 Alexa Corker1
Alexa Corker1 Maya Learmonth1
Maya Learmonth1 David M. Patrick2,3Kristine Y. DeLeon-Pennell1,4
David M. Patrick2,3Kristine Y. DeLeon-Pennell1,4 Justin P. Van Beusecum4,5*
Justin P. Van Beusecum4,5*- 1Division of Cardiology, Department of Medicine, Medical University of South Carolina, Charleston, SC, United States
- 2Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 3Department of Research Service, Tennessee Valley Healthcare Veterans Affairs (VA) Medical Center, Nashville, TN, United States
- 4Department of Research Service, Ralph H. Johnson Veterans Affairs (VA) Healthcare System, Charleston, SC, United States
- 5Division of Nephrology, Department of Medicine, Medical University of South Carolina, Charleston, SC, United States
Systemic lupus erythematosus (SLE) is a common systemic autoimmune disorder and is characterized by autoantibody formation and subsequent immune complex deposition into target organs. SLE affects nearly nine women to every one man worldwide. Patients with SLE are at an enhanced risk for cardiovascular disease (CVD) morbidity and mortality. CVD is the leading cause of death worldwide and includes heart and blood vessel disorders, cerebrovascular disease, and rheumatic heart disease. Specific mechanisms by which cardiac and vascular pathophysiology develops in patients with SLE are still not fully known. Not only do we not understand this correlation between SLE and CVD, but there is also a critical gap in scientific knowledge on the contribution of sex. In this review, we will discuss the cardiac and vascular pathological disease states that are present in some patients with SLE. More importantly, we will discuss the potential mechanisms for the role of sex and sex hormones in the development of CVD with SLE.
Introduction
While overall mortality due to complications arising from patients with systemic lupus erythematosus (SLE) has decreased with the development of new therapies in recent years, the risk of mortality from cardiovascular disease (CVD) in patients with SLE has remained steady over time (1). Patients with SLE have a 3-4 times higher risk of experiencing a CVD-associated event and mortality from these events compared with patients without SLE (2). This includes accelerated risks for hypertension, vascular dysfunction, and myocardial infarction (MI) (3, 4). Clinically, SLE predominantly affects women more than men (women versus men ratio of 9 to 1), limiting the availability of data on male SLE patients (5). Nonetheless, both male and female patients with SLE have an increased risk for CVD-related mortality (6). Studies that focus solely on men with SLE and related comorbidities are even rarer and only look at symptom prevalence and commonalities between patients with SLE within the study group (7). Despite mechanistic research being limited in men with SLE, case studies can potentially shed insight into the complexity of SLE diagnosis and CVD complications (5). This review will highlight the current knowledge of sex differences in SLE and describe potential mechanisms. To determine the pathophysiology of vascular and cardiac complications in SLE, we will also review the research gap in sexual dysmorphisms seen in CVD complications in SLE. Research into the mechanistic, symptomatic, and immunological differences between male and female patients with SLE and CVD complications may be able to highlight new therapies to lower this associated risk.
Sex differences in the pathogenesis of SLE
SLE affects multiple organs and is extremely heterogeneous in its clinical presentation across different patients. Like many other autoimmune diseases, SLE affects women more frequently than men; however, male patients with SLE tend to have a more rapid and severe disease state (8–10). This puzzling observation has led numerous studies to investigate sex differences in the development and pathogenesis of patients with SLE. Male patients with SLE experience a higher occurrence of renal involvement than women; however, progression to end-stage renal disease is not different between sex (8–10). While there are numerous mechanisms that contribute to sex differences in SLE development between male and female patients, we will discuss the role of sex hormones and chromosome complement below (Figure 1).
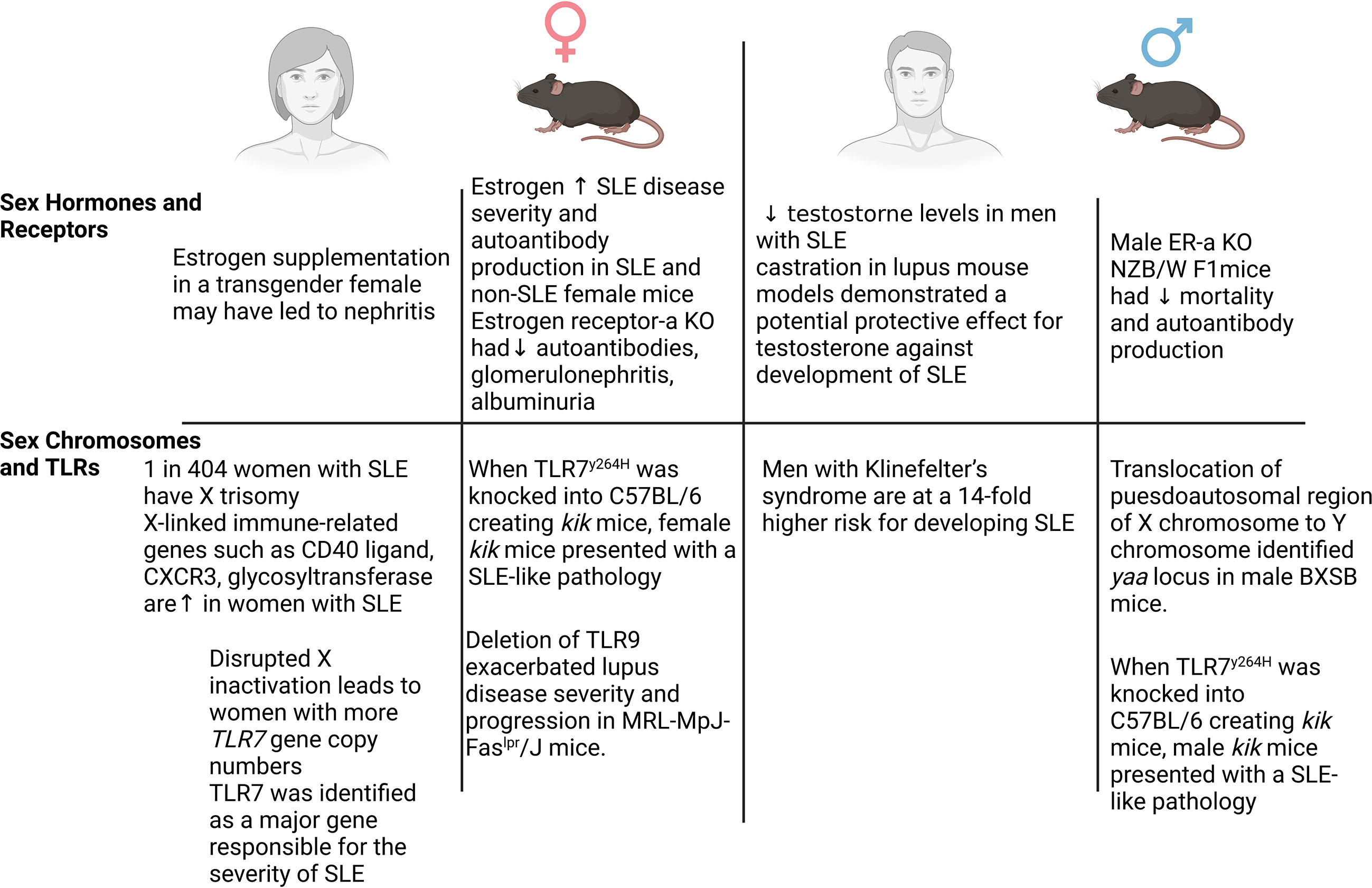
Figure 1 Sex differences in systemic lupus erythematosus (SLE). Numerous mechanisms have been demonstrated to contribute to the sex-dependent development of SLE in both patients and animal models. Sex hormones and receptors, sex chromosomes, and Toll-like receptors all have been implicated as key mechanisms in SLE. In female patients and mice (left), elevated estrogen and estrogen receptor signaling, along with the X chromosome-linked proinflammatory genes (Cxcr3, Cd40, Tlr7), contribute to the frequency and/or severity of SLE. In male patients and mice (right), an imbalance of estrogen and testosterone, elevated estrogen receptor signaling, increased copies of the X chromosome (Klinefelter’s syndrome), and TLR7 signaling contribute to the severity and development of SLE.
