
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 26 January 2023
Sec. Inflammation
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1087532
 Nienke Oskam1*†
Nienke Oskam1*† Timon Damelang1,2,3†
Timon Damelang1,2,3† Marij Streutker1Pleuni Ooijevaar-de Heer1Jan Nouta4Carolien Koeleman4
Marij Streutker1Pleuni Ooijevaar-de Heer1Jan Nouta4Carolien Koeleman4 Julie Van Coillie2,3
Julie Van Coillie2,3 Manfred Wuhrer4
Manfred Wuhrer4 Gestur Vidarsson2,3
Gestur Vidarsson2,3 Theo Rispens1
Theo Rispens1Of the four human immunoglobulin G (IgG) subclasses, IgG4 is considered the least inflammatory, in part because it poorly activates the complement system. Regardless, in IgG4 related disease (IgG4-RD) and in autoimmune disorders with high levels of IgG4 autoantibodies, the presence of these antibodies has been linked to consumption and deposition of complement components. This apparent paradox suggests that conditions may exist, potentially reminiscent of in vivo deposits, that allow for complement activation by IgG4. Furthermore, it is currently unclear how variable glycosylation and Fab arm exchange may influence the ability of IgG4 to activate complement. Here, we used well-defined, glyco-engineered monoclonal preparations of IgG4 and determined their ability to activate complement in a controlled system. We show that IgG4 can activate complement only at high antigen and antibody concentrations, via the classical pathway. Moreover, elevated or reduced Fc galactosylation enhanced or diminished complement activation, respectively, with no apparent contribution from the lectin pathway. Fab glycans slightly reduced complement activation. Lastly, we show that bispecific, monovalent IgG4 resulting from Fab arm exchange is a less potent activator of complement than monospecific IgG4. Taken together, these results imply that involvement of IgG4-mediated complement activation in pathology is possible but unlikely.
Immunoglobulin G 4 (IgG4) is the least prevalent of the four human IgG subclasses and unique in that it seems to function mainly as an anti-inflammatory, blocking antibody. In contrast to IgG1 and IgG3, the most potent subclasses in activating complement via the classical pathway and inducing complement-dependent cytotoxicity (CDC; Figure 1A), IgG4 is generally considered to be either inactive (1–4) or a poor activator (5–7). IgG4 may interfere with multimerization and complement component 1q (C1q) binding of the other subclasses, and thereby effectively block complement activation (6, 8). It has been hypothesized that the impaired ability of IgG4 to activate complement is due to a conformational change of the FG loop in the CH2 domain that may interfere with C1q binding (3, 4, 9).
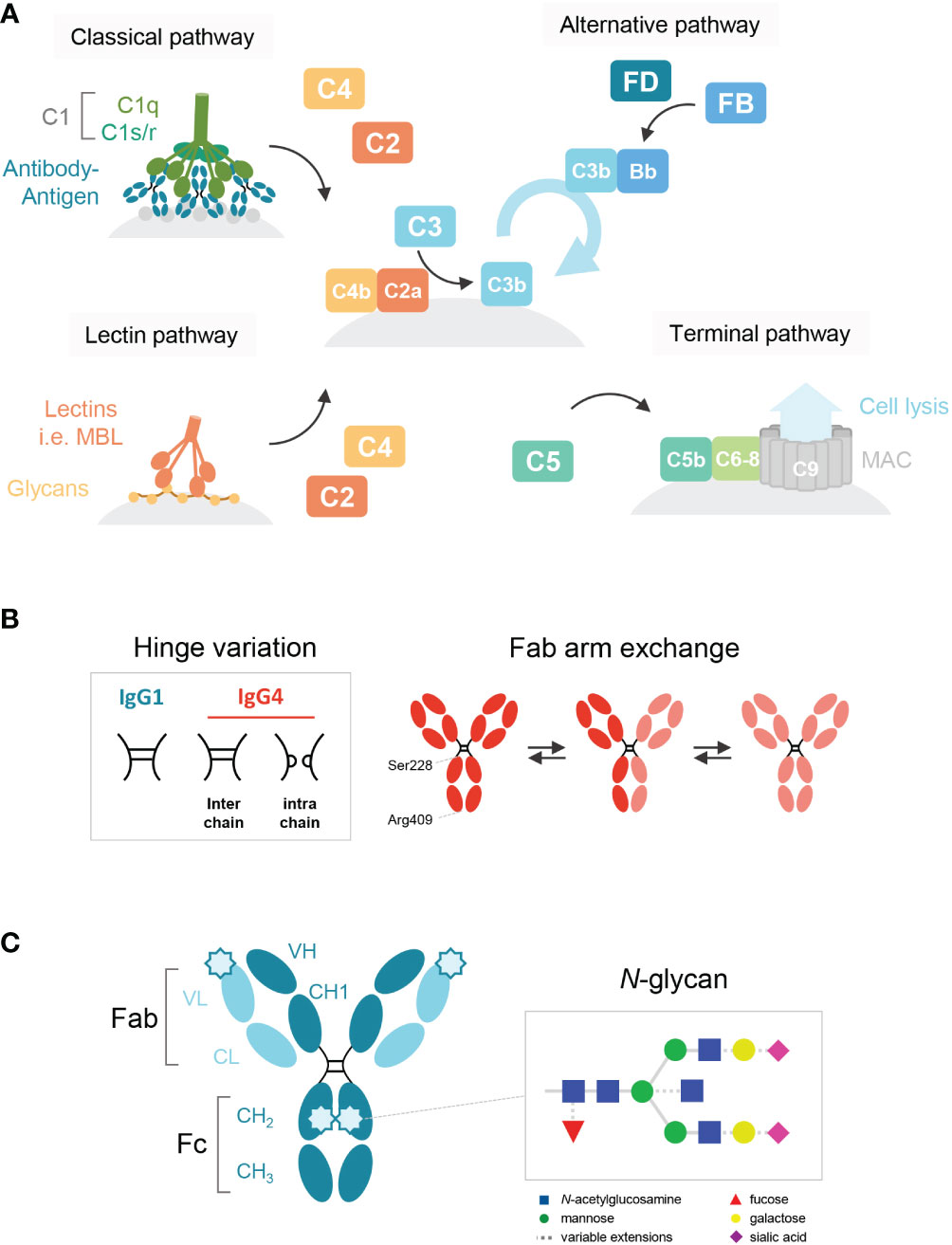
Figure 1 Schematic overview of the human IgG antibody structure and antibody-mediated complement activation. (A) Generalized overview of the complement system, which is activated through three pathways: the classical, lectin and alternative pathway. The classical pathway is activated by binding of C1q to antigen-bound IgM and IgG, which then forms the C1 complex with its proteases C1r/s. The lectin pathway is activated by the binding of lectins, such as mannose-binding lectin (MBL), to carbohydrates on cell surfaces. C3b deposition induced by these pathways or spontaneous hydrolysis of C3 can activate the alternative pathway, mediated by Factor B and D, which results in an amplification loop of C3b deposition. C3b can opsonize the target cell for phagocytosis and can lead to the assembly of the membrane attack complex with factors C5b to C9 through the terminal pathway and subsequent cell lysis. (B) A structural representation of the hinge variation and half molecule exchange of IgG4 via switch of inter-chain to intra-chain disulfide bonds at amino acid positions 228 and 409. (C) General structure of an IgG monomer and schematic representation of N-linked glycan composition of human IgG Abs. The glycans are attached to asparagine (N) at position 297 in the CH2 domain and have a biantennary heptasaccharide core (solid line) and variable extensions (dash line), such as fucose, galactose and/or sialic acid. Novel glycosylation sites can be introduced within the Fab region of matured antibodies.
Despite the consensus on the impaired ability of IgG4 to activate complement, IgG4 levels have been associated with pathologies in IgG4-related disease (IgG4-RD) and in several autoimmune disorders, such as pemphigus, autoimmune pancreatitis, and primary membranous nephropathy (pMN) (10–12). Often, deposits of both IgG4 antibodies and complement components can be detected in biopsies of affected tissues and correlate to serum level, IgG4 titers and hypocomplementemia (10–12). For example, most patients suffering from pMN (~70%) develop IgG4 autoantibodies towards PLA2R1, a secretory phospholipase A2 receptor, expressed on cells in the glomerular basement membrane of the kidney (13). Although reports vary, deposited C4d has been found in 84-100% of biopsies in pMN (10), which implicates complement activation in the pathogenesis, but its significance and relation to the presence of IgG4 autoantibodies remain debated.
This raises several questions that warrant further investigation. Even though IgG4 has been considered a poor activator of complement at best, there could be certain conditions, potentially reminiscent of in vivo deposits, in which complement activation can be observed. Furthermore, IgG4 undergoes several post-translational modifications, such as glycosylation and Fab arm exchange, that have been implicated in complement activation, but not studied in detail for IgG4 despite the importance of these modifications to its function.
Fab arm exchange is a unique aspect of IgG4 and refers to its ability to exchange half-molecules of one heavy chain-light chain pair with another IgG4 molecule (Figure 1B). Fab arm exchange leads to the formation of a bispecific antibody which remains functionally monovalent as the antibody is unable to crosslink antigens (14–17). Therefore, IgG4 is effectively unable to form large immune complexes. It has been shown for IgG1 that functional monovalence may enhance complement activation (18), but despite the importance of Fab arm exchange in IgG4 biology, its importance for complement activation has never been studied.
