
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 April 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1095267
 Jianli Zhou1†
Jianli Zhou1† Qiao Zhang1†
Qiao Zhang1† Yuzhen Zhao1†Yuchen Song2,3Yanan Leng3
Yuzhen Zhao1†Yuchen Song2,3Yanan Leng3 Moxian Chen3*
Moxian Chen3* Shaoming Zhou1*Zhaoxia Wang1*
Shaoming Zhou1*Zhaoxia Wang1*Inflammatory bowel disease (IBD) mainly includes Crohn’s disease and ulcerative colitis. These diseases have a progressive course of chronic relapse and remission and affect a large number of children and adults worldwide. The burden of IBD is rising worldwide, with levels and trends varying greatly in countries and regions. Like most chronic diseases, the costs associated with IBD are high, including hospitalizations, outpatient and emergency visits, surgeries, and pharmacotherapies. However, there is no radical cure for it yet, and its therapeutic targets still need further study. Currently, the pathogenesis of IBD remains unclear. It is generally assumed that the occurrence and development of IBD are related to the environmental factors, gut microbiota, immune imbalance, and genetic susceptibility. Alternative splicing contributes to a various diseases, such as spinal muscular atrophy, liver diseases, and cancers. In the past, it has been reported that alternative splicing events, splicing factors, and splicing mutations were associated with IBD, but there were no reports on the practical application for clinical diagnosis and treatment of IBD using splicing-related methods. Therefore, this article reviews research progress on alternative splicing events, splicing factors, and splicing mutations associated with IBD.
Inflammatory bowel disease (IBD) includes ulcerative colitis (UC), Crohn’s disease (CD), and indeterminate colitis. These diseases have a progressive course of chronic relapse and remission and affect a large number of children and adults worldwide (1). IBD can occur at any age but is most common among adolescents and young adults (2). According to age, IBD is divided into adult-onset IBD and pediatric-onset IBD. The latter includes neonatal IBD, infantile-onset IBD, very early–onset IBD (VEO-IBD; diagnosed before 6 years old), and early-onset IBD (EO-IBD; diagnosed before 10 years old) (2). The prevalence of IBD is approximately 1% in developed countries, leading to an increasing prevalence while the incidence and prevalence are also rising in developing countries (2). This rise in prevalence and increased incidence will have important global health and economic effects (2). As there is no cure for IBD, the costs are high with a mean cost of $26,555 in the first year after initial diagnosis, including hospitalizations, outpatient and emergency visits, surgeries, and pharmacotherapies (3). Therefore, there is a need to develop effective treatments for IBD.
Currently, the pathogenesis of IBD remains unclear. It is generally assumed that the occurrence and development of IBD are related to the environmental factors, gut microbiota, immune imbalance, and genetic susceptibility, which can impact a weakened gut barrier on the microbiota, leading to inappropriate intestinal immune activation (4). The genetics of IBD have benefited substantially from studying common variants in large cohorts of patients with good phenotypes and from studying rare variants in cases associated with Mendelian inheritance (5). Before being translated into protein, primary mRNA must be modified and edited. During normal splicing, a special protein and RNA complex, called the spliceosome, attaches to the mRNA. Specific RNA transcript product regions (introns) are excised, whereas flanking sequences (exons) are spliced together. This process fundamentally changes the information content of RNA transcripts directly affecting translation of genetic information into proteins. Regulation of splicing thus represents a critical step in gene expression occurring in the nucleus (Figure 1) (6). First, a branch point A nucleotide located near the 3′ splice site of the intron sequence attacks the 5′ splice site and cleaves it. The 5′ end of the intronic sequence cut is thus covalently linked to this A nucleotide, forming a branched nucleotide. Second, the 3′-OH end of the first exon sequence created in the first step is added to the beginning of the second exon sequence to cleave the RNA molecule at the 3′ splice site. Thus, two exonic sequences are linked to each other, and the intronic sequence is released as a lasso (6). At present, alternative splicing has been reported in many diseases, including spinal muscular atrophy (SMA), liver diseases, and cancers (7–9). For example, Nusinersen targets intronic splice silencer N1 to modulate splicing of survival motor neuron 2 (SMN2) exon 7, resulting in increased production of full-length SMN for SMA therapy (7).
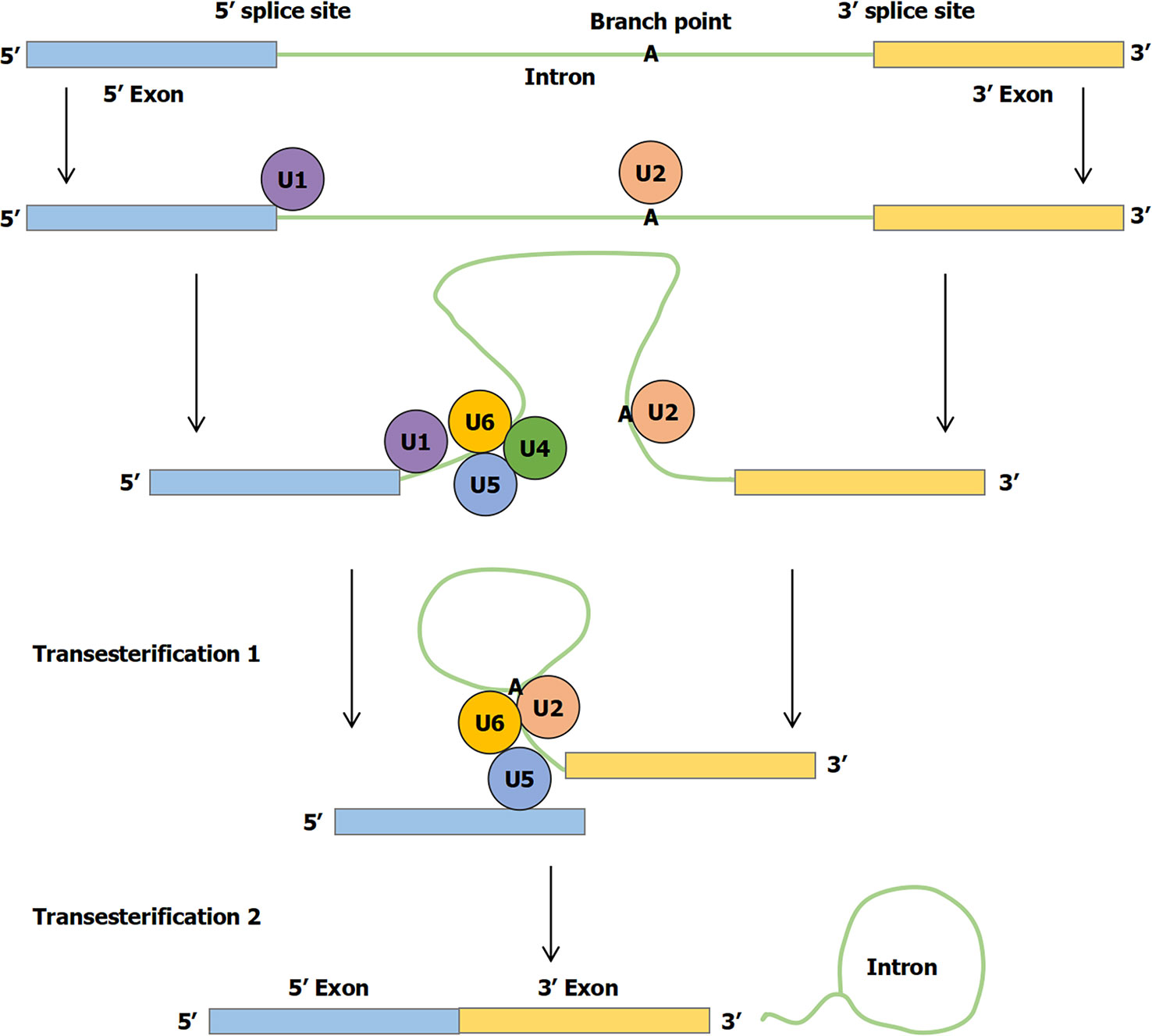
Figure 1 The mechanism of RNA splicing. First, a branch point A nucleotide located near the 3′ splice site of the intron sequence attacks the 5′ splice site and clefts it. The 5′ end of the intronic sequence cleavage is thus covalently linked to this A nucleotide, forming the branched nucleotide. Second, the 3′-OH end of the first exon sequence created in the first step is added to the beginning of the second exon sequence to cleave the RNA molecule at the 3′ splice site. Thus, the two exonic sequences are linked to each other and the intronic sequence is released as a lasso.
