
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 07 March 2023
Sec. T Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1095257
 Etienne Masle-Farquhar1,2*§
Etienne Masle-Farquhar1,2*§ Yogesh Jeelall3*†§
Yogesh Jeelall3*†§ Jacqueline White1,2Julia Bier1,2
Jacqueline White1,2Julia Bier1,2 Elissa K. Deenick1,2
Elissa K. Deenick1,2 Robert Brink1,2
Robert Brink1,2 Keisuke Horikawa3‡
Keisuke Horikawa3‡ Christopher Carl Goodnow1,4‡
Christopher Carl Goodnow1,4‡Introduction: Germline CARD11 gain-of-function (GOF) mutations cause B cell Expansion with NF-κB and T cell Anergy (BENTA) disease, whilst somatic GOF CARD11 mutations recur in diffuse large B cell lymphoma (DLBCL) and in up to 30% of the peripheral T cell lymphomas (PTCL) adult T cell leukemia/lymphoma (ATL), cutaneous T cell lymphoma (CTCL) and Sezary Syndrome. Despite their frequent acquisition by PTCL, the T cell-intrinsic effects of CARD11 GOF mutations are poorly understood.
Methods: Here, we studied B and T lymphocytes in mice with a germline Nethyl-N-nitrosourea (ENU)-induced Card11M365K mutation identical to a mutation identified in DLBCL and modifying a conserved region of the CARD11 coiled-coil domain recurrently mutated in DLBCL and PTCL.
Results and discussion: Our results demonstrate that CARD11.M365K is a GOF protein that increases B and T lymphocyte activation and proliferation following antigen receptor stimulation. Germline Card11M365K mutation was insufficient alone to cause B or T-lymphoma, but increased accumulation of germinal center (GC) B cells in unimmunized and immunized mice. Card11M365K mutation caused cell-intrinsic over-accumulation of activated T cells, T regulatory (TREG), T follicular (TFH) and T follicular regulatory (TFR) cells expressing increased levels of ICOS, CTLA-4 and PD-1 checkpoint molecules. Our results reveal CARD11 as an important, cell-autonomous positive regulator of TFH, TREG and TFR cells. They highlight T cell-intrinsic effects of a GOF mutation in the CARD11 gene, which is recurrently mutated in T cell malignancies that are often aggressive and associated with variable clinical outcomes.
Nuclear factor-κB (NF-κB) signaling downstream of the B or T cell receptor (BCR or TCR) requires the MAGUK family member Caspase Recruitment Domain-containing protein 11 (CARD11) (1) and its phosphorylation by protein kinase C (PKC) (2, 3). CARD11 forms a scaffold to recruit signaling partners B cell lymphoma/leukemia 10 (BCL10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) (4) into the CBM complex, which relays antigen receptor signals to NF-κB and activator protein 1 (AP1)-cJUN (1, 5–8).
CARD11 function is required for B and T cell immunity. In mice, germline CARD11 deficiency or loss-of-function (LOF) disrupt B cell development and humoral immunity (9–12), disrupt T cell NF-κB activation, proliferation and IL-2 production following TCR and CD28 stimulation (9–12) and perturb thymic (though not peripheral [13)] Treg development in response to TCR (13, 14) and IL-2 signaling (15). In hypomorphic Card11 “unmodulated” mice, partial reduction of TCR-NF-κB signaling causes a recessive phenotype of hyper-IgE and atopy (10) driven by reduced Treg numbers and a gradual and selective expansion of IL-4-producing TH2 cells (16). Similarly, dominant-negative heterozygous CARD11 mutations in humans skew T cells towards a TH2 phenotype and cause severe atopic disease (17, 18) as well as common variable immunodeficiency (CVID), cutaneous viral infections, lung disease and characteristics reminiscent of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) (19). Homozygous germline CARD11 truncations cause combined immunodeficiency, a developmental block at the B cell transitional stage and hypogammaglobulinemia (20, 21).
CARD11 gain-of-function (GOF) mutations also cause pathology. Heterozygous germline GOF CARD11 mutations cause B cell Expansion with NF-κB and T cell Anergy (BENTA), a rare monogenic disease characterized by B cell lymphocytosis and aspects of primary immunodeficiency including recurrent and opportunistic infections (22–29). In addition, somatic CARD11 GOF mutations recur in germinal center B cell-type (GCB) and activated B cell-like (ABC)-diffuse large B cell lymphoma (DLBCL) (30, 31), an aggressive subset of DLBCL (32) characterized by constitutive NF-κB activation (33, 34). ABC-DLBCL harbor recurrent somatic GOF mutations in the BCR-NF-κB pathway (30, 35), including CARD11 (31), and require CARD11 and the CBM complex for their survival in vitro (34). The recurrent B-lymphoma CARD11 mutations cluster in the CARD and coiled-coil (CC) domains and disrupt the intrinsic ‘auto-inhibited’ conformation of CARD11 (36), uncoupling it from activating signals and causing it to form aggregates with other CARD11 proteins, MALT1 and BCL10, and thus activate NF-κB and AP1/cJUN (31, 36, 37).
CARD11 is the fourth most mutated gene in adult T cell leukemia/lymphoma (ATL (38, 39);), a CD4 T cell neoplasm (40) that arises exclusively in individuals previously infected with Human T cell Lymphotropic Virus type 1 (HTLV-1 (41, 42);). Up to 90% of ATL (38) harbor somatic mutations in TCR-NFκB signaling molecules including PLCG1, PRKCB, VAV1 and CARD11. 24% of ATL harbor CARD11 mutations predicted to constitutively activate CARD11, and clustered in the CC domain or around the E626 hotspot in the PKC-responsive inhibitory domain (38). Moreover, 12% of ATL harbor CARD11 gene amplifications and 8% harbor small intragenic deletions in the CARD11 inhibitory domain (38). Up to 22.5% of Cutaneous T Cell Lymphomas (CTCL) harbor CARD11 amplifications (43) and up to 24% of Sezary syndrome, the aggressive form of CTCL, harbor CARD11 GOF mutations clustered in the CC domain or surrounding the E626 hotspot (44–46). PTCL are often associated with very poor outcomes (47, 48) and are thought derived from activated CD4 and TREG cells (49, 50).
The study of CARD11 mutations in B- and T-lymphomas is complicated by the many genomic alterations acquired by these cancer cells (38, 51). Previous studies have used mouse models to reveal B cell-intrinsic effects of GOF CARD11.L232LI (52), CARD11.L251P (53), CARD11.K215M or CARD11.E134G (54). These studies reported variable effects on B cells of different Card11 mutations, and the wide spectrum of GOF CARD11 mutations have diverse biochemical effects (55). The graded, variable effects of hypomorphic Card11 mutation within distinct T cell populations could not be predicted from knockout studies (16). The effects of hypermorphic CARD11 mutations are thus similarly hard to predict a priori. Collectively, the above observations highlight open questions regarding qualitative differences in NF-κB activation by CARD11 mutations, and possible discontinuity in the graded effects of CARD11 GOF mutations within different cell types. Crucially, to our knowledge, no studies have reported T cell-intrinsic effects of CARD11 GOF, despite the striking recurrence of somatic CARD11 GOF mutations in PTCL.
Here, we addressed these open questions by analyzing B and T lymphocytes in mice with a germline Card11M365K mutation identical to CARD11M365K previously identified in DLBCL (30) and modifying a conserved region of the CC domain recurrently mutated in B-lymphomas (31) and T-lymphomas (38, 39, 44–46) (Figure 1A). CARD11.M365K increased activation and proliferation of B and T lymphocytes following antigen-receptor stimulation. Card11M365K mice had increased numbers of GC B cells before and at multiple timepoints during a T cell-dependent response to immunization. Card11M365K mutation was insufficient to cause lymphoma, or B cell lymphocytosis as observed in individuals with BENTA disease. However, Card11M365K mutation caused mutant allele dose-dependent, cell-autonomous accumulation of T follicular (TFH), T regulatory (TREG) and T follicular regulatory (TFR) cells over-expressing stimulatory and inhibitory checkpoint molecules. Our findings add to our understanding of CARD11 as a critical signaling protein in lymphocytes. They reveal TFH, TREG and TFR cells as T cell populations particularly sensitive to CARD11 signaling, and help to explain the recurrence of somatic GOF CARD11 mutations in aggressive human T-lymphomas arising from CD4, TREG and TFH cells.
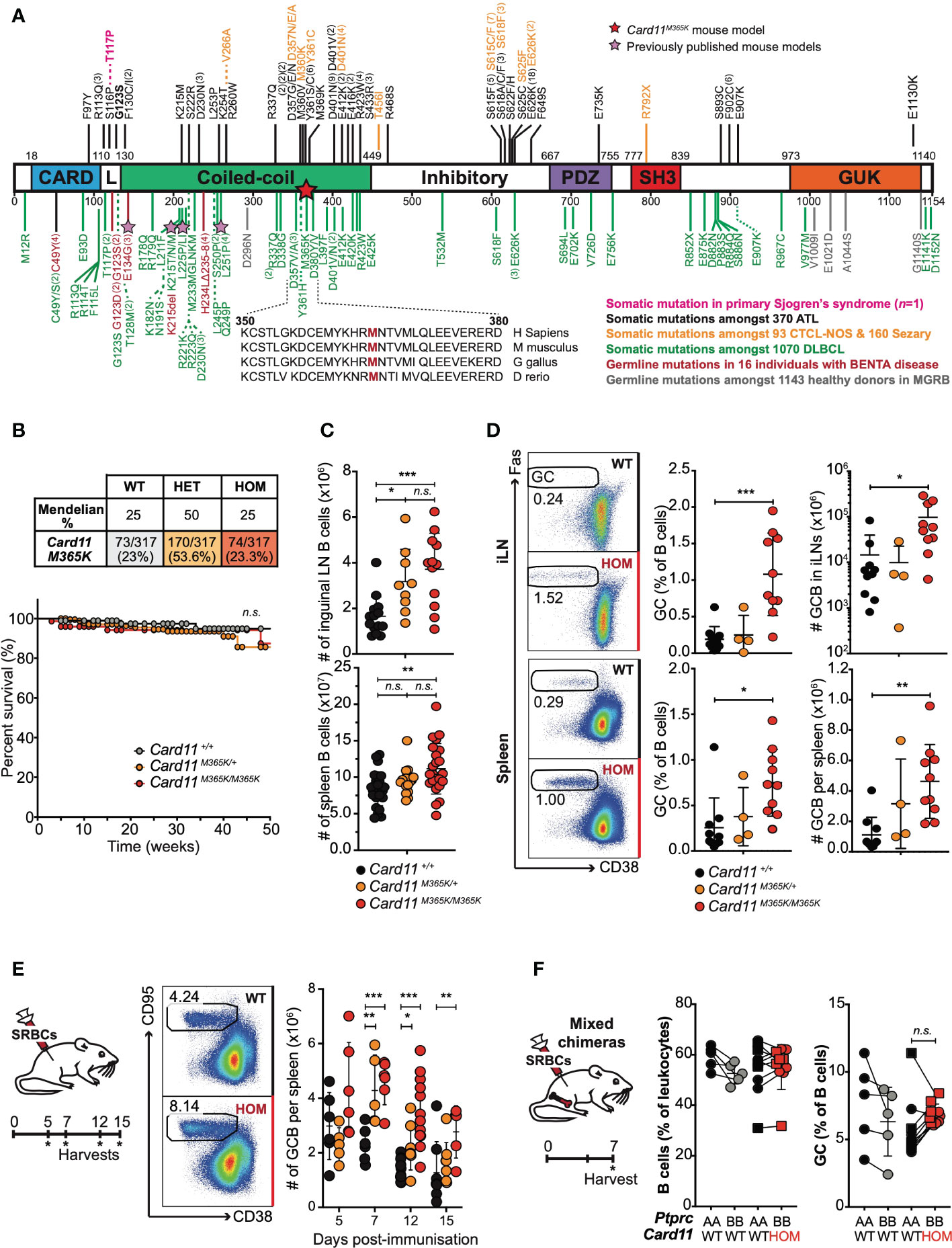
Figure 1 Gain-of-function Card11M365K germline mutant mice have increased numbers of germinal center B cells. (A). Schematic of CARD11 protein domains showing location of germline GOF mutations in healthy donors from the MGRB database (grey) or individuals with BENTA disease (red (23–29);), and somatic GOF mutations identified in adult T cell leukemia/lymphoma (ATL (38, 56–58); black), cutaneous T cell lymphoma not otherwise-specified (CTCL-NOS) or Sezary syndrome (orange (43–46, 59–61);), diffuse large B cell lymphoma (DLBCL (30, 31, 51, 62–64); green) or in one individual with primary Sjögren’s syndrome (pink (65);). Stars indicate the Card11M365K or other, previously published (52–54), mouse models of CARD11 GOF. (B). Top, Expected and observed numbers and percentages of offspring of the indicated genotypes from intercrossed heterozygous parents. Statistical testing for no difference relative to an expected 1WT:2HET:1HOM ratio p=0.4328 by Chi-Square test with n=2 degrees of freedom. Bottom, Kaplan-Meier survival curve for mice of the indicated genotypes, calculated using the product-limit method accounting for censored mice. (C). Symbols denote total number of B cells in the inguinal lymph nodes (top) or spleen (bottom) from individual mice of the indicated genotypes. (D). Left, Representative flow cytometric analysis showing the percentage of CD38low CD95+ germinal center (GC) B cells gated on splenic B cells in non-immunized mice. Right, percentage or total number of GC B cells in the inguinal lymph nodes (iLN; top) or spleen (bottom) from mice of the indicated genotypes. (E). Left: Schematic of experimental workflow. Middle: Representative flow cytometric analysis showing the percentage of GC B cells gated on splenic B cells in mice 7 days post-immunization with sheep red blood cells (SRBCs). Right: Total number per spleen of GC B cells 5, 7, 12 or 15 days post-SRBC immunization. (F). Rag1KO/KO Card11+/+ mice were irradiated and reconstituted with Ptprca/a Card11+/+ (black fill) bone marrow in a 1:1 mixture with Ptprcb/b Card11+/+ (grey fill) or Card11M365K/M365K (red fill) bone marrow, and sacrificed 7 days post-SRBC immunization. Graphs show B cells as a percentage of Ptprca/a or Ptprcb/b splenic leukocytes, or GC B cells as a percentage of Ptprca/a or Ptprcb/b B cells, in mice that received bone marrow of the indicated genotypes. (C-E). Statistical comparisons made by t-test, corrected for multiple comparisons using the Holm-Sidak method. Data are representative of n > 2 independent experiments with n > 5 mice per group. (E). Data are pooled from 3 independent experiments. Comparison made after excluding the one high outlier. (F). Statistical comparisons made by paired t-test. not significant (n.s) p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001.