Sex hormones and receptors
Diagnosis of SLE in women occurs usually when estrogen is at peak levels during a patient’s lifetime, leading one to assume that sex hormones contribute to the pathogenesis of SLE. In line with this, in a common lupus mouse model, the New Zealand black/white (NZBW F1), ovariectomy of female NZB/W F1s delays the onset of autoantibody production, suggesting the important role of female sex hormones including estrogen (11). Interestingly, in the MRL/Lpr mouse model, which affects male and female mice equally, female mice develop earlier and a more severe case of SLE compared with male mice (12). However, a clinical study by Hill et al. demonstrated that estrogen supplementation in transgender women may have resulted in lupus nephritis (13). This clinical study mimics the findings in the NZB/W F1 mouse model as treatment with androgens suppressed the development of autoimmune disease and improved overall mortality in female mice, indicating that the balance between sex hormones may play a critical role in SLE pathogenesis (14). In a non-autoimmune prone mouse (C57BL/6), estrogen treatment increased the rate of nephritis and autoantibody production, suggesting that estrogen may play a critical role through modulation of the immune system (15, 16).
While estrogen has been implicated in contributing to the development of SLE, numerous impactful studies have shed light on the role of estrogen receptors. The pioneering study showing evidence of estrogen receptor signaling in lupus comes from Bynote et al. who used the estrogen receptor-α knockout (KO) on the NZB/W F1 background (17). In this study, ER-α KO NZB/W F1 female mice had reduced development of autoantibodies, glomerulonephritis, and albuminuria and increased survival time compared with controls. Similarly, male ER-α KO NZB/W F1 mice had decreased mortality and a reduction in autoantibody production (17). In keeping with this, ER-α KO mice on the NZM2410 and B6SLE1b backgrounds exhibited reduced glomerulonephritis and proteinuria and increased survival, likely through a reduction in B- and T-cell hyperactivation (18, 19). While ER-α KO mice have provided critical evidence on the role of estrogen in the development of SLE, an ER-β KO on a lupus background has not yet been developed. Despite the lack of genetic KO, in ovariectomized female NZW/B F1 mice activated with a selective ER-β agonist, anti-DNA autoantibodies were reduced; however, albuminuria and survival were not affected (20). Together, these studies suggest that ER-α plays a predominant role in the sex-dependent development of SLE.
Men with SLE have been shown to have decreased serum testosterone levels compared with healthy controls (21). Whether this decrease in testosterone was a predisposition for SLE disease onset or if the decrease was due to chronic SLE disease was not investigated. Studies using castration in lupus mouse models demonstrated a potential protective effect of testosterone against the development of SLE (22). Despite being performed in women, clinical observational studies further corroborated the protective effects of androgens in SLE patients who displayed decreased disease burden through testosterone therapy (23, 24).
Sex chromosomes
While sex hormones have been attributed a partial role in the development of SLE, both prepubescent and postmenopausal women can develop SLE, suggesting that an alternative mechanism independent of sex hormones is also responsible for SLE pathogenesis. Sex chromosomes have been implicated to contribute to the development of SLE in both sexes. Importantly, the frequency at which rare X chromosome abnormalities occur in patients with SLE is much higher than in the general healthy population (25). Approximately 1 in 404 women with SLE has X trisomy (47, XXX), while men with Klinefelter’s syndrome (47, XXY) are at an approximately 14-fold higher risk for developing SLE (26, 27). Not only are X chromosome abnormalities linked to SLE development in male and female patients, but also disturbed X chromosome inactivation contributes to the pathogenesis of female patients with SLE. Numerous immune-related genes are contained in the X chromosome. Hewagama et al. demonstrated that X-linked immune-related genes such as the CD40 ligand, CXCR3, and glycosyltransferase (OGT) are upregulated in women with SLE (28). Interestingly, this study demonstrated that X chromosome demethylation in women played a role in the predisposition to lupus. This finding has been expanded further with additional studies demonstrating that SLE patients have significant reductions in epigenetic modifications on the inactive X and aberrant X-linked gene expression leading to abnormal autoantibody production, increased expression of type I interferon (IFN)-regulated genes, and upregulation of immune-regulatory genes on T cells (29–31).
Toll-like receptor (TLR) 7 and TLR9 are expressed on the X chromosome and have been demonstrated as potential mechanisms for the development of SLE providing an additional mechanism for promoting an inflammatory response (32). Numerous studies demonstrated that sex differences in TLR7 signaling may be due to disrupted X inactivation (46, XX) leaving women with more TLR7 gene copy numbers (32–34). This was demonstrated by the translocation of a segment near the pseudoautosomal region of the X chromosome onto the Y chromosome identifying the y-linked autoimmune accelerating locus (yaa) in male BXSB lupus mice (35). Among the key genes that were duplicated in the yaa locus, TLR7 was identified as a major gene responsible for the severity of SLE (35). Recently, Brown et al. demonstrated that both male and female mice presented with an SLE-like pathology using a human TLR7 gain-of-function mutation (TLR7y264H) mouse model. TLR7y264H was knocked into C57BL/6 creating kik mice (36). TLR7 is not the only TLR that has been implicated in the development of SLE; however, the findings are paradoxical and inconclusive. For instance, despite a positive correlation with SLE incidence, TLR9 deletion in lupus-prone mice did not reduce the severity of the disease. Moreover, TLR9 deletion led to an exacerbation of lupus in mouse models (37). In contrast, Cunningham et al. demonstrated that ER-α binding to the estrogen-responsive element led to the production of proinflammatory cytokines from TLR7/8 and TLR9 activation, suggesting that sex hormones and chromosomes provide a synchronous mechanism that likely regulates SLE pathology (38). Together, these studies suggest that X chromosome abnormalities in SLE patients likely are driving the reactivation of numerous immune-related genes and the promotion of autoimmunity.
