the role of platelets in and their interaction with the ComC in tissue inflammation and organ damage (172). Activated platelets potentiated MASP1/2 activation of the complement lectin pathway in thromboinflammation (173). C5a-dependent up-regulation of lung vascular P-selectin was reported in vitro and in vivo studies (174). Platelet-derived microparticles induced ComC activation (175) and activated platelets amplify complement-mediated lung endothelial damage by releasing serotonin, which in turn potentiates granulocyte adhesion to pulmonary endothelium (176). Mannose-binding lectin acted as an early trigger of platelet activation and vascular damage after cerebral ischemia in mice (177). Inhibiting recruitment of platelets at the sites of injured endothelial cells attenuated complement activation and lung injury in an ex vivo perfusion of porcine lung (178). It seems that complement-platelet interaction may participate in the pathophysiology of ARDS/ALI.
The emerging understanding of the pathogenic mechanisms underlying SARS-CoV-2 infection and COVID-19 progression highlight the critical role of complement-mediated neutrophil and platelet activation, endotheliopathy, and thromboinflammation (96). Recent studies in COVID-19 further demonstrate that crosstalk between complement and coagulation functions as a key trigger of COVID-19-induced ARDS. Indeed, COVID patients demonstrated systemic complement activation, which was associated with pulmonary thromboembolic events and disease severity (48, 179). Skendros et al. reported a pivotal role of complement activation in COVID-induced immunothrombosis via C5a-C5aR-NETs/TF or C3a-C3aR-thrombin-NETs/TF, while pharmacological blockade of C5aR1 or C3 disrupted NETosis-/TF-driven thrombogenicity (49).
4.3.3 Cross-talk between complement cascade and other plasma cascades in ARDS
Plasma contains four enzymatic systems: ComC, CoaC, KinC, and FibC. All relate to produce the inflammatory response. Emerging data indicate that interplay among plasma cascades fuels hyperinflammation and thrombotic microangiopathy (thromboinflammation), thereby increasing lung injury and mortality. One of the primary pathologies in ARDS is thrombogenesis as indicated by the florid fibrin deposition in the intra-alveolar space and microthrombi in the pulmonary microvasculature. Profound fibrin deposition and microthrombi in ARDS likely result from both activation of the CoaC and impairment of the FibC (180, 181). Fibrin deposition and the associated dysfunction of the FibC were evidenced from clinical studies in ARDS. Fuchs-Buder et al. found that the pro-coagulant pathway and fibrin degradation are markedly activated in ARDS patients compared to non-ARDS patients (182). Tomashefski et al. observed thrombo-emboli and macrothrombi in patients with ARDS (183). Bone et al. also found that fibrin microthrombi were present in the lungs of a majority of ARDS patients (184). The latest studies observed that hypofibrinolysis was related to increased plasminogen activator inhibitor-1 (PAI-1) presence in COVID-19 patients (185). Interestingly, increases of both tissue plasminogen activator and PAI-1 in all COVID-19 patients regardless of disease severity suggest that severe SARS-Cov2 infection may hijack normal profibrinolytic signaling and lead to a procoagulant state, including increased PAI-1 expression, prolonged PAI-1 half-life, and severe lung injury (185, 186). In addition to that, thrombocytopenia was observed in several studies and associated with ARDS mortality (187–189). Moreover, recently Lipcsey et al. presented evidence that the ComC and the KinC were strongly activated and associated with lung damage, illness severity score and mortality in critically ill patients with COVID-19 (190).These studies highlight that plasma cascades participate in ARDS development and highly affect the outcomes of ARDS.
The cross talk between the ComC and the other plasma cascades is reciprocal. In one way, during the course of inflammation, complement effectors can directly enhance coagulation system; specifically, complement works with inflammatory mediators to increase the thrombogenicity in blood. For example, in the setting of ALI/ARDS, Kambas et al. observed that C5a works with inflammatory cytokines, particularly TNF-α, to promote coagulation activation and can stimulate neutrophils from patients to produce TF resulting in the formation of hyaline membranes in the lungs of patients (191). In addition, complement can inhibit anticoagulant factors. Studies showed that C5a may stimulate PAI-1 production from mast cells and basophils (192, 193). PAI-1 is an inhibitor of fibrinolysis, the physiological process that degrades blood clots. Coagulation enzymes also regulated complement pathway activation. Huber-Lang et al. in a sepsis model showed thrombin can directly serve as C5 convertase to directly activate the complement pathway in the absence of C3 (26). Moreover, a recent study reported that intensive ComC activation (C3a, C3c and C5b-9) was positively correlated with thromboembolic events in patients with COVID-19 (48). Moreover, the crosstalk between the CoaC and the ComC is also well illustrated by the ability of certain coagulation enzymes to activate complement components (194, 195). Taken together, these insights illustrated that complement and coagulation systems can modulate each other and contribute to ARDS development via thromboinflammation and endotheliopathy (Figures 2, 3).
4.3.4 Interaction between complement and toll-like receptors
The lung is continuously exposed to a diverse array of infectious agents, foreign antigens, and host-derived danger signals. Resident macrophages, neutrophils and stromal cells of the lung constitutively express PRRs, which recognize PAMPs and DAMPs. PRRs are comprised of toll-like receptors (TLRs), complement proteins [C1q, CR1, C4BP, propoerdin, factor H, MBL, collectins and ficolins) (196, 197), RIG-I like receptors (RLRs), and NOD like receptors (NLRs).
The ComC and DAMPs are the major components of the innate immune system. However, these two major innate immune arms were described and studied primarily as separate entities. Nevertheless, an emerging body of evidence indicates close interaction and crosstalk between them in regulating the immunological processes, and the dysregulation of any of the two innate immune arms impact the other through synergistic and/or antagonistic interactions that can modulate host immune homeostasis. In particular, several studies of ComC and high mobility group box 1 (HMGB1) crosstalk via its receptors (TLRs) have recently reported that (1) C5a stimulated chemokine CXCL-10 generation in osteoclasts via the interaction between C5aR1 and TLR2 (198); (2) TLR-4 activation enhanced C5a-induced pro-inflammatory response by inhibiting the second C5a receptor (C5L2) (199); (3) C5a participated in the regulation of TLR4-induced IL-10 and IL-12 production in macrophages in response to TLR ligand stimulation (200, 201); (4) C5L2 promoted NLRP3 (NOD-, LRR- and pyrin domain-containing protein inflammasome activation and extracellular HMGB1 (eHMGB1) release from macrophages (202); (5) eHMGB1 facilitated C5a-primed neutrophil activation (203); and 6) eHMGB1 release resulted in ComC activation through binding to C1q to exacerbate sterile inflammation (61). Furthermore, other clinical data also elucidated that early eHMGB1 release after severe trauma in patients was associated with post-traumatic complement terminal pathway (CTP) activation and tissue hypoperfusion (204). Consistent with these findings, our recent studies have shown that (1) ComC activation (C3a, C5a, C5b-9, Bb) post-trauma positively correlated with plasma levels of eHMGB1 (205) and histone-complexed DNA fragments (hcDNA) in trauma patients (206); (2) blocking C5 by nomacopan reduced the release of eHMGB1 (206), whereas HMGB1 inhibition by CX-01 also reversed ComC activation in a rat model of traumatic hemorrhage-induced ALI (205); and (3) CTP inhibition by DAF reduced expression of HMGB1, RAGE, NF-kB, cytokines (IL-6, IL-12, IL-13, IL-18, GRO KC) and caspase-1, and HMGB1 nuclear translocation in blast-induced ALI in rats (207). Altogether, these findings indicate synergistic crosstalk between the ComC and DAMPs that markedly amplifies pro-inflammatory responses and may contribute to ALI. Indeed, combined inhibition of C5 and TLR co-receptor CD14 had a pronounced effect on attenuating systemic and local inflammatory response, improving pulmonary function and hemodynamics, and reducing morbidity and mortality in murine (208) and swine sepsis models (209–212). Inclusively, these data suggest that the multifaceted activation and interaction of these two cascades are of crucial importance for NI and highlight the complexity of the inflammatory response after ALI. Therefore, dual blockade might provide a general, broad-acting therapeutic regimen against ALI/ARDS where CTP and HMGB1 are improperly activated.
4.3.5 Complement-metabolism-inflammasome axis
Recent insights into ComC-mediated pathomechanisms in multiple-organ dysfunction syndrome uncover its new function in metabolism. Indeed, Denk et al. has recently demonstrated that C5a-C5aR1 engagement after septic shock induces acidosis via an increase in glucose uptake and glycolytic influx, and Na+/H+ exchanger activation in neutrophils (213), suggesting that C5a-C5aR1 is able to significantly create a micro-milieu with lactate acidosis features even in the absence of an oxygen deficit (214). Given the aforementioned low pH favors the activation of both the ComC and the CoaC, we speculate that respiratory/metabolic acidosis after ARDS may cause further ComC activation resulting in a vicious cycle with continued metabolic acidosis and inflammation, implicating that C5a may function as a metabolic switch toward acidosis and ARDS.
In addition to its pivotally canonical host protection by the classical liver-derived and blood-effective ComC, unexpected recent findings have revealed that an intracellularly active complement system, the complosome, has emerged as a centrally non-canonical regulator of the core metabolic pathways fueling immune cell activity (Figure 3). So far, little is known about the exact mechanism of the complosome in metabolic reprogramming of immune cells in tissues. However, a plausible scheme is that the complosome intercommunicates with other intracellular immune sensors such as inflammasomes and intracellular TLRs (TLR3, TLR7, TLR8, TLR9). Emerging evidence presents a common network of integrin/T cell receptor (TCR)/TLR-complosome-metabolism in the regulation of immune cell function: 1) intracellular complement synthesis and activation is triggered by integrin/TCR/TLR activation/stimulation in immune cells (215, 216); 2) Activated TCR signals shuttle C3a and C3b to the cell surface where they engage surface-expressed C3aR and CD46 respectively (215, 216); 3) C3b-CD46CYT-1 signaling triggers three key metabolic events regulating immune cell effector function: an increase in expression of nutrient transporters (GLUT1, LAT1, and CAT1) that allows to promote glucose and amino acid influx as well as late endosomal and lysosomal adaptor and mitogen activated protein kinase and mTOR activator 5 (LAMOR5)-driven mammalian target of rapamycin complex 1 (mTORC1) assembly at the lysosomes, elevation in expression of metabolic enzymes (fatty acid synthase, GAPDH, etc.), and activation of intracellular C5a-C5aR-mitochondrial ROS-inflammasome axis, which lead to the high levels of glycolysis, oxidative phosphorylation (OXPHOS), ROS production and NLRP3 inflammasome activation, resulting in IFN-γ/granzyme B-mediated CD4+ Th1 induction and cytotoxic CD8+ T cell activation, and macrophage IL-1β and IL-18 production in tissues (140, 215); 4) in resting immune cells, C3b-CD46CYT2 interaction reduces glycolysis and OXPHOS, increases cholesterol efflux and cMaf musculoaponeurotic fibrosarcoma oncogene homolog (MAF) expression, and C5a-desArg-C5L2 engagement and C1q-gC1qR signal reduce intracellular C5a-C5aR1-mtROS-NLRP3 inflammasome activity, which cause IL-10 release/Th1 contraction and decreased IL-1β/IL-18 secretion respectively (215); 5) Intracellularly generated C3a binds to intracellular C3aR on the lysosome and sustains T cell survival in the tissue through tonic mTOR activation (215). However, a mechanistic explanation for complosome-metabolism in ALI/ARDS still remains elusive and needs further research.