IgG antibodies contain a conserved N-linked glycan site, asparagine (N) at the position 297, in both CH2 domains of the Fc region (Figure 1C). The core structure of the IgG N-linked glycans comprises N-acetylglucosamine (GlcNAc) and mannose residues with possible extensions of galactose, sialic acid, core fucose, and bisecting GlcNAc (1, 19). Several studies have shown that the exact composition of the Fc glycan can either enhance or reduce the potency of IgG1 to activate complement (20–22). Galactosylation in particular has been shown to enhance IgG1 hexamerization and therefore its ability to activate complement (23). Furthermore, upon somatic hypermutation, glycosylation sites may arise within the variable regions as well (24) and levels of Fab glycosylation are elevated for IgG4 relative to other IgG subclasses (25). Although the effect of Fab glycosylation on complement activation is not entirely clear, the presence of these glycans has been shown to influence effector molecule binding and stability of the antibody (26).
Next to the classical and alternative pathways, the complement system can also be activated via the lectin pathway, which is initiated by recognition of sugars often found on pathogens by either mannose-binding lectin (MBL) or other ficolins and collectins (27, 28). Although these lectins usually bind to foreign sugars, they may also bind to aberrant glycans on human proteins. It has been suggested that IgG antibodies lacking the terminal galactose residue on their Fc-glycan could activate complement via the lectin pathway, as MBL can bind to terminal fucose, mannose and N-acetylglucosamine residues, but not galactose (29). Interestingly, in both IgG4-RD and pMN, agalactosylated IgG4 (auto)antibodies have been reported (30–33). For pMN specifically, it was recently shown that MBL could bind agalactosylated IgG4 autoantibodies towards PLA2R1 which may activate the lectin pathway (33).
In this study, we not only present conditions that allow IgG4 to activate complement, but also show for the first time the effects of differential Fc glycosylation and Fab arm exchange on the ability of IgG4 to activate complement. We demonstrate that IgG4 can only activate the classical pathway in conditions with high antigen densities and antibody concentrations, which is further enhanced by Fc galactosylation, and that agalactosylated IgG4 does not activate the lectin pathway like suggested before. Furthermore, as Fab arm exchange reduces complement activation by IgG4, it seems that this feature would limit the capacity of IgG4 to complement activation in vivo.
Recombinant IgG antibodies specific for biotin [bt; (34, 35)] and trinitrophenyl [TNP; (36)] were produced using FreeStyle™ HEK293-F cells (ThermoFisher), which were cultured in FreeStyle™ 293 Expression Medium (ThermoFisher). Using Polyethylenimine-MAX, pcDNA3.1 expression vectors encoding human κ, IgG1 and IgG4 were co-transfected under serum-free conditions into HEK293-F cells according to the manufacturer’s protocol (Invitrogen). In the case of the anti-TNP antibodies, an supplemental vector was used with a mutation of D86N in the light chain to introduce an additional glycosylation site (37). 0.9% w/v Peptone Primatone® RL (Sigma-Aldrich) was added at least 4 h post transfection. The galactose-glycovariants were produced as described previously (19). To produce low galactose-glycovariants, 0.5 mM of the galactose analogue 2-deoxy-2-fluoro-d-galactose (2FG) (Carbosynth) was added 4 h post transfection. To produce high galactose-glycovariants, 5 mM D-galactose (Sigma-Aldrich) was added to the cell culture 1 h before transfection, and cells were co-transfected with 1% DNA (of total DNA transfected) encoding the β-1,4-galactosyltransferase 1 (B4GALT1).
High mannose-glycovariants were produced using HEK293-6E cells with a MGAT1 knock-out (KO), which lack N-acetylglucosaminyl transferase I (GlcNAc-T I) (38, 39). These cells were kindly provided by Prof. H. Clausen from the Copenhagen Center for Glycomics, University of Copenhagen, Denmark. The MGAT1-KO cells were cultured in FreeStyle™ F17 Expression Medium (ThermoFisher) supplemented with 4 mM L-glutamine (Sigma-Aldrich) and 0.1% w/v Kolliphor® P 188 (Sigma-Aldrich), at 5% CO2, 37°C while shaking at 125 rpm.
Culture supernatant was harvested five or six days after transfection and filtered using a 0.2 nm Puradisc™ syringe filter (Whatman, GE Healthcare). Antibodies were purified on a HiTrap® protein G HP 1 mL column (GE Healthcare) and eluted with 0.1 M glycine pH 2.5 (Merck). The eluate was then dialyzed against 5 mM NaAc (pH 4.5). Antibody concentrations were determined by measuring A280 on a NanoDrop One spectrophotometer (ThermoFisher) and samples were stored at -20°C.
To be able to produce bispecific IgG1 antibodies, a mutation of K409R was introduced into the heavy chain of the IgG1 anti-bt clone, which allows for Fab arm exchange of IgG1 in a manner similar to IgG4. IgG1 K409R anti-bt was reacted with an in-house generated IgG1 K409R variant of adalimumab, and IgG4 anti-bt or anti-TNP with IgG4 natalizumab (TYSABRI®), both in a ratio of 1:4 (anti-bt or anti-TNP to irrelevant clone). Fab arm exchange was induced by mild reduction with 10 and 0.5 mM L-glutathione (GSH; Sigma-Aldrich) for IgG1 and IgG4 respectively, in presence of 0.1% w/v Kolliphor® P407 (BASF). Samples were incubated at 37°C for over 20 h, after which the reaction was stopped with 12 mM and 0.6 mM 2-iodoacetamide (IAM; Merck) for IgG1 and IgG4 respectively. IgG1 and IgG4 bispecific antibodies were made fresh for each experiment due to aggregation of the antibodies during storage. Bispecificity was confirmed for all clones.
The purified glycoengineered IgG1 and IgG4 were dried by vacuum centrifugation and subjected to tryptic cleavage followed by LC-MS as described previously (40, 41). The raw LC-MS spectra were converted to mzXML files and LaCyTools, an in-house developed software, was used for the alignment and targeted extraction of raw data. The analyte list for targeted extraction of the 1+, 2+, 3+ and 4+ charge states was based on manual annotation as well as on literature reports (40). The inclusion of an analyte for the final data analysis was based on quality criteria including signal-to-noise (higher than 9), isotopic pattern quality (less than 25% deviation from the theoretical isotopic pattern), and mass error (within a ±20 parts per million range) leading to a final analyte list (Table S1 and S2). Relative intensity of each glycopeptide in the final analyte list was calculated by normalization the glycopetide value to the sum of the total area of all glycopeptides included. Normalized intensities were used to calculate fucosylation, bisection, galactosylation and sialylation levels (Table S3).
To study the influence of glycosylation on the molecular weight of IgG1 and IgG4, a reduced SDS-PAGE was performed on a NuPAGE 4-12% Bis-Tris gel according to manufacturer’s protocol (Invitrogen). The gel was stained with InstantBlue® Coomassie Protein Stain (Abcam) for 1 h and the bands were visualized using a ChemiDoc™ Universal Hood III (Bio-Rad).
To assess complement activation by IgG1 and IgG4 variants, C3b deposition was measured in ELISA as described previously (42). In short, human serum albumin (HSA; Albuman, Sanquin) was either biotinylated with different concentrations (30 to 240 µM) of biotin (EZ-Link™ Sulfo-NHS-LC-Biotin No-Weigh; ThermoFisher) in PBS or TNPlated with 1 mM of 2,4,6-trinitrobenzenesulfonic acid (TNBS, Sigma-Aldrich) in 0.2 M Na2HPO4 (Merck, F173578238). Nunc MaxiSorp™ flat-bottom plates (ThermoFisher) were coated with 5 µg/mL of biotinylated HSA (HSA-bt) or 10 µg/mL of TNPlated HSA (HSA-TNP) in PBS and incubated at 4°C overnight. IgG antibodies were serially diluted in PBS supplemented with 0.1% v/v Tween®-20 and 0.2% w/v gelatin (Merck) and incubated shaking at room temperature for at least 1 h. After washing, C3b deposition was allowed for 1 hour at RT by addition of 2.5% normal human serum (NHS; pool of minimum healthy donors) in veronal buffer (3 mM barbital (Fagron), 1.8 mM sodium-barbital (Fagron), 0.146 M sodium chloride (Merck)) supplemented with 1 mM calcium chloride (CaCl2, Merck), 0.5 mM magnesium chloride (MgCl2, Merck) and 0.2% Tween-20 (VB+/+). For inhibition conditions, VB++ was supplemented with additional anti-C1q-85 (6.3 µg/mL; Sanquin (43, 44)), anti-MBL1 (0.2 µg/mL; Sanquin (45, 46)) and/or anti-complement factor D (a-FD; 0.13 µg/mL; Lampalizumab; Genentech (47, 48);) or inhouse anti- complement factor B-1 (a-FB; 24,3 µg/mL; Sanquin) in order to inhibit C1q, MBL and FD/FB respectively. All inhibitors were shown to be effective blockers of their respective route of activation (49).
Alternatively, instead of the addition of serum dilution, IgG binding to the coat was measured using mouse-anti-IgG-horseradish peroxidase (0,33 µg/mL; MH-16-1-HRP; Sanquin). Deposited C3b was detected with mouse anti-human C3-19-HRP (0.5-1 µg/mL; Sanquin (50)) and bound C1q was detected with mouse anti-human C1q-2-HRP (3 µg/mL; Sanquin (43)) and visualized using tetramethylbenzidine (TMB; Merck) mix. The reaction was stopped with 0.2 M sulfuric acid (Merck) and the optical density (OD) was measured at Δ450–540 nm using an ELISA plate reader (SynergyTM 2 Multi-Detection Microplate Reader, Biotek).
