 Yinxi Zhou†
Yinxi Zhou† Jinghua Xia†
Jinghua Xia† Shuonan Xu†
Shuonan Xu† Tao She†
Tao She† Yanning Zhang
Yanning Zhang Ying SunMiaomiao Wen
Ying SunMiaomiao Wen Tao Jiang*
Tao Jiang* Yanlu Xiong*Jie Lei*
Yanlu Xiong*Jie Lei*- Department of Thoracic Surgery, The Second Affiliated Hospital, Air Force Medicel University, Xi’an, China
The development and growth of tumors remains an important and ongoing threat to human life around the world. While advanced therapeutic strategies such as immune checkpoint therapy and CAR-T have achieved astonishing progress in the treatment of both solid and hematological malignancies, the malignant initiation and progression of cancer remains a controversial issue, and further research is urgently required. The experimental animal model not only has great advantages in simulating the occurrence, development, and malignant transformation mechanisms of tumors, but also can be used to evaluate the therapeutic effects of a diverse array of clinical interventions, gradually becoming an indispensable method for cancer research. In this paper, we have reviewed recent research progress in relation to mouse and rat models, focusing on spontaneous, induced, transgenic, and transplantable tumor models, to help guide the future study of malignant mechanisms and tumor prevention.
1 Introduction
According to the World Health Organization, there were 19.3 million new cancer cases worldwide in 2020, resulting in nearly 10 million related deaths (1). Furthermore, with the acceleration in population growth and social aging, the global cancer burden will continue to increase. It is estimated that by 2040, the number of newly diagnosed cancer patients will reach 28.4 million per year, an increase of 47% when compared with 2020 (2). Recently, new therapeutic strategies, such as immunotherapies and targeted therapies, have significantly improved the survival rates of tumor patients when compared with traditional therapies; however, there are fewer sensitive and beneficial people, and the problem of drug resistance is almost inevitable (3). Therefore, in-depth exploration of the occurrence and development mechanisms of cancer and the improvement of diagnosis and treatment strategies are of critical global importance, and consequently, this is a major focus of academic research.
Animal tumor models are valuable for experimental analysis as the animal tumors can have similar characteristics to human tumors. We can study the occurrence and development of tumors by observing animal tumor models, and study the effect of genes on tumorigenesis and development by knocking out or knocking in a certain gene, and we can also establish PDX models for drug screening and preclinical trials. Furthermore, this experimental strategy is also advantageous as these experiments are low cost, the animals have short life cycles, and it can help to avoid human experiments. Thus, the use of animal models is extremely valuable for tumor research (4). At present, based on the causes of tumor formation, we divide the animal model into four categories: spontaneous, induced, transgenic, and transplant. Among them, the spontaneous tumor model, the induced tumor model, and the transgenic model belong to the primary tumor of animals, and the transplantable tumor models include allogeneic transplantation and xenogeneic transplantation; the most used transplantable tumor model is human tumor xenografts in immunodeficient mice. Compared with non-mammals, mammals have a higher degree of similarity to humans, especially mice and rats, and the short reproduction cycle of mice greatly improves the efficiency of these experiments. The mouse genome sequencing project was completed in 2002, and it revealed that 99% of human genes exist in mice; gene homology is as high as 78.5% (5). Consequently, mice are the most commonly used animals in tumor research. This paper introduces the research progress that has been made with various human tumor models in mice and rats, and analyzes their development processes, advantages and disadvantages, applications, and future development prospects.
2 Animal models for spontaneous tumors
Spontaneous animal tumor models are utilized to investigate tumors that occur naturally in experimental animals without any conscious artificial intervention, and this occurrence type and incidence vary greatly with different species and strains of experimental animal (Figure 1). The advantage of the spontaneous animal tumor model is that the process of tumor formation is similar to that of human tumors, and the long period of tumor occurrence and development can be treated comprehensively with the intervention of many factors, and the role of genetic factors in tumorigenesis can be observed. However, the disadvantage of the spontaneous animal tumor model is that the experimental cycle is long, many experimental animals are required, and the cost is high; in addition, the spontaneous tumors of animals are often heterogeneous and there are individual growth differences, making it difficult to obtain a large number of tumor-bearing animals with uniform growth at the same time.
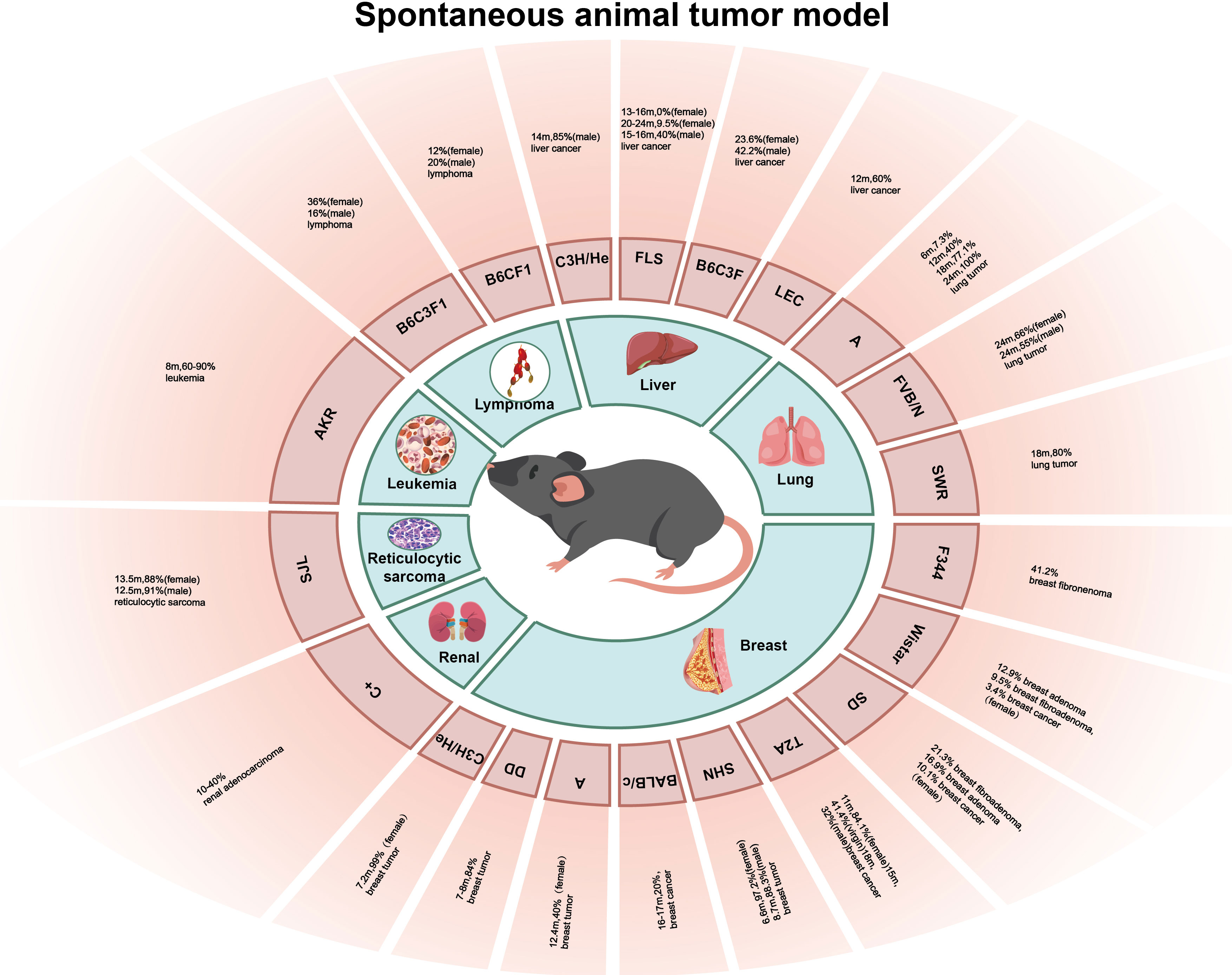
Figure 1 The incidence of various spontaneous tumors in different strains of mice (rat).
2.1 Breast tumors
The incidence and mortality of breast cancer worldwide are increasing and have surpassed lung cancer in 2020 to become the most predominant cancer in the world (1). Mice have a high incidence of breast cancer (6). The occurrence of breast tumors in mice is affected by many factors, including factors such as viruses, hormones, heredity, and feed (7). Mouse mammary tumor virus (MMTV) is the most common cause of breast cancer, which causes breast cancer and lymphoma in mice mainly by insertion mutations and clone amplification in the genome; therefore, mice carrying MMTV have a higher incidence of breast tumors (8). C3H/He mice were inbred by Strong after crossing Bagg albino female mice with a high incidence of the breast tumor strain DBA male mice in 1920. C3H/He female mice carried MMTV, and the average incidence of spontaneous breast tumors reached 99% at an average of 7.2 months, but the average age of tumorigenesis in the virgin mice was approximately 2 months older than that in postpartum mice (9). DD mice also carry MMTV, and the incidence of breast tumors is 84% at the age of 7–8 months (9). BALB/c and other mouse strains do not carry MMTV, and the incidence of breast tumors is low; in 1932, the 26th generation of mice bred by MacDowell from Bagg albino mice through inbred lines was named BALB/c mice, and the incidence of breast cancer in the BALB/c mice was only 20% at 16–17 months (9), and the incidence of breast tumors in C57BR, C57BL, AKR, and other mice without MMTV was lower (10). However, some strains of mice without MMTV had a high incidence of breast tumors, such as the A mice, SHN mice, and TA2 mice; A mice were inbred by Strong after inbreeding with the offspring of the local albino mice and Bagg albino mice in 1921, which is a highly spontaneous breast tumor strain, but the A mice do not carry MMTV, and Strain A female mice had an average breast tumor incidence of 40% at the age of 12.4 months (9). SHN mice were bred by Nagasawa et al. on the basis of the Swiss albino mice, and the incidence of breast tumors in female SHN mice was 97.2% at an average age of 6.6 months, and 88.3% in virgin female SHN mice at 8.7 months (10). In addition, the inbred strain TA2 mice bred by Sun et al. also had a high incidence of spontaneous breast cancer, and pathological analysis showed that the breast cancer cells of the TA2 mice were triple-negative. The incidence of spontaneous breast cancer reached 84.1% after 11 months in the TA2 mice, 41.4% in the virgin TA2 mice at 15 months, and 32% in the male TA2 mice at 18 months (11). Wistar, SD, F344, and other rat strains also had a high incidence of breast cancer; Wistar rats were bred by the Wistar Institute of the United States in 1907, and SD rats were bred by Wistar rats in 1925. The incidences of breast fibroadenoma, breast adenoma, and breast cancer in female SD rats were 21.3%, 16.9%, and 10.1%, respectively, while those in female Wistar rats were 12.9%, 9.5%, and 3.4%, respectively (12). F344 rats were inbred and established by Dunning in subline 344 of the Fischer strain, and the incidence of breast fibroadenoma in the F344 rats was 41.2% (13).