4.3.6 Complement in lung microbiome dysbiosis
The lung microbiome symbiosis maintains homeostasis of the lung’s immune system. Dysbiosis of gut microbiota contributes significantly to the pathogenesis of MOF including ARDS. The lung and gut microbiomes undergo profound interchangeability in critically ill patients such as those with ARDS. Indeed, compelling evidence has revealed that early lung microbiome dysbiosis characterized by the translocation of gut-associated microbes was found to correlate with alveolar and systemic inflammation and lung injury in critically ill patients with ARDS, sepsis, mechanical ventilation, trauma, ischemia-reperfusion injury, hyperoxia and aspiration (217–220). The pathogenesis of lung microbiome dysbiosis in ARDS, however, is largely unexplored. Emerging findings of the lung microbiome and its alteration in ARDS has broadened a new model of pathogenesis: an interaction network between exposure (such as sepsis, mechanical ventilation, trauma etc.), lung microbiome, and host (221). The host’s innate immune system (including complement) plays a critical role in the detection and clearance of bacteria, and complement-based recognition of pathogens or PAMPs initiates inflammation and tissue damage. Recently, there is emerging evidence that (1) SARS-CoV-2-associated ARDS in severe COVID-19 patients directly and indirectly activated the complement cascade (88, 111); (2) complement was found to participate in inflammatory tissue damage via dysregulation of host-microbiome homeostasis of the skin and periodontium (222, 223); (3) glycan of the gut fungal wall binding to MBL activates the ComC (224); (4) C5 activation induced diabetic kidney disease and C5aR blockade alleviated renal function and inflammation via partly reversing the declines of gut microbiota diversity/abundance and gut levels of short-chain fatty acids (225); (5) microorganisms frequently “highjack” complement proteins (C3d, C3b, iC3b, qC1qR, C3, C5) to gain entry into immune and non-immune cells (226); and (6) interaction between complosome and intracellular pathogens triggers mitochondrial anti-pathogen signaling which increases the expression of transcription factors (NF-κB and AP-1), which subsequently activate inflammasome and mediate a maladaptive proinflammatory response (IL-1β, IL-6, IL-18, IFN-β) (216, 226). However, the involvement of complement in lung microbiome dysbiosis and their interaction in the pathogenesis of ARDS remains poorly understood. The latest studies have highlighted the emerging important functional connection between the complosome, TLRs, metabolism, and the inflammasome axis in immune cells in regulation of host-pathogen interaction, immune cell activation, inflammation, metabolism and hemostasis (139–141, 215, 216, 226). Given that, undoubtedly it will be of significant interest to investigate the complex interplay of the complosome and lung microbiome dysbiosis and their pathogenesis in ARDS. With further insights, this area of research might lead to the development a novel, microbiome-targeted (synbiotics and fecal microbiota transplantation), and immunomodulation strategies in management of ARDS.
4.3.7 Complement activation in lung tissue repair
The role of ComC activation in the resolution of inflammation and tissue repair has been largely ignored when compared to its inflammatory aspects. In particular, accruing evidence reveals that C3a and C5a have regenerative functions on the liver, neurons, osteoblasts, and dental pulp progenitors (227–232). It is noteworthy to mention that C3a/C5a-triggered homeostasis, tonic inflammation, T cell activation, capillary leak syndrome, and edema are also necessary for the resolution of inflammation, restoration of functional tissue, and the clearance of debris and damaged cells (227, 233–236). C1q, MBL, and complement C3 fragment C3b and its receptors (CRs1-4, CD46) are important to facilitate removal of apoptotic/necrotic cells, ICs, and pathogens by enhancing phagocytosis of monocytes and macrophages and T cell homeostasis (140, 237–240). Langlois et al. measured the levels of C1rC1s-C1 inhibitor complex and the C3bP and found that the classical and alternative complement pathways are activated one day before its resolution in ARDS patients in addition to the complement activation two days prior to diagnosis of ARDS (241). Complement C3 inhibition by compstatin decreased early fibrogenic events in sepsis-induced ARDS in baboons (242), suggesting that inhibiting complement could be a potential strategy for preventing pulmonary fibrosis in ARDS patients. Furthermore, during the acute phase of ARDS, C5 exhibited a protective and anti-inflammatory role through regulating matrix metalloproteinases (MMP) and cell migration, while C5 displayed a detrimental effect during the chronic phase of ARDS likely via increasing expression of fibrogenic transforming growth factor beta1 (TGF-β) and MMP-3, revealing that C5 plays opposing roles in both inflammation and lung tissue repair (243).
In summary, acute insults (pneumonia, sepsis, trauma, inhalation, I/R injury, therapeutic approaches, etc.) with DAMPs/PAMPs can induce exaggerated ComC activation locally and systemically. The activated complementome functions as a crucial nexus to activate other systems of the immunome, coagulome, metabolome, microbiome, lipidome, genome, epigenome, proteome, and neuroendocrinome, which lead to ALI/ARDS via complementome-driven/mediated EC/AEC barrier failure, SIRS, immunothrombosis, immunometabolic dysfunction, PICS, cellular damage, dysbiosis, electrophysiological dysfunction, lung dyshomeostasis, and microcirculation dysfunction (Figure 2). This new ComC pathomechanism and its interplay with other systems provide insight into the novel development of more effective therapeutic strategies with important implications for clinical translation for ALI/ARDS patients.
5 Targeting ComC therapies in ALI and ARDS
ARDS is present in >10% of intensive care unit admissions and in nearly 25% of ventilated patients and is associated with a high mortality rate at 40%. Lung-protective mechanical ventilation strategies are still the mainstay of ARDS management. Despite more than 50 years of study, current therapeutic interventions for ARDS remain largely limited to pulmonary-supportive strategies, which fail to address the profoundly destructive impact of unchecked inflammation-driven ALI/ARDS. Despite improved understanding of the biological processes underlying the ARDS, the current lack of FDA-approved pharmacological therapies aimed at the underlying pathobiology for ALI/ARDS is a serious unmet need.
As stated above, complement activation is critically involved in the initiation and progression of ALI/ARDS. Therefore, pharmacological targeting of the ComC as well as molecules contributing to complement activation to harness immunothrombosis, alveolar–capillary barrier function, immunometabolic dysfunction and microbiome dysbiosis represents a highly promising approach for the treatment of ALI/ARDS (Figure 3).
5.1 Preclinical studies
Complement interventions for ALI/ARDS in preclinical studies are under active investigation. In the context of trauma-, hemorrhage-, sepsis-, virus- and IR-induced ALI/ARDS, the compiled evidence of preclinical experiments focusing on the role of complement in ALI/ARDS is shown in Table 5.
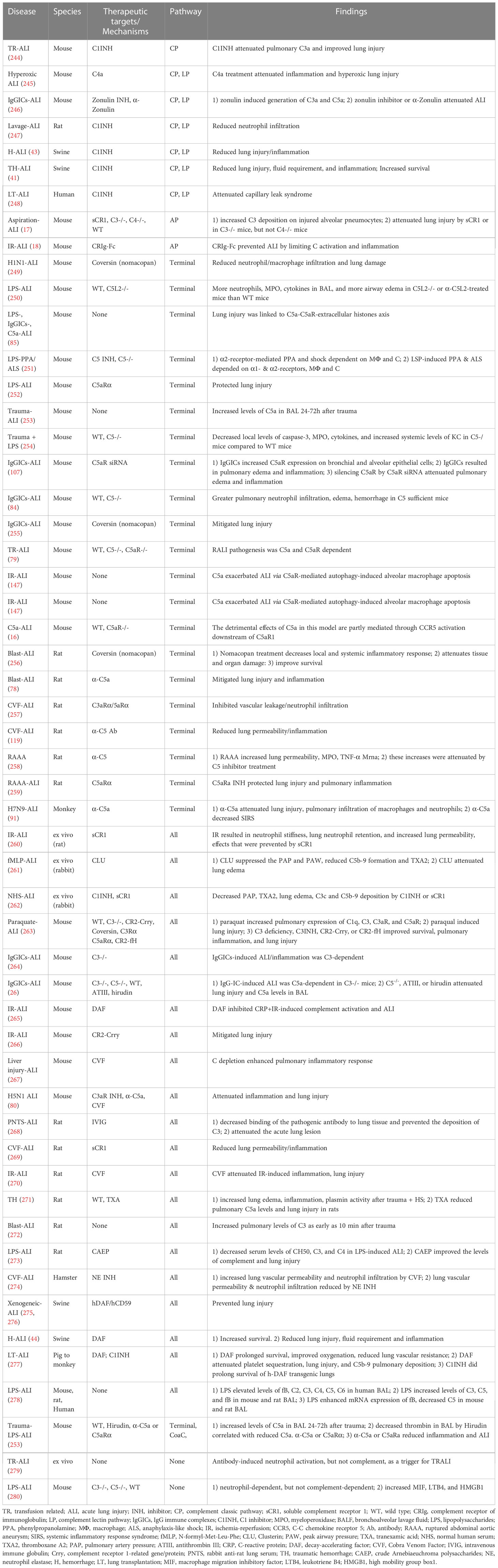
Table 5 Summary of studies in terms of the role of complement in ARDS/ALI in pre-clinical and clinical studies.
5.2 Clinical trials
Considering clinical trials of ALI/ARDS, 29 observational studies addressed complement activation after trauma, sepsis, viruses, and transplantation (Table 6). These clinical observational studies (excluding one) further demonstrated that early complement activation plays a pivotal role in the initiation and development of ALI/ARDS, and is associated with mortality. They also elucidate that complement activation is useful for prognosis and diagnosis of ALI/ARDS (Table 6). Further investigation should be carried out to test whether complement targeting pharmacotherapeutic strategies have any benefits in ALI/ARDS patients.
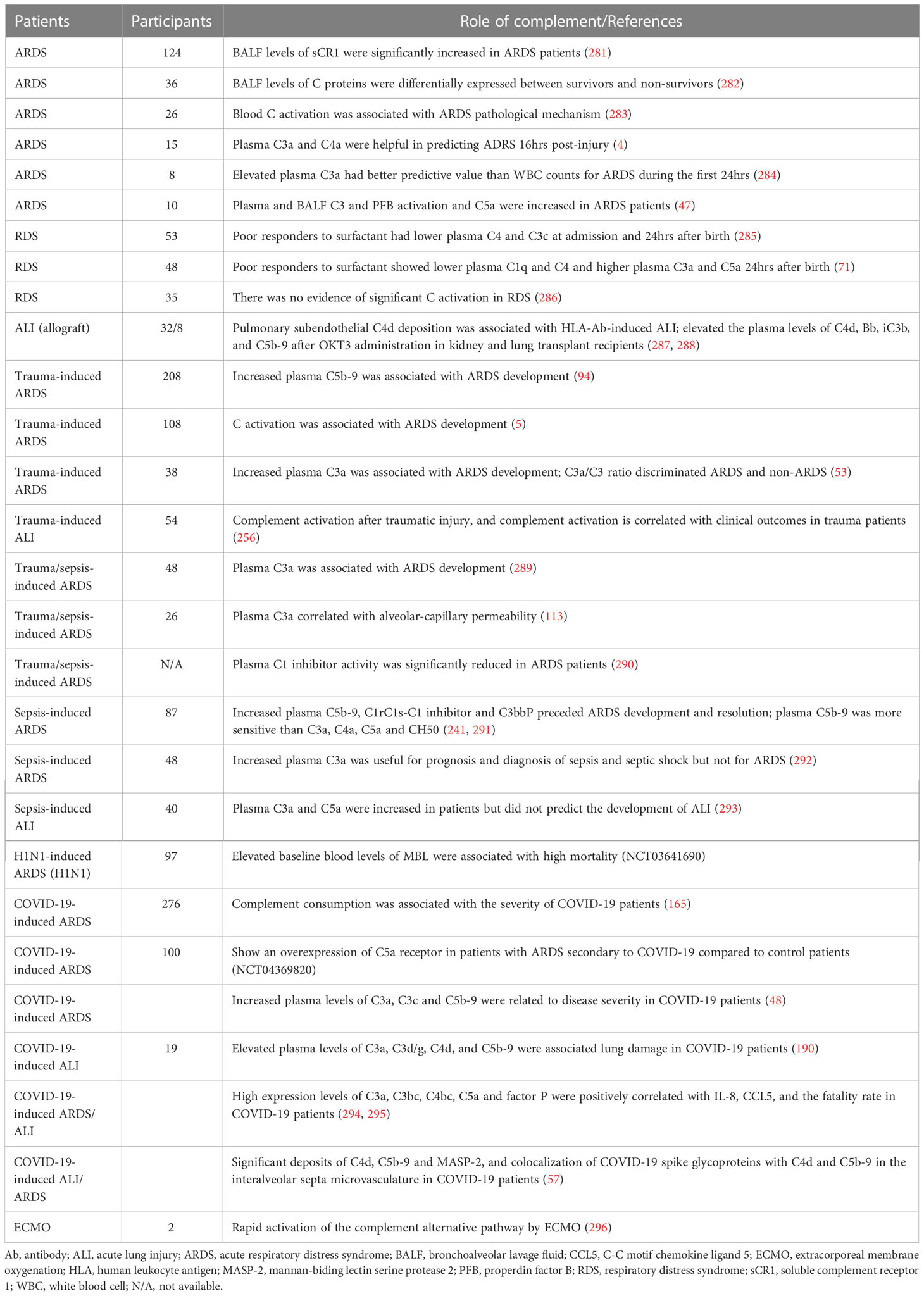
Table 6 Overview of observational clinical studies evaluating complement role in ARDS/ALI.