Mechanisms of CVD complications in SLE
It is well established that an SLE diagnosis is linked to an increased risk of CVD including atherosclerosis, coronary artery disease, cerebrovascular events, myocardial infarction, and peripheral artery disease (39–41). Compared with patients without SLE diagnosis, female patients with SLE are at risk approximately 3-4 times for experiencing a CVD event including cardiac complications (2, 6, 39). In this section, we will briefly discuss the cardiac and vascular complications that can occur in SLE (Figure 2).
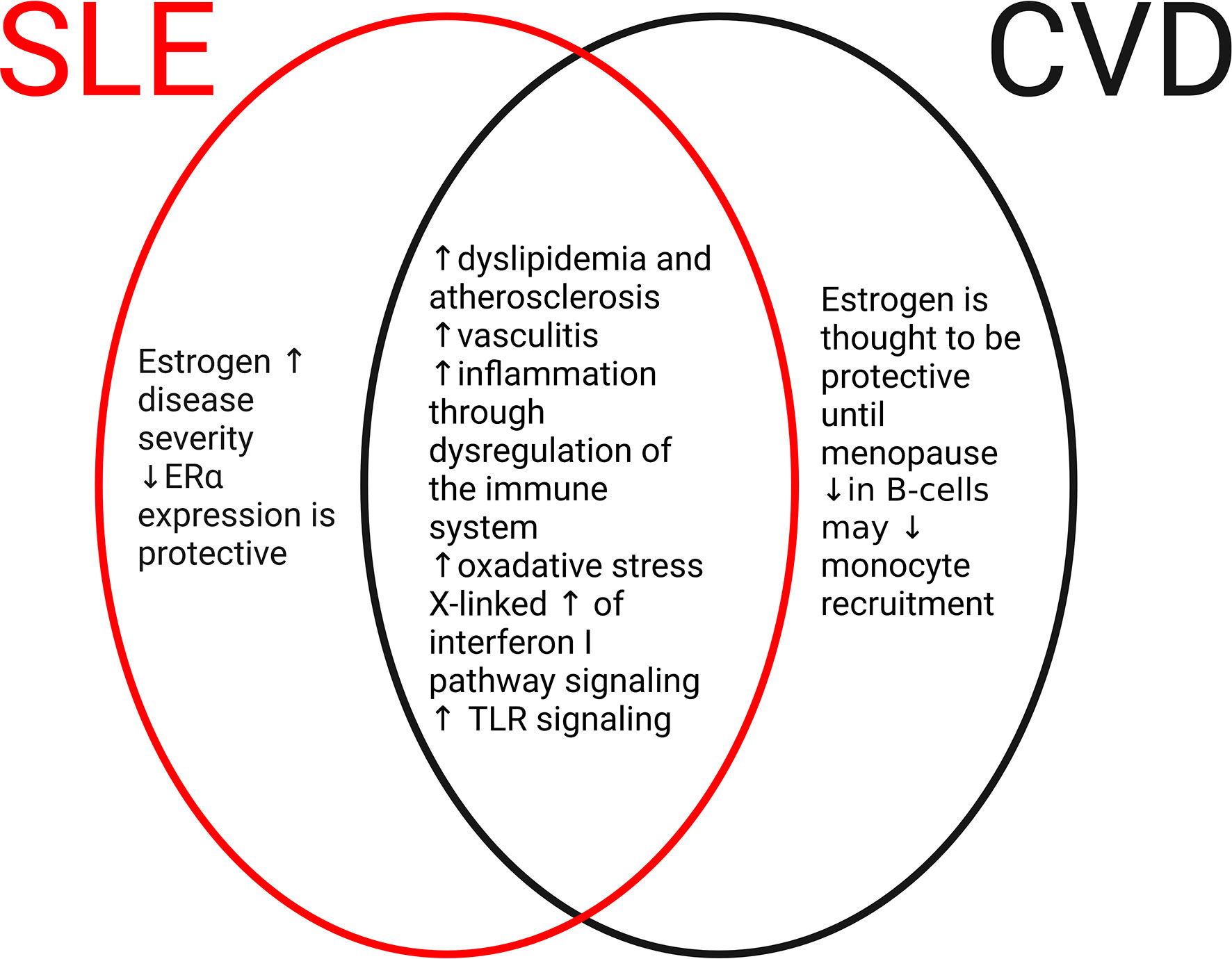
Figure 2 Common and disparate mechanisms of SLE and cardiovascular disease (CVD). In the development of SLE or CVD, there are both disparate and common mechanisms. In SLE (red oval), elevated levels of estrogen and estrogen receptor signaling promote the severity and frequency of the disease. In CVD (black oval), estrogen and receptor signaling are cardioprotective until menopause. Differences in immune cell populations including B cells and monocytes may contribute to CVD. However, numerous mechanisms are shared between SLE and CVD (middle oval) including dysregulation of the immune system, increases in oxidative stress, and development of atherosclerosis through dyslipidemia. These common pathways may contribute to CVD complications that are frequently seen in patients with SLE.
Vascular
Nearly 10% of all patients with SLE have accelerated atherosclerosis development; inflammation in small, medium, and large vessels; and impaired endothelium-dependent relaxation compared with age-matched controls (3, 42–46). Multiple clinical studies implicate dyslipidemia and clotting factors in the association between CVD and SLE (5, 43, 47). In a study published by Urowitz et al., both male and female patients with SLE had a 60% increased prevalence of dyslipidemia 3 years after study enrollment (48). In another study, young women with juvenile SLE had decreased HDL and elevated VLDL levels compared with healthy girls reaching limits comparable to age-matched healthy boy controls (49). These data indicated that SLE can shift the lipid profile of young women which may predispose them to premature CVD development, which would not otherwise be seen in healthy age-matched women (49). The opposite trend was found in young men with juvenile SLE who had elevated HDL subsets and decreased VLDL subsets compared with age-matched healthy controls. This shift in lipid profile in young men with juvenile SLE may be due to low testosterone levels that has been observed in male SLE patients (21, 50). These data present that sex differences in lipid profiles in juvenile SLE patients, along with chronic inflammation associated with juvenile SLE, may influence the risk of vascular disease.