2.2 Lung tumors
Lung cancer is one of the most common cancers and the leading cause of cancer-related death worldwide, with an estimated 2 million new cases and 1.76 million deaths annually (14). The histopathological subtypes of lung cancer include non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), which can be further divided into adenocarcinoma and squamous cell carcinoma (SCC) (15). The incidence of spontaneous lung tumors in mice is high, and the pathological types are mainly adenoma and adenocarcinoma, depending on the mouse strain. A mice and SWR mice have a higher incidence of lung tumors, followed by BALB/C mice, CBA mice, and C3H mice, which have a lower incidence of lung tumors, and the lowest incidence of lung tumors was observed in DBA and C57BL/6 mice (16). A mice are a high spontaneous lung tumor strain, and lung tumors can be detected at 3–4 weeks of age. The incidence of lung tumor was 7.3% (6/83) at 6 months of age, 40.0% (71/178) at 12 months of age, 77.1% (105/136) at 18 months, and almost 100% at 24 months (17). SWR mice were bred by Lynch et al. in 1926 using Swedish mice for inbreeding. The spontaneous rate of lung tumors in SWR mice over 18 months was 80%, and the incidence of lung tumors in mice whose parents had spontaneous lung tumors was higher than that in mice whose parents had lung tumors alone, while the incidence of lung tumors in mice whose parents had no lung tumors was lower (18), suggesting a role for genetic factors in tumorigenesis. Although the incidence of lung tumors in SWR mice is high, the long culture period limits the use of this strain in lung tumor research. FVB/N mice were established in the 1970s, and they have a high spontaneous lung tumor rate. The mean incidence of lung tumors in male and female mice at 24 months was 55% and 66%, respectively, and the fertilized egg of FVB/N mice has a large and significant pronucleus and is easy to be microinjected with DNA. Consequently, FVB/N mice have been widely used as animal tumor models (19).
2.3 Liver tumors
Primary liver cancer is the fourth leading cause of cancer death worldwide, and its incidence is increasing every year. Histologically, it can be divided into hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (20). Spontaneous liver tumors in mice often originate from hepatocytes, cholangiocarcinoma is rare, and sarcomas are even rarer. The incidence of liver tumors is different in different strains of mice, but the incidence in male mice is usually higher than that in female mice (21). Male C3H/He mice are a highly spontaneous liver tumor strain, and Heston et al. found that the incidence of liver cancer in 14-month-old male C3H/He mice could reach 85% (9). In Japanese FLS mice, inbred mice with non-obese spontaneous fatty liver developed a fatty liver shortly after being fed a normal diet, but were not visibly obese; multiple white protruding nodules appeared in the liver of mice over 12 months of age, and were histologically diagnosed as hepatocytic adenoma and HCC. The incidence of HCC in male mice was 40% at an average of 15–16 months, and that in female Japanese FLS mice was 0% (0/36) at 13–16 months and 9.5% (4/42) at 20–24 months (22). B6C3F1 mice are the first generation of female C57BL/6 and male C3H hybridizations, and the incidence of liver tumor was 42.2% in male B6C3F1 mice and 23.6% in female B6C3F1 mice (13). LEC rats are inbred mutant rats established by Joseph A. Long and Herbert M.E. Vans of the University of California, USA. Approximately 40% of LEC rats develop acute posthepatitic death 3–4 months after birth and approximately 60% of rats experience chronic hepatitis and develop HCC a year later (23, 24). Some studies have pointed out that the cause of spontaneous hepatitis and HCC in LEC rats is the excessive accumulation of copper in the liver, which produces a large amount of ROS hydroxyl radicals, followed by oxidative stress, which is similar to the development of HCC in humans. Therefore, LEC rats have been widely used in HCC models (25).
2.4 Other tumors
In 1958, Claude et al. described an animal model of renal adenocarcinoma in C+ mice. The incidence of renal adenocarcinoma in C+ mice was between 10% and 40%, and the incidence of tumors increased with age (26). The AKR mouse is a new strain established by further inbreeding of AK mice by the Rockefeller Institute in 1936; AKR mice are born with carcinogenic RNA virus, the incidence of leukemia after 8 months is approximately 60%–90%, and that of 18-month-old mice can be as high as 90% (27). The incidence of reticulocytic sarcoma in the SJL mice was high, and the incidence of reticulocytic sarcoma in 13.5-month-old female and 12.5-month-old male mice was 88% and 91%, respectively (28). Dragani et al. established hybrid mice (C57BL/6J × C3Hf) F1 (B6C3F1) and (C57BL/6J × BALB/c) F1 (B6CF1); the incidence of lymphoma in male B6C3F1 and B6CF1 mice was 16% and 20%, respectively, and that in female B6C3F1 and B6CF1 mice was 36% and 12%, respectively (21).
3 Animal models of induced tumor
The induced tumor model refers to the use of exogenous carcinogens to cause changes in the cellular genetic characteristics, resulting in the abnormal growth of active cells and the formation of tumors (Figure 2). Induction methods include physical, chemical, and biological methods, of which the chemical methods are the most extensive and effective for the induction (Table 1), and have the advantages of easy application, short experimental time, and high reproducibility (45). However, the disadvantage is a high animal mortality rate, and the time, location, and number of lesions are not uniform among individuals.
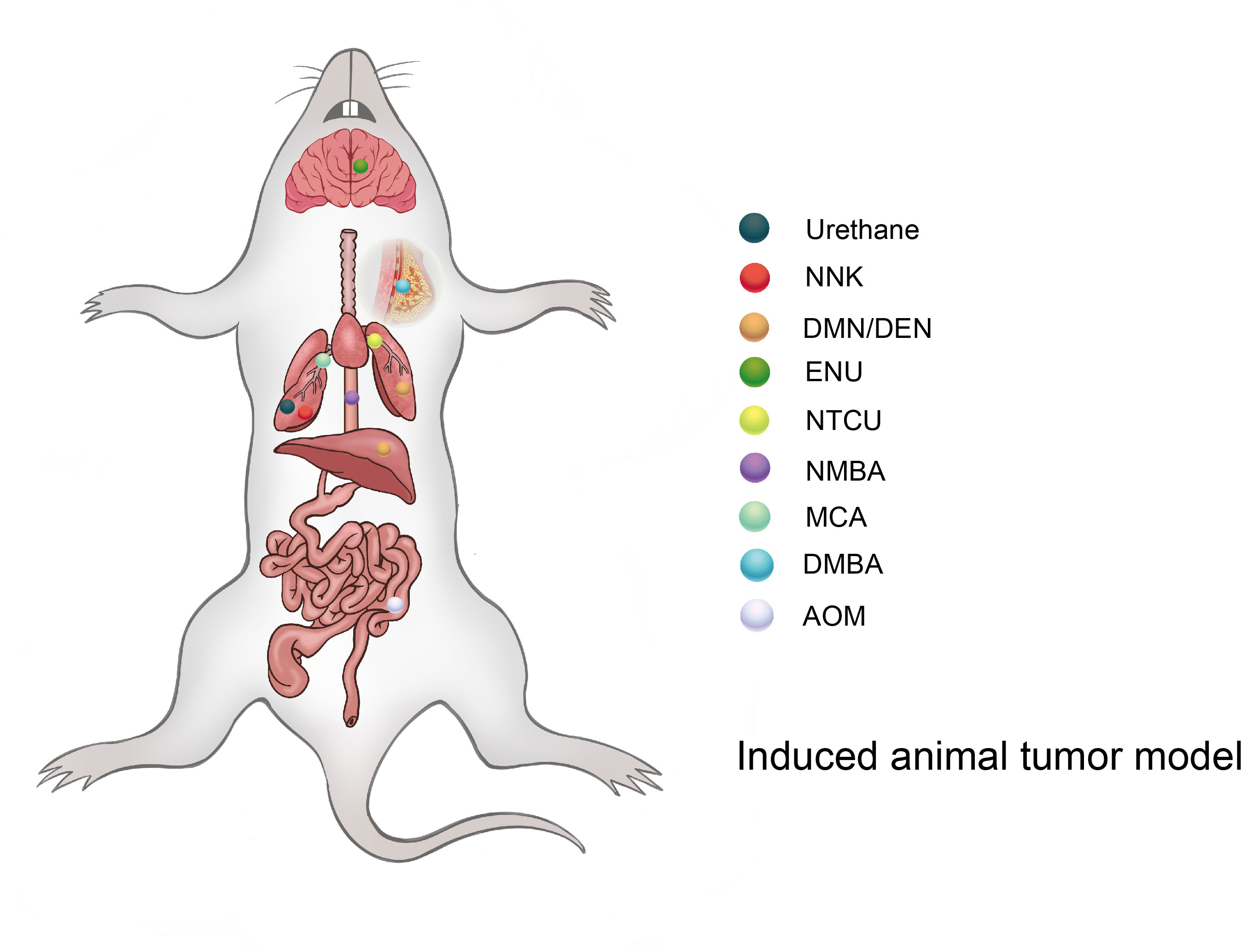
Figure 2 Different types of compounds can induce tumors in different organs of mice (rat).
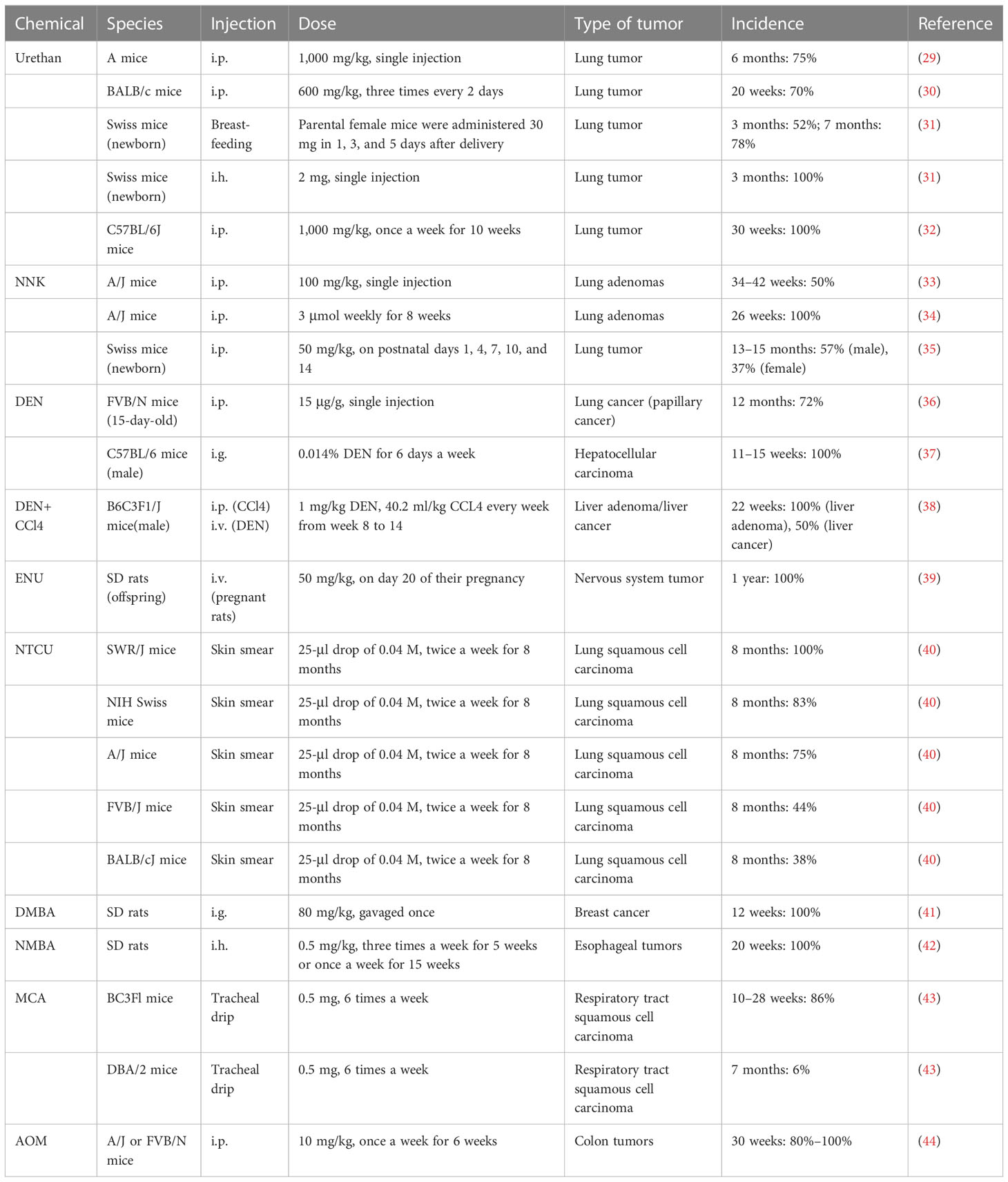
Table 1 Construction method of animal tumor model induced by chemical drugs.