In clinical practice, there are only two classes of complement inhibitors available. Plasma-derived human C1 inhibitors [Cinryze (Shire), Berinert (SCL, Behring), and Cetor (Sanquin)] and recombinant C1 inhibitor (Ruconest, Parming) are already approved for hereditary angioedema (HAE) (153, 297). C5 inhibitor Eculizumab (Soliris, Alexion) is another FDA-approved drug for use in patients with paroxysmal nocturnal hematuria (PNH) (153, 297). Previous efforts in complement drug discovery were hampered by limited molecular insights, technical challenges, and safety concerns. After years of development, there are currently more than a dozen candidate drugs covering a wide range of the ComC being evaluated in clinical trials. They demonstrate a broad spectrum of therapeutic targets, in the use of molecular entities (small molecules, peptides, proteins, antibodies, and oligonucleotides), and expending indications and off-label use such as transplantation, SIRS, sepsis, inflammatory diseases, and chronic obstructive pulmonary disease (298).
With regard to targeting complement therapy in human studies of ALI/ARDS, there are currently seven interventional trials (Table 7). All studies have been designed to address the safety and efficacy of the complement inhibition by targeting C1q/MBL, C3, or C5 in COVID-19-induced ALI/ARDS (Table 7).
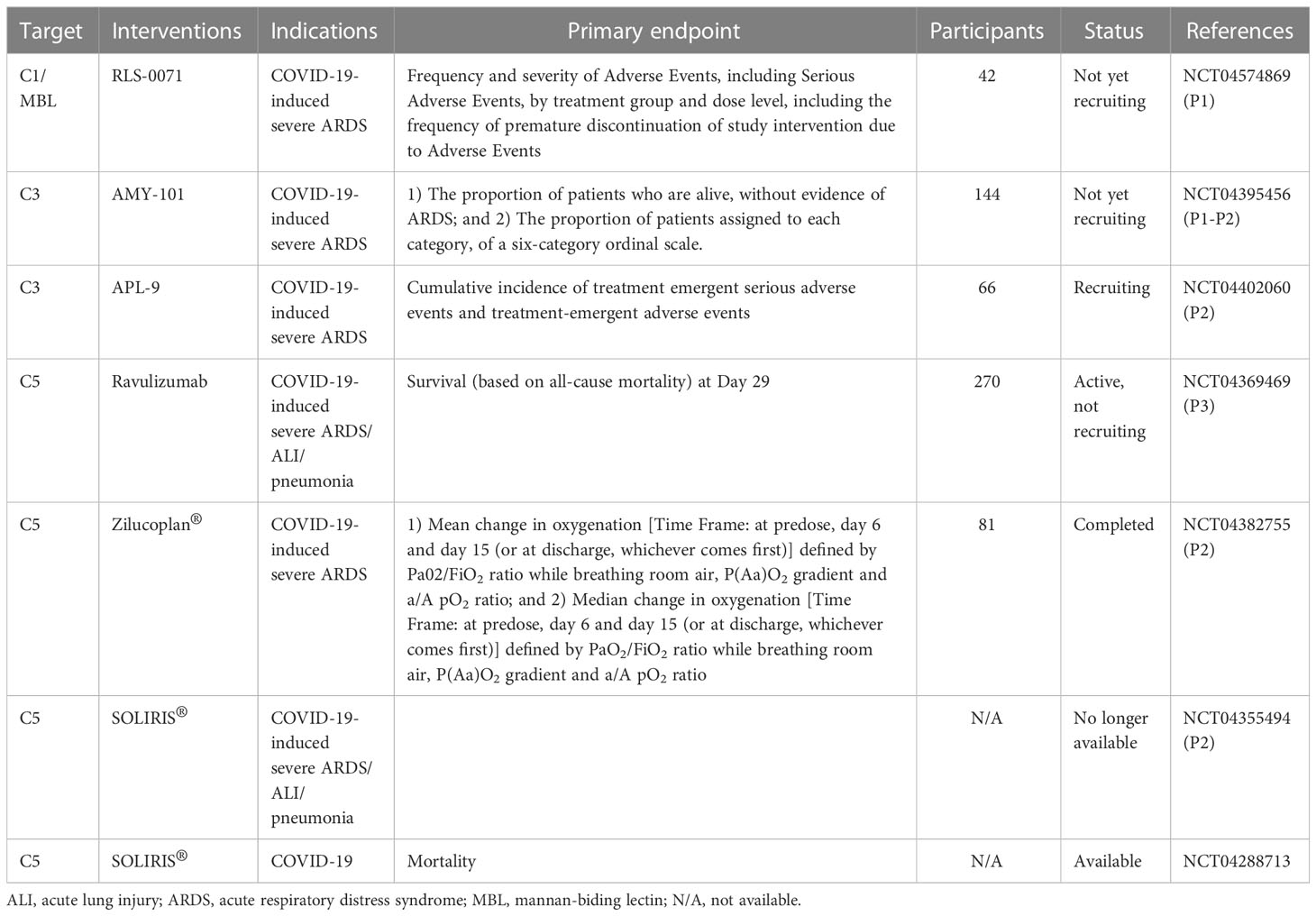
Table 7 Overview of clinical therapeutics for clinical trials in ARDS/ALI.
6 Conclusions and future directions
Although the many roles of complement-mediated pathomechanisms and various promising preclinical results of targeting complement therapies in ALI/ARDS have long been recognized, clinical trials controlling complement activation in ALI/ARDS remain rare. Despite this improved understanding, no pharmacological therapeutics targeting ComC have been approved for ALI/ARDS patients. Increasingly, ARDS have been recognized as a heterogeneous syndrome characterized by subphenotypes with distinct etiologies, pathophysiological/radiographic features, and clinical outcomes. These subphenotypes have potentially distinct ComC activation profiles (endotypes) and different responses to targeting ComC therapeutics (theratypes). Thus, there is increasing interest in better defining complement endotypes and theratypes within individual ALI/ARDS patients in order to facilitate targeting ComC personalized/precision medicine. However, several challenges still exist including (1) the lack of point-of-care diagnostic devices to timely measure the targeted complement factors or activation products and real-time monitor the dynamic change of complement activation and immune status caused by ALI/ARDS and/or post-ARDS therapeutic approaches (blood transfusion, ECMO, damage control surgery, ventilation, volume resuscitation, etc.) (214, 299); and (2) the lack of a simple prognostic/diagnostic test to identify endotypes/theratypes of ARDS because ARDS is an interpatient heterogeneous syndrome that can have a differential ComC response to the same etiology or the heterogeneous etiologies of ALI/ARDS and a different response to targeting complement therapies (300). However, the future looks much brighter due to the increasing availability of various highly effective, specific complement therapeutics developed over the last two decades targeting different components of the ComC, with different half-lives, different routes of administration, lower cost, and lower infection risk in the context of inflammation, trauma, hemorrhage, and sepsis (297, 301). Therefore, future work should focus on furthering our mechanistic understanding of ARDS endotypes/theratypes, integrating preexisting complement functions on immunome, coagulome, metabolome, microbiome, genome, epigenome, proteome, neuroendocrinome and lipidome, establishing pulmonary drug delivery, identifying specific biomarkers of ARDS and immune status, developing multiplexed point-of-care testing, and utilizing multi-omics approach to define endotypes/theratypes aimed at applying targeting complement therapies to those ARDS patients who are most likely to respond to them.
Author contributions
Conceptualization, YL and LC. Writing original draft, ZY. Writing/review/editing, YL, ZY, LC, SN, and TC. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from DoD US Army Medical Research and Development Command’s Broad Agency Announcement (BA150310 and W81XWH-19-2-0040) and Congressional Directed Medical Research Program Technology/Therapeutic Development Award (PR190914 and W81XWH-2020040).
Acknowledgments
The authors gratefully thank Dr. Subrata Haldar for critical review and editing of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AEC, Alveolar epithelial cell; ALI, cute lung injury; AP, alternative pathway; ARDS, acute respiratory distress syndrome; ASC, apoptosis-associated speck-like protein; BAL, bronchoalveolar lavage; CoaC, coagulation cascade; ComC, complement cascade; CP, classical pathway; DAF, decay-accelerating factor; DAMPs, damage-associated molecular patterns; EC, endothelial cell; FibC, fibrinolytic cascade; HMGB1, high mobility group box protein 1; KinC, kinin cascade; LP, lectin pathway; MBL, mannan-binding lectin; NETs, neutrophil extracellular traps; NETosis, neutrophil extracellular trap formation; NLRPs, NOD-, LRR-and pyrin domain-containing protein 3; OXPHOS, glycolysis, oxidative phosphorylation; PAMPs, pathogen-associated molecular patterns; PICS, persistent inflammation/immunosuppression and catabolism syndrome; ROS, reactive oxygen species; SIRS, systemic inflammatory response syndrome; TF, tissue factor; TLR, toll-like receptor.
References
1. Network TARDS. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. the acute respiratory distress syndrome network. N Engl J Med (2000) 342:1301–8. doi: 10.1056/NEJM200005043421801
2. Donahoe M. Acute respiratory distress syndrome: A clinical review. Pulm Circ (2011) 1:192–211. doi: 10.4103/2045-8932.83454
3. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio (2018) 9(5):e01753–18. doi: 10.1128/mBio.01753-18
4. Donnelly TJ, Meade P, Jagels M, Cryer HG, Law MM, Hugli TE, et al. Cytokine, complement, and endotoxin profiles associated with the development of the adult respiratory distress syndrome after severe injury. Crit Care Med (1994) 22:768–76. doi: 10.1097/00003246-199405000-00010
5. Fosse E, Pillgram-Larsen J, Svennevig JL, Nordby C, Skulberg A, Mollnes TE, et al. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury (1998) 29:509–14. doi: 10.1016/s0020-1383(98)00113-2
6. Solomkin JS, Cotta LA, Satoh PS, Hurst JM, Nelson RD. Complement activation and clearance in acute illness and injury: evidence for C5a as a cell-directed mediator of the adult respiratory distress syndrome in man. Surgery (1985) 97:668–78.
7. Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
8. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med (2005) 353:1685–93. doi: 10.1056/NEJMoa050333
9. Walkey AJ, Summer R, Ho V, Alkana P. Acute respiratory distress syndrome: epidemiology and management approaches. Clin Epidemiol (2012) 4:159–69. doi: 10.2147/CLEP.S28800
10. Shaver CM, Bastarache JA. Clinical and biological heterogeneity in acute respiratory distress syndrome: direct versus indirect lung injury. Clin Chest Med (2014) 35:639–53. doi: 10.1016/j.ccm.2014.08.004
11. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol (2011) 6:147–63. doi: 10.1146/annurev-pathol-011110-130158
12. Chudow M, Carter M, Rumbak M. Pharmacological treatments for acute respiratory distress syndrome. AACN Adv Crit Care (2015) 26:185–91. doi: 10.1097/NCI.0000000000000092
13. Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis (1977) 116:589–615. doi: 10.1164/arrd.1977.116.4.589
14. Batchinsky AI, Wyckoff R, Choi JH, Burmeister D, Jordan BS, Necsoiu C, et al. Dynamics of acute respiratory distress syndrome development due to smoke inhalation injury: Implications for prolonged field care. J Trauma Acute Care Surg (2019) 87:S91–S100. doi: 10.1097/TA.0000000000002227
15. Choi JH, Necsoiu C, Wendorff D, Jordan B, Dixon A, Roberts TR, et al. Effects of adjunct treatments on end-organ damage and histological injury severity in acute respiratory distress syndrome and multiorgan failure caused by smoke inhalation injury and burns. Burns (2019) 45:1765–74. doi: 10.1016/j.burns.2019.07.020
16. Russkamp NF, Ruemmler R, Roewe J, Moore BB, Ward PA, Bosmann M. Experimental design of complement component 5a-induced acute lung injury (C5a-ALI): a role of CC-chemokine receptor type 5 during immune activation by anaphylatoxin. FASEB J (2015) 29:3762–72. doi: 10.1096/fj.15-271635
17. Weiser MR, Pechet TT, Williams JP, Ma M, Frenette PS, Moore FD, et al. Experimental murine acid aspiration injury is mediated by neutrophils and the alternative complement pathway. J Appl Physiol (1985). (1997) 83:1090–5. doi: 10.1152/jappl.1997.83.4.1090
18. Chen J, Crispin JC, Dalle Lucca J, Tsokos GC. A novel inhibitor of the alternative pathway of complement attenuates intestinal ischemia/reperfusion-induced injury. J Surg Res (2011) 167:e131–6. doi: 10.1016/j.jss.2009.05.041
19. Gorman EA, O'Kane CM, McAuley DF. Acute respiratory distress syndrome in adults: diagnosis, outcomes, long-term sequelae, and management. Lancet (2022) 400:1157–70. doi: 10.1016/S0140-6736(22)01439-8
20. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5:18. doi: 10.1038/s41572-019-0069-0
21. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci USA (2020) 117:25018–25. doi: 10.1073/pnas.2010540117
22. Sinkovits G, Mezo B, Reti M, Muller V, Ivanyi Z, Gal J, et al. Complement overactivation and consumption predicts in-hospital mortality in SARS-CoV-2 infection. Front Immunol (2021) 12:663187. doi: 10.3389/fimmu.2021.663187
23. Siggins MK, Davies K, Fellows R, Thwaites RS, Baillie JK, Semple MG, et al. Alternative pathway dysregulation in tissues drives sustained complement activation and predicts outcome across the disease course in COVID-19. Immunology (2022) 168(3):473–92. doi: 10.1111/imm.13585
24. Sweeney RM, McAuley DF. Acute respiratory distress syndrome. Lancet (2016) 388:2416–30. doi: 10.1016/S0140-6736(16)00578-X
25. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol (2006) 176:1305–10. doi: 10.4049/jimmunol.176.3.1305
26. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med (2006) 12:682–7. doi: 10.1038/nm1419
27. Lupu F, Keshari RS, Lambris JD, Coggeshall KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res (2014) 133 Suppl 1:S28–31. doi: 10.1016/j.thromres.2014.03.014
28. Duehrkop C, Rieben R. Ischemia/reperfusion injury: effect of simultaneous inhibition of plasma cascade systems versus specific complement inhibition. Biochem Pharmacol (2014) 88:12–22. doi: 10.1016/j.bcp.2013.12.013
29. Ward PA. A plasmin-split fragment of C'3 as a new chemotactic factor. J Exp Med (1967) 126:189–206. doi: 10.1084/jem.126.2.189
30. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
31. Foley JH, Walton BL, Aleman MM, O'Byrne AM, Lei V, Harrasser M, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine (2016) 5:175–82. doi: 10.1016/j.ebiom.2016.02.011
32. Bossi F, Peerschke EI, Ghebrehiwet B, Tedesco F. Cross-talk between the complement and the kinin system in vascular permeability. Immunol Lett (2011) 140:7–13. doi: 10.1016/j.imlet.2011.06.006
33. Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest (1988) 81:1419–26. doi: 10.1172/JCI113472
34. Rothman BL, Merrow M, Bamba M, Kennedy T, Kreutzer DL. Biosynthesis of the third and fifth complement components by isolated human lung cells. Am Rev Respir Dis (1989) 139:212–20. doi: 10.1164/ajrccm/139.1.212
35. Varsano S, Kaminsky M, Kaiser M, Rashkovsky L. Generation of complement C3 and expression of cell membrane complement inhibitory proteins by human bronchial epithelium cell line. Thorax (2000) 55:364–9. doi: 10.1136/thorax.55.5.364
36. Hetland G, Johnson E, Aasebo U. Human alveolar macrophages synthesize the functional alternative pathway of complement and active C5 and C9 in vitro. Scand J Immunol (1986) 24:603–8. doi: 10.1111/j.1365-3083.1986.tb02176.x
37. Pettersen HB, Johnson E, Garred P, Hetland G, Osen SS, Mollnes TE. Quantification of non-activated (native) complement component C9 synthesized by alveolar macrophages from patients with sarcoidosis. Scand J Immunol (1990) 31:657–63. doi: 10.1111/j.1365-3083.1990.tb02816.x
38. Huber-Lang M, Younkin EM, Sarma JV, Riedemann N, McGuire SR, Lu KT, et al. Generation of C5a by phagocytic cells. Am J Pathol (2002) 161:1849–59. doi: 10.1016/S0002-9440(10)64461-6
39. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins a, d, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol (2002) 169:3978–86. doi: 10.4049/jimmunol.169.7.3978
40. Chang WC, White MR, Moyo P, McClear S, Thiel S, Hartshorn KL, et al. Lack of the pattern recognition molecule mannose-binding lectin increases susceptibility to influenza a virus infection. BMC Immunol (2010) 11:64. doi: 10.1186/1471-2172-11-64
41. Campbell JC, Li Y, van Amersfoort E, Relan A, Dubick M, Sheppard F, et al. C1 inhibitor limits organ injury and prolongs survival in swine subjected to battlefield simulated injury. Shock (2016) 46:177–88. doi: 10.1097/SHK.0000000000000677
42. Dalle Lucca JJ, Li Y, Simovic MO, Slack JL, Cap A, Falabella MJ, et al. Decay-accelerating factor limits hemorrhage-instigated tissue injury and improves resuscitation clinical parameters. J Surg Res (2013) 179:153–67. doi: 10.1016/j.jss.2012.10.017
43. Dalle Lucca JJ, Li Y, Simovic M, Pusateri AE, Falabella M, Dubick MA, et al. Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock (2012) 38:82–91. doi: 10.1097/SHK.0b013e31825a3522
44. Dalle Lucca JJ, Simovic M, Li Y, Moratz C, Falabella M, Tsokos GC. Decay-accelerating factor mitigates controlled hemorrhage-instigated intestinal and lung tissue damage and hyperkalemia in swine. J Trauma (2011) 71:S151–60. doi: 10.1097/TA.0b013e318221aa4c
45. Skidgel RA, Erdos EG. Structure and function of human plasma carboxypeptidase n, the anaphylatoxin inactivator. Int Immunopharmacol (2007) 7:1888–99. doi: 10.1016/j.intimp.2007.07.014
46. Hammerschmidt DE, Weaver LJ, Hudson LD, Craddock PR, Jacob HS. Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome. pathophysiological relevance and possible prognostic value. Lancet (1980) 1:947–9. doi: 10.1016/S0140-6736(80)91403-8
47. Robbins RA, Russ WD, Rasmussen JK, Clayton MM. Activation of the complement system in the adult respiratory distress syndrome. Am Rev Respir Dis (1987) 135:651–8. doi: 10.1164/arrd.1987.135.3.651
48. de Nooijer AH, Grondman I, Janssen NAF, Netea MG, Willems L, van de Veerdonk FL, et al. Complement activation in the disease course of coronavirus disease 2019 and its effects on clinical outcomes. J Infect Dis (2021) 223:214–24. doi: 10.1093/infdis/jiaa646
49. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
50. Kazmierowski JA, Gallin JI, Reynolds HY. Mechanism for the inflammatory response in primate lungs. demonstration and partial characterization of an alveolar macrophage-derived chemotactic factor with preferential activity for polymorphonuclear leukocytes. J Clin Invest (1977) 59:273–81. doi: 10.1172/JCI108638
51. Ishii Y, Kobayashi J, Kitamura S. Chemotactic factor generation and cell accumulation in acute lung injury induced by endotracheal acid instillation. Prostaglandins Leukot Essent Fatty Acids (1989) 37:65–70. doi: 10.1016/0952-3278(89)90188-9
52. Weigelt JA, Chenoweth DE, Borman KR, Norcross JF. Complement and the severity of pulmonary failure. J Trauma (1988) 28:1013–9. doi: 10.1097/00005373-198807000-00017
53. Zilow G, Sturm JA, Rother U, Kirschfink M. Complement activation and the prognostic value of C3a in patients at risk of adult respiratory distress syndrome. Clin Exp Immunol (1990) 79:151–7. doi: 10.1111/j.1365-2249.1990.tb05171.x
54. Zilow G, Joka T, Obertacke U, Rother U, Kirschfink M. Generation of anaphylatoxin C3a in plasma and bronchoalveolar lavage fluid in trauma patients at risk for the adult respiratory distress syndrome. Crit Care Med (1992) 20:468–73. doi: 10.1097/00003246-199204000-00006
55. Meade P, Shoemaker WC, Donnelly TJ, Abraham E, Jagels MA, Cryer HG, et al. Temporal patterns of hemodynamics, oxygen transport, cytokine activity, and complement activity in the development of adult respiratory distress syndrome after severe injury. J Trauma (1994) 36:651–7. doi: 10.1097/00005373-199405000-00009
56. Carvelli J, Demaria O, Vely F, Batista L, Chouaki Benmansour N, Fares J, et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature (2020) 588:146–50. doi: 10.1038/s41586-020-2600-6
57. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
58. Dexamethasone for COVID-19: preliminary findings. Drug Ther Bull (2020) 58:133. doi: 10.1136/dtb.2020.000045
59. Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus n protein aggravates lung injury by MASP-2-mediated complement over-activation. Signal Transduct Target Ther (2022) 7(1):318. doi: 10.1038/s41392-022-01133-5
60. Ratajczak MZ, Adamiak M, Thapa A, Bujko K, Brzezniakiewicz-Janus K, Lenkiewicz AM. NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia (2019) 33:815–25. doi: 10.1038/s41375-019-0436-6
61. Kim SY, Son M, Lee SE, Park IH, Kwak MS, Han M, et al. High-mobility group box 1-induced complement activation causes sterile inflammation. Front Immunol (2018) 9:705. doi: 10.3389/fimmu.2018.00705
62. Runza VL, Schwaeble W, Mannel DN. Ficolins: novel pattern recognition molecules of the innate immune response. Immunobiology (2008) 213:297–306. doi: 10.1016/j.imbio.2007.10.009
63. Frederiksen PD, Thiel S, Larsen CB, Jensenius JC. M-ficolin, an innate immune defence molecule, binds patterns of acetyl groups and activates complement. Scand J Immunol (2005) 62:462–73. doi: 10.1111/j.1365-3083.2005.01685.x
64. Endo Y, Iwaki D, Ishida Y, Takahashi M, Matsushita M, Fujita T. Mouse ficolin b has an ability to form complexes with mannose-binding lectin-associated serine proteases and activate complement through the lectin pathway. J BioMed Biotechnol (2012) 2012:105891. doi: 10.1155/2012/105891
65. Ma YJ, Skjoedt MO, Garred P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway–the fifth lectin pathway initiation complex. J Innate Immun (2013) 5:242–50. doi: 10.1159/000345356
66. Kenawy HI, Boral I, Bevington A. Complement-coagulation cross-talk: A potential mediator of the physiological activation of complement by low pH. Front Immunol (2015) 6:215. doi: 10.3389/fimmu.2015.00215
67. Silva MA, Mirza DF, Murphy N, Richards DA, Reynolds GM, Wigmore SJ, et al. Intrahepatic complement activation, sinusoidal endothelial injury, and lactic acidosis are associated with initial poor function of the liver after transplantation. Transplantation (2008) 85:718–25. doi: 10.1097/TP.0b013e3181663366
68. Szebeni J, Baranyi L, Savay S, Gotze O, Alving CR, Bunger R, et al. Complement activation during hemorrhagic shock and resuscitation in swine. Shock (2003) 20:347–55. doi: 10.1097/01.shk.0000082444.66379.17
69. Hecke F, Hoehn T, Strauss E, Obladen M, Sonntag J. In-vitro activation of complement system by lactic acidosis in newborn and adults. Mediators Inflamm (2001) 10:27–31. doi: 10.1080/09629350123788
70. Emeis M, Sonntag J, Willam C, Strauss E, Walka MM, Obladen M. Acidosis activates complement system in vitro. Mediators Inflamm (1998) 7:417–20. doi: 10.1080/09629359990649
71. Wagner MH, Sonntag J, Strauss E, Obladen M. Complement and contact activation related to surfactant response in respiratory distress syndrome. Pediatr Res (1999) 45:14–8. doi: 10.1203/00006450-199901000-00004
72. Sonntag J, Emeis M, Strauss E, Obladen M. In vitro activation of complement and contact system by lactic acidosis. Mediators Inflamm (1998) 7:49–51. doi: 10.1080/09629359891388
73. Sonntag J, Wagner MH, Strauss E, Obladen M. Complement and contact activation in term neonates after fetal acidosis. Arch Dis Child Fetal Neonatal Ed (1998) 78:F125–8. doi: 10.1136/fn.78.2.f125
74. Bastarache JA, Blackwell TS. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech (2009) 2:218–23. doi: 10.1242/dmm.001677
75. Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, et al. Increased C5a receptor expression in sepsis. J Clin Invest (2002) 110:101–8. doi: 10.1172/JCI15409
76. Czermak BJ, Lentsch AB, Bless NM, Schmal H, Friedl HP, Ward PA. Synergistic enhancement of chemokine generation and lung injury by C5a or the membrane attack complex of complement. Am J Pathol (1999) 154:1513–24. doi: 10.1016/S0002-9440(10)65405-3
77. Ehrnthaller C, Flierl M, Perl M, Denk S, Unnewehr H, Ward PA, et al. The molecular fingerprint of lung inflammation after blunt chest trauma. Eur J Med Res (2015) 20:70. doi: 10.1186/s40001-015-0164-y
78. Flierl MA, Perl M, Rittirsch D, Bartl C, Schreiber H, Fleig V, et al. The role of C5a in the innate immune response after experimental blunt chest trauma. Shock (2008) 29:25–31. doi: 10.1097/shk.0b013e3180556a0b
79. Strait RT, Hicks W, Barasa N, Mahler A, Khodoun M, Kohl J, et al. MHC class I-specific antibody binding to nonhematopoietic cells drives complement activation to induce transfusion-related acute lung injury in mice. J Exp Med (2011) 208:2525–44. doi: 10.1084/jem.20110159
80. Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, et al. Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol (2013) 49:221–30. doi: 10.1165/rcmb.2012-0428OC
81. Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. dependency on neutrophils and toxic oxygen metabolites. J Clin Invest. (1982) 69:1126–35. doi: 10.1172/JCI110548
82. Stimler NP, Hugli TE, Bloor CM. Pulmonary injury induced by C3a and C5a anaphylatoxins. Am J Pathol (1980) 100:327–48.
