 Kristina D. Hinman1,2,3
Kristina D. Hinman1,2,3 Sonia S. Laforce-Nesbitt4
Sonia S. Laforce-Nesbitt4 Joshua T. Cohen1Miles Mundy3
Joshua T. Cohen1Miles Mundy3 Joseph M. Bliss2,4
Joseph M. Bliss2,4 Alexander R. Horswill5
Alexander R. Horswill5 Craig T. Lefort1,2*
Craig T. Lefort1,2*- 1Division of Surgical Research, Department of Surgery, Rhode Island Hospital, Providence, RI, United States
- 2Warren Alpert Medical School, Brown University, Providence, RI, United States
- 3Graduate Program in Pathobiology, Brown University, Providence, RI, United States
- 4Department of Pediatrics, Women and Infants Hospital, Providence, RI, United States
- 5Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO, United States
Techniques for studying the clearance of bacterial infections are critical for advances in understanding disease states, immune cell effector functions, and novel antimicrobial therapeutics. Intracellular killing of Staphylococcus aureus by neutrophils can be monitored using a S. aureus strain stably expressing GFP, a fluorophore that is quenched when exposed to the reactive oxygen species (ROS) present in the phagolysosome. Here, we expand upon this method by developing a bi-fluorescent S. aureus killing assay for use in vivo. Conjugating S. aureus with a stable secondary fluorescent marker enables the separation of infected cell samples into three populations: cells that have not engaged in phagocytosis, cells that have engulfed and killed S. aureus, and cells that have viable internalized S. aureus. We identified ATTO647N-NHS Ester as a favorable dye conjugate for generating bi-fluorescent S. aureus due to its stability over time and invariant signal within the neutrophil phagolysosome. To resolve the in vivo utility of ATTO647N/GFP bi-fluorescent S. aureus, we evaluated neutrophil function in a murine model of chronic granulomatous disease (CGD) known to have impaired clearance of S. aureus infection. Analysis of bronchoalveolar lavage (BAL) from animals subjected to pulmonary infection with bi-fluorescent S. aureus demonstrated differences in neutrophil antimicrobial function consistent with the established phenotype of CGD.
Introduction
The inflammatory response to infection is a dynamic process with tissue-specific variability: leukocytes are recruited to cross endothelial barriers at different times, with specificity dependent on the cytokine environment and cellular affinity for endothelial adhesion receptors (1, 2). In response to the initial insult, resident cells such as macrophages or natural killer cells recruit innate immune cells as the first line of defense (3–5). In the acute phase, neutrophils are the most abundant infiltrating leukocyte in response to bacterial infections. This recruitment is self-amplified by neutrophil-derived signals such as interleukin-8 (IL-8) and leukotriene B4 (LTB4) (3, 6). Tight regulation over recruitment is crucial as neutrophil depletion is often fatal while overactivation leads to enzymatic and oxidative damage to the tissues involved (4, 7).
Neutrophils clear bacterial infections through numerous intra- and extracellular effector functions, depending on the species of bacteria. Staphylococcus aureus is one species of interest due to its prevalence, ability to evade the immune system, and increasing resistance to pharmaceutical anti-microbial agents. As of 2018, methicillin-resistant S. aureus (MRSA) was the leading cause of death resulting from an antibiotic-resistant pathogen (8). Neutrophils predominantly kill S. aureus via internalization into a phagolysosome (5). Within the phagolysosome, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex generates reactive oxygen species (ROS) via a series of reactions referred to as the oxidative burst (7, 9). The proposed mechanisms of bacterial killing by ROS include oxidative damage of the microbe membrane, DNA, and other cellular components (7, 10, 11). In addition, NADPH oxidase activity may modulate the potency of proteolytic enzymes that are delivered to the phagosome and contribute to microbicidal activities (10, 12).
Numerous pathologies affect the neutrophil-dependent clearance of bacteria. An impaired neutrophilic response may be the result of depleted cell counts (neutropenia), the inability to recruit cells to inflamed tissue sites, or loss of effector functions (13). Neutrophil dysfunction can be caused by a primary immunodeficiency or can occur secondary to a disease state, such as sepsis or cancer (5, 9, 14). There are sufficient experimental methods for quantifying neutrophil counts to evaluate defects in production or tissue-specific recruitment, including the characterization of surface markers by flow cytometry or morphology by cytospin. However, methods for evaluating microbe killing on a single cell level in vivo are relatively limited. Current methods depend on comparing pathogen clearance through plating tissue homogenates and counting colony forming units (CFU) ex vivo. This method is limited as it is non-specific, only providing insight into overall host defense discrepancies without identifying the specific effector cell(s) responsible for observed differences. In vitro experiments may implicate cellular function more specifically but exclude the influence of host extracellular factors and proteins that aid in the clearance of S. aureus (15). Alternatives based on live in vivo imaging require advanced technology which is expensive, time-consuming, and limited by the target tissue location (16). Therefore, we sought to develop a fluorescence-based assay to quantify in vivo neutrophil antimicrobial function on a single cell level.
Previous work by Schwartz, et al. has demonstrated in vitro that the fluorescent signal from a GFP-producing S. aureus is quenched in a ROS-dependent manner over time (17, 18). This loss of fluorescence is correlated with intracellular S. aureus viability. The mechanism of GFP bleaching is attributed to the HOCl in the phagosome, likely via chlorination of the tyrosine 66 residue in GFP (19). For a single point in time, the GFP signal alone is insufficient to distinguish cells that have not undergone phagocytosis of S. aureus from cells that have bleached the GFP fluorescent signal in conjunction with eradicating internalized S. aureus. Therefore, we screened for a secondary fluorescent dye capable of maintaining a robust fluorescent signal over time, independent of ROS production, phagolysosome activity, or S. aureus viability. We generated bi-fluorescent S. aureus by conjugating a far-red fluorescent dye to a GFP-producing USA300 S. aureus. In our study, we extensively characterize neutrophil intracellular killing of bi-fluorescent S. aureus to demonstrate the fidelity and utility of the assay for evaluating neutrophil antimicrobial function or dysfunction in vivo.
Materials and methods
Staphylococcus aureus culture and staining
Glycerol stocks of the superfolded GFP-producing USA300 strain of S. aureus were provided by Alexander Horswill and have been described (18). Liquid cultures were grown in tryptic soy broth (TSB, Sigma-Aldrich, St. Louis, MO) from the glycerol stock by incubating at 37C, shaking at 250 rpm, overnight. Before experimentation, overnight cultures were diluted and allowed to grow to the exponential phase. For staining, cultures were washed 2x in phosphate-buffered saline, pH=7.4 (PBS, Gibco, Dun Laoghaire, Co Dublin, Ireland), and brought to a concentration of 4x108 CFU/mL. S. aureus was stained for 30 minutes in the dark at a final concentration of 0.5ug/mL of ATTO647N-NHS ester (Sigma-Aldrich). To pellet, cultures were centrifuged for 5 minutes at 3000 RPM. After staining, S. aureus was washed 2x in assay buffer (Hank’s balanced salt solution/10% fetal bovine serum/20 mM HEPES). Before use in vitro, S. aureus was opsonized by incubating at 37C and shaking for 5-10 minutes in assay buffer.
Growth curves were generated by bringing bi-fluorescent or GFP S. aureus to OD600 = 0.1 in TSB. OD was measured using SmartSpec 3000 (Bio-Rad, Hercules, CA). Cultures were grown for four and a half hours, with a sample drawn every 30 minutes for the first 2 hours and then at 3 hours and 4.5 hours. At each time point, the OD600 was measured and the fluorescence of the S. aureus particles was measured using a MACSQuant Analyzer 10 flow cytometer (Miltenyi).
HoxB8 conditionally immortalized progenitor derivation, culture,and differentiation
HoxB8 conditionally immortalized neutrophil progenitors were generated as previously described (20–22). Briefly, C57BL/6 mouse bone marrow was isolated, cultured, and transduced with a tamoxifen-inducible Hoxb8 transgene. All cells were grown in Opti-Mem media supplemented with GlutaMax (Gibco), 30uM beta-mercaptoethanol (Sigma-Aldrich), 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA), 1x penicillin/streptomycin (Gibco), 1x non-essential amino acids (Gibco) at a density of ~106 cells/mL. The progenitor cell culture was supplemented with stem cell factor (SCF) and 100nM Z-4-hydroxytamoxifen (Tocris Bioscience, Bristol, UK) to induce Hoxb8 expression. SCF was derived from the supernatant of CHO cells that secrete recombinant murine SCF (a gift from Patrice Dubreuil, Centre de Recherche en Cancérologie de Marseille). For differentiation, cells were washed 3x in PBS to remove Z-4-hydroxytamoxifen induction. Cells were pelleted by centrifugation at 400xg for 3 minutes. The differentiation culture was supplemented with SCF and 20 ng/mL G-CSF (BioLegend) for 2-3 days, and then just G-CSF until use on days 5-7.
In vitro killing assay
Differentiated Hoxb8 progenitor-derived neutrophils were suspended in assay buffer at a density of 1x107 cells/mL. Samples treated with diphenyleneiodonium (DPI; Selleck Chemicals, Houston, TX) or cytochalasin D (Sigma-Aldrich) were incubated at 37°C for 20 minutes before infection. For each sample, one million cells were inoculated with 25µL of OD=0.5 S. aureus for an MOI of 20:1 for 15 minutes. Cells were then washed with warm lysostaphin 200ng/mL followed by assay buffer 1x. Cells were suspended in a final volume of 100µL of assay buffer. For killing assays, this was considered time zero. For experiments determining fluorescent intensity over time, every 20 minutes 10µL of the sample was plated in 200µL of cold PBS/1% FBS and kept on ice until analysis by the MACSQuant.
Dead S. aureus phagocytosis assay
Bi-fluorescent S. aureus was heat-inactivated at 60C for 30 minutes. To evaluate the phagocytosis of dead S. aureus, cells were inoculated and washed using the same protocol as the live S. aureus killing assay. Cells were analyzed at time zero (after lysostaphin wash) to evaluate the internalization of the heat-killed S. aureus.