3.1 Urethan
Urethan was originally used as a herbicide and was later found to inhibit cell division and, thus, utilized as a chemotherapeutic drug for leukemia (46). Urethan is structurally one of the simplest carcinogens; it is soluble in both water and lipids and was the first water-soluble carcinogen to be discovered (47). The biological effects of urethan depend on the direct inhibition of nucleic acid synthesis, especially of the pyrimidine bases, and an apparent antagonism between urea and pyrimidines also exists (47). In the 1950s, a study on a model of lung tumors induced by urethan as a carcinogen found that 100% of highly sensitive mice could induce lung tumors (48). Mice are highly sensitive to urethan, especially newborn mice, and studies have shown that the rate of clearance of the urethan in newborn mice is 1/10 of that in adult mice, and 0.5 mg/g urethan can be metabolized in adult mice within 8 h, while the same dose takes 72 h in newborn mice (49). Strain A mice have highly spontaneous lung tumor development; while >75% of mice can develop lung tumors at 18 months, 75% of A mice can develop lung tumors at 6 months after a single injection of 1,000 mg/kg urethan (29). For BALB/c mice, Koohdani et al. injected 600 mg/kg intraperitoneally with urea three times every 2 days and the incidence of lung tumors in the BALB/c mice reached 70% at 20 weeks (7/10) (30). In 1962, De Benedictis et al. induced lung tumors in Swiss mice, and the parental female mice were administered 30 mg of urea 1, 3, and 5 days after delivery; the results showed that the incidence of lung tumors in the offspring was 52% and 78% at 3 months and 7 months, respectively, and another group of offspring received a single subcutaneous injection of 2 mg in the interscapular area within 24 h after birth, and the incidence of lung tumor was 100% in 3 months (31). However, the C57BL/6 mice commonly used in genetic engineering have high resistance to lung tumors induced by urethan, and consequently, multiple injections of urethan are required to overcome their genetic resistance and induce lung cancer. C57BL/6J mice were intraperitoneally injected with 1,000 mg/kg once a week for 10 weeks, and the results showed that the incidence of lung tumors in C57BL/6J mice was close to 100% at 30 weeks (32).
3.2 NNK
NNK, also known as (methylnitrosamine)-1-(3-pyridine)-1-butanone, is one of the main chemical carcinogens in cigarette smoke (50), and it can effectively induce lung cancer in mice, rats, and hamsters (51). The α-hydroxylation of NNK catalyzed by a cytochrome 450 scan easily binds to DNA and forms DNA adducts, causing gene mutations that lead to tumor development (33, 52). Belinsky et al. showed that in 6-week-old A/J mice, a single intraperitoneal injection of 100 mg/kg NNK resulted in the development of hyperplasia along the alveolar septum after 14 weeks, and the frequency of lung adenomas in A/J mice at 34–42 weeks resulted in a significant increase to 50% (53). In another independent study, 7-week-old female A/J mice were given intraperitoneal injections of NNK 3 μmol weekly for 8 weeks, and 100% of the mice developed lung adenomas after 26 weeks (34). NNK can also induce lung cancer in offspring, as Anderson et al. gave 100 mg/kg NNK doses to female A/J mice on days 14, 16, and 18 of their pregnancies, and it was found that lung tumors occurred in 12 of the 66 offspring and 13 of the 14 female mice (54). The effects of NNK on young mice were also studied in Swiss mice. Mice were given 50 mg/kg NNK i.p. on postnatal days 1, 4, 7, 10, and 14, and the incidence of lung tumors was 57% in male mice and 37% in female mice at 13–15 months (35).
3.3 DMN/DEN
Dimethylnitrosamines (DMN) and diethylnitrosamines (DEN) are nitrosamines, which can induce various tumors in mice and rats, but predominantly liver and lung tumors. Nitrosamines are activated by CYP450 enzymes in vivo and converted into a strong alkylating agent, forming adducts in the DNA (active chemicals and cellular macromolecules form stable complexes through covalent bonds), resulting in carcinogenicity (55). Kohda et al. confirmed the mutagenicity of nitrosamines by treating Chinese hamster V79 cells with nitrosamines (56). After DEN exposure, A/J mice developed lung adenocarcinoma, 82% of which possessed KRAS mutations (57). FVB/N mice also have a high incidence of lung cancer, as Zsolt injected 15 μg/g DEN intraperitoneally into 15-day-old FVB/N mice, and the incidence of lung cancer (papillary cancer) was 72% (28/39) after 12 months. Interestingly, there were no mutations in the Kras and EGFR genes (36). In a study by Chen et al., C57BL/6 male mice were intragastrically administered 0.014% DEN for 6 days a week and given normal drinking water on the 7th day for 15 weeks, and 100% of these mice developed fibrosis (3–6 weeks), cirrhosis (7–10 weeks), and HCC (11–15 weeks) at 3–15 weeks (37). The incidence of liver tumors caused by the combination of DEN and CCl4 was significantly higher than that of DEN or CCl4 alone. Uehara et al. used 14-day-old B6C3F1/J male mice and administered 1 mg/kg DEN intravenously, and 40.2 ml/kg CCL4 was injected intraperitoneally every week from weeks 8 to 14. The incidence of liver adenoma and liver cancer was 40% and 20% at 17 weeks, and the incidence of liver adenoma and liver cancer was 100% and 50% at 22 weeks (38). In 1974, Cardesa et al. divided 8-week-old Swiss mice into two groups: subcutaneous injection of DMN or DEN 8 mg/kg; the results showed that the incidence of tumors in mice was 79% (31/39) and 87% (34/39), and the incidence of lung tumors was 67% and 61%, respectively. Lung tumors in the two groups were mainly adenocarcinoma, adenoma, and atypical adenomatoid hyperplasia (58).
3.4 ENU
Acetylnitrosourea (ENU) is a strong mutagenic agent that can cause rapid oncogenic genetic mutations in mice (59). Raju et al. reported that SD rats were administered a single intravenous injection of ENU (50 mg/kg) on day 20 of their pregnancy, and almost 100% of the offspring had central and peripheral nervous system tumors at 1 year of age (39).
3.5 NTCU
N-nitroso-tris-chloroethylurea (NTCU) is a nitrosoalkylurea compound, which has been shown to induce lung SCC in mice. Wang et al. treated eight different strains of female mice by smearing NTCU on the skin to establish a model. In their study, the back skin of 7-week-old mice was scraped, and they were injected with NTCU 25-μl drop of 0.04 M, twice a week, with a 3-day interval; 8 months later, five strains of the mice had successfully induced lung SCC in situ or lung SCC [the induction rates were as follows: SWR/J, 100% (3/3); NIH Swiss, 83% (10/12); A/J, 75% (6/8); FVB/J, 44% (4/9); and BALB/cJ, 38% (3/8)]. The other strains (AKR/J, 129/svJ, and C57BL/6J) failed to develop the carcinomas, and histological and pathological analysis showed that mouse SCC induced by NTCU had the same pathological process as human SCC, which is “normal-proliferative-metaplastic-abnormal-SCC” (40, 60).
3.6 DMBA
Dimethylbenzanthracene (DMBA) is frequently used as a model for polycyclic aromatic hydrocarbon (PAH)-induced mammary tumorigenesis because of its potent carcinogenic and immunosuppressive activities (61). Female SD rats at the age of 7 weeks were diluted with 80 mg/kg DMBA in 0.5 ml of corn oil and gavaged once, and after 12 weeks, animal models of breast cancer lesions could be established in all rats (41).
3.7 NMBA
Methyl benzylnitrosamine (NMBA) is also an important carcinogenic compound that is classified as a nitrosamine. NMBA is currently the most effective inducer of rat esophageal tumors, as they can be induced in 15 weeks or less (62). SD rats were given 0.5 mg/kg NMBA three times a week for 5 weeks or once a week for 15 weeks, and the esophageal tumor incidence was 100% at 20 weeks (42).
3.8 MCA
Methylcholanthracene (MCA) is a potent carcinogen that is often used to induce the transformation of cultured cells, and it was found that repeated intratracheal injection of MCA into BC3Fl [(C57BL×C3H)F1] and DBA/2 mice could induce respiratory tract SCC, and the induced SCC had obvious infiltration and metastasis (43). BC3F1 mice were injected with 0.5 mg of MCA six times a week, and the incidence of respiratory tract SCC was 86% at 10–28 weeks. In contrast, DBA/2 mice that received intratracheal injections of 0.5 mg of MCA four times a week resulted in 6% incidence of SCC of the respiratory tract at 7 months (43).
3.9 AOM
Azomethane (AOM) is a chemical reagent that can promote base mismatch and cause cancer through the alkylation of DNA, which is often used in colonic carcinogenicity (63); 10 mg/kg AOM was injected intraperitoneally into A/J or FVB/N mice once a week for 6 weeks, and resulted in 80%–100% of mice having spontaneous colon tumors within 30 weeks (44).
3.10 Fattening diet
Nonalcoholic fatty liver disease (NAFLD) is a condition characterized by the excessive accumulation of fat in the liver without chronic alcohol intake; it is estimated that the global prevalence rate is approximately 24% (64). NAFLD and nonalcoholic steatohepatitis (NASH) are the liver manifestations of metabolic syndrome, and some patients with NAFLD develop into NASH with associated inflammation and fibrosis, which can progress to HCC (64). Asgharpour et al. successively established a model by following a fattening diet (high fructose-glucose solution and high-fat, high-carbohydrate diet—42% calorie fat and 0.1% cholesterol) in 8- to 12-week-old male mice; 89% of the mice developed HCC at 32–52 weeks, and each mouse had three or more tumor foci (65).
3.11 Ionizing radiation
Moderate to high doses of radiation are well-established causes of cancer (66), and ionizing radiation such as x-rays, α-rays, β-rays, and γ-rays can break through genetic material and cause DNA fracture damage and gene mutations (67). Some studies have shown that irradiated mice may develop a series of malignant tumors, including sarcomas, and single high-dose radiation significantly increases the incidence of tumors when compared with fractionated radiation (68). Edmondson et al. locally irradiated the right hindlimb of C3Hf/Kam and C57BL/6J mice with a high dose (10–70 Gy) or a fractionated dose (40–80 Gy, 2 Gy per day, five times a week, for 4–8 weeks), and after 800 days, 210 tumors were induced in 788 mice. The observed tumors were primarily sarcoma (n = 201), and the occurrence frequency of cancer was low (n = 9). The incidence of tumor after single irradiation was 36.1%, and that of graded irradiation was only16.4% (68).
4 Transgenic models
In recent years, increasing evidence has shown that gene mutation is an important cause of tumorigenesis, and targeted therapies for driving genes have achieved good results in tumor patients with a specific genetic background, but the problem of drug resistance still restricts the further benefits (3). Understanding the mechanism of driving mutations in tumorigenesis and development is thus crucial. Due to the high similarity between mouse and human protein coding genes, transgenic mice have been used to study the effects of gene mutations on tumorigenesis and development since the mid-1980s (5) (Table 2). The advantages of transgenic animal tumor models are that they have great advantages in studying tumorigenesis mechanisms and tumor immune escape. However, the establishment process of transgenic animal models is long, the feeding costs are high, and it is difficult to obtain a large number of experimental animals, which hinders rapid and high-throughput research (76).