83. Larsen GL, McCarthy K, Webster RO, Henson J, Henson PM. A differential effect of C5a and C5a des arg in the induction of pulmonary inflammation. Am J Pathol (1980) 100:179–92.
84. Larsen GL, Mitchell BC, Henson PM. The pulmonary response of C5 sufficient and deficient mice to immune complexes. Am Rev Respir Dis (1981) 123:434–9. doi: 10.1164/arrd.1981.123.4.434
85. Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J (2013) 27:5010–21. doi: 10.1096/fj.13-236380
86. Bozic CR, Lu B, Hopken UE, Gerard C, Gerard NP. Neurogenic amplification of immune complex inflammation. Science (1996) 273:1722–5. doi: 10.1126/science.273.5282.1722
87. Sommerfeld O, Medyukhina A, Neugebauer S, Ghait M, Ulferts S, Lupp A, et al. Targeting complement C5a receptor 1 for the treatment of immunosuppression in sepsis. Mol Ther (2021) 29:338–46. doi: 10.1016/j.ymthe.2020.09.008
88. Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, et al. Complement as a target in COVID-19? Nat Rev Immunol (2020) 20:343–4. doi: 10.1038/s41577-020-0320-7
89. Stevens JH, O'Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, et al. Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest (1986) 77:1812–6. doi: 10.1172/JCI112506
90. Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol (2020) 215:108450. doi: 10.1016/j.clim.2020.108450
91. Sun S, Zhao G, Liu C, Fan W, Zhou X, Zeng L, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis (2015) 60:586–95. doi: 10.1093/cid/ciu887
92. Bauer M, Weyland A, Marx G, Bloos F, Weber S, Weiler N, et al. Efficacy and safety of vilobelimab (IFX-1), a novel monoclonal anti-C5a antibody, in patients with early severe sepsis or septic shock-a randomized, placebo-controlled, double-blind, multicenter, phase IIa trial (SCIENS study). Crit Care Explor (2021) 3:e0577. doi: 10.1097/CCE.0000000000000577
93. Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL napoli 2 nord experience. Eur Rev Med Pharmacol Sci (2020) 24:4040–7. doi: 10.26355/eurrev_202004_20875
94. Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, et al. Role of the alternative pathway in the early complement activation following major trauma. Shock (2007) 28:29–34. doi: 10.1097/shk.0b013e3180342439
95. Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol (2012) 946:147–59. doi: 10.1007/978-1-4614-0106-3_9
96. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol (2021) 17:46–64. doi: 10.1038/s41581-020-00357-4
97. Wang X, Sahu KK, Cerny J. Coagulopathy, endothelial dysfunction, thrombotic microangiopathy and complement activation: potential role of complement system inhibition in COVID-19. J Thromb Thrombolysis (2021) 51:657–62. doi: 10.1007/s11239-020-02297-z
98. Pandya PH, Wilkes DS. Complement system in lung disease. Am J Respir Cell Mol Biol (2014) 51:467–73. doi: 10.1165/rcmb.2013-0485TR
99. Standiford TJ, Ward PA. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl Res (2016) 167:183–91. doi: 10.1016/j.trsl.2015.04.015
100. Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase r and carboxypeptidase n. Microbiol Immunol (2002) 46:131–4. doi: 10.1111/j.1348-0421.2002.tb02669.x
101. Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature (1991) 349:614–7. doi: 10.1038/349614a0
102. Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry (2003) 42:9406–15. doi: 10.1021/bi034489v
103. Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol (2013) 305:L665–81. doi: 10.1152/ajplung.00232.2013
104. Riedemann NC, Guo RF, Sarma VJ, Laudes IJ, Huber-Lang M, Warner RL, et al. Expression and function of the C5a receptor in rat alveolar epithelial cells. J Immunol (2002) 168:1919–25. doi: 10.4049/jimmunol.168.4.1919
105. Hallgren R, Samuelsson T, Modig J. Complement activation and increased alveolar-capillary permeability after major surgery and in adult respiratory distress syndrome. Crit Care Med (1987) 15:189–93. doi: 10.1097/00003246-198703000-00001
106. Liu K, Mao YF, Zheng J, Peng ZY, Liu WW, Liu Y, et al. SC5b-9-induced pulmonary microvascular endothelial hyperpermeability participates in ventilator-induced lung injury. Cell Biochem Biophys (2013) 67:1421–31. doi: 10.1007/s12013-013-9675-8
107. Sun L, Guo RF, Gao H, Sarma JV, Zetoune FS, Ward PA. Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J (2009) 23:3808–18. doi: 10.1096/fj.09-133694
108. Chang JC. Acute respiratory distress syndrome as an organ phenotype of vascular microthrombotic disease: Based on hemostatic theory and endothelial molecular pathogenesis. Clin Appl Thromb Hemost (2019) 25:1076029619887437. doi: 10.1177/1076029619887437
109. Rambaldi A, Gritti G, Mico MC, Frigeni M, Borleri G, Salvi A, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology (2020) 225:152001. doi: 10.1016/j.imbio.2020.152001
110. Lin YC, Liao YJ, Lee YH, Tseng SF, Liu JY, Chen YS, et al. Staphylococcal phosphatidylinositol-specific phospholipase c potentiates lung injury via complement sensitisation. Cell Microbiol (2019) 21:e13085. doi: 10.1111/cmi.13085
111. Song WC, FitzGerald GA. COVID-19, microangiopathy, hemostatic activation, and complement. J Clin Invest (2020) 130:3950–3. doi: 10.1172/JCI140183
112. Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol (2005) 33:319–27. doi: 10.1165/rcmb.F305
113. Tennenberg SD, Jacobs MP, Solomkin JS. Complement-mediated neutrophil activation in sepsis- and trauma-related adult respiratory distress syndrome. clarification with radioaerosol lung scans. Arch Surg (1987) 122:26–32. doi: 10.1001/archsurg.1987.01400130032004
114. Hickey MJ, Kubes P. Intravascular immunity: the host-pathogen encounter in blood vessels. Nat Rev Immunol (2009) 9:364–75. doi: 10.1038/nri2532
115. Ward PA, Grailer JJ. Acute lung injury and the role of histones. Transl Respir Med (2014) 2:1. doi: 10.1186/2213-0802-2-1
116. Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med (1997) 155:1187–205. doi: 10.1164/ajrccm.155.4.9105054
117. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med (2000) 342:1334–49. doi: 10.1056/NEJM200005043421806
118. Bosmann M, Haggadone MD, Zetoune FS, Sarma JV, Ward PA. The interaction between C5a and both C5aR and C5L2 receptors is required for production of G-CSF during acute inflammation. Eur J Immunol (2013) 43:1907–13. doi: 10.1002/eji.201243075
119. Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, et al. Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest (1996) 98:503–12. doi: 10.1172/JCI118818
120. Carnesecchi S, Pache JC, Barazzone-Argiroffo C. NOX enzymes: potential target for the treatment of acute lung injury. Cell Mol Life Sci (2012) 69:2373–85. doi: 10.1007/s00018-012-1013-6
121. Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med (2001) 164:896–904. doi: 10.1164/ajrccm.164.5.2103040
122. Meduri GU, Annane D, Chrousos GP, Marik PE, Sinclair SE. Activation and regulation of systemic inflammation in ARDS: rationale for prolonged glucocorticoid therapy. Chest (2009) 136:1631–43. doi: 10.1378/chest.08-2408
123. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells (2020) 9(6):1383. doi: 10.3390/cells9061383
124. Bosmann M, Ward PA. Protein-based therapies for acute lung injury: targeting neutrophil extracellular traps. Expert Opin Ther Targets (2014) 18:703–14. doi: 10.1517/14728222.2014.902938
125. Grailer JJ, Ward PA. Lung inflammation and damage induced by extracellular histones. Inflamm Cell Signal (2014) 1(4):e131. doi: 10.14800/ics.131
126. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187:160–9. doi: 10.1164/rccm.201206-1037OC
127. Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol (2014) 192:5974–83. doi: 10.4049/jimmunol.1400368
128. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185:1225–34. doi: 10.1164/rccm.201201-0003OC
129. Makabe H, Kojika M, Takahashi G, Matsumoto N, Shibata S, Suzuki Y, et al. Interleukin-18 levels reflect the long-term prognosis of acute lung injury and acute respiratory distress syndrome. J Anesth (2012) 26:658–63. doi: 10.1007/s00540-012-1409-3
130. Mulligan MS, Ward PA. Immune complex-induced lung and dermal vascular injury. differing requirements for tumor necrosis factor-alpha and IL-1. J Immunol (1992) 149:331–9. doi: 10.4049/jimmunol.149.1.331
131. Aggarwal NR, King LS, D'Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol (2014) 306:L709–25. doi: 10.1152/ajplung.00341.2013
132. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
133. Gupta A, Gupta GS. Status of mannose-binding lectin (MBL) and complement system in COVID-19 patients and therapeutic applications of antiviral plant MBLs. Mol Cell Biochem (2021) 476:2917–42. doi: 10.1007/s11010-021-04107-3
134. Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol (2012) 5:605–9. doi: 10.1038/mi.2012.74
135. Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol (2005) 23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816
136. Jacobs RF, Tabor DR, Burks AW, Campbell GD. Elevated interleukin-1 release by human alveolar macrophages during the adult respiratory distress syndrome. Am Rev Respir Dis (1989) 140:1686–92. doi: 10.1164/ajrccm/140.6.1686
137. Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, et al. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet (1993) 341:643–7. doi: 10.1016/0140-6736(93)90416-e
138. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol (2011) 2:65. doi: 10.3389/fimmu.2011.00065
139. Yang J, Wise L, Fukuchi KI. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in alzheimer's disease. Front Immunol (2020) 11:724. doi: 10.3389/fimmu.2020.00724
140. Arbore G, Kemper C. A novel "complement-metabolism-inflammasome axis" as a key regulator of immune cell effector function. Eur J Immunol (2016) 46:1563–73. doi: 10.1002/eji.201546131
141. Triantafilou M, Hughes TR, Morgan BP, Triantafilou K. Complementing the inflammasome. Immunology (2016) 147:152–64. doi: 10.1111/imm.12556
142. Xie CB, Jane-Wit D, Pober JS. Complement membrane attack complex: New roles, mechanisms of action, and therapeutic targets. Am J Pathol (2020) 190:1138–50. doi: 10.1016/j.ajpath.2020.02.006
143. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
144. Dinarello CA. The C3a receptor, caspase-1, and release of IL-1beta. Blood (2013) 122:3394–5. doi: 10.1182/blood-2013-08-518282
145. Fattahi F, Grailer JJ, Parlett M, Lu H, Malan EA, Abe E, et al. Requirement of complement C6 for intact innate immune responses in mice. J Immunol (2020) 205:251–60. doi: 10.4049/jimmunol.1900801
146. Rosseau S, Hammerl P, Maus U, Walmrath HD, Schutte H, Grimminger F, et al. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol (2000) 279:L25–35. doi: 10.1152/ajplung.2000.279.1.L25
147. Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu H, et al. Complement C5a exacerbates acute lung injury induced through autophagy-mediated alveolar macrophage apoptosis. Cell Death Dis (2014) 5:e1330. doi: 10.1038/cddis.2014.274
148. Gao H, Guo RF, Speyer CL, Reuben J, Neff TA, Hoesel LM, et al. Stat3 activation in acute lung injury. J Immunol (2004) 172:7703–12. doi: 10.4049/jimmunol.172.12.7703
149. Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest (2002) 110:1823–30. doi: 10.1172/JCI16577
150. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med (1994) 150:113–22. doi: 10.1164/ajrccm.150.1.8025736
151. Steinberg J, Halter J, Schiller H, Gatto L, Nieman G. The development of acute respiratory distress syndrome after gut ischemia/reperfusion injury followed by fecal peritonitis in pigs: a clinically relevant model. Shock. (2005) 23:129–37. doi: 10.1097/01.shk.0000148053.66645.2e
152. Guo RF, Lentsch AB, Sarma JV, Sun L, Riedemann NC, McClintock SD, et al. Activator protein-1 activation in acute lung injury. Am J Pathol (2002) 161:275–82. doi: 10.1016/S0002-9440(10)64179-X
153. Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol (2007) 25:1265–75. doi: 10.1038/nbt1342
154. Harris JF, Aden J, Lyons CR, Tesfaigzi Y. Resolution of LPS-induced airway inflammation and goblet cell hyperplasia is independent of IL-18. Respir Res (2007) 8:24. doi: 10.1186/1465-9921-8-24
155. Morris PE, Glass J, Cross R, Cohen DA. Role of T-lymphocytes in the resolution of endotoxin-induced lung injury. Inflammation (1997) 21:269–78. doi: 10.1023/A:1027393715300
156. Nakajima T, Suarez CJ, Lin KW, Jen KY, Schnitzer JE, Makani SS, et al. T Cell pathways involving CTLA4 contribute to a model of acute lung injury. J Immunol (2010) 184:5835–41. doi: 10.4049/jimmunol.0903238
157. Li Q, Gu Y, Tu Q, Wang K, Gu X, Ren T. Blockade of interlukin-17 restrains the development of acute lung injury. Scand J Immunol (2015) 83(3):203–11. doi: 10.1111/sji.12408
158. Li JT, Melton AC, Su G, Hamm DE, LaFemina M, Howard J, et al. Unexpected role for adaptive alphabetaTh17 cells in acute respiratory distress syndrome. J Immunol (2015) 195:87–95. doi: 10.4049/jimmunol.1500054
159. Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol (2003) 170:2106–12. doi: 10.4049/jimmunol.170.4.2106
160. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol (2009) 183:5301–10. doi: 10.4049/jimmunol.0900995
161. Muir R, Osbourn M, Dubois AV, Doran E, Small DM, Monahan A, et al. Innate lymphoid cells are the predominant source of IL-17A during the early pathogenesis of acute respiratory distress syndrome. Am J Respir Crit Care Med (2016) 193:407–16. doi: 10.1164/rccm.201410-1782OC
162. Adamzik M, Broll J, Steinmann J, Westendorf AM, Rehfeld I, Kreissig C, et al. An increased alveolar CD4 + CD25 + Foxp3 + T-regulatory cell ratio in acute respiratory distress syndrome is associated with increased 30-day mortality. Intensive Care Med (2013) 39:1743–51. doi: 10.1007/s00134-013-3036-3
163. Yu ZX, Ji MS, Yan J, Cai Y, Liu J, Yang HF, et al. The ratio of Th17/Treg cells as a risk indicator in early acute respiratory distress syndrome. Crit Care (2015) 19:82. doi: 10.1186/s13054-015-0811-2
164. Mohamed Khosroshahi L, Rezaei N. Dysregulation of the immune response in coronavirus disease 2019. Cell Biol Int (2021) 45:702–7. doi: 10.1002/cbin.11517
165. Marcos-Jimenez A, Sanchez-Alonso S, Alcaraz-Serna A, Esparcia L, Lopez-Sanz C, Sampedro-Nunez M, et al. Deregulated cellular circuits driving immunoglobulins and complement consumption associate with the severity of COVID-19 patients. Eur J Immunol (2021) 51:634–47. doi: 10.1002/eji.202048858
166. Kwan WH, van der Touw W, Heeger PS. Complement regulation of T cell immunity. Immunol Res (2012) 54:247–53. doi: 10.1007/s12026-012-8327-1
167. Hirsh M, Dyugovskaya L, Kaplan V, Krausz MM. Response of lung gammadelta T cells to experimental sepsis in mice. Immunology (2004) 112:153–60. doi: 10.1111/j.1365-2567.2004.01854.x
168. Cravedi P, van der Touw W, Heeger PS. Complement regulation of T-cell alloimmunity. Semin Nephrol (2013) 33:565–74. doi: 10.1016/j.semnephrol.2013.08.007
169. D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest (2009) 119:2898–913. doi: 10.1172/JCI36498
170. Cheng HB, Chen RY, Wu JP, Chen L, Liang YH, Pan HF, et al. Complement C4 induces regulatory T cells differentiation through dendritic cell in systemic lupus erythematosus. Cell Biosci (2015) 5:73. doi: 10.1186/s13578-015-0052-8
171. Speth C, Rambach G, Wurzner R, Lass-Florl C, Kozarcanin H, Hamad OA, et al. Complement and platelets: Mutual interference in the immune network. Mol Immunol (2015) 67:108–18. doi: 10.1016/j.molimm.2015.03.244
172. Ioannou A, Kannan L, Tsokos GC. Platelets, complement and tissue inflammation. Autoimmunity (2013) 46:1–5. doi: 10.3109/08916934.2012.722144
173. Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost. (2016) 14:531–45. doi: 10.1111/jth.13208
174. Mulligan MS, Schmid E, Till GO, Hugli TE, Friedl HP, Roth RA, et al. C5a-dependent up-regulation in vivo of lung vascular p-selectin. J Immunol (1997) 158:1857–61. doi: 10.4049/jimmunol.158.4.1857
175. Peerschke EI, Yin W, Ghebrehiwet B. Platelet mediated complement activation. Adv Exp Med Biol (2008) 632:81–91. doi: 10.1007/978-0-387-78952-1_7
176. Jacob HS, Moldow CF, Flynn PJ, Weisdorf DJ, Vercellotti GM, Hammerschmidt DE. Therapeutic ramifications of the interaction of complement, granulocytes, and platelets in the production of acute lung injury. Ann NY Acad Sci (1982) 384:489–95. doi: 10.1111/j.1749-6632.1982.tb21395.x
177. Orsini F, Fumagalli S, Csaszar E, Toth K, De Blasio D, Zangari R, et al. Mannose-binding lectin drives platelet inflammatory phenotype and vascular damage after cerebral ischemia in mice via IL (Interleukin)-1alpha. Arterioscler Thromb Vasc Biol (2018) 38:2678–90. doi: 10.1161/ATVBAHA.118.311058
178. Pfeiffer S, Zorn GL 3rd, Zhang JP, Giorgio TD, Robson SC, Azimzadeh AM, et al. Hyperacute lung rejection in the pig-to-human model. III. platelet receptor inhibitors synergistically modulate complement activation and lung injury. Transplantation (2003) 75:953–9. doi: 10.1097/01.TP.0000058517.07194.90
179. Overmyer KA, Shishkova E, Miller IJ, Balnis J, Bernstein MN, Peters-Clarke TM, et al. Large-Scale multi-omic analysis of COVID-19 severity. Cell Syst (2021) 12:23–40 e7. doi: 10.1016/j.cels.2020.10.003
180. Sebag SC, Bastarache JA, Ware LB. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol (2011) 12:1481–96. doi: 10.2174/138920111798281171
181. Bastarache JA, Ware LB, Bernard GR. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Semin Respir Crit Care Med (2006) 27:365–76. doi: 10.1055/s-2006-948290
182. Fuchs-Buder T, de Moerloose P, Ricou B, Reber G, Vifian C, Nicod L, et al. Time course of procoagulant activity and d dimer in bronchoalveolar fluid of patients at risk for or with acute respiratory distress syndrome. Am J Respir Crit Care Med (1996) 153:163–7. doi: 10.1164/ajrccm.153.1.8542111
183. Tomashefski JF Jr., Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol (1983) 112:112–26.
184. Bone RC, Francis PB, Pierce AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med (1976) 61:585–9. doi: 10.1016/0002-9343(76)90135-2
185. Kellici TF, Pilka ES, Bodkin MJ. Therapeutic potential of targeting plasminogen activator inhibitor-1 in COVID-19. Trends Pharmacol Sci (2021) 42:431–3. doi: 10.1016/j.tips.2021.03.006
186. D'Alonzo D, De Fenza M, Pavone V. COVID-19 and pneumonia: a role for the uPA/uPAR system. Drug Discov Today (2020) 25:1528–34. doi: 10.1016/j.drudis.2020.06.013
187. Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol (2009) 40:123–34. doi: 10.1165/rcmb.2008-0241TR
188. Bone RC, Balk R, Slotman G, Maunder R, Silverman H, Hyers TM, et al. Adult respiratory distress syndrome. sequence and importance of development of multiple organ failure. the prostaglandin E1 study group. Chest (1992) 101:320–6. doi: 10.1378/chest.101.2.320
189. Mandal RV, Mark EJ, Kradin RL. Megakaryocytes and platelet homeostasis in diffuse alveolar damage. Exp Mol Pathol (2007) 83:327–31. doi: 10.1016/j.yexmp.2007.08.005
190. Lipcsey M, Persson B, Eriksson O, Blom AM, Fromell K, Hultstrom M, et al. The outcome of critically ill COVID-19 patients is linked to thromboinflammation dominated by the Kallikrein/Kinin system. Front Immunol (2021) 12:627579. doi: 10.3389/fimmu.2021.627579
191. Kambas K, Markiewski MM, Pneumatikos IA, Rafail SS, Theodorou V, Konstantonis D, et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol (2008) 180:7368–75. doi: 10.4049/jimmunol.180.11.7368
192. Wojta J, Huber K, Valent P. New aspects in thrombotic research: complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol Haemost Thromb (2003) 33:438–41. doi: 10.1159/000083842
193. Wojta J, Kaun C, Zorn G, Ghannadan M, Hauswirth AW, Sperr WR, et al. C5a stimulates production of plasminogen activator inhibitor-1 in human mast cells and basophils. Blood (2002) 100:517–23. doi: 10.1182/blood.V100.2.517
194. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
195. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi: 10.1038/ni.1923
196. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
197. Murugaiah V, Varghese PM, Beirag N, De Cordova S, Sim RB, Kishore U. Complement proteins as soluble pattern recognition receptors for pathogenic viruses. Viruses (2021) 13(5):824. doi: 10.3390/v13050824
198. Modinger Y, Rapp A, Pazmandi J, Vikman A, Holzmann K, Haffner-Luntzer M, et al. C5aR1 interacts with TLR2 in osteoblasts and stimulates the osteoclast-inducing chemokine CXCL10. J Cell Mol Med (2018) 22:6002–14. doi: 10.1111/jcmm.13873
199. Raby AC, Holst B, Davies J, Colmont C, Laumonnier Y, Coles B, et al. TLR activation enhances C5a-induced pro-inflammatory responses by negatively modulating the second C5a receptor, C5L2. Eur J Immunol (2011) 41:2741–52. doi: 10.1002/eji.201041350
200. Okazaki N, Hazeki K, Izumi T, Nigorikawa K, Hazeki O. C5a controls TLR-induced IL-10 and IL-12 production independent of phosphoinositide 3-kinase. J Biochem (2011) 149:265–74. doi: 10.1093/jb/mvq136
201. Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity (2005) 22:415–26. doi: 10.1016/j.immuni.2005.02.006
202. Yu S, Wang D, Huang L, Zhang Y, Luo R, Adah D, et al. The complement receptor C5aR2 promotes protein kinase r expression and contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages. J Biol Chem (2019) 294:8384–94. doi: 10.1074/jbc.RA118.006508
203. Wang C, Wang H, Hao J, Chang DY, Zhao MH, Chen M. Involvement of high mobility group box 1 in the activation of C5a-primed neutrophils induced by ANCA. Clin Immunol (2015) 159:47–57. doi: 10.1016/j.clim.2015.04.008
204. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care (2009) 13:R174. doi: 10.1186/cc8152
205. Yang Z, Simovic MO, Edsall PR, Liu B, Cancio TS, Batchinsky AI, et al. HMGB1 inhibition to ameliorate organ failure and increase survival in trauma. Biomolecules (2022) 1:101. doi: 10.3390/biom12010101
206. Yang Z, Le TD, Simovic MO, Liu B, Fraker TL, Cancio TS, et al. Traumatized triad of complementopathy, endotheliopathy, and coagulopathy - impact on clinical outcomes in severe polytrauma patients. Front Immunol (2022) 13:991048. doi: 10.3389/fimmu.2022.991048
207. Li Y, Yang Z, Chavko M, Liu B, Aderemi OA, Simovic MO, et al. Complement inhibition ameliorates blast-induced acute lung injury in rats: Potential role of complement in intracellular HMGB1-mediated inflammation. PloS One (2018) 13:e0202594. doi: 10.1371/journal.pone.0202594
208. Huber-Lang M, Barratt-Due A, Pischke SE, Sandanger O, Nilsson PH, Nunn MA, et al. Double blockade of CD14 and complement C5 abolishes the cytokine storm and improves morbidity and survival in polymicrobial sepsis in mice. J Immunol (2014) 192:5324–31. doi: 10.4049/jimmunol.1400341
209. Barratt-Due A, Thorgersen EB, Egge K, Pischke S, Sokolov A, Hellerud BC, et al. Combined inhibition of complement C5 and CD14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. J Immunol (2013) 191:819–27. doi: 10.4049/jimmunol.1201909
210. Hellerud BC, Orrem HL, Dybwik K, Pischke SE, Baratt-Due A, Castellheim A, et al. Combined inhibition of C5 and CD14 efficiently attenuated the inflammatory response in a porcine model of meningococcal sepsis. J Intensive Care (2017) 5:21. doi: 10.1186/s40560-017-0217-0
211. Skjeflo EW, Sagatun C, Dybwik K, Aam S, Urving SH, Nunn MA, et al. Combined inhibition of complement and CD14 improved outcome in porcine polymicrobial sepsis. Crit Care (2015) 19:415. doi: 10.1186/s13054-015-1129-9
212. Barratt-Due A, Pischke SE, Nilsson PH, Espevik T, Mollnes TE. Dual inhibition of complement and toll-like receptors as a novel approach to treat inflammatory diseases-C3 or C5 emerge together with CD14 as promising targets. J Leukoc Biol (2017) 101:193–204. doi: 10.1189/jlb.3VMR0316-132R