The CDC assay to assess terminal pathway activation by the anti-bt IgG antibodies was performed as described earlier, with minor alterations (42). To biotinylate red blood cells (RBCs), one part packed cells was reacted with seven parts of a biotinylation reagent at a final concentration of 2.5 mM Sulfo-NHS-LC-Biotin (ThermoFisher) in PBS. RBCs were incubated at 37°C for at least 1 h with serial dilutions of anti-bt IgG antibodies, 10% of NHS pool and inhibitors (indicated for each respective experiment) in veronal buffer supplemented with 0.05% w/v gelatin, 1 mM CaCl2 and 0.5 mM MgCl2. Conditions with complement inhibitors were included as described above, but with four times higher concentrations. A positive control (100% lysis) was generated by adding 0.5% w/v saponin (Merck/Millipore) to RBCs and used to normalize the data. After incubation, 40 µL of buffer was added, plates were centrifuged at 1800 rpm for 2 min and 100 µL supernatant was transferred to MaxiSorp™ plates. The OD was measured as ΔOD(412 nm) – OD(690 nm). The percentage of cell lysis for each sample was calculated as follows: % Lysis = ODsample/mean OD100% * 100.
Kruskal-Wallis test with Dunn’s multiple comparisons test was performed for statistical comparison of the IgG4 glycovariants.
To study complement activation by IgG4, C3b deposition and CDC were determined in a recombinant antibody system with anti-hapten antigens which allowed for control of antigen density. Using recombinant anti-biotin antibodies with biotinylated HSA, we found that IgG1 activated complement efficiently in all conditions tested, in contrast to IgG4 which could only activate complement at high antigen densities (HSA-bt 120 and 240 µM) and high antibody concentrations (Figure 2A). IgG4 required a roughly 100 times higher antibody concentration to reach the same OD as IgG1. Comparable results were obtained utilizing another antigen, TNP, where IgG4 was also only able to activate complement at high antigen densities and antibody concentrations (Supplementary Figure 1A). In addition, two monoclonal antibodies against protein antigens (rather than anti-hapten) were tested, anti-Betv1a (Birch pollen) and anti-idiotype Fabs (against adalimumab). No complement activation with either of these antibodies was observed, probably because sufficient densities of antibody binding were not reached (Supplementary Figure 1B, C).
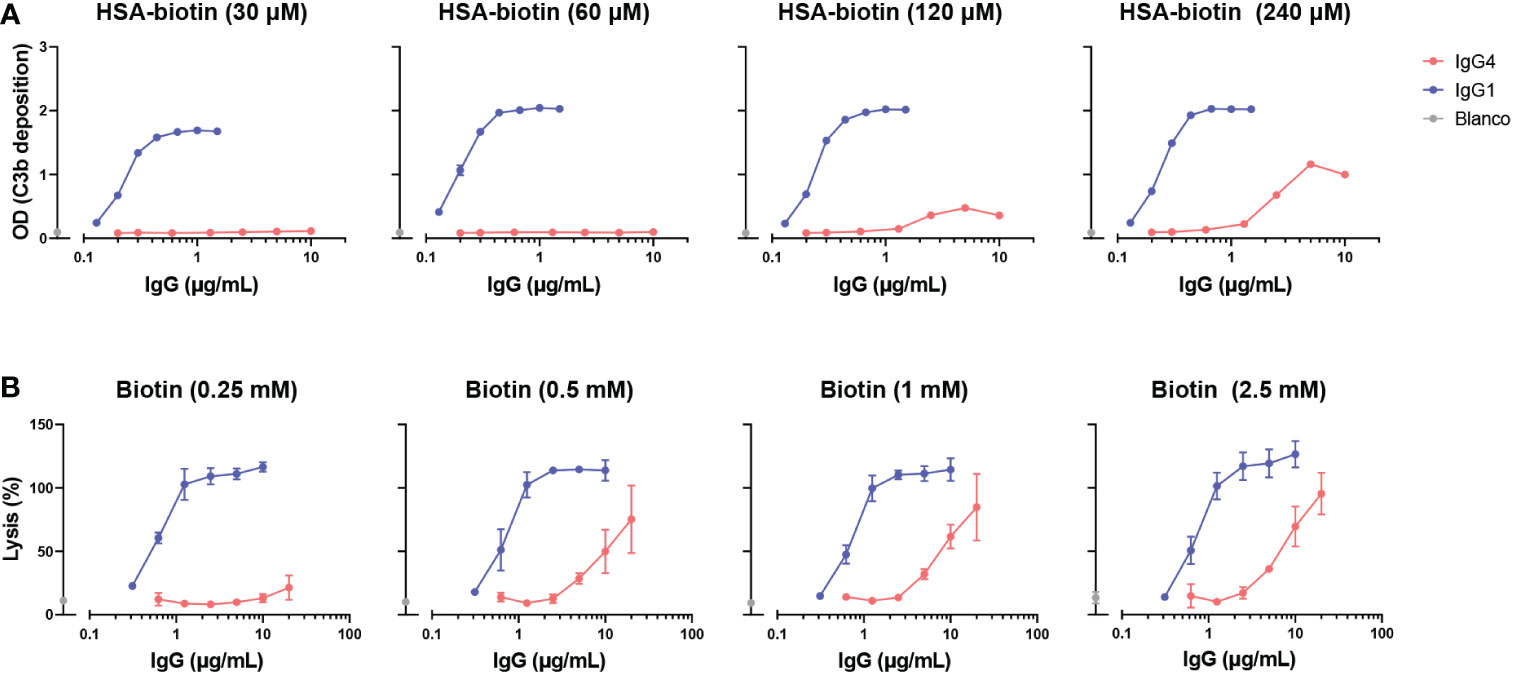
Figure 2 IgG4 activates complement at high antigen densities and antibody concentrations. (A) C3b deposition induced by anti-biotin IgG1 and IgG4 antibodies on biotinylated human serum albumin (HSA; 30, 60, 120 and 240 μM biotin; corresponding to approximately 2, 4, 8 and 50 biotins per HSA molecule respectively), in the presence of 2.5% human serum. All antibodies were tested in a two-fold serial dilution starting from 10 µg/mL for IgG4 and 2 µg/mL for IgG1 antibodies in four independent assays. (B) The capacity of biotin-specific IgG1 and IgG4 antibodies to induce complement-mediated lysis (as percentage) of biotinylated human red blood cells at various antigen densities (0.25, 0.5, 1, and 2.5 mM biotin) in the presence of 10% human serum. All antibodies were tested in a two-fold serial dilution starting from 20 µg/mL for IgG4 and 10 µg/mL for IgG1 antibodies in triplicates. The data was normalized to a 100% lysis control (RBCs with saponin).
Next, the ability of IgG4 antibodies to induce complement-dependent lysis of biotinylated RBCs was determined. In line with the results for the C3b deposition, IgG4 induced lysis at high antigen densities and antibody concentrations almost to the same extent as IgG1 antibodies (Figure 2B). However, IgG1 induced cell lysis at all conditions tested at much lower antibody concentrations. Taken together, this shows that IgG4 can activate complement to some extent, but only in conditions where both sufficient antigen and antibodies present.
It has been suggested that agalactosylated IgG4 autoantibodies can induce complement activation through the lectin pathway via MBL (33). To study this effect of galactosylation on complement activation by IgG4 in more detail, glycoengineered variants with relatively low and high levels of galactosylation were produced. These anti-biotin IgG1 and IgG4 antibodies were classified as ‘low galactose’ (LG; 10% galactose), ‘normal galactose’ (NG; 26-40%) and ‘high galactose’ (HG; 75-80%) based on their galactosylation levels determined by mass spectrometry (Supplementary Figure 2A). The antibodies were also analyzed by gel electrophoresis under reducing conditions and subtle shifts in size can be observed among the heavy chains due to varying levels of galactosylation (Supplementary Figure 2B). As a glycan control, cells deficient for N-acetylglucosaminyl transferase 1 were used to produce ‘high mannose’ (HM) variants (38, 39). An IgG1 antibody variant (referred to as IgG1 PG-LALA) with a combination of mutations that eliminate Fc-mediated effector functions, including complement was included as a “complement-dead” control (51).
The capacity of these glycovariants to activate complement was determined in C3b deposition ELISAs and CDC assays at high antigen densities (biotinylated HSA with 240 μM biotin and RBCs biotinylated at 2.5 mM respectively). At the level of C3b deposition, differences between the IgG4 glycovariants were observed, with the HG variant steadily outperforming the NG and LG variants (Figure 3A). The differences across glycoengineered IgG1 antibodies were less pronounced, but nevertheless consistent with previous results (23), as complement activation reached saturation at even low antibody concentrations. Both IgG1 and IgG4 HM variants induced the lowest levels of C3b deposition of all variants within each subclass. IgG1 PG-LALA was not able to mediate any detectable C3b deposition when tested in similar conditions as IgG4. No deposition was found with heat-inactivated serum and the binding of all glycovariants was found to be identical (Supplementary Figure 3A, C). In contrast, the differences in IgG4 galactosylation did not result in similarly clear differences at the level of cell lysis (Figure 3B). Furthermore, IgG1 PG-LALA, which is incapable of inducing complement activation, showed similar levels of cell lysis as the IgG4 variants. For both the IgG4 antibodies as well as the IgG1 PG-LALA variant, this effect appears to be predominantly complement-mediated, as heat-inactivated serum does not cause any cell lysis (Figure 3C and Supplementary Figure 3C). Red blood cells are sensitive to lysis as the formation of a single complement membrane attack complex can already cause cell lysis, especially at very high antibody concentrations and even when the antibody is considered ‘complement-dead’. Taken together, no enhanced complement activation for hypogalactosylated IgG4 was observed, on the contrary, high levels of Fc-galactosylation resulted in increased complement activation by IgG4, which is in line with observations using differently galactosylated IgG1 (20, 21, 23, 52).