Animal care and breeding
All studies were performed under the approval of the Lifespan Animal Welfare Committee (Protocol number 5017-19, Office of Laboratory Animal Welfare Assurance #A3922-01). These studies follow Public Health Service guidelines for animal care and use. The CGD mouse model was acquired from Jackson Laboratories (Bar Harbor, ME), strain B6.129S-Cybbtm1Din/J for in-house breeding. This strain was originally developed by knocking out the Cybb gene (gp91phox) to recapitulate the CGD phenotype. Female mice heterozygous for the knocked out Cybb gene (x-linked) were bred with wild-type males to yield hemizygous wild-type (WT) or knockout male progeny used in this study. Animals were housed in sterile caging until infection at 6-10 weeks old and provided access to water and standard chow ad libitum.
S. aureus pulmonary infection
S. aureus was stained as previously described and brought to an OD600 = 0.5 or 2x108 CFU/mL. One mL of S. aureus was pelleted and suspended in 100µL of normal saline for a working solution. The inoculum was further diluted to provide a bolus of 6x107CFU or 8x107CFU in 40µL. Animals were anesthetized with vaporized isoflurane (Braintree Scientific, Braintree, MA) before oropharyngeal inhalation of the inoculum.
In vivo killing assay
Twelve hours post pulmonary infection, mice were euthanized by Fatalplus (Vortech Pharmaceuticals, Dearborn, MI) and cervical dislocation. Bronchoalveolar lavage (BAL) was collected by pushing and withdrawing 1mL cold 1% FBS/PBS into the lungs via an angiocatheter (performed 3x) and then passed through a 70-micron filter (Falcon, Corning, NY). Cells were pelleted and resuspended in 2mL lysostaphin and then washed twice in 1% FBS/PBS. To determine total cell counts, the pellet was brought to 1mL before analysis. Cells were stained with PE anti-Ly6G (BioLegend) in 1% FBS/PBS for 30 minutes. Before analysis by flow cytometry, samples were washed 3x in 1%FBS/PBS. Samples were analyzed on a MACSQuant and data were analyzed with FlowJo software to determine fluorescent intensities, population percentages, and total counts.
Colony forming units assays
BAL samples were stained with PE anti-Ly6G (BioLegend) and sorted into three populations: Ly6G+ATTO647N+GFP+, Ly6G+ATTO647N+GFP-, and Ly6G+ATTO647N-GFP-. Cell sorting was performed using a FACSAria (BD). In vitro assay samples omitted the PE anti-Ly6G stain and were sorted based on ATTO647N and GFP expression. Samples were pelleted and resuspended in 1mL of pH=11 H2O. Cells were lysed by incubating and vortexing for ~5 minutes (23). Lysates were serially diluted at 1:10 and plated, 3x10µL drops per dilution onto TSB agar plates. Plates were incubated at 37C overnight. Colonies were counted and the total CFU burden of the sorted cells was determined. CFU burden was normalized to the total events sorted in each population.
Imaging
For imaging analysis of cell suspensions, cells were spun at 1500 RPM onto a superfrost plus glass slide (Fisher Scientific, Pittsburgh, PA) after phagocytosis and killing in vitro. Ex vivo analysis of lung tissue was performed through frozen sectioning of infected animals. WT animals were infected and euthanized twelve hours post-infection. The lungs were carefully removed avoiding puncture and inflated through the trachea with 0.8mL of a 2:1 mixture of optimal cutting temperature compound (OCT):PBS. The trachea was tied with a suture and the lungs were immediately frozen in liquid nitrogen. Once frozen, the tissue was then embedded in a mold with OCT (Sakura Finetek, Torrance, CA) on dry ice. Samples were sliced in a cryostat microtome at a thickness of 5-7 µm and adhered to superfrost plus glass slides. Prior to imaging, slides were fixed in cold acetone for 2 minutes and allowed to dry at room temperature.
For both types of imaging experiments, coverslips were applied with antifade mounting medium with DAPI (Vectashield, Newark, CA) and immediately imaged. Slides were imaged using a Nikon Eclipse 80i, with light provided by an X-Cite 120 lamp (Excelitas Technologies). Images were captured with a Retiga EXi Fast 1394 camera (QImaging, Burnaby, BC, Canada). Post-imaging analyses to merge images, analyze colocalization, and include scale bars were done using ImageJ.
In order to measure the colocalization of the GFP and ATTO647N fluorescence in the lung tissue, 11 images from distinct sections of lung tissue were analyzed (20x magnification). Images were collected by sectioning lungs from 3 separate animals. First, the background signal was subtracted from the GFP and ATTO647N images. The two images were compared with the “colocalization threshold” and “colocalization test” functions. Since we do not expect a relationship between the fluorescent intensity of the ATTO647N and GFP signal but do expect spatial correlation, we report Mander’s Coefficient for the GFP and ATTO647N channels relative to one another. The colocalization test was run with Costes approximation and 25 iterations of randomized pixels.
Human neutrophil infections
Human cord blood was collected following approval of the Care New England Women & Infants Hospital IRB, Project number 04-0061. To isolate white blood cells, 4mL of cord blood was lysed with red blood cell lysis buffer (BioLegend) in the dark for 20 minutes. Cells were then washed 2x in assay buffer. Cells were suspended in 1.20mL of assay buffer and 100µLwas used per sample. Samples were inoculated with 15µL of bi-fluorescent S. aureus prepared as described above. Cells were incubated for 15 minutes at 37C, washed with warm lysostaphin, and brought to a final volume of 100uL. At the given time points (0, 30, 60, and 120 minutes) 10µL of the sample was plated into 200µL of cold PBS/FBS for analysis using a MACSQuant flow cytometer.
Statistical analysis
Graphical depictions of the data represent group means and standard deviations (SD) generated using Prism 9 (GraphPad). The SD represents biological replicates across all figures except Figure 3C, in which the SD is representative of technical replicates calculating the CFU from one sample lysate for the distinct samples represented by each point on the plot. Where indicated, statistical analysis was conducted to compare groups. First, significance was calculated by the Prism 9 ordinary one-way ANOVA test. A p-value threshold of 0.05 indicated a significant difference when comparing the four groups (WT low, WT high, CGD low, CGD high) to one another using Tukey’s multiple comparisons tests. When there was no significant difference between the low and high dose for the same genotype but there was a significant difference between genotypes for each dose, this was represented graphically by grouping the genotypes under one line and indicating the asterisk for the highest multiple comparisons p-value determined by Tukey’s test. Asterisks indicate significance as follows: “ns” (not significant) for p>0.05, * for p<0.05, ** for p<0.01, *** p<0.001, and **** for p<0.0001.
Results
ATTO647N-NHS Ester fluorescently labels S. aureus
We found that staining superfolded GFP-producing S. aureus (18) with a low concentration of ATTO647N-NHS Ester generated a pure population of ATTO647N+GFP+ bi-fluorescent particles (Figure 1A). The stain did not affect S. aureus viability as the growth rate in culture was comparable to the unstained GFP-fluorescing S. aureus (Figure 1B). We predicted that with bacterial cell division, the ATTO647N mean fluorescent intensity (MFI) would decrease due to the dilution of the intracellular concentration of the dye. To determine the change in fluorescent intensity with proliferation, bi-fluorescent and GFP-fluorescent S. aureus cultures were grown from an initial optical density (OD) of 0.1. As expected, S. aureus stably expressing GFP maintained a detectable signal with culture growth, as particles remained nearly 100% GFP+ compared to a non-fluorescing, unstained S. aureus control (Figure 1C). The GFP MFI also remained robust with culture growth and was comparable between the bi-fluorescent and GFP-fluorescent groups (Figure 1D). The ATTO647N MFI began to decrease with growth in culture as the OD increased (Figure 1D). However, with the bacterial proliferation, the ATTO647N signal remained high enough to differentiate between stained and unstained S. aureus until an OD of 1.0 when the population begins to overlap with the unstained control (Figure 1E). Overall, the ATTO647N signal of conjugated S. aureus remains detectable after a substantial level of bacterial replication, without affecting S. aureus growth, making it an optimal candidate as a secondary marker for the assessment of bacterial killing by leukocytes.