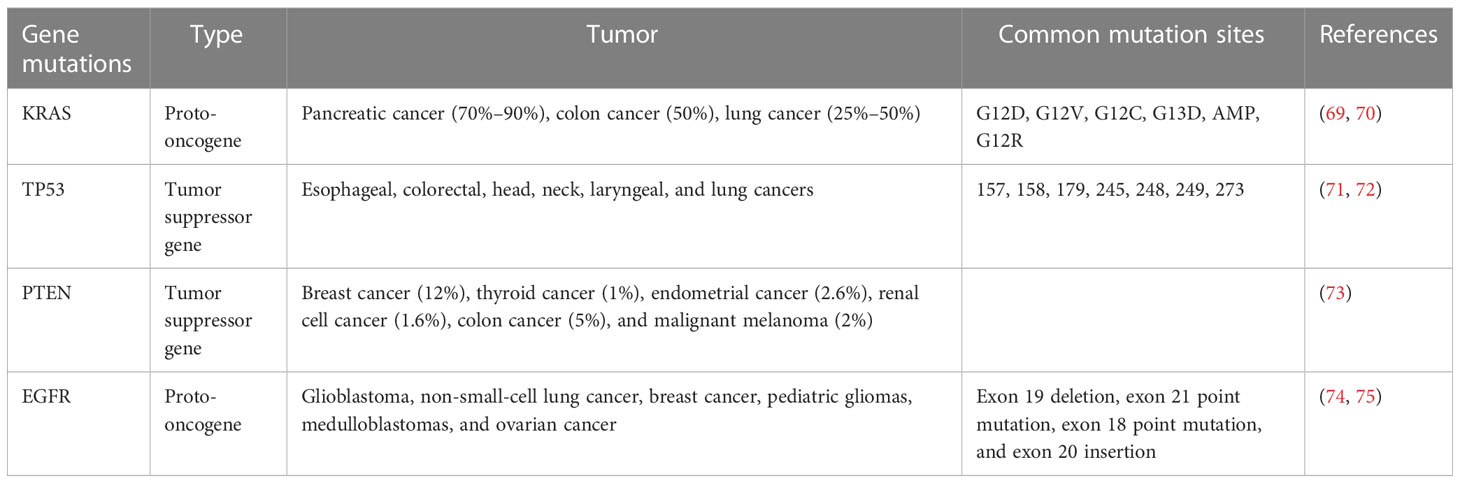
Table 2 The type of tumor caused by gene mutation.
4.1 KRAS
Approximately 30% of human tumors carry ras gene mutations, and the ras gene family includes Kras, Nras, and Hras. Kras is the most commonly mutated gene in the lung, colon, and pancreatic tumors (77), with a mutation rate of approximately 70%–90% in pancreatic cancer, 50% in colon cancer, 25%–50% in lung cancer (69), and 15%–25% in lung adenocarcinoma (78, 79). Point mutations (including G12D, G12V, G12C, G13D, AMP, and G12R) are the most common Kras mutations (70). Under normal circumstances, the activation and inactivation of Kras are finely regulated, as wild-type Kras is temporarily activated by tyrosine kinases such as active epidermal growth factor receptor (EGFR). Activated Kras can motivate downstream signaling networks to execute diverse bioactivities, and Kras is then rapidly inactivated (80). Mutant Kras proteins are uncontrollable, as they can be continuously activated in the absence of an EGFR activation signal, inducing uncontrolled cell proliferation and malignant invasion (80). Most mutations in lung cancer models are Kras dependent; Kras mutations are found in 90% of spontaneous and chemically induced lung tumors in mice, and genetically engineered Kras mice have been widely used in lung cancer research, which is very similar to the genetic and pathophysiological characteristics of human lung cancer (45, 81, 82). Johnson et al. constructed a new type of mouse with a potential KrasG12D allele (KrasLA), and mice carrying these mutations easily formed a variety of tumor types, predominantly lung tumors, and 100% of mice developed numerous kinds of lung tumors at an early stage (68). However, KrasLA mice were found to die of respiratory failure caused by lung lesions at a very young age, and the incidence of other tumors is difficult to predict, which restricts the application of this model (69). Kras gene mutations account for 6% and 18% of diffuse-type and intestinal-type gastric cancers, respectively (83). Brembeck et al. established Kras transgenic mice under the control of the cytokeratin 19 (K19) promoter; parietal cell decrease and mucous neck cell proliferation were found in 3- to 6-month-old mice (84). In pancreatic cancer, the Kras mutation occurs in the early stages of tumorigenesis and accounts for approximately 90% of pancreatic ductal adenocarcinomas; it is often combined with other classical mutations (PTEN, etc.) to induce pancreatic cancer, which will be mentioned in the following article (85).
4.2 TP53
Somatic mutations in the tumor protein P53 (TP53) gene are the most common changes in human cancers (86). The incidence of TP53 mutations in ovarian, esophageal, colorectal, head, neck, laryngeal, and lung cancers is 38%–50%, and the mutation rate is approximately 5% in primary leukemia, sarcoma, testicular cancer, malignant melanoma, and cervical cancer, and it is more common in advanced or invasive cancer subtypes (71). Most TP53 mutations are missense mutations, followed by truncated mutations, intra-frame mutations, and synonymous and uncoded mutations, which are mainly concentrated in known hot spots (the most common sites are 157, 158, 179, 245, 248, 249, and 273) (72). Normally,TP53 functions by activating or inhibiting the transcription of numerous critical genes in diverse bioactivities, including cell cycle arrest, DNA repair, metabolism, senescence, and apoptosis (86). Trp53 (TP53 is called Trp53 in mice) knockout mice are common transgenic animal models that formulate spontaneous tumors at the age of 6 months (including breast cancer, sarcoma, brain tumor, and adrenocortical carcinoma) (87). Compared with homozygous mice, mice with the Trp53 allele heterozygotes had later spontaneous tumors; the most common tumor type in homozygotes was malignant lymphoma, and the main heterozygotes were osteosarcoma and soft tissue sarcoma (88). In C57BL/6 or 129/Sv mice, the Trp53 deletion preferentially induces sarcoma and lymphoma, and the incidence of breast cancer has gradually increased from 21.4%-46.2% in the fourth generation after backcrossing with BALB/c mice for many generations (89). It has been reported that the incidence of gastric invasive adenocarcinoma in Trp53−/− mice is significantly higher than that in WT mice (90). Ralph et al. established a mouse model of neuroendocrine lung tumors by conditionally inactivating Rb1 and Trp53 in mouse lung epithelial cells, and its morphology and immunophenotype were significantly similar to those of SCLC (91). However, increasing evidence shows that the TP53 gene still has antitumor effects after the loss of these classical activities (92). Using a mutant mouse model of p53-3KR (K120R, K161R, and K162R), researchers found that Trp53 can still inhibit cancer initiation, mainly by regulating cell metabolism, despite losing the antitumor effect of inducing cell cycle arrest, apoptosis, and senescence (93).
4.3 PTEN
Pten (phosphatase and tensin homolog deleted on chromosome ten) is a classical tumor suppressor gene with a mutation rate of approximately 12% in breast cancer, 1% in thyroid cancer, 2.6% in endometrial cancer, 1.6% in renal cell cancer, 5% in colon cancer, and 2% in malignant melanoma (73). The PTEN deletion was also reported in 15% of poorly differentiated serous ovarian cancers (94), and Pten mutations were also found in 20% of endometrioid ovarian cancers (95); approximately 53% of patients with primary bladder cancer showed a decrease or deletion of the Pten protein in the cytoplasm or nucleus of their tumor cells (96). The tumor inhibitory activity of PTEN depends to a large extent on its phosphatase activity, which regulates the activity of many important cellular pathways, such as PI3K/AKT; thus, it can regulate many cell processes, including proliferation, survival, energy metabolism, cell structure, and movement (97). The current transgenic model of Pten is widely used in the study of tumorigenesis mechanisms; however, half of the Pten knockout mice died within 1 year after birth, and the rest developed a variety of tumors, including lung, breast, thyroid, endometrial, and prostate cancer, and T-cell lymphoma (98). AlbCrePten (flox/flox) mice established by Horie et al. using the Cre-loxP system showed hepatocyte specific knockouts of Pten, and AlbCrePten (flox/flox) mice showed huge liver hypertrophy and steatohepatitis, as well as triglyceride accumulation similar to human nonalcoholic steatohepatitis (NASH); at 78 weeks of age, all AlbCrePten (flox/flox) mice had liver adenomas, and 66% of AlbCrePten (flox/flox) mice had HCC (99). Yanagi et al. specifically knocked out the Pten gene in bronchiolar alveolar epithelial cells of mice under the control of doxycycline to establish SOPten (flox/flox) mice, of which 90% of the SOPten (flox/flox) offspring mice died of hypoxia shortly after birth (100). Ninety weeks later, all SOPten (flox/flox) mice born showed significant visible lung tumors: 13 tumors were adenocarcinoma and 1 tumor was SCC, as determined by histological examination (100). Russo et al. found that the Pten deletion increases cell migration, invasion, and upregulation of WNT4, which is a key regulator of Müllerian duct development during embryogenesis (101). Tsuruta et al. used the Cre-loxP system to specifically knock out the Pten gene in the urine epithelium of mice to obtain FPten (flox/flox) mice; histologically, the urine epithelium cells of the mice showed enlargement of the nucleus and cell volume, and ultimately, approximately 10% of FPten (flox/flox) mice spontaneously developed into pedicled papillary transitional cell carcinoma (TCC) (96).
4.4 EGFR
Epidermal growth factor receptor (EGFR) is a member of the HER family, which includes HER1 (erbB1, EGFR), HER2 (erbB2, NEU), HER3 (erbB3), and HER4 (erbB4) (102). Studies have shown that there is high or abnormal expression of EGFR in many solid tumors, which is related to tumor cell proliferation, angiogenesis, invasion, metastasis, and inhibition of apoptosis (103). In a study using EGFR knockout mice, the types of cells most affected by the EGFR deletion were epithelial cells and glial cells, while the types of cells overexpressing EGFR in the human tumors were epithelial cells and glial cells (104, 105). A large number of EGFR mRNA deletions have been observed in a number of neoplasms, first in glioblastoma, but recently in non-small-cell lung cancer, breast cancer, pediatric gliomas, medulloblastomas, and ovarian cancer (74). There are four main types of EGFR mutations: exon 19 deletion, exon 21 point mutation, exon 18 point mutation, and exon 20 insertion (75). The most common EGFR mutations are the exon 19 deletion mutations (19DEL) and exon 21 mutations (21L858R), followed by exon 18 G719X, exon 20 S768I, exon 21 L861Q mutation, and T790M mutation in exon 20, which are associated with acquired drug resistance in the first and second generations of EGFR-TKIs (75). EGFR knockout mice were stunted and died at different developmental stages (implantation, second trimester, or early postpartum), and mainly characterized by epithelial cell defects (including skin, lung, gastrointestinal tract, teeth, and eyelid defects), impaired intestinal proliferation, reduced stem cell area, and mucosal structure disorder (105). In order to study the role of activated EGFR mutations in lung cancer, Ohashi et al. established transgenic mice carrying EGFR mutations (five nucleotides encoding five amino acids were deleted, which was equivalent to the EGFRdelEN746-A750 mutation found in lung cancer patients) specifically in type II alveolar epithelial cells through its specific SP-C promoter and found that 9 of 47 newborn mice were positive for EGFR gene mutations, 3 mice of the positive type developed lung adenocarcinoma, only 1 mouse carrying lung adenocarcinoma could reproduce, and all of its offspring developed lung tumors after 7 weeks (106). Ohashi et al. also used EGFR-mutated mouse models to study the evolution of lung adenocarcinoma. Transgenic mice were killed at different time points for pathological examination; atypical adenomatoid hyperplasia (AAH) appeared at 3–4 weeks, diffuse bronchiolo-alveolar carcinoma (BAC) appeared at 4–5 weeks, adenocarcinoma with solid features was observed at 7 weeks, and multiple tumor nodules were observed on the lung surface (106). Ohashi et al. used a similar method to construct transgenic mice expressing EGFRL858R in type II alveolar epithelial cells; 8 mice had L858R deletion mutations in 27 newborn mice, and 2 mice with the positive mutation developed multifocal adenocarcinomas at 7 weeks. All the offspring of these two mice had BAC at 4–5 weeks and adenocarcinomas at 7 weeks (107).