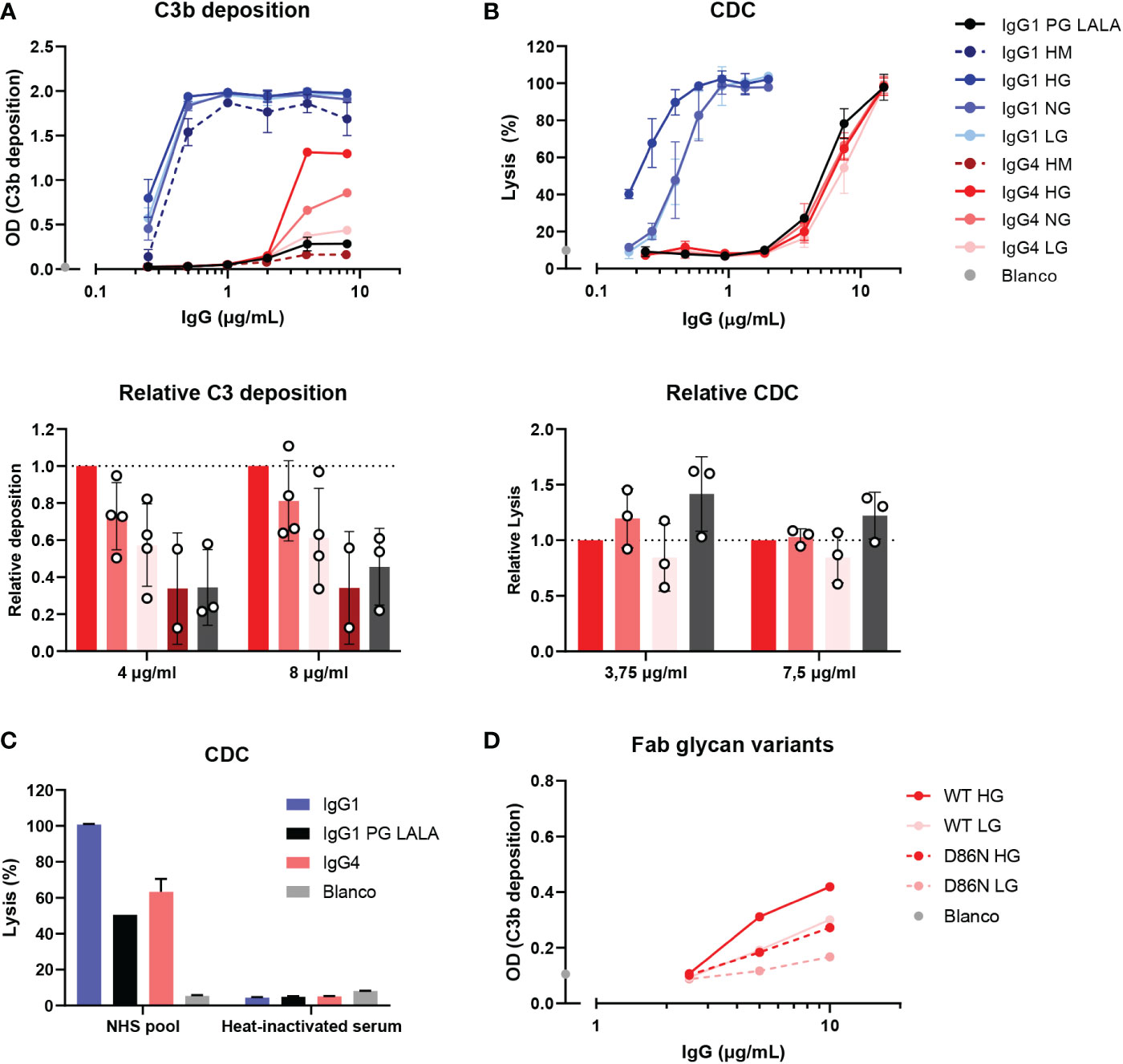
Figure 3 IgG4 glycovariants can induce C3b deposition to different extent. (A) C3b deposition induced by anti-biotin IgG1 and IgG4 glycovariants (high mannose (HM), high galactose (HG), normal galactose (NG) and low galactose (LG)), and IgG1 PG-LALA in the presence of biotinylated human serum albumin (240 μM biotin) and 2.5% human serum. Relative C3b deposition of the IgG4 glycovariants at concentrations 4 µg/mL and 8 µg/mL was normalized to IgG4 HG, which was set to 1 (dashed line), is shown as bar graphs. (B) Percentage of complement-mediated lysis of biotinylated human red blood cells (2.5 mM biotin) by the anti-biotin IgG1 and IgG4 glycovariants. The percentage of lysis was calculated based on the 100% lysis control (red blood cells with saponin). The percentage of complement-mediated lysis of the IgG4 glycovariants at concentrations 3.75 µg/mL and 7 µg/mL normalized to IgG4 HG is shown as bar graphs. Kruskal-Wallis test with Dunn’s multiple comparisons test of the IgG4 glycovariants was performed. (C) To test unspecific complement-dependent cytotoxicity (CDC), anti-biotin IgG1, IgG4 and IgG1 PG-LALA (eliminated Fc-mediated effector functions) were tested in the presence of biotinylated RBCs and 10% normal or heat-inactivated human serum. (D) C3b deposition induced by anti-trinitrophenyl (TNP) IgG4 glycovariants variants (HG and LG), which were produced as wild type IgG4, and a variant with D86N substitution in the light chain to introduce an additional glycosylation site in the Fab region. All experiments were performed at least three times, shown are representative figures for the C3b deposition and the mean ± standard deviation for the lysis experiments.
We next investigate the possible role of the Fab glycans on complement activation as they are relatively abundant on IgG4 (25). Therefore, WT IgG4 antibodies were produced against TNP (containing only an Fc glycan), and variants with a mutation in the variable region (D86N) of the light chain that introduces an additional glycosylation site within the Fab domain. These antibodies were generated as ‘low’ (10%) galactose and ‘high’ (70%) galactose variants and had Fc glycan composition levels like the anti-biotin variant as described above (Supplementary Figure 1C, D). Again, both HG variants induced more C3b deposition than the LG variants (Figure 3D), suggesting this is Fc-glycan mediated. The introduction of the additional glycan in the Fab interfered with the ability of the antibody to activate complement, as both antibodies with the additional Fab glycan performed worse than the WT variants. Antigen binding of all variants appears to be similar, indicating that this is not the cause of the decreased ability to activate complement. (Supplementary Figure 3A).
Next, we investigated which pathway of the complement system was activated by these IgG4 glycovariants. C3b deposition and RBC lysis were performed in the presence of inhibitors of C1q, MBL, and FD, to determine the contribution of the classical pathway, lectin pathway and alternative pathway, respectively. Despite the suggestion that IgG4 might activate complement via MBL (33), we found that C3b deposition by all IgG1 and IgG4 glycovariants was fully inhibited when blocking C1q, and not, or only slightly, in case of blocking MBL or FD (Figure 4A, C). This indicated that the main mode of activation of IgG4 is via the classical pathway and moreover, that this activation is irrespective of glycosylation as all variants rely on C1q for activation. Moreover, we found that C1q could also bind to IgG4, although to a lesser extent than to IgG1 (Figure 4E). Inhibition of FD decreased C3b deposition slightly compared to the condition without any inhibitor, suggesting that the alternative pathway may amplify activation via the classical pathway. As the alternative pathway requires higher serum concentrations to be studied, we also performed the experiment at 10% serum and allowed deposition at 37°C and we found similar results (Supplementary Figure 3D). No clear inhibition was found when MBL was blocked. The blocking antibody was able to completely block mannan-induced C3b deposition at equal serum concentrations, showing that the antibody is effective (Supplementary Figure 4A). Furthermore, when the assay was repeated with serum of an MBL deficient donor, IgG4 was still able to activate complement (Supplementary Figures 4A, B, D) The glycovariants were not affected differently by any of the inhibitions. Similar results at the level of cell lysis were observed: C1q inhibition significantly reduced IgG4-mediated lysis (Figures 4B, D), whereas inhibition of either MBL or FD had no impact on the induced lysis by IgG4. Furthermore, IgG4 could still induce cell lysis with serum from an MBL deficient donor (Supplementary Figure 4C).