All animals care, housing and experiments were performed in accordance with approved protocols of: (1) the ANU National University Animal Experimentation Ethics Committee, for mice on a C57BL/6 NCrl background; (2) the Garvan Institute of Medical Research/St Vincent’s Hospital Animal Ethics Committee, for mice on a C57BL/6 JAusb background. All experiments conformed to the current guidelines from the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Within independent experiments, Card11 wild-type and mutant animals were sex- and age-matched.
Card11M365K mice harbor a germline A to T nucleotide substitution at position 140,889,709 on chromosome 5, resulting in a methionine to lysine M365K substitution in the highly conserved region of the coiled-coil domain of CARD11. Card11M365K mice were obtained by exome sequencing of first-generation offspring of C57BL/6 mice exposed to N-ethyl-N-nitrosourea (ENU; databases.apf.edu.au/mutations) and bred to homozygosity on a C57BL/6 Ncrl background. Card11M365K mice were rederived onto a C57BL/6 JAusb background upon transfer from the Australian National University (ANU) Australian Phenomics Facility (APF) to Australian BioResources (ABR; MossVale, Australia).
Card11loco mice harbor 3 distinct single-nucleotide variants in Card11 introns 2, 10 and 20 that cause a complete loss of CARD11 protein expression (56). Card11loco mice were also identified by exome sequencing of first-generation offspring of C57BL/6 mice exposed to ENU (databases.apf.edu.au/mutations). The mice were bred to homozygosity and maintained on a C57BL/6 NCrl background.
C57BL/6 NCrl, C57BL/6 JAusb, B6.JSL-PtprcaPepcb (CD45.1) and B6.129S7-Rag1tm1Mom/J (Rag1KO/KO) mice were purchased from ABR.
Single-cell suspensions were prepared from mouse spleen, bone marrow, inguinal lymph nodes, peritoneal cavity and blood. 1-4 x 106 cells in PBS 2% FCS were transferred into appropriate wells of a 96-well U bottom plate. To prevent non-specific antibody binding, cells were incubated with Fc blocking antibody for 20 min at 4°C in the dark. Cells were then incubated with antibodies for 30 min, on ice and in the dark. To fix cells, they were incubated in 10% formalin (Sigma-Aldrich) for 15 min at 4°C, and washed and resuspended in PBS 2% FCS. To stain for intracellular nuclear proteins, cells were fixed and permeabilised using the manufacturer’s instructions and the eBioscience Transcription Factor Staining kit. Stained single-cell suspensions were acquired on the BD LSRFortessa™.
Where appropriate, following extracellular antibody staining, immune populations were sorted by fluorescence-activated cell sorting (FACS) on a FACS Aria III (BD Biosciences).
Antibodies used for flow cytometric study of mouse organs are listed in Table 1.

Table 1 Antibodies used for flow cytometric analyses of mouse hematopoietic cells.
To evaluate the effect of the CARD11.M365K substitution, we used a retrovirus gene transfer and culture system to introduce into primary activated B cells the following: CARD11.M365K or as controls wild-type CARD11, BENTA-associated (27–29) CARD11.G123S, CARD11.E134G or empty vector expressing EGFP only.
Briefly, replication-defective retrovirus particles were produced by the Pheonix ecotropic helper-free retrovirus packaging cell line (ATCC; CRL-3214), and transduction efficiency measured by flow cytometric measurement of EGFP expression. C57BL/6 B cells were stimulated with 10µg/mL goat anti-mouse IgM (Jackson ImmunoResearch) and 10µg/mL anti-CD40 (FGK4.5; BioXCell) for 24 hours, followed by spin-infection with retrovirus supernatant containing DOTAP (Roche). The cells were then cultured in fresh RPMI 10µg/mL anti-CD40 for 36 hours, washed with RPMI and resuspended in cRPMI at a density of 106 cells/mL.
The number of live EGFP+ cells was determined by hemocytometer counting of trypan blue–negative cells in each culture, and flow cytometric analysis of the same cells.
Approximately 20 x 106 total splenocytes were incubated for 5 min at room temperature in 1 mL RPMI-1640 (Gibco) containing Cell Trace Violet (CTV; Invitrogen) at a final concentration of 20 μM, followed by three washes in complete RPMI (RPMI-1640 containing 10% heat-inactivated fetal calf serum (HI-FCS), 2% Penicillin-Streptomycin-Glutamine (Gibco), 0.1% 50 mM 2-Mercaptoethanol). CTV-labelled splenocytes were plated at a density of 1 x 106 cells per mL and incubated for 3 to 5 days in complete RPMI alone or containing 10 μg/mL anti-CD3 and 10 μg/mL anti-CD28. Cell divisions were enumerated by flow cytometric measurements of the fluorescence intensity of CTV.
CARD11 mutations M365K and G123S were introduced into the corresponding mouse Card11 sequence using PCR-based site-directed mutagenesis. The coding sequences for Card11 and its variants were fused with the mutant ecDHFR sequence (kindly provided by Dr Wandless, Stanford university) in mammalian expression vector pcDNA3.1+ (66). HEK293 cells were transfected with expression vectors for ecDHFR-CARD11 mutants and reporter plasmids expressing firefly luciferase and Renilla luciferase under NF-kB and thymidine kinase promoters, respectively (pGL4.32 and pGL4.74 from Promega). The expression of CARD11 variants was induced by the addition of 10 mM trimethoprim (TMP). The transfected cells were lysed 5 hr after TMP addition, and luciferase activity was measured by Dual-Luciferase Reporter Assay (Promega).
Sorted Card11M365K mutant or wild-type naïve CD4 T cells were sorted to high purity by FACS, and cultured in flat bottom 96-well plates coated with 4 μg/mL anti-CD3 (BioLegend), in RPMI1640 (Life technologies) supplemented with 10% heat inactivated FCS (Life technologies), 5×10-5 M 2-ME, 0.1mM non-essential amino acids, 1mM sodium pyruvate, 10mM HEPES, 100u/mL penicillin, 100ug/mL Streptomycin, 100ug/mL Noromycin (all from Sigma) at a density of 0.5 ×106 cells/mL.
The naïve CD4 T cells were cultured for 4 days in the following polarizing conditions: Th0 (1 μg/mL anti-CD28, 5 μg/mL anti-TGFβ, 5 μg/mL anti-IL-4, 5 μg/mL anti-IFNγ); Th1 (1 μg/mL anti-CD28, 5 μg/mL anti-TGFβ, 5 μg/mL anti-IL-4, 10ng/mL IL-12); Th2 (10ng/mL IL-4, 1 μg/mL anti-CD28, 5 μg/mL anti-TGFβ, 5 μg/mL anti-IFNγ); Th17 (20ng/mL IL-6, 1ng/mL human TGFβ, 5 μg/mL anti-IFNγ, 5 μg/mL anti-IL-4, 1 μg/mL anti-CD28).
After 4 days of culture, cells were stimulated with PMA (50ng/mL) and ionomycin (375ng/mL) for 6 hrs. Brefeldin A (10 μg/mL) was added to each well after 2 hours of stimulation. Cells were harvested, washed and stained with Zombie Aqua Viability dye (BioLegend), fixed with 2% formalin, permeabilized with saponin (0.1%), and stained intracellularly with mAbs directed against TNFα, IFNγ, IL17A, IL-2, IL5, IL-4.
Age- and sex-matched Card11+/+ Rag1KO/KO C57BL/6J recipient mice were irradiated with one dose of 425 Rad from an X-ray source (X-RAD 320 Biological Irradiator, PXI). Recipient mice were then intravenously injected with 4 x 106 bone marrow cells consisting of a 1:1 mixture of Card11+/+ C57BL6 CD45.1+ (Ptprca/a) bone marrow cells and CD45.2+ (Ptprcb/b) bone marrow cells that were Card11+/+ or Card1M365K/M365K. 7 weeks were allowed for immune reconstitution before intravenous immunization of recipient mice with 2 x 108 SRBCs. The immunized chimeric mice were sacrificed 7 days post-immunization.
8-12 weeks old Card11M365K mice were sacrificed and single-cell suspensions prepared from their spleens. Splenic CD4 T cells were isolated by incubation with anti-CD4 biotin antibody and positive enrichment by manual magnetic-activated cell sorting (MACS) using LS columns (Miltenyi Biotec). 3-4 x 106 Card11+/+ or Card11M365K/M365K CD4 T cells were intravenously transferred into each recipient mouse: either into C57BL6.CD45.1+ recipients where donor cells could be isolated based on CD45.1/2 expression, or in an independent experiment into Rag1KO/KO mice that lack mature B and T cells (67). Recipient mice were treated with intraperitoneal (i.p.) injection of 200 μg anti-mouse PD-1 (clone RMP1-14; BioXCell) or rat IgG2a anti-trinitrophenol isotype control (clone 2A3; BioXCell) at days 0, 2 and 5 post-CD4 T cell transfer. Recipient mice were sacrificed 7 days post-injection, and blood and spleen harvested for analysis.
Statistical analysis of flow cytometric experiments was performed using the GraphPad Prism 6 software (GraphPad, San Diego, USA). A one-tailed unpaired Student’s t-test with Welch’s correction was used for comparisons between two normally distributed groups. An unpaired student’s t-test, corrected for multiple comparisons using the Holm-Sidak method, was used for comparisons of more than two groups. Differences between paired measurements were analyzed by paired t-test. In all graphs presented, the error bars represent the mean and standard deviation. * p < 0.05, ** p < 0.01, *** p < 0.001.
We identified the novel Card11M365K mouse strain by exome sequencing of first-generation offspring of C57BL/6 mice exposed to the mutagen N-ethyl-N-Nitrosourea (ENU). Card11M365K mutant mice carry an A to T nucleotide substitution at position 140,889,709 on Chromosome 5, resulting in a methionine to lysine change at amino acid 365 (Figure 1A).