213. Denk S, Neher MD, Messerer DAC, Wiegner R, Nilsson B, Rittirsch D, et al. Complement C5a functions as a master switch for the pH balance in neutrophils exerting fundamental immunometabolic effects. J Immunol (2017) 198:4846–54. doi: 10.4049/jimmunol.1700393
214. Karasu E, Nilsson B, Kohl J, Lambris JD, Huber-Lang M. Targeting complement pathways in polytrauma- and sepsis-induced multiple-organ dysfunction. Front Immunol (2019) 10:543. doi: 10.3389/fimmu.2019.00543
215. Kunz N, Kemper C. Complement has brains-do intracellular complement and immunometabolism cooperate in tissue homeostasis and behavior? Front Immunol (2021) 12:629986. doi: 10.3389/fimmu.2021.629986
216. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
217. Dickson RP, Schultz MJ, van der Poll T, Schouten LR, Falkowski NR, Luth JE, et al. Lung microbiota predict clinical outcomes in critically ill patients. Am J Respir Crit Care Med (2020) 201:555–63. doi: 10.1164/rccm.201907-1487OC
218. Luyt CE, Bouadma L, Morris AC, Dhanani JA, Kollef M, Lipman J, et al. Pulmonary infections complicating ARDS. Intensive Care Med (2020) 46:2168–83. doi: 10.1007/s00134-020-06292-z
219. Siwicka-Gieroba D, Czarko-Wicha K. Lung microbiome - a modern knowledge. Cent Eur J Immunol (2020) 45:342–5. doi: 10.5114/ceji.2020.101266
220. Martin-Loeches I, Dickson R, Torres A, Hanberger H, Lipman J, Antonelli M, et al. The importance of airway and lung microbiome in the critically ill. Crit Care (2020) 24:537. doi: 10.1186/s13054-020-03219-4
221. Dickson RP. The lung microbiome and ARDS. it is time to broaden the model. Am J Respir Crit Care Med (2018) 197:549–51. doi: 10.1164/rccm.201710-2096ED
222. Hajishengallis G, Maekawa T, Abe T, Hajishengallis E, Lambris JD. Complement involvement in periodontitis: Molecular mechanisms and rational therapeutic approaches. Adv Exp Med Biol (2015) 865:57–74. doi: 10.1007/978-3-319-18603-0_4
223. Ghias MH, Hyde MJ, Tomalin LE, Morgan BP, Alavi A, Lowes MA, et al. Role of the complement pathway in inflammatory skin diseases: A focus on hidradenitis suppurativa. J Invest Dermatol (2020) 140:531–6 e1. doi: 10.1016/j.jid.2019.09.009
224. Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature (2019) 574:264–7. doi: 10.1038/s41586-019-1608-2
225. Li L, Wei T, Liu S, Wang C, Zhao M, Feng Y, et al. Complement C5 activation promotes type 2 diabetic kidney disease via activating STAT3 pathway and disrupting the gut-kidney axis. J Cell Mol Med (2021) 25:960–74. doi: 10.1111/jcmm.16157
226. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
227. Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J (2006) 25:1364–74. doi: 10.1038/sj.emboj.7601004
228. Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol (2002) 169:2102–10. doi: 10.4049/jimmunol.169.4.2102
229. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med (2003) 198:913–23. doi: 10.1084/jem.20030374
230. Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells (2009) 27:2824–32. doi: 10.1002/stem.225
231. Moll G, Jitschin R, von Bahr L, Rasmusson-Duprez I, Sundberg B, Lonnies L, et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PloS One (2011) 6:e21703. doi: 10.1371/journal.pone.0021703
232. Jeanneau C, Lundy FT, El Karim IA, About I. Potential therapeutic strategy of targeting pulp fibroblasts in dentin-pulp regeneration. J Endod. (2017) 43:S17–24. doi: 10.1016/j.joen.2017.06.007
233. Hugli TE, Erickson BW. Synthetic peptides with the biological activities and specificity of human C3a anaphylatoxin. Proc Natl Acad Sci USA (1977) 74:1826–30. doi: 10.1073/pnas.74.5.1826
234. Hartmann K, Henz BM, Kruger-Krasagakes S, Kohl J, Burger R, Guhl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood (1997) 89:2863–70. doi: 10.1182/blood.V89.8.2863
235. Marder SR, Chenoweth DE, Goldstein IM, Perez HD. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des arg. J Immunol (1985) 134:3325–31. doi: 10.4049/jimmunol.134.5.3325
236. Flierman R, Daha MR. The clearance of apoptotic cells by complement. Immunobiology (2007) 212:363–70. doi: 10.1016/j.imbio.2006.11.005
237. Stienstra R, Dijk W, van Beek L, Jansen H, Heemskerk M, Houtkooper RH, et al. Mannose-binding lectin is required for the effective clearance of apoptotic cells by adipose tissue macrophages during obesity. Diabetes (2014) 63:4143–53. doi: 10.2337/db14-0256
238. Verneret M, Tacnet-Delorme P, Osman R, Awad R, Grichine A, Kleman JP, et al. Relative contribution of c1q and apoptotic cell-surface calreticulin to macrophage phagocytosis. J Innate Immun (2014) 6:426–34. doi: 10.1159/000358834
239. Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124:915–27. doi: 10.1016/j.cell.2005.12.039
240. Takizawa F, Tsuji S, Nagasawa S. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett (1996) 397:269–72. doi: 10.1016/s0014-5793(96)01197-0
241. Langlois PF, Gawryl MS. Complement activation occurs through both classical and alternative pathways prior to onset and resolution of adult respiratory distress syndrome. Clin Immunol Immunopathol (1988) 47:152–63. doi: 10.1016/0090-1229(88)90068-2
242. Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med (2015) 19:2549–63. doi: 10.1111/jcmm.12667
243. Addis-Lieser E, Kohl J, Chiaramonte MG. Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J Immunol (2005) 175:1894–902. doi: 10.4049/jimmunol.175.3.1894
244. Muller MC, Stroo I, Wouters D, Zeerleder SS, Roelofs JJ, Boon L, et al. The effect of C1-inhibitor in a murine model of transfusion-related acute lung injury. Vox Sang (2014) 107:71–5. doi: 10.1111/vox.12128
245. Jiang X, Ma Y, Yu J, Li H, Xie F. Protective effect of C4a against hyperoxic lung injury via a macrophage-dependent but not a neutrophil/lymphocyte-dependent signaling pathway. Mol Med Rep (2016) 13:1250–6. doi: 10.3892/mmr.2015.4651
246. Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang MS, Grailer JJ, et al. Zonulin as prehaptoglobin2 regulates lung permeability and activates the complement system. Am J Physiol Lung Cell Mol Physiol (2013) 304:L863–72. doi: 10.1152/ajplung.00196.2012
247. Vangerow B, Hafner D, Rueckoldt H, Marx G, Ott N, Leuwer M, et al. Effects of C1 inhibitor and r-SP-C surfactant on oxygenation and histology in rats with lavage-induced acute lung injury. Intensive Care Med (2001) 27:1526–31. doi: 10.1007/s001340101036
248. Struber M, Hagl C, Hirt SW, Cremer J, Harringer W, Haverich A. C1-esterase inhibitor in graft failure after lung transplantation. Intensive Care Med (1999) 25:1315–8. doi: 10.1007/s001340051065
249. Garcia CC, Weston-Davies W, Russo RC, Tavares LP, Rachid MA, Alves-Filho JC, et al. Complement C5 activation during influenza a infection in mice contributes to neutrophil recruitment and lung injury. PloS One (2013) 8:e64443. doi: 10.1371/journal.pone.0064443
250. Wang R, Lu B, Gerard C, Gerard NP. C5L2, the second C5a anaphylatoxin receptor, suppresses LPS-induced acute lung injury. Am J Respir Cell Mol Biol (2016) 55:657–66. doi: 10.1165/rcmb.2016-0067OC
251. Yu Z, Saito H, Otsuka H, Shikama Y, Funayama H, Sakai M, et al. Pulmonary platelet accumulation induced by catecholamines: Its involvement in lipopolysaccharide-induced anaphylaxis-like shock. Int Immunopharmacol (2017) 43:40–52. doi: 10.1016/j.intimp.2016.11.034
252. Naito M, Taguchi O, Kobayashi T, Takagi T, D'Alessandro-Gabazza CN, Matsushima Y, et al. Thrombin-activatable fibrinolysis inhibitor protects against acute lung injury by inhibiting the complement system. Am J Respir Cell Mol Biol (2013) 49:646–53. doi: 10.1165/rcmb.2012-0454OC
253. Hoth JJ, Wells JD, Jones SE, Yoza BK, McCall CE. Complement mediates a primed inflammatory response after traumatic lung injury. J Trauma Acute Care Surg (2014) 76:601–8. doi: 10.1097/TA.0000000000000129
254. Kalbitz M, Karbach M, Braumueller S, Kellermann P, Gebhard F, Huber-Lang M, et al. Role of complement C5 in experimental blunt chest trauma-induced septic acute lung injury (ALI). PloS One (2016) 11:e0159417. doi: 10.1371/journal.pone.0159417
255. Roversi P, Ryffel B, Togbe D, Maillet I, Teixeira M, Ahmat N, et al. Bifunctional lipocalin ameliorates murine immune complex-induced acute lung injury. J Biol Chem (2013) 288:18789–802. doi: 10.1074/jbc.M112.420331
256. Yang Z, Nunn MA, Le TD, Simovic MO, Edsall PR, Liu B, et al. Immunopathology of terminal complement activation and complement C5 blockade creating a pro-survival and organ-protective phenotype in trauma. Br J Pharmacol (2023) 180:422–40. doi: 10.1111/bph.15970
257. Proctor LM, Strachan AJ, Woodruff TM, Mahadevan IB, Williams HM, Shiels IA, et al. Complement inhibitors selectively attenuate injury following administration of cobra venom factor to rats. Int Immunopharmacol (2006) 6:1224–32. doi: 10.1016/j.intimp.2006.03.002
258. Harkin DW, Marron CD, Rother RP, Romaschin A, Rubin BB, Lindsay TF. C5 complement inhibition attenuates shock and acute lung injury in an experimental model of ruptured abdominal aortic aneurysm. Br J Surg (2005) 92:1227–34. doi: 10.1002/bjs.4938
259. Harkin DW, Romaschin A, Taylor SM, Rubin BB, Lindsay TF. Complement C5a receptor antagonist attenuates multiple organ injury in a model of ruptured abdominal aortic aneurysm. J Vasc Surg (2004) 39:196–206. doi: 10.1016/j.jvs.2003.07.001
260. Xiao F, Eppihimer MJ, Willis BH, Carden DL. Complement-mediated lung injury and neutrophil retention after intestinal ischemia-reperfusion. J Appl Physiol (1985) (1997) 82:1459–65. doi: 10.1152/jappl.1997.82.5.1459
261. Heller AR, Fiedler F, Braun P, Stehr SN, Bodeker H, Koch T. Clusterin protects the lung from leukocyte-induced injury. Shock (2003) 20:166–70. doi: 10.1097/01.shk.0000075569.93053.b3
262. Heller A, Kunz M, Samakas A, Haase M, Kirschfink M, Koch T. The complement regulators C1 inhibitor and soluble complement receptor 1 attenuate acute lung injury in rabbits. Shock (2000) 13:285–90. doi: 10.1097/00024382-200004000-00006
263. Sun S, Wang H, Zhao G, An Y, Guo Y, Du L, et al. Complement inhibition alleviates paraquat-induced acute lung injury. Am J Respir Cell Mol Biol (2011) 45:834–42. doi: 10.1165/rcmb.2010-0444OC
264. Baumann U, Chouchakova N, Gewecke B, Kohl J, Carroll MC, Schmidt RE, et al. Distinct tissue site-specific requirements of mast cells and complement components C3/C5a receptor in IgG immune complex-induced injury of skin and lung. J Immunol (2001) 167:1022–7. doi: 10.4049/jimmunol.167.2.1022
265. Lu X, Li Y, Simovic MO, Peckham R, Wang Y, Tsokos GC, et al. Decay-accelerating factor attenuates c-reactive protein-potentiated tissue injury after mesenteric ischemia/reperfusion. J Surg Res (2011) 167:e103–15. doi: 10.1016/j.jss.2009.10.021
266. Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, et al. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest (2005) 115:2444–53. doi: 10.1172/JCI25208
267. Glasgow SC, Kanakasabai S, Ramachandran S, Mohanakumar T, Chapman WC. Complement depletion enhances pulmonary inflammatory response after liver injury. J Gastrointest Surg (2006) 10:357–64. doi: 10.1016/j.gassur.2005.06.033
268. Dantas M, Costa RS, Barbosa JE, Graeff MS, Sarti W, De Carvalho IF. Intravenous immunoglobulin (IVIG) attenuates antibody binding in acute haemorrhagic immunopneumonitis in a rat model of complement-dependent lung injury. Clin Exp Immunol (2000) 121:139–45. doi: 10.1046/j.1365-2249.2000.01252.x
269. Mulligan MS, Yeh CG, Rudolph AR, Ward PA. Protective effects of soluble CR1 in complement- and neutrophil-mediated tissue injury. J Immunol (1992) 148:1479–85. doi: 10.4049/jimmunol.148.5.1479
270. Mao YF, Yu QH, Zheng XF, Liu K, Liang WQ, Wang YW, et al. Pre-treatment with cobra venom factor alleviates acute lung injury induced by intestinal ischemia-reperfusion in rats. Eur Rev Med Pharmacol Sci (2013) 17:2207–17.