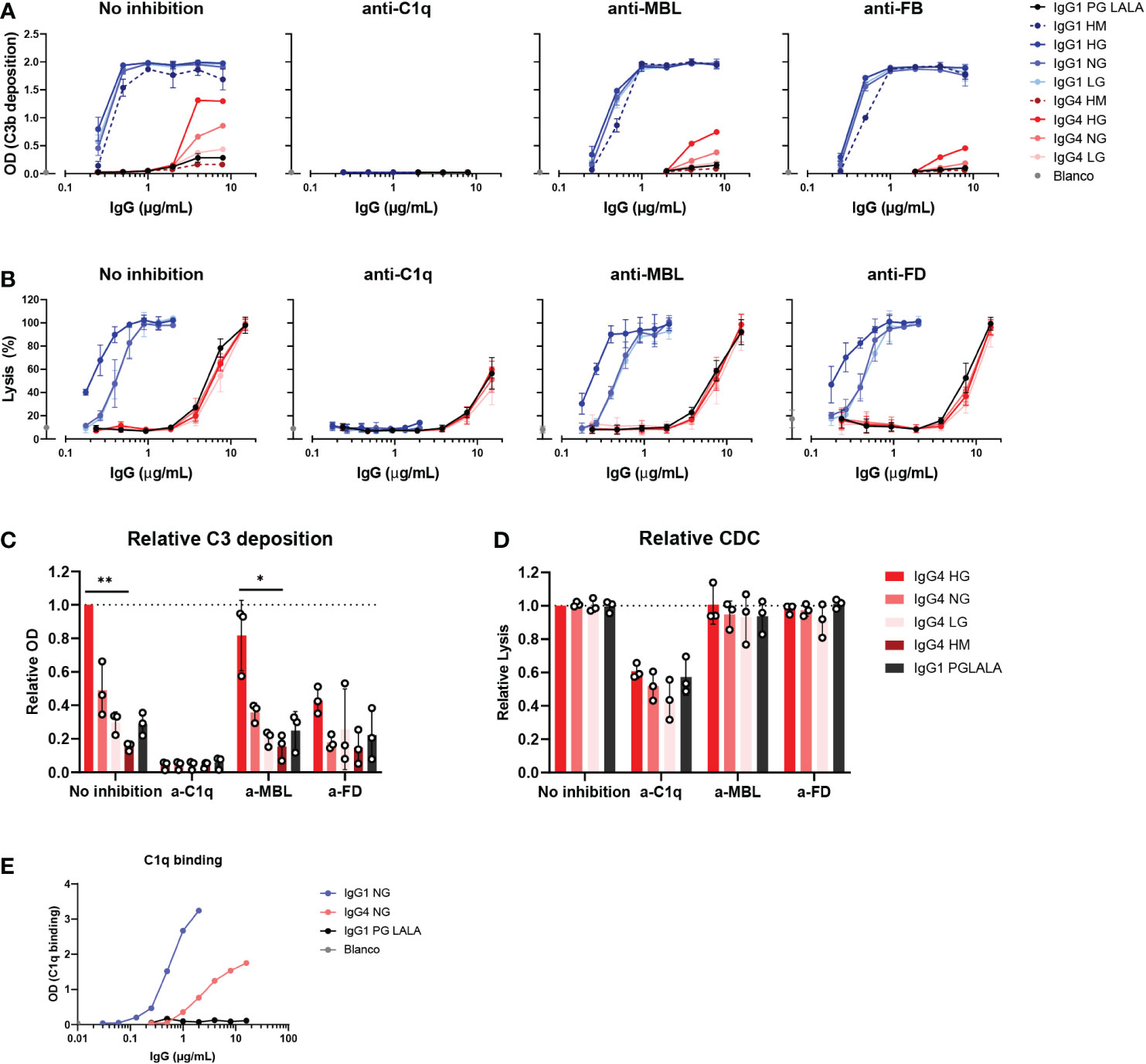
Figure 4 IgG4 activates complement via the classical pathway. (A) C3b deposition by anti-biotin IgG1, IgG4 and IgG1 PG-LALA (eliminated Fc-mediated effector functions) in presence or absence of either C1q, mannose-binding lectin (MBL), or complement factor B (FB) inhibitors, biotinylated human serum albumin (HSA; 240 μM biotin) and 2.5% human serum. Antibodies with different glycan profiles were tested: high mannose (HM), high galactose (HG), normal galactose (NH) and low galactose (LG). The molar ratios of target to inhibitor are 1:4.5 for C1q, 1:8.5 for MBL, and 1:3.5 for FB. (B) The percentage of complement-mediated lysis of biotinylated human red blood cells (2.5 mM biotin) by IgG1, IgG4 and IgG1 PG-LALA in presence or absence of C1q, MBL, or factor D (FD) inhibitors and 10% human serum. The molar ratios for target to inhibitor were 1:3 for C1q, 1:3 for MBL, and 1:2 for FD. The percentage of lysis was calculated based on the 100% lysis control (red blood cells with saponin). (C) The relative C3b deposition and (D) percentage of complement-mediated lysis of biotinylated human red blood cells induced by IgG4 glycovariants are shown as bar graphs with an antibody concentration of either (C) 8 µg/mL, or (D) 15 µg/mL. (E) To test binding of purified C1q (600 μg/mL) to the mAbs, IgG1, IgG4 and IgG1 PG-LALA were tested in the presence of biotinylated human serum albumin (HSA; 240 μM biotin). Kruskal-Wallis test with Dunn’s multiple comparisons test was performed for statistical comparison of the IgG4 glycovariants per inhibition condition: *p < 0.05; **p < 0.01. All antibodies were tested in a two-fold serial dilution starting from 8 µg/mL for C3b deposition and 15 µg/mL for CDC in at least three independent experiments.
Taken together, no role for the lectin pathway in IgG4 complement activation was found, which instead appears to mainly employ the classical pathway that was slightly enhanced by galactosylation on the level of C3b deposition.
In our previous assays, only bivalent-monospecific antibodies were used, but IgG4 antibodies can recombine half-molecules with different antigen specificities in vivo. Through this Fab arm exchange process, monovalent-bispecific IgG4 antibodies with decreased avidity for their targets are generated (15). We therefore exchanged IgG4 monoclonal anti-biotin and anti-TNP half-molecules with an irrelevant clone (natalizumab with no target in our assays) to create a bispecific antibody that would bind to biotin and TNP respectively with only one arm. In addition, a K409R mutation in the IgG1-Fc was introduced, which allowed for the IgG1 anti-biotin clone to be exchanged with an adalimumab clone (also no target in our assays) with the same mutation. For the fab arm exchange process mild reduction conditions are necessary. These antibodies were then tested for C3b deposition via ELISA, which was found to be reduced for both anti-biotin and anti-TNP bispecific IgG4 clones, but also for the bispecific anti-biotin IgG1 clone in comparison to their monospecific counterparts. The binding of the monospecific and bispecific IgG antibodies to either biotin (Figure 5A) or TNP (Figure 5B) was comparable for parental (mock condition) and Fab arm exchange clones. To conclude, this shows that Fab arm exchange greatly reduces the capacity of IgG4 to induce complement activation.
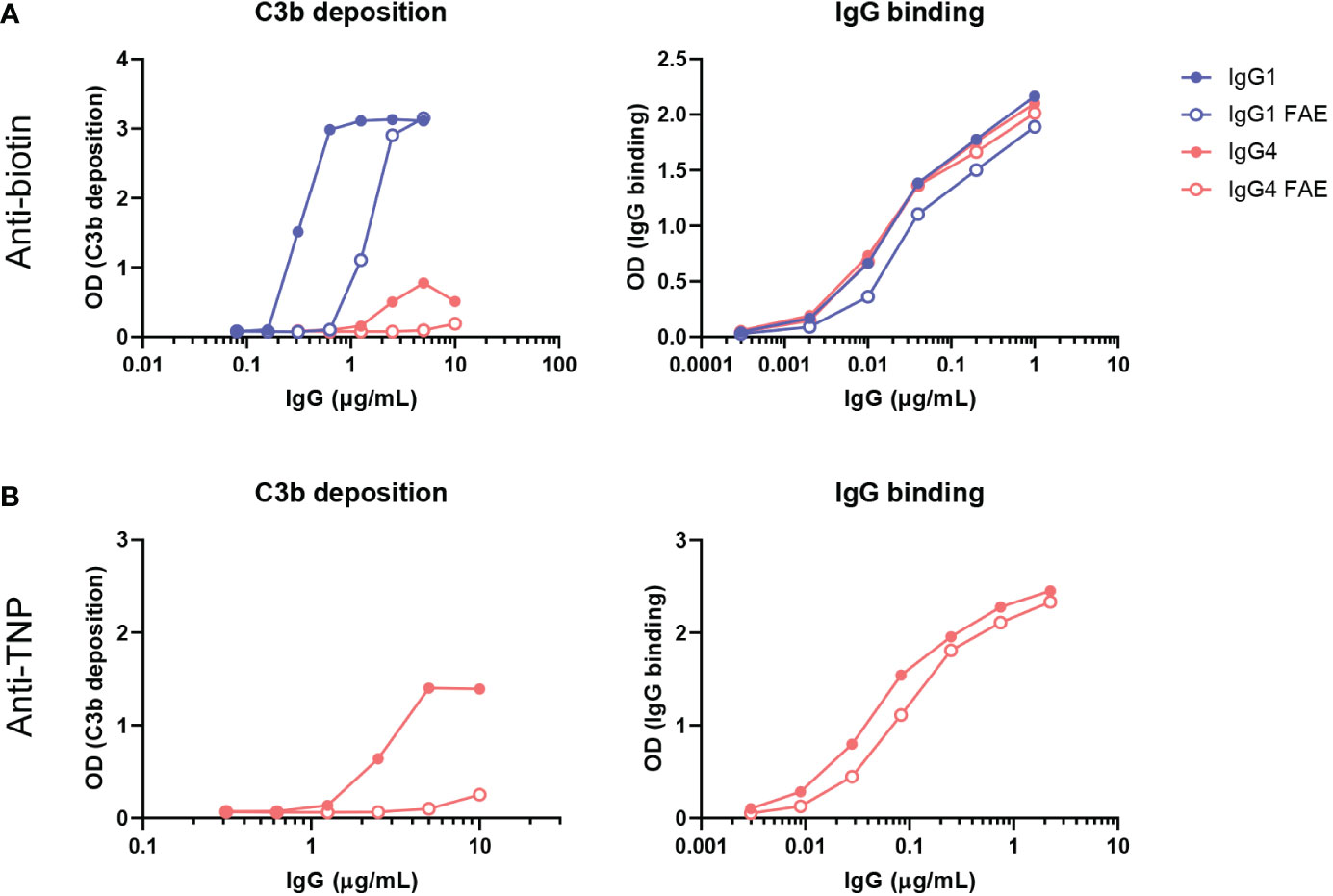
Figure 5 Fab arm exchange reduces antibody-mediated C3b deposition. Bispecific IgG1 and IgG4 variants were generated by half molecule exchanging anti-biotin and anti-trinitrophenyl (TNP) clones (both normal galactosylation) with IgG1 K409R adalimumab and IgG4 natalizumab respectively under mild reduction conditions. C3b deposition by (A) anti-biotin and (B) anti-TNP monospecific (full dots) and bispecific (empty dots) IgG1 and IgG4 in the presence of either TNPlated (1 mM 2,4,6-trinitrobenzenesulfonic acid) or biotinylated (240 μM biotin) human serum albumin and 2.5% human serum was tested. Binding to either biotin or TNP was measured for all variants (right panel). All experiments were performed at least three times, shown are representative figures.