To determine the effects in mouse B cells of Card11M365K mutation relative to known GOF Card11 mutations, we used a retroviral gene transfer and culture system to transduce Card11M365K into primary activated B cells (Supplementary Figure 1). As controls, B cells were otherwise transduced with an empty vector expressing EGFP only, expressing wild-type Card11 or Card11G123S, found in patients with BENTA disease, DLBCL and ATL (27–29, 31), or expressing Card11E134G found in several patients with BENTA disease (27–29). Card11M365K-transduced B cells expressed lower B220 and higher CD86 (Supplementary Figure 1A) cell-surface levels compared to control vector-transduced B cells, indicative of increased NF-κB activation in these cells. Card11M365K-transduced B cells expressed CD86 and B220 at levels intermediate between Card11E134G- and Card11G123S-transduced cells, and over a period of four days in culture, Card11M365K-transduced B cells accumulated in numbers intermediate between Card11E134G- and Card11G123S-transduced cells (Supplementary Figure 1B). CARD11M365K was previously shown to enhance NF-κB activity in a luciferase assay (30). To validate this and directly measure the effects of M365K and G123S mutations on NF-κB signalling, we utilized our previously published (65) luciferase reporter method. Following trimethoprim-induced expression, M365K and G123S mutant CARD11 caused a mean 5-fold and 11-fold higher induction of the NF-κB luciferase reporter, respectively, relative to that induced by wild-type CARD11 (Supplementary Figure 1C). Collectively, these results indicate that M365K mutation causes CARD11 GOF intermediate between that caused by E134G or G123S.
To test the effects on B cells of Card11M365K mutation within an otherwise normal gene, we measured survival and proliferation of splenic B cells from Card11M365K/+ relative to Card11+/+ mice. As an additional control, we included splenic B cells from homozygous Card11loco/loco mice harboring 3 distinct single-nucleotide variants that cause a complete loss of CARD11 protein expression (68). Over a period of 5 days in the absence of stimulation, the percentage of live Card11+/+ versus Card11M365K/+ B-lymphocytes decreased at the same rate, whilst live Card11loco/loco B cells decreased in frequency more rapidly (Supplementary Figure 1D). CARD11.M365K therefore does not enhance B cell survival in absence of stimulation. Similar results were obtained following stimulation with a 1 μg/mL sub-mitogenic dose of anti-IgM (Supplementary Figure 1E).
To measure proliferation following stimulation, we labelled splenic B cells with Cell Trace Violet (CTV). Relative to Card11+/+ B cells, Card11M365K/+ cells increased in size faster and Card11loco/loco cells more slowly, following stimulation with different concentrations of anti-IgM (Supplementary Figure 1E). B cells stimulated with 10 μg/mL anti-IgM divided up to 5 times and a mildly increased percentage of Card11M365K/M365K B cells divided 3 or more times relative to Card11+/+ cells, whereas 80% of Card11loco/loco B cells failed to divide at all (Supplementary Figure 1F). The mean percentage of divided cells was 70% for Card11M365K/M365K, 62% for Card11M365K/+ and 57% for Card11+/+ B cells. By contrast, only 27% of Card11loco/loco B cells had divided (Supplementary Figure 1G). Given the small number of WT CD4 T cells assessed, we were unable to conclude that these effects were statistically significant.
CARD11.M365K is thus a mild GOF protein that increases BCR-stimulated activation, survival and to a small extent proliferation.
To determine the effects of Card11M365K mutation in vivo, we analyzed Card11M365K mice on a C57BL/6 JAusb or C57BL/6 Ncrl background. All results presented herein were consistent between backgrounds and unless specified otherwise, all figures present data from C57BL/6 JAusb mice. Following inter-cross of heterozygous mutant mice, Card11M365K/+ and Card11M365K/M365K mice were detected at expected Mendelian frequencies at time of weaning and genotyping (Figure 1B). Heterozygous and homozygous mutant mice developed no obvious pathologies and had comparable weight and survival to wild-type mice over a period of up to 50 weeks (Figure 1B). Germline Card11M365K mutation therefore appears insufficient to cause overt pathology in mice.
Given the recurrence of somatic CARD11 GOF mutations in B lymphomas (31), and the effects of germline CARD11 GOF mutations on B cells in mice and humans (22–29, 52–54), we assessed B cell populations in the bone marrow, spleen and lymph nodes of wild-type and Card11M365K mutant mice. Card11+/+, Card11M365K/+ and Card11M365K/M365K mice had comparable percentages of Linneg Sca-1pos c-Kitpos (LSK) stem cells in the bone marrow (Supplementary Figure 2A). 5-7 week-old Card11M365K mutant mice had a reduced percentage of CD5pos CD11bpos CD23low CD43high peritoneal cavity B1a cells. Interestingly, this difference waned with age (Supplementary Figure 2B). Card11M365K mutant mice also had comparable percentages of bone marrow leukocytes of the B220pos B-lineage, and within these of IgMneg IgDneg precursor, IgMpos IgDint immature or IgMlow IgDpos mature recirculating B cells (Supplementary Figure 2C), and comparable percentages of precursor B cells with a CD43high CD24neg pre-pro-, CD43int CD24int pro- or CD43low CD24pos pre-B phenotype (Supplementary Figure 2C).
Notably, Card11M365K mutant mice had increased cellularity and increased percentage of B leukocytes in the spleen and inguinal lymph nodes (Figure 1C, Supplementary Figure 2D). Card11M365K mutant mice had normal numbers of CD93+ transitional and CD93- mature B cell populations (Supplementary Figure 2E), but though unimmunized, had an increased percentage and number of germinal center (GC) B cells in both spleen and lymph nodes (Figure 1D). We therefore studied the effect of Card11M365K mutation on T cell-dependent GC responses, by immunizing Card11M365K mice with sheep red blood cells (SRBCs) and sacrificing them 5, 7, 12 or 15 days later. Relative to wild-type controls, Card11M365K/M365K mice had increased numbers of B220pos CD38low CD95pos GC B cells at days 7, 12 and 15 post-immunization (Figure 1E).
To test whether CARD11.M365K drives GC B cell accumulation cell-autonomously or rather secondary to dysregulation of T cells or other hematopoietic cells, we generated mixed chimeras wherein a fraction of all hematopoietic cells had mutant Card11M365K/M365K and the remainder had wild-type Card11. Card11+/+ Rag1KO/KO mice were irradiated and transplanted with an equal mixture of Card11M365K/M365K Ptprcb/b and control Card11+/+ Ptprca/a bone marrow. As an additional control, another set of mixed chimeras received an equal mixture of Card11+/+ Ptprca/b and Card11+/+ Ptprca/a bone marrow. All chimeras were immunized with sheep red blood cells (SRBCs) and sacrificed 7 days later. Flow cytometric analysis revealed no significant difference in frequency of B cells or of germinal center B cells of Card11+/+ versus Card11M365K/M365K donor origin (Figure 1F). Card11M365K/M365K thus provides no striking cell-autonomous advantage to GC B cells, 7 days post-SRBC immunization in this model.
Based on the recurrence of somatic CARD11 GOF mutations in PTCL (38, 39, 43–46), we hypothesized that Card11M365K mutation would dysregulate T cells. Following flow cytometric analysis of T cell populations, we observed a mutant allele gene dose-dependent increase in percentage (but not total number) of CD62L- CD44+ effector memory CD8 and CD4 T cells in the spleen and lymph nodes of Card11M365K mutant relative to wild-type mice (Figure 2A). The increased fraction of effector memory CD8 T cells was not accompanied by changes in their surface expression of CX3CR1, KLRG1, NKG2D or by changes in their granularity, whereas by contrast an increased fraction of Card11-mutant effector memory CD8 T cells expressed high levels of CD69 (Supplementary Figure 3A). We observed no change in fraction of CD8 T cells expressing the cytotoxic effector molecule granzyme B (Supplementary Figure 3B). Unimmunized Card11M365K/+ and to a greater extent Card11M365K/M365K mice had an accumulation of TCRβ+ CD3+ CD4+ CXCR5high PD-1high T follicular helper (TFH)-like cells (Figure 2B). These accumulating Card11-mutant TFH-like cells expressed homogenously higher cell-surface levels of ICOS and some but not all expressed higher cell-surface levels of PD-1, relative to wild-type cells (Figure 2C). To test whether germline Card11M365K mutation also increases accumulation of TFH cells during T cell-dependent responses, we analyzed the same mice described earlier at 5, 7, 12 and 15 days post-immunization with SRBCs. Card11M365K/M365K mice had an increased frequency and total number of splenic TFH cells at days 7, 12 and 15 (Figure 2D). Interestingly, they also had a significant accumulation of TCRβ+ CD3+ CD4+ CXCR5+ PD-1+ CD25+ FoxP3+ T follicular regulatory (TFR) cells (Figure 2E). As expected, the TFH cells were Bcl-6high, ICOShigh and CD44+ and the TFR cells were FoxP3+, Bcl-6+, ICOShigh, CD44+ and Blimp-1high (Figure 2F). Similar to our observations in unimmunized mice, the accumulating Card11M365K/M365K TFH and TFR cells expressed homogeneously higher levels of ICOS relative to their Card11+/+ counterparts.
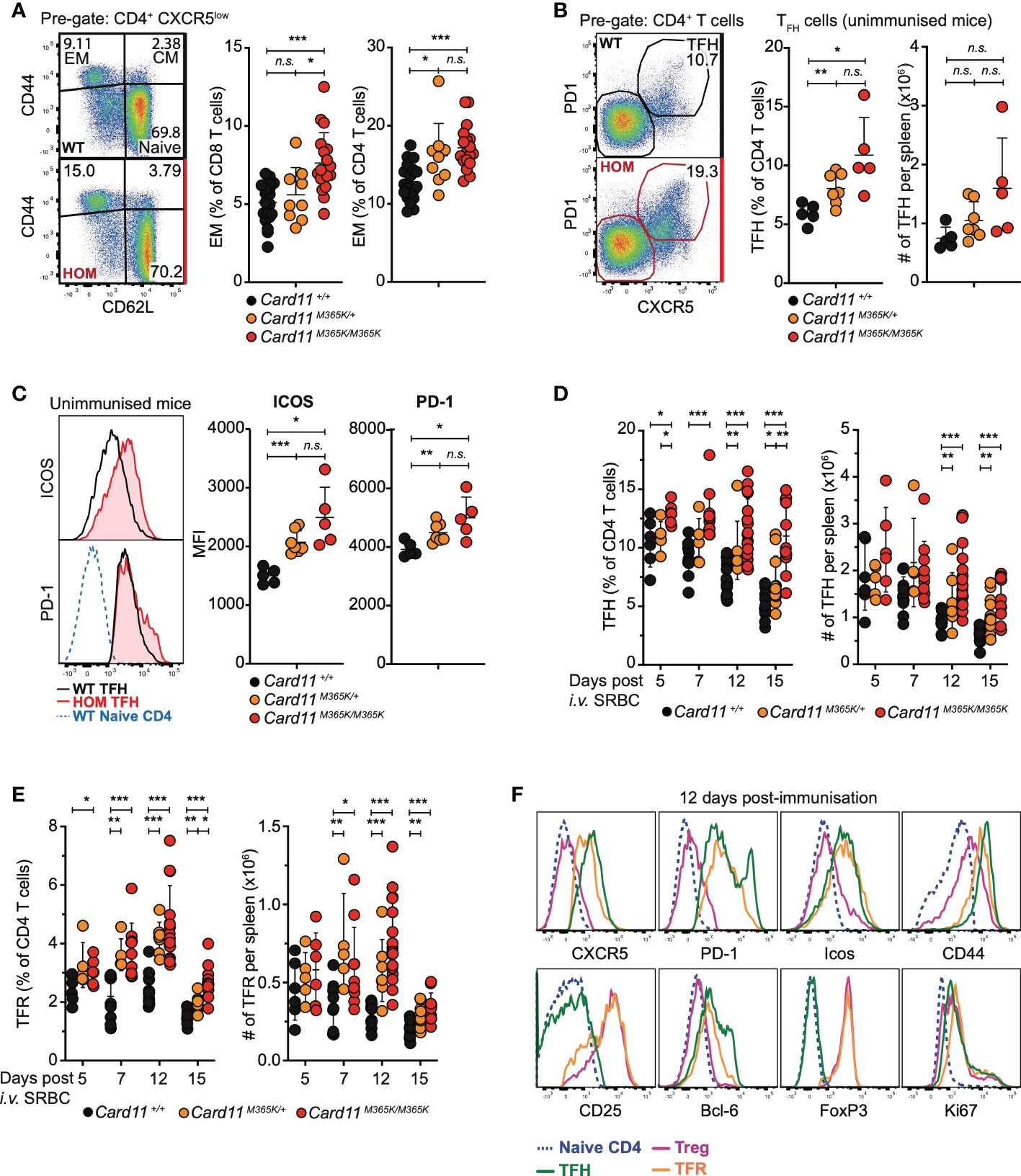
Figure 2 Germline GOF Card11M365K mutation causes accumulation of effector CD4 and CD8 T cells, T follicular and T follicular regulatory T cells. (A-C). Flow cytometric analysis of T cell populations in non-immunized mice of the indicated genotypes. (A). Left, representative flow cytometric analysis and right, percentage of CD44- CD62L+ naïve, CD44+ CD62L+ central memory (CM), and CD44+ CD62L- effector memory (EM) subsets among splenic CD8 or CD4 T cells. (B). Left, representative flow cytometric analysis and right, percentage among CD4 T cells or total number per spleen of CXCR5high PD-1high follicular helper (TFH)-like CD4 T cells. (C). Left, representative flow cytometric histograms showing distribution of ICOS (top) or PD-1 (bottom) fluorescence on Card11+/+ naïve CD4 T cells, Card11+/+ or Card11M365K/M365K TFH -like cells. Right, plots showing mean fluorescence intensity (MFI) of ICOS or PD-1 on TFH-like cells from mice of the indicated genotypes. (D-F). Flow cytometric analysis of T cell populations in mice 5, 7, 12 or 15 days post-immunization with sheep red blood cells (SRBCs). (D). FoxP3- CXCR5high PD-1high TFH cells as a percentage of CD4+ TCRβ+ T cells or as total number per spleen, in mice of the indicated genotypes. (E). CXCR5high PD-1high CD25+ FoxP3+ T follicular regulatory (TFR) cells as a percentage of CD4+ TCRβ+ T cells or as total number per spleen, in mice of the indicated genotypes. (F). Representative flow cytometric histograms showing distribution of cell-surface CXCR5, PD-1, ICOS, CD44, CD25 and intracellular Bcl-6, FoxP3, Ki-67 fluorescence in naïve CD4 (blue), TFH (green), T regulatory (TREG; magenta) or TFR (orange) cells from Card11M365K/M365K mice 12 days post-SRBC immunization. The histograms are also representative of results from Card11+/+ mice and from results 7 and 15 days post-immunization. (A-E). Statistical comparisons made by t-test, corrected for multiple comparisons using the Holm-Sidak method. Data are representative of n > 2 independent experiments with n > 4 mice per group. not significant (n.s) p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001. (A, D, E). Data are pooled from 3 independent experiments.