On the basis of the Web of Science core collection database, approximately 6–19 articles have been published in this category each year for the last decade (Figure S1A). Hot keywords include IBD, ulcerative colitis, Crohn’s disease, expression, activation, genome-wide association, pathogenesis, splicing variant and splice site mutation (Figure S1B). Some keywords are related together, such as ulcerative colitis, splicing variant, and colorectal cancer, whereas the remaining keywords are related in the other three parts (Figure S1C). In this review, we will summarize the research on alternative splicing events, splicing factors, and splicing mutations in IBD and highlight their role in diseases.
Alternative splicing is associated with the pathogenesis of IBD and may serve as a potential therapeutic target for clinical therapy. Previous studies have reported an association between alternative splicing and the pathogenesis of CD and UC (10, 11). Li’s team demonstrated a novel potential pathophysiological mechanism for alternative splicing associated with CD, presenting the first alternative splicing landscape in patients with CD, suggesting that drugs targeting alternative splicing-related genes [monoamine oxidase A (MAOA), signal transducer and activator of transcription 3 (STAT3), and interferon regulatory factor 1 (IRF1)] and splicing factors can be used for screening and treatment of patients with CD (10). They also presented a study of alternative splicing events in patients with UC, where exon deletion and substitution of the first exon were the two most significantly altered events (11). Immune response–related pathways were significantly enriched in UC-associated alternative splicing events, suggesting a strong association between dysregulation of alternative splicing and pathogenesis of UC. Moreover, histone deacetylase 6 (HDAC6) and lipase A (LIPA) had significant alternative splicing events in patients with UC involved in immune responses and inflammation, indicating that dysregulated alternative splicing of HDAC6 or LIPA may be a potential therapeutic target for patients with UC. In this section, we summarize studies on alternative splicing in adults and children with IBD (Table 1).

Table 1 Studies reporting alternative splicing in IBD of children and adults.
Alternative splicing is associated with the pathogenesis of IBD and may serve as a potential therapeutic target for IBD in adults (12–20). One study showed that the regulation of seven splicing factors (HNRPAB, DUSP11, HNRPH3, SF3B14, SFPQ, SLU7, and SFR2IP) is IBD specific (12). The expression patterns of these seven splicing factors did not share a regulatory pattern with the intron retention patterns of FGD2, PARC, and IER3, indicating that splicing factors have a potential influence on subsequent regulated intron retention in IBD pathogenesis. Years later, their team revealed a close interaction between the microbiome, gene regulation, and splicing architecture in the pathogenesis of IBD (13). Another study found that the second splicing variant of interferon gamma-antisense 1 (IFNG-AS1) is positively correlated with the severity of IBD, and a single-nucleotide polymorphism (SNP) in IFNG-AS1, rs7134599, is associated with the pathophysiology of IBD (14). Furthermore, the researchers found that two patients with IBD have 284 exons with significantly different splicing rates at exons, including CEACAM1, an established IBD risk gene, suggesting that splicing analysis derived from RNA-seq experiments may play an important role in identifying pathogenesis of IBD (15). Recently, a study provided gene expression and alternative splicing data for 445 healthy individuals showing that SNPs are enriched in disease-associated loci, including IBD, resulting in provision of the Colon Transcriptome Explorer web application (16). Another study evaluated mRNA splicing for transcription factor X-box binding protein-1 (XBP1) by quantitative reverse transcriptase real-time PCR to measure relative expression of Th1, Th17, and Treg cytokines, finding that mRNA splicing for transcription factor XBP1 may be involved in immunopathogenesis of IBD (17).
In addition to pathogenesis, alternative splicing may serve as a potential therapeutic target for IBD (18–20). One study showed that the alternatively spliced fibronectin EDA domain is expressed in patients with IBD, whereas it is barely detectable in most normal adult tissues (18). Thus, they identified the alternatively spliced EDA domain of fibronectin as a target for drug delivery applications in IBD. Moreover, different splice variants of myosin light chain kinase (MLCK) (including MLCK1 and MLCK2) could regulate paracellular and extracellular permeability in IBD (19). First-line therapy could be developed to inhibit tumor necrosis factor–like cytokine 1A–mediated MLCK2-dependent bacterial endocytosis, which may serve as a therapeutic target for patients with IBD. Autoantibodies against different splice variants of glycoprotein 2 (GP2) frequently occur in patients with IBD (20). The last study showed that autoimmunity against gut-expressed GP2 splicing variant 4 (GP2#4) resulted in enhanced adhesion of flagellar bacteria to intestinal epithelium and thus may drive the pathophysiology of IBD (20). Blocking or depleting GP2#4-directed autoantibodies with anti-idiotypic antibodies may be a new therapeutic target for IBD.
Alternative splicing is associated with the pathogenesis of CD and may serve as a potential biomarker, predictor of glucocorticoid (GC) resistance, and therapeutic target for CD in adults (10, 21–26, 33). The alternative splicing of insulin-like growth factor 1 (igf1) gene includes IGF-IEa and IGF-IEc (21). Researchers found increased IGF-IEc expression and mechano-growth factor production in patients with fibrostenotic CD, indicating that alternative splicing of igf1 may be related to pathogenesis of fibrostenotic CD (21). Li et al. reanalyzed mRNA-seq data providing a landscape of alternative splicing events in CD, suggesting that alternative splicing disorder may be a mechanism in pathogenesis of CD (10). Another study showed that mRNA expression of C-X-C motif chemokine receptor 3 (CXCR3) splice variants is detected in CD3+ peripheral blood lymphocytes (PBL) of patients with CD, indicating that CXCR3 splicing may play a significant role in pathogenesis of CD (22).
Previous studies found that serum spliceosome-associated protein 130 (SAP130) levels in patients with active CD are significantly higher than in patients with remission CD and controls and changed with different disease activities being significantly correlated with disease severity (23). Furthermore, serum SAP130 level of patients with active CD decreased after 8 weeks of exclusive enteral nutrition (EEN), demonstrating that SAP130 may be a potential biomarker for assessing the severity and clinical efficacy of EEN in adults with CD. A study showed that amount of human GC receptor (hGR) β-mRNA is significantly higher in patients with GC-resistant CD in active stage (24). IL-18 content was directly correlated with hGRβ-mRNA content in patients with active CD with GC resistance, indicating that IL-18 may be involved in the alternative splicing of hGR in patients with CD. It seemed that alternative splicing of hGR may be a predictor of GC resistance in patients with CD.
Moreover, forkhead box P3 (FOXP3) is involved in Janus kinase–signal transducer and activator of transcription (JAK-STAT) signaling pathway in Th17 lymphocytes, leading to CD (Figure 2) (33). FOXP3 is also involved in a pathway in Treg cell inhibition of IBD (Figure 2). Previous research reported that alternative splicing of FOXP3 modulated T-cell differentiation, which may be a therapeutic target for patients with CD (25). Levels of CD44v7 (one of the isoforms alternatively spliced exon of CD44) ligand osteopontin were elevated in CD, inducing IL-6 in human monocytes (26). This indicated that CD44v7 may play an important role in pathogenesis of CD and be a novel therapeutic target for CD. Another study showed that the levels of the spliced form of cylindromatosis lysine 63 deubiquitinase (sCYLD) are increased in colon tissues of patients with CD (27). Ubiquitination and nuclear translocation of SMAD family member 7 (SMAD7) were mediated by sCYLD to reduce transforming growth factor β signaling in T cells, indicating that sCYLD-SMAD7 complex may be a therapeutic target for CD.