IgG4 is generally considered a poor activator of complement, but at the same time has been linked to complement-induced fibrosis in IgG4-RD and IgG4 autoimmunity (10–12). This paradox prompted us to extensively investigate complement activation by IgG4, by searching for conditions that allowed for IgG4-induced complement activation and subsequently study how different features of IgG4 influenced this activation. These included glycosylation and Fab arm exchange using well-defined, glycoengineered, recombinant monoclonal antibodies against both protein and hapten antigens, which allowed systematic exploration of a wide range of experimental conditions.
Agalactosylated IgG has been shown to correlate with disease severity in several autoimmune diseases where complement contributes to pathology, including rheumatoid arthritis and systemic lupus erythematosus (53, 54). Agalactosylated IgG4 specifically has been implicated in IgG4-RD (31, 32) and pMN (30, 33). From there it was hypothesized that these agalactosylated IgG4 autoantibodies may be bound by MBL and could therefore activate complement via the lectin pathway (10, 12). However, PLA2R1-associated pMN can also occur in individuals with MBL deficiency (55) and IgG4 anti-PLA2R1 antibodies often co-occur with other subclasses as well (56, 57). In accordance, we found that the lectin pathway did not contribute to complement activation by IgG4 in our system. Neither the agalactosylated, nor high mannose glycovariants employed markedly different routes of activation, but also inhibition of MBL did not affect overall complement activation. Moreover, inhibition of C1q abolished complement activation, indicating that IgG4 in general, but also all tested glycovariants activate complement predominantly, if not exclusively, via the classical pathway. Lastly, variable domain glycosylation, known to be elevated in IgG4 (25), diminished rather than enhanced complement activation of IgG4. These findings are in contrast with recent findings showing that IgG4 anti-PLA2R1 antibodies from pMN patients could be bound by MBL and the suggestion that the lectin route is mainly responsible for complement activation at affected tissues (33). However, the authors also showed that upon removal of the Fc-glycan with the enzyme PGNase F, MBL was still able to bind to IgG4, which suggests that the reported binding of MBL was not dependent on the glycans on IgG4. Another recent study reported that the activation of the alternative pathway by IgG4 autoantibodies is sufficient in pMN (58), whereas the contribution of the alternative pathway was only minor in our experiments.
It has been previously described that at high concentrations of both antibody and antigen, IgG4 could induce C3 and terminal pathway activation (4, 59). We confirm that IgG4 can activate complement at high antigen densities and high antibody concentrations and show for the first time that this activation is mainly dependent on the classical pathway. This contradicts other studies which showed that IgG4 is effectively complement-dead. It is likely that these effects are missed in other studies, since IgG1 and IgG3 are much more efficient in activating complement and therefore comparable conditions with high levels of antigens and antibodies were often not included in comparative studies of IgG subclasses (21, 60, 61). The importance of the antigenic context is further illustrated by the fact that we only found complement activation by monoclonal IgG4 antibodies using haptens (biotin and TNP), but not protein antigens (Betv1a and anti-ADL fabs). This feature is likely to be the result of the high effective antigen densities accommodated by high hapten density in combination with high levels of antibodies. Having such high hapten density may possibly reflect a polyclonal antibody response targeting multiple epitopes on a protein target more closely. Whether similar conditions are met in vivo, for example within antibody deposits in IgG4-RD or pMN, remains to be investigated.
Upon recruiting antibodies to a surface via antigen binding, IgG Fc tails can cluster together to form hexamers via weak Fc-Fc interactions, mediated in part by Fc glycans, to interact with the hexagonal C1 complex (18, 62, 63). C1q has a low affinity to singular IgG molecules and requires multiple IgG-Fc tails to be in proximity and form oligomers to bind (64, 65). Despite similarities in the Fc-tails among the different IgG subclasses, IgG4 interacts poorly with C1q due to a variation in position 331, which has been shown to directly reduce binding of C1q (66, 67). Regardless, IgG4 is not completely unable to bind C1q. A fusion of the IgM tail piece to IgG4, which causes it to multimerize, greatly improved the ability of IgG4 to induce lysis (68). Similarly, this was also shown for the IgG hexamer-enhancing mutation E430G (18, 61). This suggests that although binding of C1q to IgG4 is reduced as compared to the other isotypes, this can partly be overcome by improved hexamerization.
We also studied the effect of galactosylation on IgG4 complement activation, which has previously received little attention, as most of the research on Fc glycosylation has focused on IgG1. For IgG1 it has been shown that galactosylation enhances C1q binding and downstream CDC activity (20, 21, 23, 52). Compared to IgG1, the IgG4 glycovariants in our study only showed significant differences in overall activation at the level of C3b deposition, with the high galactose variants performing better than the low galactose or high mannose variants, but the mode of activation was still similar for all variants studied. Possibly, galactosylation of IgG4 may improve its ability to hexamerize as has been shown for IgG1 (20, 21, 23, 52) and can therefore potentiate complement activation by IgG4 via the classical pathway. A slight impairment of complement activation by IgG4 was found in the presence of Fab glycans. IgG Fab glycosylation impacts antibody stability, half-life, binding characteristics, and immune complex formation (69). It has been demonstrated that IgG4 Fab glycosylation is significantly increased compared to total IgG (24). Since IgG4 antibodies are poorer activators of effector functions than other IgG subclasses, elevated levels of Fab glycan might impact the binding of IgG4 to effector molecules, such as C1q, and potentially the formation of hexamers.
Another important hallmark of IgG4 is its ability to undergo Fab arm exchange and form bispecific antibodies (14–16), which has not been considered in any of the previous studies which used mainly recombinant monoclonal antibodies (4, 21, 59–61). The Fab arm exchange of IgG4 may have a physiological role as natural bi-specific molecules cannot cross-link antigen or elicit lymphoid responses, may dampen inflammatory responses (16, 70) and potentially reduce complement activation. Most of the normal human plasma IgG4 molecules are bispecific (71, 72). It has been shown that within the total serum of healthy inndividuals, the hybrid IgG4λ/κ antibodies fraction accounts for up to 33%. Most likely the remaining IgG4 with the same light chain on both Fab arms may also have undergone half-molecule exchange at some point (72). Bispecific IgG4 molecules are widespread in normal serum and possibly represents the typical configuration and even therapeutical IgG4 molecules can undergo Fab arm exchange with serum IgG4 (73). Therefore, studies relying on monovalent antibodies may not be reflective of the in vivo situation. Here we show that bispecific IgG4 induces less C3b deposition than monospecific IgG4, which is particularly relevant for interpreting other studies on IgG4 complement activation that use monospecific antibody preparations. Interestingly, it has been previously shown that a monovalent IgG1 antibody outperformed its bivalent monospecific counterpart in CDC experiments (18, 74). However, for another IgG1 antibody tested, both the monospecific and bispecific antibodies performed similarly (18), suggesting the effect may also be dependent on specific antigen-antibody interactions, like we have seen in our studies. This indicates that the Fab arm exchange of IgG4 molecules may contribute to the limited IgG4-mediated complement activation in vivo.
IgG4 is believed to have mainly neutralizing and anti-inflammatory functions due to limited complement fixation and crosslinking, therefore it has been exploited for the development of therapeutic antibodies that are intended to block their target, often including a stabilizing S228P mutation to minimize in vivo Fab arm exchange. Currently IgG4 antibodies account for approximately 13% of all therapeutic monoclonal antibodies approved or in development (75). Two examples are the humanized kappa IgG4 antibodies pembrolizumab and nivolumab, which are both programmed death receptor-1 (PD1)-blocking antibodies (76, 77). However, our results show that the ability of monospecific IgG4 to activate complement should be taken into consideration when developing new therapeutics binding membrane-bound targets that are administered at high concentrations. As a blocking antibody, a monovalent (bispecific) monoclonal antibody might be considered if unwanted complement activation might be an issue.
In summary, we demonstrate that although IgG4 is a weak trigger for complement activation, it cannot be considered completely ‘complement-dead’, since it does activate complement, but only at high antigen densities and high antibody concentrations, and only via the classical pathway. In addition, galactosylation has a subtle enhancing impact on the capacity of IgG4 antibodies to activate complement, whereas Fab arm exchange, a hallmark of IgG4, has the opposite effect. These findings imply that pathogenic role of IgG4 in diseases such as IgG4-RD or pMN via recruitment of the complement system is possible, but unlikely.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All authors contributed to the article and approved the submitted version. Designing research studies: NO, TD, TR, GV. Conducting experiments and data acquisition: NO, TD, MS, PO-H, JN, CK, MW. Analyzing data: NO, TD, MS, JC, TR. Writing the manuscript: NO, TD, TR.