Unimmunized Card11M365K/+ and to a greater extent Card11M365K/M365K mice had a mutant allele dose-dependent increase accumulation of TREG cells (Figure 3A), of phenotype TCRβ+ CD3+ CD4+ CD25+ FoxP3+ and having first excluded CXCR5high PD-1high TFH-like or TFR-like cells. The accumulating Card11-mutant TREGS expressed homogeneously higher levels of ICOS but also of CTLA-4, and higher levels of CD69 and CD44 (Figure 3B), and the Card11-mutant mice had a significant accumulation of TREG cells with a CD62L- CD44+ effector memory-like phenotype (Figure 3C). Similarly, Card11M365K mice on a C57BL/6 Ncrl background had a significant increase in percentage and total number per spleen of CD44high and PD-1high CD4 and CD8 T cells (Supplementary Figures 3A-C) and of TFH-like and TREG cells, which were by contrast significantly reduced in Card11loco/loco mice (Supplementary Figures 3D, E).
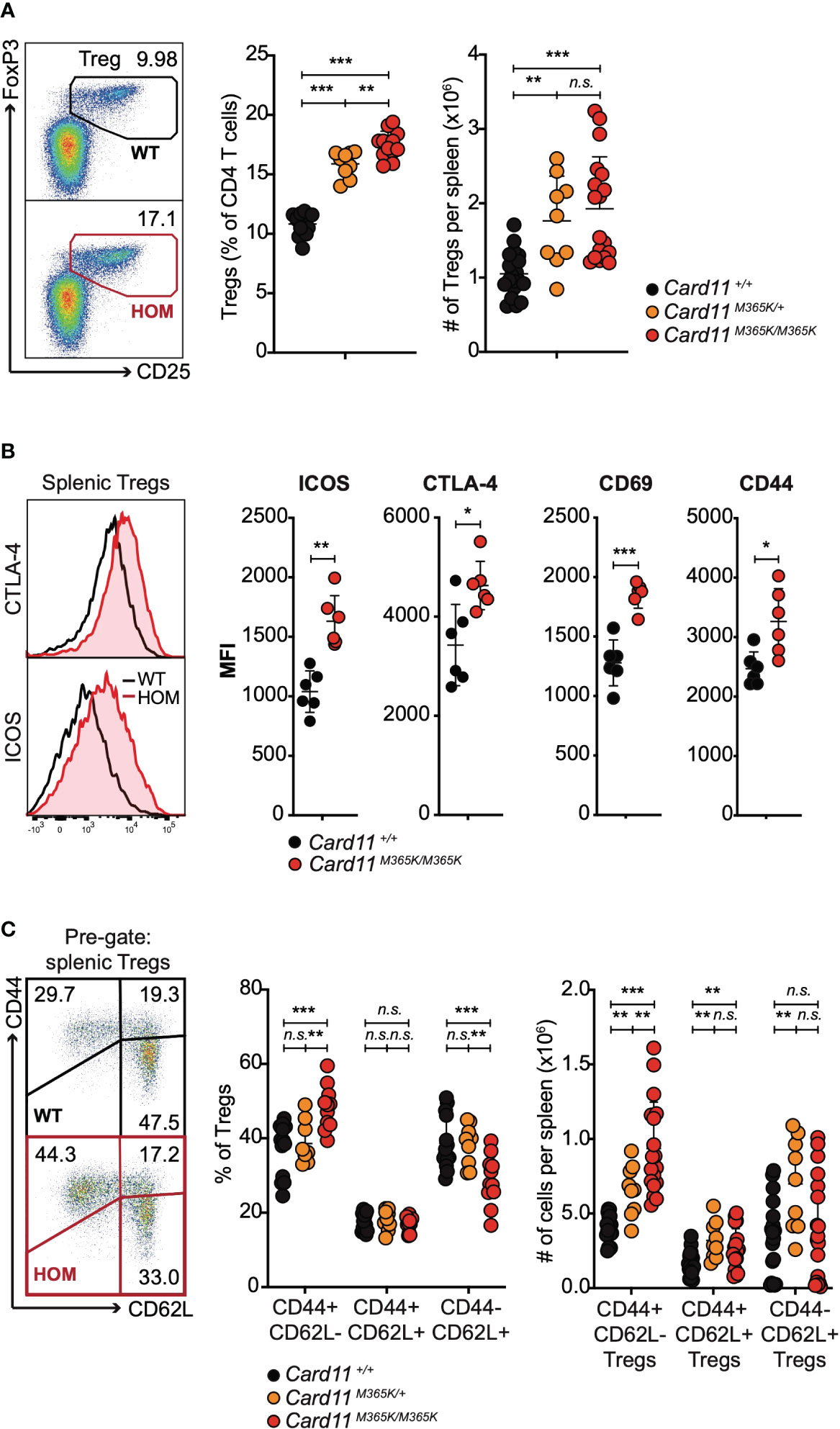
Figure 3 Germline GOF Card11M365K mutation causes accumulation of CD62Lneg CD44high ICOShigh CTLA-4high T regulatory cells. (A). Left, representative flow cytometric analysis and right, percentage amongst CD4 T cells or total number per spleen of TCRβ+ CD4+ CD25+ FoxP3+ T regulatory (TREG) cells, in mice of the indicated genotypes. (B). Left, representative flow cytometric histograms showing distribution of intracellular CTLA-4 and cell-surface ICOS fluorescence and right, plots of mean fluorescence intensity (MFI) of ICOS, CTLA-4, CD69 or CD44, for splenic TREGS from mice of the indicated genotypes. (C) Left, representative flow cytometric analysis and right, percentage amongst TREGS or total number per spleen of CD44- CD62L+, CD44+ CD62L+ and CD44+ CD62L- TREGS, in mice of the indicated genotypes. (A-C). Statistical comparisons made by t-test, corrected for multiple comparisons using the Holm-Sidak method. Data are representative of n > 2 independent experiments with n > 4 mice per group. not significant (n.s) p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001. (A, C) Data are pooled from 3 independent experiments.
Given the above findings, we tested whether germline Card11M365K mutation alters early T cell development in the thymus. Thymus cellularity was similar in Card11M365K/+ but mildly decreased in Card11M365K/M365K relative to Card11+/+ mice (Figure 4A). Card11-mutant mice had a significantly increased percentage of CD25+ FoxP3+ TREGS among CD4 single-positive (SP) cells, but no change in total number of thymic TREGS, relative to wild-type mice (Figure 4B). Cell-surface Neuropilin-1 (NRP1), CCR6 and CD24 were used to identify peripherally induced versus newly developed or recirculating thymus-derived TREGS (68–70). We observed no change in percentage (or total number) of thymus-derived NRP1+, thymus-derived nascent CCR6- CD24+ or recirculating CCR6+ CD24- TREGS in Card11-mutant relative to wildtype mice (Figure 4C). Card11 wild-type and mutant mice also had comparable frequencies and numbers of CD4- CD8- double-negative (DN), CD4+ CD8+ double-positive (DP), CD4+ single-positive (SP) and CD8+ SP thymocytes (Figure 4D), and of CD44+ CD25- DN1, CD44+ CD25+ DN2, CD44- CD25+ DN3 and CD44- CD25- DN4 early progenitors (Figure 4E). Thymic T cell development thus appears overtly normal in Card11M365K mutant mice.