4.5 Combined mutations
Tumors are often caused by multiple gene mutations, the most common of which is the activation of oncogenes and inactivation of tumor suppressor genes (108). Kras and TP53 are common combined gene mutations in human cancer, and KP mice with both Kras and Trp53 mutations are the most classic transgenic model; all KP mice developed primary lung tumors. Six weeks after the lung lesions of mice developed from atypical adenomatous hyperplasia to lung adenomas, the tumors of these mice showed a high degree of nuclear atypia, causing interstitial connective tissue hyperplasia, invasiveness, and metastasis (109). Kras mutations were found in 95% of pancreatic ductal adenocarcinomas. KPC mice were triple mutants with aKrasLSL-G12D, p53LoxP, and Pdx1-CreER for tamoxifen-inducible pancreatic ductal adenocarcinoma. Pancreatic ductal metaplasia and pancreatic intraepithelial neoplasia occurred in KPC mice at 8–10 weeks, and invasive pancreatic ductal adenocarcinoma occurred at 14–16 weeks, and metastasized to the liver, lung, and peritoneum (110). Combined mutations of Kras and Pten are also common in human cancers, and the Ptf1aCre-ERTM, KrasLSL-G12D, and Ptenflox, tamoxifen-inducible triple mutant strain (KPP) may be useful as a model for pancreatic adenocarcinoma (PDA)-induced cachexia-a wasting syndrome characterized by the pronounced loss of skeletal and cardiac muscle and adipose tissues (111). Mutation activation of BRAF is the earliest and most common genetic change in human melanoma; mice specifically expressing BRAF(V600E) showed benign melanocyte proliferation, but did not develop melanoma after 15–20 months, and BRAF(V600E) expression combined with PTEN gene silencing could induce malignant melanoma and metastasis to lymph nodes and lungs (112). The Rb1 deletion, TP53 deletion, and Myc amplification are all common mutations in SCLC (113). RPM mice carried three gene mutations: Rb1fl/fl, Trp53fl/fl, and MycLSL/LSL; 100% of these mice developed SCLC after 6–8 weeks (113). It is worth noting that these common classical gene mutations are often combined with new gene mutations to study the function of these new genes in carcinogenesis.
5 Transplantable animal tumor models
Human tumor generated mouse models are established by transplanting human tumor cells and/or tissues of research interest into recipient animals (almost always possessing immune function deficiencies) (Figure 3). The advantage is that most types of human tumors can establish transplantable tumor models in immunodeficient animals, and under the same inoculation conditions, the growth rate of animals is the same, the difference in tumor formation rate is small, and the inoculation tumor formation rate is high. The disadvantage is that the recipient host animal needs to be in an immunodeficient state requiring special housing in an aseptic environment, which is expensive to maintain. Furthermore, not all cell types of human tumors can be successfully established in rodent models and the stroma of the human tumor tissue obtained may contain the components of the recipient animals. It is worth noting that the key to developing an optimal transplanted tumor model lies in the immune status of the host (immunodeficiency state) and the composition of the graft (containing important or all components of the tumor).
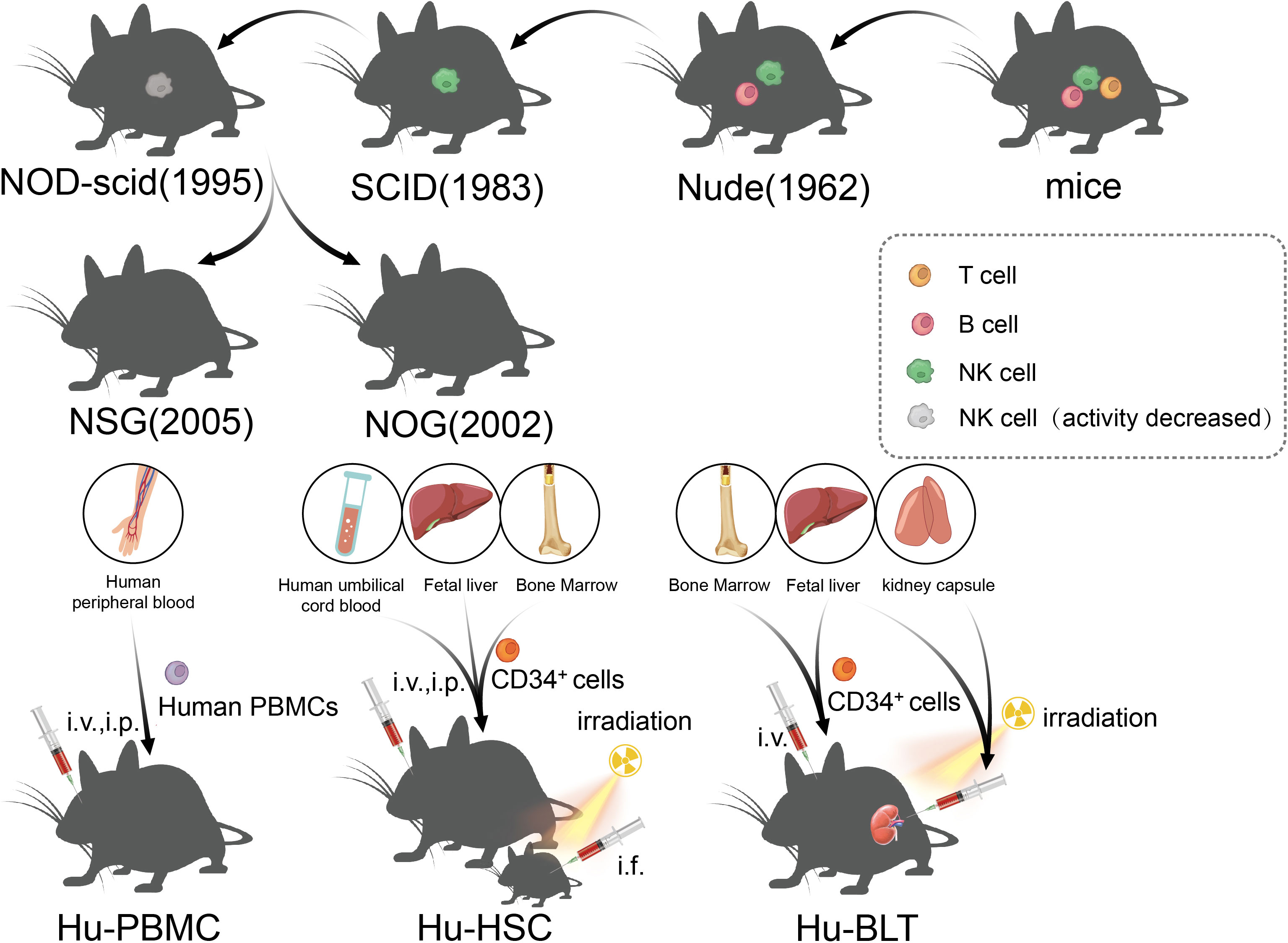
Figure 3 The development of immunodeficient mice and the construction method of the humanized mice model.
5.1 Immunodeficient mice
The immune status of the host has undergone many improvements, which is the key to the development of a successful transplant tumor model. Transplantable tumors are divided into allogeneic transplantation and xenogeneic transplantation, and the most used is the xenotransplantation of human tumors. Due to the existence of immune rejection, wild mice cannot be used for xenotransplantation of human tumor. However, human tumors can be implanted in immunodeficient mice, and the tumors can maintain the histological, immunological, and biological characteristics of original tumors. Nude mice were first identified by Grist in 1962, and they were called nude mice as they are hairless. Flanagan found that nude mice lack thymus and T lymphocytes, and thus, they lacked T cell-mediated immune responses and could be used as recipients for human tumor xenotransplantation (114). However, because nude mice still have B cells and NK, they are not suitable for use as hosts for lymphoma and leukemia, which limits the extensive application of nude mice in relation to transplanted tumors. Severe combined immunodeficiency (SCID) mice were first reported by Bosma in 1983 (115). SCID mice are more severely immunodeficient than nude mice, and the mutant genes of the SCID mice were identified in 1996. The maturation defects of B lymphocytes and T lymphocytes in SCID mice were caused by a point mutation in the Prkdc (protein kinase, DNA-activated, catalytic subunit) gene, which causes the affected lymphocytes to die prior to maturation or be cleared by the macrophages, granulocytes, and NK cells in the body (116, 117). However, it is usually difficult to establish a successful model for lymphoma and metastatic tumors in SCID mice because of leakage (some SCID mice will restore the function of some T and B lymphocytes with age) (118). In addition, NK cells and other innate immunoreactive substances in SCID mice were present at high levels, which limits the success rate of transplantation, such as human hematopoietic stem cell (HSC) transplantation (119). Subsequently, the establishment of NOD/SCID mice ushered in a new breakthrough in transplanted animal tumors, which are a type of spontaneous type 1 diabetic mice, caused by T lymphocytes infiltrating and destroying islets, as well as complement loss and impaired function of NK, macrophages, and dendritic cells (120, 121). NOD/SCID mice were established through the hybridization of NOD and SCID mice, and NOD/SCID mice will not develop type 1 diabetes, as they lack an adaptive immune system and the loss of effector T cells; moreover, due to extensive defects in innate and adaptive immunity (low activity of NK cells and loss of T- and B-cell function), NOD/SCID mice have become a stable and excellent animal model for human HSCs and human solid tumor transplantation (122). At the beginning of the 21st century, scientists introduced IL-2Ry mutations on the basis of NOD/SCID mice, resulting in NSG and NOG mice, which, in addition to being the mice with the highest degree of immunodeficiency at present, could also be used to construct humanized immune system mice to study the antitumor effects of chimeric antigen receptor T-cell immunotherapy (CAR-T) and immune checkpoint inhibitors (ICIs) (123), and will be discussed in detail later.
5.2 PDX
Transplant components have also been the focus of transplantable tumor model research in recent years, and in the past, we transplanted tumor cell lines into animals, which are simpler and easier to use, but the defects cannot be ignored: first, this tumor model cannot fully represent the unique characteristics of each cancer patient; furthermore, this model cannot reconstruct the remaining non-tumor cell components in the tumor tissues, which plays an important role in tumorigenesis and development. Consequently, the patient-derived xenotransplantation (PDX) model has been actively generated and applied, and the PDX model can directly implant cancerous tissues from the patient’s tumor into immunodeficient mice, thus preserving both the cell–cell interaction and the tumor microenvironment (124). The PDX model retains the characteristics of the primary patient tumor, including the gene expression profile and drug response, and offers great advantages for drug screening, biomarker development, and the evaluation of therapeutic effects (125). The PDX model usually takes approximately 6 months to 2 years to establish, and the success rate varies (10%–90%), depending on the tumor source and disease characteristics (125). Specifically, invasive, recurrent, and the transplantation rates tend to be higher for highly metastatic tumors (126). Gastrointestinal tumors (such as colon and pancreatic cancers) have higher transplantation success rates than other cancers, and the transplantation rate in breast cancer is low (127). The success rates of the PDX model is as follows: colon cancer [63.5% (54/85) in nude mice and 87% (74/85) in NOD/SCID mice] (128, 129), pancreatic cancer [61% (42/69) in nude mice and 67% (8/12) in SCID mice] (130, 131), and breast cancer [12.5% (25/200) in nude mice and 27% (13/49) in NOD/SCID mice] (132, 133).