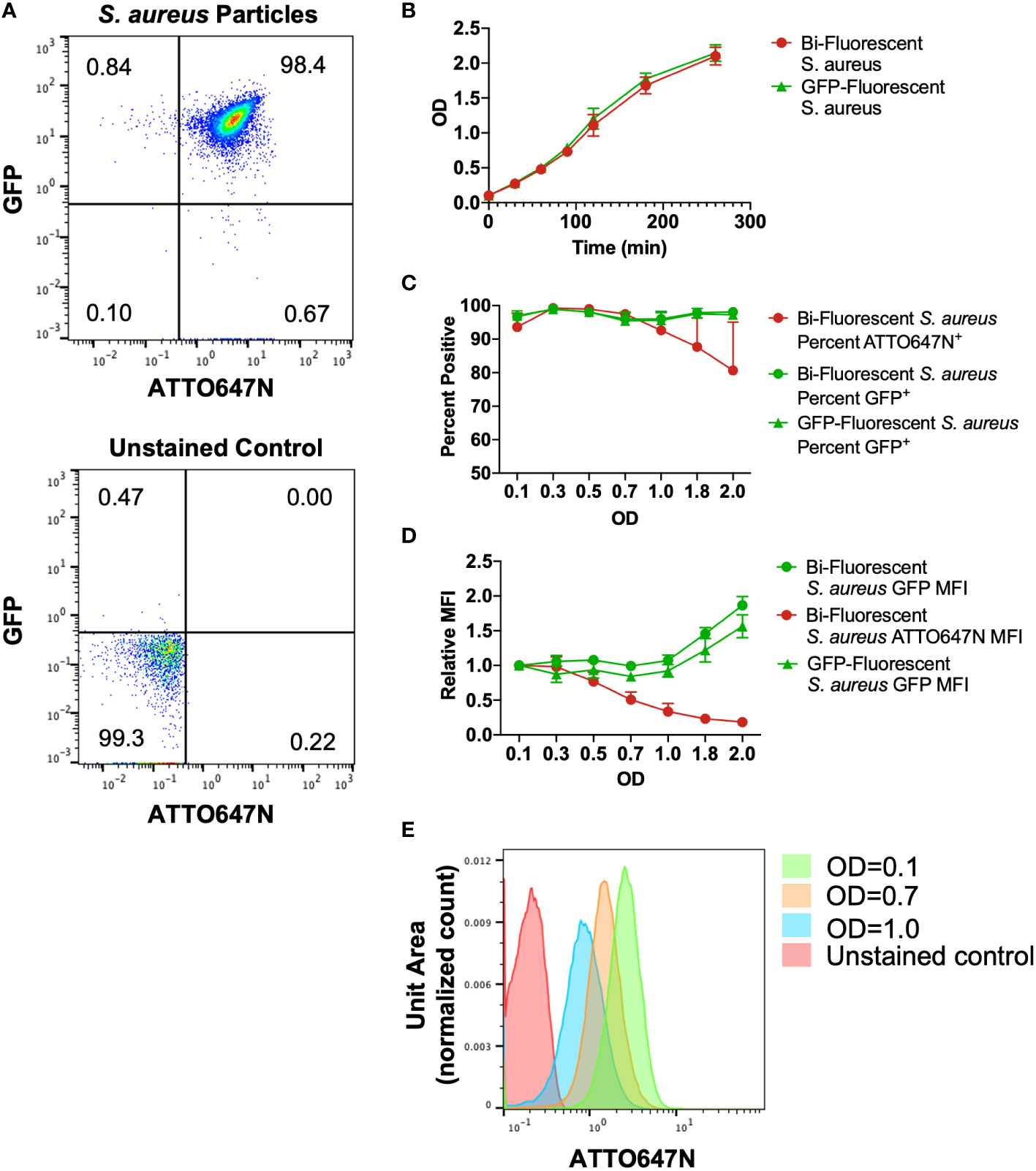
Figure 1 Characterization of cultured bi-fluorescent S. aureus. (A) Staining GFP-S. aureus with 200ng/mL ATTO647N-NHS Ester generates a pure population of bi-fluorescent S. aureus detectable by flow cytometry. (B) The growth curve of S. aureus with or without secondary ATTO647N-NHS Ester stain was indistinguishable. (C) The percent of particles gated as GFP+ or ATTO647N+ relative to unstained control demonstrates a loss of detection of the ATTO647N signal with bacterial replication. (D) Geometric mean fluorescent intensity (MFI) of GFP or ATTO647N signal relative to the signal at the start of growth in culture demonstrates robust GFP MFI and loss of ATTO647N signal with bacterial replication (n=3, performed across three independent experiments). (E) Representative shifts in the ATTO647N fluorescent intensity towards the unstained control with increasing OD, with overlap visualized at OD=1.0.
ATTO647N-NHS ester fluorescence is resistant to intraphagosomal bleaching
Conditional ectopic expression of transcription factor Hoxb8 in murine bone marrow stem cells produces a progenitor cell line capable of exponential expansion while Hoxb8 is induced or neutrophil differentiation through Hoxb8 withdrawal (22). Differentiation of Hoxb8 progenitors derived from a wild-type (WT) C57BL/6 mouse into mature neutrophils (HB8 neutrophils) was confirmed through characterization of surface markers. Differentiation of progenitors into neutrophils was confirmed by increased Ly6G expression and loss of cKit expression by HB8 neutrophils (Supplemental Figures 1A, B). HB8 neutrophils produce ROS and have microbial killing capacity in vitro (20), making them a useful screening tool for the effects of phagocytosis and intracellular killing of the bi-fluorescent S. aureus. For preliminary experiments, the bi-fluorescent S. aureus was phagocytosed by HB8 neutrophils in vitro, treated with lysostaphin to remove residual extracellular S. aureus (24), and changes in fluorescence were monitored over time as intracellular killing occurred (Figure 2A). Treating cells with cytochalasin D before inoculation to prevent phagocytosis by disrupting the actin cytoskeleton blocked neutrophils from acquiring the GFP and the ATTO647N signal, confirming that the detected fluorescence is predominantly acquired by internalizing S. aureus (Supplemental Figures 2A, B).
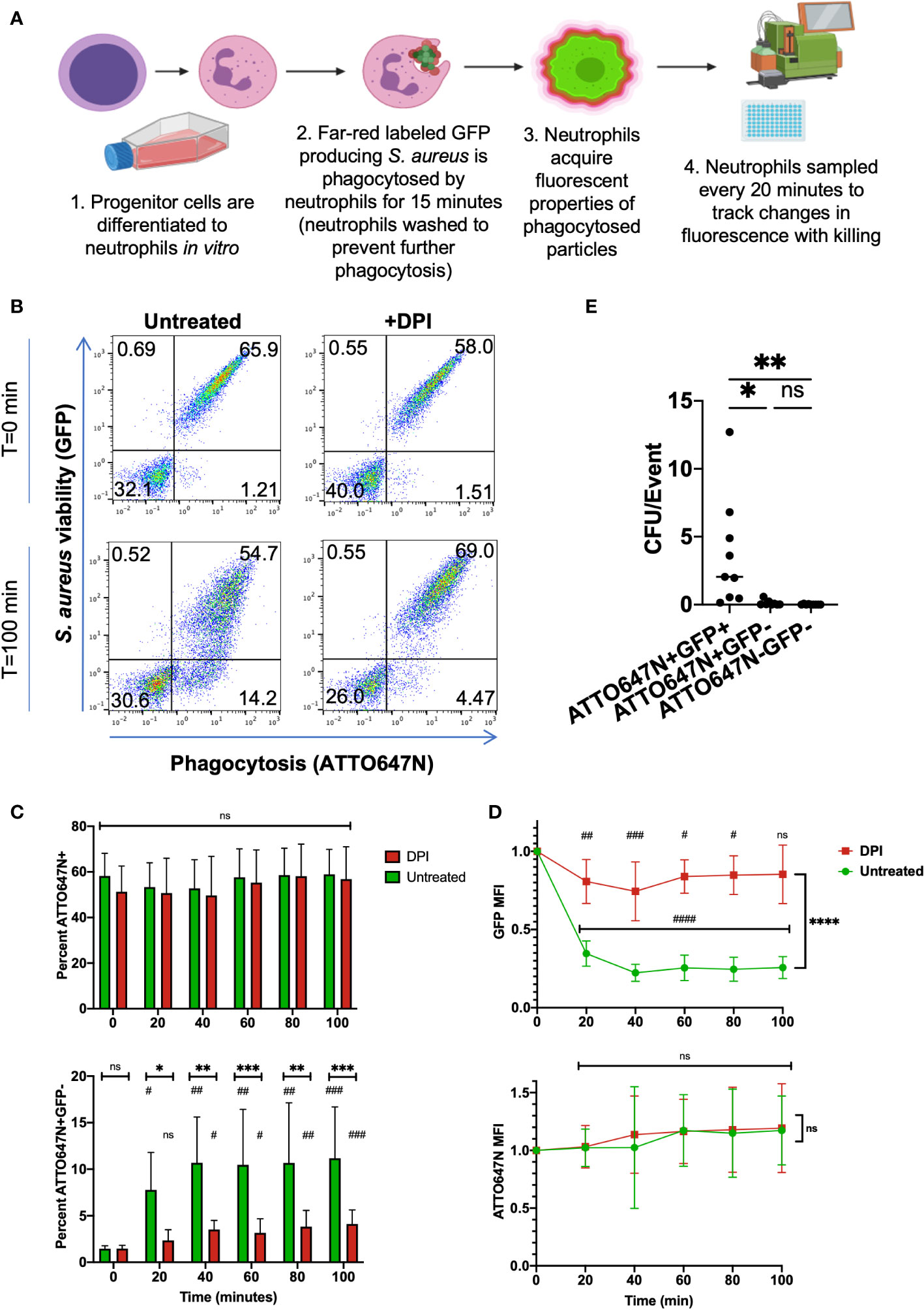
Figure 2 In vitro killing assay of bi-fluorescent S. aureus. (A) Schematic of in vitro screening process of bi-fluorescent S. aureus. (B) Immediately following phagocytosis, HB8 neutrophils are either ATTO647N-GFP- or ATTO647N+GFP+. After 100 minutes, an ATTO647N+GFP- population forms in the untreated group but not the DPI-treated group. (C) The percent of total ATTO647N+ HB8 neutrophils remains constant over time with no significant difference between untreated and DPI groups, or between the initial time point and subsequent time points within each group. Meanwhile, the percent of ATTO+GFP- cells increases over time within each group (statistical significance indicated by #) and these changes are significantly greater in the untreated cells relative to the DPI treated cells at each time point (statistical significance indicated by *) (D) Quantification of GFP MFI within the ATTO647N+ events shows a dramatic loss of fluorescence over time in the untreated cells compared to the DPI treated cells and compared to the initial time point (the statistical significance between each time point relative to the initial time point is indicated by #; statistical significance between the untreated and DPI group is indicated by *). Meanwhile, the ATTO647N MFI remains consistent in both groups (n=9, performed across three independent experiments). (E) Bacterial burden from sorted cell lysates from both DPI treated and untreated cells, normalized to the total number of sorted events (n=9, performed across two independent experiments). “ns” (not significant) for p>0.05, * for p<0.05, ** for p<0.01, *** p<0.001, and **** for p<0.0001.
At the initial time point following phagocytosis, the HB8 neutrophils with engulfed S. aureus acquired the bi-fluorescent properties, as measured by flow cytometry (Figure 2B). The percentage of ATTO647N+ events was stable over time despite changes in the percentage of GFP+ events (Figure 2C). As expected, the percentage of GFP+ events and GFP MFI decreased over time in HB8 neutrophils, suggesting successful intracellular S. aureus killing (Figure 2D). Importantly, these metrics remained the same over time in the ATTO647N channel (Figure 2D). HB8 neutrophils treated with diphenyleneiodonium (DPI), to inhibit the generation of ROS, exhibit a robust GFP signal that is maintained over time (Figures 2B–D). The bleaching of the GFP signal, but not the ATTO647N signal, suggests that the latter fluorophore is resistant to quenching by neutrophils and remains stable in the conditions of the active phagosome.