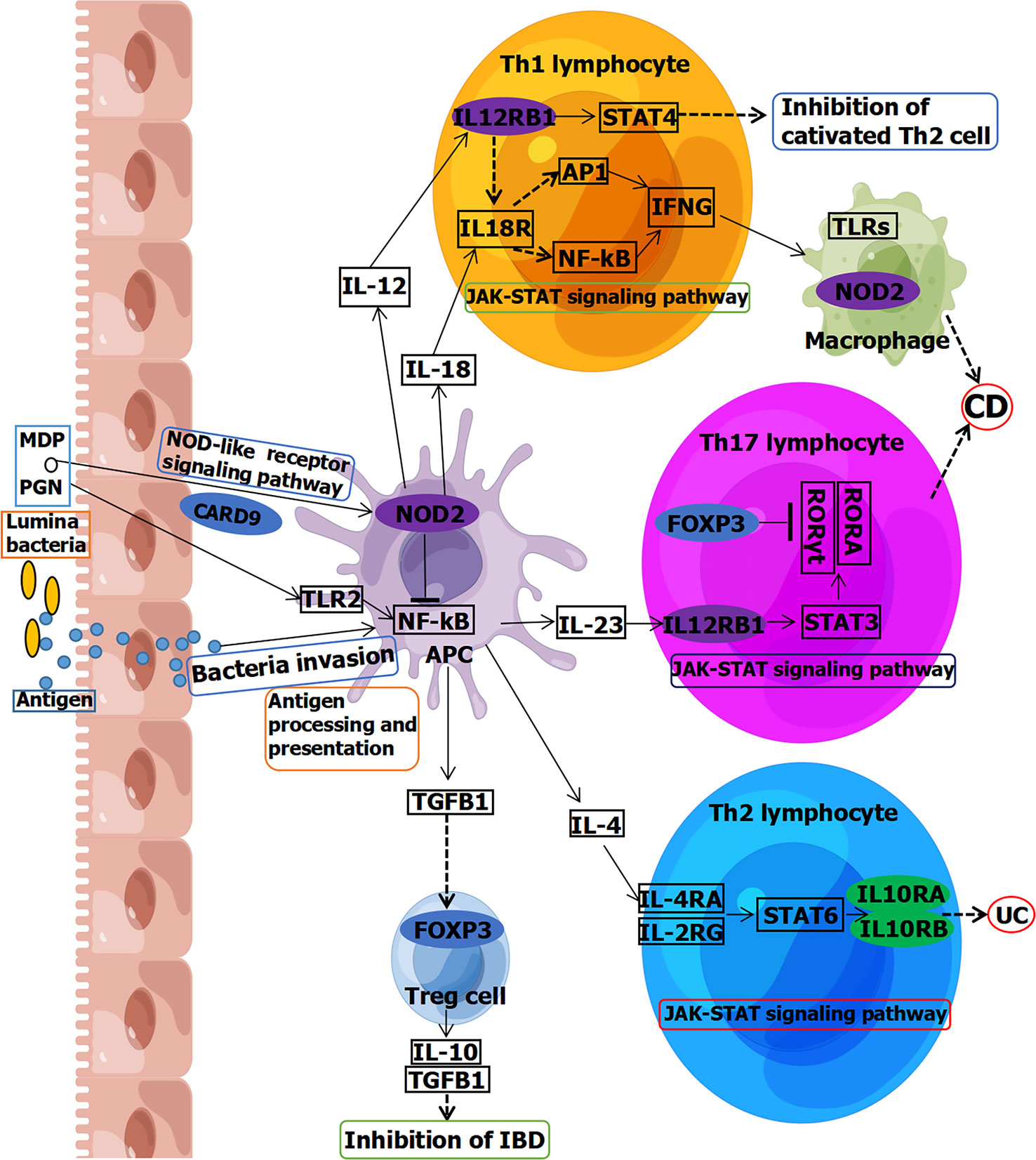
Figure 2 Partial signaling pathways of IBD associated with splicing, including CD and UC. Genes circled in ovals are involved in splicing. Blue is reported in adults. Green is reported in children. Purple is reported both in children and adults. MDP, muramyldipeptide; PGN, peptidoglycans; TLR, Toll-like receptor; NF-kB, nuclear factor–kappa B; APC, antigen-presenting cell; IFNG, gamma interferon; STAT, signal transducer and activator of transcription; JAK, Janus kinase; RORγt, retinoic acid–related orphan receptor gamma; RORA, retinoic acid–related orphan receptor alpha; TGFB1, transforming growth factor beta 1; FOXP3, forkhead box P3; CARD9, caspase recruitment domain family member 9; IL12RB1, interleukin-12 receptor subunit beta 1; NOD2, nucleotide-binding oligomerization domain containing 2; IL10RA, interleukin-10 receptor subunit alpha; IL10RB, interleukin-10 receptor subunit beta. This figure was drawn using Figdraw (ID: YYSUS3f0fd).
Alternative splicing is also associated with pathogenesis of UC and may serve as a valuable biomarker for clinical diagnosis and therapy in adults (11, 28–31). Alternative splicing events, gene expression, and pathways were altered in patients with UC with long and short disease durations, linking alternative splicing to pathogenesis of UC (28). Receptor protein tyrosine phosphatase sigma (PTPRS-encoding PTPσ) knockout mice may spontaneously develop mild colitis (29). Presence of some SNPs was associated with novel splicing that removed a third immunoglobulin-like domain (exon 9) from extracellular portion of PTPσ, potentially altering dimerization or ligand recognition. This indicated that splicing of the PTPRS gene may also be related to pathogenesis of UC. Furthermore, splicing of XBP1 in patients with UC with costimulation was greater than those with single stimulation (30). This indicated that splicing of XBP1 may contribute to pathogenesis of UC. Moreover, a previous study showed that some genes (POLR2A, CDC42, PIK3R1, RAC1, SRC, and MAPK1) are mainly enriched in intercellular items related to cell junction, cell adhesion, actin cytoskeleton, transmembrane receptor signaling pathways, and intracellular items related to RNA splicing, metabolism, and localization (31). These results suggested that these genes related to splicing may be associated with pathogenesis of adults with UC and serve as diagnostic and therapeutic targets of UC. In addition, abnormal regulation of alternative splicing may play a key role in pathogenesis and serve as a potential therapeutic target of UC (11).
Currently, only a few studies have reported on relationship between alternative splicing and IBD in children (15, 31, 32). Researchers found that two patients with IBD have 284 exons with significantly different splicing rates at exons, including CEACAM1, an established IBD risk gene (15). This suggested that splicing analysis derived from RNA-seq experiments may play an important role in identifying pathogenesis of IBD. Synonymous variants of IL10RA have been reported to affect RNA splicing in pediatric patients with CD, which may be associated with pathogenesis of children with CD (32). Because genes (POLR2A, CDC42, PIK3R1, RAC1, SRC, and MAPK1) were mainly enriched in intercellular items related to cell junction, cell adhesion, actin cytoskeleton, transmembrane receptor signaling pathways, and intracellular items related to RNA splicing, metabolism, and localization, splicing of above genes may be associated with pathogenesis of UC, serving as potential biomarkers for clinical diagnosis and therapy in children (31). Alternative splicing has been studied more in adult IBD than in children, indicating a need for further studies on alternative splicing in pediatric IBD.
There is considerable evidence that diseases are associated with abnormal expression of splicing factors (34–37). Many splicing factors have been reported to be expressed in patients with IBD (Table 2) (10–12). Regulatory patterns of seven splicing factors (HNRPAB, DUSP11, HNRPH3, SF3B14, SFPQ, SLU7, and SFR2IP) observed in a study were specific to IBD rather than solely dependent on inflammation (12). SF3B14 and SFPQ showed upregulation in patients with CD but not in those with UC, indicating that the splicing factor expression levels of UC and CD are different and play different roles in different disease subtypes, suggesting that a mechanism of this aspect may serve as a diagnostic marker for IBD in the future.