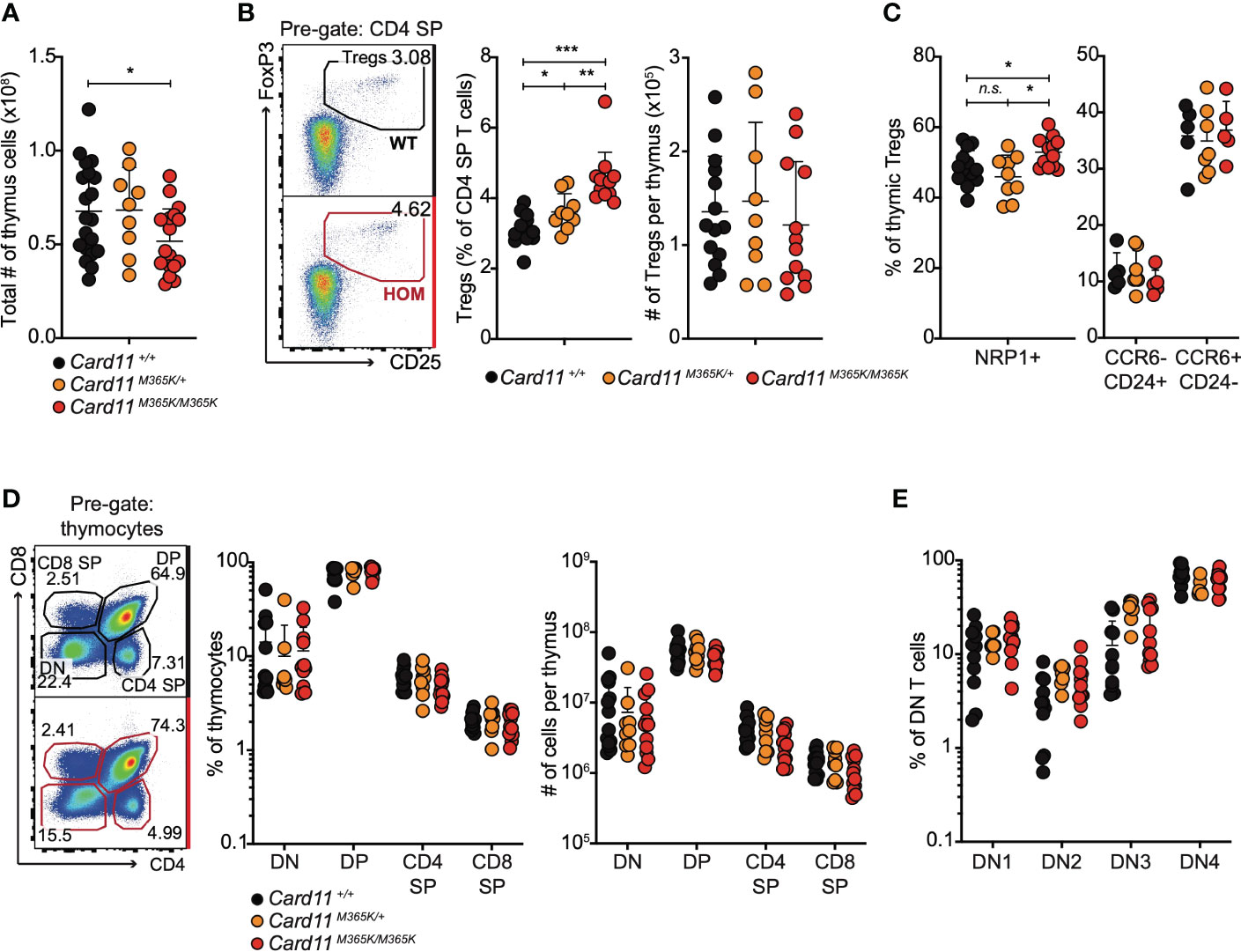
Figure 4 Card11M365K mutant mice have normal numbers of thymic T cell precursors, single positive CD4 and CD8 T cells and T regulatory cells. (A). Total number of cells per thymus from individual mice of the indicated genotypes. (B). Left, representative flow cytometric analysis and right, percentage amongst CD4 single-positive (SP) T cells or total number per thymus of CD25+ FoxP3+ CD4+ T regulatory (TREG) cells, in mice of the indicated genotypes. (C). Percentage of NRP1+ thymic TREGS (left) or of CCR6- CD24+ or CCR6+ CD24- thymic TREGS (right), in mice of the indicated genotypes. (D). Left, representative flow cytometric analysis and right, percentage amongst thymocytes or total number per thymus of CD4- CD8- double-negative (DN), CD4+ CD8+ double-positive (DP), CD4+ SP or CD8+ SP T cells, in mice of the indicated genotypes. (E). Percentage of DN T cells with a CD25- CD44+ DN1, CD25+ CD44+ DN2, CD25+ CD44- DN3 or CD25- CD44- DN4 phenotype, in mice of the indicated genotypes. Statistical comparisons made by t-test, corrected for multiple comparisons using the Holm-Sidak method. Data are representative of n > 2 independent experiments with n > 4 mice per group. not significant (n.s) p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001. (A, B) Data are pooled from 3 independent experiments.
Collectively, the above results demonstrate that germline CARD11 gain-of-function causes over-accumulation in the periphery of activated CD8 and CD4 T cells, TFH, TFR and TREG cells expressing increased levels of activating and inhibitory checkpoint molecules.
To test whether Card11M365K acts cell-autonomously to dysregulate CD8 and CD4 T cells, we analyzed mixed chimeric mice containing Card11+/+ CD45.1+ bone marrow-derived hematopoietic cells and CD45.2+ bone marrow-derived hematopoietic cells that were either Card11+/+ or Card11M365K/M365K. Within individual chimeric mice, there was a significant increase in frequency of Ptprcb/b Card11M365K/M365K relative to Ptprca/a Card11+/+ CD4 effector memory (EM), TFH, TREG and CD8 EM cells – whereas no such difference was observed between Ptprcb/b Card11+/+ and Ptprca/a Card11+/+ cells (Figure 5A). Card11M365K/M365K thus provides a cell-intrinsic advantage to effector CD8 and CD4 T cells, TFH and TREG cells. The accumulating Card11M365K/M365K TREGS had a significant cell-intrinsic increase in levels of cell-surface ICOS and CD44 and of intracellular CTLA-4 (Figures 5B, C). Within TREG cells, and reminiscent of observations in germline Card11-mutant mice, Card11M365K/M365K mutation caused significant cell-autonomous accumulation of CD62L- CD44+ TREGS (Figure 5D).
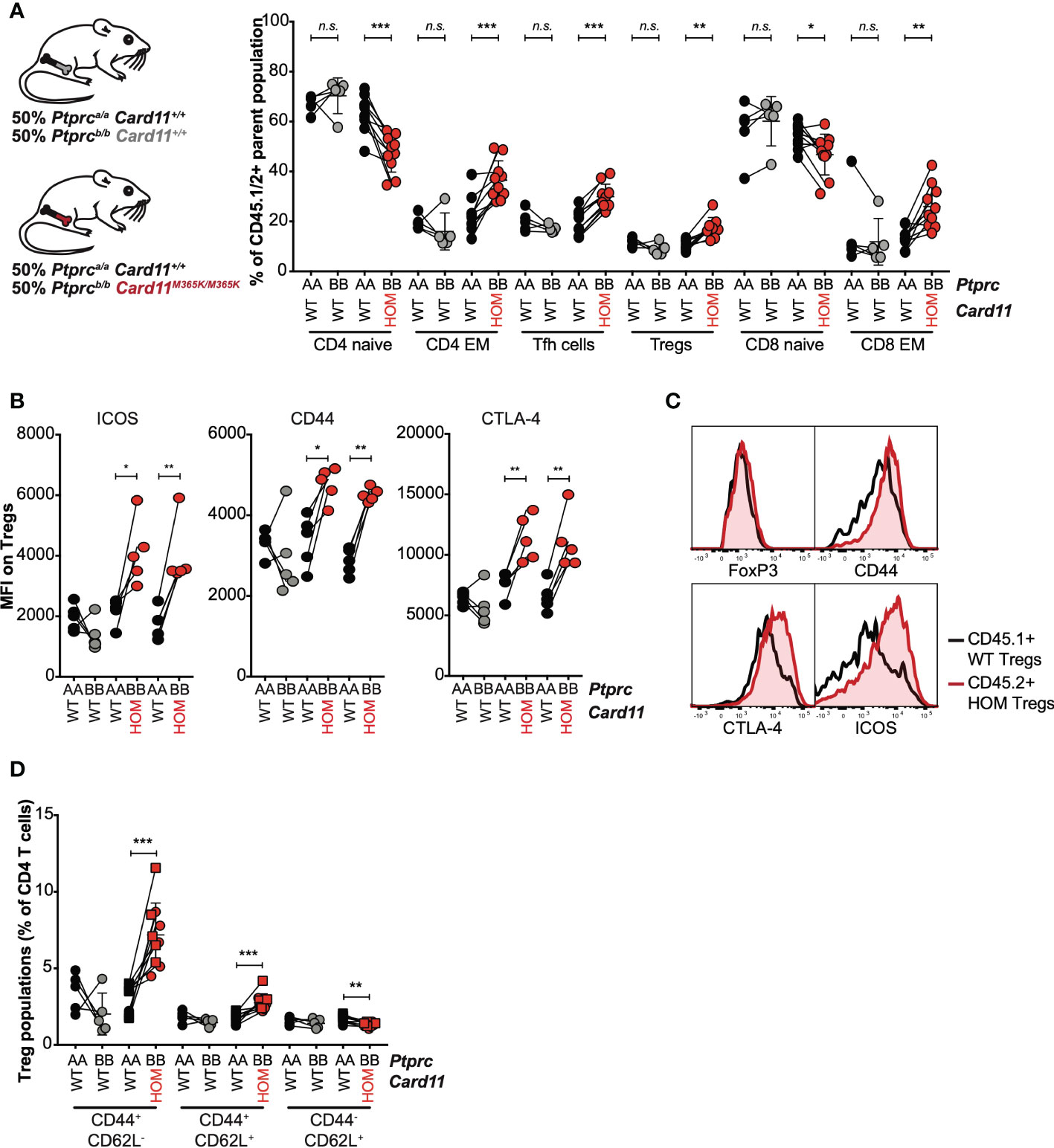
Figure 5 GOF Card11M365K/M365K mutation provides a cell-intrinsic advantage to activated CD8 and CD4 T cells, T follicular helper-like and T regulatory cells. (A-D). Mixed chimeras were generated by irradiating Rag1KO/KO Card11+/+ mice and reconstituting them with Ptprca/a Card11+/+ (black fill) bone marrow in a 1:1 mixture with Ptprcb/b Card11+/+ (grey fill; 1 donor) or Card11M365K/M365K (red fill; 2 separate donors) bone marrow. These mixed chimeras were sacrificed 7 days post-immunization with SRBCs. (A). Percentage, within the corresponding Ptprca/a or Ptprcb/b parent population, of CD44- CD62L+ naïve and CD44+ CD62L- effector memory (EM) CD4 T cells, CXCR5high PD-1high T follicular helper (TFH) and CD25+ FoxP3+ T regulatory (TREG) CD4 T cells, and naïve and EM CD8 T cells. (B). ICOS, CD44 and CTLA-4 mean fluorescence intensity (MFI) for Ptprca/a versus Ptprcb/b TREGS of the indicated genotypes (recipients received bone marrow from n=1 Ptprcb/b Card11+/+ donor and n=2 Ptprcb/b Card11M365K/M365K donors). (C). Representative flow cytometric histograms showing distribution of intracellular FoxP3 and CTLA-4 and cell-surface CD44 and ICOS fluorescence, for splenic Ptprca/a Card11+/+ (black line) versus Ptprcb/b Card11M365K/M365K (red line) TREGS. (D). CD44+ CD62L-, CD44+ CD62L+ and CD44- CD62L+ Ptprca/a or Ptprcb/b TREGS, as a percentage of splenic CD4 T cells. Statistical comparisons made by paired t-test. not significant (n.s) p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001.
Given the above findings, we tested whether Card11M365K mutation increases T cell responses to TCR, CD28 or high-affinity IL-2 receptor stimulation, which engage pathways crucial to the differentiation, survival and proliferation of effector T cells and TREGS. Following CTV labelling and 3 days of stimulation with anti-CD3 and anti-CD28 in vitro, the mean percentage of divided cells was 74% for Card11M365K/M365K, 68% for Card11M365K/+ and 58% for Card11+/+ CD4 T cells, and 94%, 90% and 87% for CD8 T cells of the respective genotypes. By contrast, only 12% and 24% for Card11loco/loco CD4 and CD8 T cells, respectively (Supplementary Figure 4A). Card11M365K mutation also increased and Card11loco/loco mutation decreased the size and cell-surface CD25 and PD-1 levels of stimulated CD4 and CD8 T cells (Supplementary Figure 4B, C). Given the small number of WT CD4 T cells assessed, we were unable to conclude that these effects were statistically significant. Our flow cytometric analysis of Ki67 expression revealed an increased fraction of Ki67+ T cells within the spleens of Card11M365K/M365K mutant mice (Supplementary Figure 4D). Thus, GOF CARD11.M365K caused increased activation and a mild increase in proliferation of CD4 and CD8 T cells following TCR stimulation and CD28 co-stimulation. By contrast, Card11M365K/M365K mutation had no effect on STAT5 phosphorylation following IL-2 stimulation (Supplementary Figure 4E). To determine the effects of CARD11 GOF on CD4 T cell differentiation in vitro, we purified naïve CD4 T cells from wild-type and mutant mice by fluorescence-activated cell sorting (FACS), and incubated them for 4 days in conditions that skew towards T helper 0 (TH0), TH1, TH2 or TH17 differentiation. At day 4, we observed increased frequencies of Card11M365K/M365K relative to Card11+/+ IL-4+ and IL-5+ TH2-like cells (Supplementary Figure 4F). These results indicate that weak CARD11 GOF may skew naïve CD4 T cells towards TH2 differentiation in response to TCR and cytokine stimulation.
Collectively, these results demonstrate that GOF CARD11 increases T cell activation and proliferation following TCR and CD28 stimulation and provides a cell-intrinsic advantage to activated CD8 and CD4 T cells, TFH, TREG and TFR cells expressing increased levels of checkpoint molecules ICOS and PD-1.