The lifetime prevalence of vasculitis in SLE patients is estimated to be as high as 56% with large studies showing a prevalence of 11% to 36% (51, 52). The consequences of vasculitis range from cutaneous manifestations to life-threatening organ dysfunction. Interestingly, the territories typically associated with lupus vasculitis include cutaneous vessels, renal small vessels, coronary arteries, and vessels of the brain; however, pulmonary and gastrointestinal involvement is less common (53). Clinically, it is important to separate inflammatory vasculitis from antiphospholipid syndrome which may present with similar manifestations. Vasculitis in SLE is characterized by inflammatory cell infiltration into the vessel that is initiated by the formation or deposition of immune complexes into the vessel wall (51). Interestingly, patients with vasculitis are younger and primarily male, suggesting the importance of biological sex in the pathogenesis of SLE-associated vasculitis (51). While the complete mechanisms of the development of vascular dysfunction and inflammation in SLE are unknown, both basic research and clinical studies have identified several key contributors.
Similarly, anticardiolipin antibodies and antiphospholipid syndrome have been observed in SLE patients (between 30% and 40%) and are known risk factors for clotting disorders (47). While there have not been many studies evaluating sex differences, a cohort study conducted by Bayraktar et. al., investigating the clinical spectrum of catastrophic antiphospholipid syndrome in patients with and without SLE, demonstrated that at the time of disease presentation, patients with SLE were more likely to be female and younger, have cerebral and pancreatic involvement, receive corticosteroids and cyclophosphamide, and have a higher risk of mortality after adjusting for age, sex, organ involvement, and treatment than those without SLE (54). In a separate study, greater than 55% of SLE patients presented with antiphospholipid antibodies and organ damage at a 15-year follow-up. Interestingly, in this study, older men were associated with an increased risk of organ damage, in contrast to the findings of Bayraktar et al. It is likely that these observed differences were due to differences in disease stage and possibly due to the sample sizes of men versus women (55). Importantly, these key contributors are mostly demonstrated in female patients, and future studies including male patients with SLE are needed to investigate true sex-dependent mechanisms.
Cardiac
Cardiac-related mortality is significantly increased in both male and female patients with SLE than in non-SLE patients (6). A commonality between cardiac disease and SLE is a dysregulation of the immune system. This has been demonstrated by numerous groups where aberrant antigen-presenting cells (macrophages, dendritic cells, and B cells) and T-cell activation promote both CVD complications and SLE disease progression (56–59). Increased inflammatory activity and oxidative stress in SLE patients are associated with an increased risk for cardio-specific pathologies including myocarditis, atherosclerosis, and MI (40, 60). Whether this dysregulation is due to overreactive immune cells or sex hormone levels is unknown, both of which may be implicated in SLE pathogenesis.
While SLE may not affect the observed sex differences in the type of vascular obstruction, premature atherosclerosis is a severe risk factor for SLE patients (61, 62). Women with lupus between 35 and 44 years old were over 50 times more likely to have an MI than women of similar age in the Framingham Offspring Study (rate ratio = 52.43, 95% CI: 21.6-98.5) (62). With advancements in treatment and increased life expectancy of SLE patients, CVD has become a more prevalent health issue in SLE patients. More recently, Tornvall et al. investigated 4,192 patients with SLE and 41,892 non-SLE age-matched control patients (83.2% women in each cohort). During the 20-year follow-up, the incidence of MI was 9.6 (95% CI: 8.9–10.5) and 4.9 (95% CI:4.8-5.1) events/1,000 person-years in patients with SLE and controls, respectively (63). Even still, the impact of this problem has been understudied and underrecognized. Future studies should focus on the aggressive management of hypercholesterolemia and other possible risk factors for plaque formation. In addition, early detection of atherosclerosis may provide an opportunity for therapeutic intervention.
The role of sex hormones and chromosomes in CVD complications in SLE
Sex hormones
While we acknowledge that sex-dependent mechanisms in the pathogenesis of SLE are more complex than these key mechanisms, similar mechanisms have been implicated in CVD alone and SLE alone. Sex hormones have been implicated to influence CVD prevalence and prognosis through multiple mechanisms including their effects on the immune system. Serum from SLE patients with higher levels of estrogen positively correlates to SLE disease severity (64). In non-SLE patients, a high estrogen:testosterone ratio has been considered cardioprotective; however, due to the findings from the Women’s Health Initiative (WHI), hormone therapy has been demonstrated to have a complex pattern of risks and benefits with younger women having a greater benefit than older women (age 70 to 79), who had a trend toward increased risk for adverse CVD events (65–71). While there is a clear link between estrogen signaling and SLE with the loss of ER-α being protective, the role of estrogen in CVD progression in SLE patients is not as clear. Estrogen signaling has been shown to be attenuated with age in animal models of hypertension and myocarditis likely due to a decrease in ER-α levels which has been shown to stimulate vasoconstriction and modulate immune cell activation highlighting a divergent mechanism compared with SLE literature (72, 73).
One potential mechanism for estrogen loss of protection in SLE versus non-SLE settings may be due to the cell type most affected in these two disease states as patients with SLE are more likely to have B-cell dysregulation which is not as common when evaluating non-SLE patients. Estrogen signaling via ER-α acts in a B-cell intrinsic manner to promote the development of autoantibodies (74). Taking what we know from the SLE studies, we can extrapolate that the adverse effects of estrogen via ER-α in the setting of CVD may be due to an exacerbation of the immune response which is a known critical player in multiple cardiovascular pathologies. Future studies further evaluating the commonalities in addition to the differences between these two disease settings may provide additional evidence for the increased risk of CVD in patients with SLE.
Male sex is a traditional risk factor for CVD, and with SLE, there has been some indication that there is a compounded risk of CVD in men. Whether this is due to sex hormones or some other mechanisms is unknown. The literature on testosterone and the regulation of inflammation is mixed with some studies indicating testosterone has anti-inflammatory properties while others indicate the opposite (75, 76). Early studies have shown that a decrease in testosterone increased circulating inflammatory cytokine expression and the readdition of testosterone protected human and mouse models that demonstrate deficiency (77). Due to the known correlation between prolonged or exacerbated immune response and CVD, it is reasonable to conclude that altered testosterone levels in men with SLE may be facilitating poor prognosis after CVD incidence.
Sex hormones, more specifically testosterone, have been found to influence dyslipidemia which is a risk factor for CVD. In a retrospective cross-sectional study including 442 men and 2,122 women, dyslipidemia prevalence was increased in men and women with SLE and increased testosterone (78). In men, the prevalence of dyslipidemia and testosterone levels were dependent on age, whereas dyslipidemia in women and the effect of testosterone were dependent on menopausal status (78). There is a need for further investigation of the role of sex hormones and how they contribute to CVD complications in patients with SLE and in mouse models of SLE.