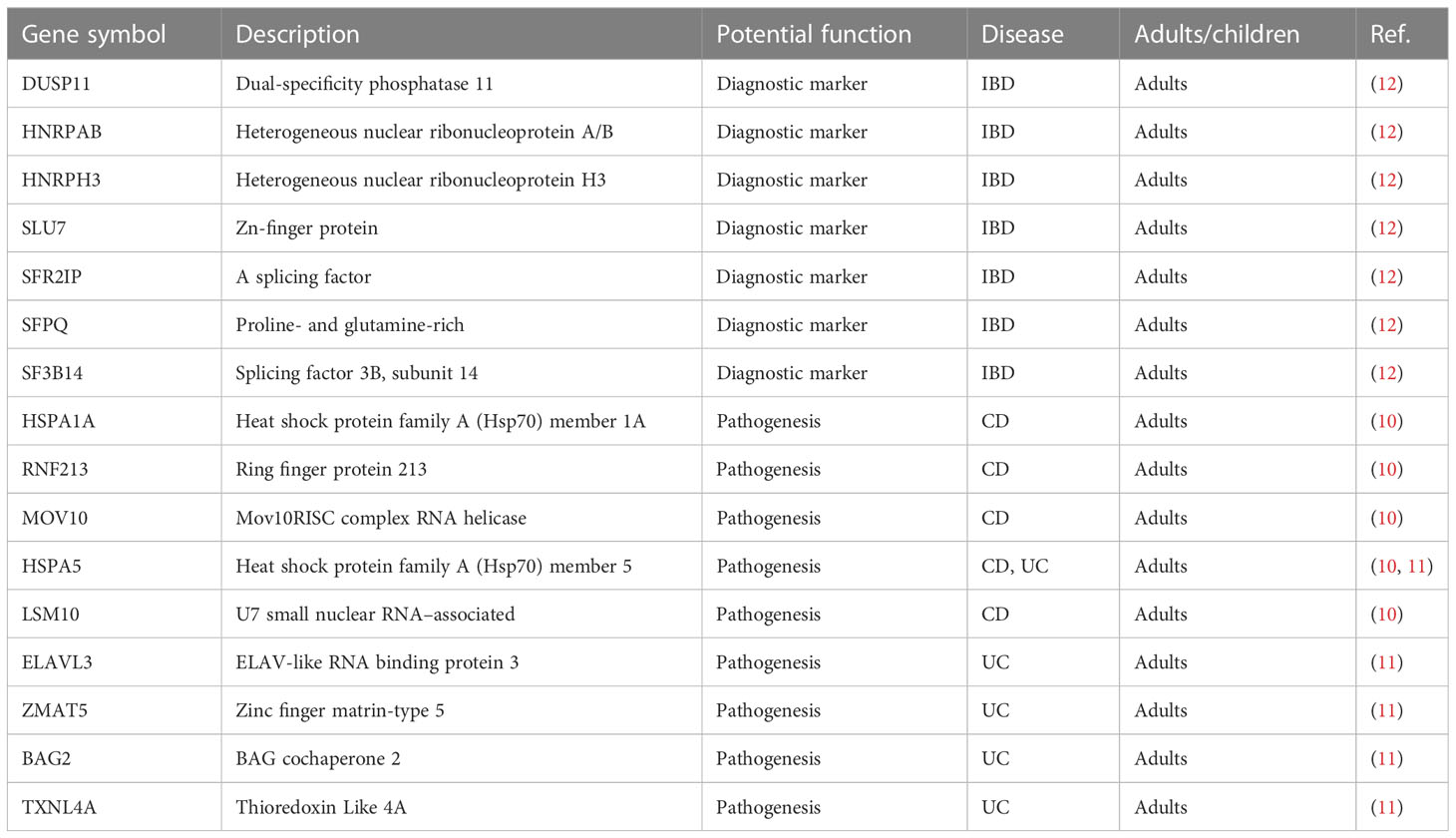
Table 2 Studies reporting alterations of RNA splicing factor expression in IBD.
Splicing factors have also been reported in two subtypes of IBD (10, 11). One study identified five splicing factors (HSPA1A, RNF213, MOV10, HSPA5, and LSM10) that are significantly differentially expressed in patients with CD, playing a key role in the regulation of alternative splicing events in patients with CD (10). Another study also identified five splicing factors (ELAVL3, ZMAT5, BAG2, HSPA5, and TXNL4A) that are significantly differentially expressed in patients with UC, closely related to change in immune response in patients with UC (11). These studies revealed potential splicing factors in mechanism of alternative splicing changes in patients with CD or UC.
To date, more than 200 genes affecting or associated with IBD have been reported, with a total of more than 600 mutation sites reported in the Human Gene Mutation Database (v2022.6) (38, 39). According to genetic classification, there are currently 31 types of IBD, including IBD1 (OMIM #266600), IBD2 (OMIM #601458), and IBD3 (OMIM #604519) (40). Currently, 24 mutations of splicing or associated with splicing have been reported in 16 genes associated with IBD. Among them, genes associated with splicing mutations in adult IBD include CARD9, CUL2, IL12RB1, MSH2, NCF4, and NOD2. In pediatric IBD, they include ANKZF1, CD40LG, CTLA4, IL10RA, IL10RB, IL12RB1, NCF4, NOD2, SKIV2L, STXBP3, TTC37, TTC7A, and WAS (Figure 3, Table 3). Splicing mutations can result in complete exon skipping, intron retention, or the introduction of a new splice site within an exon or intron (72). However, mutations that do not destroy or create splice sites can also activate preexisting pseudosplice sites. These variants can also affect the fine balance of isoforms arising from alternatively spliced exons and thus cause disease.

Figure 3 The structure of the 16 genes associated with IBD (including CD and UC). Red: the mutation sites of splicing or resulting in splicing. Blue, green, orange, purple, and black: the domain locations of every gene from Prosite or Pfam. ZF, zinc finger; AR, ankyrin repeat; CARD, caspase recruitment domain; MutS, DNA mismatch repair protein MutS; SH3, Src homology 3; NACHT, the NAIP, CIITA, HET-E, and TP1 domain with NTPase activity; DSH, dynein stalk head; TPR1, tetratricopeptide repeat 1; WH1, Wiskott–Aldrich syndrome protein homology 1; EVH1, enabled vasodilator-stimulated protein homology 1; CRIB, Cdc42 and Rac interactive binding domain; WH2, Wiskott–Aldrich syndrome protein homology 2. The full names of these genes are shown in Table 3.

Table 3 Mutations of splice or associated with splicing have been reported in IBD of children and adults.
Caspase recruitment domain family member 9 (CARD9), located on chromosome 9q34.3, is a susceptibility gene for IBD that encodes a connexin that integrates signals downstream from pattern recognition receptors and plays a key role in immune responses against microbes (73). To date, it has been reported that a splicing variant of CARD9 (c.1434+1G>C) may affect IBD risk in adults (41–43). CARD9 is involved in the nucleotide-binding domain (NOD)–like receptor signaling pathway, which is involved in the occurrence of IBD (Figure 2) (74). Except for CARD9, there are many susceptibility genes for IBD (75). However, only some of them with splicing mutations are associated with IBD. Meanwhile, cullin 2 (CUL2), located on chromosome 10p11.21, is a protein coding gene, and its splicing site (c.1675-5T>C) has also been reported to affect IBD risk (42).
Furthermore, interleukin-12 receptor subunit beta 1 (IL12RB1), located on chromosome 19p13.11, has a 3′ splicing site mutation (c.1791+2T>G) that has been reported to be associated with pathogenesis of adults with CD (44). IL12RB1 is involved in JAK-STAT signaling pathway in both Th1 and Th17 lymphocytes, leading to CD (Figure 2) (76). MutS homolog 2 (MSH2), located on chromosome 2p21-p16.3, has a splicing variant (c.2006-6T>C) that has been reported to be associated with an excess risk of colorectal cancer for patients with UC (46). Neutrophil cytosolic factor 4 (NCF4), located on chromosome 22q12.3, encodes the p40phox protein and is another susceptibility gene for IBD (77, 78). The splicing variant of NCF4 (c.32+1258T>C) has been reported to be involved in the pathogenesis of CD (47, 48).
Moreover, nucleotide-binding oligomerization domain containing 2 (NOD2, OMIM #605956), located on chromosome 16q12.1, is associated with CD (IBD1, OMIM #266600) (53). NOD2 signaling in macrophages is strongly associated with increased bacterial susceptibility in CD (79). NOD2 is involved in the signaling pathway in both antigen-presenting cells (APCs) and macrophages of CD (Figure 2) (33, 76, 80, 81). A splicing variant of NOD2 (c.2798+158C>T) has been reported to affect CD risk (53, 54). Another splice site of NOD2 (c.74-7T>A) may be associated with the pathogenesis of CD (50, 51). In total, seven splicing mutations from six genes (CARD9, CUL2, IL12RB1, MSH2, NCF4, and NOD2) have been reported to be related to IBD (including CD and UC) in adults (Table 3). They may be involved in pathogenesis or influenced disease risk. NOD2 is directly related to the genetic typing of IBD (IBD1), and further studies on these splicing sites will be of great help to the pathogenesis, diagnosis, and treatment of IBD1.
Ankyrin repeat and zinc finger domain containing 1 (ANKZF1), located on chromosome 2q35, is associated with infantile-onset IBD (56). The splicing variant of ANKZF1 (c.262-97G>A) has been reported to be involved in the pathogenesis of infantile-onset IBD (56). In their study, four infantile-onset patients with IBD carried ANKZF1 mutations on one or two alleles. When only IBD with onset before 6 months of age was considered, the mutation frequency of ANKZF1 in their study population was 100%, indicating that ANKZF1 is indeed important in the pathogenesis of infantile-onset IBD. CD40 ligand (CD40LG), located on chromosome Xq26, is associated with VEO-IBD and EO-IBD (57). The splicing variant of CD40LG (c.410-2A>T) may affect the treatment of VEO-IBD and EO-IBD (58). Meanwhile, cytotoxic T-lymphocyte associated–protein 4 (CTLA4), located on chromosome 2q33, is associated with pediatric IBD (58). The splicing variant of CTLA4 (c.457+1G>A) may affect the treatment of pediatric IBD through regulatory T cells and immune modulation (58). Whereas IL12RB1 is associated with CD in adults, it is also associated with IBD in children (45). The variant of IL12RB1 (c.684C>T) plays a very important role in the splicing process and might be a diagnostic and therapeutic target for VEO-IBD (45). Furthermore, another gene, NCF4, is associated with CD in adults and with IBD in children. Another splicing variant of NCF4 (c.32+1258T>C) may be involved in the pathogenesis of VEO-IBD (Figure 4) (49).