This work was supported by the Dutch Arthritis Foundation grant 17-2-404. The work of TD was funded by Genmab. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
We thank Esther de Boer, Mieke Brouwer and Dr Richard Pouw (Sanquin Research and Landsteiner Laboratory, Department of Immunopathology, Academic Medical Center, Amsterdam, The Netherlands) for their technical guidance and complement insight. We also thank Prof. H. Clausen (Copenhagen Center for Glycomics, University of Copenhagen, Denmark) for providing HEK293-6E cells with a MGAT1 knock-out (KO) to generate the high mannose IgG glycovariants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1087532/full#supplementary-material
1. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: From structure to effector functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
2. Damelang T, Rogerson SJ, Kent SJ, Chung AW. Role of IgG3 in infectious diseases. Trends Immunol (2019) 40:197–211. doi: 10.1016/j.it.2019.01.005
3. Lilienthal G-M, Rahmöller J, Petry J, Bartsch YC, Leliavski A, Ehlers M. Potential of murine IgG1 and human IgG4 to inhibit the classical complement and fcγ receptor activation pathways. Front Immunol (2018) 9:958. doi: 10.3389/fimmu.2018.00958
4. Tao MH, Canfield SM, Morrison SL. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH-terminal sequence of the CH2 domain. J Exp Med (1991) 173:1025–8. doi: 10.1084/jem.173.4.1025
5. Lighaam LC, Rispens T. The immunobiology of immunoglobulin G4. Semin Liver Dis (2016) 36:200–15. doi: 10.1055/s-0036-1584322
6. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of complement activation by IgG4 antibodies. Clin Exp Immunol (1986) 64:415–22.
7. Ishizaka T, Ishizaka K, Salmon S, Fudenberg H. Biologic activities of aggregated gamma-globulin. 8. aggregated immunoglobulins of different classes. J Immunol (1967) 99:82–91. doi: 10.4049/jimmunol.99.1.82
8. Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: An odd antibody. Clin Exp Allergy (2009) 39:469–77. doi: 10.1111/j.1365-2222.2009.03207.x
9. Davies AM, Rispens T, Ooijevaar-De Heer P, Gould HJ, Jefferis R, Aalberse RC, et al. Structural determinants of unique properties of human IgG4-fc. J Mol Biol (2014) 426:630–44. doi: 10.1016/j.jmb.2013.10.039
10. Reinhard L, Stahl RAK, Hoxha E. Is primary membranous nephropathy a complement mediated disease? Mol Immunol (2020) 128:195–204. doi: 10.1016/j.molimm.2020.10.017
11. Dainichi T, Chow Z, Kabashima K. IgG4, complement, and the mechanisms of blister formation in pemphigus and bullous pemphigoid. J Dermatol Sci (2017) 88:265–70. doi: 10.1016/j.jdermsci.2017.07.012
12. Kawa S. The immunobiology of immunoglobulin G4 and complement activation pathways in IgG4-related disease. Curr Top Microbiol Immunol (2017) 401:61–73. doi: 10.1007/82_2016_39
13. Beck LH, Bonegio RGB, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med (2009) 361:11–21. doi: 10.1056/NEJMoa0810457
14. Labrijn AF, Rispens T, Meesters J, Rose RJ, den Bleker TH, Loverix S, et al. Species-specific determinants in the IgG CH3 domain enable fab-arm exchange by affecting the noncovalent CH3–CH3 interaction strength. J Immunol (2011) 187:3238–46. doi: 10.4049/jimmunol.1003336
15. Rispens T, Davies AM, Ooijevaar-de Heer P, Absalah S, Bende O, Sutton BJ, et al. Dynamics of inter-heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic fab arm exchange. J Biol Chem (2014) 289:6098–109. doi: 10.1074/jbc.M113.541813
16. van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic fab arm exchange. Science (2007) 317:1554–7. doi: 10.1126/science.1144603
17. Aalberse RC, Schuurman J, Van Ree R. The apparent monovalency of human IgG4 is due to bispecificity. Int Arch Allergy Immunol (1999) 118:187–9. doi: 10.1159/000024062
18. Diebolder CA, Koning RI, Koster AJ, Rosati S, Wilson IA, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Sci (80- ) (2014) 343:1260–3. doi: 10.1126/science.1248943
19. Dekkers G, Plomp R, Koeleman CAM, Visser R, Von Horsten HH, Sandig V, et al. Multi-level glyco-engineering techniques to generate IgG with defined fc-glycans. Sci Rep (2016) 6:1–12. doi: 10.1038/srep36964
20. Dekkers G, Treffers L, Plomp R, Bentlage AEH, de Boer M, Koeleman CAM, et al. Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of fc-receptor- and complement-mediated-effector activities. Front Immunol (2017) 8:877. doi: 10.3389/fimmu.2017.00877
21. Peschke B, Keller CW, Weber P, Quast I, Lünemann JD. Fc-galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front Immunol (2017) 8:646. doi: 10.3389/fimmu.2017.00646
22. Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, et al. Sialylation of IgG fc domain impairs complement-dependent cytotoxicity. J Clin Invest (2015) 125:4160–70. doi: 10.1172/JCI82695
23. van Osch TLJ, Nouta J, Derksen NIL, van Mierlo G, van der Schoot CE, Wuhrer M, et al. Fc galactosylation promotes hexamerization of human IgG1, leading to enhanced classical complement activation. J Immunol (2021) 207:1545–54. doi: 10.4049/jimmunol.2100399
24. Van De Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, Van Schie KA, Kruithof S, Berkowska MA, et al. Adaptive antibody diversification through n-linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci U.S.A. (2018) 115:1901–6. doi: 10.1073/pnas.1711720115
25. Koers J, Derksen NIL, Ooijevaar-de Heer P, Nota B, van de Bovenkamp FS, Vidarsson G, et al. Biased n -glycosylation site distribution and acquisition across the antibody V region during b cell maturation. J Immunol (2019) 202:2220–8. doi: 10.4049/jimmunol.1801622
26. van de Bovenkamp FS, Derksen NIL, van Breemen MJ, de Taeye SW, Ooijevaar-de Heer P, Sanders RW, et al. Variable domain n-linked glycans acquired during antigen-specific immune responses can contribute to immunoglobulin G antibody stability. Front Immunol (2018) 9:740. doi: 10.3389/fimmu.2018.00740
27. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 – the “Swiss army knife” of innate immunity and host defense. Immunol Rev (2016) 274:33–58. doi: 10.1111/imr.12500
28. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement–MBL and beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
29. Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med (1995) 1:237–43. doi: 10.1038/nm0395-237
30. Chinello C, de Haan N, Capitoli G, Trezzi B, Radice A, Pagani L, et al. Definition of IgG subclass-specific glycopatterns in idiopathic membranous nephropathy: Aberrant IgG glycoforms in blood. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23094664
31. Culver EL, van de Bovenkamp FS, Derksen NIL, Koers J, Cargill T, Barnes E, et al. Unique patterns of glycosylation in immunoglobulin subclass G4-related disease and primary sclerosing cholangitis. J Gastroenterol Hepatol (2019) 34:1878–86. doi: 10.1111/jgh.14512
32. Konno N, Sugimoto M, Takagi T, Furuya M, Asano T, Sato S, et al. Changes in n-glycans of IgG4 and its relationship with the existence of hypocomplementemia and individual organ involvement in patients with IgG4-related disease. PloS One (2018) 13:1–21. doi: 10.1371/journal.pone.0196163
33. Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1-associated membranous nephropathy. J Clin Invest (2021) 131. doi: 10.1172/JCI140453
34. Kohen F, Bagci H, Barnard G, Bayer EA, Gayer B, Schindler DG, et al. Preparation and properties of anti-biotin antibodies. Methods Enzymol (1997) 279:451–63. doi: 10.1016/s0076-6879(97)79049-9
35. Bagçi H, Kohen F, Kusçuoglu U, Bayer EA, Wilchek M. Monoclonal anti-biotin antibodies simulate avidin m the recognition of biotin. FEBS Lett (1993) 322:47–50. doi: 10.1016/0014-5793(93)81108-C
36. Kruijsen D, Einarsdottir HK, Schijf MA, Coenjaerts FE, van der Schoot EC, Vidarsson G, et al. Intranasal administration of antibody-bound respiratory syncytial virus particles efficiently primes virus-specific immune responses in mice. J Virol (2013) 87:7550–7. doi: 10.1128/jvi.00493-13
37. van de Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, Rispens T. The enzymatic removal of immunoglobulin variable domain glycans by different glycosidases. J Immunol Methods (2019) 467:58–62. doi: 10.1016/j.jim.2019.02.005
38. Narimatsu Y, Joshi HJ, Nason R, Van Coillie J, Karlsson R, Sun L, et al. An atlas of human glycosylation pathways enables display of the human glycome by gene engineered cells. Mol Cell (2019) 75:394–407.e5. doi: 10.1016/j.molcel.2019.05.017
39. Narimatsu Y, Joshi HJ, Yang Z, Gomes C, Chen YH, Lorenzetti FC, et al. A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology (2018) 28:295–305. doi: 10.1093/glycob/cwx101
40. Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Sci (80- ) (2021) 371. doi: 10.1126/science.abc8378
41. Pongracz T, Nouta J, Wang W, van Meijgaarden KE, Linty F, Vidarsson G, et al. Immunoglobulin G1 fc glycosylation as an early hallmark of severe COVID-19. eBioMedicine (2022) 78:103957. doi: 10.1016/j.ebiom.2022.103957
42. Oskam N, Ooijevaar-de Heer P, Derksen NIL, Kruithof S, de Taeye SW, Vidarsson G, et al. At Critically low antigen densities, IgM hexamers outcompete both IgM pentamers and IgG1 for human complement deposition and complement-dependent cytotoxicity. J Immunol (2022) 209(1):16–25. doi: 10.4049/jimmunol.2101196
43. McGrath FDG, Brouwer MC, Arlaud GJ, Daha MR, Hack CE, Roos A. Evidence that complement protein C1q interacts with c-reactive protein through its globular head region. J Immunol (2006) 176:2950–7. doi: 10.4049/jimmunol.176.5.2950
44. Rijkers M, Schmidt D, Lu N, Kramer CSM, Heidt S, Mulder A, et al. Anti-HLA antibodies with complementary and synergistic interaction geometries promote classical complement activation on platelets. Haematologica (2019) 104:403–16. doi: 10.3324/haematol.2018.201665
45. Bultink IEM, Hamann D, Seelen MA, Hart MH, Dijkmans BAC, Daha MR, et al. Deficiency of functional mannose-binding lectin is not associated with infections in patients with systemic lupus erythematosus. Arthritis Res Ther (2006) 8:1–10. doi: 10.1186/ar2095
46. Kilpatrick DC. Isolation of human mannan binding lectin, serum amyloid p component and related factors from Cohn fraction III. Transfus Med (1997) 7:289–94. doi: 10.1046/j.1365-3148.1997.d01-40.x
47. Loyet KM, Hass PE, Sandoval WN, Morando A, Liu P, Shatz W, et al. In vivo stability profiles of anti-factor d molecules support long-acting delivery approaches. Mol Pharm (2019) 16:86–95. doi: 10.1021/acs.molpharmaceut.8b00871
48. Katschke KJ, Wu P, Ganesan R, Kelley RF, Mathieu MA, Hass PE, et al. Inhibiting alternative pathway complement activation by targeting the factor d exosite. J Biol Chem (2012) 287:12886–92. doi: 10.1074/jbc.M112.345082
49. De Boer ECW, Thielen AJF, Langereis JD, Kamp A, Brouwer MC, Oskam N, et al. The contribution of the alternative pathway in complement activation on cell surfaces depends on the strength of classical pathway initiation. Clin Transl Immunol (in press).