Sex chromosomes
To evaluate the effects of sex chromosomes in CVD pathology, the most common animal model used is the four-core genotypes (FCG) mouse model. To generate the FCG mouse, the testis-determining gene Sry is removed and replaced on an autosome resulting in a gonadal type model not controlled by the sex chromosomes (79). Using this model, the X chromosome has been implicated in promoting injury due to elevated expression of inflammatory genes including the CD40 ligand, interleukin-1 receptor-associated kinase 1, and FoxP3 along with genes that promote apoptosis, lipid oxidation, and generation of oxygen-derived free radicals (33, 80, 81). These findings are similar to what has been observed in SLE patients which, as discussed above, have demonstrated that upregulation of X-linked genes dysregulates the immune response. This loss in the balance regulating the immune response in SLE patients would likely exacerbate the response to CVD, further promoting disease pathology. The X-linked upregulation of interferon I pathway signaling associated with SLE patients has also been linked to poor atherosclerosis outcomes through potentiation of foam cells, extracellular traps, endothelial dysfunction, and pro-injurious dendritic cell phenotypes (82–84). Interestingly, self-DNA released from either dying cells or neutrophil extracellular traps in atherosclerotic lesions has been hypothesized to stimulate autoimmune activation of dendritic cells aggravating atherosclerosis lesion formation, implicating a potential overlapping mechanism between SLE and atherosclerosis (83). In the case of MI in the non-SLE setting, B-cell depletion has been shown to be beneficial due to an attenuation of monocyte recruitment via Toll-like receptor activation and B-cell secretion of Ccl7 (85). Autoantibody formation against myocardial epitopes has not yet been studied in this context; however, it is likely that SLE would play a big role in the post-MI response.
There have been numerous TLRs implicated in the development of CVD alone especially TLR2, TLR4, and TLR9 (38, 86, 87). Interestingly, there is very little known about the role of TLRs in the development of CVD complications in SLE, even though TLRs play an important role in the pathogenesis of SLE. Recently, Elshikha et al. reported that TLR7 activation with resiquimod (R848) promoted the development of CVD complications in B6SLE123 mice (88). After TLR7 agonism with R848, B6SLE123 mice developed a CVD pathology including microvascular inflammation and myocytolysis of the heart (88). CD45+ leukocyte infiltration into the heart was higher in female mice compared with males; however, no other sex differences were further investigated (88). Without full evaluation, this could lead the scientific community to assume that there were no sex differences in TLR7 agonism in B6SLE123 mice in the development of CVD complications highlighting the need for clear transparency when presenting findings. Although a direct investigation of sex differences in CVD complications in SLE was not conducted, there are clear associations between TLR signaling, specifically TLR7 and TLR9, in the development of both SLE and CVD independently. Future studies need to be conducted in both sexes.
Future directions
Current data suggest that men with SLE have a higher predisposition and more severe disease presentation of CVD compared with women with SLE; however, the mechanisms behind this remain unclear (10, 89, 90). As CVD is the leading cause of death worldwide, there is critical importance to address this large gap in scientific knowledge and begin to unwind the key players that link these two disease states (Figure 2). One critical question that remains unanswered is are the drivers of cardiac complications without concomitant SLE the same or different than those with concomitant SLE? As discussed in the sections above, SLE disease severity in both men and women is linked to a higher risk of negative cardiovascular outcomes with one potential mechanism being chronic inflammation. Under non-SLE settings, studies have indicated that despite similarities in the pathological endpoint, the pathway and signaling mechanisms that stimulate disease progression differ between the sexes (91). Whether or not this is true in SLE patients is not known; however, it can be assumed that due to sex differences in SLE pathology, the pathway to CVD progression also likely differs. Interrogating signaling mechanisms that may differ in men versus women with SLE could provide invaluable information on patient-specific therapeutics. Answering this fundamental question from a basic science and translational research standpoint could have vast clinical implications for the future of patients with CVD complications and SLE.
Conclusion
Despite SLE being associated with a significantly increased risk for the development of CVD, there is still much to be learned regarding the mechanisms that drive SLE contributions in CVD pathology. There is limited clinical, translational, and even fewer basic science research investigating sex differences in CVD complications with SLE. Moreover, much of the current literature only includes women with SLE and female mouse models, providing little to no insight into the sex-dependent mechanisms of CVD complications in SLE. There is a clear scientific and clinical gap in the understanding of the physiological and pathophysiological mechanisms of the development of CVD complications in men with SLE. Future studies should focus on characterizing sex difference-specific mechanisms in the development of cardiac and vascular complications in SLE. It is also imperative that clinical studies include male patients with SLE to begin to investigate the role of sex and sex hormones as biological variables in CVD complications in SLE. These approaches may lead to a better understanding of this complex disease process which help identify potential preventatives or therapeutic treatments to reduce or eliminate CVD morbidity and mortality in SLE.
Author contributions
AC and JPVB prepared the figures. AC, ML, DMP, KYD-P, and JPVB drafted the manuscript. AC, KYD-P, and JB edited the manuscript. AC, ML, DMP, KYD-P, and JPVB approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the American Heart Association (IPA35260039 to KYD-P), National Institutes of Health (UL1TR001450 to KYD-P, R25GM113278 to ML, and T32GM132055 to AC), MUSC Core Centers for Clinical Research Project (awarded to JPVB), MUSC College of Medicine Program Project (grant given to JPVB), and the Biomedical Laboratory Research and Development Service of the VA Office of Research and Development (award nos. IK2BX005376 to DP, IK2BX003922 to KYD-P, and IK2BX005605 to JPVB).