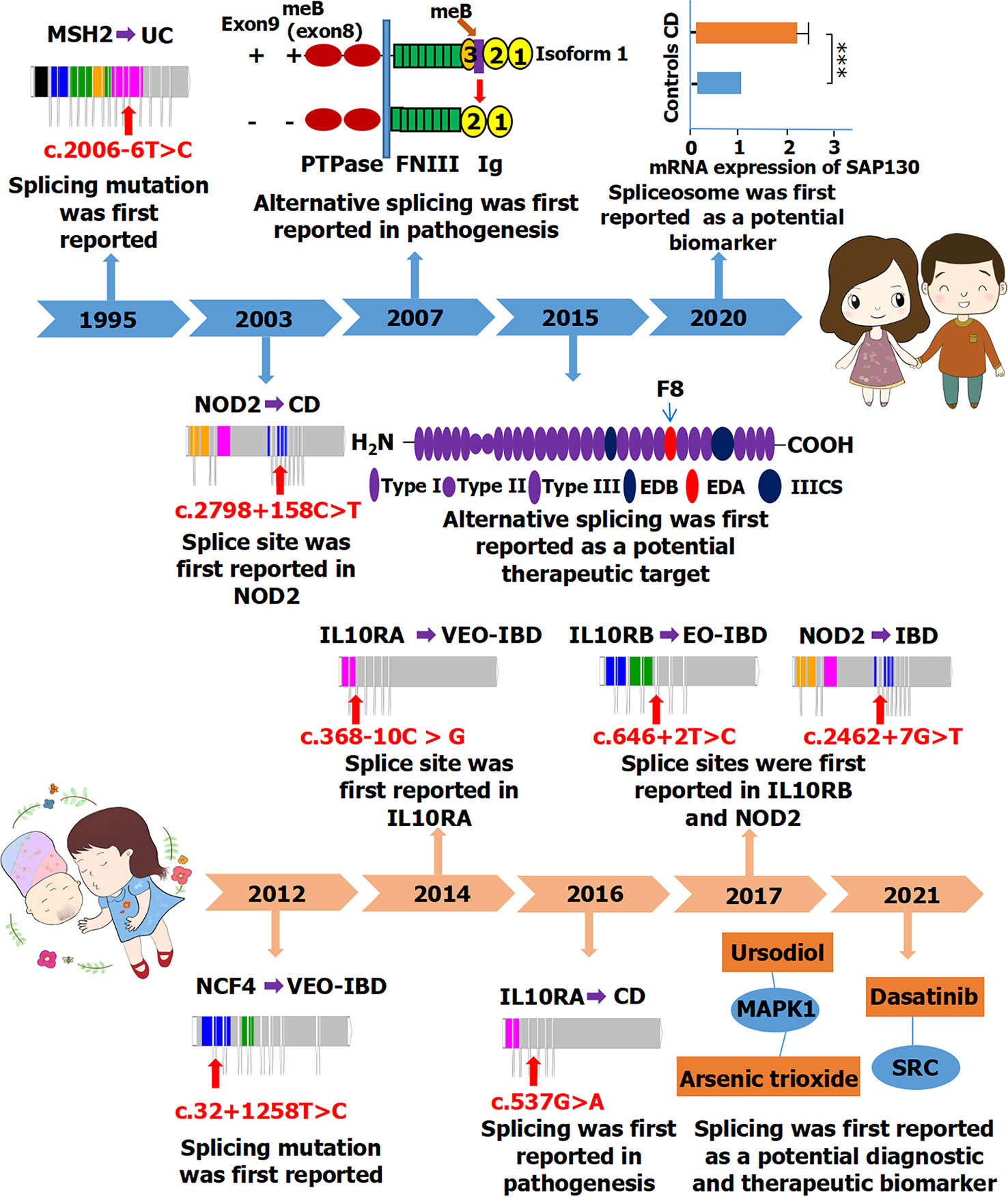
Figure 4 A timeline of alternative splicing and splicing mutations reported in IBD (including CD and UC) between adults (Top) and children (Bottom). PTPase, protein-tyrosine phosphatase; FNIII, fibronectin type III; SAP130, spliceosome-associated protein 130; ***P < 0.001; IIICS, type III connecting segment; MAPK1, mitogen-activated protein kinase 1; SRC, proto-oncogene tyrosine-protein kinase Src. The full names of those genes are shown in Table 3.
In addition, ski2-like RNA helicase (SKIV2L), located on chromosome 6p21, is associated with VEO-IBD and UC in children (67, 68). Whereas the splicing variant of SKIV2L (c.354+5G>A) may be associated with the pathogenesis of UC in children, another splicing variant (c.355-2A>C) may be involved in the pathogenesis of VEO-IBD (67, 68). Syntaxin binding protein 3 (STXBP3), located on chromosome 1p13.3, is associated with VEO-IBD (69). A splicing variant of STXBP3 (c.1029+1G>A) may be associated with the pathogenesis of VEO-IBD (70). Tetratricopeptide repeat domain 7A (TTC7A), located on chromosome 2p21, is also associated with VEO-IBD (70). Two splicing variants of TTC7A (c.844-1G>T and c.1204-2A>G) may be involved in the pathogenesis of VEO-IBD (70). Tetratricopeptide repeat domain 37 (TTC37), located on chromosome 5q15, is associated with VEO-IBD and EO-IBD (57). A splicing variant of TTC37 (c.4497-1G>A) may affect the treatment of VEO-IBD and EO-IBD (57). Wiskott–Aldrich syndrome (WAS), located on chromosome Xp11.4-p11.21, is associated with EO-IBD and VEO-IBD (66, 71). Splicing variants of WAS (c.463+8G>A and c.360+1G>C) may be involved in the pathogenesis of EO-IBD and VEO-IBD, respectively (66, 71).
Moreover, whereas NOD2 is associated with CD (IBD1) in adults, it is also associated with IBD1 in children (52, 71). Three splicing variants of NOD2 (c.74-7T>A, c.2798+158C>T, and c.2462+7G>T) may be involved in the pathogenesis of IBD1 in children (52, 55). Interleukin-10 receptor subunit beta (IL10RB, OMIM #123889), located on chromosome 21q22.11, is associated with IBD25 (OMIM #612567) (82). A splicing variant of IL10RB (c.646+2T>C) may be involved in the pathogenesis of EO-IBD (IBD25) (66). Interleukin-10 receptor subunit alpha (IL10RA, OMIM #146933), located on chromosome 11q23, is associated with IBD28 (OMIM #613148) (59). Splicing variant of IL10RA (c.188+1G>A) may be a pluripotency marker for IBD28 (59). A synonymous variant in IL10RA (c.537G>A) may be associated with the pathogenesis of IBD28 by affecting RNA splicing (32, 60–64). Another splicing variant of IL10RA (c.368-10C>G) may also be associated with the pathogenesis of VEO-IBD (IBD28) (65). Both IL10RA and IL10RB are involved in the JAK-STAT signaling pathway in Th2 lymphocytes, leading to UC (Figure 2) (76).
In the signaling pathway, some genes are involved in the NOD-like receptor signaling pathway, Toll-like receptor signaling pathway, and JAK-STAT signaling pathway connected with CD, including CARD9, NOD2, and IL12RB1, whereas IL10RA and IL10RB are involved in the JAK-STAT signaling pathway connected with UC (Figure 2). Compared with adults, more mutations and genes have been reported in children. This may be attributed to the fact that patients with monogenic IBD before the age of 6 years account for the majority of cases (63.4%), whereas adolescent and adult-onset monogenic IBD account for one-third of cases (83). Approximately, 20 splicing mutations in 13 genes (ANKZF1, CD40LG, CTLA4, IL10RA, IL10RB, IL12RB1, NCF4, NOD2, SKIV2L, STXBP3, TTC37, TTC7A, and WAS) have been reported to be associated with IBD in children (Table 3). Among them, splicing mutations of IL12RB1, NCF4, and NOD2 have been reported both in adults and pediatric IBD. Some may play a role in pathogenesis, whereas others may act as markers or affect treatment. Some may even serve as diagnostic or therapeutic targets. In children, there were two synonymous variants that affected RNA splicing, which could participate in pathogenesis or serve as diagnostic or therapeutic targets.