Notably, PD-1 acts as a tumor suppressor in CD4 T cells (71), but PD-1 checkpoint therapy significantly worsens disease progression in some (72) but not all (73) individuals with ATL. ATL, which are thought to arise from effector and/or FoxP3+ CD4 T cells (74, 75), harbor recurrent somatic GOF CARD11 mutations (38, 39). To test whether PD-1 restrains the over-accumulation of Card11M365K mutant CD4 T cells in vivo, we adapted a workflow used by Wartewig et al. to demonstrate that PD-1 inhibition synergises with ITK-SYK fusion to cause lethal CD4 T cell lymphoproliferation (71). We adoptively transferred 4 x 106 Ptprcb/b CD4 T cells that were either Card11+/+ or Card11M365K/M365K, into Ptprca/a Card11+/+ C57BL/6 recipient mice. We injected the recipient mice with anti-PD-1 monoclonal antibody (mAb) or isotype control mAb at days 1, 3, 5 and 6, and sacrificed them at day 7 post-adoptive transfer (Supplementary Figure 5A). Consistent with our mixed chimera results, a higher fraction of Card11-mutant relative to wild-type CD4 T cells had an effector memory phenotype (Supplementary Figure 5B). Relative to control mAb-treated mice, anti-PD-1-treated mice had similar total numbers of cells per spleen (Supplementary Figure 5C). Amongst mice that received Card11M365K/M365K CD4 T cells, anti-PD-1 treatment increased the total number of donor-derived CD4 T cells, and resulted in a trend towards increased number of donor-derived effector memory CD4 T cells (Supplementary Figure 5D, E). These results indicate that PD-1 inhibition is insufficient to cause lymphoma or lethal lymphoproliferation of Card11M365K/M365K CD4 T cells, but that PD-1 may play a role in restraining the accumulation of Card11M365K/M365K CD4 T cells in vivo.
The findings here reveal that gain-of-function mutation of a conserved CARD11 residue, located in the coiled-coil domain recurrently mutated in B- and T-lymphomas, caused cell-autonomous accumulation of effector CD8 and CD4 T cells, and particularly of TFH, TREG and TFR cells that are critical to coordinating and regulating adaptive immune responses. Germline Card11M365K GOF caused accumulation of spontaneous GC B cells and increased GC response to immune challenge but caused no dramatic B cell lymphocytosis as observed in individuals with BENTA disease. Our results demonstrate that CARD11 GOF perturbs T cells by increasing their activation and proliferation downstream of TCR or co-stimulatory receptor signaling. By revealing that GOF CARD11 drives aberrant expression of checkpoint molecules including PD-1 and ICOS, a known positive regulator of TFH, TREG and TFR cells, the findings here indicate that GOF CARD11 mutations perturb T lymphocytes by dysregulating not only TCR-NFκB signaling but also co-stimulatory signaling. These results highlight likely effects of acquired GOF CARD11 mutations that are strikingly recurrent in aggressive human PTCL derived from effector, follicular and regulatory CD4 T cells.
The mild increase in B cell numbers in Card11M365K mutant mice contrasts with lethal B cell lymphoproliferation upon B cell-conditional Card11L232LI mutation (Card11L225LI in the original publication (52);). This latter phenotype is also absent from mice with a Card11E134G or Card11K215M (54) or Card11L251P mutation (53). Card11K215M creates a cell-intrinsic advantage whereas Card11E134G creates a cell-intrinsic disadvantage for GC B cells (54), and Card11L251P acts primarily to alter GC kinetics (53). The variable effects on B cells of different CARD11 mutations may relate to qualitative differences in their effect on NF-κB signaling activity (31, 55). In a luciferase reporter system in 293T cells, CARD11 p.M365K increased the transcription of a NF-κB target gene to levels above wild-type CARD11, but below CARD11 p.L251P (30). Our results, based on a NF-κB luciferase assay, B220 and CD86 expression and survival of B cells transduced with CARD11WT, CARD11G123S, CARD11E134G and CARD11M365K, indicate that CARD11M365K leads to weak GOF intermediate between that caused by CARD11E134G and CARD11G123S.
The Card11M365K mutant mouse strain was generated by ENU mutagenesis, which enabled us to study the effects of Card11 GOF mutation in an otherwise normal gene, as opposed to expression of mutant Card11 cDNA from a heterologous promoter and locus (52, 53, 76). In this context, CARD11M365K was insufficient to cause B cell malignancy or the striking B cell lymphocytosis seen in individuals with BENTA disease. This contrast may relate to differing CARD11 mutations, as discussed above and given that CARD11M365K has not been identified in the germline of children with BENTA disease. Alternatively, the contrast may relate to the specific-pathogen free environment of the Card11M365K mice or to differences between human and mouse lymphocytes. Like germline GOF CARD11 mutations, BTK, NFKB1 or NFKB2 deficiency have different consequences in humans relative to mice. In humans, they cause profound loss of transitional and mature naïve B cells (77–80) but in mice they cause a less drastic decrease (81–84), indicating that human B cells may be more strongly dependent on BTK-NF-κB signaling. Several hypotheses may explain the profound increase in transitional and mature B cells caused by germline GOF CARD11 mutations in humans but not mice. (i) Card11 mRNA increases 10-fold between pre-B cells and immature IgM+ B cells and transitional B cells in mice (Immgen Database). It is possible that CARD11 mRNA and protein are more strongly expressed in human relative to mouse naïve B cells, beyond a threshold where GOF in the protein dysregulates proliferation and survival. Nevertheless, previous publications (52, 54, 76) and our in vitro data indicate that GOF CARD11 can provide a cell-intrinsic advantage to mouse B cells. (ii) CARD11 mRNA or protein may be down-regulated in mouse B cells as an adaptive response to GOF CARD11 signaling that does not function in human B cells. (iii) Human B cells may be less able to induce counter-regulatory processes acting downstream or upstream from CARD11 (i.e. induction of NFKBIA or TNAIP3). Future studies comparing CARD11, NFKBIA and TNFAIP3 protein levels in normal and CARD11-mutant human and mouse transitional and mature B cells may help to distinguish between these alternatives.
T cells from human BENTA patients carrying CARD11 GOF mutations typically proliferate less than healthy controls in response to anti-CD3/CD28 stimulation, a difference linked to a mildly anergic phenotype associated with poor IL-2 expression by CARD11-mutant human T cells (29). By contrast, Card11M365K mutant mouse T cells had a mild proliferative advantage relative to Card11 wild-type mouse T cells. In addition to the considerations discussed above, it is possible that secondary effects that are visible in humans over time may not be visible in mice at 8-12 weeks of age. (i) These secondary effects may be pathological, as seen in CTLA4 deficiency in humans, which results in loss of B cells even though B cells mostly lack CTLA4 expression. Affected patients have relatively normal B cell numbers prior to developing pathology but start losing B cells when they develop the syndrome (85). (ii) These secondary effects may be compensatory, as seen in transgenic B cells expressing chimeric IgMG receptors containing the IgG tail segment. These cells adopt a gene expression profile of anergy, but this occurs secondary to their down-regulation of cell-surface receptor (86).
The skewing of towards Th2 differentiation of Card11M365K/M365K CD4 T cells is interesting, given that Th2 skewing occurs in humans with loss-of-function or dominant negative CARD11 mutations. We cannot exclude the possibility that CARD11.M365K results in “blended” GOF and LOF effects, as previously observed in BENTA disease (26). Mice homozygous for the hypomorphic Card11unmodulated mutation develop penetrant, spontaneous atopy and dermatitis with age (10), caused by partial reduction in effector T cell accumulation but also partial TREG deficiency leading to progressive, selective TH2 accumulation and subsequent IgE production (16). In that context, hypomorphic Card11 mutation produces outcomes that could not be predicted from null alleles, through unequal titration of opposing effects within different T cell subsets (16). Similarly, unequal effects of hypermorphic mutations in different lymphocyte populations may contribute to the variable B and T cell pathologies in humans and mice with germline and somatic CARD11 GOF mutations.
In addition to cell-intrinsic effects, B cell homeostasis may be perturbed by CARD11 GOF within CD4 T cells. Previous publications studied Card11L251P (53) and Card11L232LI (52) expressed in B cells only, whilst T cell populations from germline Card11E134G and Card11K215M mutant mice were not reported (54). In Card11M365K mutant mice, the accumulation of splenic TFH cells at days 7, 12 and 15, but not at day 5 post-immunization, correlated with accumulation of splenic GC B cells at days 7, 12 and 15, but not at day 5. Card11M365K mutant TFH cells expressed homogeneously increased levels of cell-surface ICOS, and both TFH accumulation and increased ICOS expression (87) are known to drive GC B cell accumulation. Card11M365K/M365K mutation caused cell-autonomous accumulation of ICOShigh TFH cells, but also of TFR cells that can act to suppress the GC response (88–90). The relative, and possibly graded, effects of Card11 mutation in TFH versus TFR cells, and in turn on B cell homeostasis, are difficult to distinguish without TFH or TFR-specific CARD11 GOF models. Nevertheless, our data raise the possibility that CARD11 GOF CD4 T cells may perturb B cells in individuals with germline or somatic CARD11 GOF mutations. Individuals with BENTA disease have normal numbers of circulating CD4 and CD8 T cells (25, 29), but to our knowledge no detailed T cell immunophenotyping has been reported. Future studies should assess T cell populations in humans and mouse models with different germline or T cell-restricted CARD11 mutations.
With regards to T cell lymphoma, our results reveal likely cell-intrinsic effects of the somatic GOF CARD11 mutations that recur in up to 30% of ATL (38, 39), CTCL and Sezary Syndrome (43–46) and at lesser frequency in angioimmunoblastic T cell lymphoma (AITL) (91). CARD11 and PRKCB mutations are positively correlated in ATL (38), suggesting that NF-κB activating mutations may synergize in driving ATL. The striking recurrence of mutations modifying the TCR/NF-κB pathway highlights its importance in PTCLs including ATL (38, 39, 56–58) and CTCL or Sezary syndrome (43–46, 59–61, 92, 93). One limitation of our study is that CARD11M365K has not been identified in PTCL or CTCL. Nevertheless, CARD11M365K modifies a conserved region of the CC domain recurrently mutated in PTCL (Figure 1). ATL, CTCL and AITL are thought to arise from activated, TFH-like and/or TREG-like CD4 T cells (50, 94–96), and Card11M365K mutation causes cell-autonomous accumulation of activated, TFH, TREG and TFR CD4 T cells. In addition, Card11M365K/M365K caused over-expression of stimulatory and inhibitory receptors ICOS, CTLA-4 and PD-1, and increased activation, proliferation and PD-1 expression by mutant T cells following TCR and CD28 stimulation. Activating CD28 mutations recur in 10-11% of AITL (97, 98), and in-frame fusions involving CD28, CTLA4 and ICOS recur in 7% of ATL along with CD28 focal gains and missense mutations, all of which result in continuous or prolonged co-stimulatory signaling (38). When expressed in mice on a Tet2-/- background, the RHOAG17V mutation identified in 70% of AITL (99–101) results in T cell lymphomas that partially require ICOS and PI3K signaling for their proliferation and survival (102). The cell-intrinsic increase of ICOS and CTLA-4 expression on Card11M365K mutant CD4 and TREG cells indicates that CARD11 GOF may contribute to CD4 T cell dysregulation not just via TCR-NFκB but also via PI3K signaling. ICOS expression increases the accumulation of TFH cells but also of TREG and TFR cells (103), such that ICOS over-expression on expanded CARD11M365K mutant TFH, TREG and TFR cells may further their accumulation. By contrast, increased CTLA-4 on the surface of these Card11-mutant cells may limit their accumulation (104).
In addition to ICOS and CTLA-4, Card11M365K mutation increased PD-1 expression by CD4 T cells, in vivo and following TCR/CD28 stimulation ex vivo. Parallel observations could be drawn by future studies testing the association of CARD11 mutations with increased PD-1 or ICOS expression on human T-lymphoma cells. PDCD1 (encoding PD-1) is increased in CD4 malignancies with gene signatures of dysregulated TCR signaling (71). PD-1 acts as a tumor suppressor in CD4 T cells and PDCD1 alterations, most commonly focal deletions, recur in 10-20% of CTCL, 36% of Sezary syndrome and 26% of ATL (71). Consistent with the effects of PD-1 in inhibiting TCR signaling and also CD28 co-stimulation (105), PD-1 inhibition mildly increased Card11M365K/M365K CD4 T cell accumulation in vivo, but was nevertheless insufficient to cause CD4 lymphoma or lymphoproliferation. This contrasts with the lethal lymphoproliferation of CD4 T cells expressing an ITK-SYK fusion upon their exposure to anti-PD-1 monoclonal antibody (71). This dichotomy may point to a threshold of CARD11 or NF-κB GOF required for synergy with PD-1 LOF to drive CD4 lymphoproliferation. The acquisition of different somatic driver gene mutations (e.g. ITK-SYK fusion versus intermediate CARD11 GOF mutation) may explain why PD-1 inhibition accelerates disease progression in some (72) but not all (73) cases of ATL.