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wolf VL, Taylor EB, Ryan MJ. Cyclophosphamide treatment for hypertension and renal injury in an experimental model of systemic lupus erythematosus. Physiol Rep (2019) 7:e14059. doi: 10.14814/phy2.14059
2. Ajeganova S, Hafstrom I, Frostegard J. Patients with SLE have higher risk of cardiovascular events and mortality in comparison with controls with the same levels of traditional risk factors and intima-media measures, which is related to accumulated disease damage and antiphospholipid syndrome: a case-control study over 10 years. Lupus Sci Med (2021) 8. doi: 10.1136/lupus-2020-000454
3. Wilhelm AJ, Major AS. Accelerated atherosclerosis in SLE: Mechanisms and prevention approaches. Int J Clin Rheumtol (2012) 7:527–39. doi: 10.2217/ijr.12.46
4. Zeller CB, Appenzeller S. Cardiovascular disease in systemic lupus erythematosus: The role of traditional and lupus related risk factors. Curr Cardiol Rev (2008) 4:116–22. doi: 10.2174/157340308784245775
5. do Socorro Teixeira Moreira Almeida M, da Costa Arcoverde J, Barros Jacobino MN, Coimbra Neto AR. Male Systemic lupus erythematosus, an overlooked diagnosis. Clin Pract (2011) 1:e103. doi: 10.4081/cp.2011.e103
6. Bartels CM, Buhr KA, Goldberg JW, Bell CL, Visekruna M, Nekkanti S, et al. Mortality and cardiovascular burden of systemic lupus erythematosus in a US population-based cohort. J Rheumatol (2014) 41:680–7. doi: 10.3899/jrheum.130874
7. Dey D, Ofori E, Hutton-Mensah KA, Akutek MLK, Okine R, Amoaba I, et al. Clinical characteristics of males with systemic lupus erythematosus (SLE) in an inception cohort of patients in Ghana. Ghana Med J (2019) 53:2–7. doi: 10.4314/gmj.v53i1.1
8. Schwartzman-Morris J, Putterman C. Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol (2012) 2012:604892. doi: 10.1155/2012/604892
9. Nusbaum JS, Mirza I, Shum J, Freilich RW, Cohen RE, Pillinger MH, et al. Sex differences in systemic lupus erythematosus: Epidemiology, clinical considerations, and disease pathogenesis. Mayo Clin Proc (2020) 95:384–94. doi: 10.1016/j.mayocp.2019.09.012
10. Ramirez Sepulveda JI, Bolin K, Mofors J, Leonard D, Svenungsson E, Jonsen A, et al. Sex differences in clinical presentation of systemic lupus erythematosus. Biol Sex Differ (2019) 10:60. doi: 10.1186/s13293-019-0274-2
11. Roubinian JR, Talal N, Greenspan JS, Goodman JR, Siiteri PK. Effect of castration and sex hormone treatment on survival, anti-nucleic acid antibodies, and glomerulonephritis in NZB/NZW F1 mice. J Exp Med (1978) 147:1568–83. doi: 10.1084/jem.147.6.1568
12. Gao HX, Sanders E, Tieng AT, Putterman C. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus-prone mice. J Neuroimmunol (2010) 229:112–22. doi: 10.1016/j.jneuroim.2010.07.020
13. Hill BG, Hodge B, Misischia R. Lupus nephritis in a transgender woman on cross-sex hormone therapy: A case for the role of oestrogen in systemic lupus erythematosus. Lupus (2020) 29:1807–10. doi: 10.1177/0961203320946372
14. Roubinian JR, Talal N, Greenspan JS, Goodman JR, Siiteri PK. Delayed androgen treatment prolongs survival in murine lupus. J Clin Invest (1979) 63:902–11. doi: 10.1172/JCI109390
15. Verthelyi D, Ansar Ahmed S. Characterization of estrogen-induced autoantibodies to cardiolipin in non-autoimmune mice. J Autoimmun (1997) 10:115–25. doi: 10.1006/jaut.1996.0121
16. Verthelyi D, Ahmed SA. 17 beta-estradiol, but not 5 alpha-dihydrotestosterone, augments antibodies to double-stranded deoxyribonucleic acid in nonautoimmune C57BL/6J mice. Endocrinology (1994) 135:2615–22. doi: 10.1210/endo.135.6.7988450
17. Bynote KK, Hackenberg JM, Korach KS, Lubahn DB, Lane PH, Gould KA. Estrogen receptor-alpha deficiency attenuates autoimmune disease in (NZB x NZW)F1 mice. Genes Immun (2008) 9:137–52. doi: 10.1038/sj.gene.6364458
18. Graham JH, Yoachim SD, Gould KA. Estrogen receptor alpha signaling is responsible for the female sex bias in the loss of tolerance and immune cell activation induced by the lupus susceptibility locus Sle1b. Front Immunol (2020) 11:582214. doi: 10.3389/fimmu.2020.582214
19. Svenson JL, EuDaly J, Ruiz P, Korach KS, Gilkeson GS. Impact of estrogen receptor deficiency on disease expression in the NZM2410 lupus prone mouse. Clin Immunol (2008) 128:259–68. doi: 10.1016/j.clim.2008.03.508
20. Li J, McMurray RW. Effects of estrogen receptor subtype-selective agonists on autoimmune disease in lupus-prone NZB/NZW F1 mouse model. Clin Immunol (2007) 123:219–26. doi: 10.1016/j.clim.2007.01.008
21. Mackworth-Young CG, Parke AL, Morley KD, Fotherby K, Hughes GR. Sex hormones in male patients with systemic lupus erythematosus: A comparison with other disease groups. Eur J Rheumatol Inflammation (1983) 6:228–32.
22. Viselli SM, Stanziale S, Shults K, Kovacs WJ, Olsen NJ. Castration alters peripheral immune function in normal male mice. Immunology (1995) 84:337–42.
23. van Vollenhoven RF. Dehydroepiandrosterone for the treatment of systemic lupus erythematosus. Expert Opin Pharmacother (2002) 3:23–31. doi: 10.1517/14656566.3.1.23
24. van Vollenhoven RF, Park JL, Genovese MC, West JP. And McGuire JL. a double-blind, placebo-controlled, clinical trial of dehydroepiandrosterone in severe systemic lupus erythematosus. Lupus (1999) 8:181–7. doi: 10.1191/096120399678847588
25. Sharma R, Harris VM, Cavett J, Kurien BT, Liu K, Koelsch KA, et al. Rare X chromosome abnormalities in systemic lupus erythematosus and sjogren's syndrome. Arthritis Rheumatol (2017) 69:2187–92. doi: 10.1002/art.40207
26. Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, et al. Klinefelter's syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum (2008) 58:2511–7. doi: 10.1002/art.23701
27. Barbosa FB, Sinicato NA, Julio PR, Londe AC, Marini R, Gil-da-Silva-Lopes VL, et al. Trisomy X in a patient with childhood-onset systemic lupus erythematosus. J Transl Autoimmun (2020) 3:100043. doi: 10.1016/j.jtauto.2020.100043
28. Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E, et al. Overexpression of X-linked genes in T cells from women with lupus. J Autoimmun (2013) 41:60–71. doi: 10.1016/j.jaut.2012.12.006
29. Pyfrom S, Paneru B, Knox JJ, Cancro MP, Posso S, Buckner JH, et al. The dynamic epigenetic regulation of the inactive X chromosome in healthy human b cells is dysregulated in lupus patients. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2024624118
30. Odhams CA, Roberts AL, Vester SK, Duarte CST, Beales CT, Clarke AJ, et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in systemic lupus erythematosus. Nat Commun (2019) 10:2164. doi: 10.1038/s41467-019-10106-2
31. Syrett CM, Paneru B, Sandoval-Heglund D, Wang J, Banerjee S, Sindhava V, et al. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight (2019) 4. doi: 10.1172/jci.insight.126751
32. Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U.S.A. (2010) 107:15838–43. doi: 10.1073/pnas.1001337107
33. Brooks WH, Renaudineau Y. Epigenetics and autoimmune diseases: the X chromosome-nucleolus nexus. Front Genet (2015) 6:22. doi: 10.3389/fgene.2015.00022
34. Hagen SH, Henseling F, Hennesen J, Savel H, Delahaye S, Richert L, et al. Heterogeneous escape from X chromosome inactivation results in sex differences in type I IFN responses at the single human pDC level. Cell Rep (2020) 33:108485. doi: 10.1016/j.celrep.2020.108485
35. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U.S.A. (2006) 103:9970–5. doi: 10.1073/pnas.0603912103
36. Brown GJ, Canete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
37. Tilstra JS, John S, Gordon RA, Leibler C, Kashgarian M, Bastacky S, et al. And shlomchik MJ. b cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest (2020) 130:3172–87. doi: 10.1172/JCI132328
38. Cunningham MA, Wirth JR, Naga O, Eudaly J, Gilkeson GS. Estrogen receptor alpha binding to ERE is required for full Tlr7- and Tlr9-induced inflammation. SOJ Immunol (2014) 2. doi: 10.15226/soji.2014.00107
39. Katz G, Smilowitz NR, Blazer A, Clancy R, Buyon JP, Berger JS. Systemic lupus erythematosus and increased prevalence of atherosclerotic cardiovascular disease in hospitalized patients. Mayo Clin Proc (2019) 94:1436–43. doi: 10.1016/j.mayocp.2019.01.044
40. Kostopoulou M, Nikolopoulos D, Parodis I, Bertsias G. Cardiovascular disease in systemic lupus erythematosus: Recent data on epidemiology, risk factors and prevention. Curr Vasc Pharmacol (2020) 18:549–65. doi: 10.2174/1570161118666191227101636
41. Lu X, Wang Y, Zhang J, Pu D, Hu N, Luo J, et al. Patients with systemic lupus erythematosus face a high risk of cardiovascular disease: A systematic review and meta-analysis. Int Immunopharmacol (2021) 94:107466. doi: 10.1016/j.intimp.2021.107466
42. Sacre K, Escoubet B, Pasquet B, Chauveheid MP, Zennaro MC, Tubach F, et al. Increased arterial stiffness in systemic lupus erythematosus (SLE) patients at low risk for cardiovascular disease: a cross-sectional controlled study. PloS One (2014) 9:e94511. doi: 10.1371/journal.pone.0094511
43. Dziedzic-Oleksy H, Mazurek A, Bugala K, Perricone C, Drabik L, Plazak W. Arterial stiffness and atherosclerosis in systemic lupus erythematosus patients. Reumatologia (2022) 60:165–72. doi: 10.5114/reum.2022.117836
44. Svensson C, Eriksson P, Bjarnegard N, Jonasson H, Stromberg T, Sjowall C, et al. Impaired microcirculation and vascular hemodynamics in relation to macrocirculation in patients with systemic lupus erythematosus. Front Med (Lausanne) (2021) 8:722758. doi: 10.3389/fmed.2021.722758
45. Barnes JN, Nualnim N, Sugawara J, Sommerlad SM, Renzi CP, Tanaka H. Arterial stiffening, wave reflection, and inflammation in habitually exercising systemic lupus erythematosus patients. Am J Hypertens (2011) 24:1194–200. doi: 10.1038/ajh.2011.143
46. Cypiene A, Kovaite M, Venalis A, Dadoniene J, Rugiene R, Petrulioniene Z, et al. Arterial wall dysfunction in systemic lupus erythematosus. Lupus (2009) 18:522–9. doi: 10.1177/0961203308099625
47. Misita CP, Moll S. Antiphospholipid antibodies. Circulation (2005) 112:e39–44. doi: 10.1161/CIRCULATIONAHA.105.548495
48. Urowitz MB, Gladman D, Ibanez D, Fortin P, Sanchez-Guerrero J, Bae S, et al. Systemic lupus international collaborating c. accumulation of coronary artery disease risk factors over three years: data from an international inception cohort. Arthritis Rheum (2008) 59:176–80. doi: 10.1002/art.23353
49. Robinson GA, Peng J, Peckham H, Radziszewska A, Butler G, Pineda-Torra I, et al. Sex hormones drive changes in lipoprotein metabolism. iScience (2021) 24:103257. doi: 10.1016/j.isci.2021.103257
50. Mok CC, Lau CS. Profile of sex hormones in male patients with systemic lupus erythematosus. Lupus (2000) 9:252–7. doi: 10.1191/096120300680198926
51. Pyrpasopoulou A, Chatzimichailidou S, Aslanidis S. Vascular disease in systemic lupus erythematosus. Autoimmune Dis (2012) 2012:876456. doi: 10.1155/2012/876456
52. Barile-Fabris L, Hernandez-Cabrera MF, Barragan-Garfias JA. Vasculitis in systemic lupus erythematosus. Curr Rheumatol Rep (2014) 16:440. doi: 10.1007/s11926-014-0440-9
53. D'Cruz D. Vasculitis in systemic lupus erythematosus. Lupus (1998) 7:270–4. doi: 10.1191/096120398678920082
54. Bayraktar UD, Erkan D, Bucciarelli S, Espinosa G, Asherson R, Catastrophic Antiphospholipid Syndrome Project G. The clinical spectrum of catastrophic antiphospholipid syndrome in the absence and presence of lupus. J Rheumatol (2007) 34:346–52.
55. Taraborelli M, Leuenberger L, Lazzaroni MG, Martinazzi N, Zhang W, Salmon J, et al. FRI0408 Antiphospholipid antibodies and the risk of damage accrual in systemic lupus erythematosus. Annals of the Rheumatic Diseases (2015) 74(2):574.