Taken together, these results suggest that ATTO647N fluorescence identifies the population of cells that have undergone phagocytosis of S. aureus and GFP fluorescence indicates which of those cells contain viable S. aureus. Therefore, we can group cells that have encountered the bi-fluorescent S. aureus into three categories: ATTO647N+GFP+ cells which have viable intracellular S. aureus, ATTO647N+GFP- cells which have non-viable intracellular S. aureus, and ATTO647N-GFP- cells which have not phagocytosed S. aureus.
Fluorescent properties of neutrophils reflect intracellular S. aureus viability
To validate that the ATTO647N and GFP fluorescence associated with neutrophils represent the expected intracellular S. aureus viability, we exposed neutrophils to bi-fluorescent S. aureus and then sorted cells within the three quadrants described above. After allowing 120 minutes for intracellular killing of ingested S. aureus, both untreated and DPI-treated HB8 neutrophils were sorted, lysed, and plated on solid media to quantify the number of viable S. aureus. Cells were lysed using deionized water with a pH of 11 to release all viable intracellular bacteria and ensure that S. aureus colonies were not under-represented due to impaired dispersal on the agar plate (23). The number of S. aureus colony forming units (CFUs) was normalized to the number of events (HB8 neutrophils) that were sorted from the bulk population. Both the ATTO647N+GFP- and ATTO647N-GFP- sorted cell populations yielded negligible CFU (Figure 2E). Meanwhile, the sorted ATTO647N+GFP+ cells contained significantly more CFUs confirming the presence of viable intracellular S. aureus (Figure 2E). These data also validate that the quenching of GFP is a reliable marker for neutrophils that have completed intracellular killing of S. aureus.
Next, we investigated whether the GFP MFI was related to intracellular S. aureus viability. Microscopic imaging of HB8 neutrophils after 120 minutes of intracellular killing in suspension provides insight into the variability of neutrophil phagocytosis and killing (Figures 3A, B). Within the bulk population, there are HB8 neutrophils that have phagocytosed different quantities of S. aureus particles, as indicated by the broad range of ATTO647N fluorescent intensity by flow cytometry (Figure 2B) and visualized microscopically (Figures 3A, B). Accordingly, in the untreated HB8 neutrophil population some cells have both viable (GFP+) and non-viable (GFP-) particles internalized and potentially within the same phagolysosome (Figure 3B). Meanwhile, based on GFP fluorescence, nearly all S. aureus is near completely viable DPI-treated cells (Figure 3B). These images, in conjunction with the findings of Schwartz, et al., establish a positive relationship between GFP fluorescence and S. aureus cocci within neutrophils suggesting that GFP MFI may provide insight into the intracellular S. aureus burden of individual cells or bulk populations. We expect that cells or populations with a higher MFI have a greater bacterial burden despite some degree of variability in the fluorescent intensity of the original S. aureus population (Figure 1A).
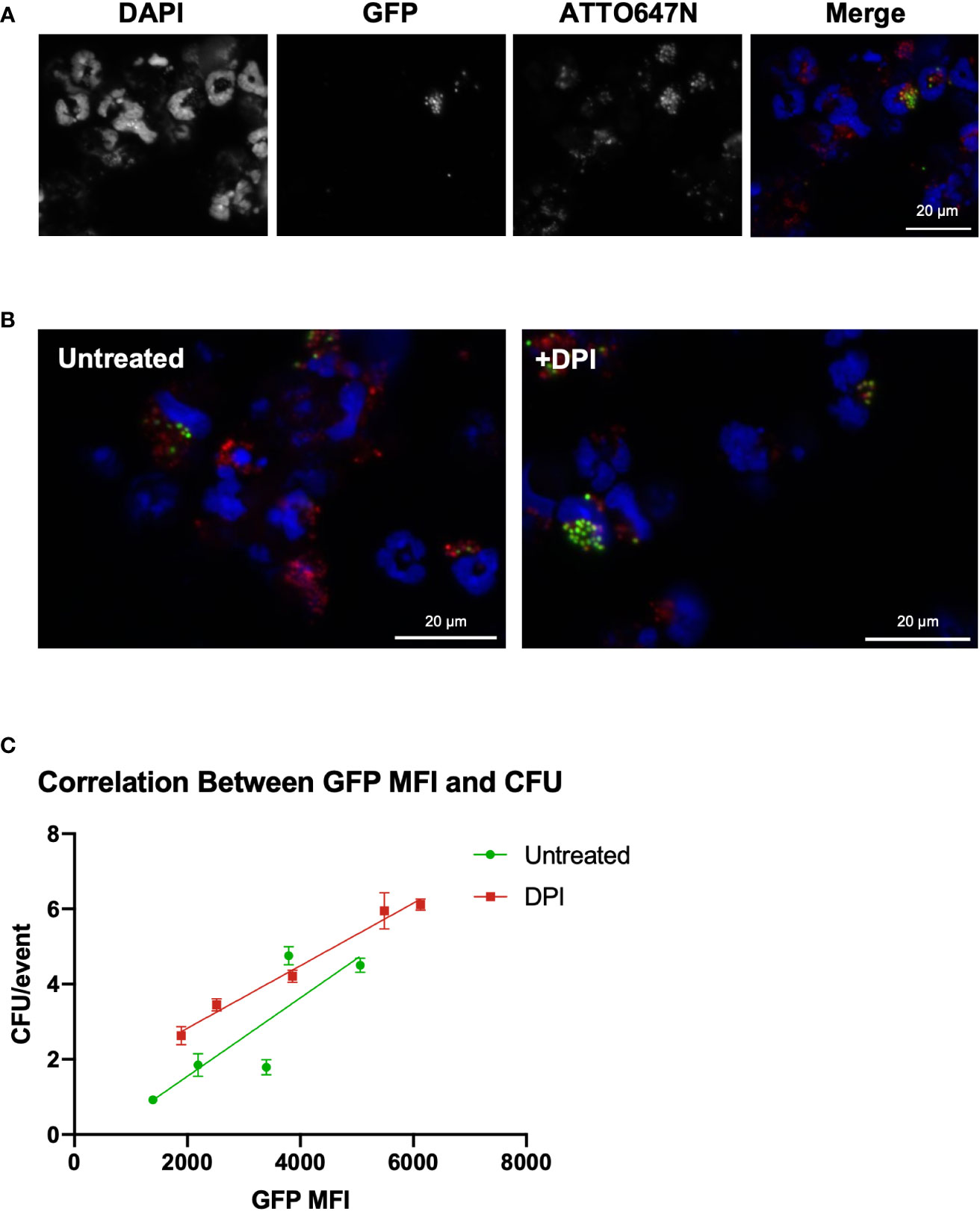
Figure 3 GFP fluorescent intensity correlates with the number of viable internalized S. aureus cocci. (A) Imaging of HB8 neutrophils after 2 hours of killing in vitro (40x). (B) GFP/ATTO647N merged images for untreated and DPI treated HB8 neutrophils (40x). (C) Correlation between GFP MFI and viable intracellular bacterial burden (CFU representative of plating lysate in triplicate).
To generate cell populations with various MFIs, we inoculated HB8 neutrophils with an increasing multiplicity of infection and sorted the ATTO647N+GFP+ population of neutrophils 120 minutes after allowing the intracellular killing to occur. As expected, we found that the cell populations with higher GFP MFI had a greater intracellular bacterial burden (Figure 3C). We also wanted to determine whether the GFP MFI of DPI-treated neutrophils was proportionately representing viable intracellular bacterial burden, a key question given that GFP quenching is ROS-dependent (17). We found the correlation between GFP MFI and CFUs from the DPI-treated sample was comparable to that of the control, supporting the notion that GFP MFI is a robust parameter for quantifying intracellular bacterial burden (Figure 3C).
Application of bi-fluorescent S. aureus to demonstrate neutrophil dysfunction
Chronic granulomatous disease (CGD) is marked by neutrophil dysfunction due to a loss of function of the NADPH oxidase responsible for superoxide production in the phagolysosome. Since S. aureus killing is dependent on NADPH oxidase activity, hosts with CGD are disproportionately susceptible to S. aureus infections (7). Loss-of-function mutations in the x-linked CYBB gene encoding the gp91phox subunit are the most common cause of CGD. The widely used murine CGD model harbors knockout of the Cybb gene leading to loss of ROS production by neutrophils. CGD mice recapitulate the human disease well, including the impaired clearance of S. aureus infections (25). Therefore, we used this disease model to assess the in vivo use of the bi-fluorescent S. aureus to quantify neutrophil killing capacity.
We harvested samples from wild-type (WT) and CGD mice twelve hours after pulmonary infection with a low (6x107 CFU) or high (8x107 CFU) inoculum of bi-fluorescent S. aureus. Since the ATTO647N signal dilutes with significant bacterial replication, we recognize that bi-fluorescent S. aureus is primarily applicable to analyzing the acute response to infection. Bronchoalveolar lavages (BAL) were collected and stained with anti-Ly6G to identify the neutrophils via flow cytometry (Figure 4A). Very few Ly6G- events were GFP+ or ATTO647N+ indicating that neutrophils were the predominant cell type responsible for phagocytosis of S. aureus in the airspaces at this time point (Supplemental Figure 3).