Unlike other PTCL, ATL requires HTLV-1 infection (41, 106, 107). The variable, often long, latent phase between HTLV-1 infection and ATL diagnosis implicated additional environmental or genetic events in ATL pathogenesis, and led to the discovery of TCR/NF-κB pathway genes and CARD11 as recurrently mutated in ATL (38, 39). Notably, HTLV-1 viral proteins TAX and HBZ increase NF-κB activation and survival (108, 109), and HTLV-1 has tropism for FoxP3+ CD4 T cells (109–111). Given that Card11M365K mutation increases NF-κB activation and creates a cell-intrinsic advantage for TREGS, the cell-intrinsic effects of CARD11 mutations and of HTLV-1 infection may cooperate in driving ATL pathogenesis.
There is a striking paucity of information on the cell-intrinsic effects of somatic CARD11 GOF mutations in PTCL, which are heterogeneous and often aggressive malignancies associated with poor clinical outcomes (112). The above findings reveal cell-intrinsic effects of a CARD11 GOF protein within T cells. They highlight the need to study T cells in humans with germline CARD11 GOF mutations and BENTA disease, and in mouse models with PTCL hotspot CARD11 mutations. Our findings further highlight the crucial role played by CARD11 in lymphocytes and the possible therapeutic utility of developing small molecule inhibitors targeting CARD11.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Garvan Institute of Medical Research/St Vincent’s Hospital Animal Ethics Committee; ANU National University Animal Experimentation Ethics Committee.
EM-F and YJ designed and performed the experiments. JW, RB and KH designed and/or performed transduction experiments. JB and ED performed the Th differentiation assay. EM-F, YJ, KH and CG interpreted experiments and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Health and Medical Research Council (NHMRC) Program (1113904, to C.C.G.) and Fellowship (1081858, to C.C.G.) grants and by The Bill and Patricia Ritchie Foundation.
We thank the Australian Phenomics Facility (Australian National University), the Garvan-Weizmann Center for Clinical Genomics (Garvan Institute of Medical Research) and the Garvan Biological Testing Facility, for providing technical services.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1095257/full#supplementary-material
1. Bertin J, Wang L, Guo Y, Jacobson MD, Poyet JL, Srinivasula SM, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa b. J Biol Chem (2001) 276(15):11877–82. doi: 10.1074/jbc.M010512200
2. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity (2005) 23(6):561–74. doi: 10.1016/j.immuni.2005.09.014
3. Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, et al. Phosphorylation of CARMA1 plays a critical role in T cell receptor-mediated NF-kappaB activation. Immunity (2005) 23(6):575–85. doi: 10.1016/j.immuni.2005.10.007
4. Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat Rev Immunol (2006) 6(11):799–812. doi: 10.1038/nri1944
5. Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev (2009) 228(1):199–211. doi: 10.1111/j.1600-065X.2008.00749.x
6. Gaide O, Martinon F, Micheau O, Bonnet D, Thome M, Tschopp J. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-kappaB activation. FEBS Lett (2001) 496(2-3):121–7. doi: 10.1016/S0014-5793(01)02414-0
7. McAllister-Lucas LM, Inohara N, Lucas PC, Ruland J, Benito A, Li Q, et al. Bimp1, a MAGUK family member linking protein kinase c activation to Bcl10-mediated NF-kappaB induction. J Biol Chem (2001) 276(33):30589–97. doi: 10.1074/jbc.M103824200
8. Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: Nucleation-induced filamentous assembly. Mol Cell (2013) 51(6):766–79. doi: 10.1016/j.molcel.2013.08.032
9. Egawa T, Albrecht B, Favier B, Sunshine MJ, Mirchandani K, O’Brien W, et al. Requirement for CARMA1 in antigen receptor-induced NF-kappa b activation and lymphocyte proliferation. Curr Biol (2003) 13(14):1252–8. doi: 10.1016/S0960-9822(03)00491-3
10. Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity (2003) 18(6):751–62. doi: 10.1016/S1074-7613(03)00141-9
11. Newton K, Dixit VM. Mice lacking the CARD of CARMA1 exhibit defective b lymphocyte development and impaired proliferation of their b and T lymphocytes. Curr Biol (2003) 13(14):1247–51. doi: 10.1016/S0960-9822(03)00458-5
12. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity (2003) 18(6):763–75. doi: 10.1016/S1074-7613(03)00148-1
13. Barnes MJ, Krebs P, Harris N, Eidenschenk C, Gonzalez-Quintial R, Arnold CN, et al. Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PloS Biol (2009) 7(3):e51. doi: 10.1371/journal.pbio.1000051
14. Molinero LL, Yang J, Gajewski T, Abraham C, Farrar MA, Alegre ML. CARMA1 controls an early checkpoint in the thymic development of FoxP3+ regulatory T cells. J Immunol (2009) 182(11):6736–43. doi: 10.4049/jimmunol.0900498
15. Lee AJ, Wu X, Cheng H, Zhou X, Cheng X, Sun SC. CARMA1 regulation of regulatory T cell development involves modulation of interleukin-2 receptor signaling. J Biol Chem (2010) 285(21):15696–703. doi: 10.1074/jbc.M109.095190
16. Altin JA, Tian L, Liston A, Bertram EM, Goodnow CC, Cook MC. Decreased T-cell receptor signaling through CARD11 differentially compromises forkhead box protein 3-positive regulatory versus T(H)2 effector cells to cause allergy. J Allergy Clin Immunol (2011) 127(5):1277–85 e5. doi: 10.1016/j.jaci.2010.12.1081
17. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet (2017) 49(8):1192–201. doi: 10.1038/ng.3898
18. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol (2018) 141(5):1818–30 e2. doi: 10.1016/j.jaci.2017.06.047
19. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. J Allergy Clin Immunol (2019) 143(4):1482–95. doi: 10.1016/j.jaci.2018.08.013
20. Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol (2013) 131(5):1376–83 e3. doi: 10.1016/j.jaci.2013.02.012
21. Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol (2013) 131(2):477–85 e1. doi: 10.1016/j.jaci.2012.11.050
22. Arjunaraja S, Nose BD, Sukumar G, Lott NM, Dalgard CL, Snow AL. Intrinsic plasma cell differentiation defects in b cell expansion with NF-kappaB and T cell anergy patient b cells. Front Immunol (2017) 8:913. doi: 10.3389/fimmu.2017.00913
23. Arjunaraja S, Snow AL. Gain-of-function mutations and immunodeficiency: at a loss for proper tuning of lymphocyte signaling. Curr Opin Allergy Clin Immunol (2015) 15(6):533–8. doi: 10.1097/ACI.0000000000000217
24. Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 mutation in a patient with severe congenital b cell lymphocytosis. J Clin Immunol (2015) 35(1):32–46. doi: 10.1007/s10875-014-0106-4
25. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild b-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol (2015) 136(3):819–21 e1. doi: 10.1016/j.jaci.2015.03.008
26. Desjardins M, Arjunaraja S, Stinson JR, Dorjbal B, Sundaresan J, Niemela J, et al. A unique heterozygous CARD11 mutation combines pathogenic features of both gain- and loss-of-Function patients in a four-generation family. Front Immunol (2018) 9:2944. doi: 10.3389/fimmu.2018.02944
27. Gupta M, Aluri J, Desai M, Lokeshwar M, Taur P, Lenardo M, et al. Clinical, immunological, and molecular findings in four cases of b cell expansion with NF-kappaB and T cell anergy disease for the first time from India. Front Immunol (2018) 9:1049. doi: 10.3389/fimmu.2018.01049
28. Outinen T, Syrjanen J, Rounioja S, Saarela J, Kaustio M, Helminen M. Constant b cell lymphocytosis since early age in a patient with CARD11 mutation: A 20-year follow-up. Clin Immunol (2016) 165:19–20. doi: 10.1016/j.clim.2016.02.002
29. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital b cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831
30. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large b-cell lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
31. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large b cell lymphoma. Science (2008) 319(5870):1676–9. doi: 10.1126/science.1153629
32. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large b-cell lymphoma identified by gene expression profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
33. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated b cell-like diffuse large b cell lymphoma cells. J Exp Med (2001) 194(12):1861–74. doi: 10.1084/jem.194.12.1861
34. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature (2006) 441(7089):106–10. doi: 10.1038/nature04687
35. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active b-cell-receptor signalling in diffuse large b-cell lymphoma. Nature (2010) 463(7277):88–92. doi: 10.1038/nature08638
36. Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry (2010) 49(38):8240–50. doi: 10.1021/bi101052d
37. Jattani RP, Tritapoe JM, Pomerantz JL. Cooperative control of caspase recruitment domain-containing protein 11 (CARD11) signaling by an unusual array of redundant repressive elements. J Biol Chem (2016) 291(16):8324–36. doi: 10.1074/jbc.M115.683714
38. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet (2015) 47(11):1304–15. doi: 10.1038/ng.3415
39. Kataoka K, Iwanaga M, Yasunaga JI, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood (2018) 131(2):215–25. doi: 10.1182/blood-2017-01-761874
40. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood (1977) 50(3):481–92. doi: 10.1182/blood.V50.3.481.481
41. Robert-Guroff M, Nakao Y, Notake K, Ito Y, Sliski A, Gallo RC. Natural antibodies to human retrovirus HTLV in a cluster of Japanese patients with adult T cell leukemia. Science (1982) 215(4535):975–8. doi: 10.1126/science.6760397
42. Seiki M, Hattori S, Hirayama Y, Yoshida M. Human adult T-cell leukemia virus: complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc Natl Acad Sci U S A. (1983) 80(12):3618–22. doi: 10.1073/pnas.80.12.3618
43. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet (2015) 47(9):1011–9. doi: 10.1038/ng.3356
44. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and sezary syndrome. Nat Genet (2015) 47(12):1465–70. doi: 10.1038/ng.3442
45. Prasad A, Rabionet R, Espinet B, Zapata L, Puiggros A, Melero C, et al. Identification of gene mutations and fusion genes in patients with sezary syndrome. J Invest Dermatol (2016) 136(7):1490–9. doi: 10.1016/j.jid.2016.03.024
46. Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet (2015) 47(12):1426–34. doi: 10.1038/ng.3444
47. Katsuya H, Ishitsuka K, Utsunomiya A, Hanada S, Eto T, Moriuchi Y, et al. Treatment and survival among 1594 patients with ATL. Blood (2015) 126(24):2570–7. doi: 10.1182/blood-2015-03-632489
48. Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, et al. Cutaneous lymphoma international consortium study of outcome in advanced stages of mycosis fungoides and sezary syndrome: Effect of specific prognostic markers on survival and development of a prognostic model. J Clin Oncol (2015) 33(32):3766–73. doi: 10.1200/JCO.2015.61.7142
49. Shimauchi T, Kabashima K, Tokura Y. Adult T-cell leukemia/lymphoma cells from blood and skin tumors express cytotoxic T lymphocyte-associated antigen-4 and Foxp3 but lack suppressor activity toward autologous CD8+ T cells. Cancer Sci (2008) 99(1):98–106. doi: 10.1111/j.1349-7006.2007.00646.x
50. Uchiyama T, Hori T, Tsudo M, Wano Y, Umadome H, Tamori S, et al. Interleukin-2 receptor (Tac antigen) expressed on adult T cell leukemia cells. J Clin Invest. (1985) 76(2):446–53. doi: 10.1172/JCI111992
51. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large b-cell lymphoma. Nat Genet (2011) 43(9):830–7. doi: 10.1038/ng.892
52. Knies N, Alankus B, Weilemann A, Tzankov A, Brunner K, Ruff T, et al. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant b-cell proliferation via cooperative NF-kappaB and JNK activation. Proc Natl Acad Sci U S A. (2015) 112(52):E7230–8. doi: 10.1073/pnas.1507459112