271. Wu X, Dubick MA, Schwacha MG, Cap AP, Darlington DN. Tranexamic acid attenuates the loss of lung barrier function in a rat model of polytrauma and hemorrhage with resuscitation. Shock (2017) 47:500–5. doi: 10.1097/SHK.0000000000000758
272. Rieber J, Schmitt J, Warth A, Muley T, Kappes J, Eichhorn F, et al. Outcome and prognostic factors of multimodal therapy for pulmonary large-cell neuroendocrine carcinomas. Eur J Med Res (2015) 20:64. doi: 10.1186/s40001-015-0158-9
273. Ou YY, Jiang Y, Li H, Zhang YY, Lu Y, Chen DF. Polysaccharides from arnebia euchroma ameliorated endotoxic fever and acute lung injury in rats through inhibiting complement system. Inflammation (2017) 40:275–84. doi: 10.1007/s10753-016-0478-0
274. Hagio T, Nakao S, Matsuoka H, Matsumoto S, Kawabata K, Ohno H. Inhibition of neutrophil elastase activity attenuates complement-mediated lung injury in the hamster. Eur J Pharmacol (2001) 426:131–8. doi: 10.1016/s0014-2999(01)01191-8
275. Yeatman M, Daggett CW, Lau CL, Byrne GW, Logan JS, Platt JL, et al. Human complement regulatory proteins protect swine lungs from xenogeneic injury. Ann Thorac Surg (1999) 67:769–75. doi: 10.1016/s0003-4975(99)00049-1
276. Daggett CW, Yeatman M, Lodge AJ, Chen EP, Van Trigt P, Byrne GW, et al. Swine lungs expressing human complement-regulatory proteins are protected against acute pulmonary dysfunction in a human plasma perfusion model. J Thorac Cardiovasc Surg (1997) 113:390–8. doi: 10.1016/S0022-5223(97)70337-4
277. Poling J, Oezkur M, Kogge K, Mengel M, Niemann H, Winkler M, et al. Hyperacute rejection in ex vivo-perfused porcine lungs transgenic for human complement regulatory proteins. Transpl Int (2006) 19:225–32. doi: 10.1111/j.1432-2277.2006.00267.x
278. Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, et al. Complement levels and activity in the normal and LPS-injured lung. Am J Physiol Lung Cell Mol Physiol (2007) 292:L748–59. doi: 10.1152/ajplung.00127.2006
279. Sachs UJ, Hattar K, Weissmann N, Bohle RM, Weiss T, Sibelius U, et al. Antibody-induced neutrophil activation as a trigger for transfusion-related acute lung injury in an ex vivo rat lung model. Blood (2006) 107:1217–9. doi: 10.1182/blood-2005-04-1744
280. Rittirsch D, Flierl MA, Day DE, Nadeau BA, McGuire SR, Hoesel LM, et al. Acute lung injury induced by lipopolysaccharide is independent of complement activation. J Immunol (2008) 180:7664–72. doi: 10.4049/jimmunol.180.11.7664
281. Hamacher J, Sadallah S, Schifferli JA, Villard J, Nicod LP. Soluble complement receptor type 1 (CD35) in bronchoalveolar lavage of inflammatory lung diseases. Eur Respir J (1998) 11:112–9. doi: 10.1183/09031936.98.11010112
282. Bhargava M, Viken K, Wang Q, Jagtap P, Bitterman P, Ingbar D, et al. Bronchoalveolar lavage fluid protein expression in acute respiratory distress syndrome provides insights into pathways activated in subjects with different outcomes. Sci Rep (2017) 7:7464. doi: 10.1038/s41598-017-07791-8
283. Chen X, Shan Q, Jiang L, Zhu B, Xi X. Quantitative proteomic analysis by iTRAQ for identification of candidate biomarkers in plasma from acute respiratory distress syndrome patients. Biochem Biophys Res Commun (2013) 441:1–6. doi: 10.1016/j.bbrc.2013.09.027
284. Gama de Abreu M, Kirschfink M, Quintel M, Albrecht DM. White blood cell counts and plasma C3a have synergistic predictive value in patients at risk for acute respiratory distress syndrome. Crit Care Med (1998) 26:1040–8. doi: 10.1097/00003246-199806000-00025
285. Turker G, Koksal N. Complement 4 levels as early predictors of poor response to surfactant therapy in respiratory distress syndrome. Am J Perinatol (2005) 22:149–54. doi: 10.1055/s-2005-865021
286. Cat R, Rosario NA, de Messias IT, Resener TD, Kirschfink M. Evaluation of complement activation in premature newborn infants with hyaline membrane disease. Eur J Pediatr (1993) 152:205–8. doi: 10.1007/BF01956145
287. Ionescu DN, Girnita AL, Zeevi A, Duquesnoy R, Pilewski J, Johnson B, et al. C4d deposition in lung allografts is associated with circulating anti-HLA alloantibody. Transpl Immunol (2005) 15:63–8. doi: 10.1016/j.trim.2005.05.001
288. Vallhonrat H, Williams WW, Cosimi AB, Tolkoff-Rubin N, Ginns LC, Wain JC, et al. In vivo generation of C4d, bb, iC3b, and SC5b-9 after OKT3 administration in kidney and lung transplant recipients. Transplantation (1999) 67:253–8. doi: 10.1097/00007890-199901270-00011
289. Kellermann W, Frentzel-Beyme R, Welte M, Jochum M. Phospholipase a in acute lung injury after trauma and sepsis: its relation to the inflammatory mediators PMN-elastase, C3a, and neopterin. Klin Wochenschr (1989) 67:190–5. doi: 10.1007/BF01711352
290. Carvalho AC, DeMarinis S, Scott CF, Silver LD, Schmaier AH, Colman RW. Activation of the contact system of plasma proteolysis in the adult respiratory distress syndrome. J Lab Clin Med (1988) 112:270–7.
291. Langlois PF, Gawryl MS. Accentuated formation of the terminal C5b-9 complement complex in patient plasma precedes development of the adult respiratory distress syndrome. Am Rev Respir Dis (1988) 138:368–75. doi: 10.1164/ajrccm/138.2.368
292. Stove S, Welte T, Wagner TO, Kola A, Klos A, Bautsch W, et al. Circulating complement proteins in patients with sepsis or systemic inflammatory response syndrome. Clin Diagn Lab Immunol (1996) 3:175–83. doi: 10.1128/cdli.3.2.175-183.1996
293. Weinberg PF, Matthay MA, Webster RO, Roskos KV, Goldstein IM, Murray JF. Biologically active products of complement and acute lung injury in patients with the sepsis syndrome. Am Rev Respir Dis (1984) 130:791–6. doi: 10.1164/arrd.1984.130.5.791
294. Hamed ME, Naeem A, Alkadi H, Alamri AA, AlYami AS, AlJuryyan A, et al. Elevated expression levels of lung complement anaphylatoxin, neutrophil chemoattractant chemokine IL-8, and RANTES in MERS-CoV-Infected patients: Predictive biomarkers for disease severity and mortality. J Clin Immunol (2021) 41(7):1607–20. doi: 10.1007/s10875-021-01061-z
295. Nossent EJ, Schuurman AR, Reijnders TDY, Saris A, Jongerius I, Blok SG, et al. Pulmonary procoagulant and innate immune responses in critically ill COVID-19 patients. Front Immunol (2021) 12:664209. doi: 10.3389/fimmu.2021.664209
296. Vallhonrat H, Swinford RD, Ingelfinger JR, Williams WW, Ryan DP, Tolkoff-Rubin N, et al. Rapid activation of the alternative pathway of complement by extracorporeal membrane oxygenation. ASAIO J (1999) 45:113–4. doi: 10.1097/00002480-199901000-00025
297. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
298. Ricklin D, Barratt-Due A, Mollnes TE. Complement in clinical medicine: Clinical trials, case reports and therapy monitoring. Mol Immunol (2017) 89:10–21. doi: 10.1016/j.molimm.2017.05.013
299. Huber-Lang MS, Ignatius A, Kohl J, Mannes M, Braun CK. Complement in trauma-traumatised complement? Br J Pharmacol (2021) 178:2863–79. doi: 10.1111/bph.15245
300. Reilly JP, Calfee CS, Christie JD. Acute respiratory distress syndrome phenotypes. Semin Respir Crit Care Med (2019) 40:19–30. doi: 10.1055/s-0039-1684049
301. Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov (2015) 14:857–77. doi: 10.1038/nrd4657
Glossary
Keywords: ARDS, complementome, multiome, connectome, pathobiology, therapeutics, clinical trial, COVID-19
Citation: Yang Z, Nicholson SE, Cancio TS, Cancio LC and Li Y (2023) Complement as a vital nexus of the pathobiological connectome for acute respiratory distress syndrome: An emerging therapeutic target. Front. Immunol. 14:1100461. doi: 10.3389/fimmu.2023.1100461
Received: 16 November 2022; Accepted: 27 February 2023;
Published: 17 March 2023.
Edited by:
Yu Cao, Sichuan University, ChinaReviewed by:
Marion Russier, Genmab (Netherlands), NetherlandsPeter A. Ward, University of Michigan, United States
Copyright © 2023 Yang, Nicholson, Cancio, Cancio and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yansong Li, eXNsaTkxNkBnbWFpbC5jb20=
†These authors share senior authorship