Animal models also play an important role in drug development and screening. In the past, the average success rate of translating animal research into human clinical trials was approximately 8%, and <5% of antineoplastic drugs were approved to enter the market (134, 135). Although the accuracy of the PDX model on drug efficacy and drug resistance was up to 90% (136), the traditional PDX model takes a long time to construct; owing to this, it cannot quickly reflect the drug sensitivity of patients and cannot meet clinical needs. China Lidi Biological has developed a new rapid PDX drug sensitivity detection technique (OncoVee ®MiniPDX) for screening clinically related programs of cancer, which uses patient-derived tumor cells arranged in hollow fiber capsules after 7 days of subcutaneous culture. The active morphology and pharmacokinetics of the tumor cells in MiniPDX capsules were evaluated systematically, and the morphological and histopathological characteristics of the tumor cells in MiniPDX capsules were consistent with those of the PDX model and primary tumor (137).
5.3 Humanized mice model
Recently, ICI and CAR-T have enabled new breakthroughs in the treatment of tumors (138, 139). Although PDX, which depends on immunodeficient mice, has been widely used in the research of tumor immunity and the development of new therapies, the lack of models for the human immune system and tumor immune microenvironment limits the study of immune mechanisms and transformation of immunotherapy to a great extent. The humanized mouse model is a mouse model that reconstructs the human immune system by implanting human hematopoietic cells, lymphocytes, or tissues into immunodeficient mice, which can effectively reconstruct the human immune system and better simulate the characteristics of human immunity (140). From the earliest nude mice to the later SCID and NOD/SCID mice, the success rate of human cell implantation has been low either due to the existence of innate immunity or because of high sensitivity to radiation and the limited life cycle. Owing to this, all these models limited the application of immune system-humanized mice in practical research to some extent. In NOG mice, mature T/B cells and NK cells are lacking, complement activity is decreased, and the function of macrophages and dendritic cells is impaired; therefore, NOG mice are an ideal model for human immune cell transplantation (141). Subsequently, the IL-2 receptor γ mutation was further developed. NSG mice had a complete IL-2 receptor γ chain-invalid allele, similar to NOG mice, with a loss of T/B and NK cells, lack of complement activity, and defects in macrophages and dendritic cells (142). In addition, other highly immunodeficient mice, such as BRG and BRGS mice, have been established as humanized mouse models that lack T/B cells and NK cells (143). Although NOG and NSG mice are very useful humanized models, they still lack the ability to reconstruct myeloid and NK cells, and a new generation of super immunodeficiency models HuNOG-EXL and HuNSG-SGM3 have been developed, which can express some growth factors to promote myeloid regeneration (142, 144). According to the method of human immune system reconstruction, the humanized mouse model can be divided into three categories: Hu-PBMCs (humanized-peripheral blood mononuclear cells), Hu-HSCs (humanized-hematopoietic stem cells), and Hu-BLTs (humanized-bone marrow, liver, thymus) (145). The Hu-PBMC model is a simple and economical humanized mouse model in which mature lymphocytes from peripheral blood mononuclear cells (PBMCs) are injected into immunodeficient host mice through intraperitoneal (i.p.) or intravenous (i.v.) injection (145). This method was first described in CB17-SCID mice in 1988 and has been widely used to study the human immune response in autoimmunity and infectious diseases (117). However, graft-versus-host disease (GvHD) occurs in the Hu-PBMC model in 2–3 weeks, and the survival time is short (146). At present, this model is often used to study the activation of human effector T cells and to evaluate immunosuppressive drugs; in the Hu-HSC mouse model, human CD34+ HSCs from human umbilical cord blood, adult bone marrow, or fetal liver were injected into adult mice (intravenous or intrafemoral) or newborn mice (intracardiac or intrahepatic) (147, 148), and sublethal irradiation of host mice is needed to eliminate HSC and promote the transfer of human HSC (145). Fetal liver and umbilical cord blood are the most commonly used sources of human CD34+ HSCs, which are easier to colonize in immunodeficient mice than that in adult HSCs. Although this method can produce a variety of HSCs in adult mice, the number of T cells produced is small and the model does not possess functional immune cells (149). In newborn mice (less than 4 weeks old), good human cell transplantation can be obtained, and T cells, B cells, macrophages, NK cells, and DCs can be produced (150). At present, it is believed that the Hu-HSC model can establish the human innate immune system and lymphocytes, with little or no occurrence of GvHD, which can be used for long-term research; however, owing to the species differences between humans and mice, there is a lack of human cytokines in mice, and the development of human stem cells in mice is limited (149). The method of establishing Hu-BLT is to transplant human fetal liver and thymus tissue into the renal capsule of adult immunodeficient recipient mice after sublethal irradiation, and at the same time, fetal liver or bone marrow-derived CD34+ HSCs from the same individual is injected intravenously (i.v.) into recipient mice (151). The Hu-BLT model is often used to study adaptive immune responses, such as HIV infection. However, the incidence of GvHD in the Hu-BLT model is higher than that in other CD34+ HSC transplantation models (145), and complex and precise surgical procedures are required, thus limiting the application of the Hu-BLT model in the research and development of tumor immune drugs.
6 Prospects
The use of animal models to develop human healthcare can be traced back to the 6th century BC (152). During this time, there have been significant developments in biotechnology and animal models that have contributed to the development of mechanisms of disease and drug discovery. Since the establishment of the earliest spontaneous animal tumor model, we have obtained a great deal of information about tumor generation and progression. However, we have gradually eliminated this method because of its randomness, high cost, and long cycle of tumorigenesis. Then, we used chemical drugs to induce tumors in mice to establish tumor models and study the carcinogenic effect of a certain factor. In fact, induced tumor is the most widely used method for establishing animal tumor models at present, due to the low technical requirements and cost, most of the laboratories can perform this biotechnique. With the development of the transgenic animal model and the PDX model, we have made great progress in understanding specific gene function, building animal models of human diseases, and evaluating the safety and effectiveness of new drugs, and it is hopeful that it will be a bright light for the study of animal tumor models in the future.
However, they still have significant limitations in modeling human cancer, which is mainly reflected in the immune microenvironment and tumor microenvironment, or the more subtle differences caused by species-specific differences. Although we have developed the PDX model and humanized model to reduce these differences, it is urgent that we develop and establish more effective animal tumor models to facilitate more detailed investigation of the tumor development process.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Natural Science Foundation of China (82002421), the Young Talent Program of Tangdu Hospital, Health Research Fund of Shaanxi Province (2021B004), and the Discipline Innovation Development Plan Project of Tangdu Hospital (2021LCYJ005).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Sengupta R, Zaidi SK. AACR cancer progress report 2021: Discovery science driving clinical breakthroughs. Clin Cancer Res (2021) 27(21):5757–9. doi: 10.1158/1078-0432.CCR-21-3367
3. Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria JC. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discovery (2021) 11(4):874–99. doi: 10.1158/2159-8290.CD-20-1638
4. Robinson NB, Krieger K, Khan FM, Huffman W, Chang M, Naik A, et al. The current state of animal models in research: A review. Int J Surg (2019) 72:9–13. doi: 10.1016/j.ijsu.2019.10.015
5. Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature (2002) 420(6915):520–62. doi: 10.1038/nature01262
6. Zeng L, Li W, Chen CS. Breast cancer animal models and applications. Zool Res (2020) 41(5):477–94. doi: 10.24272/j.issn.2095-8137.2020.095
7. Holland JF, Pogo BG. Mouse mammary tumor virus-like viral infection and human breast cancer. Clin Cancer Res (2004) 10(17):5647–9. doi: 10.1158/1078-0432.CCR-04-1234
8. Parisi F, Freer G, Mazzanti CM, Pistello M, Poli A. Mouse mammary tumor virus (MMTV) and MMTV-like viruses: An in-depth look at a controversial issue. Viruses (2022) 14(5):977. doi: 10.3390/v14050977
9. Heston WE, Vlahakis G. Mammary tumors, plaques, and hyperplastic alveolar nodules in various combinations of mouse inbred strains and the different lines of the mammary tumor virus. Int J Cancer (1971) 7(1):141–8. doi: 10.1002/ijc.2910070116
10. Nagasawa H, Yanai R, Taniguchi H, Tokuzen R, Nakahara W. Two-way selection of a stock of Swiss albino mice for mammary tumorigenesis: establishment of two new strains (SHN and SLN). J Natl Cancer Inst (1976) 57(2):425–30. doi: 10.1093/jnci/57.2.425
11. Sun B, Zhang S, Zhang D, Li Y, Zhao X, Luo Y, et al. Identification of metastasis-related proteins and their clinical relevance to triple-negative human breast cancer. Clin Cancer Res (2008) 14(21):7050–9. doi: 10.1158/1078-0432.CCR-08-0520
12. He YN, Zhang SC, Zhang HM. [Pathologic changes of spontaneous tumors in sprague-dawley and wistar rats]. Zhonghua Bing Li Xue Za Zhi (2017) 46(4):249–54. doi: 10.3760/cma.j.issn.0529-5807.2017.04.007
13. Haseman JK, Hailey JR, Morris RW. Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: a national toxicology program update. Toxicol Pathol (1998) 26(3):428–41. doi: 10.1177/019262339802600318
14. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet (2021) 398(10299):535–54. doi: 10.1016/S0140-6736(21)00312-3
15. Bade BC, Dela Cruz CS. Lung cancer 2020: Epidemiology, etiology, and prevention. Clin Chest Med (2020) 41(1):1–24. doi: 10.1016/j.ccm.2019.10.001
16. Tuveson DA, Jacks T. Modeling human lung cancer in mice: similarities and shortcomings. Oncogene (1999) 18(38):5318–24. doi: 10.1038/sj.onc.1203107
17. Shimkin MB, Stoner GD. Lung tumors in mice: application to carcinogenesis bioassay. Adv Cancer Res (1975) 21:1–58. doi: 10.1016/S0065-230X(08)60970-7
18. Lynch CJ. Studies on the relation between tumor susceptibility and heredity : Iii. spontaneous tumors of the lung in mice. J Exp Med (1926) 43(3):339–55. doi: 10.1084/jem.43.3.339
19. Mahler JF, Stokes W, Mann PC, Takaoka M, Maronpot RR. Spontaneous lesions in aging FVB/N mice. Toxicol Pathol (1996) 24(6):710–6. doi: 10.1177/019262339602400606
20. Dasgupta P, Henshaw C, Youlden DR, Clark PJ, Aitken JF, Baade PD. Global trends in incidence rates of primary adult liver cancers: A systematic review and meta-analysis. Front Oncol (2020) 10:171. doi: 10.3389/fonc.2020.00171
21. Dragani TA, Sozzi G, Della Porta G. Spontaneous and urethan-induced tumor incidence in B6C3F1 versus B6CF1 mice. Tumori (1984) 70(6):485–90. doi: 10.1177/030089168407000603
22. Soga M, Kishimoto Y, Kawamura Y, Inagaki S, Makino S, Saibara T. Spontaneous development of hepatocellular carcinomas in the FLS mice with hereditary fatty liver. Cancer Lett (2003) 196(1):43–8. doi: 10.1016/S0304-3835(03)00213-1
23. Yoshida MC, Masuda R, Sasaki M, Takeichi N, Kobayashi H, Dempo K, et al. New mutation causing hereditary hepatitis in the laboratory rat. J Hered (1987) 78(6):361–5. doi: 10.1093/oxfordjournals.jhered.a110416
24. Li Y, Togashi Y, Sato S, Emoto T, Kang JH, Takeichi N, et al. Spontaneous hepatic copper accumulation in long-Evans cinnamon rats with hereditary hepatitis. a model of wilson's disease. J Clin Invest (1991) 87(5):1858–61. doi: 10.1172/JCI115208
25. Marquez A, Villa-Trevino S, Gueraud F. The LEC rat: a useful model for studying liver carcinogenesis related to oxidative stress and inflammation. Redox Rep (2007) 12(1):35–9. doi: 10.1179/135100007X162220
26. Claude A. [Morphology and organization of nuclear constituents in a case of renal carcinoma in the mouse]. C R Seances Soc Biol Fil (1961) 252:4186–8.