Alternative splicing may be related to pathogenesis, biomarker for diagnosis, and therapeutic targets for both adults and children with IBD. The study of alternative splicing has been reported more in adults, but children have more splicing mutations and genes associated with IBD. Splicing was first reported in adults with IBD in 1995, whereas studies on splicing in children with IBD only began in the last decade (Figure 4). Some splicing factors have been reported to be involved in regulating alternative splicing events in adult patients with IBD. Thus far, no related splicing factors have been reported in children; therefore, research should be strengthened in this regard.
A total of 24 mutations of splicing or associated with splicing in 16 genes have been reported to be associated with IBD in children or adults. Whereas seven splicing mutations from six genes have been reported to be related with IBD (including CD and UC) in adults, 20 splicing mutations in 13 genes have been reported to be associated with IBD in children. Splicing mutation sites and genes are much more frequently reported in children. Importantly, three of these genes are associated with IBD1, IBD25, and IBD28, including NOD2, IL10RB, and IL10RA, respectively.
Nevertheless, there are no validated splicing events that have a causative component of IBD. There are no validated splicing events that are just a consequence of IBD. No splicing events are employed routinely in the therapy of IBD. There are no validated or approved biomarkers of IBD that employ splicing events and are used by clinicians. No splicing-related events are employed routinely for the direct benefit of the patient with IBD. However, studies have shown that some alternative splicing may serve as potential biomarkers or therapeutic targets for IBD (Table 1) (18–20, 23, 25–27, 31). We suggest that the study of alternative splicing in IBD can be strengthened in the future, especially in the study of three genes (NOD2, IL10RB, and IL10RA). At present, research on alternative splicing in IBD is still limited. Future studies are needed to understand the role of alternative splicing in IBD and to find new pathogenesis, diagnostic, and therapeutic targets for children and adults with IBD. If biomarkers and therapeutic targets for IBD can be found in this field, then it will provide convenience for the diagnosis and treatment of IBD.
JZ, QZ, YZ, YS, YL, MC, SZ, and ZW designed and drafted the article. MC, SZ, and ZW reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Science Technology and Innovation Committee of Shenzhen (2021N062-JCYJ20210324115408023 and No. JCYJ20210324115213036) and Guangdong High-level Hospital Construction Fund of Shenzhen Children’s Hospital (No. ynkt2021-zz38).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1095267/full#supplementary-material
Supplementary Table 1 | Bibliometric analysis of the studies on alternative splicing and inflammatory bowel disease (IBD) (including Crohn’s disease (CD) and ulcerative colitis (UC)) based on the web of science core collection database. A. Histogram with Alternative splicing, IBD (including CD and UC) as keywords for the last decade (2012-2021), based on the Web of Science core collection database. Approximately 6-19 articles have been published in this category each year for the last decade. B. Heatmap of bibliometric analysis, the number of documents with “INFLAMMATORY-BOWEL-DISEASE” “ULCERATIVE-COLITIS” “CROHNS-DISEASE” “EXPRESSION” “ACTIVATION” “GENOME-WIDE ASSOCIATION” “PATHOGENESIS” “MECHANISMS” “SPLICE VARIANT” “SPLICE-SITE MUTATION” as the keywords in the Web of Science core collection database. Color saturation indicates the strength of citation keyword. C. Network diagram for bibliometric analysis of alternative splicing in studies of IBD (including CD and UC) based on the Web of Science core collection database. Nodes represent unique keywords; node size is proportional to the number of references; node colors indicate modules. Some keywords are related together, such as Ulcerative colitis, splicing variant and colorectal cancer, while the remaining keywords are related in other three parts. Note: NF kappa B, nuclear factor- kappa B; ER stress, endoplasmic reticulum stress.
1. GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Gastroenterol Hepatol (2020) 5(1):17–30. doi: 10.1016/S2468-1253(19)30333-4
2. Agrawal M, Spencer EA, Colombel JF, Ungaro RC. Approach to the management of recently diagnosed inflammatory bowel disease patients: a user's guide for adult and pediatric gastroenterologists. Gastroenterology (2021) 161(1):47–65. doi: 10.1053/j.gastro.2021.04.063
3. Park KT, Ehrlich OG, Allen JI, Meadows P, Szigethy EM, Henrichsen K, et al. The cost of inflammatory bowel disease: an initiative from the crohn's & colitis foundation. Inflammation Bowel Dis (2020) 26(1):1–10. doi: 10.1093/ibd/izz104
4. Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc (2019) 94(1):155–65. doi: 10.1016/j.mayocp.2018.09.013
5. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature (2020) 578(7796):527–39. doi: 10.1038/s41586-020-2025-2
6. Blencowe BJ. Alternative splicing: new insights from global analyses. Cell (2006) 126(1):37–47. doi: 10.1016/j.cell.2006.06.023
7. Mercuri E, Pera MC, Scoto M, Finkel R, Muntoni F. Spinal muscular atrophy - insights and challenges in the treatment era. Nat Rev Neurol (2020) 16(12):706–15. doi: 10.1038/s41582-020-00413-4
8. Zhou JL, Zhao YZ, Wang SS, Chen MX, Zhou S, Chen C. RNA Splicing: a versatile regulatory mechanism in pediatric liver diseases. Front Mol Biosci (2021) 8:725308. doi: 10.3389/fmolb.2021.725308
9. Stanley RF, Abdel-Wahab O. Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat Cancer (2022) 3(5):536–46. doi: 10.1038/s43018-022-00384-z
10. Li D, Liang Y, Lu J, Tan Y. An alternative splicing signature in human crohn's disease. BMC Gastroenterol (2021) 21(1):420. doi: 10.1186/s12876-021-02001-2
11. Li D, Tan Y. Dysregulation of alternative splicing is associated with the pathogenesis of ulcerative colitis. BioMed Eng Online (2021) 20(1):121. doi: 10.1186/s12938-021-00959-4
12. Häsler R, Kerick M, Mah N, Hultschig C, Richter G, Bretz F, et al. Alterations of pre-mRNA splicing in human inflammatory bowel disease. Eur J Cell Biol (2011) 90(6-7):603–11. doi: 10.1016/j.ejcb.2010.11.010
13. Häsler R, Sheibani-Tezerji R, Sinha A, Barann M, Rehman A, Esser D, et al. Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut (2017) 66(12):2087–97. doi: 10.1136/gutjnl-2016-311651
14. Rankin CR, Shao L, Elliott J, Rowe L, Patel A, Videlock E, et al. The IBD-associated long noncoding RNA IFNG-AS1 regulates the balance between inflammatory and anti-inflammatory cytokine production after T-cell stimulation. Am J Physiol Gastrointest Liver Physiol (2020) 318(1):G34–40. doi: 10.1152/ajpgi.00232.2019
15. Berger K, Somineni H, Prince J, Kugathasan S, Gibson G. Altered splicing associated with the pathology of inflammatory bowel disease. Hum Genomics (2021) 15(1):47. doi: 10.1186/s40246-021-00347-y
16. Díez-Obrero V, Dampier CH, Moratalla-Navarro F, Devall M, Plummer SJ, Díez-Villanueva A, et al. Genetic effects on transcriptome profiles in colon epithelium provide functional insights for genetic risk loci. Cell Mol Gastroenterol Hepatol (2021) 12(1):181–97. doi: 10.1016/j.jcmgh.2021.02.003
17. Iacomino G, Rotondi Aufiero V, Iannaccone N, Melina R, Giardullo N, De Chiara G, et al. IBD: role of intestinal compartments in the mucosal immune response. Immunobiology (2020) 225(1):151849. doi: 10.1016/j.imbio.2019.09.008