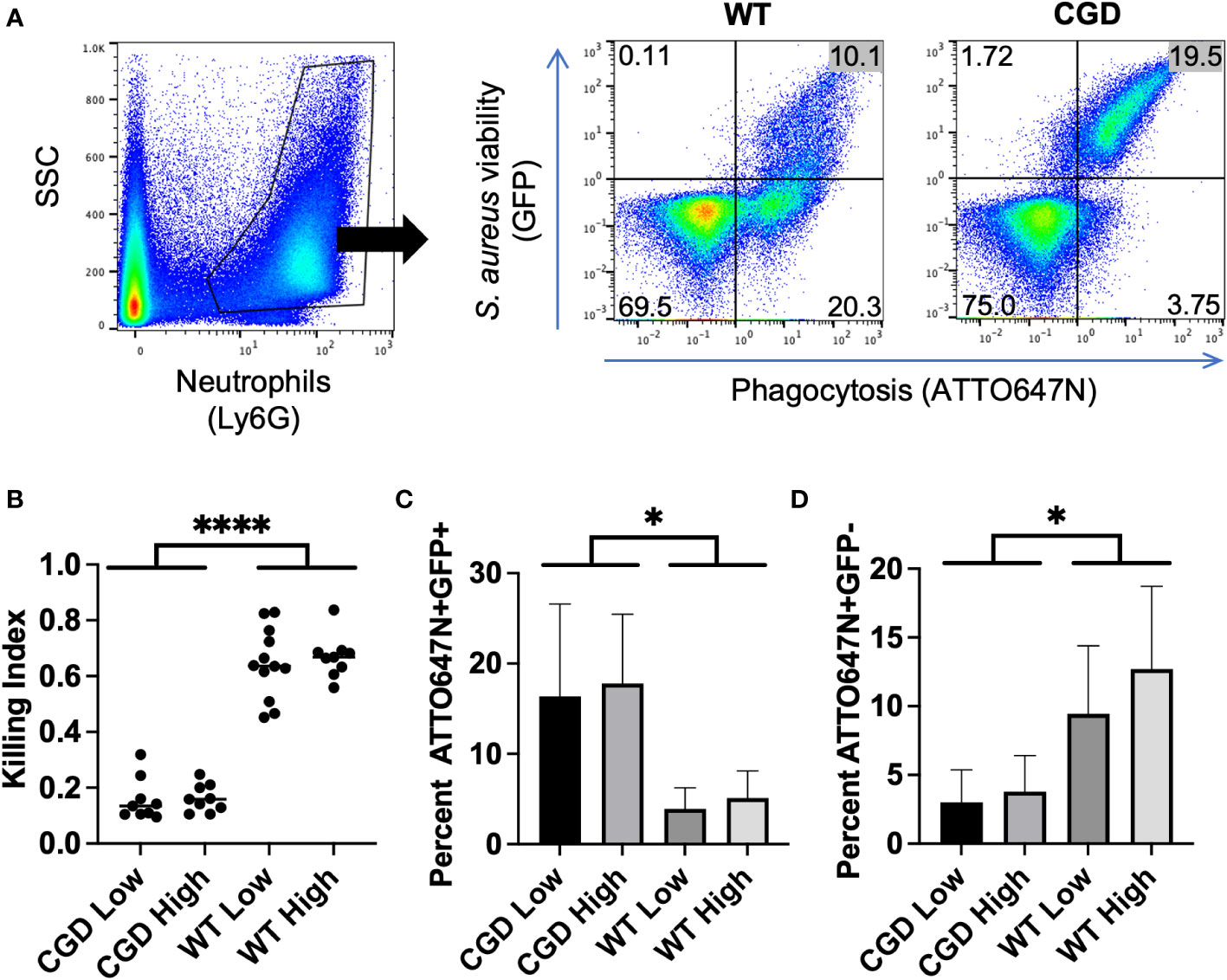
Figure 4 BAL from WT and CGD animals infected with bi-fluorescent S. aureus. (A) Gating on Ly6G+ events within the BAL enables the separation of neutrophils based on GFP and ATTO647N fluorescence. (B) The ratio of ATTO647N+GFP- cells to all ATTO647N+ cells represents the neutrophils that have completely killed the S. aureus compared to those which have undergone phagocytosis: the killing index. (C) Percent of neutrophils that are ATTO647N+GFP+ indicated the intracellular burden of viable S. aureus. (D) Percent of neutrophils that are ATTO647N+GFP- indicates the population of cells that have completed killing (n=8-11 animals per group, from 7 independent experiments). * for p<0.05 and **** for p<0.0001.
After gating for Ly6G+ events to identify neutrophils within the BAL, we were able to assess their S. aureus killing capacity based on the GFP and ATTO647N fluorescence associated with neutrophils (Figure 4A). Since the percent of neutrophils that engage in phagocytosis varies between experiments, we calculated a killing index defined as the percent of neutrophils that had completed killing relative to the percentage of neutrophils engaged in phagocytosis (ATTO+GFP-/ATTO+GFP+/-). Based on this metric, populations with a ratio closer to 1 represent greater success in killing S. aureus relative to populations closer to 0. We observed that the killing index of CGD neutrophils was significantly lower than that of the WT controls (Figure 4B).
The fraction of BAL neutrophils within each ATTO647N/GFP quadrant also behaved as expected based on genotype. The percentage of neutrophils containing viable S. aureus (ATTO647N+GFP+) was greater for the CGD animals than for the WT animals (Figure 4C). Since CGD leads to increased recruitment of neutrophils to the BAL (6, 25), we also compared the total number of neutrophils within each gate. Again, the CGD host trended towards a higher total neutrophil count compared to WT mice (Supplemental Figure 4). This demonstrates a larger intracellular burden of viable S. aureus in CGD BAL neutrophils, accompanied by a diminished population of ATTO647N+GFP- neutrophils that had completed killing S. aureus (Figure 4D). We speculate that the small population of ATTO647N+GFP- cells in the CGD animals may represent the phagocytosis of dead S. aureus or the successful employment of non-oxidative mechanisms to kill S. aureus. In vitro, we observed that both WT and CGD neutrophils readily ingest heat-killed S. aureus and acquire ATTO647N fluorescence (Supplemental Figure 5). It is also possible that non-oxidative killing mechanisms are effective in cells with a low level of S. aureus phagocytosis, as the ATTO647N+GFP- neutrophil population in CGD mice has a significantly lower ATTO647N mean fluorescent intensity compared to the same population from WT mice (Figure 5A).
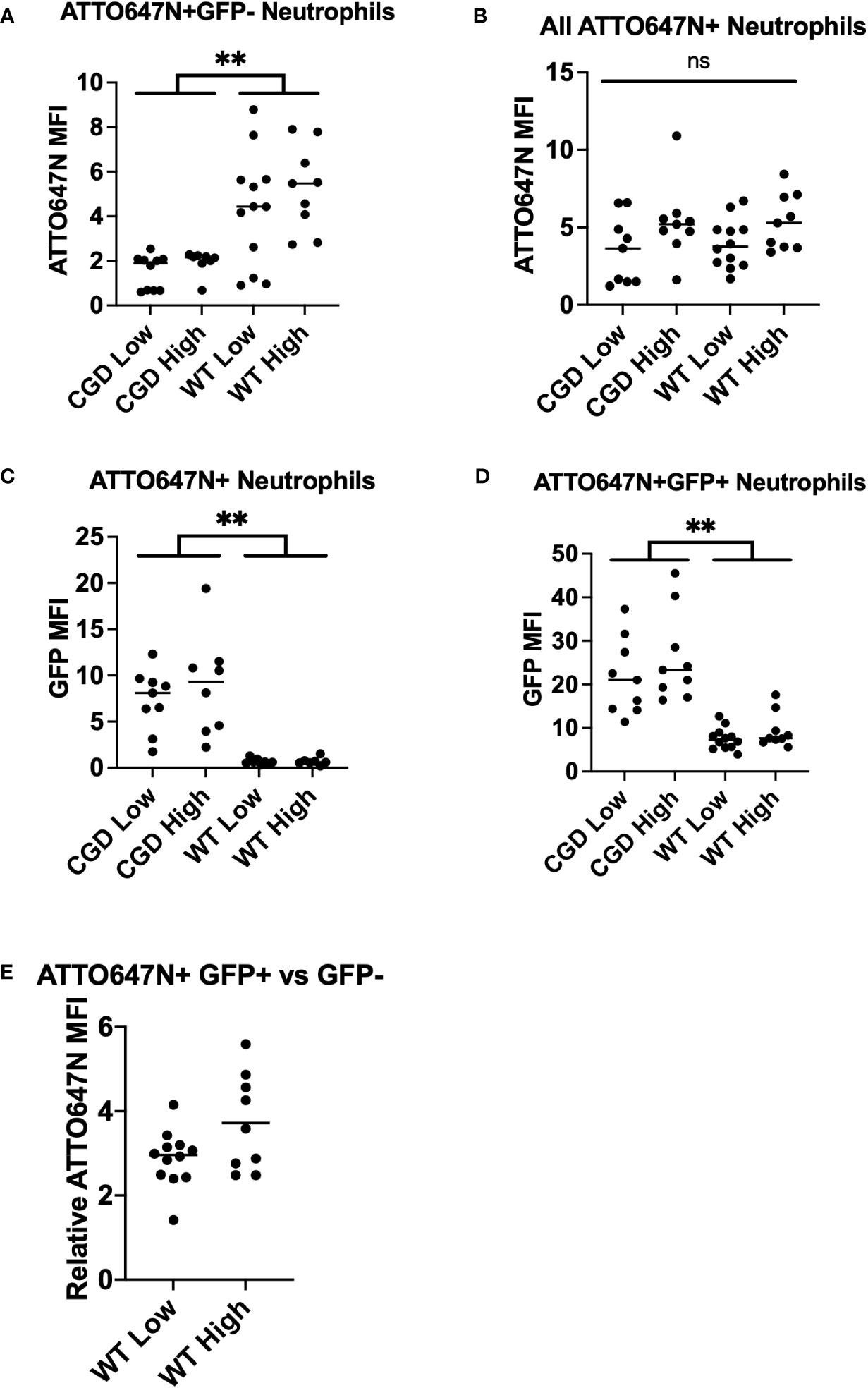
Figure 5 Analysis of BAL neutrophil subpopulation fluorescent intensities. (A) WT neutrophils have increased ATTO647N MFI for neutrophils that have completed killing, indicating clearance of larger bacterial burdens on a per-cell basis. (B) The neutrophil ATTO647N MFI was comparable between groups implying similar levels of phagocytosis. (C) The GFP MFI for all ATTO647N+ events demonstrate an increased bacterial burden in phagocytosing CGD neutrophils relative to WT neutrophils. (D) GFP MFI of the ATTO647M+GFP+ population demonstrates the same trend of increased intensity in CGD animals. (E) The relative ATTO647N MFI was calculated by dividing the ATTO647N MFI of ATTO647N+GFP+ events by ATTO647N+GFP- events (n=8-11 animals per group, from 7 independent experiments). “ns” (not significant) for p>0.05, ** for p<0.01.