50. Hack CE, Paardekooper J, Smeenk RJ, Abbink J, Eerenberg AJ, Nuijens JH. Disruption of the internal thioester bond in the third component of complement (C3) results in the exposure of neodeterminants also present on activation products of C3. Anal monoclonal antibodies J Immunol (1988) 141:1602–9. doi: 10.4049/jimmunol.141.5.1602
51. Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel (2016) 29:457–66. doi: 10.1093/protein/gzw040
52. Wei B, Gao X, Cadang L, Izadi S, Liu P, Zhang HM, et al. Fc galactosylation follows consecutive reaction kinetics and enhances immunoglobulin G hexamerization for complement activation. MAbs (2021) 13:1–10. doi: 10.1080/19420862.2021.1893427
53. Dekkers G, Rispens T, Vidarsson G. Novel concepts of altered immunoglobulin G galactosylation in autoimmune diseases. Front Immunol (2018) 9:553. doi: 10.3389/fimmu.2018.00553
54. Cobb BA. The history of IgG glycosylation and where we are now. Glycobiology (2021) 30:202–13. doi: 10.1093/GLYCOB/CWZ065
55. Bally S, Debiec H, Ponard D, Dijoud F, Rendu J, Fauré J, et al. Phospholipase A2 receptor-related membranous nephropathy and mannan-binding lectin deficiency. J Am Soc Nephrol (2016) 27:3539–44. doi: 10.1681/ASN.2015101155
56. Lateb M, Ouahmi H, Payré C, Brglez V, Zorzi K, Dolla G, et al. Anti-PLA2R1 antibodies containing sera induce in vitro cytotoxicity mediated by complement activation. J Immunol Res (2019) 2019. doi: 10.1155/2019/1324804
57. Segawa Y, Hisano S, Matsushita M, Fujita T, Hirose S, Takeshita M, et al. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr Nephrol (2010) 25:1091–9. doi: 10.1007/s00467-009-1439-8
58. Manral P, Caza TN, Storey AJ, Beck LH, Borza D. The alternative pathway is necessary and sufficient for complement activation by anti-THSD7A autoantibodies, which are predominantly IgG4 in membranous nephropathy. Front Immunol (2022) 13:952235. doi: 10.3389/fimmu.2022.952235
59. Garred P, Michaelsen TE, Aase A. The IgG subclass pattern of complement activation depends on epitope density and antibody and complement concentration. Scand J Immunol (1989) 30:379–82. doi: 10.1111/j.1365-3083.1989.tb01225.x
60. Brüggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med (1987) 166:1351–61. doi: 10.1084/jem.166.5.1351
61. Zwarthoff SA, Widmer K, Kuipers A, Strasser J, Ruyken M, Aerts PC, et al. C1q binding to surface-bound IgG is stabilized by C1r 2 s 2 proteases. Proc Natl Acad Sci (2021) 118:e2102787118. doi: 10.1073/pnas.2102787118
62. Wang G, de Jong RN, van den Bremer ETJ, Beurskens FJ, Labrijn AF, Ugurlar D, et al. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol Cell (2016) 63:135–45. doi: 10.1016/j.molcel.2016.05.016
63. Ugurlar D, Howes SC, De Kreuk B, Koning RI, De Jong RN, Beurskens FJ, et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. ACS Nano (2018) 797:794–7. doi: 10.1126/science.aao4988
64. Strasser J, De Jong RN, Beurskens FJ, Schuurman J, Parren PWHI, Hinterdorfer P, et al. Weak fragment crystallizable (Fc) domain interactions drive the dynamic assembly of IgG oligomers upon antigen recognition. ACS Nano (2020) 14:2739–50. doi: 10.1021/acsnano.9b08347
65. Strasser J, De Jong RN, Beurskens FJ, Wang G, Heck AJR, Schuurman J, et al. Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano Lett (2019) 19:4787–96. doi: 10.1021/acs.nanolett.9b02220
66. Tao BM, Smith RIF, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specitic differences in complement activation. J Exp Med (1993) 178:661–7. doi: 10.1084/jem.178.2.661
67. Brekke OH, Michaelsen TE, Aase A, Sandin RH, Sandlie I. Human IgG isotype-specific amino acid residues affecting complement-mediated cell lysis and phagocytosis. Eur J Immunol (1994) 24:2542–7. doi: 10.1002/eji.1830241042
68. Smith RI, Coloma MJ, Morrison SL. Addition of a mu-tailpiece to IgG results in polymeric antibodies with enhanced effector functions including complement-mediated cytolysis by IgG4. J Immunol (1995) 154:2226–36. doi: 10.4049/jimmunol.154.5.2226
69. van de Bovenkamp FS, Hafkenscheid L, Rispens T, Rombouts Y. The emerging importance of IgG fab glycosylation in immunity. J Immunol (2016) 196:1435–41. doi: 10.4049/jimmunol.1502136
70. Hansen K, Ruttekolk IR, Glauner H, Becker F, Brock R, Hannus S. The in vitro biological activity of the HLA-DR-binding clinical IgG4 antibody 1D09C3 is a consequence of the disruption of cell aggregates and can be abrogated by fab arm exchange. Mol Immunol (2009) 46:3269–77. doi: 10.1016/j.molimm.2009.07.031
71. Schuurman J, Van Ree R, Perdok GJ, Van Doorn HR, Tan KY, Aalberse RC. Normal human immunoglobulin G4 is bispecific: It has two different antigen-combining sites. Immunology (1999) 97:693–8. doi: 10.1046/j.1365-2567.1999.00845.x
72. Young E, Lock E, Ward DG, Cook A, Harding S, Wallis GLF. Estimation of polyclonal IgG4 hybrids in normal human serum. Immunology (2014) 142:406–13. doi: 10.1111/imm.12265
73. Shapiro RI, Plavina T, Schlain BR, Pepinsky RB, Garber EA, Jarpe M, et al. Development and validation of immunoassays to quantify the half-antibody exchange of an IgG4 antibody, natalizumab (Tysabri ®) with endogenous IgG4. J Pharm BioMed Anal (2011) 55:168–75. doi: 10.1016/j.jpba.2011.01.006
74. Watts HF, Anderson VA, Cole VM, Stevenson GT. Activation of complement pathways by univalent antibody derivatives with intact fc zones. Mol Immunol (1985) 22:803–10. doi: 10.1016/0161-5890(85)90146-4
75. Tang Y, Cain P, Anguiano V, Shih JJ, Chai Q, Feng Y. Impact of IgG subclass on molecular properties of monoclonal antibodies. MAbs (2021) 13:1–12. doi: 10.1080/19420862.2021.1993768
76. Kwok G, Yau TCC, Chiu JW, Tse E, Kwong Y. Pembrolizumab ( keytruda ). Hum Vaccin Immunother (2016) 12:2777–89. doi: 10.1080/21645515.2016.1199310
Keywords: antibodies, glycoengineering, fab arm exchange, IgG4-related disease, primary membranous nephropathy, complement activation
Citation: Oskam N, Damelang T, Streutker M, Ooijevaar-de Heer P, Nouta J, Koeleman C, Van Coillie J, Wuhrer M, Vidarsson G and Rispens T (2023) Factors affecting IgG4-mediated complement activation. Front. Immunol. 14:1087532. doi: 10.3389/fimmu.2023.1087532
Received: 02 November 2022; Accepted: 16 January 2023;
Published: 26 January 2023.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Dorin-Bogdan Borza, Meharry Medical College, United StatesCopyright © 2023 Oskam, Damelang, Streutker, Ooijevaar-de Heer, Nouta, Koeleman, Van Coillie, Wuhrer, Vidarsson and Rispens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nienke Oskam, bi5vc2thbUBzYW5xdWluLm5s
†These authors contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.