27. Kawashima K, Ikeda H, Hartley JW, Stockert E, Rowe WP, Old LJ. Changes in expression of murine leukemia virus antigens and production of xenotropic virus in the late preleukemic period in AKR mice. Proc Natl Acad Sci USA (1976) 73(12):4680–4. doi: 10.1073/pnas.73.12.4680
28. Owens MH, Bonavida B. Immune functions characteristic of SJL/J mice and their association with age and spontaneous reticulum cell sarcoma. Cancer Res (1976) 36(3):1077–83.
29. Cowen PN. Some studies on the action of urethane on mice. Br J Cancer (1947) 1(4):401–5. doi: 10.1038/bjc.1947.39
30. Koohdani F, Sasani F, Mohammad K, Mehdipour P. Comparison of ki-67 antigen expression and K-ras mutation in lung tumours induced by urethane in mice. Singapore Med J (2009) 50(7):729–33.
31. De Benedictis G, Maiorano G, Chiecobianchi L, Fiore-Donati L. Lung carcinogenesis by urethane in newborn, suckling, and adult Swiss mice. Br J Cancer (1962) 16:686–9. doi: 10.1038/bjc.1962.78
32. Sozio F, Schioppa T, Sozzani S, Del Prete A. Urethane-induced lung carcinogenesis. Methods Cell Biol (2021) 163:45–57. doi: 10.1016/bs.mcb.2020.09.005
33. Hecht SS, Carmella SG, Kenney PM, Low SH, Arakawa K, Yu MC. Effects of cruciferous vegetable consumption on urinary metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in singapore chinese. Cancer Epidemiol Biomarkers Prev (2004) 13(6):997–1004. doi: 10.1158/1055-9965.997.13.6
34. Hecht SS, Isaacs S, Trushin N. Lung tumor induction in A/J mice by the tobacco smoke carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and benzo[a]pyrene: a potentially useful model for evaluation of chemopreventive agents. Carcinogenesis (1994) 15(12):2721–5. doi: 10.1093/carcin/15.12.2721
35. Anderson LM, Hecht SS, Kovatch RM, Amin S, Hoffmann D, Rice JM. Tumorigenicity of the tobacco-specific carcinogen 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone in infant mice. Cancer Lett (1991) 58(3):177–81. doi: 10.1016/0304-3835(91)90097-2
36. Mervai Z, Egedi K, Kovalszky I, Baghy K. Diethylnitrosamine induces lung adenocarcinoma in FVB/N mouse. BMC Cancer (2018) 18(1):157. doi: 10.1186/s12885-018-4068-4
37. Chen Q, You X, Yang W, Jiang S, Lai J, Zhang H, et al. Survival of endogenous hepatic stem/progenitor cells in liver tissues during liver cirrhosis. Life Sci (2020) 241:117121. doi: 10.1016/j.lfs.2019.117121
38. Uehara T, Ainslie GR, Kutanzi K, Pogribny IP, Muskhelishvili L, Izawa T, et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol Sci (2013) 132(1):53–63. doi: 10.1093/toxsci/kfs342
39. Raju NR, Yaeger MJ, Okazaki DL, Lovell K, Koestner A. Immunohistochemical characterization of rat central and peripheral nerve tumors induced by ethylnitrosourea. Toxicol Pathol (1990) 18(1Pt 1):18–23. doi: 10.1177/019262339001800103
40. Wang Y, Zhang Z, Yan Y, Lemon WJ, LaRegina M, Morrison C, et al. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res (2004) 64(5):1647–54. doi: 10.1158/0008-5472.CAN-03-3273
41. Karnam KC, Ellutla M, Bodduluru LN, Kasala ER, Uppulapu SK, Kalyankumarraju M, et al. Preventive effect of berberine against DMBA-induced breast cancer in female sprague dawley rats. BioMed Pharmacother (2017) 92:207–14. doi: 10.1016/j.biopha.2017.05.069
43. Nettesheim P, Hammons AS. Induction of squamous cell carcinoma in the respiratory tract of mice. J Natl Cancer Inst (1971) 47(3):697–701.
44. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc (2007) 2(8):1998–2004. doi: 10.1038/nprot.2007.279
45. Malyla V, Paudel KR, Shukla SD, Donovan C, Wadhwa R, Pickles S, et al. Recent advances in experimental animal models of lung cancer. Future Med Chem (2020) 12(7):567–70. doi: 10.4155/fmc-2019-0338
46. Field KJ, Lang CM. Hazards of urethane (ethyl carbamate): a review of the literature. Lab Anim (1988) 22(3):255–62. doi: 10.1258/002367788780746331
47. Mirvish SS. The carcinogenic action and metabolism of urethan and n-hydroxyurethan. Adv Cancer Res (1968) 11:1–42. doi: 10.1016/s0065-230x(08)60386-3
48. Malkinson AM, Beer DS. Major effect on susceptibility to urethan-induced pulmonary adenoma by a single gene in BALB/cBy mice. J Natl Cancer Inst (1983) 70(5):931–6.
49. Mirvish SS. The metabolism of n-hydroxyurethane in relation to its carcinogenic action: conversion into urethane and an n-hydroxyurethane glucuronide. Biochim Biophys Acta (1966) 117(1):1–12. doi: 10.1016/0304-4165(66)90146-2
50. Liu Y, Yin T, Feng Y, Cona MM, Huang G, Liu J, et al. Mammalian models of chemically induced primary malignancies exploitable for imaging-based preclinical theragnostic research. Quant Imaging Med Surg (2015) 5(5):708–29. doi: 10.3978/j.issn.2223-4292.2015.06.01
51. Ge GZ, Xu TR, Chen C. Tobacco carcinogen NNK-induced lung cancer animal models and associated carcinogenic mechanisms. Acta Biochim Biophys Sin (Shanghai) (2015) 47(7):477–87. doi: 10.1093/abbs/gmv041
52. Kiyohara C, Yoshimasu K, Takayama K, Nakanishi Y. NQO1, MPO, and the risk of lung cancer: a HuGE review. Genet Med (2005) 7(7):463–78. doi: 10.1097/01.gim.0000177530.55043.c1
53. Belinsky SA, Devereux TR, Foley JF, Maronpot RR, Anderson MW. Role of the alveolar type II cell in the development and progression of pulmonary tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the A/J mouse. Cancer Res (1992) 52(11):3164–73.
54. Anderson LM, Hecht SS, Dixon DE, Dove LF, Kovatch RM, Amin S, et al. Evaluation of the transplacental tumorigenicity of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in mice. Cancer Res (1989) 49(14):3770–5.
55. Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther (1996) 71(1-2):57–81. doi: 10.1016/0163-7258(96)00062-9
56. Huang Kohda K, Anjo T, Mochizuki M, Kawazoe Y, Okada M. Mutagenicity of alpha-acetoxy derivatives of n-methyl-N-benzylnitrosamine and N,N-dibenzylnitrosamine in V79 cells: A comparison between methylating and benzylating mutagens. Gan (1982) 73(4):517–21.
57. You M, Wang Y, Lineen AM, Gunning WT, Stoner GD, Anderson MW. Mutagenesis of the K-ras protooncogene in mouse lung tumors induced by n-ethyl-N-nitrosourea or n-nitrosodiethylamine. Carcinogenesis (1992) 13(9):1583–6. doi: 10.1093/carcin/13.9.1583
58. Cardesa A, Pour P, Althoff J, Mohr U. Comparative studies of neoplastic response to a single dose of nitroso compounds. 4. the effect of dimethyl- and diethyl-nitrosamine in Swiss mice. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol (1974) 81(3-4):229–33. doi: 10.1007/BF00305023
59. Salinger AP, Justice MJ. Mouse mutagenesis using n-Ethyl-N-Nitrosourea (ENU). CSH Protoc (2008) 2008:pdb prot4985. doi: 10.1101/pdb.prot4985
60. Wang Y, Zhang Z, Garbow JR, Rowland DJ, Lubet RA, Sit D, et al. Chemoprevention of lung squamous cell carcinoma in mice by a mixture of Chinese herbs. Cancer Prev Res (Phila) (2009) 2(7):634–40. doi: 10.1158/1940-6207.CAPR-09-0052
61. Jung KJ, Wallig MA, Singletary KW. Purple grape juice inhibits 7,12-dimethylbenz[a]anthracene (DMBA)-induced rat mammary tumorigenesis and in vivo DMBA-DNA adduct formation. Cancer Lett (2006) 233(2):279–88. doi: 10.1016/j.canlet.2005.03.020
62. Beer DG, Stoner GD. Clinical models of chemoprevention for the esophagus. Hematol Oncol Clin North Am (1998) 12(5):1055–77. doi: 10.1016/S0889-8588(05)70041-1
63. Papanikolaou A, Shank RC, Delker DA, Povey A, Cooper DP, Rosenberg DW. Initial levels of azoxymethane-induced DNA methyl adducts are not predictive of tumor susceptibility in inbred mice. Toxicol Appl Pharmacol (1998) 150(1):196–203. doi: 10.1006/taap.1998.8393
64. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol (2018) 15(1):11–20. doi: 10.1038/nrgastro.2017.109
65. Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol (2016) 65(3):579–88. doi: 10.1016/j.jhep.2016.05.005
66. Abalo KD, Rage E, Leuraud K, Richardson DB, Le Pointe HD, Laurier D, et al. Early life ionizing radiation exposure and cancer risks: systematic review and meta-analysis. Pediatr Radiol (2021) 51(1):45–56. doi: 10.1007/s00247-020-04803-0
68. Edmondson EF, Hunter NR, Weil MM, Mason KA. Tumor induction in mice after localized single- or fractionated-dose irradiation: Differences in tumor histotype and genetic susceptibility based on dose scheduling. Int J Radiat Oncol Biol Phys (2015) 92(4):829–36. doi: 10.1016/j.ijrobp.2015.03.002
69. Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature (2001) 410(6832):1111–6. doi: 10.1038/35074129
70. Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov (2022) 12(4):924–37. doi: 10.1158/2159-8290.CD-21-1331
71. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat (2007) 28(6):622–9. doi: 10.1002/humu.20495
72. Saleh MM, Scheffler M, Merkelbach-Bruse S, Scheel AH, Ulmer B, Wolf J, et al. Comprehensive analysis of TP53 and KEAP1 mutations and their impact on survival in localized- and advanced-stage NSCLC. J Thorac Oncol (2022) 17(1):76–88. doi: 10.1016/j.jtho.2021.08.764
73. Ngeow J, Eng C. PTEN in hereditary and sporadic cancer. Cold Spring Harb Perspect Med (2020) 10(4):a036087. doi: 10.1101/cshperspect.a036087
74. Voldborg BR, Damstrup L, Spang-Thomsen M, Poulsen HS. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol (1997) 8(12):1197–206. doi: 10.1023/A:1008209720526
75. Santos EDS, Nogueira KAB, Fernandes LCC, Martins JRP, Reis AVF, Neto JBV, et al. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int J Pharm (2021) 592:120082. doi: 10.1016/j.ijpharm.2020.120082
76. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov (2006) 5(9):741–54. doi: 10.1038/nrd2110
77. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res (2020) 80(14):2969–74. doi: 10.1158/0008-5472.CAN-19-3682
78. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res (2008) 14(18):5731–4. doi: 10.1158/1078-0432.CCR-08-0646
79. Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res (2002) 62(23):6997–7000.
80. Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann Oncol (2021) 32(9):1101–10. doi: 10.1016/j.annonc.2021.06.001
81. Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res (2012) 72(10):2457–67. doi: 10.1158/0008-5472.CAN-11-2612
82. Malkinson AM. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp Lung Res (1998) 24(4):541–55. doi: 10.3109/01902149809087385
83. Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell (2004) 5(2):121–5. doi: 10.1016/S1535-6108(04)00033-9
84. Brembeck FH, Moffett J, Wang TC, Rustgi AK. The keratin 19 promoter is potent for cell-specific targeting of genes in transgenic mice. Gastroenterology (2001) 120(7):1720–8. doi: 10.1053/gast.2001.24846
85. Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther (2016) 168:29–42. doi: 10.1016/j.pharmthera.2016.09.003
86. Levine AJ. Spontaneous and inherited TP53 genetic alterations. Oncogene (2021) 40(41):5975–83. doi: 10.1038/s41388-021-01991-3
87. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature (1992) 356(6366):215–21. doi: 10.1038/356215a0
88. Harvey M, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet (1993) 5(3):225–9. doi: 10.1038/ng1193-225
89. Machida Y, Sudo Y, Uchiya N, Imai T. Increased susceptibility to mammary carcinogenesis and an opposite trend in endometrium in Trp53 heterozygous knockout female mice by backcrossing the BALB/c strain onto the background C3H strain. J Toxicol Pathol (2019) 32(3):197–203. doi: 10.1293/tox.2018-0057
90. Ohgaki H, Fukuda M, Tohma Y, Huang H, Stoica G, Tatematsu M, et al. Effect of intragastric application of n-methylnitrosourea in p53 knockout mice. Mol Carcinog (2000) 28(2):97–101. doi: 10.1002/1098-2744(200006)28:2<97::AID-MC5>3.0.CO;2-O
91. Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell (2003) 4(3):181–9. doi: 10.1016/S1535-6108(03)00220-4
92. Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol (2021) 85:4–32. doi: 10.1016/j.semcancer.2021.03.010
93. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell (2012) 149(6):1269–83. doi: 10.1016/j.cell.2012.04.026
94. Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discovery (2012) 2(1):56–67. doi: 10.1158/2159-8290.CD-11-0170
95. Cho KR. Ovarian cancer update: lessons from morphology, molecules, and mice. Arch Pathol Lab Med (2009) 133(11):1775–81. doi: 10.5858/133.11.1775
96. Tsuruta H, Kishimoto H, Sasaki T, Horie Y, Natsui M, Shibata Y, et al. Hyperplasia and carcinomas in pten-deficient mice and reduced PTEN protein in human bladder cancer patients. Cancer Res (2006) 66(17):8389–96. doi: 10.1158/0008-5472.CAN-05-4627
97. Worby CA, Dixon JE. Pten. Annu Rev Biochem (2014) 83:641–69. doi: 10.1146/annurev-biochem-082411-113907
98. Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired fas response and autoimmunity in pten+/- mice. Science (1999) 285(5436):2122–5. doi: 10.1126/science.285.5436.2122
99. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest (2004) 113(12):1774–83. doi: 10.1172/JCI20513
100. Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest (2007) 117(10):2929–40. doi: 10.1172/JCI31854
101. Russo A, Czarnecki AA, Dean M, Modi DA, Lantvit DD, Hardy L, et al. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene (2018) 37(15):1976–90. doi: 10.1038/s41388-017-0097-8
102. Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell (2002) 110(6):669–72. doi: 10.1016/S0092-8674(02)00966-2
103. Kumar R, George B, Campbell MR, Verma N, Paul AM, Melo-Alvim C, et al. HER family in cancer progression: From discovery to 2020 and beyond. Adv Cancer Res (2020) 147:109–60. doi: 10.1016/bs.acr.2020.04.001
104. Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation (2007) 75(9):770–87. doi: 10.1111/j.1432-0436.2007.00238.x
105. Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science (1995) 269(5221):234–8. doi: 10.1126/science.7618085
106. Ohashi K, Rai K, Fujiwara Y, Osawa M, Hirano S, Takata K, et al. Induction of lung adenocarcinoma in transgenic mice expressing activated EGFR driven by the SP-c promoter. Cancer Sci (2008) 99(9):1747–53. doi: 10.1111/j.1349-7006.2008.00875.x
107. Ohashi K, Takigawa N, Osawa M, Ichihara E, Takeda H, Kubo T, et al. Chemopreventive effects of gefitinib on nonsmoking-related lung tumorigenesis in activating epidermal growth factor receptor transgenic mice. Cancer Res (2009) 69(17):7088–95. doi: 10.1158/0008-5472.CAN-08-4205
108. Iranzo J, Martincorena I, Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proc Natl Acad Sci USA (2018) 115(26):E6010–9. doi: 10.1073/pnas.1803155115
109. Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res (2005) 65(22):10280–8. doi: 10.1158/0008-5472.CAN-05-2193
110. Maddipati R, Stanger BZ. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discovery (2015) 5(10):1086–97. doi: 10.1158/2159-8290.CD-15-0120
111. Talbert EE, Cuitiño MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer-induced cachexia. Cell Rep (2019) 28(6):1612–1622.e4. doi: 10.1016/j.celrep.2019.07.016
112. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. Braf(V600E) cooperates with pten loss to induce metastatic melanoma. Nat Genet (2009) 41(5):544–52. doi: 10.1038/ng.356
113. Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell (2017) 31(2):270–85. doi: 10.1016/j.ccell.2016.12.005
114. Flanagan SP. 'Nude', a new hairless gene with pleiotropic effects in the mouse. Genet Res (1966) 8(3):295–309. doi: 10.1017/s0016672300010168
115. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature (1983) 301(5900):527–30. doi: 10.1038/301527a0
116. McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science (1988) 241(4873):1632–9. doi: 10.1126/science.241.4873.1632
117. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature (1988) 335(6187):256–9. doi: 10.1038/335256a0
118. Shigematsu T, Wolf RE, Granger DN. T-Lymphocytes modulate the microvascular and inflammatory responses to intestinal ischemia-reperfusion. Microcirculation (2002) 9(2):99–109. doi: 10.1080/mic.9.2.99.109
119. Miao M, Masengere H, Yu G, Shan F. Reevaluation of NOD/SCID mice as NK cell-deficient models. BioMed Res Int 2021 (2021) p:8851986. doi: 10.1155/2021/8851986
120. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu (1980) 29(1):1–13. doi: 10.1538/expanim1978.29.1_1
121. Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol (1992) 51:285–322. doi: 10.1016/S0065-2776(08)60490-3
122. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol (1995) 154(1):180–91. doi: 10.4049/jimmunol.154.1.180
123. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol (2007) 7(2):118–30. doi: 10.1038/nri2017
124. Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat (2018) 50(1):1–10. doi: 10.4143/crt.2017.307
125. Okada S, Vaeteewoottacharn K, Kariya R. Application of highly immunocompromised mice for the establishment of patient-derived xenograft (PDX) models. Cells (2019) 8(8):889. doi: 10.3390/cells8080889
126. Collins AT, Lang SH. A systematic review of the validity of patient derived xenograft (PDX) models: the implications for translational research and personalised medicine. PeerJ (2018) 6:e5981. doi: 10.7717/peerj.5981
127. Murayama T, Gotoh N. Patient-derived xenograft models of breast cancer and their application. Cells (2019) 8(6):621. doi: 10.3390/cells8060621
128. Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res (2012) 18(19):5314–28. doi: 10.1158/1078-0432.CCR-12-0372
129. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov (2011) 1(6):508–23. doi: 10.1158/2159-8290.CD-11-0109
130. Mattie M, Christensen A, Chang MS, Yeh W, Said S, Shostak Y, et al. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia (2013) 15(10):1138–50. doi: 10.1593/neo.13922
131. Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, et al. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin Cancer Res (2011) 17(17):5793–800. doi: 10.1158/1078-0432.CCR-11-0341
132. Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res (2007) 13(13):3989–98. doi: 10.1158/1078-0432.CCR-07-0078
133. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breastcancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med (2011) 17(11):1514–20. doi: 10.1038/nm.2454
134. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res (2014) 6(2):114–8.
135. DiMasi JA, Reichert JM, Feldman L, Malins A. Clinical approval success rates for investigational cancer drugs. Clin Pharmacol Ther (2013) 94(3):329–35. doi: 10.1038/clpt.2013.117
136. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol (2012) 9(6):338–50. doi: 10.1038/nrclinonc.2012.61
137. Zhang F, Wang W, Long Y, Liu H, Cheng J, Guo L, et al. Characterization of drug responses of mini patient-derived xenografts in mice for predicting cancer patient clinical therapeutic response. Cancer Commun (Lond) (2018) 38(1):60. doi: 10.1186/s40880-018-0329-5
138. Portuguese AJ, Tykodi SS, Blosser CD, Gooley TA, Thompson JA, Hall ET. Immune checkpoint inhibitor use in solid organ transplant recipients: A systematic review. J Natl Compr Canc Netw (2022) 20(4):406–416.e11. doi: 10.6004/jnccn.2022.7009
139. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol (2020) 13(1):86. doi: 10.1186/s13045-020-00910-5
140. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol (2012) 12(11):786–98. doi: 10.1038/nri3311
141. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood (2002) 100(9):3175–82. doi: 10.1182/blood-2001-12-0207
142. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol (2005) 174(10):6477–89. doi: 10.4049/jimmunol.174.10.6477
143. Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci USA (2011) 108(32):13224–9. doi: 10.1073/pnas.1101398108
144. Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved b cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun Inflamm Dis (2016) 4(4):427–40. doi: 10.1002/iid3.124
145. De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol (2018) 39(9):748–63. doi: 10.1016/j.it.2018.07.001
146. King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol (2009) 157(1):104–18. doi: 10.1111/j.1365-2249.2009.03933.x
147. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science (2004) 304(5667):104–7. doi: 10.1126/science.1093933
148. Holyoake TL, Nicolini FE, Eaves CJ. Functional differences between transplantable human hematopoietic stem cells from fetal liver, cord blood, and adult marrow. Exp Hematol (1999) 27(9):1418–27. doi: 10.1016/S0301-472X(99)00078-8
149. Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, et al. The analysis of the functions of human b and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice). Int Immunol (2009) 21(7):843–58. doi: 10.1093/intimm/dxp050
150. Brehm MA, Bortell R, Diiorio P, Leif J, Laning J, Cuthbert A, et al. Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD-Rag1null IL2rgammanull Ins2Akita mice. Diabetes (2010) 59(9):2265–70. doi: 10.2337/db10-0323
151. Xia J, Hu Z, Yoshihara S, Li Y, Jin CH, Tan S, et al. Modeling human leukemia immunotherapy in humanized mice. EBioMedicine (2016) 10:101–8. doi: 10.1016/j.ebiom.2016.06.028
Keywords: tumor, immune microenvironment, animal model, spontaneity, induced, transgene, PDX, immunodeficiency
Citation: Zhou Y, Xia J, Xu S, She T, Zhang Y, Sun Y, Wen M, Jiang T, Xiong Y and Lei J (2023) Experimental mouse models for translational human cancer research. Front. Immunol. 14:1095388. doi: 10.3389/fimmu.2023.1095388
Received: 11 November 2022; Accepted: 20 February 2023;
Published: 10 March 2023.
Edited by:
Rajat Varma, AstraZeneca, United StatesReviewed by:
Michael W. Melkus, Texas Tech University Health Science Center Amarillo, United StatesVirginia Brancato, Italian Institute of Technology (IIT), Italy
Copyright © 2023 Zhou, Xia, Xu, She, Zhang, Sun, Wen, Jiang, Xiong and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Lei, bGVpamllbWRAMTYzLmNvbQ==; Yanlu Xiong, eGlvbmcyMUBmbW11LmVkdS5jbg==; Tao Jiang, amlhbmd0YW9jaGVzdEAxNjMuY29t
†These authors have contributed equally to this work