Having shown that the GFP MFI of neutrophils that have taken up S. aureus corresponds to the viable intracellular burden of S. aureus, we performed further analyses of BAL neutrophils from WT and CGD mice that were infected with bi-fluorescent S. aureus. We observed a similar relationship in vivo as was established in vitro, suggesting that BAL neutrophils with a greater GFP MFI contain a greater S. aureus bacterial burden (Figure 3C; Supplemental Figure 6). Comparing the ATTO647N MFI for all ATTO647N+ events, we observed no differences across groups (Figure 5B), implying that the degree of phagocytosis is comparable between the WT and CGD neutrophils. We first looked at the GFP MFI of all neutrophils involved in phagocytosis to understand the overall population activity. The CGD ATTO647N+ neutrophils had a significantly higher GFP MFI compared to the analogous population of neutrophils in WT animals, further supporting the conclusion that the population of neutrophils with viable intracellular S. aureus is greater in the CGD animals (Figure 5C). Next, we analyzed the GFP MFI of only the cells with viable intracellular S. aureus (ATTO647N+GFP+) and again observed a greater GFP MFI for CGD neutrophils relative to WT (Figure 5D). We recognize that the analysis of ATTO647N+GFP+ may be biased if the GFP MFI of cells with a lower intracellular burden are more efficient at killing (and transitioning to the GFP- population), which may lead to a higher GFP signal in the remaining population of cells. However, MFI information from ATTO647N+GFP+ neutrophils is valuable to understanding the state of cells which are unable to complete ROS-dependent intracellular killing of S. aureus. In BAL neutrophils from WT mice, we also observe that the ATTO647N MFI is higher in the ATTO647N+GFP+ population than in the ATTO647N+GFP- population (Figure 5E), supporting the observations of others that neutrophils with increased intracellular S. aureus burden have decreased intracellular killing (17).
Bi-fluorescent S. aureus allows localization within the airspace in vivo
Analyses to probe the localization of cells laden with S. aureus within the context of the surrounding tissue environment provide insight into the dynamics of the host response. To visualize bi-fluorescent S. aureus in vivo, we imaged frozen sections from WT mice twelve hours post-infection. Merging DAPI, ATTO647N, and GFP images enabled the identification and localization of viable and nonviable S. aureus (Figures 6A–D). We again often observed viable cocci with larger clumps or clusters compared to the non-viable ones (Figure 6D).
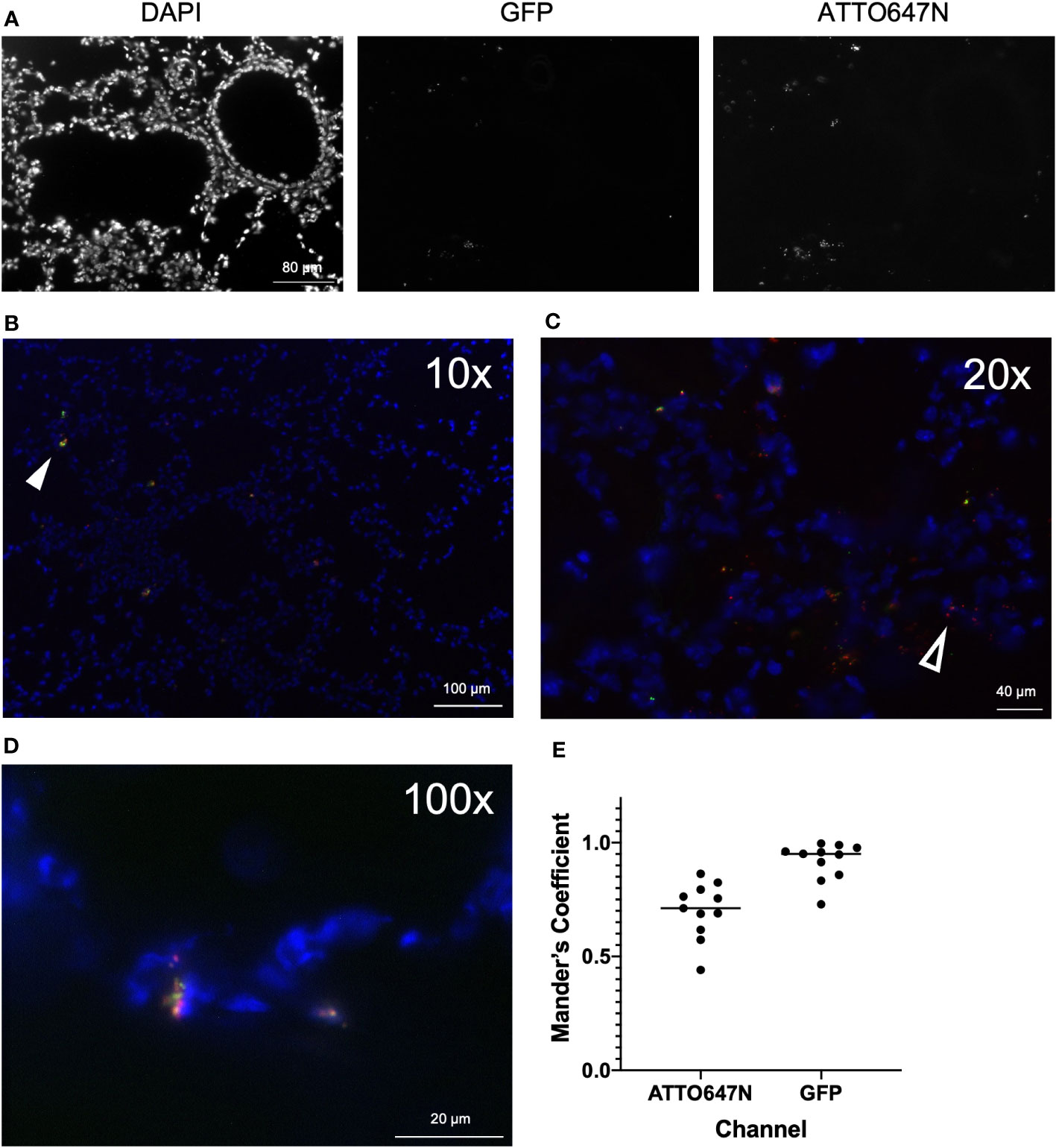
Figure 6 Imaging of airways post-infection with bi-fluorescent S. aureus. (A) Frozen lung sections were prepared from WT mice (n=3) 12 hours post-infection with bi-fluorescent S. aureus to visualize the infection in the tissue. Unmerged DAPI, GFP, and ATTO647N images show localization of airway cells, viable S. aureus, and total S aureus, respectively (20x). Merged images at (B) 10x (C) 20x and (D) 100x visualize both killed (red, open arrowhead) and viable (yellow, arrowhead) S. aureus within the tissue. (E) Colocalization analysis of GFP and ATTO647N images (n=11) show high overlap of the GFP signal with the ATTO647N signal (Mander’s Coefficient ~1) and incomplete overlap of the ATTO647N signal with the GFP signal (Mander’s Coefficient <1).
Visualization of infected lung tissue by microscopy emphasizes the presence of cocci throughout, as opposed to analysis of killing on a single-cell level by flow cytometry. Images were analyzed to determine the degree of colocalization of the GFP and ATTO647N fluorescent signal. Mander’s coefficient was calculated to quantify spatial overlap for GFP and ATTO647N. We expected that Mander’s coefficient for the GFP channel would be equivalent to one, since anywhere GFP-expressing viable S. aureus is detected, the ATTO647N fluorescent signal should also be present. As anticipated, the average Mander’s coefficient for the GFP channel was 0.92 +/- 0.08 (Figure 6E). The Mander’s coefficient in the ATTO647N channel ranged from 0.44 to 0.86 (Figure 6E). This value roughly represents the proportion of cocci that are still viable, marked by the overlapping GFP signal. This proportion is higher compared to the proportion of cells with viable internalized S. aureus measured by the killing index or GFP fluorescence intensity measured by flow cytometry (Figures 4, 5). However, the Mander’s coefficient is representative of all cocci, both intracellular and extracellularly located, while the flow cytometry analysis only measures intracellular killing. This difference also highlights the differences in looking at S. aureus killing on a per-cell basis versus a per-cocci basis and further supports the notion that cells heavily loaded with cocci may have impaired killing. Finally, differences in the killing index and Mander’s coefficient may represent differences in the killing capacity of cells found in the BAL as opposed to whole tissue.
Bi-fluorescent S. aureus is killed by human leukocytes
Animal models of infection have obvious benefits for studying disease in vivo. However, findings in the murine host do not always translate to human pathophysiology. To investigate whether the assay we have extensively characterized using murine neutrophils translates to human cells, we conducted a screen of the bi-fluorescent S. aureus in human leukocytes. Cells were obtained from freshly drawn umbilical cord blood samples that were subjected to red blood cell lysis and then inoculated with bi-fluorescent S. aureus to characterize changes in fluorescence over time.
The use of the ATTO647N-Ester conjugation of S. aureus to identify cells that have undergone S. aureus phagocytosis translates well to human cells, as we observed that leukocytes maintained a robust ATTO647N MFI over time (Figure 7A). Again, the GFP fluorescence intensity decreased over time, suggesting intracellular killing of S. aureus within the untreated group. These findings were confirmed via microscopic imaging of cells two hours after allowing phagocytosis and intracellular killing to occur in suspension. Many cells contain particles with ATTO647N fluorescence (red) in the absence of GFP (Figure 7B). The areas which did have viable S. aureus, determined by co-localized ATTO647N and GFP (yellow), were predominantly present in cells that had likely had multiple phagolysosomes based on their distribution. The DPI-treated samples largely showed viable S. aureus within cells (Figure 7D). Interestingly, the GFP MFI decreased more than expected—though not to the degree of the untreated cells—in the DPI-treated cells (Figures 7A, E). This apparent decrease in GFP fluorescence intensity was also accompanied by a slight decrease in percent GFP+ events within the ATTO647N+ population (Figure 7C). Taken together, these data indicate that bi-fluorescent S. aureus reports its internalization and eradication by human leukocytes.