18. Bootz F, Schmid AS, Neri D. Alternatively spliced EDA domain of fibronectin is a target for pharmacodelivery applications in inflammatory bowel disease. Inflammation Bowel Dis (2015) 21(8):1908–17. doi: 10.1097/MIB.0000000000000440
19. Pai YC, Weng LT, Wei SC, Wu LL, Shih DQ, Targan SR, et al. Gut microbial transcytosis induced by tumor necrosis factor-like 1A-dependent activation of a myosin light chain kinase splice variant contributes to IBD. J Crohns Colitis. (2020) 15(2):258–72. doi: 10.1093/ecco-jcc/jjaa165
20. Derer S, Brethack AK, Pietsch C, Jendrek ST, Nitzsche T, Bokemeyer A, et al. Inflammatory bowel disease-associated GP2 autoantibodies inhibit mucosal immune response to adherent-invasive bacteria. Inflammation Bowel Dis (2020) 26(12):1856–68. doi: 10.1093/ibd/izaa069
21. Li C, Vu K, Hazelgrove K, Kuemmerle JF. Increased IGF-IEc expression and mechano-growth factor production in intestinal muscle of fibrostenotic crohn's disease and smooth muscle hypertrophy. Am J Physiol Gastrointest Liver Physiol (2015) 309(11):G888–99. doi: 10.1152/ajpgi.00414.2014
22. Manousou P, Kolios G, Drygiannakis I, Pyrovolaki K, Bourikas L, Papadaki HA, et al. Expression of a splice variant of CXCR3 in crohn's disease patients; indication for a lymphocyte–epithelial cell interaction. J Gastroenterol Hepatol (2008) 23(12):1823–33. doi: 10.1111/j.1440-1746.2008.05486.x
23. Gong W, Guo K, Zheng T, Fang M, Xie H, Li W, et al. Preliminary exploration of the potential of spliceosome-associated protein 130 for predicting disease severity in crohn's disease. Ann N Y Acad Sci (2020) 1462(1):128–38. doi: 10.1111/nyas.14240
24. Towers R, Naftali T, Gabay G, Carlebach M, Klein A, Novis B. High levels of glucocorticoid receptors in patients with active crohn's disease may predict steroid resistance. Clin Exp Immunol (2005) 141(2):357–62. doi: 10.1111/j.1365-2249.2005.02846.x
25. Mailer RK, Joly AL, Liu S, Elias S, Tegner J, Andersson J. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep (2015) 5:14674. doi: 10.1038/srep14674
26. Wittig BM, Sabat R, Holzlöhner P, Witte-Händel E, Heilmann K, Witte K, et al. Absence of specific alternatively spliced exon of CD44 in macrophages prevents colitis. Mucosal Immunol (2018) 11(3):846–60. doi: 10.1038/mi.2017.98
27. Tang Y, Reissig S, Glasmacher E, Regen T, Wanke F, Nikolaev A, et al. Alternative splice forms of CYLD mediate ubiquitination of SMAD7 to prevent TGFB signaling and promote colitis. Gastroenterology (2019) 156(3):692–707.e7. doi: 10.1053/j.gastro.2018.10.023
28. Low END, Mokhtar NM, Wong Z, Raja Ali RA. Colonic mucosal transcriptomic changes in patients with long-duration ulcerative colitis revealed colitis-associated cancer pathways. J Crohns Colitis. (2019) 13(6):755–63. doi: 10.1093/ecco-jcc/jjz002
29. Muise AM, Walters T, Wine E, Griffiths AM, Turner D, Duerr RH, et al. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol (2007) 17(14):1212–8. doi: 10.1016/j.cub.2007.06.013
30. Li N, Wang XM, Jiang LJ, Zhang M, Li N, Wei ZZ, et al. Effects of endoplasmic reticulum stress on the expression of inflammatory cytokines in patients with ulcerative colitis. World J Gastroenterol (2016) 22(7):2357–65. doi: 10.3748/wjg.v22.i7.2357
31. Xiu MX, Liu YM, Chen GY, Hu C, Kuang BH. Identifying hub genes, key pathways and immune cell infiltration characteristics in pediatric and adult ulcerative colitis by integrated bioinformatic analysis. Dig Dis Sci (2021) 66(9):3002–14. doi: 10.1007/s10620-020-06611-w
32. Oh SH, Baek J, Liany H, Foo JN, Kim KM, Yang SC, et al. A synonymous variant in IL10RA affects RNA splicing in paediatric patients with refractory inflammatory bowel disease. J Crohns Colitis. (2016) 10(11):1366–71. doi: 10.1093/ecco-jcc/jjw102
33. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res (2019) 2019:7247238. doi: 10.1155/2019/7247238
34. López-Cánovas JL, Del Rio-Moreno M, García-Fernandez H, Jiménez-Vacas JM, Moreno-Montilla MT, Sánchez-Frias ME, et al. Splicing factor SF3B1 is overexpressed and implicated in the aggressiveness and survival of hepatocellular carcinoma. Cancer Lett (2021) 496:72–83. doi: 10.1016/j.canlet.2020.10.010
35. Goulielmaki E, Tsekrekou M, Batsiotos N, Ascensão-Ferreira M, Ledaki E, Stratigi K, et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for r-loop processing. Nat Commun (2021) 12(1):3153. doi: 10.1038/s41467-021-23505-1
36. Gárate-Rascón M, Recalde M, Jimenez M, Elizalde M, Azkona M, Uriarte I, et al. Splicing factor SLU7 prevents oxidative stress-mediated hepatocyte nuclear factor 4α degradation, preserving hepatic differentiation and protecting from liver damage. Hepatology (2021) 74(5):2791–807. doi: 10.1002/hep.32029
37. Wang Y, Yan Q, Mo Y, Liu Y, Wang Y, Zhang S, et al. Splicing factor derived circular RNA circCAMSAP1 accelerates nasopharyngeal carcinoma tumorigenesis via a SERPINH1/c-myc positive feedback loop. Mol Cancer (2022) 21(1):62. doi: 10.1186/s12943-022-01502-2
38. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet (2017) 136(6):665–77. doi: 10.1007/s00439-017-1779-6
39. Hagihara Y, Yoshimatsu Y, Mikami Y, Takada Y, Mizuno S, Kanai T. Epigenetic regulation of T helper cells and intestinal pathogenicity. Semin Immunopathol (2019) 41(3):379–99. doi: 10.1007/s00281-019-00732-9
40. Zhang ZZ, Zhang Y, He T, Sweeney CL, Baris S, Karakoc-Aydiner E, et al. Homozygous IL37 mutation associated with infantile inflammatory bowel disease. Proc Natl Acad Sci U S A. (2021) 118(10):e2009217118. doi: 10.1073/pnas.2009217118
41. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet (2011) 43(11):1066–73. doi: 10.1038/ng.952
42. Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PloS Genet (2013) 9(9):e1003723. doi: 10.1371/journal.pgen.1003723
43. Huang H, Fang M, Jostins L, Umićević Mirkov M, Boucher G, Anderson CA, et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature (2017) 547(7662):173–8. doi: 10.1038/nature22969
44. Khoshnevisan R, Nekooei-Marnany N, Klein C, Kotlarz D, Behnam M, Ostadi V, et al. IL-12Rβ1 deficiency corresponding to concurrency of two diseases, mendelian susceptibility to mycobacterial disease and crohn's disease. J Clin Tuberc Other Mycobact Dis (2019) 17:100123. doi: 10.1016/j.jctube.2019.100123
45. Dara N, Nemati S, Teimourian S, Imanzadeh F, Hosseini A, Tajalli S, et al. Diagnostic challenges in the early onset of inflammatory bowel disease: a case report. Int J Mol Cell Med (2018) 7(4):251–7. doi: 10.1038/nature22969
46. Brentnall TA, Rubin CE, Crispin DA, Stevens A, Batchelor RH, Haggitt RC, et al. A germline substitution in the human MSH2 gene is associated with high-grade dysplasia and cancer in ulcerative colitis. Gastroenterology (1995) 109(1):151–5. doi: 10.1016/0016-5085(95)90280-5
47. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi: 10.1038/ng2032
48. Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal crohn's disease in a population-based cohort. Genes Immun (2008) 9(6):561–5. doi: 10.1038/gene.2008.49
49. Muise AM, Xu W, Guo CH, Walters TD, Wolters VM, Fattouh R, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut (2012) 61(7):1028–35. doi: 10.1136/gutjnl-2011-300078
50. King K, Sheikh MF, Cuthbert AP, Fisher SA, Onnie CM, Mirza MM, et al. Mutation, selection, and evolution of the crohn disease susceptibility gene CARD15. Hum Mutat (2006) 27(1):44–54. doi: 10.1002/humu.20264
51. Rivas MA, Avila BE, Koskela J, Huang H, Stevens C, Pirinen M, et al. Insights into the genetic epidemiology of crohn's and rare diseases in the ashkenazi Jewish population. PloS Genet (2018) 14(5):e1007329. doi: 10.1371/journal.pgen.1007329