56. Viola M, de Jager SCA, Sluijter JPG. Targeting inflammation after myocardial infarction: A therapeutic opportunity for extracellular vesicles? Int J Mol Sci (2021) 22. doi: 10.3390/ijms22157831
57. Zhang Q, Wang L, Wang S, Cheng H, Xu L, Pei G, et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther (2022) 7:78. doi: 10.1038/s41392-022-00925-z
58. Gottschalk TA, Tsantikos E, Hibbs ML. Pathogenic inflammation and its therapeutic targeting in systemic lupus erythematosus. Front Immunol (2015) 6:550. doi: 10.3389/fimmu.2015.00550
59. Podolska MJ, Biermann MH, Maueroder C, Hahn J, Herrmann M. Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflammation Res (2015) 8:161–71. doi: 10.2147/JIR.S70325
60. Svenungsson E, Jensen-Urstad K, Heimburger M, Silveira A, Hamsten A, de Faire U, et al. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation (2001) 104:1887–93. doi: 10.1161/hc4101.097518
61. Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med (2003) 349:2407–15. doi: 10.1056/NEJMoa035611
62. Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA Jr., Jansen-McWilliams L, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the framingham study. Am J Epidemiol (1997) 145:408–15. doi: 10.1093/oxfordjournals.aje.a009122
63. Tornvall P, Goransson A, Ekman J, Jarnbert-Pettersson H. Myocardial infarction in systemic lupus erythematosus: Incidence and coronary angiography findings. Angiology (2021) 72:459–64. doi: 10.1177/0003319720985337
64. Cai Z, Xie C, Qiao W, Fei X, Guo X, Liu H, et al. The role of estrogen membrane receptor (G protein-coupled estrogen receptor 1) in skin inflammation induced by systemic lupus erythematosus serum IgG. Front Immunol (2017) 8:1723. doi: 10.3389/fimmu.2017.01723
65. Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ (2017) 8:33. doi: 10.1186/s13293-017-0152-8
66. Xu Y, Arenas IA, Armstrong SJ, Plahta WC, Xu H, Davidge ST. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc Res (2006) 69:836–44. doi: 10.1016/j.cardiores.2005.11.031
67. Meyer MR, Haas E, Barton M. Gender differences of cardiovascular disease: new perspectives for estrogen receptor signaling. Hypertension (2006) 47:1019–26. doi: 10.1161/01.HYP.0000223064.62762.0b
68. Murphy E. Estrogen signaling and cardiovascular disease. Circ Res (2011) 109:687–96. doi: 10.1161/CIRCRESAHA.110.236687
69. Mahmoodzadeh S, Leber J, Zhang X, Jaisser F, Messaoudi S, Morano I, et al. Cardiomyocyte-specific estrogen receptor alpha increases angiogenesis, lymphangiogenesis and reduces fibrosis in the female mouse heart post-myocardial infarction. J Cell Sci Ther (2014) 5:153. doi: 10.4172/2157-7013.1000153
70. Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE. And stampfer MJ. a prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med (2000) 133:933–41. doi: 10.7326/0003-4819-133-12-200012190-00008
71. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA (2002) 288:321–33. doi: 10.1001/jama.288.3.321
72. Wynne FL, Payne JA, Cain AE, Reckelhoff JF, Khalil RA. Age-related reduction in estrogen receptor-mediated mechanisms of vascular relaxation in female spontaneously hypertensive rats. Hypertension (2004) 43:405–12. doi: 10.1161/01.HYP.0000111833.82664.0c
73. Koenig A, Buskiewicz I, Huber SA. Age-associated changes in estrogen receptor ratios correlate with increased female susceptibility to coxsackievirus B3-induced myocarditis. Front Immunol (2017) 8:1585. doi: 10.3389/fimmu.2017.01585
74. Tabor DE, Gould KA. Estrogen receptor alpha promotes lupus in (NZBxNZW)F1 mice in a b cell intrinsic manner. Clin Immunol (2017) 174:41–52. doi: 10.1016/j.clim.2016.10.011
75. Ben-Batalla I, Vargas-Delgado ME, von Amsberg G, Janning M, Loges S. Influence of androgens on immunity to self and foreign: Effects on immunity and cancer. Front Immunol (2020) 11:1184. doi: 10.3389/fimmu.2020.01184
76. Bianchi VE. The anti-inflammatory effects of testosterone. J Endocr Soc (2019) 3:91–107. doi: 10.1210/js.2018-00186
77. Mohamad NV, Wong SK, Wan Hasan WN, Jolly JJ, Nur-Farhana MF, Ima-Nirwana S, et al. The relationship between circulating testosterone and inflammatory cytokines in men. Aging Male (2019) 22:129–40. doi: 10.1080/13685538.2018.1482487
78. Self A, Zhang J, Corti M, Esani M. Correlation between sex hormones and dyslipidemia. American Society for Clinical Laboratory Science (2019) ascls.119.002071. doi: 10.29074/ascls.119.002071
79. Arnold AP, Chen X. What does the "four core genotypes" mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol (2009) 30:1–9. doi: 10.1016/j.yfrne.2008.11.001
80. Li J, Chen X, McClusky R, Ruiz-Sundstrom M, Itoh Y, Umar S, et al. The number of X chromosomes influences protection from cardiac ischaemia/reperfusion injury in mice: one X is better than two. Cardiovasc Res (2014) 102:375–84. doi: 10.1093/cvr/cvu064
81. Stamova B, Tian Y, Jickling G, Bushnell C, Zhan X, Liu D, et al. The X-chromosome has a different pattern of gene expression in women compared with men with ischemic stroke. Stroke (2012) 43:326–34. doi: 10.1161/STROKEAHA.111.629337
82. Li J, Fu Q, Cui H, Qu B, Pan W, Shen N, et al. Interferon-alpha priming promotes lipid uptake and macrophage-derived foam cell formation: A novel link between interferon-alpha and atherosclerosis in lupus. Arthritis Rheum (2011) 63:492–502. doi: 10.1002/art.30165
83. Doring Y, Manthey HD, Drechsler M, Lievens D, Megens RT, Soehnlein O, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation (2012) 125:1673–83. doi: 10.1161/CIRCULATIONAHA.111.046755
84. Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15:623–5. doi: 10.1038/nm.1959
85. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med (2013) 19:1273–80. doi: 10.1038/nm.3284
86. Van Beusecum JP, Zhang S, Cook AK, Inscho EW. Acute toll-like receptor 4 activation impairs rat renal microvascular autoregulatory behaviour. Acta Physiol (Oxf) (2017) 221:204–20. doi: 10.1111/apha.12899
87. Feng Y, Chao W. Toll-like receptors and myocardial inflammation. Int J Inflam (2011) 2011:170352. doi: 10.4061/2011/170352
88. Elshikha AS, Teng XY, Kanda N, Li W, Choi SC, Abboud G, et al. TLR7 activation accelerates cardiovascular pathology in a mouse model of lupus. Front Immunol (2022) 13:914468. doi: 10.3389/fimmu.2022.914468
89. Crosslin KL, Wiginton KL. Sex differences in disease severity among patients with systemic lupus erythematosus. Gend Med (2011) 8:365–71. doi: 10.1016/j.genm.2011.10.003
90. Riveros Frutos A, Casas I, Rua-Figueroa I, Lopez-Longo FJ, Calvo-Alen J, Galindo M, et al. Systemic lupus erythematosus in Spanish males: A study of the Spanish rheumatology society lupus registry (RELESSER) cohort. Lupus (2017) 26:698–706. doi: 10.1177/0961203316673728
Keywords: lupus, sex differences, cardiovascular complications, vascular, cardiac
Citation: Corker A, Learmonth M, Patrick DM, DeLeon-Pennell KY and Van Beusecum JP (2023) Cardiac and vascular complications in lupus: Is there a role for sex? Front. Immunol. 14:1098383. doi: 10.3389/fimmu.2023.1098383
Received: 14 November 2022; Accepted: 07 March 2023;
Published: 29 March 2023.
Edited by:
Trine N Jorgensen, Case Western Reserve University, United StatesReviewed by:
George Anthony Robinson, University College London, United KingdomCoziana Ciurtin, University College London, United Kingdom
Copyright © 2023 Corker, Learmonth, Patrick, DeLeon-Pennell and Van Beusecum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin P. Van Beusecum, dmFuYmV1c2VAbXVzYy5lZHU=