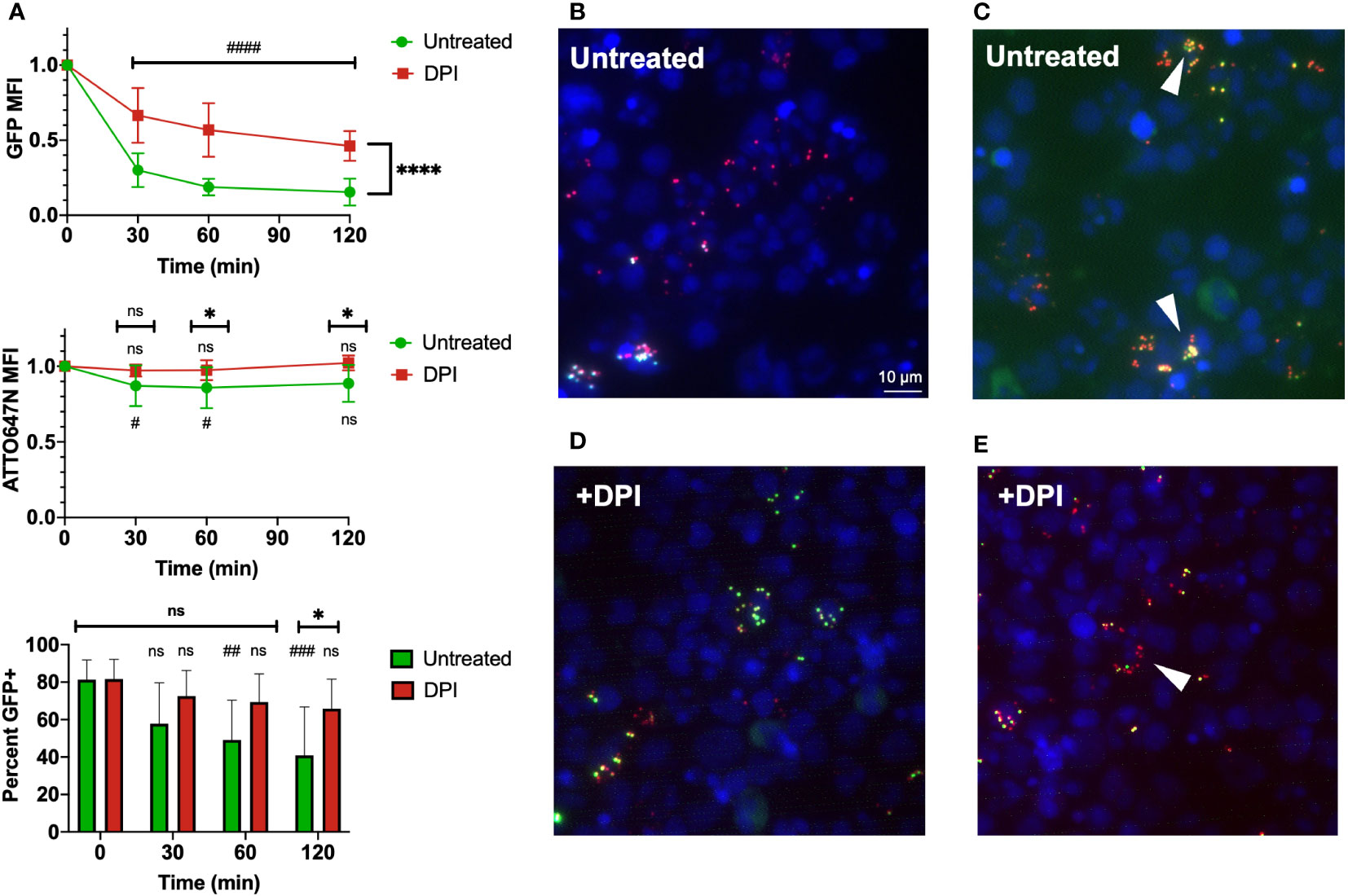
Figure 7 Use of bi-fluorescent S. aureus to model infection in human neutrophils. (A) Human leukocytes inoculated with bi-fluorescent S. aureus maintain the ATTO647N fluorescent signal while quenching the GFP fluorescent intensity over time based on intensity and percent GFP+ cells, statistical differences between groups indicated with a * and differences within each group relative to the initial time point indicated with a # (n=8, performed across three independent experiments). (B) Imaging (60x) performed two hours after human cells were infected in vitro demonstrates cells with few cocci broadly kill the internalized S. aureus and fluoresce as ATTO647N+ (red). (C) Cells that have viable S. aureus internalized ATTO647N+GFP+ (yellow, arrowheads) often have internalized larger clumps of S. aureus. (D) DPI treated human cells largely have viable (yellow) internalized S. aureus regardless of cocci internalized. (E) Some DPI treated cells still quench the GFP fluorescent signal (red, arrowhead). “ns” (not significant) for p>0.05, * for p<0.05 and **** for p<0.0001.
Discussion
The rapid and voluminous response of neutrophils to S. aureus infection is critical to halt its progression from its initial site. During the acute stages of the innate immune response, there is a complex orchestration of cellular host defense as the inflammatory environment develops. Early events can quickly shift the microbial burden towards containment and eradication, or, in cases of inadequate immunity, towards the expansion and dissemination of infection. Understanding the innate immune response to S. aureus as it occurs at tissue sites requires innovative techniques for measuring cell function against microbes. In this study, we have further developed a method to detect and quantify the intracellular eradication of S. aureus and demonstrate its utility in vivo and at the resolution of individual cells.
The use of bi-fluorescent S. aureus to quantify its intracellular killing in vivo provides convenient insight into the mechanism and dynamics of neutrophil phagocytosis and microbicidal activity of the phagosome. Single-cell analysis of killing activity provides a valuable tool for better understanding the role of cellular subpopulations in bacterial clearance in vivo. The relevant applications of this adapted technique include neutrophil heterogeneity studies in pathological and non-pathological states (26–31). Despite a growing understanding of the existence of different subpopulations of neutrophils, much remains unknown as to the relation between characterized differences and influence on function. Tsuda et al. previously identified three subsets of neutrophils in the context of susceptibility to S. aureus infection. Despite variable surface markers and cytokine production profiles, subsets were described to have similar in vitro MRSA killing capabilities (29). However, when subject to the in vivo environment these neutrophil subsets may respond differently to the cytokines they produce or their ability to crosstalk with other immune cells, modulating their bactericidal activity. Characterization of the neutrophil subpopulation which readily engulfs and kills S. aureus—as identified by the ATTO+GFP- population in the assay we characterize here—may provide insight for developing therapies that support or augment innate immune function.
In the study that first characterized GFP-expressing S. aureus as a tool for quantifying its intracellular viability, Schwartz et al. described amplified resistance of S. aureus to eradication as the number of cocci within the neutrophil increased in vitro (17). While validating the use of this assay, we observed a similar trend in vivo as neutrophils with viable intracellular S. aureus were predominantly those that had engulfed a greater number of S. aureus. The relatively large population of neutrophils that have not undergone phagocytosis but are present in the BAL provokes questions as to why some cells engulf more S. aureus than they can efficiently kill while others do not internalize any. In studies of competitive phagocytosis, others have described phagocytosis as a “nonrandom” event, with a subset of neutrophils more likely to undergo phagocytosis (32). There is even apparent variability within a single neutrophil, as the production of HOCl within the multiple phagolysosomes of the same cell can occur with widely disparate lag time after formation (26). S. aureus has also evolved to evade or neutralize host defenses. For example, the production of superoxide dismutase by S. aureus converts the free radicals in the phagolysosome to water and oxygen (15). Therefore, targeting neutrophils to improve their killing capacity through modulating their degree of phagocytosis or intracellular ROS production may improve clearance of infection.
We anticipate that in extending the capability of a S. aureus killing assay with single-cell resolution will also enable further pursuit of questions that remain in understanding the mechanisms by which neutrophils eradicate microbes intracellularly. There has been debate as to the precise role of the biochemical pathways and products of the NADPH oxidase in the phagosome. Specifically, there are questions about whether ROS are responsible for direct toxicity towards microbes or confer their antimicrobial capacity predominantly by buffering the phagolysosome for optimal non-oxidative killing activity (e.g., by serine proteases) (10, 12). Our results correlating intracellular CFU and GFP MFI are consistent with the idea that HOCl production, as reported by GFP bleaching, is tightly correlated with the killing of internalized S. aureus. However, further investigation to delineate the necessity of proteolytic enzymes is warranted and having a means to quantify both S. aureus burden and viability will provide additional insight to solve questions that remain.
Within this study, we included a low and high dose of S. aureus, distinguished as being non-lethal and lethal doses, respectively, in CGD mice. The activity of the neutrophils that are recruited to the infected airspaces of the lung was independent of dose, as there were no statistical differences in the fraction of neutrophils that had completed S. aureus killing or the neutrophil GFP MFI between mice of the same genotype receiving the different initial bolus of S. aureus. This indicates that the activity of neutrophils was dependent on genotype, but not necessarily on potential differences in the environment during the acute response to low versus the high burden of infection. As pathologic resistance to conventional antimicrobial agents steadily increases, methods such as this bi-fluorescent killing assay to study the in vivo dynamics of cell-mediated antimicrobial activity will be of the utmost importance to enhance the pursuit of innovative therapeutic development.
Relatedly, it is important to understand how the use of biological or pharmaceutical interventions to treat infection occurs in conjunction with the host’s immune response. For example, antibiotics that interfere with the effector functions of immune cells may in turn have detrimental effects despite potent antimicrobial activity in vitro (33, 34). Conversely, treatments can have a synergistic effect or support the immune system’s intrinsic response leading to better outcomes than expected based on in vitro results (35–37). Therefore, when testing and developing treatments, in vivo models of infection should be prioritized. This is particularly relevant for therapies targeting in vivo evasion tactics of S. aureus, which employs numerous mechanisms to downregulate inflammation, avoid internalization, neutralize anti-microbial factors, and induce neutrophil cytolysis (15, 38). Multiple factors produced by S. aureus within the phagolysosome neutralize the antimicrobial components generated by the neutrophil. Anti-virulence therapies are gaining traction but also need to be understood in the context of the host response (39, 40). Consequently, methods to understand and quantify changes in immune-mediated microbial clearance are vital for developing and testing these much-needed new approaches to antimicrobial therapy.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board, Care New England Women & Infants Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by Lifespan Animal Welfare Committee, Rhode Island Hospital.