52. Girardelli M, Basaldella F, Paolera SD, Vuch J, Tommasini A, Martelossi S, et al. Genetic profile of patients with early onset inflammatory bowel disease. Gene (2018) 645:18–29. doi: 10.1016/j.gene.2017.12.029
53. Sugimura K, Taylor KD, Lin YC, Hang T, Wang D, Tang YM, et al. A novel NOD2/CARD15 haplotype conferring risk for crohn disease in ashkenazi jews. Am J Hum Genet (2003) 72(3):509–18. doi: 10.1086/367848
54. Chua KH, Hilmi I, Ng CC, Eng TL, Palaniappan S, Lee WS, et al. Identification of NOD2/CARD15 mutations in Malaysian patients with crohn's disease. J Dig Dis (2009) 10(2):124–30. doi: 10.1111/j.1751-2980.2009.00374.x
55. Andreoletti G, Shakhnovich V, Christenson K, Coelho T, Haggarty R, Afzal NA, et al. Exome analysis of rare and common variants within the NOD signaling pathway. Sci Rep (2017) 7:46454. doi: 10.1038/srep46454
56. van Haaften-Visser DY, Harakalova M, Mocholi E, van Montfrans JM, Elkadri A, Rieter E, et al. Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J Biol Chem (2017) 292(19):7904–20. doi: 10.1074/jbc.M116.772038
57. Lega S, Pin A, Arrigo S, Cifaldi C, Girardelli M, Bianco AM, et al. Diagnostic approach to monogenic inflammatory bowel disease in clinical practice: a ten-year multicentric experience. Inflammation Bowel Dis (2020) 26(5):720–7. doi: 10.1093/ibd/izz178
58. Crowley E, Warner N, Pan J, Khalouei S, Elkadri A, Fiedler K, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology (2020) 158(8):2208–20. doi: 10.1053/j.gastro.2020.02.023
59. Ma Y, Zhang H, Zhang S, Dong R, Yang X, Li Y, et al. An integration-free iPSC line (SDQLCHi012-a) derived from a patient with inflammatory bowel disease- 28 carrying compound heterozygote mutations in IL10RA gene. Stem Cell Res (2019) 41:101577. doi: 10.1016/j.scr.2019.101577
60. Yanagi T, Mizuochi T, Takaki Y, Eda K, Mitsuyama K, Ishimura M, et al. Novel exonic mutation inducing aberrant splicing in the IL10RA gene and resulting in infantile-onset inflammatory bowel disease: a case report. BMC Gastroenterol (2016) 16:10. doi: 10.1186/s12876-016-0424-5
61. Suzuki T, Sasahara Y, Kikuchi A, Kakuta H, Kashiwabara T, Ishige T, et al. Targeted sequencing and immunological analysis reveal the involvement of primary immunodeficiency genes in pediatric IBD: a Japanese multicenter study. J Clin Immunol (2017) 37(1):67–79. doi: 10.1007/s10875-016-0339-5
62. Lv H, Qiao B, Fang L, Yang L, Wang Q, Wu S, et al. Neonatal crohn's disease with oral ulcer as the first symptom caused by a compound heterozygote mutation in IL-10RA: a case report. Hereditas (2019) 156:38. doi: 10.1186/s41065-019-0114-8
63. Nemati S, Teimourian S, Tabrizi M, Najafi M, Dara N, Imanzadeh F, et al. Very early onset inflammatory bowel disease: investigation of the IL-10 signaling pathway in Iranian children. Eur J Med Genet (2017) 60(12):643–9. doi: 10.1016/j.ejmg.2017.08.016
64. Peng K, Qian X, Huang Z, Lu J, Wang Y, Zhou Y, et al. Umbilical cord blood transplantation corrects very early-onset inflammatory bowel disease in Chinese patients with IL10RA-associated immune deficiency. Inflammation Bowel Dis (2018) 24(7):1416–27. doi: 10.1093/ibd/izy028
65. Murugan D, Albert MH, Langemeier J, Bohne J, Puchalka J, Järvinen PM, et al. Very early onset inflammatory bowel disease associated with aberrant trafficking of IL-10R1 and cure by T cell replete haploidentical bone marrow transplantation. J Clin Immunol (2014) 34(3):331–9. doi: 10.1007/s10875-014-9992-8
66. Petersen BS, August D, Abt R, Alddafari M, Atarod L, Baris S, et al. Targeted gene panel sequencing for early-onset inflammatory bowel disease and chronic diarrhea. Inflammation Bowel Dis (2017) 23(12):2109–20. doi: 10.1097/MIB.0000000000001235
67. Ashton JJ, Andreoletti G, Coelho T, Haggarty R, Batra A, Afzal NA, et al. Identification of variants in genes associated with single-gene inflammatory bowel disease by whole-exome sequencing. Inflammation Bowel Dis (2016) 22(10):2317–27. doi: 10.1097/MIB.0000000000000890
68. Kammermeier J, Drury S, James CT, Dziubak R, Ocaka L, Elawad M, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease–evaluation and prospective analysis. J Med Genet (2014) 51(11):748–55. doi: 10.1136/jmedgenet-2014-102624
69. Ouahed J, Kelsen JR, Spessott WA, Kooshesh K, Sanmillan ML, Dawany N, et al. Variants in STXBP3 are associated with very early onset inflammatory bowel disease, bilateral sensorineural hearing loss and immune dysregulation. J Crohns Colitis. (2021) 15(11):1908–19. doi: 10.1093/ecco-jcc/jjab077
70. Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology (2014) 146(4):1028–39. doi: 10.1053/j.gastro.2014.01.015
71. Esmaeilzadeh H, Bordbar MR, Dastsooz H, Silawi M, Fard MAF, Adib A, et al. A novel splice site mutation in WAS gene in patient with wiskott-Aldrich syndrome and chronic colitis: a case report. BMC Med Genet (2018) 19(1):123. doi: 10.1186/s12881-018-0647-0
72. Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet (2005) 42(10):737–48. doi: 10.1136/jmg.2004.029538
73. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22(6):598–605. doi: 10.1038/nm.4102
74. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol (2006) 6(1):9–20. doi: 10.1038/nri1747
75. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet (2015) 47(9):979–86. doi: 10.1038/ng.3359
76. Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in crohn's disease. Front Immunol (2021) 12:622934. doi: 10.3389/fimmu.2021.622934
77. Hong M, Ye BD, Yang SK, Jung S, Lee HS, Kim BM, et al. Immunochip meta-analysis of inflammatory bowel disease identifies three novel loci and four novel associations in previously reported loci. J Crohns Colitis. (2018) 12(6):730–41. doi: 10.1093/ecco-jcc/jjy002
78. Wu PB, Dai JF, Wang Q, Zhang G, Tan SY, Li M, et al. Association between NCF4 rs4821544T/C polymorphism and inflammatory bowel disease risk in Caucasian: a meta-analysis. Inflammation Res (2015) 64(10):825–31. doi: 10.1007/s00011-015-0866-1
79. Tsay TB, Chang CJ, Chen PH, Hsu CM, Chen LW. Nod2 mutation enhances NF-kappa b activity and bacterial killing activity of macrophages. Inflammation (2009) 32(6):372–8. doi: 10.1007/s10753-009-9145-z
80. Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest (2007) 117(3):514–21. doi: 10.1172/JCI30587
81. Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and crohn's disease. Gut (2008) 57(12):1682–9. doi: 10.1136/gut.2007.135053
82. Yazdani R, Moazzami B, Madani SP, Behniafard N, Azizi G, Aflatoonian M, et al. Candidiasis associated with very early onset inflammatory bowel disease: first IL10RB deficient case from the national Iranian registry and review of the literature. Clin Immunol (2019) 205:35–42. doi: 10.1016/j.clim.2019.05.007
Keywords: alternative splicing, Crohn’s disease, inflammatory bowel disease, splicing factor, splicing mutation, transcriptome, ulcerative colitis
Citation: Zhou J, Zhang Q, Zhao Y, Song Y, Leng Y, Chen M, Zhou S and Wang Z (2023) The regulatory role of alternative splicing in inflammatory bowel disease. Front. Immunol. 14:1095267. doi: 10.3389/fimmu.2023.1095267
Received: 11 November 2022; Accepted: 31 March 2023;
Published: 21 April 2023.
Edited by:
Satoshi Tanaka, Kyoto Pharmaceutical University, JapanReviewed by:
Robert Häsler, University Medical Center Schleswig-Holstein, GermanyCopyright © 2023 Zhou, Zhang, Zhao, Song, Leng, Chen, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoxia Wang, d2FuZ3poYW94aWExOTY5QDE2My5jb20=; Shaoming Zhou, emhvdXNtMTVkQGFsaXl1bi5jb20=; Moxian Chen, Y214MjAwOTkyMDczNEBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.