53. Wray-Dutra MN, Chawla R, Thomas KR, Seymour BJ, Arkatkar T, Sommer KM, et al. Activated CARD11 accelerates germinal center kinetics, promoting mTORC1 and terminal differentiation. J Exp Med (2018) 215(9):2445–61. doi: 10.1084/jem.20180230
54. Wei Z, Zhang Y, Chen J, Hu Y, Jia P, Wang X, et al. Pathogenic CARD11 mutations affect b cell development and differentiation through a noncanonical pathway. Sci Immunol (2019) 4(41). doi: 10.1126/sciimmunol.aaw5618
55. Meitlis I, Allenspach EJ, Bauman BM, Phan IQ, Dabbah G, Schmitt EG, et al. Multiplexed functional assessment of genetic variants in CARD11. Am J Hum Genet (2020) 107(6):1029–43. doi: 10.1016/j.ajhg.2020.10.015
56. Vicente C, Cools J. The genomic landscape of adult T cell leukemia/lymphoma. Nat Genet (2015) 47(11):1226–7. doi: 10.1038/ng.3428
57. Nagata Y, Kontani K, Enami T, Kataoka K, Ishii R, Totoki Y, et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood (2016) 127(5):596–604. doi: 10.1182/blood-2015-06-644948
58. Kogure Y, Kataoka K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci (2017) 108(9):1719–25. doi: 10.1111/cas.13303
59. Vaque JP, Gomez-Lopez G, Monsalvez V, Varela I, Martinez N, Perez C, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood (2014) 123(13):2034–43. doi: 10.1182/blood-2013-05-504308
60. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in sezary syndrome. Nat Commun (2015) 6:8470. doi: 10.1038/ncomms9470
61. Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and sezary syndrome identifies recurrent alterations in TNFR2. Nat Genet (2015) 47(9):1056–60. doi: 10.1038/ng.3370
62. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large b-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. (2012) 109(10):3879–84. doi: 10.1073/pnas.1121343109
63. Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large b-cell lymphoma. Proc Natl Acad Sci U S A. (2013) 110(4):1398–403. doi: 10.1073/pnas.1205299110
64. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse Large b cell lymphoma. Cell (2017) 171(2):481–94 e15. doi: 10.1016/j.cell.2017.09.027
65. Singh M, Jackson KJL, Wang JJ, Schofield P, Field MA, Koppstein D, et al. Lymphoma driver mutations in the pathogenic evolution of an iconic human autoantibody. Cell (2020) 180 878–894. doi: 10.1016/j.cell.2020.01.029
66. Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol (2010) 17(9):981–8. doi: 10.1016/j.chembiol.2010.07.009
67. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature b and T lymphocytes. Cell (1992) 68(5):869–77. doi: 10.1016/0092-8674(92)90030-G
68. Policheni A, Horikawa K, Milla L, Kofler J, Bouillet P, Belz GT, et al. CARD11 is dispensable for homeostatic responses and suppressive activity of peripherally induced FOXP3(+) regulatory T cells. Immunol Cell Biol (2019) 97(8):740–52. doi: 10.1111/imcb.12268
69. Hu DY, Yap JY, Wirasinha RC, Howard DR, Goodnow CC, Daley SR. A timeline demarcating two waves of clonal deletion and Foxp3 upregulation during thymocyte development. Immunol Cell Biol (2016) 94(4):357–66. doi: 10.1038/icb.2015.95
70. Cowan JE, McCarthy NI, Anderson G. CCR7 controls thymus recirculation, but not production and emigration, of Foxp3(+) T cells. Cell Rep (2016) 14(5):1041–8. doi: 10.1016/j.celrep.2016.01.003
71. Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature (2017) 552(7683):121–5. doi: 10.1038/nature24649
72. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T-cell leukemia-lymphoma after PD-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947–8. doi: 10.1056/NEJMc1803181
73. Ishitsuka K, Utsunomiya A, Ishida T. PD-1 inhibitor therapy in adult T-cell leukemia-lymphoma. N Engl J Med (2018) 379(7):695. doi: 10.1056/NEJMc1807852
74. Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, et al. Expression of FoxP3, a key molecule in CD4CD25 regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br J Haematol (2004) 126(1):81–4. doi: 10.1111/j.1365-2141.2004.04999.x
75. Kohno T, Yamada Y, Akamatsu N, Kamihira S, Imaizumi Y, Tomonaga M, et al. Possible origin of adult T-cell leukemia/lymphoma cells from human T lymphotropic virus type-1-infected regulatory T cells. Cancer Sci (2005) 96(8):527–33. doi: 10.1111/j.1349-7006.2005.00080.x
76. Jeelall YS, Wang JQ, Law HD, Domaschenz H, Fung HK, Kallies A, et al. Human lymphoma mutations reveal CARD11 as the switch between self-antigen-induced b cell death or proliferation and autoantibody production. J Exp Med (2012) 209(11):1907–17. doi: 10.1084/jem.20112744
77. Conley ME. B cells in patients with X-linked agammaglobulinemia. J Immunol (1985) 134(5):3070–4. doi: 10.4049/jimmunol.134.5.3070
78. de Weers M, Brouns GS, Hinshelwood S, Kinnon C, Schuurman RK, Hendriks RW, et al. B-cell antigen receptor stimulation activates the human bruton’s tyrosine kinase, which is deficient in X-linked agammaglobulinemia. J Biol Chem (1994) 269(39):23857–60. doi: 10.1016/S0021-9258(19)51014-6
79. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-kappaB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol (2016) 36(6):533–40. doi: 10.1007/s10875-016-0306-1
80. Lougaris V, Moratto D, Baronio M, Tampella G, van der Meer JWM, Badolato R, et al. Early and late b-cell developmental impairment in nuclear factor kappa b, subunit 1-mutated common variable immunodeficiency disease. J Allergy Clin Immunol (2017) 139(1):349–52 e1. doi: 10.1016/j.jaci.2016.05.045
81. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective b cell development and function in btk-deficient mice. Immunity (1995) 3(3):283–99. doi: 10.1016/1074-7613(95)90114-0
82. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa b leads to multifocal defects in immune responses. Cell (1995) 80(2):321–30. doi: 10.1016/0092-8674(95)90415-8
83. Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med (1998) 187(2):147–59. doi: 10.1084/jem.187.2.147
84. Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and b cell-mediated immune responses. J Exp Med (1998) 187(2):185–96. doi: 10.1084/jem.187.2.185
85. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
86. Horikawa K, Martin SW, Pogue SL, Silver K, Peng K, Takatsu K, et al. Enhancement and suppression of signaling by the conserved tail of IgG memory-type b cell antigen receptors. J Exp Med (2007) 204(4):759–69. doi: 10.1084/jem.20061923
87. Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature (2007) 450(7167):299–303. doi: 10.1038/nature06253
88. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and bcl-6 suppress germinal center reactions. Nat Med (2011) 17(8):983–8. doi: 10.1038/nm.2426
89. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
90. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187(9):4553–60. doi: 10.4049/jimmunol.1101328
91. Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood (2016) 128(11):1490–502. doi: 10.1182/blood-2016-02-698977
92. Sekulic A, Liang WS, Tembe W, Izatt T, Kruglyak S, Kiefer JA, et al. Personalized treatment of sezary syndrome by targeting a novel CTLA4:CD28 fusion. Mol Genet Genomic Med (2015) 3(2):130–6. doi: 10.1002/mgg3.121
93. Woollard WJ, Pullabhatla V, Lorenc A, Patel VM, Butler RM, Bayega A, et al. Candidate driver genes involved in genome maintenance and DNA repair in sezary syndrome. Blood (2016) 127(26):3387–97. doi: 10.1182/blood-2016-02-699843
94. Chen S, Ishii N, Ine S, Ikeda S, Fujimura T, Ndhlovu LC, et al. Regulatory T cell-like activity of Foxp3+ adult T cell leukemia cells. Int Immunol (2006) 18(2):269–77. doi: 10.1093/intimm/dxh366
95. de Leval L, Rickman DS, Thielen C, Reynies A, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood (2007) 109(11):4952–63. doi: 10.1182/blood-2006-10-055145
96. de Leval L, Savilo E, Longtine J, Ferry JA, Harris NL. Peripheral T-cell lymphoma with follicular involvement and a CD4+/bcl-6+ phenotype. Am J Surg Pathol (2001) 25(3):395–400. doi: 10.1097/00000478-200103000-00015
97. Lee SH, Kim JS, Kim J, Kim SJ, Kim WS, Lee S, et al. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T-cell lymphoma patients. Haematologica (2015) 100(12):e505–7. doi: 10.3324/haematol.2015.133074
98. Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia (2016) 30(5):1062–70. doi: 10.1038/leu.2015.357
99. Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(4):371–5. doi: 10.1038/ng.2916
100. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171–5. doi: 10.1038/ng.2872
101. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166–70. doi: 10.1038/ng.2873
102. Cortes JR, Ambesi-Impiombato A, Couronne L, Quinn SA, Kim CS, da Silva Almeida AC, et al. RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell (2018) 33(2):259–73 e7. doi: 10.1016/j.ccell.2018.01.001
103. Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, et al. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol (2008) 180(2):774–82. doi: 10.4049/jimmunol.180.2.774
104. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006
105. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
106. Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. (1982) 79(6):2031–5. doi: 10.1073/pnas.79.6.2031
107. Yoshikai Y, Anatoniou D, Clark SP, Yanagi Y, Sangster R, Van den Elsen P, et al. Sequence and expression of transcripts of the human T-cell receptor beta-chain genes. Nature (1984) 312(5994):521–4. doi: 10.1038/312521a0
108. Munoz E, Courtois G, Veschambre P, Jalinot P, Israel A. Tax induces nuclear translocation of NF-kappa b through dissociation of cytoplasmic complexes containing p105 or p100 but does not induce degradation of I kappa b alpha/MAD3. J Virol (1994) 68(12):8035–44. doi: 10.1128/jvi.68.12.8035-8044.1994
109. Kannian P, Yin H, Doueiri R, Lairmore MD, Fernandez S, Green PL. Distinct transformation tropism exhibited by human T lymphotropic virus type 1 (HTLV-1) and HTLV-2 is the result of postinfection T cell clonal expansion. J Virol (2012) 86(7):3757–66. doi: 10.1128/JVI.06900-11
110. Michaelsson J, Barbosa HM, Jordan KA, Chapman JM, Brunialti MK, Neto WK, et al. The frequency of CD127low expressing CD4+CD25high T regulatory cells is inversely correlated with human T lymphotrophic virus type-1 (HTLV-1) proviral load in HTLV-1-infection and HTLV-1-associated myelopathy/tropical spastic paraparesis. BMC Immunol (2008) 9:41. doi: 10.1186/1471-2172-9-41
111. Satou Y, Utsunomiya A, Tanabe J, Nakagawa M, Nosaka K, Matsuoka M. HTLV-1 modulates the frequency and phenotype of FoxP3+CD4+ T cells in virus-infected individuals. Retrovirology (2012) 9:46. doi: 10.1186/1742-4690-9-46
Keywords: CARD11, mutation, gain-of-function, regulatory T cell, follicular T cell, germinal center, lymphoproliferation, lymphoma
Citation: Masle-Farquhar E, Jeelall Y, White J, Bier J, Deenick EK, Brink R, Horikawa K and Goodnow CC (2023) CARD11 gain-of-function mutation drives cell-autonomous accumulation of PD-1+ ICOShigh activated T cells, T-follicular, T-regulatory and T-follicular regulatory cells. Front. Immunol. 14:1095257. doi: 10.3389/fimmu.2023.1095257
Received: 11 November 2022; Accepted: 23 February 2023;
Published: 07 March 2023.
Edited by:
Niklas Beyersdorf, Julius Maximilian University of Würzburg, GermanyReviewed by:
Miguel A. Piris, University Hospital Fundación Jiménez Díaz, SpainCopyright © 2023 Masle-Farquhar, Jeelall, White, Bier, Deenick, Brink, Horikawa and Goodnow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Etienne Masle-Farquhar, ZS5tYXNsZS1mYXJxdWhhckBnYXJ2YW4ub3JnLmF1; Yogesh Jeelall, eW9nZXNoLmplZWxhbGxAZ21haWwuY29t
†Present address: Yogesh Jeelall, Royal Perth Hospital, Perth, Western Australia
‡These authors have contributed equally to this work and share last authorship
§These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.