Author contributions
KH: Designing research studies, conducting experiments, analyzing data, writing the manuscript. SL-N: Cord blood collection. JC: Designing research studies, conducting experiments, analyzing data. MM: Designing research studies, conducting experiments, analyzing data. JB: Supervising the project, designing research studies. AH: Developing GFP-Staphylococcus aureus strain. CL: Supervising the project, designing research studies, conducting experiments, analyzing data, writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by awards from the National Institutes of Health (GM124911 to CL; T32GM065085 supported JC).
This study also received funding from Brown Physicians, Inc. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
We thank Dr. Paul Ekert and Dr. Patrice Dubreuil for kindly providing reagents/resources. We greatly appreciate the assistance of Kevin Carlson at the Brown University Flow Cytometry and Sorting Facility. Schematic diagrams were created using BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1089111/full#supplementary-material
References
1. Garrood T, Lee L, Pitzalis C. Molecular mechanisms of cell recruitment to inflammatory sites: general and tissue-specific pathways. Rheumatol (Oxford) (2006) 45:250–60. doi: 10.1093/rheumatology/kei207
2. Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood (2022) 139:2130–44. doi: 10.1182/blood.2021012295
3. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
4. Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest (2019) 129:2629–39. doi: 10.1172/JCI124616
5. Rigby KM, DeLeo FR. Neutrophils in innate host defense against staphylococcus aureus infections. Semin Immunopathol (2012) 34:237–59. doi: 10.1007/s00281-011-0295-3
6. Song Z, Huang G, Chiquetto Paracatu L, Grimes D, Gu J, Luke CJ, et al. NADPH oxidase controls pulmonary neutrophil infiltration in the response to fungal cell walls by limiting LTB4. Blood (2020) 135:891–903. doi: 10.1182/blood.2019003525
7. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
8. Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of staphylococcus aureus. Virulence (2021) 12:547–69. doi: 10.1080/21505594.2021.1878688
9. Dinauer MC. Primary immune deficiencies with defects in neutrophil function. Hematol Am Soc Hematol Educ Program (2016) 2016:43–50. doi: 10.1182/asheducation-2016.1.43
10. Segal AW. How neutrophils kill microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
11. Zeng MY, Miralda I, Armstrong CL, Uriarte SM, Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol (2019) 34:27–38. doi: 10.1111/omi.12252
12. Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by k+ flux. Nature (2002) 416:291–7. doi: 10.1038/416291a
13. Fine N, Tasevski N, McCulloch CA, Tenenbaum HC, Glogauer M. The neutrophil: Constant defender and first responder. Front Immunol (2020) 11:571085. doi: 10.3389/fimmu.2020.571085
14. Bodey GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med (1966) 64:328–40. doi: 10.7326/0003-4819-64-2-328
15. Greenlee-Wacker M, DeLeo FR, Nauseef WM. How methicillin-resistant staphylococcus aureus evade neutrophil killing. Curr Opin Hematol (2015) 22:30–5. doi: 10.1097/MOH.0000000000000096
16. Zinn KR, Chaudhuri TR, Szafran AA, O'Quinn D, Weaver C, Dugger K, et al. Noninvasive bioluminescence imaging in small animals. ILAR J (2008) 49:103–15. doi: 10.1093/ilar.49.1.103
17. Schwartz J, Leidal KG, Femling JK, Weiss JP, Nauseef WM. Neutrophil bleaching of GFP-expressing staphylococci: probing the intraphagosomal fate of individual bacteria. J Immunol (2009) 183:2632–41. doi: 10.4049/jimmunol.0804110
18. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. agr-dependent interactions of staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun (2010) 2:546–59. doi: 10.1159/000319855
19. Espey MG, Xavier S, Thomas DD, Miranda KM, Wink DA. Direct real-time evaluation of nitration with green fluorescent protein in solution and within human cells reveals the impact of nitrogen dioxide vs. peroxynitrite mechanisms. Proc Natl Acad Sci U.S.A. (2002) 99:3481–6. doi: 10.1073/pnas.062604199
20. Cohen JT, Danise M, Hinman KD, Neumann BM, Johnson R, Wilson ZS, et al. Engraftment, fate, and function of HoxB8-conditional neutrophil progenitors in the unconditioned murine host. Front Cell Dev Biol (2022) 10:840894. doi: 10.3389/fcell.2022.840894
21. Cohen JT, Danise M, Machan JT, Zhao R, Lefort CT. Murine myeloid progenitors attenuate immune dysfunction induced by hemorrhagic shock. Stem Cell Rep (2021) 16:324–36. doi: 10.1016/j.stemcr.2020.12.014
22. Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods (2006) 3:287–93. doi: 10.1038/nmeth865
23. Decleva E, Menegazzi R, Busetto S, Patriarca P, Dri P. Common methodology is inadequate for studies on the microbicidal activity of neutrophils. J Leukoc Biol (2006) 79:87–94. doi: 10.1189/jlb.0605338
24. Kim JH, Chaurasia AK, Batool N, Ko KS, Kim KK. Alternative enzyme protection assay to overcome the drawbacks of the gentamicin protection assay for measuring entry and intracellular survival of staphylococci. Infect Immun (2019) 87:e00119-19. doi: 10.1128/IAI.00119-19
25. Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet (1995) 9:202–9. doi: 10.1038/ng0295-202
26. Albrett AM, Ashby LV, Dickerhof N, Kettle AJ, Winterbourn CC. Heterogeneity of hypochlorous acid production in individual neutrophil phagosomes revealed by a rhodamine-based probe. J Biol Chem (2018) 293:15715–24. doi: 10.1074/jbc.RA118.004789
27. Rosales C. Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol (2018) 9:113. doi: 10.3389/fphys.2018.00113
28. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through mac-1. J Clin Invest (2012) 122:327–36. doi: 10.1172/JCI57990
29. Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant staphylococcus aureus. Immunity (2004) 21:215–26. doi: 10.1016/j.immuni.2004.07.006
30. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
31. Filep JG, Ariel A. Neutrophil heterogeneity and fate in inflamed tissues: implications for the resolution of inflammation. Am J Physiol Cell Physiol (2020) 319:C510–C32. doi: 10.1152/ajpcell.00181.2020
32. Hellebrekers P, Hietbrink F, Vrisekoop N, Leenen LPH, Koenderman L. Neutrophil functional heterogeneity: Identification of competitive phagocytosis. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.01498
33. Kristian SA, Timmer AM, Liu GY, Lauth X, Sal-Man N, Rosenfeld Y, et al. Impairment of innate immune killing mechanisms by bacteriostatic antibiotics. FASEB J (2007) 21:1107–16. doi: 10.1096/fj.06-6802com
34. Berti A, Rose W, Nizet V, Sakoulas G. Antibiotics and innate immunity: A cooperative effort toward the successful treatment of infections. Open Forum Infect Dis (2020) 7:ofaa302. doi: 10.1093/ofid/ofaa302
35. Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, et al. Nafcillin enhances innate immune-mediated killing of methicillin-resistant staphylococcus aureus. J Mol Med (Berl) (2014) 92:139–49. doi: 10.1007/s00109-013-1100-7
36. Lin L, Nonejuie P, Munguia J, Hollands A, Olson J, Dam Q, et al. Azithromycin synergizes with cationic antimicrobial peptides to exert bactericidal and therapeutic activity against highly multidrug-resistant gram-negative bacterial pathogens. EBioMedicine (2015) 2:690–8. doi: 10.1016/j.ebiom.2015.05.021
37. Ulloa ER, Uchiyama S, Gillespie R, Nizet V, Sakoulas G. Ticagrelor increases platelet-mediated staphylococcus aureus killing, resulting in clearance of bacteremia. J Infect Dis (2021) 224:1566–69. doi: 10.1093/infdis/jiab146
38. Spaan AN, Surewaard BG, Nijland R, van Strijp JA. Neutrophils versus staphylococcus aureus: a biological tug of war. Annu Rev Microbiol (2013) 67:629–50. doi: 10.1146/annurev-micro-092412-155746
39. Ford CA, Hurford IM, Cassat JE. Antivirulence strategies for the treatment of staphylococcus aureus infections: A mini review. Front Microbiol (2020) 11:632706. doi: 10.3389/fmicb.2020.632706
40. Fleitas Martinez O, Cardoso MH, Ribeiro SM, Franco OL. Recent advances in anti-virulence therapeutic strategies with a focus on dismantling bacterial membrane microdomains, toxin neutralization, quorum-sensing interference and biofilm inhibition. Front Cell Infect Microbiol (2019) 9:74. doi: 10.3389/fcimb.2019.00074
Keywords: infection, Staphylococcus aureus, phagocytosis, reactive oxygen species, neutrophil, flow cytometry
Citation: Hinman KD, Laforce-Nesbitt SS, Cohen JT, Mundy M, Bliss JM, Horswill AR and Lefort CT (2023) Bi-fluorescent Staphylococcus aureus infection enables single-cell analysis of intracellular killing in vivo. Front. Immunol. 14:1089111. doi: 10.3389/fimmu.2023.1089111
Received: 03 November 2022; Accepted: 03 January 2023;
Published: 23 January 2023.
Edited by:
Carrie Ambler, Durham University, United KingdomReviewed by:
Tamás Laskay, University of Lübeck, GermanyKristian Juul-Madsen, Aarhus University, Denmark
Copyright © 2023 Hinman, Laforce-Nesbitt, Cohen, Mundy, Bliss, Horswill and Lefort. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig T. Lefort, Y3JhaWdfbGVmb3J0QGJyb3duLmVkdQ==